

ABSTRACT
Constant technological advancement enabled the production of therapeutic monoclonal antibodies (mAbs) and will continue to contribute to their rapid expansion. Compared to small-molecule drugs, mAbs have favorable characteristics, but also more complex pharmacokinetics (PK), e.g., target-mediated nonlinear elimination and recycling by neonatal Fc-receptor. This review briefly discusses mAb biology, similarities and differences in PK processes across species and within human, and provides a detailed overview of allometric scaling approaches for translating mAb PK from preclinical species to human and extrapolating from adults to children. The approaches described here will remain vital in mAb drug development, although more data are needed, for example, from very young patients and mAbs with nonlinear PK, to allow for more confident conclusions and contribute to further growth of this field. Improving mAb PK predictions will facilitate better planning of (pediatric) clinical studies and enable progression toward the ultimate goal of expediting drug development.
KEYWORDS: monoclonal antibody, allometric scaling, allometry, interspecies scaling, translation, extrapolation, pre-clinical, pediatric, paediatric
Introduction
Technological advancement in the past few decades enabled engineering and manufacturing of therapeutic monoclonal antibodies (mAbs), which gained attention due to their favorable characteristics in comparison to typical small-molecule drugs. For example, mAbs exhibit high target specificity, and, due to their limited off-target toxicity, they are usually relatively safe.1 Therapeutic mAbs are a rapidly expanding drug class, and by the end of 2019 over 80 antibody-based therapeutics have been approved in the EU or US (mainly in cancer indications and for autoimmune diseases); with approximately 80 in late-stage clinical studies and at least 550 in early drug development.2 Although the pharmacokinetics (PK) and pharmacodynamics (PD) of mAbs in preclinical species and adults have been previously described, knowledge about the characteristics of mAbs in the pediatric population is lacking. The recent increase of pediatric drug development (also owing to recent regulations promoting pediatric development3–5) could help, as it has contributed to more pediatric mAb PK (and PD) data becoming available.1,6,7 Until recently, over 15 mAbs were licensed for use in the pediatric population, with palivizumab even approved in neonates from 35 gestation weeks onwards.8,9 Still, challenges, specific to pediatric trials, such as ethical concerns, scarcity of available patients, and operational/practical difficulties (e.g., slow and lengthy recruitment) may all contribute to a delay in mAb treatment being approved for pediatric diseases, with median delays around 6 years.8 To facilitate the design of the studies and so decrease the delay in the availability of treatments, a better understanding of the mAb PK, PD, and how they are related, is vital, especially for pediatric patients. Once these are well understood, the methodologies to describe and extrapolate their PK (and PD) can be further developed and evaluated. Then, allometric scaling approaches can be used more efficiently to facilitate drug development.10,11
Allometric scaling is a commonly used approach for translating animal PK knowledge to human (adults) and for extrapolating PK from adults to pediatrics. For small-molecule drugs, standard allometric approaches have been established, and mostly agreed on.7,12,13 Despite the relatively complex PK behavior of mAbs (e.g., they undergo target-mediated nonlinear elimination and recycling by neonatal Fc-receptor), several methodologies have been proposed and rather successfully used throughout the years for scaling from preclinical species to human,14 although there is still no agreement on how exactly to best scale. An approach that would also allow using fewer animals, accelerate early mAb development, and be more cost-effective, would be beneficial. For the pediatric population, experience and guidance on scaling approaches are lacking, although the field is developing fast.8,10,15 Additionally, there is limited information available on what allometric exponent to use, especially for the youngest pediatric patients, and whether to estimate it, or use a fixed value. Interestingly, there are also different values of exponents suggested and used in preclinical and clinical settings, and authors may refer to the same approach (e.g., simple allometry), but mean different things,16–20 which can cause confusion.
We aimed to provide an overview of the current state of mAbs PK scaling in both preclinical and clinical settings, and also summarize and compare the allometric scaling approaches in both settings head-to-head. Moreover, suggestions and recommendations for how allometric scaling could be improved/applied in the future are given, with knowledge gaps identified. We first introduce mAb PK and PD processes and their biology, then summarize different allometric scaling approaches used in preclinical and clinical mAb drug development, and conclude with a detailed overview of the current state in both. Despite the term children being strictly defined as pediatric subjects between ages of 2 to 11 years,21 hereafter this term will be used in the broader sense, i.e., the entire pediatric population.
Specific characteristics of mAbs
Structure and biology of mAbs
A mAb is a large protein molecule with a molecular weight of approximately 150 kDa, composed of two identical heavy and two light chains that are covalently linked by a disulfide bond, with an overall shape resembling the letter Y. Two antigen-binding fragments (Fabs) on the “head” of a mAb are responsible for binding to a specific antigen target. A crystallizable fragment (Fc) in the “tail” region binds to Fc receptors on effector cell types, and is responsible for disposition through the interaction with the neonatal Fc receptor (FcRn) on various cell types.22
In vitro generation of mAbs became possible after the hybridoma technology was invented in 1975,23 with the first mAbs being mainly murine antibodies. Recently, benefiting from the advancement in technologies that allow incorporation of human sequences, humanized and human mAbs have become the main focus, as they are less immunogenic compared to murine mAbs.24 Large-scale manufacturing of antibody therapeutics takes place primarily in bacterial or mammalian cells and yeast systems. Chinese hamster ovary cells are most often used as a platform for mass production of mAbs, due to their capacity to produce correctly folded and complex post-translational modification that is compatible to human.25 The mAb manufacturing process is complex and needs to be carefully controlled to ensure product consistency.
Recently, novel derivatives of mAbs have been developed, including bi- (BsAbs) and multi-specific antibodies (MsAbs),26 antibody fragments, antibody-drug conjugates (ADCs),27 and radiolabelled antibodies,28 with several of them approved for clinical use.29 The novel mAb formats were designed to increase drug specificity, and potentially lead to higher efficacy and increased safety. For example, an ADC comprises a highly specific mAb connected with a linker to a small molecule (e.g., potent cytotoxic drug), which is released once an ADC is internalized in a tumor, thereby killing the tumor cells with a much higher specificity.27 BsAbs/MsAbs have two/more antigen-binding regions that bind to two/more different targets, or different epitopes on the same target, which also increases their specificity and therapeutic potential.26
Pharmacokinetics of mAbs
Due to their large molecular size, relative polarity – resulting in low permeability through gastrointestinal mucosa – and high susceptibility to enzymatic degradation in the gastrointestinal tract, mAbs are typically administered via parental administration. While intravenous (i.v.) administration is the most common, subcutaneous (s.c.) and intramuscular (i.m.) administration are more convenient for patients and thus are also widely used.30 Following s.c. or i.m. injection, a mAb is taken up via the lymphatic system, with the bioavailability generally in the range 40–80%,31,32 and then slowly absorbed into the systemic circulation, i.e., the typical time to the maximal concentration (Tmax) is around 1 week.33 For mAbs given s.c., a mono-exponential decay is usually observed (as compared to a bi-exponential decay with the i.v. administration), as the first (fast) distribution phase is often difficult to observe.34
Large molecular size and hydrophilic character of mAbs not only affect their absorption, but also their distribution into tissues, limiting and slowing down their transfer across cell membranes.9,35 Antibodies distribute from the blood to the interstitial space of tissues mainly through convective transport, and not diffusion like small-molecule drugs. Due to their physicochemical characteristics, mAbs are mainly confined to the vascular and extracellular fluids, and therefore exhibit relatively small volumes of distribution,9,34 with the volumes of the central compartment (Vc) (in adults) for most mAbs around 2–3 L.9,14 The typical volume of the peripheral compartment (Vp) is often reported of the similar size as Vc,9,34 and the steady state volume of distribution (Vss) in the range of ~4-12 L.9,14 MAbs that have a target in the tissue compartment can have a larger volume of distribution, particularly if they exhibit high affinity for a specific target.35
MAbs predominantly undergo intracellular proteolytic catabolism via lysosomal degradation and are broken down into peptide fragments and amino acids.14 Nonspecific mAb uptake into cells can be via pinocytosis, or via endocytosis, following mAb interaction with the Fc receptors on the cell surface. This leads to a linear clearance, which is typically non-saturable at therapeutic concentrations. The interaction with FcRn “rescues” the mAb from lysosomal degradation (i.e., the mAb is recycled), thus prolonging its half-life.36 MAbs can also be taken up into cells by specific receptor-mediated endocytosis, which occurs after mAbs binds to their tissue antigen target (via the complimentary-determinant region of the Fabs); this pathway is nonlinear, as saturation of receptors decreases mAb clearance (CL), and is thus referred to as target-mediated drug disposition (TMDD).32,37 At high(er) mAb concentrations, when the target receptors are saturated, the process becomes linear. The disposition and elimination of a mAb through TMDD depends on the target expression level, mAb concentration, affinity of the antibody for the target receptor, and the mAb-target internalization rate.35 While nonlinear CL can be relatively variable across mAbs, linear CL is more comparable,8,9,34 with the serum half-life for a typical mAb of around 3 weeks.32,38
Unlike small molecules or small antibody fragments, mAbs are too large to be filtered via the kidney (50–60 kDa cutoff) and are therefore not eliminated in the urine, except in pathologic conditions.38 Similarly, antibodies do not undergo cytochrome P450 (CYP)-mediated hepatic metabolism due to their large size and lack of access to intracellular enzymes; therefore, they do not share elimination pathways with small-molecule drugs. Hence, drug–drug interactions for concomitant administration of mAbs and small molecules are usually not expected and also not assessed, unless mAbs are involved in the regulation of pro-inflammatory cytokines (which can affect the CYP system) or are ADCs.34,39 As nonspecific CL pathways (via proteolysis) are generally not saturable at therapeutic doses of mAbs, interactions with other therapeutic proteins are also not expected. CL of mAbs may be altered, if therapeutics that affect the receptor density or compete for mAb binding sites are co-administered.32
Body size (usually represented by body weight) is the most often reported patient characteristic that affects the PK of mAbs with linear CL, while other demographic features or organ function are seldom mentioned.32 For mAbs that (also) exhibit nonlinear elimination, the additional patient factors that can affect the CL are usually the expression of the target antigen/receptor density, receptor shedding, and the disease (status).32 The size of a mAb might also affect its CL, with larger mAbs having slower CL.40,41
Since mAbs are produced in biological systems, they can be antigens themselves and have the potential to induce immunogenicity.32 Although most antibody therapeutics currently in development are either humanized or human to reduce immunogenicity, other factors including amino acid modification, structural changes, mAb chemical properties, glycosylation pattern, product purity, and also patient characteristics or administration route can affect immunogenicity.42 Development of anti-drug antibodies (ADAs) can influence mAb PK by enhancing mAb CL (and thereby shortening serum half-life) or, conversely, prolonging serum half-life, thereby possibly affecting their efficacy and/or safety. Immunogenicity in animal models is typically not predictive of immunogenicity in the human situation, so animal PK data impacted by ADA should be carefully evaluated before translating to humans.43 One should also note that ADA results are assay-dependent and may not be comparable across different (clinical) studies.44,45
Newer mAb formats might have more complex PK compared to a traditional mAb. For example, PK properties of a BsAbs/MsAbs can be complicated by potential contribution of TMDD from various targets. After all targets are saturated by the circulating BsAb/MsAb, the Fc portion plays a dominant role in the distribution and elimination, and BsAb/MsAb will exhibit similar PK properties to a monospecific mAb.46 PK properties of an ADC can be similarly complex: as the mAb represents most of the weight of an ADC, its cellular Fc receptor binding and target binding play a major role in determining the PK of an ADC. Other factors, such as rate of cytotoxic release, stability of the linker, and the conjugation site of the cytotoxin, may also affect the PK and/or stability of an ADC.29
The main mAb characteristics, described above, compared to typical small-molecule drugs, are summarized in Table 1. A more detailed description of the absorption, distribution, metabolism, and elimination processes of mAbs is outside the scope of this review, but such information may be found elsewhere.14,32,34,35,38
Table 1.
A summary of pharmacokinetic and other characteristics of typical therapeutic monoclonal antibodies (mAbs), and key differences with small-molecule drugs14,32,33,35,47–49.
| mAbs (IgG type) | Small-molecule drugs | |
|---|---|---|
| Size | Large molecules, typically around 150,000 Da | <1000 Da |
| Chemical characteristics | Hydrophilic character | Hydrophilic or lipophilic |
| Production | In cell lines; identical copies are not possible, the structure is difficult to completely characterize | Chemical synthesis; identical copies obtained from batch to batch; chemical structure defined |
| Administration | Parenteral: usually intravenous (i.v.), but also intramuscular (i.m.) or subcutaneous (s.c.) | Mostly oral, but can also be parenteral/other |
| Absorption | Uptake (after s.c. or i.m. injection) via the lymphatic system, with ~40-80% bioavailability; Tmax ~1 week after s.c. or i.m. administration | Bioavailability (after non i.v. administration) – depends on drug’s chemical properties |
| Distribution into tissues | Slow and limited transfer across cell membranes;Convection primary mechanism for transport from vasculature to the interstitial space of tissues;Relatively small volume of distribution (V) (Vc = ~2-3 L, Vss = ~4-12 L);Do not bind to plasma proteins | Diffusion often main mechanism for transport;Can bind to tissues and exhibit large V;Can bind to plasma proteins |
| Metabolism/Elimination | Primarily by proteolytic catabolism via lysosomes: mAbs are metabolized to peptides and amino acids;Two major elimination pathways:
|
Most often metabolized by hepatic enzymes from the cytochrome P450 family (e.g., CYP3A4), and/or renally eliminated;Usually linear elimination, and relatively short half-life;Do not bind to FcRn |
| Main factors influencing elimination | Body size, target expression, disease (status), immunogenicity;Often not significant: gender, ethnicity, organ function | Body size and other patient/disease factors, organ function, age |
| Drug-drug interactions (DDI) | Metabolic DDIs rare and not expected, since mAbs do not share elimination pathways with small-molecule drugs; the nonspecific clearance pathways usually not saturable at therapeutic doses | Can occur often, especially with other small molecules/food |
| Immunogenicity | Can induce immune response and generation of anti-drug antibodies | Usually not immunogenic |
| Adverse events/safety | Relatively safe due to limited off-target toxicity | Off-target toxicities possible |
Animal specific differences and similarities to human mAb pharmacokinetics
Due to the sequence homology between non-human primates and human, most mAb-based therapeutics can bind to human and cynomolgus monkey targets similarly, i.e., their target-binding epitopes and in vitro binding affinities are comparable.14,50 In addition to target binding, binding to FcRn and tissue cross-reactivity are also comparable. Hence, the disposition and elimination pathway of mAbs are typically similar between monkeys and human,17,50 and mAbs with linear elimination in monkeys also often exhibit the same in humans.51 However, it should be noted that this may not be the case if, for example, the target concentration/tissue distribution between cynomolgus monkey and human differs, or if much higher doses are used in monkeys (as is common in toxicology studies). Most mAbs do not bind to rodent antigens well; thus, rodent species are less relevant for nonclinical PK studies compared to monkeys.14 Recently, it was found that human FcRn transgenic mice might be an exception, as their CL was in good correlation with human mAb CL;52 this is discussed below in the section “Allometric scaling in the context of preclinical mAb development”. An overview of typical body weights, brain weights, and maximum life potential of preclinical species, commonly used when scaling animal PK to human, is given in Table 2.
