

Abstract
Purpose
Lung cancer is one of the leading causes of deaths in the United States, but currently available therapies for lung cancer are associated with reduced efficacy and adverse side effects. Small interfering RNA (siRNA) can knock down the expression of specific genes and result in therapeutic efficacy in lung cancer. Recently, mTOR siRNA has been shown to induce apoptosis in NSCLC cell lines but its use is limited due to poor stability in biological conditions.
Methods
In this study, we modified an aminoglyocisde-derived cationic poly (amino-ether) by introducing a thiol group using Traut’s reagent to generate a bio-reducible modified–poly (amino-ether) (mPAE). The mPAE polymer was used to encapsulate mTOR siRNA by nanoprecipitation method, resulting in the formation of stable and bio-reducible nanoparticles (NPs) which possessed an average diameter of 114 nm and a surface charge of approximately +27 mV.
Results
The mTOR siRNA showed increased release from the mTS-mPAE NPs in the presence of 10 mM glutathione (GSH). The polymeric mTS-mPAE-NPs were also capable of efficient gene knockdown (60 and 64%) in A549 and H460 lung cancer cells, respectively without significant cytotoxicity at 30 μg/ml concentrations. The NPs also showed time-dependent cellular uptake for up to 24 h as determined using flow cytometry. Delivery of the siRNA using these NPs also resulted in significant inhibition of A549 and H460 cell proliferation in vitro, respectively.
Conclusions
The results demonstrate that the mPAE polymer based NPs show strong potential for siRNA delivery to lung cancer cells. It is anticipated that future modification can help improve the efficacy of nucleic acid delivery, leading to higher inhibition of lung cancer growth in vitro and in vivo.
Keywords: bioreducible, drug delivery, lung cancer, mTOR, nanomedicine, nanoparticles, polymer, siRNA
INTRODUCTION
Lung cancer is the leading cause of new cancer cases and cancer-related deaths in the United States (US) and worldwide [1]. It is mainly divided into two types, namely, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [2]. In particular, NSCLC is the most common type of lung cancer and a major cause of death throughout the world [3]. The American Cancer Society estimates around 222,500 new cases and 155,870 deaths due to lung cancer in 2017 [4]. Various treatment options for the treatment of NSCLC, including chemotherapy, radiation, and surgery [5] are currently available, and chemotherapy is still the mainstay of treatment for NSCLC [6]. Conventional chemotherapeutic agents are usually administered via the intravenous (i.v.) route. However, the major drawbacks with i.v. administration of therapeutic agents is their widespread distribution throughout the body via the bloodstream, affecting both malignant and rapidly dividing normal cells (e.g. cells in the bone marrow) [7]. In addition, most chemotherapeutic agents used to treat lung cancer cells can kill non-dividing cells, and also lead to adverse side effects such as hair loss, fatigue, infection and other problems [8, 9].
Extensive efforts have been made to design new drugs that selectively target precise pathways driving the development of NSCLC, but many attractive cancer targets remain difficult to target specifically [10]. Recently, it was shown that abnormal activation of the Akt/mTOR pathway is commonly observed in the NSCLC cases [11]. The deregulation of the Akt/PI3K/mTOR pathway is known to have contributed to the development and maintenance of NSCLC. Various negative regulators act as tumor suppressors and have been found to be mutated in NSCLC suggesting a role of the mTOR pathway in lung cancer [12]. mTOR inhibitors have been used to treat NSCLC, however, they cause adverse effects due to an unwanted effect on normal cells [13, 14]. Thus, the newer type of drugs is needed to treat NSCLC. In that regard, mTOR siRNA has been used recently to successfully inhibit cell growth in NSCLC cells in addition to inducing apoptosis [15].
RNA interference (RNAi)-induced gene knockdown is an exciting, naturally occurring cellular pathway that targets mRNA transcripts for cleavage in a sequence-specific manner [16] This pathway can be exploited to target mTOR gene and alter cellular behavior with the delivery of short interfering RNA (siRNA) complementary to the mRNA transcript of the gene of interest [17]. Furthermore, mTOR siRNA has been used recently to successfully inhibit cell growth in NSCLC cells [15]. In particular, short interfering RNA (siRNA)-based therapies are a promising alternative approach because they can theoretically silence expression of any oncogene [18, 19]. However, at the cellular level, the therapeutic potential of siRNA cannot be realized without efficient delivery to its site of action in the cytoplasm [2, 20, 21].
Bioreducible polymers have been used for the delivery of nucleic acids for cancer therapy [22]. In particular, one of the main features of bioreducible polymers is the ease of encapsulation of the nucleic acids and show triggered release of therapeutic agent under presence of glutathione (GSH) in the cytoplasm. The formed nanoparticles enables to the protection of the nucleic acids [23, 24]. Among bioreducible polymers, the classes that form disulfide bonds upon oxidation to encapsulate the nucleic acid (siRNA, miRNA, DNA) and impart stability to the NPs, are attractive in delivery applications. These bioreducible polymers are based on alternative mechanisms of degradation [25]. For instance, bioreducible polymers containing thiol groups that easily form disulfide linkages upon oxidation can be degraded in response to the redox potential present inside the cells and release the nucleic acid inside the cytoplasm [26].
Recently, we generated a cationic polymer library by addition polymerization reaction between diglycidyl ethers and amines [27]. This strategy was further extended to generate a library of biodegradable aminoglycoside-derived polymers and lipopolymers for the delivery of plasmid DNA [28–30]. One such polymer that specifically showed very high transfection efficiency is the poly(amino ethers) (PAE) (Mw 3804), synthesized by reacting neomycin with diglycidyl ether at a molar ratio of 1:2 [31]. The group further explored the use of aminoglycoside-derived polymers by using them to functionalize gold nanorods (GNRs) [32] to generate PAE-functionalized GNR (PAE-GNR) nanoassemblies, which were generated by depositing polyelectrolyte multilayers on cetyltrimethyl ammonium bromide (CTAB)-based gold nanorods (CTAB-GNRs). The PAE-GNR nanoassemblies demonstrated significantly higher transgene expression efficacy and lower or comparable cytotoxicity compared to PEI-25 (branched 25 kDa polyethylene imine)-functionalized GNRs (PEI-GNRs) and also demonstrated gene silencing activity [33, 34].
In order to exploit the intracellular reducing environment, which would result in a faster release of the payload, we introduced a thiol group (−SH) to transform the PAE into a bioreducible polymer. The novelty of PAE polymer is that the development of cationic biodegradable aminoglycoside based polymer and introduction of thiol groups in PAE polymer yielding disulfide bond for the delivery of siRNA against lung cancer. This is the foremost investigation of development of disulfide bond forming stable PAE polymeric siRNA loaded nanocarriers for lung cancer. We have screened the developed bioreducible polymer for the delivery of mTOR siRNA against lung cancer. The advantages of bioreducible PAE polymeric nanoparticles include, biodegradability, efficient encapsulation of siRNA, cellular internalization via cationic charge, and cytoplasmic triggered release of loaded siRNA. The thiol group in the polymer can form a disulfide group under oxidative conditions and result in the formation of stable nanoparticles which can then be reduced inside cells, resulting in release of the siRNA payload. The thiol group can be introduced by using a simple and fast one-step reaction with iminothiolane hydrochloride (Traut’s reagent) while retaining the net positive charge due to the introduction of the −NH2 group. We hypothesize that a delivery system based on siRNA encapsulated in a bioreducible mPAE polymeric nanoparticles will be cleaved by cytoplasmic glutathione (GSH) followed by triggered release of siRNA inside the cytoplasm leading to effective gene silencing. We demonstrate the potential of this mPAE polymer for the delivery of mTOR siRNA in NSCLC cell lines (A549 and H460), resulting in gene silencing and cell death.
MATERIALS
The poly (amino ether) polymer was generated as described in previous reports [28]. SMARTpool: ON-TARGETplus mTOR siRNA (mTOR siRNA was obtained with the following target sequences: GGCCAUAGCUAGCCUCAUA, CAAAGGACUUCGCCCAUAA, GCAGAAUUGUCAAGGGAUA, and CCAAAGCACUACACUACAA), DHARMAfect 1 Transfection Reagent and ON-TARGETplus non-targeting pool (GCACUUACUUCGUGUCCGA, AGUCAUAACCUUACCAGAU, GGUAGUAUGAUGGGUGUUU, UCAUCUUGUUCAUUGGAAU) were obtained from Dharmacon(GE Dharmacon USA). PEI was obtained from Spectrum Chemicals (New Brunswick, NJ). AccuBlue high sensitivity dsDNA quantification kit was obtained from Biotium Inc. (Fremont, CA), mTOR (7C10) Rabbit mAb and β-Actin (13E5) Rabbit mAb were obtained from Cell signaling Technology. Goat anti-Rabbit IgG (H&L) Secondary Antibody, DYLight 800 4X was obtained from Thermofisher Scientific USA. Immobilon-FL PVDF, 0.45 μm membranes were obtained from EMD Millipore USA. All Other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
METHODS
Synthesis of the Bioreducible Modified-PAE (mPAE)
The PAE polymer was synthesized by ring-opening polymerization method described previously [35, 36]. Briefly, sulfate-free aminoglycoside (neomycin) was reacted with glycerol diglycidylether (GDE) in 1:2.2 M ratio in a solvent mixture of water and N,N-dimethylformamide (DMF) (1.5:1) for 5 h at 60°C. The product obtained upon precipitation using acetone was further purified by dialysis using a 3500 molecular weight cutoff (MWCO) membrane in order to remove unreacted aminoglycoside. The soluble fractions from the dialyzed material were lyophilized to obtain the polymer product. Henceforth, neomycin-RDE is referred to as PAE. The modified bioreducible PAE (mPAE) was synthesized by a simple one-step reaction with Traut’s reagent (Fig. 1) as described previously [37]. Briefly, 10 g of PAE was dissolved in 1 ml of ddH20 (double distilled water) and was allowed to react with 1.85 g (5-times molar excess) of 2-iminothiolane hydrochloride (IH) at room temperature for 15 h [38]. The product was dialyzed extensively using a dialysis membrane (MWCO = 1000 Da) against 5 mM HCl for 24 h followed by dialysis against 1 mM HCl solution for 24 h. The purified mPAE was lyophilized and stored at −20°C for future use.
Fig. 1. Illustrated Scheme for modification of polyamino-ether (PAE):
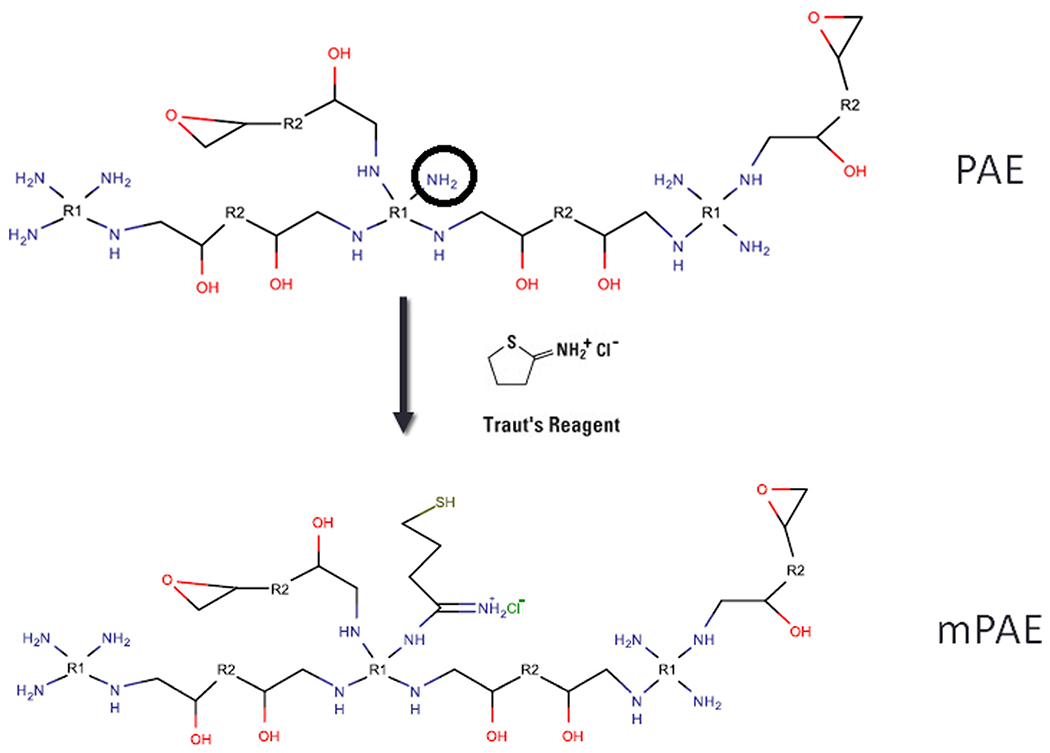
The PAE polymer was dissolved in water and allowed to react with iminothiolane hydrochloride overnight. The resulting modified PAE or mPAE was collected after extensive dialysis and freeze dried for future use. The modification of amino groups (−-NH2) is shown as representative scheme. In this polymer, R1: neomycin R2: glycerol diglycidyl ether. Please note that this schematic is only an illustration and not an exact representation of how the polymerization proceeds.
Thiol Group Estimation
To confirm the introduction of sulfhydryl groups in the mPAE, Ellman’s reagent test was performed. The assay was performed according to the manufacturer’s protocol as described before [39]. Briefly, a set of test tubes, each containing 50 μl of Ellman’s reagent solution and 2.5 mL of reaction buffer (0.1 M sodium phosphate, pH 8.0, containing 1 mM EDTA) were prepared. To the above test tubes, 250 μl of unknown (PAE and mPAE) are added and incubated for 15 min at room temperature. The absorbance was measured spectrophotometrically using UV-1800 Spectrophotometer (Shimadzu) at 412 nm and compared between PAE and mPAE. All data are presented as the mean ± SD (one standard deviation), n = 3.
