

Abstract
Aims
Deregulation of mTOR (mammalian target of rapamycin) signalling occurs in diabetes, which exacerbates injury following myocardial infarction (MI). We therefore investigated the infarct-limiting effect of chronic treatment with rapamycin (RAPA, mTOR inhibitor) in diabetic mice following myocardial ischaemia/reperfusion (I/R) injury and delineated the potential protective mechanism.
Methods and results
Adult male diabetic (db/db) or wild-type (WT) (C57) mice were treated with RAPA (0.25 mg/kg/day, intraperitoneal) or vehicle (5% DMSO) for 28 days. The hearts from treated mice were subjected to global I/R in Langendorff mode. Cardiomyocytes, isolated from treated mice, were subjected to simulated ischaemia/reoxygenation (SI/RO) to assess necrosis and apoptosis. Myocardial infarct size was increased in diabetic heart following I/R as compared to WT. Likewise, enhanced necrosis and apoptosis were observed in isolated cardiomyocytes of diabetic mice following SI/RO. Treatment with RAPA reduced infarct size as well as cardiomyocyte necrosis and apoptosis of diabetes and WT mice. RAPA increased STAT3 phosphorylation and miRNA-17/20a expression in diabetic hearts. In addition, RAPA restored AKT phosphorylation (target of mTORC2) but suppressed S6 phosphorylation (target of mTORC1) following I/R injury. RAPA-induced cardioprotection against I/R injury as well as the induction of miR-17/20a and AKT phosphorylation were abolished in cardiac-specific STAT3-deficient diabetic mice, without alteration of S6 phosphorylation. The infarct-limiting effect of RAPA was obliterated in cardiac-specific miRNA-17-92-deficient diabetic mice. The post-I/R restoration of phosphorylation of STAT3 and AKT with RAPA were also abolished in miRNA-17-92-deficient diabetic mice. Additionally, RAPA suppressed the pro-apoptotic prolyl hydroxylase (Egln3/PHD3), a target of miRNA-17/20a in diabetic hearts, which was abrogated in miRNA-17-92-deficient diabetic mice.
Conclusion
Induction of STAT3-miRNA-17-92 signalling axis plays a critical role in attenuating MI in RAPA-treated diabetic mice. Our study indicates that chronic treatment with RAPA might be a promising pharmacological intervention for attenuating MI and improving prognosis in diabetic patients.
Keywords: Apoptosis, Diabetes, Egln3/PHD3, Ischaemia/reperfusion, miR-17-92, mTOR, STAT3
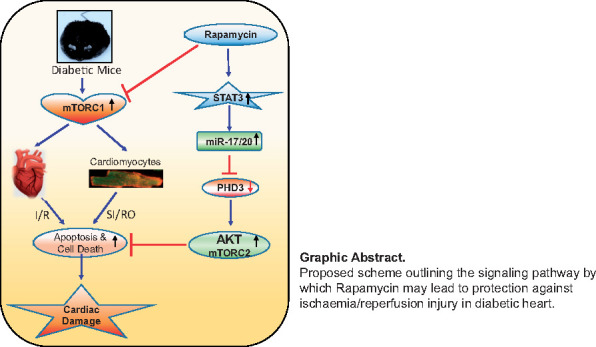
Graphical Abstract
Graphical Abstract.
Time for primary review: 19 days
1. Introduction
Type 2 diabetes (T2D) imposes a substantial risk for the incidence of cardiovascular disease, including multivessel coronary artery disease, congestive heart failure, and acute myocardial infarction (MI).1 Besides the prevalence of coronary artery disease among patients with diabetes, those who suffer an acute MI have poorer prognosis than non-diabetic patients.1,2 Clinical studies indicate nearly 68% of the mortalities observed in T2D are associated with cardiovascular complications.3 Evidence demonstrates aberrant activation of mTOR (mammalian target of rapamycin) signalling in metabolically active organs, including the heart of both genetic and diet-induced animal models of obesity and metabolic disorders.4,5 Since a sustained hyperactivation of mTOR is consistently reported in diabetic myocardium,6–8 there are ongoing efforts to pharmacologically target mTOR signalling in protecting against diabetes-associated cardiovascular diseases.