Table 2.
Preclinical species commonly used in pharmacokinetic translation to human with their typical body weights, brain weights and maximum life potential (MLP)53–55.
| Species | Body weight (kg) | Brain weight (g) | Maximum life potentiala (years) |
|---|---|---|---|
| Mouse | 0.020–0.025 | 0.36–0.42 | ~2.7–3 years |
| Rat | 0.25 | 1.74–1.8 | ~4.5–4.7 years |
| Cynomolgus monkey | 3.75 | 42.4–63 | ~18-24 years |
| Rhesus monkey | 5–6 | 76.8–90 | ~20-25 years |
| Human | 70 | 1400–1530 | ~90 years |
Pediatric mAb pharmacokinetics
The PK of mAbs is primarily affected by body weight,33,57 but additional age-associated changes in the physiological processes and the receptor system may further affect the disposition and elimination of mAbs. This is especially true for the youngest children, i.e., neonates and infants, whose characteristics, such as body composition, membrane permeability, and plasma protein concentration, are the most different to adult.9,33,58
Infants appear to exhibit a faster lymph flow compared to adults, which can lead to an increased rate of absorption after s.c. or i.m. administration of mAbs.6 This was observed by Robbie et al. studying palivizumab PK in adult and infant patients, who found that the typical absorption rate constant after i.m. administration was 0.373/day in adults and 1.01/day in 12.3-month infants.59 Similar behavior was also observed in other mAb studies.60 However, despite the differences in the absorption rate, the bioavailability was very similar between both populations, i.e., 0.73 in adults and 0.69 in infants.59 This could be due to the greater extent of pre-systemic mAb elimination in children,6 but more studies are needed to confirm this assumption.
Additionally, during growth and development, the relative total body water decreases from ~80% at birth until it reaches a plateau at approximately 60% at around 9 months of age.58 The fraction of the extracellular water is also higher in infants, and therefore they can appear to have a larger volume of distribution (V), normalized by body weight, compared to adults.6,9,58 Furthermore, extravasation of antibodies into tissues can occur more rapidly in infants because they have larger capillary surface area per volume unit and a larger proportion of “leaky” tissues, where capillary permeability is higher relative to the body size, compared to the adult population.6 However, it is not yet clear if the theoretically expected faster and greater mAb distribution in neonates and infants also results in clinically meaningful differences, compared to adults.33
Infants and young children might also have lower expression levels of FcRn, with relatively high endogenous IgG concentrations reported,6 additionally competing for FcRn binding. Together, this may lead to less mAb being “recycled” and increase the mAb elimination via intracellular lysosomal degradation,6 although this is also still to be observed in the clinic.15,33
Potential differences in the age-associated expression of mAb targets, binding affinity and turnover rates may also affect mAb distribution and CL.33 More clinical data and further studies are warranted to assess these hypotheses and facilitate a better description and prediction of mAb PK in pediatric patients.
In order to provide an overview of the allometric scaling landscape in preclinical and clinical mAb drug development, we screened the PubMed database (https://pubmed.ncbi.nlm.nih.gov/) in June 2020 to identify relevant publications involving monoclonal or therapeutic antibodies/antibody/protein(s), and any type of allometric scaling. Additionally, we manually checked the references of the identified papers for more potentially useful publications. Below we first describe all allometric scaling approaches used throughout the years for allometrically scaling preclinical and clinical mAb PK data, using non-compartmental or compartmental modeling techniques (also summarized in Table 3). Then we provide specific details on these approaches (such as, the discussion and evidence for different values of the allometric exponent) in preclinical and clinical settings (an overview is also given in Supplementary tables S1-S3).
Table 3.
An overview of the commonly used allometric scaling approaches for monoclonal antibodies (mAbs) with the typical values of the allometric weight exponents for clearance
| Scaling method | Description a | Comments | Preclinical setting | Clinical setting |
|---|---|---|---|---|
| Simple allometric scaling | Plot log clearance (CL) vs log weight (WT), fit linear regression and estimate the slope and the intercept to predict human CL | Requires at least 3 preclinical species; needs subjects/species with large WT range16,61 | The value of the estimated CL exponent often approximately 0.7516,41 | Not used |
| Simple allometric scaling with brain weight (BrWT) correction | Plot log CL*BrWT vs log WT plot, fit linear regression line and estimate the slope and the intercept, then divide by human BrWT to predict human CL | Requires at least 3 preclinical species; per the rule of exponents (ROE) brain weight correction should be applied when the estimated CL exponent >1.62 Some argue that no/other corrections might be better.17,41,63,64 | The BrWT correction factor usually <160 | Not used |
| Simplified allometry: Using a fixed exponent and single species (usually monkey) data | Predict human CL directly from the animal CL; the exponent is fixed | Advantageous as simple and only one preclinical species needed | The exponent is usually 0.75–0.8517,18,41,51,52 (or up to ~0.9 for mAbs with membrane-bound targets19,53) | Not used |
| Dedrick plot: elementary/complex Dedrick plot; species time-invariant method | Scale the concentration and time axes of the pharmacokinetic (PK) profile, then obtain CL estimates from the scaled PK curve | Provides the plasma PK profile in human; monkey typically used as the single animal species to scale from | Can use estimated exponents from simple allometry, but typically the exponents are fixed: “clearance” exponents commonly range 0.75–0.917,65,66 | Not used |
| Population PK(/pharmacodynamic (PD))analysis with allometric scaling | All concentration – time data analyzed simultaneously with a nonlinear mixed-effects model; possible to account for nonlinear CL. Allometric scaling of PK parameters included in the model. | PK profile and PK parameter estimates are obtained; in case of nonlinear CL, also parameters of the (approximated) target-mediated drug disposition (TMDD) model. PK parameters usually scaled to a typical 70-kg adult. | Currently not that frequently used, except in cases of nonlinear elimination; exponent for linear CL often ~0.75;51,67–70 PD parameters of the full/approximated TMDD models usually not scaled, except Vmax (exponent of 0.75) in the Michaelis-Menten (MM) model51,69–72 | A fixed exponent of 0.75 recommended and often used on linear CL;8,59,73 scaling PK in case of nonlinearities might be possible by allometrically scaling Vmax of the MM model, and keeping Km the same (although so far limited data)8,74 |
afor equations see text
Allometric scaling: description and types
Allometric scaling is a simple and commonly applied empirical approach used to describe the relationship between body size (most often represented by body weight) and a physiological variable, such as CL, or any other PK parameter.
The relationship between body weight and mAb CL is nonlinear, and described with a power function with some controversy around which value of the exponent should be applied. However, for allometrically scaling a mAb’s V, there is a general agreement: it scales linearly with weight, meaning that the exponent is 1.8,9,14,17,75 This has physiological rationale because mAbs mainly distribute to vascular and extracellular space, which scale linearly with weight in animals and human.17 Although most researchers use a fixed value of the exponent, some estimate it (often to around one) either with simpler allometric scaling approaches or with a population approach.62,76 However, if the population weight range is not large enough (e.g., only having data from adults), it might appear as if V does not scale linearly with body weight,8,77 which is why the exponent should rather be fixed to 1. Due to the reasons mentioned above, scaling of V is not the focus of this review, but rather allometric approaches for scaling CL.
Simple allometry
The simplest and most traditional allometric scaling approach is referred to as simple allometry. Although this empirical method was first used already in the 1930s,78 it was only applied to therapeutic proteins in 1991, when Mordenti et al. showed that the PK of biologics also follow the same nonlinear size-related physiologic relationship.16 Nowadays, it is still often used, albeit only for interspecies scaling. It is not applicable to clinical data due to relatively small weight ranges and often sparse PK data in the pediatric trials. To obtain a prediction of human CL with this approach, one needs to estimate the relationship between body weight (WT) and CL by fitting a linear regression line to a logarithm of CL versus logarithm of WT plot (Equation 1). The mAb CL values are usually obtained by performing non-compartmental analysis (NCA), and using at least 3 animal species.13,62 The slope of the linear regression (b) is the allometric exponent, and the intercept (a) is the coefficient (Equations 1 and 2).
| (Equation 1) |
| (Equation 2) |
The value of the exponent b is typically <1,16,78 meaning that CL increases slower than weight, i.e., heavier animals have relatively lower CL (per unit of body weight), compared to smaller animals. Sometimes, however, the exponent is estimated at >1,62,79 indicating that this method is not the most appropriate. To address this, but still use simple allometry, mathematical corrections to decrease and thus improve the mAb human CL prediction were suggested.
Simple allometry with correction factors
Correction factors, most commonly based on species’ brain weight or maximal life potential (MLP), are applied by simply multiplying the predicted CL with a constant.80 This constant, however, does not have a biologic rationale, and is not related to the mAb drug properties.14,80 Correction factors are usually estimated as <1 (the value depends on the (combination of) animal species used in the simple allometry80), so expectedly they decrease the value of CL, improving its prediction.61,80
To help determine when to use which correction, the rule of exponents (ROE)13 was proposed. According to the ROE, simple allometric scaling should be performed with at least 3 animal species, and the choice of the appropriate correction is guided by the value of the estimated allometric exponent. For mAbs, it was recommended that if the value of the exponent was below 1, no correction was required, and if it was above 1, one needed to factor in brain weight.62 The MLP correction was (initially) determined to be necessary only for small-molecule drugs (with weight exponents above 0.7 and below 1),62 supporting the hypothesis that it is related to CYP oxidation rate,41 therefore it will not be discussed in depth here. However, more recently, some researchers found that this correction could also be useful for mAbs,17,81 as detailed in the following preclinical section.
To correct for the brain weight, brain weight of the animals needs to be included on the log(CL) versus log(WT) plot by multiplying the values of CL with the corresponding animal brain weight (Table 2). Then, a prediction of human CL is obtained as shown in Equation 3. Human (adult) brain weight of around 1.5 kg is typically used (Table 2).
| (Equation 3) |
A similar principle is true for the MLP correction, only that MLP is used instead of the brain weight.
Recently, another correction factor was suggested for improving the human CL prediction of large molecules with simple allometry, i.e., antigen concentration (Concantigen) (Equation 4).63,64
| (Equation 4) |
As TMDD is driven by the antigen concentration, this approach was proposed as potentially useful for scaling CL of mAbs with nonlinear PK,63 but more work is needed to confirm this.
Using a fixed allometric scaling exponent and single species data
Using data from a single preclinical species and a fixed allometric weight exponent (Equation 5) to predict human mAb CL is an approach that is preferred and recommended by many,17,18,53 and was shown to improve mAb CL predictions, compared to the above mentioned approaches.17 Due to its simplicity, it is often referred to as “simplified allometry”.14,53 It is mostly only used for translation to human (and not for extrapolation to children), with the animal CL typically determined by the NCA approach. Non-human primates, such as (cynomolgus) monkeys are the suggested species to be used in this approach, due to the comparable PK processes to human.14
| (Equation 5) |
The main advantages of this approach are its simplicity, the need for only one preclinical species, and that it can rather accurately predict human mAb CL.17 Reducing the number of animals used is advantageous from the ethical perspective, and it saves resources, thereby shortening early drug development.17 Using rodents to extrapolate to humans, often as one of the species in the simple allometry, can also create bias in mAb PK predictions because of the differences in binding affinity to rodent vs human FcRn. Similarly to the simple scaling approach, this approach is also only appropriate for mAbs with linear elimination, as it can otherwise underpredict mAb CL at low doses, when the receptors are not yet saturated.82
Dedrick plots
In 1970 Dedrick et al. found that one can synchronize the PK curves of methotrexate in mouse, rat, monkey, dog and human by normalizing the chronological time to their species-specific physiological time; this was achieved by scaling the time axis with the weight exponent of 0.2583 (Equation 6). Boxenbaum and Ronfeld corroborated this by observing that species with shorter MLP (which is correlated with smaller weight) have faster “biologic clocks”, compared to larger species.84 For the PK curves to become superimposable, in addition to the time axis, the y axis also needs to be normalized (Equation 7).14,85,86 Allometric exponents c and d are usually referred to as the V and CL exponent, respectively (Equations 6–9). The applicability and predictive value of this approach was later also shown for mAbs by Ling et al. using data from several mAbs.53
| (Equation 6) |
| (Equation 7) |
Although in the Dedrick scaling approach the PK parameters are not directly scaled, but instead the concentration – time profile, body weight is still used for scaling; therefore, it is regarded as a variation of allometric scaling. The typical animal species used in this approach is (cynomolgus) monkey. This approach is, however, not used for pediatric scaling. Equations 8 and 9 summarize how to directly obtain human time and concentration from monkey data.14,87 Once the scaled human PK profile is obtained, the PK parameters are estimated either by NCA or by using a compartmental approach.
| (Equation 8) |
| (Equation 9) |
There are 4 main types of Dedrick plots, depending on how the allometric exponents are obtained and whether they are fixed or estimated:14,37,84 1) most commonly used Dedrick plot, or species-invariant time method, is an empirical approach where both exponents are fixed: allometric exponent c is fixed to 1, and d to a value <1; 2) elementary Dedrick plot: where the weight exponent c is fixed to 1, and the exponent d is based on an estimate from simple allometry using at least 3 preclinical species, with the time unit in this plot being called kallynochron; 3) complex Dedrick plot: where both allometric exponents (c and d) are estimated with simple allometry and the unit of time is apolysichron; this approach is less often used, as it is generally accepted that V is proportional to body weight; and 4) rarely used “advanced” Dedrick plot: where exponents c and d are obtained with simple allometry, but corrected for the MLP, and the unit of time is dienetichron.