Sulforhodamine B (SRB) Cytotoxicity Assay
To evaluate the cytotoxicity of mPAE and PEI polymers in A549 and H460 cells, in vitro cytotoxicity assay was performed. For this experiment, the A549 and H460 cells were plated in 96-well plates at a density 5 × 103 and 6 × 103 cells per well, respectively and allowed to grow overnight. The cells were then incubated with 2.5, 5, 10, 25, 50, 100 and 200 μg/ml of mPAE and PEI (25 kDa branched) polymers for 48 h in RPMI medium (supplemented with 10% FBS). After the incubation period, the polymer-containing media was removed and replaced with fresh RPMI media (200 μl). The cell viability (%) was evaluated using a modified SRB assay as described before [40]. Briefly, 100 μl of cold 10% w/v trichloroacetic acid (TCA) was added to each well and the 96 well plates were incubated at 4°C for 1 h. After incubation, the plates were washed 4–5 times with ddH20 and excess water was removed using paper towels. The plates were allowed to air-dry at room temperature (20-25°C). After fixing, 100 μl of 0.057% (w/v in 1% acetic acid) SRB solution was added to each well. After 30 min, the plate was quickly rinsed 4 times with 1% (v/v) acetic acid to remove the unbound dye. The plates were allowed to dry at room temperature. After the drying step, 200 μl of 10 mM Tris base solution (pH 10.5) was added to each well and allowed to incubate for 5 min in the gyratory shaker to dissolve the dye. The absorbance of SRB was measured at 564 nm using a plate reader (Synergy H1 Biotek, H1MFD). The cell viability (%) was calculated according to the following formula. All data are presented as the mean ± SD (standard deviation).
| (1) |
Acid Base Titration
Acid-base titration was performed to evaluate and compare the buffering capacity of the mPAE and PEI polymers. The experiment was performed as described previously [41, 42]. The solution of mPAE and PEI was prepared in diethylpyrocarbonate (DEPC) treated RNase free water with a concentration of 1.5 mM of protonable nitrogens in 150 mM NaCl. The pH of the polymer solutions (10 ml) was adjusted to 4.5 using 0.5 M HCl. This solution was titrated with 0.01 M NaOH to pH 7.5 using a pH meter (Mettler Toledo Seven Multi). The solution of 150 mM NaCl solution was used as a reference for this procedure. All data were presented as the mean ± SD (one Standard Deviation).
Formulation of mTOR-siRNA-mPAE NPs (mTS-mPAE-NPs)
The mTOR siRNA encapsulated NPs were prepared using the nanoprecipitation method (Fig. 2) [43, 44]. Briefly, 1 mg of the mPAE polymer and mTOR siRNA (2 μg) was dissolved in the DEPC-treated RNase free water for 1 h. The above solution was added dropwise (0.2 ml/min) to 1 ml of 70% v/v acetone to obtain mTOR-siRNA-mPAE-NP suspension. The NP suspension was allowed to stir overnight at 200 rpm at RT to evaporate the acetone. To the above suspension, 2 μl of 40% glyoxal was added to cross-link the sulfhydryl groups and form stable disulfide cross-linked NPs. The NPs were dialyzed extensively against DEPC treated RNase free water using a dialysis membrane (MWCO: 50kD) to remove any unreacted glyoxal and obtain the mTS-mPAE-NPs.
Fig. 2. Scheme for the formation of disulfide cross-linked mTS-mPAE-NPs.
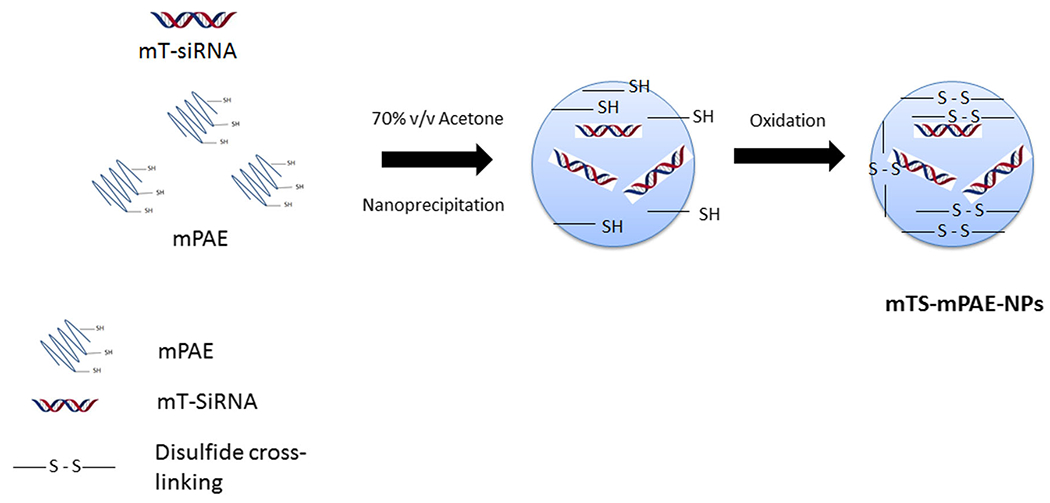
The NPs are formed by a nanoprecipitation method. The first step involves the formation of mPAE-mTOR siRNA nanoparticles with thiol groups. In the second step, the mTS-mPAE-NPs are formed after cross-linking via oxidation with glyoxal. The final formulation was collected by centrifugation for future studies.
Agarose Gel Retardation Assay
mTOR siRNA Encapsulation Ability
To determine the concentration of mPAE required to fully encapsulate mTOR siRNA, gel retardation assay was performed. For this experiment, mTS-mPAE-NPs were formed by the nanoprecipitation method as described above with mPAE polymer to mTOR siRNA weight (w/w) ratios ranging from 300:1 to 1:1 based on siRNA (weight 500 ng) in DEPC-treated RNase free water. The NPs were collected following centrifugation of the nanoparticle suspension. The gel retardation assay was performed by loading a 20 μl aliquot of the sample together with 4 μl of loading dye (6X) New England Biolabs on a 1% agarose gel, prepared in 1X Tris-acetic acid-EDTA (TAE) buffer. Electrophoresis was carried out in TAE buffer (40 mM tris(hydroxymethyl) aminomethane, 20 mM acetic acid, 10 mM EDTA, pH 8,0) supplemented with 1.25 μM ethidium bromide at 100 V for 1 h using Biorad PowerPac 300 Electrophoresis instrument. After electrophoresis, pictures were taken on a Biorad multi-image ChemiDoc XRS UV illumination and analyzed using Biorad Image lab software version 3.0.
Serum Protection Assay and Glutathione-Mediated Release
To evaluate the mTOR siRNA protection ability of mTS-mPAE-NPs and enhanced glutathione (GSH) mediated release, NPs were prepared with mPAE: siRNA w/w ratio of 45:1 using the nanoprecipitation method. The ratio of NP formulation was selected on the basis of their smaller particle size, net positive charge and high entrapment efficiency of the formulation. A volume of 200 μl of siRNA-loaded mPAE NPs (containing 5 μg of siRNA) was incubated at 37°C with an equal volume of Roswell Park Memorial Institute (RPMI) medium supplemented with 10% FBS and GSH 10 mM (together and separately) for a period of 4 h at 37°C incubator, respectively. Naked siRNA was used as a control and treated in the same manner. The NPs was incubated with RNAse for 2 h and then subsequently with GSH after the removal of nuclease by centrifugation. After each time interval, 40 μl of the mixture was removed and incubated in a batch incubator at 60°C for 3 min to terminate serum activity. The samples were electrophoresed using gel electrophoresis protocol as described in the above procedure. After electrophoresis, pictures were taken on a Biorad multi-image ChemiDoc XRS+ UV illumination and analyzed using Biorad Image lab software version 3.0.
Dynamic Light Scattering (Particle Size) and Zeta Potential Measurement
Particle size distribution and the zeta potential of the mTS-mPAE-NPs prepared at weight ratios of 30:1, 45:1 and 60:1 (mPAE: siRNA) were measured using NICOMP 380ZLS (Port Richey, FL). The mTS-mPAE-NPs were formed in DEPC-treated RNase-free water by nanoprecipitation method as described above. The NP suspension was diluted using distilled water to a final volume of 600 μl before measurement. For zeta potential determination, the NP suspension (200 μl) was diluted to 2 ml and the measurement was performed using NICOMP 380ZLS.
Entrapment Efficiency (%) and Loading Capacity (%)
The entrapment efficiency of mTOR siRNA in mTS-mPAE-NPs was measured using AccuBlue high sensitivity dsDNA quantification kit (Biotium). Based on the results obtained from the gel retardation assay of different weight ratios of mPAE and mTOR siRNA, three different weight ratios (30:1, 45:1, and 60:1) were selected to determine the entrapment efficiency (%) and loading capacity (%). Briefly, the mTS-mPAE-NPs suspension was subjected to centrifugation (13,000 x g at 10°C for 30 min) to separate the mTOR siRNA. The free siRNA in the supernatant obtained from centrifugation was quantified by using the AccuBlue high sensitivity dsDNA quantification kit (Biotium) as described in the manufacturer’s protocol. The measurement was performed at the excitation-emission wavelength of 485/530 nm with a plate reader (Synergy H1 Biotek, H1MFD) [45]. The unknown concentration of siRNA in the supernatant was calculated based on the standard curve of siRNA at different concentrations of 0.05, 0.5, 1, 2,5 and 10 μM (equivalent to 0.65, 6.5, 13, 26, 65 and 130 ng/μl of mTOR siRNA). The entrapment efficiency (%) and loading capacity (%) was calculated by using the following equations.
| (2) |
| (3) |
In Vitro mTOR siRNA Release Study
The concentration of glutathione (GSH) in the cytoplasm (0.5–10 mM) is substantially higher compared to the extracellular environments (2–20 μM) [46, 47] as a result of which, the bio-reducible NPs are expected to release significantly higher amounts of mTOR siRNA in the cytoplasm. To perform in vitro release study, mTS-mPAE-NPs (containing 1 μg equivalent mTOR siRNA) was incubated in 1 ml of 0.1 M PBS solution (pH 7.4) containing 10 mM and 10 μM GSH at 37°C for a period of 7 d. After incubation for certain time period interval (1, 2, 3, 4, 5, 6 and 7 d), 100 μl of the media was taken out. The released siRNA was separated from the sample by centrifugation at 13,000 x g for 15 min using a Vivaspin filter membrane (50 kDa) at 37°C. The released siRNA was measured using AccuBlue high sensitivity dsDNA quantification kit (Biotium). As determined previously, a standard curve of mTOR siRNA was plotted to determine the unknown concentration in the supernatant. The cumulative release of mTOR siRNA (%) was calculated as described previously [48].
Cell Proliferation Assay
To evaluate the effects of mTOR siRNA on cell proliferation, mTS-mPAE-NPs were incubated in A549 and H460 cells lines and cell viability (%) was calculated. A549 and H460 cells were plated into 96-well plates at a density 5 × 103 and 6 × 103 cells per well, respectively and allowed to grow overnight. The cells were then transfected with mTOR siRNA or scrambled siRNA (SS) using Dharmafect (Dharmacon, GE lifesciences USA) (siRNA concentration 50 nM) according to the manufacturer’s protocol for a period of 48 h. The cells were also transfected with mTOR siRNA and SS siRNA (control) containing mPAE NPs (siRNA concentration 50 and 100 nM) for 48 h. The NPs prepared at a weight ratio of 45:1 (20 μl and 10 μl) were mixed with 180 μl of RPMI (supplemented with 10% FBS) and allowed to incubate with the A549 and H460 cells for a period of 48 h. The final concentration of mPAE polymer achieved on incubation of mTS-mPAE-NPs was 30 μg/ml and 60 μg/ml to incubate 50 nM and 100 nM siRNA concentration respectively.
The cell viability (%) following treatment with the mTOR siRNA was evaluated using a modified SRB assay according to the protocol as described in the above section before [40]. The cell viability (%) was calculated according to the formula described above in Eq. 1. All data were presented as the mean ± SD (Standard Deviation).
Determination of Cellular Uptake of mTS-mPAE-NPs Using Flow-Cytometry Analysis
To investigate the cellular uptake, mTS-mPAE-NPs were prepared at the weight ratio of 45:1 (mPAE: siRNA) and flow cytometry analysis was performed. To impart fluorescence to the NPs, fluorescein isothiocyanate (FITC) was attached (mTS-mPAE-FITC-NPs) according to the procedure discussed before [49, 50]. In brief, 100 μg/ml of mTS-mPAE-NPs were allowed to react with 2 times molar excess of FITC solution (1 mg/ml in DMSO) overnight in 1 ml of ddH20. The unreacted FITC was removed by dialyzing using a 15 kDa MWCO membrane against 50 ml of ddH20 and mTS-mPAE-FITC-NPs were collected for further experiments.