Randomized studies of diabetic patients have shown an advantage with implantation of rapamycin (RAPA; Sirolimus®, a specific mTOR inhibitor) eluting stents, compared to bare-metal stents and paclitaxel-eluting stents in preventing coronary restenosis and reducing adverse cardiac events.9–11 We reported the beneficial effect of RAPA treatment in improving metabolic status and preventing cardiac dysfunction in T2D mice through attenuation of oxidative stress as well as alteration of contractile and glucose metabolic proteins.12 Notably, diabetic hearts showed decreased level of activated STAT3, which was associated with insulin resistance and dilated cardiomyopathy.13,14 We also showed that acute pre-treatment with RAPA protects against myocardial ischaemia–reperfusion (I/R) injury in non-diabetic mice through unique cardioprotective signalling including phosphorylation of ERK, STAT3, and endothelial NOS, in concert with increased Bcl-2 to Bax ratio and inactivation of GSK-3β.15 Moreover, RAPA infusion at the onset of reperfusion in diabetic heart provided protection through STAT3-AKT signalling pathway.16 However, the chronic effects of RAPA treatment in protection against I/R injury are currently unknown.
Another interesting aspect relevant to mTOR signalling is the role of miR-17 and miR-20a, since there is a strong interaction between STAT3 and miR-17/20a17,18 Following microRNA array analysis, we identified that miR-17 and miR-20a were significantly elevated in diabetic heart with chronic RAPA treatment. Interestingly, miR-17-92 cluster is known to be indispensable for the cardiac development and its deletion leads to neonatal lethality due to severe anomalies of the cardiovascular system.19 Moreover, cadiomyocyte-specific overexpression of miR-17-92 protects the adult mouse heart from MI.20 However, the potential relationship of STAT3, miR-17-92 and mTOR signalling in cardioprotection following chronic treatment of RAPA against I/R injury in diabetic mice is currently unknown. To address this question, we used cardiac-specific conditional miR-17-92-cluster deficient as well as STAT3-deficient mice following induction of diabetes with high-fat diet (HFD). Our results suggest that STAT3-miR17/20 signalling axis plays a critical role in attenuating MI in RAPA-treated diabetic mice.
2. Methods
2.1 Animals
Adult male leptin receptor null, homozygous db/db mice (strain: BKS.Cg-Dock7m+/+Leprdb/J) and their corresponding wild-type (WT) mice (C57/BL/6J) were purchased from the Jackson Laboratories (Bar Harbor, ME, USA). Inducible, cardiac-specific STAT3 deficient mice (MCM TG: STAT3flox/flox) and WT non-transgenic control mice (MCM NTG: STAT3flox/flox) were created by interbreeding STAT3flox/flox mice with tamoxifen-inducible MCM TG mice.16,21 The breeding pair of tamoxifen-inducible cardiac-specific miR-17-92-deficient mice (miR-17-92-def: MCM TG: miR17-92flox/flox) and their WT littermates (WT: MCM NTG: miR17-92flox/flox) were provided by Dr Yaoliang Tang, Augusta University. STAT3-deficient, miR-17-92-defficient, and their corresponding WT male mice (8–10 weeks) were fed with HFD (containing 60% fat calories, Bio-Serv Company, NJ, USA) for 16 weeks to induce obesity and diabetes (glucose >200 mg/dL), after which they received tamoxifen (20 mg/kg/day i.p.) for 5 days.16 The animal and experiments protocols were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University and conducted the Guide for the care and use of Laboratory Animals for Biomedical Research published by the National Institutes of Health (No. 85-23, revised 1996).
2.2 Experimental groups and treatment protocol
Adult (16–18 weeks) male db/db and C57BL/6J mice were treated daily for 28 days with RAPA (0.25 mg/kg i.p.) or an equivalent volume of vehicle (5% DMSO, i.p.) as control (Supplementary material online, Figure S1). After establishing diabetes (glucose level > 200 mg/dL) (Supplementary material online, Figures S2 and S6), HFD-fed STAT3-deficient, miR-17-20-deficient mice and their WT mice were treated with RAPA (0.25 mg/kg/d, i.p.) for 28 days following tamoxifen treatment. Mice were randomized into groups on the first day of the treatment and the experimenters were blind to group assignment and outcome assessment.