The main advantage of the Dedrick plots over the simplified allometric scaling is that beside the prediction of the PK parameters, the entire (multiexponential) PK profile is also obtained, and can be plotted for different doses,14 provided that the mAb elimination is linear (or nonlinear CL is saturated). Whether this methodology can also be applied to mAbs with nonlinear PK is not yet clear.14
Population modeling approach
The approaches mentioned above are mainly based on obtaining CL and V estimates from the non-compartmental analysis, and are primarily used for preclinical translation of mAb PK to human. The nonlinear mixed-effects modeling (NLMEM) approach is the preferred approach for analyzing mAb clinical data;8,9,34 however, its use is also increasing for preclinical mAb data analysis.51,67,68,71,88–91 Unlike NCA, the population modeling approach is a compartmental analysis, with typically a 2-compartmental model used for i.v. administration, and 1-compartmental model for s.c. administration,34 but compartments have no physiological meaning. In addition to describing and quantifying the typical PK (and PD) parameters, the population approach can also provide an estimate of the variability from the central tendency in the population (i.e., variability between subjects), and so-called residual unexplained variability, i.e., variability coming from several sources, such as an assay.92 Covariates (i.e., subject-specific factors) that influence the PK can also be identified and their effect quantified, which can help explain some of the variability observed in mAb PK.9,47 Body weight is typically included by allometrically scaling the PK parameters, such as CL (Equation 10 exemplifies extrapolation to children), to account for the size differences between patients and/or populations, with b commonly fixed (details provided in the following sections).
| (Equation 10) |
The population approach can also deal with sparse data and data below the limit of quantification, all of which can otherwise affect PK parameter estimates.93 Furthermore, once an adequate model is established, simulation of PK profiles for different dosing/patient scenarios is enabled, facilitating planning of future (pediatric) studies;1,6,8,94,95 thereby this approach is also encouraged by the regulatory agencies.4
Unlike the NCA approach, NLMEM approach supports inclusion of linear (with typically 1 or 2 compartment PK model) and nonlinear elimination (with a mechanistic TMDD part), making it appropriate for analysis of data from mAbs that are eliminated via TMDD and exhibit nonlinear PK.35,96 If data from a rich-enough sampling scheme and wide concentration range are available (e.g., from several dosing levels), thus enabling reliable estimation of the TMDD model parameters, a full TMDD model is advocated14,97 (Figure 1). However, often the concentration range is insufficient, and/or the sampling schedule provides only limited information on the initial fast PK processes; therefore, the full TMDD model is frequently simplified as the quasi-equilibrium (QE), the quasi-steady state (QSS), or the Michaelis–Menten (MM) approximation.14,82,97,98 The QE approximation is often used,71,72,89,99 since the process of mAb binding with its receptor is generally much quicker compared to other processes (such as, mAb or mAb-target complex elimination). The equilibrium dissociation constant (Kd) is thus used instead of separately estimating the binding, or association (Kon), and dissociation (Koff) rates, with Kd = Koff/Kon.97,98 For mAbs with not insignificant mAb-target complex internalization rates (Kint), relative to Koff, the QE assumption may not be valid.97 In such cases the QSS approximation can be applied,96 where the free mAb, the target antigen and the mAb-target complex are assumed to reach steady-state concentrations much faster compared to other processes,90 and binding (i.e. Kon) is balanced by the sum of the dissociation (Koff) and internalization processes (Kint).97 The simplest TMDD approximation is the MM approach (Figure 2), which is suitable for systems where Kint is rapid enough for mAb elimination via the mAb-target complex to play a significant role, even at low target concentrations.97 This approximation can be appropriate for cases where, for example, the target antigen concentration is small, compared to the free mAb concentration, or when the targets are completely saturated.97 In the MM model, only two parameters need to be estimated: the maximum elimination rate (Vmax = Kint * Rtot, where Rtot is the total concentration of the target) and the MM constant (Km = (Kint + Koff)/Kon).97 An implicit assumption in the MM model is that Rtot is invariant with time (and thus Vmax is also constant97).
Figure 1.
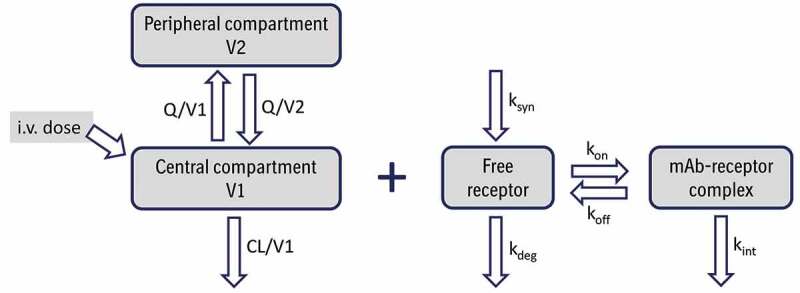
Schematic representation of a 2-compartment population PK model with a full target-mediated drug disposition model for an intravenously (i.v.) administered monoclonal antibody (mAb). CL and Q are central and inter-compartmental clearances, V1 and V2 are central and peripheral volumes, respectively, Ksyn is target production rate constant, Kdeg is target degradation rate constant, Kon is association/binding rate constant, Koff is dissociation rate constant, and Kint is mAb-target complex internalization rate constant. Adapted from.96
Figure 2.
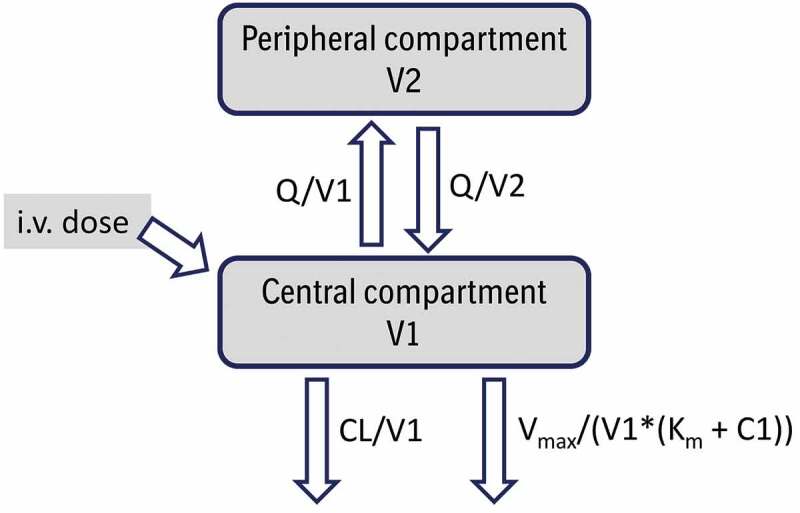
Schematic representation of a 2-compartment population PK model with nonlinear elimination described by Michaelis-Menten approximation for an intravenously (i.v.) administered monoclonal antibody. CL and Q are central and inter-compartmental clearances, V1 and V2 are central and peripheral volumes, respectively, Vmax is the maximum rate of nonlinear elimination and Km the Michaelis-Menten constant; C1 is concentration in the central compartment.96,97 Adapted from Wang et al.14
Antigen density, affinity/binding of the mAb to the target antigen, and turnover rates might differ in animals and humans, and might not be proportional to size; thus, these factors should be considered when translating PK and PD of mAbs to humans, or extrapolating to children, especially for mAbs with nonlinear CL.1,14,32
Mechanistic approaches for scaling mAb pharmacokinetics
Although fully mechanistic modeling approaches, such as, physiologically based PK (PBPK) modeling, are not within the scope of this review, a short summary is nevertheless provided below, since they are useful for mAb PK translation/extrapolation, and allometric scaling is often used within the PBPK models too.
While the classical compartmental models are mostly empirical, and based on in vivo PK data, (full) PBPK models include mechanistic mAb species- and drug-specific characteristics obtained from the literature and in vitro/vivo experiments.11,15 These mathematical models are thus quite complex, comprising several compartments that have physiological meaning (i.e., representing organs or tissues), and with many differential equations describing the movements (e.g., blood or lymph flows) between the compartments.15,63 This enables prediction of mAb concentrations and/or receptor occupancy in, for example, tumor tissues, determining the effects of drug or system factors on mAb PK, and exploring different administration routes.37,100,101 PBPK models may also be used to predict mAb in vivo PK behavior from only in vitro information,102 which can be beneficial in early drug development to, for example, exclude mAbs with unwanted PK characteristics. Additionally, PBPK models can account for the differences in pediatric and adult disease pathology,1,101 and e.g., FcRn ontogeny,101,103 and can therefore also be useful for predicting pediatric PK (and dose),8,10,95,101,104–106 especially for very young patients. However, despite first PBPK models for large molecules and mAbs being developed already in the 1980s and early 1990s, respectively,107,108 followed by several other (full and minimal) PBPK models, they mostly focus on translation of mAb PK from animals to humans.11,100,108–112 Thus far, this approach has not been commonly used for clinical mAb PK data and only limited (minimal) PBPK models are available for the pediatric population10,103,104 or established for pediatric mAb regulatory submissions.15 This may be partly due to the lack of information regarding the age-related changes in mAb PK and ontogeny of, for example, transporters, and would need to improve for these models to develop further, enable confident predictions,10,15,104 and be used to support regulatory submissions for pediatric patients.95,101
When allometrically scaling physiological parameters (such as CL, or rate constants) within PBPK models, often “typical” values of weight exponents (i.e., 0.75, or −0.25, respectively) are used,10,109,111,113 or estimated to approximately that value.103,112
While PBPK models can be valuable in some cases, they are complex and require large amounts of detailed data; therefore, their development (and calibration/validation) can be lengthy and difficult,11,14,37,51,101 likely contributing to them being used less commonly compared to empirical population PK models.15 The selection of the type of the model to use should depend on a specific question, for example, population modeling may be useful in the majority of cases (especially for clinical mAb PK data),101 but as PBPK models can provide additional, otherwise unavailable information, or include (receptor) ontogeny, they would be especially applicable in cases where this is required.
Allometric scaling in the context of preclinical mAb development
Interspecies scaling, or translation of mAb PK from animal species to human, has an ultimate goal to facilitate the design of first-in-human (FIH) studies. Knowing the differences and similarities in mAb PK (and PD) processes between these species is key for successful PK scaling.14
Preclinical studies usually include rodent and non-rodent species. Among species that are, or were, used for human PK prediction are mouse, rat, guinea pig, rabbit, sheep, dog, cynomolgus monkey, chimpanzee, baboon;14,53,62,114 with mouse, rat and cynomolgus monkey being the most common. When using only a single species, monkeys are recommended,14,87 due to the abovementioned reasons. Recently, however, transgenic mice that have human FcRn expressed are also gaining attention.115–117 For example, Tam et al. found that for 7 mAbs exhibiting mainly linear PK there was a strong correlation between the CL of human FcRn transgenic mouse (Tg32) and human CL.116 The potential of human FcRn Tg mouse model was further confirmed in a study of 27 mAbs by Avery et al., with Tg32 line proving especially valuable as CL from these mice correlated with human CL even better than CL from non-human primates.52 Although transgenic mice show potential, more work is needed, with more mAbs investigated to improve the understanding of these mice and be able to confirm their role in human mAb PK prediction. If confirmed useful, they could be used before the studies in monkeys are performed, to screen mAbs (based on their PK) or help to optimize studies in the monkey, thereby decreasing monkey usage in early research.52,116,117
Especially for mAbs with linear elimination, several relatively simple allometric approaches are widely used and were shown rather accurate for predicting human PK parameters from preclinical species. This could be because proteolytic enzymes involved in nonspecific mAb elimination are universal throughout animal species,41 or because mAbs are normally not eliminated via complex renal or hepatic pathways.37 Simple allometric approaches are, however, typically not appropriate for analyzing preclinical PK data from mAbs with nonlinear elimination, and more sophisticated approaches are required.9,14 Additional factors that can make animal-to-human mAb PK translation challenging include species-specific immunogenicity, FcRn binding, bioavailability after extravascular administration, antigen density and its binding to the mAb, and receptor occupancy. These should be taken into account by, for example, including them into a PKPD model.14,37,50 Modeling approaches may also be helpful in addressing other potential difficulties in preclinical translation, such as small sample sizes, inter-animal variability, and assay specificities. When scaling to human, most often a standard human weight of 70 kg is assumed, but sometimes also lower114,118 or higher weight68,77 is used.
A detailed overview of the scaling methods together with the values of the allometric exponents used in the preclinical mAb development is given below. Studies in which several mAbs (or large-molecule drugs) were used to compare or investigate allometric scaling approaches are summarized in Supplementary table S1. Sometimes somewhat higher values of the allometric CL exponent than the “typical” 0.75 are suggested for mAb interspecies scaling, i.e., the value that has been associated with interspecies scaling of basal metabolism,78 oxygen consumption and passive renal filtration, and is often used for scaling CL of small molecules. However, this exponent may also reflect the rate of various cellular processes critical for the intracellular uptake and catabolism of mAbs, as these processes are likely linked to basic cellular functions. Still, considering that a significant component of nonspecific fluid pinocytosis is mediated by the capillary endothelium, a higher allometric exponent of 0.85 could perhaps also be justified, as it has been associated with interspecies scaling of organ weights, and, via inference, capillary surface area per gram tissue due to strict spatial requirements for oxygen and nutrient diffusion. Since likely both of these factors contribute to the catabolism of antibodies, this might explain the range of observed CL exponents used across different preclinical studies.
Multiple species scaling approaches
After the research initially focused on small-molecule drugs, Mordenti et al. applied simple allometric scaling to preclinical and human data from 5 large-molecule drugs (6–98 kDa).16 The estimated weight exponents on CL were around 0.75 (mean: 0.75, median: 0.74, range: 0.65–0.84), and prediction fold-errors (with human data excluded) ranged 0.7–1.04,16 indicating that the allometric weight exponent on CL of ~0.75 is also appropriate for therapeutic proteins. Further work by Mahmood included 1 mAb targeting vascular endothelial growth factor (VEGF) and 14 other macro molecules, and, based on simple allometry (from at least 3 animal species), the exponents were estimated between 0.644 and 1.287 (0.795 for anti-VEGF).61 Later, additional research likewise showed that the estimated exponents are usually approximately 0.75 (based on n = 36 macromolecule drugs).41 Simple allometric scaling with CL information from ≥3 preclinical species has been commonly used with mostly satisfactory results,41,53 although sometimes human CL data are lacking, making the evaluation impossible.114,119
When allometric exponents are >1, simple allometry overpredicts mAb CL and thus ROE with brain weight correction was suggested, albeit this was initially based on somewhat limited data with only 4 therapeutic proteins having the allometric exponent >1.61 This approach was later explored in antibodies (n = 9, of which 6 had human CL values available for comparison, but not all were full-length mAbs), where CL exponents from simple allometry were 0.534–1.299.62 However, brain weight correction was again suggested based on a limited sample size (i.e., improved accuracy of CL prediction for 1 mAb).62 Deng et al. further found that the aforementioned ROE for mAbs can be useful, as it improved CL predictions for all 4 mAbs (with exponent >1), but also proposed the MLP correction when the weight exponents are between 0.71 and 1, although it only decreased the prediction errors in 2 of 5 mAbs.17 Additional work showed that MLP correction might be helpful, but it included only one mAb (anti-VEGF) and the estimated allometric exponent was outside the suggested range (1.03).81 Conversely, Huh et al. concluded that no MLP or brain weight correction was needed for large molecules.41 Using data from 4 mAbs, Wang et al. more recently found that correcting for antigen concentration outperformed simple allometry and both standard corrections, resulting in relatively accurate CL predictions (maximal percentage error 74%).63 However, it is not clear if ROE was applied or the corrections were used regardless of the value of the estimated weight exponent. The potential of the antigen concentration correction method was later also confirmed for another protein therapeutic.64
Single species scaling approaches
Despite the usefulness of the simple allometry (with corrections) being demonstrated (albeit often with rather limited datasets), single-species scaling with a fixed allometric exponent is often the preferable approach for translating mAb PK to human.17,18,53 However, the best value of the allometric weight exponent for mAbs (with linear elimination) has been historically controversial, ranging from 0.75 to 0.85 (or higher for mAbs with membrane-bound targets). For example, Ling et al. found that based on PK data from 14 mAbs, the exponent of 0.85 and 0.90 provided the best results for mAbs with soluble vs membrane-bound antigen targets (the latter at high doses), respectively.53 Oitate et al., however, investigated 24 mAbs and observed that a lower exponent of 0.79 is better for mAbs with soluble targets, although confirmed the trend of larger allometric exponents for mAbs with membrane-bound antigens (0.96).19 Not stratifying by the antigen target, Deng et al. found that allometric exponent of 0.85 provided the best CL predictions for the investigated mAbs (n = 13).17 Conversely, Wang et al. analyzed data from 12 mAbs and fusion proteins and reported that the exponent of 0.80 gave the most accurate predictions of human CL.18 Furthermore, using data from 10 mAbs with linear CL, Dong et al. compared the (standard) fixed exponent of 0.75 with exponents up to 0.90, and reported that, although the exponent of 0.85 or 0.90 decreased the CL prediction errors in some cases, in 4 of 10 mAbs it made the predictions worse, with errors up to 3-fold.51 Based on that, they concluded that there are no advantages of using an allometric exponent of 0.85/0.90 over 0.75 for scaling mAb CL data.51 This was confirmed by Avery et al. (n = 27 mAbs), who determined the best value of the exponent as 0.80, but observed that the exponent of 0.75 gave almost identical results for the training and test sets of mAbs (15/27 mAbs).52 Moreover, Huh et al. also corroborated that the optimal weight exponent (with the smallest average fold error) for 53 large-molecule drugs was between 0.75 and 0.80.41 If human FcRn transgenic mice are used in single-species scaling, Avery et al. determined (based on data from 15 mAbs) that a higher exponent than in monkeys might be needed (0.93).52
The second approach where only single species (i.e., monkey) data are used to predict human mAb PK is the Dedrick approach, which was shown to be able to reasonably predict human mAb PK by Deng et al. using data from 13 mAbs and a fixed “clearance” exponent.17 There is also no agreement on the value of the allometric exponent in this method, but it mostly ranges between 0.75 and 0.90.17,53,65,66 Oitate et al. investigated which type of Dedrick plot is superior, and determined that for the 12 studied mAbs the complex Dedrick plot was slightly better than the elementary, although the results were very similar.87
While the Dedrick plot and single-species allometric scaling both include one species, and can provide satisfactory results, the second is often preferred, as it is more straightforward.14,53,86 Additionally, non-monkey species might not be appropriate for the Dedrick scaling approach.120 Despite many researchers noting the predictive value of using single species approaches, they are mainly recommended for mAbs with linear PK,18 or mAbs with nonlinear PK at doses where the targets are saturated or nonlinear CL represents only a small part of the elimination.51,53 For mAbs with substantial TMDD, or at lower doses, and thus with nonlinear PK, approaches that take into account the TMDD process should be applied.14,69
Population modeling approach
Recently, pharmacometric (or, modeling and simulation) approaches are becoming more common in the preclinical drug development,67,68,91 especially for mAbs with nonlinear elimination.69,88–90 They can provide valuable information, and so support the design and/or dose selection for the clinical studies.