Cellular uptake was assessed using a BD ACCURI C6 PLUS flow cytometer at a minimum of 10 × 103 cells gated per sample. FITC conjugated mTS-mPAE-NPs were formed according to the procedure above. A549 and H460 cells (2 × 105 cells/well) were seeded into 6-well plate in RPMI medium containing 10% FBS and incubated for 48 h. When the confluence reached ~70–80%, the media in each well was removed and the cells were incubated with mTS-mPAE-FITC-NPs 200 μl and 1800 μl of RPMI media (supplemented with 10% FBS). After certain time intervals (30 min, 2 h, and 24 h), the cells were washed three times with PBS and trypsinized for further processing. The cells were collected by centrifugation for 4 min at 3000 g. The collected cells were suspended in 100 μl of PBS and measured for cellular uptake by BD ACCURI C6 PLUS flow cytometer. Analysis of cellular uptake was performed by using Flowjo 7.6.1 single cell analysis software. At least 10,000 cells were analyzed for each experiment. The flow cytometry data were presented as frequency distribution histograms. The uptake of maximum fluorescence NCs was set as 100%, and the relative uptake of different treatments was presented as a percentage. The mean cell fluorescence was normalized to that of the untreated cells. All experiments were repeated at least three times.
mTOR Gene Silencing Efficiency Analysis by Western Blot Analysis
Western blot analysis was performed in order to evaluate the mTOR gene silencing efficiency of mTOR siRNA-loaded mTS-mPAE-NPs in A549 and H460 cells. A549 and H460 cells were plated into 6-well plates at 2 × 105 and 2.5 × 105 cells per well, respectively and allowed to grow overnight. The cells were then transfected with mTOR siRNA or scrambled siRNA (SS) using Dharmafect (Dharmacon, GE lifesciences USA) (siRNA concentration 50 nM) for 48 h. The cells were also transfected with mTOR siRNA and SS siRNA containing MP-NPs (siRNA concentration 50 nM) for 48 h. The prepared NPs (200 μl) at the weight ratio of 45:1 were mixed with 1800 μl of RPMI (supplemented with 10% FBS) and allowed to incubated with the cells for a period of 48 h to achieve a final concentration of 30 μg/ml.
The cells were collected and washed thrice with ice-cold PBS and then lysed by the cocktail containing RIPA buffer (cell signaling technology, Danvers, USA), protease inhibitor and phosphatase inhibitor using ultrasonication method (Qsonica Sonicators USA) for 3 s. The lysates were then centrifuged at 13,000 g at 4°C for 10 min. The supernatant was collected and the total protein concentration was determined using bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). To confirm the mTOR protein (289 kDa) levels in A549 and H460 cell lines, 20 μg of protein were mixed with equal volumes of loading buffer (2X Laemlli sample buffer, 5% β-mercaptoethanol) and loaded in each lane and separated by an 8% SDS-polyacrylamide gel electrophoresis. Electrophoresis was conducted in the running buffer (25 mM Tris base, 192 mM glycine, 0.1% SDS). For the first 45 min, the run was conducted under 90 V then it was increased to 130 V for 90 min. The proteins were then transferred to a PVDF membrane using Mini Trans-Blot® Electrophoretic Transfer Cell (Bio- Rad) at 300 constant mA. The PVDF membrane was then blocked with 5% milk in Tris-buffered saline with 0.1% tween 20(TBS-T) for 1 h at room temperature by stirring in a gyratory shaker to prevent nonspecific binding of the antibodies.
The PVDF membrane was then incubated with primary mTOR (7C10) Rabbit antibody (1:1000 dilution Cell Signaling Technology) in 5% BSA in TBST at 4°C overnight. The membrane was washed three times with TBST, followed by incubation with anti-Rabbit antibody [1: 10,000 Goat anti-Rabbit IgG (H&L)] secondary Antibody and imaged with Odyssey imaging system (LI-COR Biosciences). The membrane was washed three times with TBST and was incubated with β-Actin (13E5) rabbit antibody (1:1000 dilution, Cell Signaling Technology) prepared in 5% BSA in TBST for 3–4 h. The membrane was then washed three times followed by incubation with an anti-rabbit antibody (1: 10,000 Goat anti-Rabbit IgG (H&L) Secondary Antibody) and imaged using Odyssey imaging (LI-COR Biosciences). The quantitative amount of mTOR gene silencing was calculated using the densitometric analysis (Image J software) with β-Actin as the loading control.
Statistical Analyses
Results are expressed as means ± standard errors. Significant differences between two groups were determined using Student’s t-test and differences among multiple groups were determined using Analysis of Variance (ANOVA). We performed statistical analysis with GraphPad Prism Version 5 software. The differences were considered statistically significant at a p-value <0.05 and p < 0.01 in all cases.
RESULTS
Synthesis and Characterization of Thiolated Polymer (mPAE)
The one-step reaction of PAE with Traut’s reagent resulted in the successful introduction of a thiol group, to produce mPAE [37, 51]. The introduction of the thiol group was confirmed using Ellman’s reagent. The Ellman’s test is based on the reaction of chromogenic reagent 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) with free thiol groups present in the polymer to produce one molecule of 5-thio-2-nitrobenzoic acid (TNB) [52]. The reaction of the mPAE free thiol with the highly oxidizing disulfide bond of DTNB will result in the release of one molecule of TNB. In particular, for every free thiol group, there is a release of one TNB group which can be detected spectrophotometrically. We found that the absorbance of the mPAE polymer was significantly greater (***p < 0.001) than the absorbance of parent PAE polymer.
mPAE Polymers Were less Cytotoxic than PEI-25
A549 and H460 cells were seeded and transfected with mPAE and PEI polymers as described in the experimental section. Cytotoxicity is an important evaluation parameter to determine the safety of gene delivery vehicle [53]. To ensure the safety of the polymeric vehicles being used in this study, the cytotoxicity of mPAE and 25kD PEI was investigated in A549 and H460 cells using the SRB assay. As shown in Fig. 3, mPAE and PEI exhibited cytotoxicity for both A549 and H460 lung cancer cells in a dose-dependent way. Fundamentally, with the increase of mPAE and PEI concentration, the cytotoxicity increased accordingly in all tested cell lines. Compared with 5, 10 and 25 μg/ml concentrations of 25-kD PEI, mPAE had no apparent cytotoxicity on A549 and H460 cell lines. The lethal dose 50 (LD50) of the two polymers is summarized in Table I. Lethal dose 50 (LD50) of the PEI was approximately 30 ± 5.4 μg/ml and 25 ± 5.2 μg/ml for A549 and H460 cells, respectively which was lower (2.33 times and 2.6 times) than the LD50 for the mPAE polymer (75 ± 6.2 μg/ml and 70 ± 3.4 μg/ml for A549 and H460 cells respectively). This suggests that on a weight basis, the mPAE polymer possesses less cytotoxicity compared to the PEI polymer. This property can be exploited to safely deliver genes to the lung cancer cells.
Fig. 3. Cytotoxicity analysis of mPAE and PEI polymers.
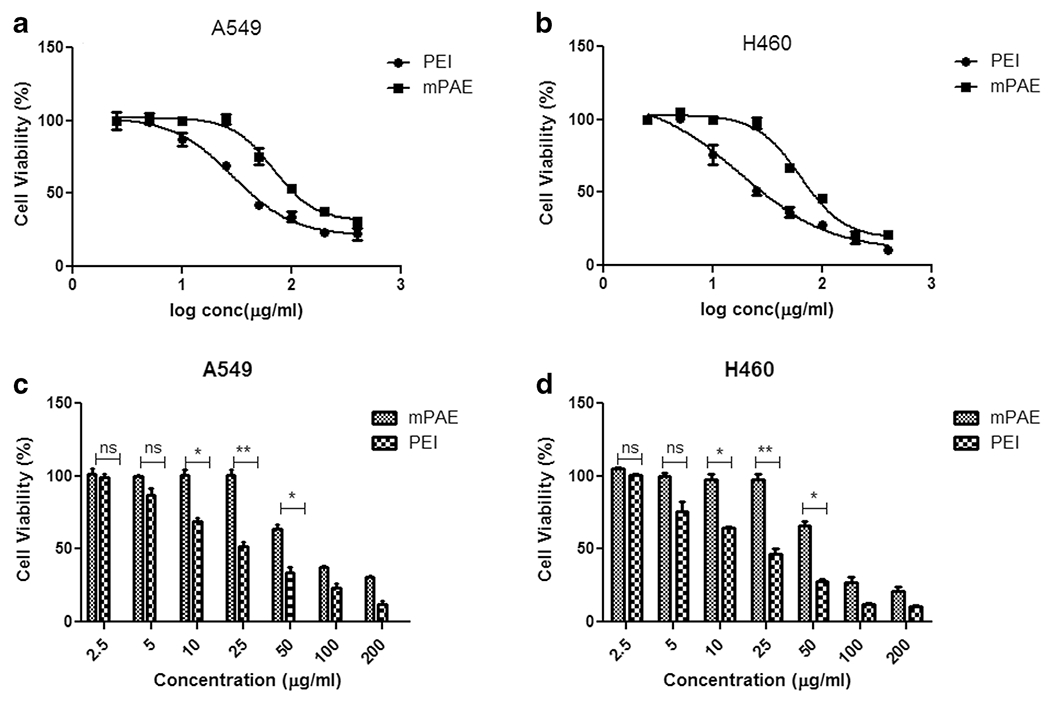
(a) Cell viability (%) of “mPAE and PEI” polymers at various log concentrations in (μg/ml) A549 cell line (b) Cell viability (%) of “mPAE and PEI” polymers at various log concentrations (μ/ml) in H460 cell line (c) Cell viability (%) after treatment of mPAE and PEI at concentrations (2.5, 5, 10, 25, 50, 100 and 200 μg/ml) in A549 cell line. and (d) Cell viability (%) after treatment of mPAE and PEI at concentrations (2.5, 5, 10, 25, 50, 100 and 200 μg/ml) in H460 cell lines. The cytotoxicity was determined using SRB assay after 48 h polymer exposure (n = 3, error bars represent standard deviation).
Table I. LD50 Values of PEI and mPAE Polymers in A549 and H460 cells.
(n = 3, ± Represent Standard Deviation)
| A549 cells (μg/ml) | H460 cells (μg/ml) | |
|---|---|---|
| PEI-LD50 | 30 ± 5.4 | 25 ± 5.2 |
| mPAE – LD50 | 70 ± 6.2 | 65 ± 3.4 |
mPAE Polymers Possess Superior Buffering Capacity
The buffering capacity of the cationic polymer is responsible for the release of encapsulated siRNA from the endosome [54, 55]. To evaluate the potential of mPAE polymer to escape endosome via proton sponge effect, buffering capacity of mPAE and PEI polymers was assessed by measuring the change in pH of the solution by titrating with 0.01 M NaOH. From the titration graph presented in Fig. 4, it was observed that there was a small change in pH in mPAE curve compared to PEI curve when the same amount of 0.01 M NaOH was added to the polymer solution during titration. To change the pH 4.5 to pH 7.5 of the polymeric solution, a significant higher 5.8 ± 0.8 ml of 0.01 M NaOH was required for mPAE solution compared to 4.63 ml ± 1.2 ml 0.01 M NaOH for PEI solution (Fig. 4, *p < 0.034). This indicates that mPAE polymer’s resistance to change in pH is higher than PEI, which further indicates that the mPAE has higher buffering capacity and high tendency to be protonated compared to PEI [56]. A polymer with high buffering capacity would undergo a small change in pH when the same amount of NaOH was added to the polymer solution during titration. The mPAE would be able to undergo proton sponge effect upon cell internalization in the acidic endosomal compartments of the cells thereby releasing the loaded siRNA in the cytoplasm.
Fig. 4.
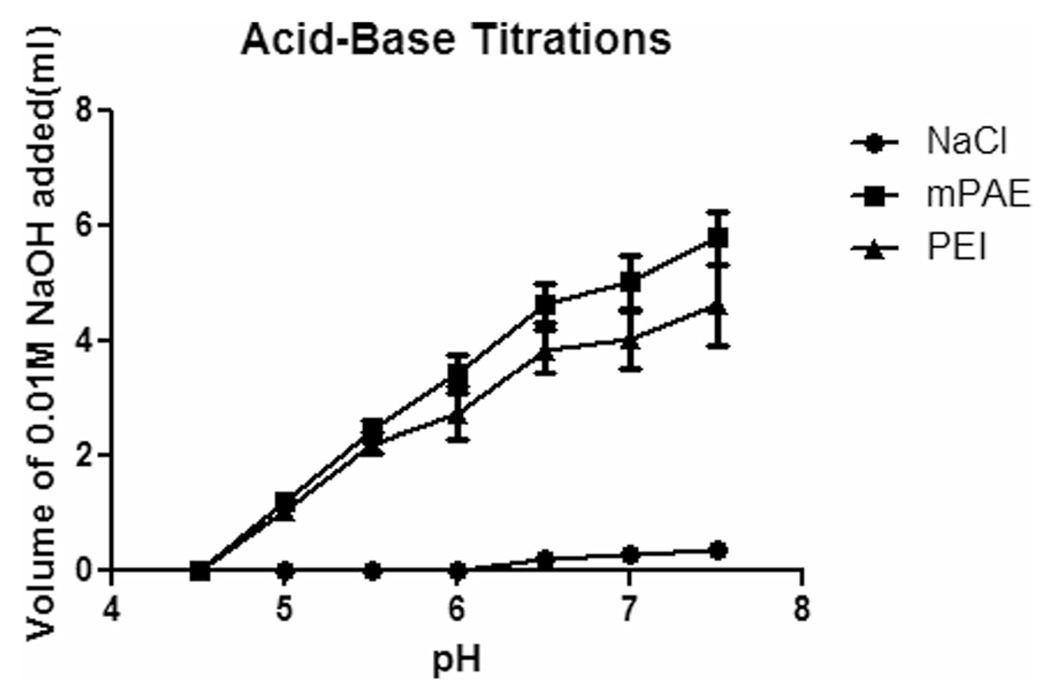
Determination of the buffering capacity of mPAE and branched PEI (Mw =25,000 g/mol) by acid-base titration; NaCl were used as control. Solutions were titrated with 0.01 M NaOH from pH 4.5–7.5. (n = 3, error bars represent standard deviation).
mTS-mPAE-NP Encapsulated mTOR siRNA at 25:1 and Higher Polymer:siRNA Weight Ratios
We further investigated the ability of the mPAE polymer to encapsulate and form NPs with mTOR siRNA, as a function of weight ratio (w/w) of polymer weight to mTOR siRNA by gel electrophoresis method combined with an AccuBlue high sensitivity dsDNA quantification kit. The NPs were prepared by the nanoprecipitation method. We performed a gel retention assay at weight ratios of 1:1–300:1 of mPAE polymer to siRNA. The siRNA band is seen to disappear in Lane 4 of the gel (Fig. 5a) as the polymer to siRNA w/w ratio increases from 25:1 and higher, indicating that the siRNA was largely encapsulated within the mTS-mPAE-NP (compare lanes 5–7: mTS-MPAE-NP formed at w/w ratio of 100:1–300:1, with lane 2–4: mTS-mPAE-NP formed at w/w ratio of 1:1–25:1). The entrapment efficiency calculated by image J software of the mPAE NPs to siRNA is seen to be below (< 60%) of the polymer to siRNA w/w ratio of 25:1 (Fig. 5b). However, at weight ratios higher than 100:1, the siRNA is completely encapsulated in the NPs and 100% entrapment is achieved.