2.3 Ischaemia/reperfusion and infarct size measurement
After anaesthetizing mice with sodium pentobarbital (Nembutal® sodium solution; 100 mg/kg i.p.), the isolated heart was perfused for 20 min of stabilization and then subjected to 30 min of no-flow global ischaemia and 60 min of reperfusion in Langendorff perfusion system15,22 (detailed in Supplementary material online, Figure S1). Cardiac function was recorded throughout the period. After the end of reperfusion, the sections of heart were stained by 10% tetrazolium chloride (TTC) and the infarct size was determined by computer morphometry using image J software (NIH Bethesda, MD. USA) in a blinded fashion.
2.4 Isolation of adult mouse cardiomyocytes and simulated ischaemia/reoxygenation (SI/RO) protocol
The ventricular cardiomyocytes were isolated using an enzymatic technique as previously reported23 (detailed in Supplementary material online, Figure S1). Cardiomyocytes were subjected to SI for 40 min and RO for 1 h or 18 h.23
2.5 Measurement of cell death and mitochondrial membrane potential
Cardiomyocyte necrosis was assessed by trypan blue exclusion assay following 40 min SI and 1 h of RO. Mitochondrial membrane potential was assessed using cationic lipophilic probe JC-1 (BD Biosciences MitoScreen Kit, San Jose, CA, USA).23 Apoptosis was determined after 40 min SI and 18 h of RO using TUNEL staining kit (BD Biosciences, San Jose, CA, USA) as previously reported.23
2.6 Western blot
Total soluble protein was extracted from the whole heart tissue with lysis buffer (Cell Signaling, MA, USA). Protein (50 µg) from each sample was separated by SDS-PAGE and transferred onto nitrocellulose membrane.22–24 The membrane was incubated overnight with primary antibody at a dilution 1:1000 for each of the respective proteins (phospho-tyrosine705-STAT3 and STAT3, phospho-Ser473-AKT and AKT, phospho-Ser235/236-S6 and S6, Bcl-2 and Bax from Cell Signaling, MA, USA PHD3 and GAPDH from Santa Cruz Biotechnology, TX, USA). The membrane was washed and incubated with horseradish peroxidase conjugated secondary antibody (1:2000 dilutions) for 1 h. The blots were developed using a chemiluminescent system (ECL Plus; PerkinElmer, Inc. MA, USA).
2.7 Real-time PCR
Total RNA was isolated from mouse heart using miRNeasy kit (Qiagen, Germantown, MD, USA). cDNA was synthesized using high capacity cDNA synthesis kit (Applied Biosystems, CA, USA) with random hexamer as primer. The cycle conditions were: 25°C for 10 min; 37°C for 120 min; 85°C for 5 min. For miRNA cDNA purpose, 10 ng of total RNA was subjected to reverse complement strand synthesis with microRNA specific stem loop primer using microRT reverse transcription kit (Applied Biosystems, CA, USA) according to manufacturer’s protocol. Following were the cycle conditions: 16°C for 30 min; 42°C for 30 min; and 85°C for 5 minutes. Information of sequence of miRNAs and accession numbers are presented in Supplementary material online, Table S1. The expression of Egln3/PHD3 (Assay ID-Mm01286844_m1), miR-17 (Assay ID-002308), miR-20a (Assay ID-000580), and miR-92 (Assay ID-000430) were quantified by real-time PCR using amplicon specific TaqMan assay probe and primer (Applied Biosystems, CA, USA) in CFX96–C1000 Touch Thermal Cycler system (Bio-Rad, Hercules, CA, USA). The PCR conditions were: 50°C for 2 min; 95°C for 5 min and 39 cycles of 95°C for 3 s and 60°C for 1 min. GAPDH (Assay ID-Mm99999915_g1) was used as housekeeping control for mRNA, U6 (Assay ID-001973) or Sno-202 (Assay ID-001232) were used for control of miRNA expression. The relative quantification of gene expression was analysed by using 2-ΔΔCT method and normalized to their respective controls and expressed as fold increase.