When analyzing preclinical PK data from mAbs with linear elimination, typically a 2-compartment PK model is used, with weight exponents on central CL and inter-compartmental clearance (Q) mostly around 0.75–0.85 (and around 1 on volume parameters). For example, Haraya et al. analyzed cynomolgus monkey and human data from 24 mAbs showing linear PK and determined that the optimal allometric scaling exponents for the PK parameters were 0.8 for CL, and 0.75 for Q.68 Betts et al. used a population PK approach to analyze data from 27 mAbs exhibiting linear PK and found similar values of the exponents: weight exponent on CL was estimated as 0.81 (95% confidence interval: 0.77–0.85) when only monkey and human data were used, and around 0.89 when data from transgenic mice were also included; the weight exponent on Q was between 0.57 and 0.67.67 Biliouris et al., however, scaled monkey PK data of an anti-blood dendritic cell antigen 2 (BDCA2) mAb to human and found that somewhat higher fixed exponents on CL and Q of 0.85 gave accurate PK predictions.91
An example of translational scaling for a mAb with nonlinear elimination is a full PK-TMDD model for an anti-activin receptor-like kinase 1 (ALK1) mAb by Luu et al.69 The authors estimated the monkey PK parameters by fitting a 2-compartment population PK model to monkey PK data, but used fixed (experimentally determined or obtained from literature) values for the monkey TMDD parameters, i.e., binding affinity (Kon, Koff), Kint, and target degradation rate (Kdeg). Apart from the PK, baseline receptor concentration (R0) was also estimated. To translate into human, (linear) monkey PK parameters were scaled allometrically with a fixed exponent of −0.25 on rate constants (i.e., equivalent to 0.75 on clearances and 1 on volumes), and Vc was assumed to equal human plasma volume.69 Human TMDD parameters were not scaled, but obtained experimentally, or from the literature (Kdeg). Human R0 was assumed to be the same as in monkeys. The model was found adequate, as the predicted human PK parameters (e.g., CL) and PK exposure metrics (i.e., area under the curve (AUC) and maximal concentration (Cmax)) were <2-fold different to the observed values.69 Although there are also other examples of the full TMDD model,88 its approximations are more common. In the QE approximation, an exponent of −0.25 is also often used for the PK rate constants.69,71,72,88,89,99 Vugmeyster et al. additionally compared the CL exponent of 0.85 versus 0.75 and found that it did not result in any significant changes of the dose projections; however, there were no human data available to confirm the human PK prediction.88 PD parameters of the QE approximation are also typically not scaled, but assumed the same across species or obtained experimentally,69,71,72,88,89,99 although there are some differences in the work published so far. For example, in the analysis by Park et al., Kd was obtained experimentally in vitro, but target-related parameters were scaled; the weight exponent used for the CL of the target was 0.75, and 0.85 for the rate of input of binding targets.89 The QSS approximation is less common, compared to the QE approximation, and, for example, Roepcke et al. applied the QSS approach to translate monkey PK to human; however, no observed human data were available for comparison,90 hence it is difficult to assess the model. The MM approach (i.e., the simplest TMDD approximation) was used, for example, by Dong et al. to scale monkey data from 6 mAbs with nonlinear PK to human.51 Again, the same typical allometric PK exponents were used as mentioned for the full TMDD model, and 0.75 also on Vmax.51 Based on in vitro similarities in target-binding characteristics or equal target protein sequences, Km was presumed the same for human as in monkeys,51 a common approach when using the MM model.70 When compared to the observed human data, the predictions of PK metrics, such as Cmax and AUC, were acceptable at mAb concentrations, where the targets were saturated, but, at low mAb concentrations, the predictions of the PK metrics proved challenging, with, for example, Cmaxs consistently being overpredicted (although the majority were still within the 2-fold error).51 This might indicate that a full TMDD model, or the QE/QSS approximation is needed.69,71 For the bispecific mAb MCLA-128, the MM model was also used (with allometric exponents of 0.75 on CL, Q and Vmax, and 1 on Vc and Vp), but no human data were available to be able to evaluate the predictions.70
Selection of starting and therapeutic human dose
Interspecies PK scaling (based on approaches outlined above) is used to determine the human efficacious dose and the starting mAb dose in clinical trials. To establish the human therapeutic dose for mAbs, human PK, predicted from preclinical species, is typically combined with PD information (e.g., biomarker modulation data, or tumor growth inhibition120,121) to determine a dose that provides a sustained desired pharmacological effect in human. Understanding and predicting the PKPD relationship is therefore key, and PKPD modeling can be a useful tool to facilitate this.14
When determining the starting dose for mAbs in FIH trials, the primary focus is on safety, followed by finding a dose that also produces the intended pharmacological activity (i.e., pharmacologically active dose). Relevant preclinical species need to be selected too, and an appropriate safety factor applied.122 Several approaches exist to determine a starting dose for FIH trials, including:123–126 1) the no observed adverse effect level (NOAEL), i.e., the highest dose that does not significantly increase adverse events in animal studies, compared to a placebo group; 2) the highest non-severely toxic dose (HNSTD), i.e., the highest dose that does not result in irreversible, lethal or life-threatening toxicities in preclinical studies; and 3) the minimal anticipated biological effect level (MABEL), i.e., the dose or exposure/concentration required to produce the minimal pharmacological activity (meaning, biological effect or its surrogate, such as receptor occupancy) in preclinical and clinical systems.124 PKPD (and PBPK) modeling is a useful approach to determine MABEL, as it can integrate preclinical pharmacology data (with or without animal toxicology information) and extrapolate it to human.124 It can also be used for deriving receptor occupancy.126 The MABEL approach was suggested to help prevent future tragic events (as occurred with the first administration of TGN1412),127,128 and should be used especially for high-risk medicinal products.127 It may also be helpful when mAb toxicology studies do not provide useful information, for example, when the species used in the toxicology studies are not pharmacologically relevant.129
These approaches can be used separately or in combination to determine the optimal starting dose.35,126 There is no consensus on the best approach; often it depends on the mAb mechanism of action and the available preclinical toxicology, pharmacology, and PK data.35,123 Several guidance documents from the Food and Drug Administration and European Medicines Agency also exist on this topic.122,125,130,131 And recently, information for specific formats and modalities such as ADCs,132 immune activating mAbs including immune checkpoint modulators,133 and CD3-targeting bispecific constructs,129 has been published. For the latter, it was suggested that a FIH dose corresponding to 10–30% of the pharmacological activity is acceptable, but the approaches based on receptor occupancy, HNSTD or NOAEL are not acceptable.129 This highlights the fact that approaches for determining the starting dose for FIH studies should be carefully evaluated and selected for each mAb.
Although for simplicity doses are often directly scaled from preclinical species to human, approaches that involve scaling mAb PK can improve prediction of doses for FIH trials, by taking into account potential differences between species, TMDD, and immunogenicity;37,62 therefore when possible, all relevant information should be included in an appropriate modeling approach.125 Traditionally, body-weight dosing was used for FIH studies involving mAbs,35 but lately, fixed dosing was suggested as appropriate for FIH studies.77 This is because the PK variability that body-weight dosing would reduce is moderate, compared to the observed PD variability.77 Additionally, a flat dosing approach is preferable from the practical perspective.77 However, for the trials beyond FIH studies, other dosing strategies might be more appropriate.
Allometric scaling in the context of clinical mAb development
The main goal of using allometry in clinical development is either to describe the pediatric and/or adult PK or to extrapolate PK information from adults to the pediatric patients, and so support the design of the pediatric clinical studies, help decide on a starting dose, or optimize a dosing/sampling regimen.3,4,21,95 It may also be used to waive certain unnecessary pediatric trials, and hence avoid ethical and practical concerns that can occur with pediatric trials.8,10,57 Allometric scaling approaches are thus regularly used for pediatric PK extrapolation, and are applicable to small-molecule drugs20,95,134 and also mAbs.1,7,15,47 Allometric weight scaling of clinical PK data is typically done within a population PK approach, due to the abovementioned advantages, namely the ability to analyze sparse data (common in adult trials beyond Phase 1 and typical for pediatric studies), and possibility to identify and include covariates that affect PK, facilitating (pediatric) dose selection.15,95
The most commonly included covariate in population PK models analyzing mAb clinical data is body weight (or sometimes body surface area (BSA)),1,33,75 with suggested reference adult weight of 70 kg,7,12 although this is not always used.8 However, despite weight also being the main demographic factor explaining the (size-related) PK differences between the adult and pediatric patients, age might additionally need to be included to describe the maturational differences, especially for the very young patients (i.e., infants and neonates). Age can be included with a sigmoidal/asymptotic maturation function, guided by the sum of gestational and postnatal age.7,59 Other covariates, typically identified in models analyzing pediatric (pooled with adult) data, can include ADA status/titer,9,75,135,136 which can affect efficacy and/or safety,35,75 albumin concentration, ethnicity, disease (status),9,136 and, sometimes, sex or co-medication.47,73,137 The numbers of ADA positive patients are often low, which might be the reason for frequently not identifying ADA status as a covariate with a significant effect on the mAb CL.137,138 Also, using ADA titer instead of ADA status might be preferred, as it was previously observed that only high ADA titers from ADA-positive patients affected CL, but not the low titers.59 More PK data from (premature) neonates and infants are needed to be able to make a conclusion regarding the factors affecting the PK in this population specifically.9,75 The same is true for mAbs with TMDD.1,9 However, it should also be noted that, while many covariates may be statistically significant and therefore included in the population PK model, some might not have a meaningful clinical relevance,139 meaning that they would not need to be taken into account when optimizing/adjusting a dosing regimen.1
Although PBPK modeling is currently not typically used for analyzing/extrapolating clinical mAb data,15 it might be desirable to use it, in addition to population PK modeling, when extrapolating to the very young pediatric patients (especially for first-in-child trials), as it may increase the confidence of PK predictions.8
Population modeling approach
Although allometric weight exponents are often estimated in adult population PK models, it was recently found that, to describe adult mAb PK data, the use of fixed standard values of the allometric exponents (i.e., 0.75 for clearances, 1 for volumes) is appropriate.8 This sounds reasonable, as due to the small weight range typically observed in the adult population, the values of the allometric exponents can sometimes be very low (<0.5),8,77,106,139 which is not in line with what is known about mAb PK and human biology and further supports fixing the value of the exponents. A recent review of population PK models for therapeutic mAbs (and a few fusion proteins) that are on the market, which included 75 PK models (50 of them with linear elimination) of 55 different drugs, developed based mainly on adult data (n = 55), but also pooled data from adult and pediatric patients (n = 12), and only pediatric patients (n = 8), showed that although the median allometric exponent for CL was 0.72, for Vc it was 0.68,8 i.e., much lower than expected. The low value of the exponent on V might be due to limited sampling around Cmax (since because of the long half-lives of mAbs sparse sampling was frequently used, with mostly only trough samples collected),8 not supporting its estimation. Especially when extrapolating to children, the exponent on V should be fixed to 1, rather than using adult estimates. This was confirmed using concentration-time data from 5 mAbs with linear PK, where using a fixed V exponent (to 1) resulted in better predictions of Cmax, compared to using the adult estimates.8
An agreement on how to analyze pediatric mAb data (or pooled adult and pediatric data), or how to extrapolate from adults to children, has been lacking, and both fixed and estimated values of the allometric exponents have been used – published PK models built using pooled pediatric and adult mAb PK data are discussed below, and summarized in Supplementary table S2. Supplementary table S3 provides an overview of models built on pediatric data only. Recently, use of fixed standard allometric values (i.e., 0.75 on CL and 1 on V parameters) was found appropriate, also for analyzing pediatric mAb data,8,33 although no definite conclusion could be made for pediatric patients below 6 years, as only limited data were available for these ages.8 However, studies that included data below that age also often used the same approach59,73,101 (detailed below).