Fig. 5.
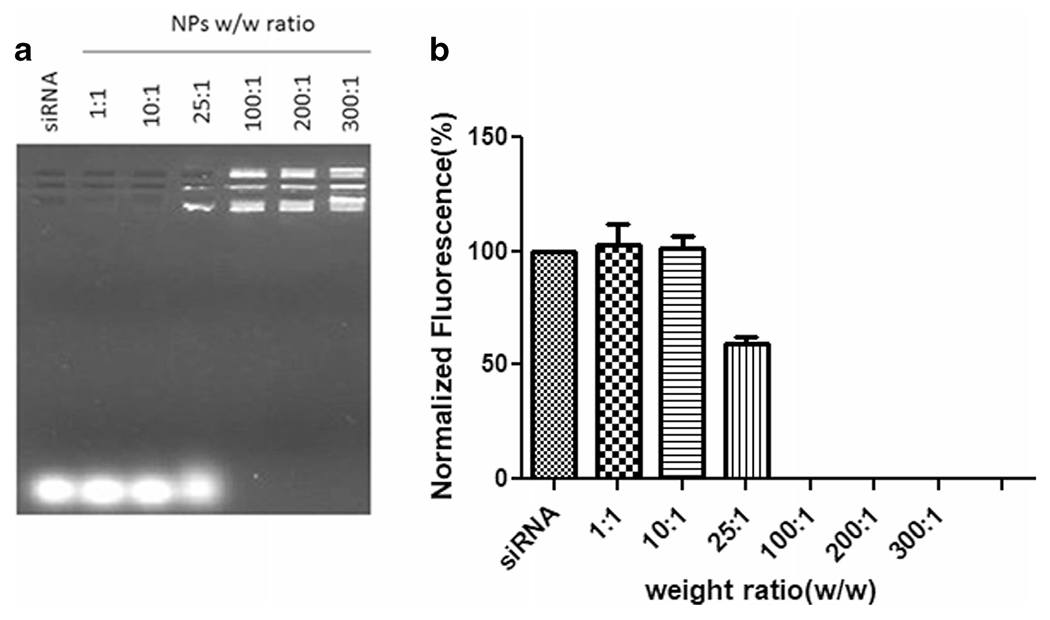
(a) Agarose gel electrophoresis result of the mTS-mPAE nanoparticles. NPs formed from polymer to siRNA at w/w ratio of 1:1, 10:1, 25:1, 100:1, 200:1 and 300:1. Samples were prepared at room temperature before being loaded onto a 1.0% w/v agarose gel (75 V, 45 min). (b) Densitometric analysis of the siRNA bands performed using Image J software. The experiments were repeated at least thrice. The best representative images are shown.
mTS-mPAE-NPs Are Nano-Sized with a Positive Surface Charge
It is essential to optimize the physicochemical properties (particle size and surface charge) of mTS-mPAE-NPs to achieve high efficiency for mTOR siRNA delivery to NSCLC cells. Therefore, particle size and zeta potential of the mTS-mPAE-NPs were studied. Based on the gel retardation assay studies three different weight ratios of mPAE to mTOR siRNA-based mTS-mPAE-NPs were investigated at constant siRNA weight. As shown in Table II, the hydrodynamic diameters of the NPs were between 112 nm and 122 nm for polymer to siRNA ratios ranging from 30:1 to 60:1. The hydrodynamic diameter of the NPs increased steadily from 112 nm to 122 nm with increasing ratio from 30:1 to 60:1, which is in good agreement with previous reports [57–59]. In addition, the relative homogeneous size distributions of mTS-mPAE-NPs are unimodal with a polydispersity index of 0.35 as shown in Fig. S1.
Table II. Characterization of the mTS-mPAE-NPs.
Hydrodynamic Diameter, Zeta Potential, Entrapment Efficiency (%) and Loading Capacity (%) of mTS-mPAE NPs at Different mPAE: siRNA Weight Ratios (30:1, 45:1 and 60:1)
| Hydrodynamic diameter(nm) | Zeta potential(mV) | mPAE: siRNA ratio | Entrapment efficiency (%) | Loading capacity (%) | mTOR siRNA loading (μg/mg nanoparticles) |
|---|---|---|---|---|---|
| 112.9 ± 8.2 | −2.8 ± 1.5 | 30:1 | 55.5 ± 2.8 | 1.83 ± 0.86 | 18.3 ± 8.6 |
| 114.2 ± 7.4 | +26.5 ± 5.3 | 45:1 | 99.9 ± 3.5 | 2.25 ± 0.34 | 22.2 ± 3.4 |
| 121.5 ± 6.5 | +32.2 ± 4.3 | 60:1 | 100 ± 4.2 | 1.66 ± 0.54 | 16.7 ± 5.4 |
The surface charge of the NPs was also determined at various weight ratios (Table II). The zeta potential for all the NPs remained in the range of +26.5 – +32.2 mV except for the ratio of 30:1, where stable NPs of mTOR siRNA and mPAE NPs were not formed completely resulting in the negative surface charge. The concentration of mPAE in mTS-mPAE-NPs also influenced the surface charge of nanoparticles (Table II). Increasing the concentration of mPAE in nanoparticles increased the zeta potential of nanoparticles possibly because of the cationic charge of mPAE. We also found similar results with the gel electrophoresis experiment Fig. 5., wherein the increase in polymer concentration resulted in complete encapsulation (beyond 100:1) thus increasing the zeta potential. The relative surface charge of the mTS-mPAE-NPs (45:1 ratio) also showed a unimodal distribution as shown in Fig. S2. The three ratios of 30:1, 45:1 and 60:1 were selected on the basis of gel electrophoresis experiment (between 25:1 and 100:1) Fig. 5. For enhanced cellular uptake of mTOR loaded mPAE NPs, positive surface charge and small particle size (< 150 nm) is necessary for reaching and binding to anionic cells surface of the NSCLC cells. We believe that the nano-sized mTS-mPAE-NPs charged positively have a potential as an efficiency mTOR siRNA delivery carrier.
mTS-mPAE-NP Protected mTOR siRNA against Nuclease Degradation
To achieve maximum delivery efficiency, the NP carrier must protect siRNA from degradation by nucleases in the biological systems. To evaluate the serum protection property of mPAE polymer, serum stability test was carried out for mTS-mPAE-NPs in 10% FBS Fig. 6a. As can be seen in lane 2, the presence of RNase in 10% FBS resulted in nuclease-mediated degradation of the siRNA and thus disappearance of the siRNA band. In comparison to naked siRNA, lane 5 shows that mTS-mPAE-NP effectively protected siRNA from nuclease degradation as a brighter and dense band was observed in the NP well. mTOR siRNA encapsulated in the crosslinked NPs remained intact upon the treatment of RNase compared with the naked siRNA indicating that NPs was able to prevent nucleolytic degradation. The NPs were able to protect the siRNA from RNase degradation as can be seen in the lane 6 with lower intensity siRNA band. Glutathione-mediated cleavage of the siRNA resulted in a release of the protected siRNA from the mTS-mPAE-NPs. The mTS-mPAE-NP was incubated with GSH final concentration (10 mM) to verify intracellular release of mTOR siRNA by agarose gel electrophoresis. As shown in Fig. 6b (Lane-7) around 55 ± 3.6% of the siRNA release can be observed at elevated GSH concentrations of 10 mM. This result suggests that bio-reducible nanoparticles can protect the siRNA against nuclease degradation as well as reductively disassemble by GSH to release the mTOR siRNA. The Lane 8 further shows that the mPAE-NP were able to protect the mTOR siRNA from degradation by nucleases and around 48% of mTOR siRNA was released from the NPs. The bands were not as intense as the non-treated free siRNA (control) because some siRNA might remain within the nanoparticles (could not be stained by ethidium bromide) or be degraded in small quantity.
Fig. 6.
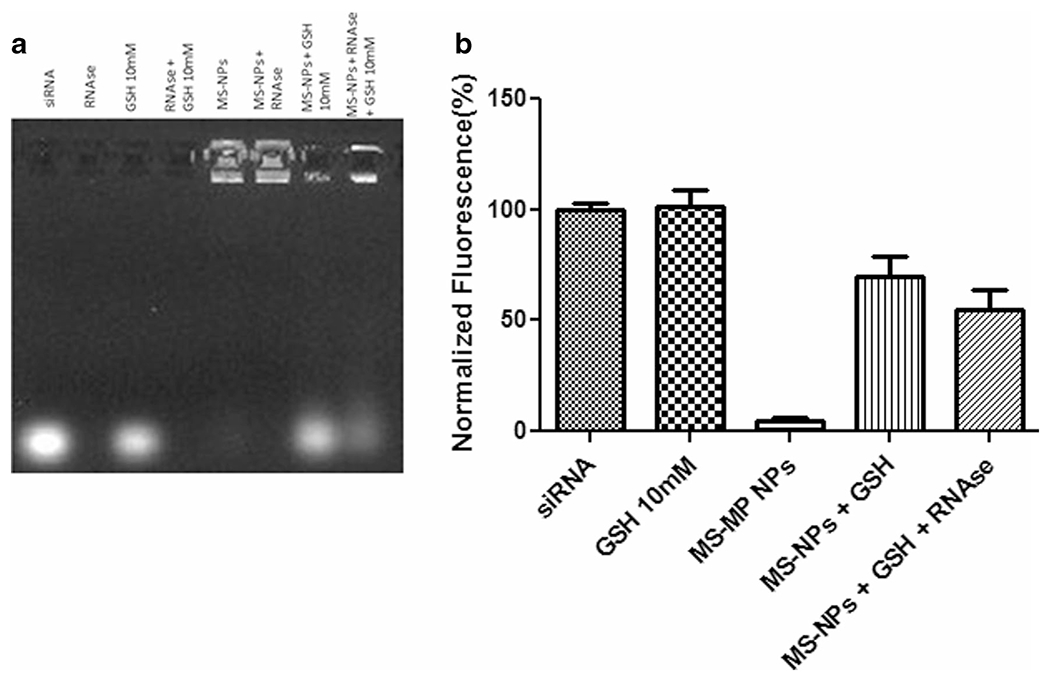
(a) Serum stability and glutathione-mediated siRNA release from mTS-mPAE NPs using gel retardation assay. A Gel electrophoresis of mTS-mPAE NPs prepared at weight ratio 45:1 with (mPAE: mTOR siRNA). The NPs were incubated with 10%FBS containing RNase and glutathione 10 mM for a period of 4 h at 37°C. Samples were prepared at room temperature before being loaded onto a 1% w/vol agarose gel (75 V, 45 min). (b) Densitometric analysis of the siRNA band using Image J analysis software. Abbreviations: RNase – 10%FBS containing RNase, GSH – Glutathione, MS -NPs – mTOR siRNA – mPAE NPs. The experiments were repeated at least thrice. The best representative images are shown.
mTS-mPAE-NPs Has High mTOR siRNA Entrapment Efficiency
Based on the results obtained from the gel retention assay, three different w/w ratios of mTS-mPAE-NPs were prepared (30:1, 45:1 and 60:1). The free siRNA was separated from the NP suspension by centrifugation using vivaspin column (MWCO size 50kD). The concentrations of siRNA were then measured using the AccuBlue high sensitivity dsDNA quantification kit by plotting a standard curve of mTOR siRNA at 0.65, 6.5 13, 26, 65 and 130 ng/μl as shown in Supplementary Fig. S3. The results of the entrapment efficiency (%) are shown in Table II. siRNA entrapment efficiency in the range of 55.5 and 100% was achieved for mTS-mPAE-NPs. In general, the entrapment efficiency decreased with a decrease in the weight ratio from 30:1 to 60:1 as shown in Table II. The higher concentrations of cationic polymer mPAE allowed better binding with the negatively charged mTOR siRNA resulting in almost 100% entrapment efficiencies for ratios 45:1 and higher. On the basis of entrapment efficiency, loading capacity (%) was also calculated as shown in Table II. The most optimum loading capacity was found for the polymer: mTOR siRNA weight ratio of 45:1 which indicates that for 100 μg of polymer 2.2 μg of siRNA (2.2%) can be entrapped or encapsulated.
Glutathione-Mediated Triggered Release of mTOR siRNA
The in vitro release profiles of siRNA-loaded mTS-mPAE-NPs were investigated for 7 d in PBS at pH 7.4 with varied glutathione (GSH) concentration. GSH is an intracellular reducing agent that is present in higher concentration inside the cells (2-10 mM) and significantly lower levels in the extracellular environment (2-20 μM) [20]. The release of siRNA was divided into 2 stages on the basis of release rate, which was calculated as the slope of the release profile. In the first stage (end of day 1), siRNA-loaded mTS-mPAE-NPs showed rapid release (58 ± 3%) in the presence of excess GSH (10 mM) while the siRNA was released at a slower rate (18.5 ± 1.7%) in lower GSH concentration (10 μM). At day 1 Fig. 7b, there was a significant increase in the release of siRNA due to presence of the excess GSH (10 mM) compared to lower GSH concentration (10 μM) (p < 0.001). The second stage can be seen of as similar in both GSH concentrations with a steady release of siRNA from the NPs Fig. 7b. After a period of 7 d around 70% siRNA was released in presence of 10 mM GSH, compared to around 42% in 10 μM GSH. The initial burst release of siRNA from mTS-mPAE-NPs in PBS pH 7.4 10 mM GSH was expected due to the cleavage of disulfide bonds present in the mTS-mPAE-NPs compared to less or almost no cleavage of disulfide bonds in the mTS-mPAE-NPs in presence of lower concentration of GSH (10 μM). In the second stage, siRNA was released at a sustained constant rate from mTS-mPAE-NPs for up to 7 d. The sustained slow release of siRNA from the NPs at both conditions after day 1 can be attributed to the ionic interactions between the siRNA and the cationic polymer (mPAE) NPs.