2.8 Cardiac function by echocardiography
Cardiac contractile function was measured before and after treatment with RAPA using the Vevo 770TM imaging system (VisualSonics, Inc., Toronto, Canada). Isoflurane (2.5%) was used to anaesthetize the mice prior to echocardiography. Cardiac functional parameters included measurement of left ventricular (LV) end-diastolic diameter (LVEDD) and LV end-systolic diameter (LVESD). LV fractional shortening was calculated as (LVEDD−LVESD)/LVEDD*100.12
2.9 Data analysis and statistics
Data are presented as mean ± S.E. The differences between more than two groups were analysed with one-way analysis of variance followed by Student–Newman–Keuls post hoc test for pairwise comparison using Graphpad Prism 8 (Graphpad Software, La Jolla, CA, USA). P < 0.05 was considered to be statistically significant.
3. Results
3.1 Rapamycin protects diabetic heart against I/R injury
Infarct size was significantly higher in hearts of db/db mice (38.43 ± 3.6%) as compared to C57 mice (27.75 ± 1.02%) following I/R (n = 5, P < 0.001 vs. C57, Figure 1A). Treatment with RAPA reduced infarct size both in C57 (11.74 ± 1.2%) and db/db mice (11.29 ± 1.8%) (n = 5, P < 0.001 vs. db/db). Cardiac function measured as rate-force product (product of heart rate and ventricular developed force and presented as % of pre-ischaemic baseline) was improved in RAPA-treated C57 and db/db hearts as compared to vehicle-treated db/db and C57 mice (n = 5, P < 0.01 vs. C57 and db/db, Figure 1A). The post-ischaemic coronary flow rate remained unchanged between groups (Supplementary material online, Figure S8A).
Figure 1.

Cardiac protection following rapamycin treatment in diabetic mice. (A) Representative images of TTC stained heart sections (Scale indicates 5 mm), myocardial infarct size in control and rapamycin (RAPA)-treated C57 and db/db mice following ischaemia/reperfusion (I/R) injury. n = 5 mice/group; *P < 0.001 vs. others and αP < 0.0001 vs. controls. Product of heart rate and ventricular developed force. n = 5; *P < 0.01 vs. controls. (B) Cardiomyocyte necrosis was measured by trypan blue staining following 40 min of simulated ischaemia (SI) and 1 h of reoxygenation (RO). (C) Representative images and quantitative data of cardiomyocytes apoptosis following 40 min SI and 18 h RO. (D) Representative images of JC-1 staining and quantitative data of JC-1 aggregate (red)/monomer (green) ratio. n = 4; *P < 0.001 vs. controls, αP < 0.05 vs. C57+SI/RO, βP < 0.001 vs. C57+SI/RO and db/db+SI/RO. Scale indicates 100 µm. Data are presented as mean ± S.E. Statistics: one-way ANOVA.
3.2 Rapamycin reduces necrosis and apoptosis in cardiomyocytes
Necrosis and apoptosis following SI/RO were significantly elevated in primary cardiomyocytes isolated from db/db mice as compared to C57 mice (Figure 1B and C). Mitochondrial membrane potential assessed by JC-1 aggregate/monomer ratio was also significantly reduced in cardiomyocytes of db/db mice as compared to C57 mice (Figure 1D). Cardiomyocytes isolated from RAPA-treated diabetic mice showed significant reduction in necrosis and apoptosis as well as induction of JC-1 aggregate/monomer ratio following SI/RO as compared to vehicle-treated cardiomyocytes isolated from db/db and C57 mice (Figure 1).
3.3 Rapamycin induces STAT3 phosphorylation in diabetic heart
The ratio of phospho-STAT3 (Tyr-705) to total STAT3 was reduced in the hearts of db/db mice when compared with C57 mice (Figure 2A). RAPA treatment significantly increased phosphorylation of STAT3 in the hearts of db/db mice (n = 3; P < 0.05 vs. others). Concomitantly, the ratio of STAT3 to GAPDH was high in the hearts of db/db when compared with C57 mice.