Population PK analyses that included neonatal and infant mAb PK data are very rare. Additionally, there are no studies that would include a sufficient number of subjects below 2 years of age and subjects of other pediatric ages up to adults, which would cover the whole human weight range, and facilitate identification and/or evaluation of allometric exponents. Robbie et al. analyzed infant and adult palivizumab PK data, but there were no data between these age groups.59 In their 2-compartment model with linear elimination, body weight was included on all PK parameters with fixed (0.75, 1) allometric scaling, and additionally a postmenstrual-age-guided maturation function, to account for the age differences in patients ≤2 years was used.59 Body weight was the main covariate affecting the PK, but ADA titer (over 80), chronic lung disease, and race were also identified as statistically significant, although they did not explain much of the between-subject variability.59 Another study that looked at pediatric (n = 69 patients, 0.6–17 years) and adult mAb PK data (n = 18) found that the PK and covariate relationships were similar between both populations: allometric exponents on CL were estimated as 0.795 in pediatric and 0.808 in the adult population.136 In this study, age was not found to further affect the PK (once weight was included in the model), but only 2 patients were below 2 years of age.136
Other population models developed to analyze mAb PK data from adults and children mostly included older children and adolescents. Fasanmade et al. investigated infliximab data from 112 children (6–17 years old, median age 13 years) and 580 adult patients and found that weight (together with baseline serum albumin concentration, immune response status, and concurrent immunomodulator usage) was sufficient to describe infliximab PK, and age was not required.137 This result is not unexpected given the age of the pediatric patients in the study. In their model, CL per body weight was modeled, which was reflected in the (unusual) values of the allometric weight exponents: −0.313 on CL, −0.233 on Vc and −0.588 on Vp; no weight relationship was included on Q.137 Lowe et al. analyzed omalizumab nonlinear PK data from allergic asthma patients, with 787 patients below 12 years of age, and 1288 patients 12–79 years old.140 They found that CLs best scaled with the weight exponent of 0.914, and Vs with the exponent of 1.05.140 In a previous study of omalizumab (n = 1781 adolescent and adult patients), the weight exponent on free omalizumab CL was similarly estimated at 1.0 (and 0.828 on the V), but on the CL of the mAb-target complex it was 0.671.141 Further identified studies, including both pediatric and adult patients, mostly estimated the allometric weight exponent on CL, and found values of 0.695,142 0.595,143 0.698,144 and 0.813.145 In none of the cases age was found to further affect the PK of the mAbs, likely due to the lack of the very young patients. In studies that included pediatric patients and young adults (up to 19/21 years), the allometric exponent on CL parameters was either fixed to 0.7573,129 or estimated as 0.823 on CL.138 In both studies no additional effects of age on CL or V parameters were found after weight differences were accounted for, albeit both studies included only a limited number of very young pediatric patients.73,138 A population analysis of alemtuzumab PK in patients aged 0.2–19 years, however, used an allometric exponent that varied with the weight of the patient.146 The exponent decreased with increasing age (and thus, weight) and was estimated between 3.69 and 0.41.146 Studies that only included pediatric patients either used fixed values, most often of 0.75 on CL and 1 on V,101,147 or estimated the allometric exponents: e.g., 0.48 for CL and 0.904 for V,135 or 0.84 for CL and 0.720 for V.148
Some studies found that instead of using total body weight to describe the changes in pediatric mAb CL, ideal body weight might be a better body-size descriptor;149 or resting energy expenditure, which is thought to reflect the age dependence in mAb CL.147 Both these studies were, however, based on a very limited sample size, so the results should be further investigated. Another recent suggestion is to scale pediatric mAb CL by using different values of the allometric weight exponent (ranging between 1.2 and 0.75), depending on the pediatric age group.150 However, these cutoffs were arbitrary determined, and in some cases it can be difficult to evaluate the predictions, as often children of wide age groups were pooled together with only one summary CL value available.150
Pediatric population PK studies of mAbs with nonlinear elimination are uncommon. To describe, or extrapolate, the nonlinear PK of mAbs in, or to, pediatric patients, the MM approximation76,146 or a more mechanistic TMDD model/approximation describing the mAb-target binding and turnover dynamics are most often applied.138,140–142 When a comparison was performed, the MM approximation was shown to give similar results to the full TMDD model.106 Although more data from mAbs with nonlinear PK are needed, especially for infants and neonates, it is currently suggested that extrapolating nonlinear mAb CL to pediatrics may be possible by using the MM approximation and allometrically scaling the Vmax, but keeping Km the same as in adults.8,74 PBPK models might also be useful in the case of complex nonlinear mAb elimination.101
Pediatric dose selection
Determining a dose for first-in-child trials is challenging, as it needs to be safe, but also effective to not expose children to toxic or ineffective treatment. To achieve that, differences in mAb PK between adult and pediatric patients need to be appropriately considered. Considering that body weight is the main covariate affecting mAb PK, historically dose scaling from adult population to children was mainly done by taking body size into account using one of the following 4 methods:7,8,57,73 1) weight-based approach: adult dose is scaled linearly with weight (i.e., mg/kg dosing); 2) BSA-based approach: adult dose is scaled with BSA (i.e., mg/m2 dosing), which is similar to using an allometric exponent of 2/3; 3) a hybrid approach: children above some weight/age threshold get fixed adult dose, and for children below the threshold the adult dose is scaled with one of the aforementioned approaches; or 4) a tiered approach: a different fixed dose (or sometimes body-weight-adjusted dose33) is used for each body weight/age tier.
Although using allometric weight scaling to determine the dose accounts for the nonlinear changes with weight and could be appropriate, it is not used for the pediatric dosing for any of the approved mAbs, likely due to its presumed clinical impracticality.33 Knowing that mAb CL does not scale linearly with weight, the mg/kg dosing is likely to cause children with low weights/ages to receive a possibly ineffective dose, compared to the adult population.75 However, given the generally wide therapeutic index of mAbs, this approach may be adequate for older pediatric patients (e.g., above 40 kg), who often receive the same (fixed or mg/kg) dose as adults.57,151,152 This was confirmed by performing simulations of pediatric (≥40 kg) and adult mAb exposure, which showed clinically insignificant differences.8,57
Instead of scaling the dose directly, approaches (outlined above), such as population PK modeling with allometric scaling or PBPK modeling should be used to match safe and effective mAb exposures observed in the adult patients.1,6–8,57,94,101 Additionally, when designing a dosing regimen for younger pediatric patients (e.g., <2 years), accounting for maturation is warranted,6,9,33,75 albeit the evidence is currently still limited. The modeling approaches are also suggested by the regulatory authorities, and consequently often used nowadays in the pediatric studies.3,4,15 Furthermore, using modeling and simulation can be helpful in designing optimal sampling schemes, which is particularly important for pediatric patients, who can only provide a limited number of samples;153 and can be useful for mAbs with nonlinear elimination due to TMDD.106 Other body size descriptors, such as ideal body weight, might also be considered, to account for some variability between patients who are over- or underweight.149,154
Future perspectives and recommendations
The significance and role of pharmacometric modeling and simulation techniques in drug development will continue to grow, both for analyses of preclinical and clinical data. Knowing how to best scale preclinical mAb PK data to facilitate FIH studies, and also how to then use adult mAb PK data to help guide pediatric study design is thus of paramount importance. To help increase the confidence of the abovementioned approaches, we also identified some relevant knowledge gaps.
Although non-modeling approaches, such as simple allometry and single-species scaling using monkey mAb PK data are still commonly used (for preclinical mAb PK data) and might be appropriate in some cases, modeling (and simulation) approaches are slowly gaining recognition, as outlined above. Using modeling may not only enable better planning of FIH studies by, for example, accounting for TMDD and improving human PK predictions,69,91 but, as an initial (nonlinear or PKPD) model would already be developed, it may accelerate the overall drug development, also once clinical data become available. Additionally, modeling may support decreased sampling frequency, or the sample size of animals needed for a preclinical mAb PK evaluation. The value of the allometric exponent used preclinically is still a matter of discussion, and the rationale for choosing either value of the exponent has been historically often based on limited data, as mentioned. However, as there is no biological reason that a CL relationship with weight would be different within different weight (nonobese) human and animal species (at least monkeys), it would be reasonable to use the same exponent value (0.75) across all species (and apply a modeling approach). The allometric weight exponent of 0.75 on CL is within the range of values used preclinically (Table 3 and S1), and is the typical value used clinically.7,8,12 Using any exponent between 0.75 and 0.90 to scale preclinical (monkey) data to human also only changes the CL prediction <2-fold, considered nonsignificant (Figure 3). Additionally, when analyzing pediatric mAb data, it would be useful to apply the same reference body weight (i.e., 70 kg), as this would facilitate comparisons across studies, and different patient weights, including adult, thus better characterizing mAb PK and contributing to faster pediatric drug development.7,9 Approaches such as PBPK modeling are currently still rarely used for pediatric mAb data and would also benefit from further evaluation and more informative data, especially in the very young patients.15,104
Figure 3.
Relationship between clearance and body weight for allometric exponents ranging from 0.75 to 0.90. Human clearance and body weight were fixed to 0.2 L/day and 70 kg, respectively
In general, more clinical PK data from the youngest patients receiving mAb treatment should be collected, and if possible, throughout the whole pediatric age range, to be able to explore the allometric weight relationship throughout the entire human weight range, and gain valuable information on the effects of age on the mAb PK and/or PKPD relationship.8,15,104 Sampling schemes should be designed with slow elimination of mAbs in mind to be able to correctly describe CL and capture potential effects of changes in receptor expression after repeated dosing.53 Optimized PK sampling could also enable capturing potential TMDD,14 and detecting ADAs.1
Knowledge is also lacking for mAbs with nonlinear elimination, and target concentration should be quantified and explored (for a wide dose range) in animals and human to be able to determine its possible effect on the mAb systemic and target site exposure.14,69 Recording information with regards to disease status/severity, where feasible, might also be beneficial. Identifying possible differences between preclinical species and human in terms of target expression and binding would contribute to improved human dose prediction, especially at the lower (starting) doses.51 Collecting more/better data would also clarify whether TMDD differs between adult and pediatric patients,9,106 and would help confirm the appropriate scaling methods for such mAbs.
Most mAbs induce ADAs,35 and it is currently unclear whether immunogenicity differs between adult and pediatric population,1,33 so this should be investigated further. To facilitate the comparison of results across studies and eliminate the high dependency of results on the assay type, assays to detect and evaluate ADAs need to first be improved and standardized.1,44,45 As immune response cannot usually be extrapolated to human, ADA-positive animals are typically excluded (or only data points prior to development of ADAs are kept) when extrapolating preclinical PK to human.53 However, already preclinically, a modeling approach could alternatively be used to capture ADAs in animal species, such as monkeys, and learn about the possible effects of ADAs on the mAb PK,14,155 which could lead to improved safety and efficacy of mAbs in FIH clinical studies.
With most of the work focusing on scaling mAb PK, much is still unclear about how mAb PD scales, and reports involving PD scaling in large-molecule drugs are scarce.72,91,156 PD parameters, related to efficacy and sensitivity (i.e., maximal effect (Emax) and drug concentration that produces 50% of the maximal effect (EC50), respectively), are often considered similar in preclinical species and human,14,82 although EC50 can also be scaled to account for changes in the receptor number and density.91 To describe the changes in the physiological turnover processes, it might be appropriate to allometrically scale the turnover rate constant.14,82 The PD similarities/differences between adults and children are also not yet well characterized.1 With more information regarding the PD, and how PK and PD are related, PKPD models could be established and used for extrapolation, instead of only relying on PK exposure matching.
Although there is no apparent reason to expect otherwise, more studies are needed to confirm if the same scaling approaches as for “classical” mAbs can be used for the new formats, such as ADCs and bi-/multi-specific antibodies. Currently, there is a lack of agreement, i.e., while some found that allometric exponent on CL of ~0.75 is appropriate for ADCs,144,157,158 others found higher values (~1) to fit better.158–160 Similarly, different CL exponents are currently being used for bi-/multi-specific antibodies.70,161–163
The use and role of transgenic mice that have human FcRn expressed might also increase in the future, although they may not be appropriate for all purposes (e.g., if a mAb does not react with the target receptor in the mouse). Using rodents to (partially) replace now commonly used nonhuman primates to determine human PK would be more ethical, as it may decrease/eliminate the use of monkeys in research, it could help reduce the development time and costs, and make it more practical, as mice are generally easier to handle.52,117 Currently, however, data are lacking to be able to fully evaluate the appropriateness of these mice in preclinical mAb development.
Conclusions
With the constant advancement in mAb engineering and manufacturing technologies, mAb-based therapeutics will continue to grow and remain one of the most prevalent drug classes. The allometric scaling approaches discussed herein will remain critically important in the mAb drug development, specifically for designing and conducting FIH and pediatric clinical studies. The direct comparison of the use of allometry in preclinical and clinical settings presented here highlights the current status in both, enables identification of knowledge gaps and can enable the field to advance faster. In particular, more data from very young patients and mAbs with nonlinear elimination are needed to be able to make more confident conclusions. With more and more data becoming available, these approaches will continue to evolve, and we will likely see a growing recognition and expanding use of pharmacometric modeling approaches in both settings, especially in the preclinical research. This would help incorporate and increase knowledge regarding e.g., the TMDD, ADA effects, and age-related effects, and lead to better mAb PK predictions thereby improving FIH/pediatric dose determination, which will facilitate and ultimately accelerate drug development.
Supplementary Material
Funding Statement
No specific sources of funding were used to assist the preparation of this review.