Fig. 7.
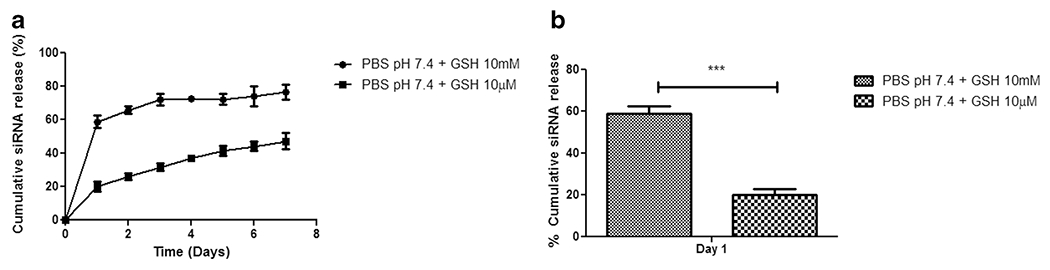
In vitro release of siRNA from the nanoparticles. (a) Profile of siRNA release from mTS-mPAE-NPs prepared at a weight ratio of 45:1 in presence of GSH 10 mM and GSH 10 μM for a period of 7 d (a) and 1 d (b). (n = 3, error bars represent standard deviation).
mTS-mPAE-NPs Showed Increased Cellular Uptake in A549 and H460 Cells
In many cases, nucleic acid delivery vehicles encounter with low transfection efficiency as a consequence of low cellular uptake [37]. Hence, the efficacy of cellular uptake of the vehicles was determined using flow cytometry experiment. The mTS-mPAE NPs were prepared by the nanoprecipitation method as described above at a weight ratio of 45:1(mPAE: mTOR siRNA). The average hydrodynamic diameter of the NPs was about 114 nm and zeta potential was around +26 ± 4.2 mV. Cellular uptake of mTS-mPAE NPs was evaluated by using flow cytometry in A549 and H460 cell lines. The mTS-mPAE-NPs were conjugated with FITC to determine their cellular uptake following incubation with cells at various time periods (0.5 h, 2 h and 24 h). As shown in Fig. 8a and b, the cellular uptake efficiency of mTS-mPAE-NPs at a fixed mTOR siRNA concentration (equivalent to 50 nM mTOR siRNA) was time dependent in A549, and H460 cells. Figure 8c shows relative cellular uptake of the cells that have taken up NPs. The mTOR siRNA encapsulated NPs showed quantified cellular uptake (%) 34.18 ± 3.2 (0.5 h), 49.6 ± 3.6 (2 h), 95.44 ± 7.6 (24 h) and 40.42 ± 45 (0.5 h), 54.88 ± 2.98 (2 h), 100.15 ± 4.77 (24 h) in A549 and H460 cells, respectively. Nearly 50% of the NPs were inside the A549 and H460 cells after 2 h of transfection with NPs. As mentioned before, the mTS-mPAE NPs are nano-sized and possess positive charge which enables the NPs to get entry into the cells. The NPs did not show any significant (p > 0.05) increase in the cellular uptake upon further incubation at 36 h (result not shown here).
Fig. 8. Cellular uptake analysis of mTS-mPAE-NPs analyzed using flow cytometry.
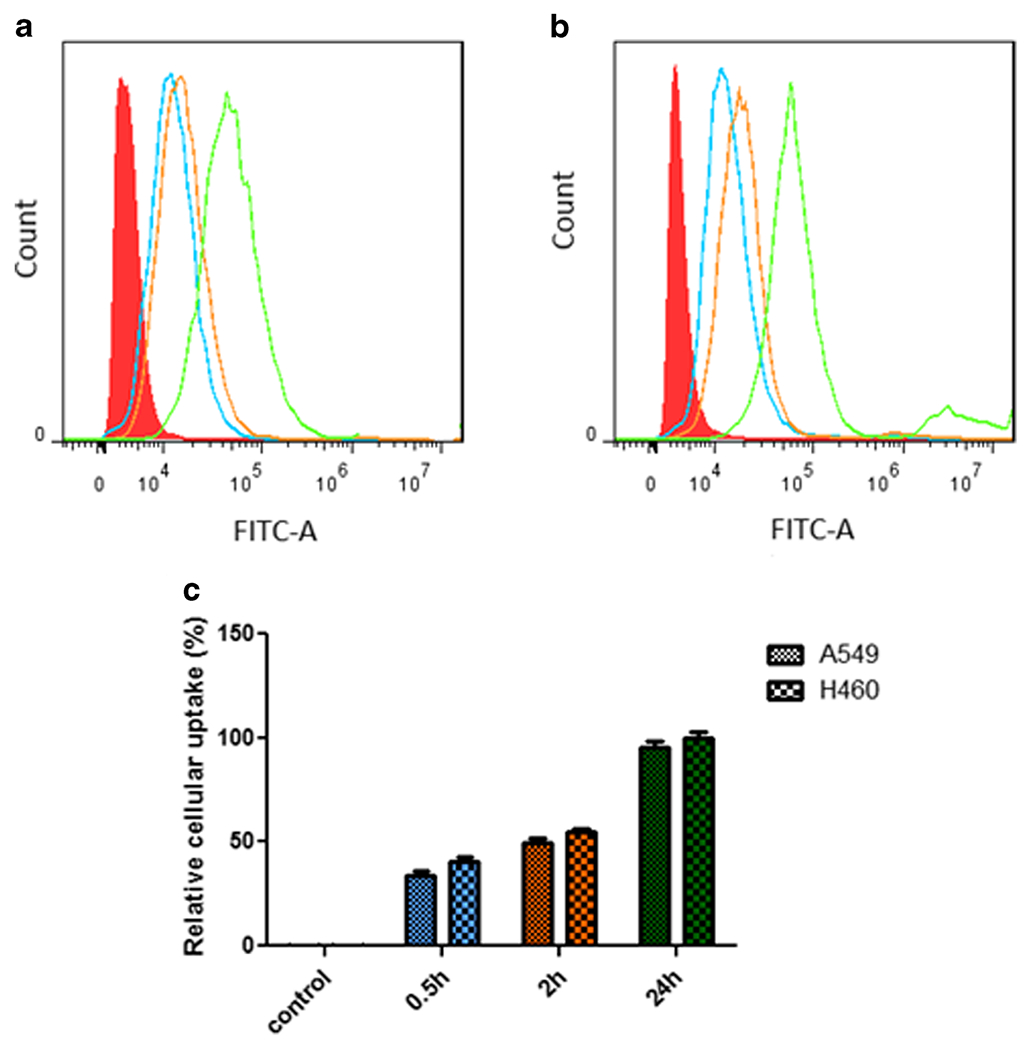
mTS-mPAE NPs were formed at a weight ratio of 45:1 siRNA (1.33 μg). FITC was conjugated to the surface of NPs using a one-step reaction before the flow cytometry experiment to form mTS-mPAE-FITC-NPs. After certain time intervals (0.5 h, 2 h and 24 h) the cells were washed trypsinized and collected to perform the flow cytometry experiment. The fluorescence intensity of MS-MP-FITC-NPs in (a) A549, (b) H460 cells (2 × 105 cell/well). (c) Bar graph representing the mean percentages of cellular uptake. The results are presented as mean ± SD (n = 3). Analysis of cellular uptake was performed by using Flowjo 7.6.1 single cell analysis software.
mTS-mPAE NPs Demonstrate High mTOR Gene Silencing Efficiency in Human NSCLC Cells
We investigated the mTOR silencing efficiency of mTS-mPAE NPs (using 50 nM siRNA) in human lung cancer cell lines A549 and H460 cells. The mTOR gene silencing ability was measured at the fixed weight ratio of 45:1 (mPAE: mTOR siRNA) the scrambled siRNA based mPAE NPs was also prepared as control. As shown in Fig. 9a and c the negative control (SS-mPAE NPs) did not show gene silencing effects in the A549 and H460 cell lines. Meanwhile, the expression of the mTOR protein was significantly decreased after treatment with mTS-mPAE-NPs in both A549 and H460 cells (**p < 0.01) Fig. 9a and c. Representative bands show the expression of mTOR protein (upper band) β-actin (lower bands). As shown in Fig. 9b and d the relative mTOR protein expression expressed as histograms (%) in mTS-mPAE NPs treated A549 and H460 cell lines (normalized to the β-actin expression) was significantly less (~60 and 64% mTOR knockdown) than SS-mPAE NPs (*p < 0.05). We found that the mTOR siRNA loaded NPs efficiently silenced the mTOR gene by using 50 nM siRNA concentrations compared to using Dharmafect (Transfection reagent) at 50 nM siRNA.
Fig. 9. Silencing efficiency of MS-DF (mTOR-siRNA Dharmafect transfection reagent complex) transfection reagent and mTS-mPAE NPs at 50 nM concentration.
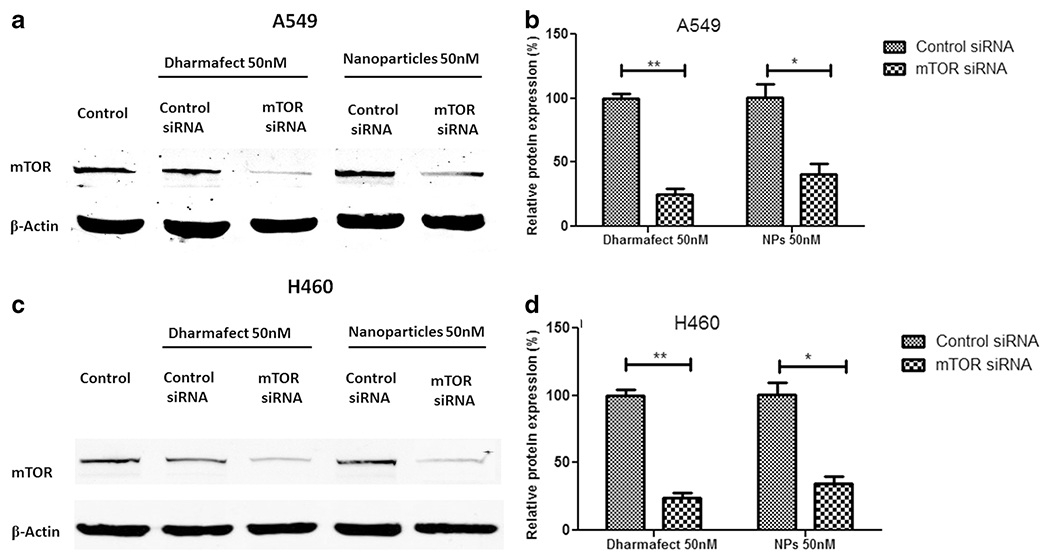
Expression of mTOR protein determined using Western blot in A549 and H460 cells (2× 105 cell/well). mTS-MPAE NPs/mTOR siRNA and SS-MP NPS/scrambled siRNA prepared at weight ratio 45:1 (siRNA concentration 50 nM or 1.33μg per well) (a) Western blot showed a marked reduction in the mTOR protein levels after treatment with mTS-mPAE NPS at 50 nM concentrations in A549 cells. (c) Western blot showed a marked reduction in the mTOR protein levels after treatment with mTS-mPAE NPS at 50 nM concentrations in H460 cells. (b) mTOR protein silencing was calculated using the densitometric analysis and β-Actin as loading control in A549 cells and (d) mTOR protein silencing was calculated using the densitometric analysis and β-Actin as loading control in H460 cells. The results are presented as mean ± SD (n = 3). (* p < 0.05, ** p < 0.01).
mTS-mPAE-NPs Induced Cell Growth Inhibition against NSCLC Cell Lines
mTOR siRNA-inhibition by siRNA has been reported to induce cell apoptosis [60, 61]. We performed cell growth inhibition studies to evaluate cell growth inhibitory property of the mTS-mPAE-NPs in A549 and H460 cells. Figure 10b and c shows the cell viability (%) of mTS-mPAE-NPs over the negative scrambled siRNA control based NPs (SS-mPAE NPs) and placebo NPs in both A549 and H460 cells. The cell viability (%) with 50 nM of siRNA concentrations of mTS-mPAE-NPs was significantly lower (p < 0.05) compared to both SS-mPAE-NPs and placebo NPs alone in A549 and H460 cells. The 100 nM siRNA concentration mTS-mPAE-NPs also induced significant cell growth inhibition compared to the controls. However, the placebo NPs and SS-mPAE NPs were cytotoxic to the cells and resulted in significant toxicity (*p < 0.05) to the cells themselves. These results demonstrate that the mTS-mPAE NPs induced inhibition was significantly larger than the inhibition observed in SS-mPAE NPs and placebo control treated A549 and H460 cells. The 50 nM of Dharmafect mTOR siRNA resulted in 36 and 42% decrease in cell viability in A549 and H460 cells respectively (Fig. 10a and d). In comparison, the 50 nM concentration of mTS-mPAE NPs resulted in only 31 and 32% of cell growth inhibition of after 48 h treatment in A549 and H460 cells, respectively.
Fig. 10.