Figure 2.

Role of STAT3 in hearts of diabetic mice following rapamycin treatment. (A) Representative immunoblots for p-STAT3, total STAT3, and GAPDH expression in whole heart of C57 and db/db mice following 28 days of RAPA treatment. Densitometry analysis of immunoblots for the ratio of p-STAT3/STAT3 (*P < 0.05 vs. others) and the ratio of STAT3/GAPDH (*P < 0.05 vs. C57-Control; n = 3). (B) Myocardial infarct size following ischaemia/reperfusion in STAT3-deficient diabetic mice. High-fat diet (HFD)-fed WT and STAT3-deficient mice were treated with RAPA for 28 days prior to evaluation of I/R injury. Upper panel: representative images of heart sections following TTC staining (Scale indicates 5 mm). Lower panels: quantitative data of infarct size following I/R injury (*P < 0.001 vs. other) and rate-force product (*P < 0.01 vs. other; n = 5). (C) Isolated cardiomyocyte necrosis was determined by trypan blue staining following 40 min SI and 1 h RO (n = 4–8). (D) Representative pictures of TUNEL staining (scale indicates 100 µm) and quantitative data of cardiomyocyte apoptosis following 40 min SI and 18 h RO (n = 4; *P < 0.001 vs. controls, αP < 0.001 vs. both SI/RO and βP < 0.05 vs. WT-SI/RO). Statistics: one-way ANOVA.
3.4 STAT3 knock-down abolishes infarct-limiting effect of rapamycin
We previously demonstrated a 67% decrease in cardiac STAT3 expression in cardiac-specific STAT3 deficient mice (MCM TG: STAT3flox/flox) following tamoxifen treatment as compared to WT littermates (MCM NTG: STAT3flox/flox).16 In the current study, we established diabetes in STAT3-deficient and WT mice after feeding HFD for 16 weeks (Supplementary material online, Figure S2). The blood glucose levels in both STAT3-deficient and WT mice remained elevated up to 120 min after glucose administration (i.p.), indicating glucose intolerance (Supplementary material online, Figure S2C and D). Treatment with RAPA reduced myocardial infarct size in HFD-fed WT mice following I/R as compared to non-treated mice (n = 5, P < 0.001 vs. control, Figure 2B). Cardiac function (rate-force product) was also restored in RAPA-treated HFD-fed WT mice (n = 5, P < 0.01 vs. control, Figure 2B). However, the infarct-sparing effect of RAPA was blunted in HFD-fed STAT3-deficient mice (Figure 2B). In addition, the restoration of post-ischaemic cardiac function with RAPA was abolished in STAT3-deficient mice. The post-ischaemic coronary flow rate did not change between groups (Supplementary material online, Figure S8B).
RAPA treatment had significant impact in reducing body weight, liver weight and plasma glucose level in HFD-fed WT mice, but not in the corresponding STAT3-deficient mice (Supplementary material online, Figure S3). Interestingly, the HFD-fed STAT3-deficient mice developed hypertrophy as indicated by higher ratio of heart weight to tibia length as compared to WT mice (n = 5, P < 0.001 vs. WT, Supplementary material online, Figure S3C), which was attenuated after RAPA treatment.
RAPA treatment did not alter LV ejection fraction (EF), heart rate, LVEDD, and LVESD between groups (Supplementary material online, Figure S4). Notably, there was a significant increase in anterior wall diastolic thickness (AWDT) in STAT3-deficient mice as compared to WT mice supporting hypertrophic response in STAT3-deficient mice (Supplementary material online, Figure S4A). RAPA treatment decreased AWDT as well as anterior wall systolic thickness (AWST) in STAT3-deficient mice, while having no effect on wall thickness in WT mice.
3.5 Rapamycin-induced cardiomyocyte protection is blunted in STAT3-deficient mice
Necrosis and apoptosis were significantly reduced following SI/RO in cardiomyocytes from HFD-fed WT mice treated with RAPA as compared to non-treated mice (Figure 2C and D). Treatment with RAPA also improved mitochondrial membrane potential following SI/RO in cardiomyocytes from HFD-fed WT mice (Supplementary material online, Figure S9). However, such protective effect of RAPA in cardiomyocytes was blunted in HFD-fed STAT3-deficient mice.