Abbreviations
ADAanti-drug antibody
ADCantibody-drug conjugate
ALK1activin receptor-like kinase 1
AUCarea under the curve
BDCA2blood dendritic cell antigen 2
BrWTbrain weight
BSAbody surface area
BsAbsbispecific antibodies
CLclearance
Cmaxmaximal concentration
Concantigenantigen concentration
CYPcytochrome P450
DDIdrug-drug interaction
EC50drug concentration that produces 50% of the maximum effect
Emaxmaximum effect
FcRnneonatal Fc receptor
FIHfirst in human
HNSTDhighest non-severely toxic dose
i.m.intramuscular
i.v.intravenous
Kdequilibrium dissociation constant
Kdegtarget degradation rate constant
KintmAb-target internalization rate constant
KmMichaelis-Menten constant
Koffdissociation rate constant
Konbinding/association rate constant
Ksyntarget production rate constant
mAbtherapeutic monoclonal antibody
MABELminimal anticipated biological effect level
MLPmaximal life potential
MMMichaelis-Menten
MsAbsmultispecific antibodies
NCAnon-compartmental analysis
NLMEMnonlinear mixed-effects modelling
NOAELno observed adverse effect level
PBPKphysiologically based pharmacokinetic
PDpharmacodynamics
PKpharmacokinetics
Qinter-compartmental clearance
QEquasi-equilibrium
QSSquasi-steady state
R0baseline receptor concentration
Rtottotal receptor concentration
ROErule of exponents
s.c.subcutaneous
Tmaxtime to maximal concentration
TMDDtarget-mediated drug disposition
Vvolume of distribution
Vcvolume of the central compartment
VEGFvascular endothelial growth factor
Vmaxmaximum elimination rate
Vpvolume of the peripheral compartment
Vsssteady state volume of distribution
WTweight
Disclosure statement
The authors are employees of Boehringer Ingelheim but have no conflicts of interest that might be relevant to the content of this review.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Xu Z, Davis HM, Zhou H.. Rational development and utilization of antibody-based therapeutic proteins in pediatrics. Pharmacol Therapeut. 2013;137(2):225–22. doi: 10.1016/j.pharmthera.2012.10.005. PMID: 23092685. [DOI] [PubMed] [Google Scholar]
- 2.Kaplon H, Muralidharan M, Schneider Z, Reichert JM.. Antibodies to watch in 2020. Mabs. 2019;12:1703531. doi: 10.1080/19420862.2019.1703531. PMID: 31847708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . General clinical pharmacology considerations for pediatric studies for drugs and biological products. Guidance for Industry, 2014. [accessed02June 2021]. Available from:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-clinical-pharmacology-considerations-pediatric-studies-drugs-and-biological-products
- 4.EMA Committee for Medicinal Products for Human Use (CHMP) . Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. 2006. [accessed02June 2021]. Available from: https://www.ema.europa.eu/documents/scientific-guideline/guideline-role-pharmacokinetics-development-medicinal-products-paediatric-population_en.pdf
- 5.U. S. Food and Drug Administration, Department of Health and Human Services. Best Pharmaceuticals for Children Act and Pediatric Research Equity Act . Status Report to Congress, 2016. [accessed30Apr 2021]. Available from:https://www.fda.gov/science-research/pediatrics/best-pharmaceuticals-children-act-and-pediatric-research-equity-act
- 6.Malik P, Edginton A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin Drug Met. 2018;14(6):585–99. doi: 10.1080/17425255.2018.1482278. PMID: 29806953. [DOI] [PubMed] [Google Scholar]
- 7.Germovsek E, Barker CIS, Sharland M, Standing JF. Pharmacokinetic–pharmacodynamic modeling in pediatric drug development, and the importance of standardized scaling of clearance. Clin Pharmacokinet. 2019;58(1):39–52. doi: 10.1007/s40262-018-0659-0. PMID: 29675639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Langevin BA, Zhou H, Xu Z. Model‐aided adults‐to‐children pharmacokinetic extrapolation and empirical body size‐based dosing exploration for therapeutic monoclonal antibodies—is allometry a reasonable choice?. J Clin Pharmacol. 2020;60:1573–84. doi: 10.1002/jcph.1677. PMID: 32578225. [DOI] [PubMed] [Google Scholar]
- 9.Edlund H, Melin J, Parra-Guillen ZP, Kloft C. Pharmacokinetics and pharmacokinetic–pharmacodynamic relationships of monoclonal antibodies in children. Clin Pharmacokinet. 2015;54(1):35–80. doi: 10.1007/s40262-014-0208-4. PMID: 25516414. [DOI] [PubMed] [Google Scholar]
- 10.Pan X, Stader F, Abduljalil K, Gill KL, Johnson TN, Gardner I, Jamei M. Development and application of a physiologically-based pharmacokinetic model to predict the pharmacokinetics of therapeutic Proteins from full-term neonates to adolescents. Aaps J. 2020;22:76. doi: 10.1208/s12248-020-00460-1. PMID: 32449129. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Cao Y, Jusko WJ. Across-species scaling of monoclonal antibody pharmacokinetics using a minimal PBPK model. Pharm Res. 2015;32(10):3269–81. doi: 10.1007/s11095-015-1703-5. PMID: 25939552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci. 2013;102:2941–52. doi: 10.1002/jps.23574. PMID: 23650116. [DOI] [PubMed] [Google Scholar]
- 13.Mahmood I. Interspecies scaling: predicting clearance of anticancer drugs in humans. A comparative study of three different approaches using body weight or body surface area. Eur J Drug Metab Pharmacokinet. 1996;21(3):275–78. doi: 10.1007/BF03189726. PMID: 8980928. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Iyer S, Fielder PJ, Davis JD, Deng R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: comparative evaluation of prediction approaches in early drug development. Biopharm Drug Dispos. 2016;37:51–65. doi: 10.1002/bdd.1952. PMID: 25869767. [DOI] [PubMed] [Google Scholar]
- 15.Liu XI, Dallmann A, Wang YM, Green DJ, Burnham JM, Chiang B, Wu P, Sheng M, Lu K, Anker JN. Monoclonal antibodies and fc‐fusion Proteins for pediatric use: dosing, immunogenicity, and modeling and simulation in data submitted to the US food and drug administration. J Clin Pharmacol. 2019;59:1130–43. doi: 10.1002/jcph.1406. PMID: 30865317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mordenti J, Chen SA, Moore JA, Ferraiolo BL, Green JD. Interspecies scaling of clearance and volume of distribution data for five therapeutic Proteins. Pharm Res. 1991;8(11):1351–59. doi: 10.1023/a:1015836720294. PMID: 1798669. [DOI] [PubMed] [Google Scholar]
- 17.Deng R, Iyer S, Theil F-P, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data. Mabs. 2011;3(1):61–66. doi: 10.4161/mabs.3.1.13799. PMID: 20962582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W, Prueksaritanont T. Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm Drug Dispos. 2010;31:253–63. doi: 10.1002/bdd.708. PMID - 20437464. [DOI] [PubMed] [Google Scholar]
- 19.Oitate M, Masubuchi N, Ito T, Yabe Y, Karibe T, Aoki T, Murayama N, Kurihara A, Okudaira N, Izumi T. Prediction of human pharmacokinetics of therapeutic monoclonal antibodies from simple allometry of monkey data. Drug Metab Pharmacokinet. 2011;26(4):423–30. doi: 10.2133/dmpk.dmpk-11-rg-011. PMID: 21606605. [DOI] [PubMed] [Google Scholar]
- 20.Wu Q, Peters SA. A retrospective evaluation of allometry, population pharmacokinetics, and physiologically‐based pharmacokinetics for pediatric dosing using clearance as a surrogate. Cpt Pharmacometrics Syst Pharmacol. 2019;8:220–29. doi: 10.1002/psp4.12385. PMID: 30762304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.European Medicines Agency . ICH topic E 11 clinical investigation of medicinal products in the paediatric population. 2001. [accessed02June 2021]. Available from:https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-1.pdf
- 22.Roopenian DC, Akilesh S. FcRn: the neonatal fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–25. doi: 10.1038/nri2155. PMID: 17703228. [DOI] [PubMed] [Google Scholar]
- 23.Freysdottir J. Production of monoclonal antibodies. Methods in Molecular Biology. 2000:267–79. doi: 10.1385/1-59259-076-4:267. PMID: 21337095. [DOI] [PubMed] [Google Scholar]
- 24.Lu R-M, Hwang Y-C, Liu IJ, Lee -C-C, Tsai H-Z, Li H-J, Wu H-C. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. PMID: 31894001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kildegaard HF, Baycin-Hizal D, Lewis NE, Betenbaugh MJ. The emerging CHO systems biology era: harnessing the ‘omics revolution for biotechnology. Curr Opin Biotech. 2013;24(6):1102–07. doi: 10.1016/j.copbio.2013.02.007. PMID: 23523260. [DOI] [PubMed] [Google Scholar]
- 26.Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Dev Ther. 2018;12:195–208. doi: 10.2147/dddt.s151282. PMID: 29403265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody–drug conjugates: the last decade. Pharm. 2020;13:245. doi: 10.3390/ph13090245. PMID: 32937862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayne MC, Illidge TM. Antibody therapy of lymphoma. Expert Opin Pharmaco. 2005;2:953–61. doi: 10.1517/14656566.2.6.953. PMID: 11585011. [DOI] [PubMed] [Google Scholar]
- 29.Paci A, Desnoyer A, Delahousse J, Blondel L, Maritaz C, Chaput N, Mir O, Broutin S. Pharmacokinetic/pharmacodynamic relationship of therapeutic monoclonal antibodies used in oncology: part 1, monoclonal antibodies, antibody-drug conjugates and bispecific T-cell engagers. Eur J Cancer. 2020;128:107–18. doi: 10.1016/j.ejca.2020.01.005. PMID: 32037061. [DOI] [PubMed] [Google Scholar]
- 30.Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. Biodrugs. 2018;32(5):425–40. doi: 10.1007/s40259-018-0295-0. PMID: 30043229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–68. doi: 10.1002/jps.20178. PMID: 15389672. [DOI] [PubMed] [Google Scholar]
- 32.Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies. Biodrugs. 2010;24(1):23–39. doi: 10.2165/11530560-000000000-00000. PMID: 20055530. [DOI] [PubMed] [Google Scholar]
- 33.Temrikar ZH, Suryawanshi S, Meibohm B. Pharmacokinetics and clinical pharmacology of monoclonal antibodies in pediatric patients. Pediatr Drugs. 2020;22(2):199–216. doi: 10.1007/s40272-020-00382-7. PMID: 32052309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. doi: 10.2165/11531280-000000000-00000. PMID: 20608753. [DOI] [PubMed] [Google Scholar]
- 35.Liu L. Pharmacokinetics of monoclonal antibodies and fc-fusion proteins. Protein Cell. 2018;9(1):15–32. doi: 10.1007/s13238-017-0408-4. PMID: 28421387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuo TT, Aveson VG. Neonatal fc receptor and IgG-based therapeutics. Mabs. 2011;3:422–30. doi: 10.4161/mabs.3.5.16983. PMID: 22048693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma V, McNeill JH. To scale or not to scale: the principles of dose extrapolation. Brit J Pharmacol. 2009;157(6):907–21. doi: 10.1111/j.1476-5381.2009.00267.x. PMID: 19508398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. Cpt Pharmacometrics Syst Pharmacol. 2017;6:576–88. doi: 10.1002/psp4.12224. PMID: 28653357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) . Drug-drug interaction assessment for therapeutic Proteins guidance for industry. 2020. [accessed22April 2021]. Available from:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/drug-drug-interaction-assessment-therapeutic-proteins-guidance-industry
- 40.Li Z, Krippendorff B-F, Shah DK. Influence of molecular size on the clearance of antibody fragments. Pharm Res. 2017;34(10):2131–41. doi: 10.1007/s11095-017-2219-y. PMID: 28681164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huh Y, Smith DE, Feng MR. Interspecies scaling and prediction of human clearance: comparison of small- and macro-molecule drugs. Xenobiotica. 2011;41(11):972–87. doi: 10.3109/00498254.2011.598582. PMID: 21892879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deehan M, Garcês S, Kramer D, Baker MP, Rat D, Roettger Y, Kromminga A. Managing unwanted immunogenicity of biologicals. Autoimmun Rev. 2015;14(7):569–74. doi: 10.1016/j.autrev.2015.02.007. PMID: 25742758. [DOI] [PubMed] [Google Scholar]
- 43.Swanson SJ, Bussiere J. Immunogenicity assessment in non-clinical studies. Curr Opin Microbiol. 2012;15(3):337–47. doi: 10.1016/j.mib.2012.05.015. PMID: 22770538. [DOI] [PubMed] [Google Scholar]
- 44.Gress K, Bass JA, Funk RS, Morrow RP, Hasenkamp R, Shakhnovich V. Facing real-world challenges of immunogenicity in pediatric inflammatory bowel disease. Front Immunol. 2020;11:1148. doi: 10.3389/fimmu.2020.01148. PMID: 32582213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strand V, Balsa A, Al-Saleh J, Barile-Fabris L, Horiuchi T, Takeuchi T, Lula S, Hawes C, Kola B, Marshall L. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. Biodrugs. 2017;31(4):299–316. doi: 10.1007/s40259-017-0231-8. PMID: 28612180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uchida N, Sambe T, Yoneyama K, Fukazawa N, Kawanishi T, Kobayashi S, Shima M. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633–41. doi: 10.1182/blood-2015-06-650226. PMID: 26626991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59. doi: 10.2165/11535960-000000000-00000. PMID: 20818831. [DOI] [PubMed] [Google Scholar]
- 48.Jang GR, Harris RZ, Lau DT. Pharmacokinetics and its role in small molecule drug discovery research. Medical Care Research and Review. 2001;5:382–96. doi: 10.1002/med.1015. PMID: 11579439. [DOI] [PubMed] [Google Scholar]
- 49.Ferri N, Bellosta S, Baldessin L, Boccia D, Racagni G, Corsini A. Pharmacokinetics interactions of monoclonal antibodies. Pharmacol Res. 2016;111:592–99. doi: 10.1016/j.phrs.2016.07.015. PMID: 27438459. [DOI] [PubMed] [Google Scholar]
- 50.Deng R, Jin F, Prabhu S, Iyer S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development?. Expert Opin Drug Met. 2012;8(2):141–60. doi: 10.1517/17425255.2012.643868. PMID: 22248267. [DOI] [PubMed] [Google Scholar]
- 51.Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu C-P, Stouch BJ, Hurh E, Gibbs M. Quantitative prediction of human pharmacokinetics for monoclonal antibodies. Clin Pharmacokinet. 2011;50(2):131–42. doi: 10.2165/11537430-000000000-00000. PMID: 21241072. [DOI] [PubMed] [Google Scholar]
- 52.Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan -Y-Y, Dowty ME, Zhang M, Zhang Y, Cheng A. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. Mabs. 2016;8(6):1–15. doi: 10.1080/19420862.2016.1193660. PMID: 27232760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ling J, Zhou H, Jiao Q, Davis HM. Interspecies scaling of therapeutic monoclonal antibodies: initial look. J Clin Pharmacol. 2009;49(12):1382–402. doi: 10.1177/0091270009337134. PMID: 19837907. [DOI] [PubMed] [Google Scholar]
- 54.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;7:1093–95. doi: 10.1023/a:1018943613122. PMID: 8378254. [DOI] [PubMed] [Google Scholar]
- 55.Boxenbaum H, Fertig JB. Scaling of antipyrine intrinsic clearance of unbound drug in 15 mammalian species. Eur J Drug Metab Ph. 1984;9:177–83. doi: 10.1007/bf03189622. PMID: 6745307. [DOI] [PubMed] [Google Scholar]
- 56.Sacher GA.Relationship of lifespan to brain weight and body weight in mammals. In: Wolstenholme GEW, O’Connor M, editors. Ciba Foundation Colloquia on Ageing. Vol. 5. The Lifespan of Animals. . Boston, London, UK: Little Brown and Co; 1959. p. 115–41. doi: 10.1002/9780470715253.ch9. [DOI] [Google Scholar]