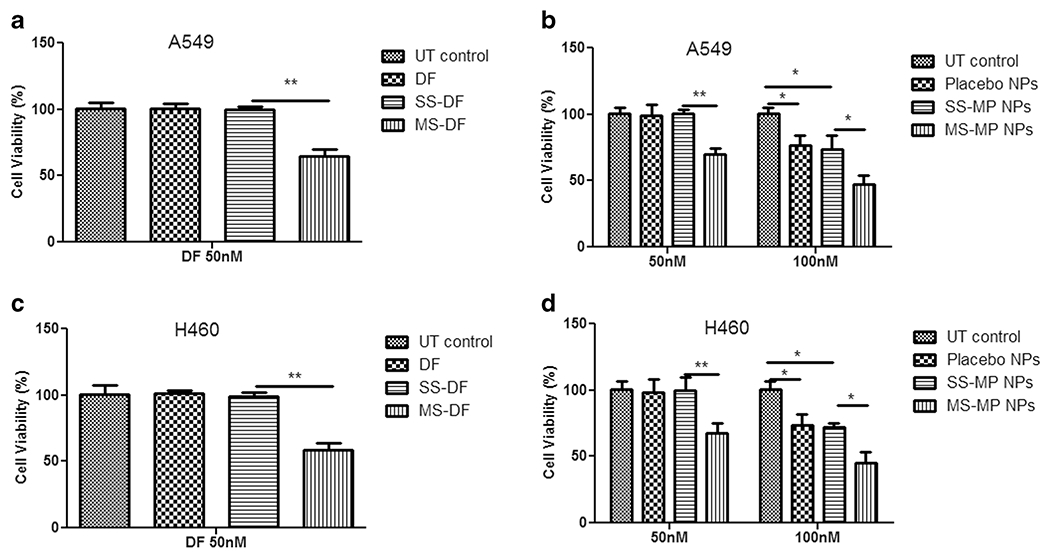
Cell viability (%) of mTOR siRNA with commercial transfection reagent Dharmafect (DF) and mPAE based NPs in A549 cells (a and b) and H460 cells (c and d) seeded at density of (5 × 103 cell/well and 6 × 104 cells / well for A549 and H460 cells respectively) at 48 h incubation. mTS-mPAE-NPs(mTOR-siRNA – mPAE nanoparticles) and SS-mPAE-NPs (Scrambled-siRNA – mPAE nanoparticles) prepared at 45:1 weight ratios were incubated with cells at a concentration of 30 and 60 μg/ml of mPAE polymer to achieve 50 nM and 100 nM siRNA concentration, respectively. Figure (a) and (c) indicates the cell viability (%) of the A549 and H460 cells after transfection with scrambled siRNA and mTOR siRNA using DF reagent. Figure (b) indicates the cell viability (%) of the scrambled siRNA and mTOR siRNA encapsulated mPAE NPs in A549 cells and (d) indicates the cell viability (%) of the scrambled siRNA and mTOR siRNA encapsulated mPAE NPs in H460 cells The results are presented as mean ± SD (n = 3). A value of p less than 0.05 was accepted to be significant (**p < 0.01, *p < 0.05) and the statistics were performed with Student’s t test.
DISCUSSION
The ultimate goal of the present study was the development of a bioreducible delivery system capable of delivering mTOR siRNA to lung cancer cells. Non-specific drug deliveries using non-targeted drug delivery decreases the therapeutic efficacy of drugs and increase the risk of toxic effects, which remain a major challenge in the treatment of cancer [62]. Thus, targeted delivery of mTOR siRNA to lung cancer cells is critical for efficient delivery of nucleic acid drugs provide anti-tumor effects. The mPAE NPs are passively targeted systems. The cationic NPs were reported to be internalized by direct translocation [63]. The passively targeted cationic mPAE NPs are expected to accumulate in the tumor tissue via enhanced permeation and retention (EPR) effect. In this study, we have successfully developed thiolated PAE (mPAE) by a one-step reaction with Traut’s reagent. We used –SH group attached mPAE to encapsulate mTOR siRNA and deliver to the NSCLC cell lines. Kommareddy et al. prepared thiolated gelatin with the 2-iminothiolane HCl reagent using a similar approach and used the thiolated cells [37, 64]. We selected 2-iminothiolane concentration and the introduction rate based on this previously reported study [37]. We used polyaminoethers as the backbone to be converted to bioreducible polymer for effective gene delivery. These polymers exhibited effective transgene delivery capabilities and thus provide a strong advantage compared to conventional bioreducible systems with only bioreducible nature. The novelty of these PAE polymers is the presence of aminoglycosides as the backbone which provides the biodegradability and several amino groups to be easily converted to bioreducible polymer.
Bioreducible polymers have been extensively studied during past decades due to their attractive structural advantages [25, 65]. They possess unique features of high stability in extracellular physiological condition and immediate cleavage of disulfide linkages in a reductive intracellular environment, lower cytotoxicity due to the avoidance of accumulation of the high molecular weight polycations and triggering controlled delivery of genetic materials from the polyplexes. Further, to confirm the siRNA release ability from the NPs through the cleaved disulfide bond of mTS-mPAE, GSH was used to simulate the intracellular reducing environment. As shown in Figs. 6 and 7, the cleavage of disulfide-linked mTS-mPAE-NPs with GSH resulted in increased siRNA release in presence of 10 mM GSH. This significant release can be beneficial for improved siRNA release on reaching the cytosol and thus improved gene silencing efficiency. The differential level of GSH provides a high-redox potential that provides a highly reducing environment inside the cells for disulfide bonds [66]. The high-redox potential gradient between the extracellular and intracellular environments provides the ability for disulfide cleavage inside the cells resulting in fast siRNA release from the cationic mPAE based siRNA NPs [47, 67]. Others have also reported a similar mechanism of improved gene silencing efficiency with increased GSH mediated siRNA release in the cytoplasm from cationic polymeric NPs [68–71]. In a recent study by Breunig et al., disulfide-linked PEI-based siRNA delivery showed an improved release of siRNA in the cytosol and resulted in improved accessibility of siRNA for the gene silencing complex. Once released from the NPs, the double-stranded siRNA is loaded into the RISC complex and it results in degradation of the mRNA.
In addition to bioreducibility, the polymer must also demonstrate low cytotoxicity. To evaluate this property, we performed a cell cytotoxicity study in A549 and H460 cells to compare the LD50 values of mPAE and PEI. Results suggest that the mPAE polymer exhibited less cytotoxicity (LD50 70 ± 6.2 and 65 ± 3.4 μg/ml) compared to the PEI polymer (LD50–30 ± 5.4 and 25 ± 5.2 μg/ml) in A549 and H460 cells, respectively Fig. 3. Cationic polymers like PEI have been extensively studied for their high gene transfection efficiency to deliver nucleic acids such as DNA [72–74], siRNA [75–78] miRNA [79, 80] both in vivo and in vitro. However, their use is limited due to a significant toxicity which causes significant cell damage to the normal cells due to various cellular mechanisms [81, 82]. In a recent study by Moghimi et al., PEI-induced membrane damage and initiated apoptosis in different cell lines including Jurkat T cells, umbilical vein endothelial cells, and THLE3 hepatocyte-like cells [83]. In addition, recently a study by Beyerle et al., showed that PEI-25 kDa caused severe toxicity at concentrations of 50 μg/ml and higher, rendering the cell viability levels below detection [53]. Thus, newer, less cytotoxic, biodegradable polymers which possess high gene transfection efficiency as well as better safety than polymers like PEI are needed. This study demonstrates the relatively less cytotoxicity of mPAE polymer to PEI polymer.
To effectively escape from the endosome, polymers must possess high buffering capacity in the range of pH 4.5 to pH 7.5 [41]. Polymers containing primary and secondary amines groups such as native PEI and mPAE exhibited considerable buffering capacity over a wide pH range of 4.5 to 7.5 due to a proton sponge effect [84]. The buffering capacity of mPAE polymers and PEI determined by the acid-base titration method, show that mPAE polymer possesses a higher proton capturing tendency compared to PEI alone Fig. 4. This may be due to the presence of the different ratios of primary, secondary and tertiary amine groups present in the mPAE and PEI polymer. In a similar study, Singh et al. concluded that the lower the amine groups present in the polymers, the lower the buffering capacity [41]. The presence of amine group along with other moieties contribute to the endo/lysosomal membrane destabilization [85]. Pinchon et al., clearly demonstrated in a study using confocal microscopy the protonated amine groups in PEI helped in membrane destabilization of the acidic components and deliver the DNA inside the cytosol. This study clearly demonstrated the effectiveness of the mPAE polymer’s buffering capacity compared to the PEI. This provides a rationale for the formulation of siRNA encapsulated mPAE-based NPs to deliver mTOR siRNA to the NSCLC cells for therapeutic effect by endosomal escape mechanism. The cationic NPs were reported to be internalized by direct translocation [63]. The cationic mPAE showed superior buffering capacity compared to PEI (Fig. 4, *p < 0.034). Thus, it is expected that the mPAE will exert the proton sponge effect mediated endosomal escape.
Various studies have shown that at certain weight ratios the positively charged cationic polymer encapsulates the siRNA and form stable NPs [86–88]. We formulated the NPs with mPAE polymer and mTOR siRNA to deliver to NSCLC cell lines (A549 and H460). The encapsulation of mTOR siRNA to form mTS-mPAE-NP is via binding and subsequently coating by the positively charged cationic mPAE to form nanoparticles. A recent study by Shen et al., showed that Fe3O4@SiO2 coated with cationic PEI resulted in the successful encapsulation of VEGF siRNA at weight ratios of 30:1 and higher [89]. In another study, Read et al. recently reported successful nucleic acid encapsulation by versatile reducible polycation (consisting of histidine and polylysine residues) (RPC) based NPs at various weight ratios (4–7% w/w) [90]. Agarose gel electrophoresis study confirmed that DNA was partially retained by RPC at (w/w) ratio of 2 and completely retained at w/w ratio of 4 and higher [20]. Similarly, in our study from gel electrophoresis of mTOR siRNA and mPAE polymer at different weight ratios, we found that at a weight ratio of 25:1 and higher the siRNA was being successfully encapsulated in the MP-NP (Fig. 5). Further, the % encapsulation efficiency experiment proved that at a concentration of 45:1 and higher we were able to successfully encapsulate 100% of the mTOR siRNA Table II. Further increasing the concentration of polymer will lead to unwanted polymer related cytotoxicity and thus it was necessary to use optimum amount of mPAE polymer to encapsulate the mTOR siRNA. Also by taking into consideration of the cytotoxicity and the entrapment efficiency, a weight ratio of 45:1 was selected for further experiments. Thus, the data indicates that mPAE polymer was capable of successfully binding and encapsulating mTOR siRNA and could be employed as a gene delivery carrier to NSCLC cells. In addition, the strong positive charged nanoparticles interact with negatively charged serum proteins leading to their rapid removal from the circulation by macrophages and the reticuloendothelial system (RES). The interaction can be avoided to certain extent by introducing a polyethylene glycol molecule on the surface of positively charged nanoparticle, which renders it a stealth property [91].
RNases degrade siRNA in biological systems [92], but nanoparticles can protect siRNA against degradation by these nucleases. In this study, we also showed that at an mPAE polymer to mTOR siRNA weight ratio of 45:1, the NPs successfully protect the mTOR siRNA against nuclease degradation. We found that at least 45% of the siRNA was released following the incubation with RNase and GSH for a period of 4 h each at 37°C (Fig. 6). The systemic administration of siRNA is required to achieve therapeutic effect for cancer treatment. However, several obstacles limit the systemic administration of therapeutic siRNA including the extremely low plasma half-life of siRNA due to degradation by serum nucleases [93]. The determination of stability of siRNA therapeutics in serum-containing media is important, mainly due to the reported interaction of siRNA therapeutics with blood before the distribution to target tissues [94]. Several studies have shown the potential of cationic polymer based NPs to successfully protect the therapeutic siRNA against serum nuclease-mediated degradation [88]. In a recent study by Shen et al., PEI-based nanoparticles were shown to successfully protect siRNA from degradation by serum enzymes at a weight ratio of 15:1 (polymer: siRNA) [88]. In another study, by Mokhtarieh et al., cationic lipids based NPs successfully protected the siRNA against RNase A degradation for a period of 4 h [95]. The circulating NPs must extravasate through the leakycapillaries to reach and accumulate in the tumor tissues [96, 97]. Therefore, in present study, we focused to generate the NPs with a size range of <150 nm. For all three formulations prepared, nanoparticle diameter remained between 111 nm and 125 nm, which fall in the appropriate size range for efficient cellular uptake [44, 98]. The surface charge also falls in the range for efficient cellular uptake (+27 mV) as reported previously with the cationic polymer based NPs.
Free siRNA cannot passively diffuse across the cell membrane due to its high molecular weight, hydrophilicity and an overall net negative charge [99]. In such cases, cationic polymers based NPs can facilitate their enhanced association with the anionic cell membrane and subsequent internalization by one of the several internalization mechanisms [99]. In this study, we found an increase in mTOR siRNA loaded mTS-mPAE-FITC-NPs cellular uptake with increasing incubation time up to 24 h (Fig. 8a and b). This mPAE-sIRNA NP system relies on the passive targeting to the cancer cells. As mentioned above, the mTS-mPAE-NPs prepared at a weight ratio of 45:1, (mPAE: mTOR siRNA) formed a nano-sized particle and has a positive surface charge. The small size (~115 nm) and a positive surface charge (~ +27 mV) is a very important characteristic for the efficient siRNA delivery into the cells as observed in this study. Several studies reported that small sized and positively charged carriers can facilitate better uptake by the cell in comparison with both large sized and negatively charged carriers [100–103]. The nano-sized particles can preferentially accumulate at the tumor site via the EPR effect and tend to circulate for longer times when injected via intravenous injection [103]. Various other studies demonstrated the ability of a cationic polymer to form nanoparticles and increase the cellular uptake. In a recent study, PAM-ABP polymer formed compact nanoparticles which showed enhanced cellular uptake of about 5 fold more than ABP at a weight ratio 5 [104]. Generally, cationic polymers are well known in assisting enhanced adsorptive endocytosis, membrane permeability and facilitating nuclear localization [105, 106]. This study further confirms the ability of the bioreducible polymer to escape the endosomes and release the nucleic acid in the cytoplasm.