3.6 Role of STAT3 in mTOR signalling in diabetic hearts
The phosphorylation of STAT3 was significantly reduced following I/R injury in hearts of HFD-fed WT mice with simultaneous increase in the total-STAT3 level (Figure 3A–C). Treatment with RAPA restored post-I/R phosphorylation of STAT3 (Figure 3A and B). RAPA also salvaged post-I/R phosphorylation of AKT (Ser473, as marker of mTORC2) in hearts of HFD-fed WT mice (Figure 3A and D). However, treatment with RAPA failed to restore the phosphorylation of STAT3 and AKT in hearts of HFD-fed STAT3-deficient mice. Nonetheless, total-AKT was significantly higher in RAPA-treated hearts of STAT3-deficient mice (Figure 3A and E). Interestingly, the significant induction of post-I/R phosphorylation of ribosomal protein S6 (by mTORC1) was inhibited in RAPA-treated hearts of both HFD-fed WT and STAT3-deficient mice (Figure 3A and F). Taken together, these data indicate that STAT3 regulates mTORC2 activity in RAPA-treated diabetic hearts without interfering with mTORC1 activity.
Figure 3.

Phosphorylation of STAT3, AKT, and S6 in hearts of diabetic WT and STAT3-deficient mice following I/R injury. High-fat-diet (HFD)-fed WT and STAT3-deficient mice were treated with RAPA for 28 days. (A) Representative immunoblots of phospho-STAT3, STAT3, Phospho-AKT, AKT, Phospho-S6, S6 and GAPDH in hearts of WT, and STAT3-deficient mice following I/R injury. (B–G) Densitometry analysis of the ratios of phosphorylated (p) to total protein, total protein to GAPDH (n = 3; one-way ANOVA).
3.7 Regulation of miR-17/20a expression with rapamycin treatment
Using microRNA array (LC Sciences, LLC, Houston, TX, USA), we identified that miR-17 and miR-20a were significantly elevated in diabetic hearts following chronic RAPA treatment (Supplementary material online, Figure S5). Real-time PCR confirmed significant induction of miR-17 and -20a, which are parts of miR-17-92 cluster, both in the heart (Figure 4A and B) and cardiomyocytes (Figure 4C and D) of RAPA-treated db/db mice. RAPA also induced miR-17 and miR-20a in the hearts of HFD-fed WT mice, but the induction of miR-17 and -20a were abolished in HFD-fed STAT3-deficient mice (Figure 4E). miR-92 expression remained unchanged between groups.
Figure 4.

Effect of rapamycin treatment on expression of miR-17 and miR-20a. C57 and db/db mice were treated with RAPA for 28 days. (A and B) Expression of miRNAs in the heart of C57 and db/db; (C and D) Expression of miRNAs in cardiomyocytes from wild-type and db/db mice. The quantitative expression of miR-17 and miR-20a were normalized to U6 small RNA. n = 3; *P < 0.05 vs. other (one-way ANOVA). (E) Expression of miR-17, miR-20a, and miR-92 in hearts from high-fat diet (HFD)-fed MCM NTG: STAT3flox/flox (WT) and MCM TG: STAT3flox/flox (STAT3-deficient) mice following RAPA treatment. The expression of miRNA was normalized to Sno-202. n = 6; *P < 0.0001 vs. other (one-way ANOVA).