- 57.Yang N, Xu MC, Yao Z. Evaluation of weight thresholds for pediatric patients to use adult dosage of therapeutic monoclonal antibodies. J Clin Pharmacol. 2019;59:1309–18. doi: 10.1002/jcph.1434. PMID: 31050000. [DOI] [PubMed] [Google Scholar]
- 58.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology - drug disposition, action, and therapy in infants and children. N Engl J Med. 2003:1157–67. doi: 10.1056/NEJMra035092. PMID: 13679531. [DOI] [PubMed] [Google Scholar]
- 59.Robbie GJ, Zhao L, Mondick J, Losonsky G, Roskos LK. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob Agents Chemother. 2012;56(9):4927–36. doi: 10.1128/aac.06446-11. PMID: 22802243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ternant D, Paintaud G, Trachtman H, Gipson DS, Joy MS. A possible influence of age on absorption and elimination of adalimumab in focal segmental glomerulosclerosis (FSGS). Eur J Clin Pharmacol. 2016;72:253–55. doi: 10.1007/s00228-015-1973-1. PMID: 26521258. [DOI] [PubMed] [Google Scholar]
- 61.Mahmood I. Interspecies scaling of protein drugs: prediction of clearance from animals to humans. J Pharm Sci. 2004;93(1):177–85. doi: 10.1002/jps.10531. PMID: 14648647. [DOI] [PubMed] [Google Scholar]
- 62.Mahmood I. Pharmacokinetic allometric scaling of antibodies: application to the first‐in‐human dose estimation. J Pharm Sci. 2009;98:3850–61. doi: 10.1002/jps.21682. PMID: 19177515. [DOI] [PubMed] [Google Scholar]
- 63.Wang L, Qiang W, Cheng Z. Allometric scaling of therapeutic monoclonal antibodies using antigen concentration as a correction factor: application to the human clearance prediction. J Pharm Sci. 2016;105(3):1335–40. doi: 10.1016/j.xphs.2015.12.021. PMID: 26886347. [DOI] [PubMed] [Google Scholar]
- 64.Deng Y, Hu L, Qiang W, Cheng Z, Wang L, Wang X. TNF‐α level affects etanercept clearance: TNF‐α concentration as a new correction factor of allometric scaling to predict individual etanercept clearances in patients with ankylosing spondylitis. Clin Exp Pharmacol P. 2018;45:643–51. doi: 10.1111/1440-1681.12924. PMID: 29436715. [DOI] [PubMed] [Google Scholar]
- 65.Xiang H, Bender BC, Reyes AE, Merchant M, Jumbe NLS, Romero M, Davancaze T, Nijem I, Mai E, Young J. Onartuzumab (MetMAb): using nonclinical pharmacokinetic and concentration–effect data to support clinical development. Clin Cancer Res. 2013;19:5068–78. doi: 10.1158/1078-0432.ccr-13-0260. PMID: 23894056. [DOI] [PubMed] [Google Scholar]
- 66.Giragossian C, Vage C, Li J, Pelletier K, Piché-Nicholas N, Rajadhyaksha M, Liras J, Logan A, Calle RA, Weng Y. Mechanistic investigation of the preclinical pharmacokinetics and interspecies scaling of PF-05231023, a fibroblast growth factor 21–antibody Protein conjugate. Drug Metab Dispos. 2015;43(6):803–11. doi: 10.1124/dmd.114.061713. PMID: 25805881. [DOI] [PubMed] [Google Scholar]
- 67.Betts A, Keunecke A, TJv S, PHvd G, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: a comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. Mabs. 2018;10:1–55. doi: 10.1080/19420862.2018.1462429. PMID: 29634430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haraya K, Tachibana T, Nezu J. Quantitative prediction of therapeutic antibody pharmacokinetics after intravenous and subcutaneous injection in human. Drug Metab Pharmacokinet. 2017;32(4):208–17. doi: 10.1016/j.dmpk.2017.05.002. PMID: 28734646. [DOI] [PubMed] [Google Scholar]
- 69.Luu KT, Bergqvist S, Chen E, Hu-Lowe D, Kraynov E. A model-based approach to predicting the human pharmacokinetics of a monoclonal antibody exhibiting target-mediated drug disposition. J Pharmacol Exp Ther. 2012;341(3):702–08. doi: 10.1124/jpet.112.191999. PMID: 22414855. [DOI] [PubMed] [Google Scholar]
- 70.Schultink AHMdV, Doornbos RP, Bakker ABH, Bol K, Throsby M, Geuijen C, Maussang D, Schellens JHM, Beijnen JH, Huitema ADR. Translational PK-PD modeling analysis of MCLA-128, a HER2/HER3 bispecific monoclonal antibody, to predict clinical efficacious exposure and dose. Invest New Drug. 2018;36:1006–15. doi: 10.1007/s10637-018-0593-x. PMID: 29728897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh AP, Krzyzanski W, Martin SW, Weber G, Betts A, Ahmad A, Abraham A, Zutshi A, Lin J, Singh P. Quantitative prediction of human pharmacokinetics for mAbs exhibiting target-mediated disposition. Aaps J. 2015;17:389–99. doi: 10.1208/s12248-014-9690-8. PMID: 25445845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kagan L, Abraham AK, Harrold JM, Mager DE. Interspecies scaling of receptor-mediated pharmacokinetics and pharmacodynamics of type I interferons. Pharm Res. 2010;27(5):920–32. doi: 10.1007/s11095-010-0098-6. PMID: 20232116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han K, Peyret T, Quartino A, Gosselin NH, Gururangan S, Casanova M, Merks JHM, Massimino M, Grill J, Daw NC. Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Brit J Clin Pharmaco. 2016;81:148–60. doi: 10.1111/bcp.12778. PMID: 26345283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Standing JF. Understanding and applying pharmacometric modelling and simulation in clinical practice and research. Brit J Clin Pharmaco. 2017;83:247–54. doi: 10.1111/bcp.13119. PMID: 27567102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Wei X, Bajaj G, Barrett JS, Meibohm B, Joshi A, Gupta M. Challenges and considerations for development of therapeutic proteins in pediatric patients. J Clin Pharmacol. 2015;55(S3):S103–S115. doi: 10.1002/jcph.382. PMID: 25707958. [DOI] [PubMed] [Google Scholar]
- 76.Bhattacharya I, Manukyan Z, Chan P, Heatherington A, Harnisch L. Application of quantitative pharmacology approaches in bridging pharmacokinetics and pharmacodynamics of domagrozumab from adult healthy subjects to pediatric patients with duchenne muscular disease. J Clin Pharmacol. 2018;58(3):314–26. doi: 10.1002/jcph.1015. PMID: 29023829. [DOI] [PubMed] [Google Scholar]
- 77.Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico-Beyer LA, Gupta M, Tang M, Allison DE, Lu D. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51(2):119–35. doi: 10.2165/11596370-000000000-00000. PMID: 22257150. [DOI] [PubMed] [Google Scholar]
- 78.Kleiber M. Body size and metabolism. Hilgardia. 1932;6(11):315–53. doi: 10.3733/hilg.v06n11p315. [DOI] [Google Scholar]
- 79.Tang H, Mayersohn M. A global examination of allometric scaling for predicting human drug clearance and the prediction of large vertical allometry. J Pharm Sci. 2006;95:1783–99. doi: 10.1002/jps.20481. PMID: 16795013. [DOI] [PubMed] [Google Scholar]
- 80.Tang H, Mayersohn M. A mathematical description of the functionality of correction factors used in allometry for predicting human drug clearance. Drug Metab Dispos. 2005;33:1294–96. doi: 10.1124/dmd.105.004135. PMID: 15919851. [DOI] [PubMed] [Google Scholar]
- 81.Lee WS, Shim SR, Lee SY, Yoo JS, Cho SK. Preclinical pharmacokinetics, interspecies scaling, and pharmacokinetics of a phase I clinical trial of TTAC-0001, a fully human monoclonal antibody against vascular endothelial growth factor 2. Drug Des Dev Ther. 2018;12:495–504. doi: 10.2147/dddt.s150241. PMID: 29563774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diao L, Meibohm B. Tools for predicting the PK/PD of therapeutic proteins. Expert Opin Drug Met. 2015;11:1115–25. doi: 10.1517/17425255.2015.1041917. PMID: 25936400. [DOI] [PubMed] [Google Scholar]
- 83.Dedrick RL, Bischoff KB, Zaharko DS. Interspecies correlation of plasma concentration history of methotrexate (NSC-740). Cancer Chemother Rep 1970;54:95–101. PMID: 5003581.. [PubMed]
- 84.Boxenbaum H, Ronfeld R. Interspecies pharmacokinetic scaling and the dedrick plots. Am J Physiol. 1983;245:R768–75. doi: 10.1152/ajpregu.1983.245.6.R768. PMID: 6660320. [DOI] [PubMed] [Google Scholar]
- 85.Dedrick RL, Bischoff KB, Zaharko DS. Interspecies correlation of plasma concentration history of methotrexate (NSC-740). Cancer Chemother Rep. 1970;54:95–101. PMID: 5003581. [PubMed] [Google Scholar]
- 86.Offman E, Edginton AN. Contrasting toxicokinetic evaluations and interspecies pharmacokinetic scaling approaches for small molecules and biologics: applicability to biosimilar development. Xenobiotica. 2012;43:561–69. doi: 10.3109/00498254.2012.744113. PMID: 23244626. [DOI] [PubMed] [Google Scholar]
- 87.Oitate M, Nakayama S, Ito T, Kurihara A, Okudaira N, Izumi T. Prediction of human plasma concentration-time profiles of monoclonal antibodies from monkey data by a species-invariant time method. Drug Metab Pharmacokinet. 2012;27(3):354–59. doi: 10.2133/dmpk.dmpk-11-sh-059. PMID: 22146109. [DOI] [PubMed] [Google Scholar]
- 88.Vugmeyster Y, Rohde C, Perreault M, Gimeno RE, Singh P. Agonistic TAM-163 antibody targeting tyrosine kinase receptor-B: applying mechanistic modeling to enable preclinical to clinical translation and guide clinical trial design. Mabs. 2013;5(3):373–83. doi: 10.4161/mabs.23826. PMID: 23529133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park WS, Han S, Lee J, Hong T, Won J, Lim Y, Lee K, Byun HY, Yim DS. Use of a target‐mediated drug disposition model to predict the human pharmacokinetics and target occupancy of GC1118, an anti‐epidermal growth factor receptor antibody. Basic Clin Pharmacol. 2017;120:243–49. doi: 10.1111/bcpt.12675. PMID: 27637171. [DOI] [PubMed] [Google Scholar]
- 90.Roepcke S, Plock N, Yuan J, Fedyk ER, Lahu G, Zhao L, Smithson G. Pharmacokinetics and pharmacodynamics of the cytolytic anti‐CD38 human monoclonal antibody TAK‐079 in monkey – model assisted preparation for the first in human trial. Pharmacol Res Perspectives. 2018;6:e00402. doi: 10.1002/prp2.402. PMID: 29864242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Biliouris K, Nestorov I, Naik H, Dai D, Xiao G, Wang Q, Pellerin A, Rabah D, Lesko LJ, Trame MN. A pre-clinical quantitative model predicts the pharmacokinetics/pharmacodynamics of an anti-BDCA2 monoclonal antibody in humans. J Pharmacokinet Phar. 2018;45:817–27. doi: 10.1007/s10928-018-9609-6. PMID: 30377889. [DOI] [PubMed] [Google Scholar]
- 92.Ette EI, Williams PJ. Population pharmacokinetics I: background, concepts, and models. Ann Pharmacother. 2004;38(10):1702–06. doi: 10.1345/aph.1d374. PMID: 15328391. [DOI] [PubMed] [Google Scholar]
- 93.Dubois A, Gsteiger S, Balser S, Pigeolet E, Steimer JL, Pillai G, Mentré F. Pharmacokinetic similarity of biologics: analysis using nonlinear mixed‐effects modeling. Clin Pharmacol Ther. 2012;91:234–42. doi: 10.1038/clpt.2011.216. PMID: 22205196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mulugeta Y, Barrett JS, Nelson R, Eshete AT, Mushtaq A, Yao L, Glasgow N, Mulberg AE, Gonzalez D, Green D. Exposure matching for extrapolation of efficacy in pediatric drug development. J Clin Pharmacol. 2016;56:1326–34. doi: 10.1002/jcph.744. PMID: 27040726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mehrotra N, Bhattaram A, Earp JC, Florian J, Krudys K, Lee JE, Lee JY, Liu J, Mulugeta Y, Yu J. Role of quantitative clinical pharmacology in pediatric approval and labeling. Drug Metab Dispos. 2016;44(7):924–33. doi: 10.1124/dmd.116.069559. PMID: 27079249. [DOI] [PubMed] [Google Scholar]
- 96.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Phar. 2001;28:507–32. doi: 10.1023/a:1014414520282. PMID: 11999290. [DOI] [PubMed] [Google Scholar]
- 97.Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Phar. 2008;35:573–91. doi: 10.1007/s10928-008-9102-8. PMID: 19005743. [DOI] [PubMed] [Google Scholar]
- 98.Mager DE, Krzyzanski W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm Res. 2005;22(10):1589–96. doi: 10.1007/s11095-005-6650-0. PMID: 16180117. [DOI] [PubMed] [Google Scholar]
- 99.Kagan L, Zhao J, Mager DE. Interspecies pharmacokinetic modeling of subcutaneous absorption of rituximab in mice and rats. Pharm Res. 2014;31(12):3265–73. doi: 10.1007/s11095-014-1416-1. PMID: 24852895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Phar. 2012;39:67–86. doi: 10.1007/s10928-011-9232-2. PMID: 22143261. [DOI] [PubMed] [Google Scholar]
- 101.Malik PRV, Edginton AN. Physiologically‐based pharmacokinetic modeling vs. allometric scaling for the prediction of infliximab pharmacokinetics in pediatric patients. Cpt Pharmacometrics Syst Pharmacol. 2019;8:835–44. doi: 10.1002/psp4.12456. PMID: 31343836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones HM, Zhang Z, Jasper P, Luo H, Avery LB, King LE, Neubert H, Barton HA, Betts AM, Webster R. A physiologically‐based pharmacokinetic model for the prediction of monoclonal antibody pharmacokinetics from in vitro data. Cpt Pharmacometrics Syst Pharmacol. 2019;8:738–47. doi: 10.1002/psp4.12461. PMID: 31464379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hardiansyah D, Ng CM. Effects of the FcRn developmental pharmacology on the pharmacokinetics of therapeutic monoclonal IgG antibody in pediatric subjects using minimal physiologically-based pharmacokinetic modelling. Mabs. 2018;10:1–13. doi: 10.1080/19420862.2018.1494479. PMID: 29969360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ince I, Solodenko J, Frechen S, Dallmann A, Niederalt C, Schlender J, Burghaus R, Lippert J, Willmann S. Predictive pediatric modeling and simulation using ontogeny information. J Clin Pharmacol. 2019;59(S1):S95–S103. doi: 10.1002/jcph.1497. PMID: 31502689. [DOI] [PubMed] [Google Scholar]
- 105.Gill KL, Gardner I, Li L, Jamei M. A bottom-up whole-body physiologically based pharmacokinetic model to mechanistically predict tissue distribution and the rate of subcutaneous absorption of therapeutic Proteins. Aaps J. 2016;18:156–70. doi: 10.1208/s12248-015-9819-4. PMID: 26408308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zheng S, Gaitonde P, Andrew MA, Gibbs MA, Lesko LJ, Schmidt S. Model‐based assessment of dosing strategies in children for monoclonal antibodies exhibiting target‐mediated drug disposition. Cpt Pharmacometrics Syst Pharmacol. 2014;3:1–10. doi: 10.1038/psp.2014.38. PMID: 25271939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Covell DG, Barbet J, Holton OD, Black CD, Parker RJ, Weinstein JN. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab’)2, and Fab’ in mice. Cancer Res. 1986;46:3969–78. PMID: 3731067. [PubMed] [Google Scholar]
- 108.Baxter LT, Zhu H, Mackensen DG, Jain RK. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 1994;54:1517–28. PMID: 8137258. [PubMed] [Google Scholar]
- 109.Glassman PM, Balthasar JP. Physiologically-based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J Pharmacokinet Phar. 2016;43:427–46. doi: 10.1007/s10928-016-9482-0. PMID: 27377311. [DOI] [PubMed] [Google Scholar]
- 110.Davda JP, Jain M, Batra SK, Gwilt PR, Robinson DH. A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. Int Immunopharmacol. 2008;8(3):401–13. doi: 10.1016/j.intimp.2007.10.023. PMID: 18279794. [DOI] [PubMed] [Google Scholar]