To effectively perform their action of gene knockdown, nucleic acid (siRNA) must be delivered into the cancer cell and released into the cytoplasm. Transfection of mTOR siRNA using mTS-mPAE-NPs at a polymer concentration of 30 μg/ml and mTOR siRNA concentration of 50 nM produced mTOR gene silencing monitored by Western blotting two days’ post-transfection. Approximately 60 and 64% knockdown was observed with at 50 nM concentration of mTOR siRNA with mTS-mPAE-NPs when compared with control SS-MP-NPs in A549 and H460 cells, respectively (Fig. 9). Thus, these data confirm that mTS-mPAE-NPs suppressed the targeted mTOR gene specifically through RNAi mechanism. There may be two factors that might contribute to the mTOR gene silencing efficiency observed in this study. First the improved siRNA release due to the degradable disulfide linkage present in the mTS-mPAE-NPs. Secondly, degradation of the biodegradable cationic polymer in the presence of enzymes, resulting in deprotonation and loss of siRNA binding ability and thus release into the cytoplasm [107].
A significant role for mTOR is also to promote cell growth and proliferation by regulating protein synthesis in NSCLC cell lines [60]. Therefore, it can be conceived that mTOR knockdown may control or alter cell proliferation to some extent [108]. Suppression of cell proliferation is shown after A549 and H460 cell transfection with mTOR siRNA via mTS-mPAE-NPs (Fig. 9b and d). Nearly 31 and 32% decrease in cell viability in after mTS-mPAE-NPs treatment is observed compared to SS-mPAE-NPs transfections, supporting that mTOR function specifically, not cytotoxicity was responsible for the tumor cell growth inhibition. Several studies have reported delivery of mTOR siRNA to different cell lines (fibroblasts and human pulmonary arterial cells - PASMC) with the help of NPs which resulted in reduced cell proliferation [109, 110]. You et al., administered mTOR siRNA in a DNA nanotubes to the pulmonary artery smooth muscle cells and found that mTOR siRNA resulted in 47% reduction in cell viability after 48 h treatment [110]. In another study, Takahashi et al., treated fibroblasts with mTOR siRNA loaded branched cationic polyethyleneimine (bPEI) nanoparticles. The mTOR siRNA knockdown by the bPEI nanoparticles resulted in 70% inhibition of fibroblast proliferation [109]. We also observed significant cancer cell growth inhibition (in vitro) with higher concentrations of mTS-mPAE-NPs (mTOR siRNA 100 nM) compared to SS-mPAE-NPs, however, the negative control based NPs also showed significant cytotoxicity to the cells (*p < 0.05). Unfortunately, such higher concentrations of NPs resulted in significant cytotoxicity in both A549 and H460 cells. As indicated by the SRB assay results obtained using placebo NPs and SS-MP-NPs, concentrations higher than 30 μg/ml reduced the cell viability below 80% and are thus not suitable to load mTOR siRNA for further investigation. Consequently, the proposed mTS-mPAE NPs provided for an effective intracellular delivery of mTOR siRNA in the required compartments of cancer cells and delivered components were capable of performing their major functions – cell growth inhibition and silencing of targeted genes specifically in lung tumor cells.
The polymer mPAE possess several advantages compared to conventional bioreducible system. In addition to possessing the bioreducible property, they also undergoes proton sponge effect. This lead to a faster release of the siRNA, which is an advantage over several other bioreducible polymers.
CONCLUSIONS
In this study, we have developed and accessed the potential of bioreducible cationic polymer mPAE as an mTOR siRNA delivery carrier for lung cancer therapy. This targeted delivery of mTOR siRNA will allow the decreased probability of treatment failure and reduced adverse effects. The mPAE polymer was found to be less cytotoxic than the gold standard of cationic polymer based gene delivery carrier, i.e. PEI. The mPAE polymer formed nanoscale (114 nm), positively charged (+27 mV) particles when complexed with mTOR siRNA and the mTOR siRNA were prevented from degradation by the nuclease action. The release of mTOR siRNA was also accelerated in presence of 10 mM GSH. The mTS-mPAE-NPs showed inhibition of cell proliferation at an siRNA concentration of 50 nM. In addition, the mTS-mPAE-NPs showed increase cellular uptake due to cationic nature of the polymer and effective mTOR gene silencing. Our results demonstrated that the newer cationic mPAE polymer consisting of disulfide bonds is an effective mTOR siRNA carrier for lung cancer therapy applications. While our results are exciting, the nanoparticles were found to be toxic at higher concentrations limiting the ability to deliver higher concentrations of siRNA. Our results also provide an avenue for developing poly (ethylene glycol) (PEG)ylated receptor targeted NPs for lung cancer treatment using mTOR siRNA to achieve serum stability, targeting effect and limiting the cytotoxicity to the visceral organs.
Supplementary Material
ACKNOWLEDGMENTS AND DISCLOSURES
Dr. Gandhi acknowledges the Daniel K. Inouye College of Pharmacy, University of Hawaii at Hilo, Hilo, HI 96720, USA, for providing graduate assistantship. The authors also acknowledge the Department of Pharmaceutics and Drug Delivery, School of Pharmacy, University of Mississippi, University, MS, USA for providing start-up support to Dr. Chougule’s lab. Dr. Rege’s group is grateful to the NIH/NIGMS (Grant 1R01GM093229-01A1) and the Arizona Biomedical Research Commission (ABRC; Grant number ADHS14–082980) for partial financial support of this study. The authors report no conflicts of interest.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s11095-018-2460-z) contains supplementary material, which is available to authorized users.
REFERENCES
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. [DOI] [PubMed] [Google Scholar]
- 2.Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nichols L, Saunders R, Knollmann FD. Causes of death of patients with lung cancer. Arch Pathol Lab Med. 2012;136:1552–7. [DOI] [PubMed] [Google Scholar]
- 4.Key Statistics for Lung Cancer, in, 2017. http://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2017.html. Assessed 1 Jul 2017
- 5.Gadgeel SM, Ramalingam SS, Kalemkerian GP. Treatment of lung cancer. Radiol Clin N Am. 2012;50:961–74. [DOI] [PubMed] [Google Scholar]
- 6.Artal Cortés Á, Calera Urquizu L, Hernando Cubero J. Adjuvant chemotherapy in non-small cell lung cancer: state-of-the-art. Translational Lung Cancer Research. 2015;4:191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Links M, Brown R. Clinical relevance of the molecular mechanisms of resistance to anti-cancer drugs. Expert Rev Mol Med. 1999;1999:1–21. [DOI] [PubMed] [Google Scholar]
- 8.Moding EJ, Kastan MB, Kirsch DG. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov. 2013;12:526–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56:1649–59. [DOI] [PubMed] [Google Scholar]
- 10.Kozielski KL, Tzeng SY, De Mendoza BA, Green JJ. Bioreducible cationic polymer-based nanoparticles for efficient and environmentally triggered cytoplasmic siRNA delivery to primary human brain cancer cells. ACS Nano. 2014;8:3232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobashi Y, Watanabe Y, Miwa C, Suzuki S, Koyama S. Mammalian target of rapamycin: a central node of complex signaling cascades. Int J Clin Exp Pathol. 2011;4:476–95. [PMC free article] [PubMed] [Google Scholar]
- 12.Goschzik T, Gessi M, Denkhaus D, Pietsch T. PTEN mutations and activation of the PI3K/Akt/mTOR signaling pathway in papillary tumors of the pineal region. J Neuropathol Exp Neurol. 2014;73:747–51. [DOI] [PubMed] [Google Scholar]
- 13.Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12:177–86. [DOI] [PubMed] [Google Scholar]
- 14.Sadowski K, Kotulska K,Jóźwiak S. Management of side effects of mTOR inhibitors in tuberous sclerosis patients. Pharmacol Rep. 2016;68:536–42. [DOI] [PubMed] [Google Scholar]
- 15.Matsubara H, Sakakibara K, Kunimitsu T, Matsuoka H, Kato K, Oyachi N, et al. Non-small cell lung carcinoma therapy using mTOR-siRNA. Int J Clin Exp Pathol. 2012;5:119–25. [PMC free article] [PubMed] [Google Scholar]
- 16.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. [DOI] [PubMed] [Google Scholar]
- 17.Kuwabara PE, Coulson A. RNAi – prospects for a general technique for determining gene function. Parasitol Today. 2000;16(8): 347–49. [DOI] [PubMed] [Google Scholar]
- 18.Li SD, Chono S, Huang L. Efficient oncogene silencing and metastasis inhibition via systemic delivery of siRNA. Mol Ther. 2008;16:942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng L, Li J, Wang Y, Qian C, Chen Y, Zhang Q et al. Combination of siRNA-directed Kras oncogene silencing and arsenic-induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer. Nanomedicine. 2014;10: 463–72. [DOI] [PubMed] [Google Scholar]
- 20.Fresta M, Villari A, Puglisi G, Cavallaro G. 5-fluorouracil: various kinds of loaded liposomes: encapsulation efficiency, storage stability and fusogenic properties. Int J Pharm. 1993;99:145–56. [Google Scholar]
- 21.Youngren-Ortiz SR, Gandhi NS, Espana-Serrano L, Chougule MB. Aerosol delivery of siRNA to the lungs. Part 1: rationale for gene delivery systems. Kona. 2016;33:63–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee H, Kim Y-H. Nanobiomaterials for pharmaceutical and medical applications. Arch Pharm Res. 2014;37:1–3. [DOI] [PubMed] [Google Scholar]
- 23.Lee YS, Kim SW. Bioreducible polymers for therapeutic gene delivery. J Control Release. 2014;190:424–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kosuge H, Sherlock SP, Kitagawa T, Dash R, Robinson JT, Dai H, et al. Near infrared imaging and photothermal ablation of vascular inflammation using single-walled carbon nanotubes. J Am Heart Assoc. 2012;1:e002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Son S, Namgung R, Kim J, Singha K, Kim WJ. Bioreducible polymers for gene silencing and delivery. Acc Chem Res. 2012;45:1100–12. [DOI] [PubMed] [Google Scholar]
- 26.Kim T-i, Kim SW. Bioreducible polymers for gene delivery. React Funct Polym. 2011;71:344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barua S, Joshi A, Banerjee A, Matthews D, Sharfstein ST, Cramer SM, et al. Parallel synthesis and screening of polymers for nonviral gene delivery. Mol Pharm. 2008;6:86–97. [DOI] [PubMed] [Google Scholar]
- 28.Gosnell H, Kasman LM, Potta T, Vu L, Garrett-Mayer E, Rege K, et al. Polymer-enhanced delivery increases adenoviral gene expression in an orthotopic model of bladder cancer. J Control Release. 2014;176:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miryala B, Zhen Z, Potta T, Breneman CM, Rege K. Parallel synthesis and quantitative structure–activity relationship (QSAR) modeling of aminoglycoside-derived Lipopolymers for transgene expression. ACS Biomaterials Science & Engineering. 2015;1: 656–68. [DOI] [PubMed] [Google Scholar]
- 30.Miryala B, Feng Y, Omer A, Potta T, Rege K. Quaternization enhances the transgene expression efficacy of aminoglycoside-derived polymers. Int J Pharm. 2015;489:18–29. [DOI] [PubMed] [Google Scholar]
- 31.Ramos J, Rege K. Transgene delivery using poly(amino ether)-gold nanorod assemblies. Biotechnol Bioeng. 2012;109:1336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramos J, Rege K. Comparative investigation of polymeric and nanoparticle vehicles for transgene delivery. Nano LIFE. 2016;06: 1641001. [Google Scholar]
- 33.Ramos J, Rege K. Poly(aminoether)-gold Nanorod assemblies for shRNA plasmid-induced gene silencing. Mol Pharm. 2013;10: 4107–19. [DOI] [PubMed] [Google Scholar]
- 34.Huang H-C, Barua S, Kay DB, Rege K. Simultaneous enhancement of Photothermal stability and gene delivery efficacy of gold Nanorods using polyelectrolytes. ACS Nano. 2009;3:2941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Godeshala S, Nitiyanandan R, Thompson B, Goklany S, Nielsen DR, Rege K. Folate receptor-targeted aminoglycoside-derived polymers for transgene expression in cancer cells. Bioengineering & Translational Medicine. 2016;1:220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miryala B, Godeshala S, Grandhi TSP, Christensen MD, Tian Y, Rege K. Aminoglycoside-derived amphiphilic nanoparticles for molecular delivery. Colloids Surf B: Biointerfaces. 2016;146: 924–37. [DOI] [PubMed] [Google Scholar]
- 37.Kommareddy S, Amiji M. Preparation and evaluation of thiol-modified gelatin nanoparticles for intracellular DNA delivery in response to glutathione. Bioconjug Chem. 2005;16:1423–32. [DOI] [PubMed] [Google Scholar]
- 38.King TP, Li Y, Kochoumian L. Preparation of protein conjugates via intermolecular disulfide bond formation. Biochemistry. 1978;17:1499–506. [DOI] [PubMed] [Google Scholar]
- 39.Riddles PW, Blakeley RL, Zerner B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—a reexamination. Anal Biochem. 1979;94:75–81. [DOI] [PubMed] [Google Scholar]
- 40.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006; 1:1112–6. [DOI] [PubMed] [Google Scholar]
- 41.Singh B, Maharjan S, Park TE, Jiang T, Kang SK, Choi YJ, et al. Tuning the buffering capacity of polyethylenimine with glycerol molecules for efficient gene delivery: staying in or out of the endosomes. Macromol Biosci. 2015;15:622–35. [DOI] [PubMed] [Google Scholar]
- 42.Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther. 2013;21:149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alshamsan A. Nanoprecipitation is more efficient than emulsion solvent evaporation method to encapsulate cucurbitacin I in PLGA nanoparticles. Saudi Pharmaceutical Journal. 2014;22: 219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24:67–75. [DOI] [PubMed] [Google Scholar]
- 45.AccuBlue high sensitivity dsDNA quantification kit (Biotium), in, www.biotium.com, 2016.