3.8 Role of miR-17-92 cluster in rapamycin-induced cardioprotection
The critical role of miR-17-92 in RAPA-induced cardioprotection in diabetic mice was examined using HFD-fed αMHC-MerCreMer-miR-17-92-deficient mice. Body weight and glucose levels were significantly increased after 16 weeks of feeding with HFD (Supplementary material online, Figure S6A and B). However, there was no significant difference in body weight or glucose level between WT and miR-17-92-deficient mice. Diabetes-induced glucose intolerance in both HFD-fed WT and miR-17-20-deficient mice was confirmed by elevated level of glucose after 120 min of administration of glucose (Supplementary material online, Figure S6C and D). Real-time PCR confirmed significant reduction of miR-17 and miR-20 after 5 days of tamoxifen treatment in the heart of HFD-fed miR-17-92-deficient mice, but not in WT mice (Supplementary material online, Figure S6E and F). RAPA treatment reduced body weight and glucose level of HFD-fed WT mice, which were not discernible in HFD-fed miR-17-92-def mice (Supplementary material online, Figure S7A and B). Heart weight did not change with RAPA treatment between groups (Supplementary material online, Figure S7C). RAPA significantly improved cardiac function in HFD-fed WT mice (EF: 58.63 ± 1.92%) as compared to HFD-fed miR-17-92-def mice (EF: 49.63 ± 2.38%) (Supplementary material online, Figure S7D and E).
HFD-fed miR-17-92-deficient mice had higher infarct size as compared to WT following I/R injury (n = 5; P < 0.05 vs. WT control, Figure 5A and B). RAPA treatment reduced infarct size in HFD-fed WT mice following I/R as compared to vehicle-treated WT mice (n = 5, P < 0001 vs. WT control; Figure 5A and B). The infarct-limiting effect of RAPA was blunted in HFD-fed miR-17-92-deficient mice (n = 6). In addition, cardiac function (rate-force product) was improved in RAPA-treated HFD-fed WT mice as compared to vehicle-treated WT mice following I/R (n = 8; P < 0.01 vs. WT control, Figure 5C). However, there was no improvement of post-ischaemic recovery in RAPA treated HFD-fed miR-17-92-deficient mice (n = 9).
Figure 5.

Exacerbation of myocardial injury in diabetic mice with miR-17-92 deficiency (A) Representative images of TTC stained heart sections (scale bar indicates 5 mm), (B) infarct size (n = 5–6; *P < 0.05 vs. WT-control and **P < 0.0001 vs. others). Scale indicates 5 mm. (C) Rate force product (n = 8–9; *P < 0.01 vs. WT-control and αP < 0.001 vs. WT-control and WT-RAPA) of HFD-fed WT and miR-17-92-deficient mice following I/R injury. Statistics: one-way ANOVA.
3.9 Regulation of the pro-apoptotic protein prolyl hydroxylase (Egln3/PHD3) with rapamycin
Bioinformatics analysis with the miRanda algorithm using miRTarBase tool predicted Egln3/PHD3 as a direct target of miR-17 and miR-20a (Supplementary material online, Figure S10). Figure demonstrates that miR-17 and miR-20a contain the complementary sequence (7-mer seed region) in the 3′-UTR of Egln3/PHD3. RAPA treatment reduced the expression and transcript level of the pro-apoptotic protein prolyl hydroxylase (Egln3/PHD3), a target of miR-17/20a, in diabetic hearts (Figure 6A). Moreover, the expression of Egln3/PHD3 was increased following myocardial I/R injury in diabetic WT as well as miR-17-92-deficient mice (Figure 6B). Treatment with RAPA suppressed the expression of Egln3/PHD3 in the hearts of diabetic WT mice, but such an effect was abolished in diabetic miR-17-92-deficient mice (Figure 6B). These results also resonated with the expression of apoptosis modifying proteins. The post-I/R induction of pro-apoptotic protein, Bax, and the ratio of Bax to Bcl-2 was inhibited by RAPA treatment in the hearts of diabetic WT mice (Figure 6C), but not in miR-17-92-deficient diabetic mice.
Figure 6.

Effect of rapamycin on expression of prolyl hydroxylase 3 and apoptosis modifying proteins in diabetic heart. Wild-type (C57) and diabetic (db/db) mice were treated with RAPA for 28 days. (A) Expression of prolyl hydroxylase 3 (PHD3) protein (western blot and quantitative bar diagram on left; *P < 0.05 vs db/db) and transcript level (on right; *P < 0.005 vs others). n = 3; (B) The expression of PHD3 in the hearts of RAPA treated HFD-fed WT and miR-17-92-deficient mice after I/R injury (n = 5; *P < 0.05 vs. Controls and αP < 0.05 vs. WT-I/R). (C) The expression of Bcl-2 and Bax in RAPA-treated HFD-fed WT and miR-17-92-deficient mice after I/R injury (n = 5). Statistics: one-way ANOVA.