- 111.Chen X, DuBois DC, Almon RR, Jusko WJ. Characterization and interspecies scaling of rhTNF-α pharmacokinetics with minimal physiologically-based pharmacokinetic (mPBPK) models. Drug Metab Dispos. 2017;45:798–806. doi: 10.1124/dmd.116.074799. PMID: 28411279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cao Y, Jusko WJ. Applications of minimal physiologically-based pharmacokinetic models. J Pharmacokinet Phar. 2012;39:711–23. doi: 10.1007/s10928-012-9280-2. PMID: 23179857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baxter LT, Zhu H, Mackensen DG, Butler WF, Jam RK. Biodistribution of monoclonal antibodies: scale-up from mouse to human using a physiologically based pharmacokinetic model. Cancer Res. 1995;55:4611–22. PMID: 7553638. [PubMed] [Google Scholar]
- 114.Vugmeyster Y, Szklut P, Tchistiakova L, Abraham W, Kasaian M, Xu X. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of humanized monoclonal anti-IL-13 antibodies with different IL-13 neutralization mechanisms. Int Immunopharmacol. 2008;8(3):477–83. doi: 10.1016/j.intimp.2007.12.004. PMID: 18279802. [DOI] [PubMed] [Google Scholar]
- 115.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Khabbaz HA, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18(12):1759–69. doi: 10.1093/intimm/dxl110. PMID: 17077181. [DOI] [PubMed] [Google Scholar]
- 116.Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. Mabs. 2013;5(3):397–405. doi: 10.4161/mabs.23836. PMID: 23549129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dostalek M, Prueksaritanont T, Kelley RF. Pharmacokinetic de-risking tools for selection of monoclonal antibody lead candidates. Mabs. 2017;9(5):756–66. doi: 10.1080/19420862.2017.1323160. PMID: 28463063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lin YS, Nguyen C, Mendoza J-L, Escandon E, Fei D, Meng YG, Modi NB. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J Pharmacol Exp Ther. 1999;288:371–78. PMID: 9862791. [PubMed] [Google Scholar]
- 119.Kelley SK, Gelzleichter T, Xie D, Lee WP, Darbonne WC, Qureshi F, Kissler K, Oflazoglu E, Grewal IS. Preclinical pharmacokinetics, pharmacodynamics, and activity of a humanized anti‐CD40 antibody (SGN‐40) in rodents and non‐human primates. Brit J Pharmacol. 2006;148:1116–23. doi: 10.1038/sj.bjp.0706828. PMID: 16847437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duconge J, Castillo R, Crombet T, Alvarez D, Matheu J, Vecino G, Alonso K, Beausoleil I, Valenzuela C, Becquer MA. Integrated pharmacokinetic–pharmacodynamic modeling and allometric scaling for optimizing the dosage regimen of the monoclonal ior EGF/r3 antibody. Eur J Pharm Sci. 2004;21(2–3):261–70. doi: 10.1016/j.ejps.2003.10.015. PMID: 14757498. [DOI] [PubMed] [Google Scholar]
- 121.Singh AP, Shah DK. Application of a PK-PD modeling and simulation-based strategy for clinical translation of antibody-drug conjugates: a case study with trastuzumab emtansine (T-DM1). Aaps J. 2017;19:1054–70. doi: 10.1208/s12248-017-0071-y. PMID: 28374319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Guidance for industry. estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. 2005. [accessed02June 2021]. Available from:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers
- 123.Hansen AR, Cook N, Ricci MS, Razak A, Tourneau CL, McKeever K, Roskos L, Dixit R, Siu LL, Hinrichs MJ. Choice of starting dose for biopharmaceuticals in first‐in‐human phase I cancer clinical trials. Oncol. 2015;20:653–59. doi: 10.1634/theoncologist.2015-0008. PMID: 25964306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Muller PY, Milton M, Lloyd P, Sims J, Brennan FR. The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr Opin Biotech. 2009;20(6):722–29. doi: 10.1016/j.copbio.2009.10.013. PMID: 19896825. [DOI] [PubMed] [Google Scholar]
- 125.EMA Committee for Medicinal Products for Human Use (CHMP) . Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. 2017. [accessed02June 2021]. Available from:https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational_en.pdf [DOI] [PMC free article] [PubMed]
- 126.Mishra A, Sarangi SC, Reeta K. First-in-human dose: current status review for better future perspectives. Eur J Clin Pharmacol. 2020;76(9):1237–43. doi: 10.1007/s00228-020-02924-x. PMID: 32488334. [DOI] [PubMed] [Google Scholar]
- 127.EMA Committee for medicinal products for human use (CHMP) . Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal products. 2007. [accessed02June 2021]. Available from:https://www.ema.europa.eu/documents/scientific-guideline/draft-guideline-requirements-first-man-clinical-trials-potential-high-risk-medicinal-products-first_en.pdf
- 128.European Medicines Agency . Press release ‘First-in-man’ clinical trials guideline released for public consultation. 2007. [accessed02June 2021]. Available from:https://www.ema.europa.eu/documents/press-release/first-man-clinical-trials-guideline-released-public-consultation_en.pdf
- 129.Saber H, Valle PD, Ricks TK, Leighton JK. An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul Toxicol Pharm. 2017;90:144–52. doi: 10.1016/j.yrtph.2017.09.001. PMID: 28887049. [DOI] [PubMed] [Google Scholar]
- 130.EMA Committee for Medicinal Products for Human Use (CHMP) . ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. 2009. [accessed02June 2021]. Available from:https://www.ema.europa.eu/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-marketing-authorisation_en.pdf
- 131.U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) . Guidance for industry. S9 nonclinical evaluation for anticancer pharmaceuticals. 2010. [accessed30April 2021]. Available from:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s9-nonclinical-evaluation-anticancer-pharmaceuticals
- 132.Saber H, Leighton JK. An FDA oncology analysis of antibody-drug conjugates. Regul Toxicol Pharm. 2015;71(3):444–52. doi: 10.1016/j.yrtph.2015.01.014. PMID: 25661711. [DOI] [PubMed] [Google Scholar]
- 133.Saber H, Gudi R, Manning M, Wearne E, Leighton JK. An FDA oncology analysis of immune activating products and first-in-human dose selection. Regul Toxicol Pharm. 2016;81:448–56. doi: 10.1016/j.yrtph.2016.10.002. PMID: 27743776. [DOI] [PubMed] [Google Scholar]
- 134.Germovsek E, Barker CIS, Sharland M, Standing JF. Scaling clearance in paediatric pharmacokinetics: all models are wrong, which are useful?. Brit J Clin Pharmaco. 2017;83:777–90. doi: 10.1111/bcp.13160. PMID: 27767204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sharma S, Eckert D, Hyams JS, Mensing S, Thakkar RB, Robinson AM, Rosh JR, Ruemmele FM, Awni WM. Pharmacokinetics and exposure–efficacy relationship of adalimumab in pediatric patients with moderate to severe crohn’s disease. Inflamm Bowel Dis. 2015;21:783–92. doi: 10.1097/mib.0000000000000327. PMID: 25723614. [DOI] [PubMed] [Google Scholar]
- 136.Shemesh CS, Chanu P, Jamsen K, Wada R, Rossato G, Donaldson F, Garg A, Winter H, Ruppel J, Wang X. Population pharmacokinetics, exposure-safety, and immunogenicity of atezolizumab in pediatric and young adult patients with cancer. J Immunother Cancer. 2019;7(1):314. doi: 10.1186/s40425-019-0791-x. PMID: 31753029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fasanmade AA, Adedokun OJ, Blank M, Zhou H, Davis HM. Pharmacokinetic properties of infliximab in children and adults with crohn’s disease: a retrospective analysis of data from 2 phase III clinical trials. Clin Ther. 2011;33:946–64. doi: 10.1016/j.clinthera.2011.06.002. PMID: 21741088. [DOI] [PubMed] [Google Scholar]
- 138.Sun H, Van LM, Floch D, Jiang X, Klein UR, Abrams K, Sunkara G. Pharmacokinetics and pharmacodynamics of canakinumab in patients with systemic juvenile idiopathic arthritis. J Clin Pharmacol. 2016;56(12):1516–27. doi: 10.1002/jcph.754. PMID: 27119439. [DOI] [PubMed] [Google Scholar]
- 139.Yee KL, Kleijn HJ, Kerbusch T, Matthews RP, Dorr MB, Garey KW, Wrishko RE. Population pharmacokinetics and pharmacodynamics of bezlotoxumab in adults with primary and recurrent clostridium difficile infection. Antimicrob Agents Chemother. 2019. doi: 10.1128/aac.01971-18. PMID: 30455246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lowe PJ, Renard D. Omalizumab decreases IgE production in patients with allergic (IgE‐mediated) asthma; PKPD analysis of a biomarker, total IgE. Brit J Clin Pharmaco. 2011;72:306–20. doi: 10.1111/j.1365-2125.2011.03962.x. PMID: 21392073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lowe PJ, Tannenbaum S, Gautier A, Jimenez P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE‐mediated) asthma. Brit J Clin Pharmaco. 2009;68:61–76. doi: 10.1111/j.1365-2125.2009.03401.x. PMID: 19660004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chakraborty A, Tannenbaum S, Rordorf C, Lowe PJ, Floch D, Gram H, Roy S. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1β monoclonal antibody. Clin Pharmacokinet. 2012;51(6):e1–e18. doi: 10.2165/11599820-000000000-00000. PMID: 22550964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ahamadi M, Freshwater T, Prohn M, Li CH, DPd A, Greef R, Elassaiss‐Schaap J, Kondic A, Stone JA. Model‐based characterization of the pharmacokinetics of pembrolizumab: a humanized anti–PD‐1 monoclonal antibody in advanced solid tumors. Cpt Pharmacometrics Syst Pharmacol. 2017;6:49–57. doi: 10.1002/psp4.12139. PMID: 27863186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li H, Han TH, Hunder NN, Jang G, Zhao B. Population pharmacokinetics of brentuximab vedotin in patients with CD30‐expressing hematologic malignancies. J Clin Pharmacol. 2017;57:1148–58. doi: 10.1002/jcph.920. PMID: 28513851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xu Y, Adedokun OJ, Chan D, Hu C, Xu Z, Strauss RS, Hyams JS, Turner D, Zhou H. Population pharmacokinetics and exposure‐response modeling analyses of golimumab in children with moderately to severely active ulcerative colitis. J Clin Pharmacol. 2019;59:590–604. doi: 10.1002/jcph.1353. PMID: 30536638. [DOI] [PubMed] [Google Scholar]
- 146.Admiraal R, Zijde CMJ-V, Silva JMF, Knibbe CAJ, Lankester AC, Boelens JJ, Hale G, Etuk A, Wilson M, Adams S. Population pharmacokinetics of alemtuzumab (campath) in pediatric hematopoietic cell transplantation: towards individualized dosing to improve outcome. Clin Pharmacokinet. 2019;58:1609–20. doi: 10.1007/s40262-019-00782-0. PMID: 31131436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee SK. Resting energy expenditure and the clearance of therapeutic Proteins in pediatric subjects. Pharmacology. 2014;93(5–6):225–28. doi: 10.1159/000362562. PMID: 25012840. [DOI] [PubMed] [Google Scholar]
- 148.Gupta A, Pouliquen I, Austin D, Price RG, Kempsford R, Steinfeld J, Bradford ES, Yancey SW. Subcutaneous mepolizumab in children aged 6 to 11 years with severe eosinophilic asthma. Pediatr Pulm. 2019;54:1957–67. doi: 10.1002/ppul.24508. PMID: 31502421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Turner DC, Navid F, Daw NC, Mao S, Wu J, Santana VM, Neel M, Rao B, Willert JR, Loeb DM. Population pharmacokinetics of bevacizumab in children with osteosarcoma: implications for dosing. Clin Cancer Res. 2014;20:2783–92. doi: 10.1158/1078-0432.ccr-13-2364. PMID: 24637635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mahmood I. Prediction of clearance of monoclonal and polyclonal antibodies and non-antibody Proteins in children: application of allometric scaling. Antibodies. 2020;9(3):40. doi: 10.3390/antib9030040. PMID: 32764408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mohamed MEF, Rakhmanina N, Hassan HE. Inclusion of adolescents with adults in phase 3 clinical trials: overview of the current state and a call for action. J Clin Pharmacol. 2020. doi: 10.1002/jcph.1591. PMID: 32119133. [DOI] [PubMed] [Google Scholar]
- 152.U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Oncology Center of Excellence (OCE) . Considerations for the inclusion of adolescent patients in adult oncology clinical trials. 2019. [accessed02June 2021]. Available from:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-inclusion-adolescent-patients-adult-oncology-clinical-trials
- 153.Roberts JK, Stockmann C, Balch A, Yu T, Ward RM, Spigarelli MG, Sherwin CMT. Optimal design in pediatric pharmacokinetic and pharmacodynamic clinical studies. Pediatr Anesth. 2015;25:222–30. doi: 10.1111/pan.12575. PMID: 25580772. [DOI] [PubMed] [Google Scholar]
- 154.Eleveld DJ, Proost JH, Absalom AR, Struys MMRF. Obesity and allometric scaling of pharmacokinetics. Clin Pharmacokinet. 2011;50:751–53. doi: 10.2165/11594080-000000000-00000. PMID: 21973272. [DOI] [PubMed] [Google Scholar]
- 155.Gómez-Mantilla JD, Trocóniz IF, Parra-Guillén Z, Garrido MJ. Review on modeling anti-antibody responses to monoclonal antibodies. J Pharmacokinet Phar. 2014;41:523–36. doi: 10.1007/s10928-014-9367-z. PMID: 25027160. [DOI] [PubMed] [Google Scholar]
- 156.Mager DE, Woo S, Jusko WJ. Scaling pharmacodynamics from in vitro and preclinical animal studies to humans. Drug Metab Pharmacokinet. 2009;1:16–24. doi: 10.2133/dmpk.24.16. PMID: 19252333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Deng R, Zhou C, Li D, Cai H, Sukumaran S, Carrasco-Triguero M, Saad O, Nazzal D, Lowe C, Ramanujan S. Preclinical and translational pharmacokinetics of a novel THIOMAB™ antibody-antibiotic conjugate against staphylococcus aureus. Mabs. 2019;11:1–13. doi: 10.1080/19420862.2019.1627152. PMID: 31219754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bouillon‐Pichault M, Brillac C, Amara C, Nicolazzi C, Fagniez N, Fau JB, Koiwai K, Ziti‐Ljajic S, Veyrat‐Follet C. Translational model‐based strategy to guide the choice of clinical doses for antibody–drug conjugates. J Clin Pharmacol. 2017;57:865–75. doi: 10.1002/jcph.869. PMID: 28138963. [DOI] [PubMed] [Google Scholar]
- 159.Li C, Zhang C, Deng R, Leipold D, Li D, Latifi B, Gao Y, Zhang C, Li Z, Miles D. Prediction of human pharmacokinetics of antibody–drug conjugates from nonclinical data. Clin Transl Sci. 2019;12:534–44. doi: 10.1111/cts.12649. PMID: 31115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Haddish-Berhane N, Shah DK, Ma D, Leal M, Gerber H-P, Sapra P, Barton HA, Betts AM. On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: a PK/PD approach. J Pharmacokinet Phar. 2013;40:557–71. doi: 10.1007/s10928-013-9329-x. PMID: 23933716. [DOI] [PubMed] [Google Scholar]
- 161.Ferl GZ, Reyes A, Sun LL, Cheu M, Oldendorp A, Ramanujan S, Stefanich EG. A preclinical population pharmacokinetic model for anti‐CD20/CD3 T‐cell‐dependent bispecific antibodies. Clin Transl Sci. 2018;11:296–304. doi: 10.1111/cts.12535. PMID: 29351372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Campagne O, Delmas A, Fouliard S, Chenel M, Chichili GR, Li H, Alderson R, Scherrmann J-M, Mager DE. Integrated pharmacokinetic/pharmacodynamic model of a bispecific CD3xCD123 DART® molecule in nonhuman primates: evaluation of activity and impact of immunogenicity. Clin Cancer Res. 2018;24:2631–41. doi: 10.1158/1078-0432.ccr-17-2265. PMID: 29463552. [DOI] [PubMed] [Google Scholar]
- 163.Chen X, Haddish‐Berhane N, Moore P, Clark T, Yang Y, Li H, Xuan D, Barton HA, Betts AM, Barletta F. Mechanistic projection of first‐in‐human dose for bispecific immunomodulatory P‐Cadherin LP‐DART: an integrated PK/PD modeling approach. Clin Pharmacol Ther. 2016;100:232–41. doi: 10.1002/cpt.393. PMID: 27170541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.