- 46.Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–92. [DOI] [PubMed] [Google Scholar]
- 48.Fu Y, Kao WJ. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opinion on Drug Delivery. 2010;7:429–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hara K, Tsujimoto H, Huang CC, Kawashima Y, Ando R, Kusuoka O, et al. Ultrastructural and Immunohistochemical studies on uptake and distribution of FITC-conjugated PLGA nanoparticles administered Intratracheally in rats. J Toxicol Pathol. 2012;25:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu CW, Lin WJ. Polymeric nanoparticles conjugate a novel heptapeptide as an epidermal growth factor receptor-active targeting ligand for doxorubicin. Int J Nanomedicine. 2012;7: 4749–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2:3–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–7. [DOI] [PubMed] [Google Scholar]
- 53.Beyerle A, Irmler M, Beckers J, Kissel T, Stoeger T. Toxicity pathway focused gene expression profiling of PEI-based polymers for pulmonary applications. Mol Pharm. 2010;7:727–37. [DOI] [PubMed] [Google Scholar]
- 54.Liang W, Lam JKW. Endosomal escape pathways for non-viral nucleic acid delivery systems. In: Ceresa B, editor. Molecular regulation of endocytosis. Rijeka: InTech; 2012. p. Ch. 17. [Google Scholar]
- 55.Shi J, Schellinger JG, Johnson RN, Choi JL, Chou B, Anghel EL, et al. Influence of histidine incorporation on buffer capacity and gene transfection efficiency of HPMA-co-oligolysine brush polymers. Biomacromolecules. 2013;14:1961–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tseng WC, Tang CH, Fang TY. The role of dextran conjugation in transfection mediated by dextran-grafted polyethylenimine. The Journal of Gene Medicine. 2004;6:895–905. [DOI] [PubMed] [Google Scholar]
- 57.Shim MS, Kwon YJ. Acid-responsive linear polyethylenimine for efficient, specific, and biocompatible siRNA delivery. Bioconjug Chem. 2009;20:488–99. [DOI] [PubMed] [Google Scholar]
- 58.Mok H, Lee SH, Park JW, Park TG. Multimeric small interfering ribonucleic acid for highly efficient sequence-specific gene silencing. Nat Mater. 2010;9:272–8. [DOI] [PubMed] [Google Scholar]
- 59.Kim J, Kim SW, Kim WJ. PEI-g-PEG-RGD/small interference RNA polyplex-mediated silencing of vascular endothelial growth factor receptor and its potential as an anti-angiogenic tumor therapeutic strategy. Oligonucleotides. 2011;21:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ekman S, Wynes MW, Hirsch FR. The mTOR pathway in lung cancer and implications for therapy and biomarker analysis. J Thorac Oncol. 2012;7:947–53. [DOI] [PubMed] [Google Scholar]
- 61.Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90:197–207. [DOI] [PubMed] [Google Scholar]
- 62.Dang L, Liu J, Li F, Wang L, Li D, Guo B, et al. Targeted delivery Systems for Molecular Therapy in skeletal disorders. Int J Mol Sci. 2016;17:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin J, Alexander-Katz A. Cell membranes open “doors” for cationic nanoparticles/biomolecules: insights into uptake kinetics. ACS Nano. 2013;7:10799–808. [DOI] [PubMed] [Google Scholar]
- 64.Kommareddy S, Amiji M. Poly(ethylene glycol)-modified thiolated gelatin nanoparticles for glutathione-responsive intracellular DNA delivery. Nanomedicine. 2007;3:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee D, Lee YM, Jeong C, Lee J, Kim WJ. Bioreducible Guanidinylated Polyethylenimine for efficient gene delivery. ChemMedChem. 2014;9:2718–24. [DOI] [PubMed] [Google Scholar]
- 66.Wardman P, Dennis MF, Stratford MR, White J. Extracellular: intracellular and subcellular concentration gradients of thiols. Int J Radiat Oncol Biol Phys. 1992;22:751–4. [DOI] [PubMed] [Google Scholar]
- 67.Carilho Torrao RBD, Dias IHK, Bennett SJ, Dunston CR, Griffiths HR. Healthy ageing and depletion of intracellular glutathione influences T cell membrane thioredoxin-1 levels and cytokine secretion. Chemistry Central Journal. 2013;7:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Breunig M, Hozsa C, Lungwitz U, Watanabe K, Umeda I, Kato H, et al. Mechanistic investigation of poly(ethylene imine)-based siRNA delivery: disulfide bonds boost intracellular release of the cargo. J Control Release. 2008;130:57–63. [DOI] [PubMed] [Google Scholar]
- 69.Muthiah M, Che HL, Kalash S, Jo J, Choi SY, Kim WJ, et al. Formulation of glutathione responsive anti-proliferative nanoparticles from thiolated Akt1 siRNA and disulfide-crosslinked PEI for efficient anti-cancer gene therapy. Colloids Surf B: Biointerfaces. 2015;126:322–7. [DOI] [PubMed] [Google Scholar]
- 70.Shen Y, Wang J, Li Y, Tian Y, Sun H, Ammar O, et al. Co-delivery of siRNA and paclitaxel into cancer cells by hyaluronic acid modified redox-sensitive disulfide-crosslinked PLGA-PEI nanoparticles. RSC Adv. 2015;5:46464–79. [Google Scholar]
- 71.Tai Z, Wang X, Tian J, Gao Y, Zhang L, Yao C, et al. Biodegradable Stearylated peptide with internal disulfide bonds for efficient delivery of siRNA in vitro and in vivo. Biomacromolecules. 2015;16:1119–30. [DOI] [PubMed] [Google Scholar]
- 72.Erbacher P, Bettinger T, Brion E, Coll JL, Plank C, Behr JP, et al. Genuine DNA/polyethylenimine (PEI) complexes improve transfection properties and cell survival. J Drug Target. 2004;12:223–36. [DOI] [PubMed] [Google Scholar]
- 73.Longo PA, Kavran JM, Kim MS, Leahy DJ. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 2013;529:227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hou S, Ziebacz N, Wieczorek SA, Kalwarczyk E, Sashuk V, Kalwarczyk T, et al. Formation and structure of PEI/DNA complexes: quantitative analysis. Soft Matter. 2011;7:6967–72. [Google Scholar]
- 75.Hobel S, Aigner A. Polyethylenimine (PEI)/siRNA-mediated gene knockdown in vitro and in vivo. Methods Mol Biol. 2010;623:283–97. [DOI] [PubMed] [Google Scholar]
- 76.Knight M, Miller A, Liu Y, Scaria P, Woodle M, Ittiprasert W. Polyethyleneimine (PEI) mediated siRNA gene silencing in the Schistosoma mansoni snail host, Biomphalaria glabrata. PLoS Negl Trop Dis. 2011;5:e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang J-H, Tachibana Y, Kamata W, Mahara A, Harada-Shiba M, Yamaoka T. Liver-targeted siRNA delivery by polyethylenimine (PEI)-pullulan carrier. Bioorg Med Chem. 2010;18:3946–50. [DOI] [PubMed] [Google Scholar]
- 78.Lee S-Y, Huh MS, Lee S, Lee SJ, Chung H, Park JH, et al. Stability and cellular uptake of polymerized siRNA (poly-siRNA)/polyethylenimine (PEI) complexes for efficient gene silencing. J Control Release. 2010;141:339–46. [DOI] [PubMed] [Google Scholar]
- 79.Ibrahim AF, Weirauch U, Thomas M, Grunweller A, Hartmann RK, Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011;71:5214–24. [DOI] [PubMed] [Google Scholar]
- 80.Meneksedag-Erol D, Tang T, Uludag H. Probing the effect of miRNA on siRNA-PEI Polyplexes. J Phys Chem B. 2015;119: 5475–86. [DOI] [PubMed] [Google Scholar]
- 81.Florea BI, Meaney C, Junginger HE, Borchard G. Transfection efficiency and toxicity of polyethylenimine in differentiated Calu-3 and nondifferentiated COS-1 cell cultures. AAPS pharm Sci. 2002;4:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Breunig M, Lungwitz U, Liebl R, Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc Natl Acad Sci. 2007;104:14454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11: 990–5. [DOI] [PubMed] [Google Scholar]
- 84.Nimesh S, Goyal A, Pawar V, Jayaraman S, Kumar P, Chandra R, et al. Polyethylenimine nanoparticles as efficient transfecting agents for mammalian cells. J Control Release. 2006;110:457–68. [DOI] [PubMed] [Google Scholar]
- 85.Pichon C, Goncalves C, Midoux P. Histidine-rich peptides and polymers for nucleic acids delivery. Adv Drug Deliv Rev. 2001;53:75–94. [DOI] [PubMed] [Google Scholar]
- 86.Mehrotra S, Lee I, Chan C. Multilayer mediated forward and patterned siRNA transfection using linear-PEI at extended N/P ratios. Acta Biomater. 2009;5:147–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mishra S, Vaughn AD, Devore DI, Roth CM. Delivery of siRNA silencing Runx2 using a multifunctional polymer-lipid nanoparticle inhibits osteogenesis in a cell culture model of heterotopic ossification. Integr Biol (Camb). 2012;4:1498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shen J, Kim HC, Mu C, Gentile E, Mai J, Wolfram J, et al. Multifunctional gold nanorods for siRNA gene silencing and photothermal therapy. Advanced Healthcare Materials. 2014;3: 1629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li T, Shen X, Chen Y, Zhang C, Yan J, Yang H, et al. Polyetherimide-grafted Fe(3)O(4)@SiO2(2) nanoparticles as theranostic agents for simultaneous VEGF siRNA delivery and magnetic resonance cell imaging. Int J Nanomedicine. 2015;10: 4279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Read ML, Singh S, Ahmed Z, Stevenson M, Briggs SS, Oupicky D, et al. A versatile reducible polycation-based system for efficient delivery of a broad range of nucleic acids. Nucleic Acids Res. 2005;33:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yoshino K, Nakamura K, Terajima Y, Kurita A, Matsuzaki T, Yamashita K, et al. Comparative studies of irinotecan-loaded polyethylene glycol-modified liposomes prepared using different PEG-modification methods. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2012;1818:2901–7. [DOI] [PubMed] [Google Scholar]
- 92.Conti DS, Brewer D, Grashik J, Avasarala S, da Rocha SRP. Poly(amidoamine) dendrimer Nanocarriers and their aerosol formulations for siRNA delivery to the lung epithelium. Mol Pharm. 2014;11:1808–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ambardekar VV, Han H-Y, Varney ML, Vinogradov SV, Singh RK, Vetro JA. The modification of siRNA with 3′ cholesterol to increase nuclease protection and suppression of native mRNA by select siRNA Polyplexes. Biomaterials. 2011;32:1404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barnaby SN, Lee A, Mirkin CA. Probing the inherent stability of siRNA immobilized on nanoparticle constructs. Proc Natl Acad Sci U S A. 2014;111:9739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mokhtarieh AA, Cheong S, Kim S, Chung BH, Lee MK. Asymmetric liposome particles with highly efficient encapsulation of siRNA and without nonspecific cell penetration suitable for target-specific delivery. Biochim Biophys Acta Biomembr. 2012;1818:1633–41. [DOI] [PubMed] [Google Scholar]
- 96.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–6. [PubMed] [Google Scholar]
- 97.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–6. [PubMed] [Google Scholar]
- 98.Green JJ, Langer R, Anderson DG. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc Chem Res. 2008;41:749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dandekar P, Jain R, Keil M, Loretz B, Koch M, Wenz G, et al. Enhanced uptake and siRNA-mediated knockdown of a biologically relevant gene using cyclodextrin polyrotaxane. J Mater Chem B. 2015;3:2590–8. [DOI] [PubMed] [Google Scholar]
- 100.Park SC, Nam JP, Kim YM, Kim JH, Nah JW, Jang MK. Branched polyethylenimine-grafted-carboxymethyl chitosan co-polymer enhances the delivery of pDNA or siRNA in vitro and in vivo. Int J Nanomedicine. 2013;8:3663–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim TI, Ou M, Lee M, Kim SW. Arginine-grafted bioreducible poly(disulfide amine) for gene delivery systems. Biomaterials. 2009;30:658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen L, McCrate JM, Lee JC, Li H. The role of surface charge on the uptake and biocompatibility of hydroxyapatite nanoparticles with osteoblast cells. Nanotechnology. 2011;22:105708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nam HY, Nam K, Lee M, Kim SW, Bull DA. Dendrimer type bio-reducible polymer for efficient gene delivery. J Control Release. 2012;160:592–600. [DOI] [PubMed] [Google Scholar]
- 105.Morris VB, Sharma CP. Enhanced in-vitro transfection and biocompatibility of L-arginine modified oligo (–alkylaminosiloxanes)-graft-polyethylenimine. Biomaterials. 2010;31:8759–69. [DOI] [PubMed] [Google Scholar]
- 106.Lu S, Morris VB, Labhasetwar V. Codelivery of DNA and siRNA via arginine-rich PEI-based polyplexes. Mol Pharm. 2015;12: 621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Savic R, Luo L, Eisenberg A, Maysinger D. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science (New York, NY). 2003;300:615–8. [DOI] [PubMed] [Google Scholar]
- 108.Gridelli C, Maione P, Rossi A. The potential role of mTOR inhibitors in non-small cell lung Cancer. Oncologist. 2008;13: 139–47. [DOI] [PubMed] [Google Scholar]
- 109.Takahashi H, Wang Y, Grainger DW. Device-based local delivery of siRNA against mammalian target of rapamycin (mTOR) in a murine subcutaneous implant model to inhibit fibrous encapsulation.J Control Release. 2010;147:400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.You Z, Qian H, Wang C, He B, Yan J, Mao C, et al. Inhibition of DNA nanotube-conjugated mTOR siRNA on the growth of pulmonary arterial smooth muscle cells. Data in Brief. 2015;5:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.