3.10 Role of miR-17-92 in mTOR signalling in RAPA-treated diabetic hearts
The restoration of post-I/R phosphorylation of STAT3 and AKT in hearts of RAPA-treated HFD-fed WT mice were abolished in HFD-fed miR-17-92-deficient mice (Figure 7A, B, D), while the inhibition of phosphorylation of S6 with RAPA treatment was not hindered (Figure 7A, F). These results suggest that miR-17-92 plays a critical role in regulating phosphorylation of STAT3 and mTORC2 activity in RAPA-treated diabetic hearts following I/R injury.
Figure 7.

Phosphorylation of STAT3, AKT, and S6 in hearts of diabetic WT and miR-17-92-deficient mice following I/R injury. High-fat diet (HFD)-fed WT and miR-17-92-deficient mice were treated with RAPA for 28 days. (A) Representative immunoblots of phospho-STAT3, STAT3, Phospho-AKT, AKT, Phospho-S6, S6 and GAPDH in hearts of WT, and STAT3-deficient mice following I/R injury. (B–G) Densitometry analysis of the ratios of phosphorylated (p) to total protein, total protein to GAPDH (n = 3).
4. Discussion
The increasing prevalence of diabetes-associated cardiovascular disease is becoming a global challenge. There is compelling evidence for the role of mTOR activation in the pathogenesis of insulin resistance in T2D.25,26 mTOR has two functionally distinct complexes referred to as mTOR complex 1 (mTORC1) and mTORC2. RAPA preferentially inhibits mTORC1, although recent studies suggest that prolonged treatment with the drug also inhibits mTORC2 thereby leading to insulin resistance.27 We recently demonstrated that chronic treatment with a ‘cardioprotective’ dose of RAPA (0.25 mg/kg/day) preferentially inhibits mTORC1 in the diabetic hearts, without interfering with mTORC2.12 This treatment strategy improved the metabolic status of diabetic mice and prevented cardiac dysfunction. In addition, specific mTORC1 inhibition by PRAS40 (proline rich AKT substrate of 40 kDa) prevented the development of diabetic cardiomyopathy and improved metabolic profile in diabetic mice.28 In the present study, we show that the myocardial infarct size is higher in diabetic mice following I/R injury than in non-diabetic mice. Remarkably, chronic treatment with the same dose of RAPA (0.25 mg/kg/day, i.p.) showed a similar protective effect against I/R injury in diabetic hearts as in non-diabetic hearts. Moreover, the post-ischaemic cardiac function as measured by rate-force product was maintained after RAPA treatment in both diabetic and non-diabetic hearts. No alterations in coronary flow was observed before or after I/R among the groups suggesting that the protective effect of RAPA was independent of improvement in coronary endothelial function. At the cellular level, RAPA prevented necrosis and apoptosis, and preserved mitochondrial membrane potential in adult primary cardiomyocytes isolated from diabetic as well as WT mice chronically treated with RAPA. Taken together, our data indicate that diabetes does not blunt the cardioprotective effect of RAPA.
4.1 Role of STAT3 in cardioprotection
We previously demonstrated that STAT3 signalling plays an essential role in RAPA-induced cardioprotection against I/R injury in non-diabetic mice.15 Prior studies also revealed that the activation of STAT3 contributes to adaptation of the heart to ischaemic stress (preconditioning) via pro-survival signalling cascades or inhibition of pro-apoptotic proteins.29,30 Constitutive cardiomyocyte-restricted deletion of STAT3 increases apoptosis and infarct size after I/R14,21 and abolishes cardioprotection with ischaemic post-conditioning or pharmacological preconditioning.31,32 However, the effect of diabetes on cardiac STAT3 phosphorylation/activation may be inconsistent due to substantial differences in the method of induction, type, severity, and duration of diabetes. It has been shown that in the diabetic heart, the phosphorylation/activation of STAT3 is decreased,33 which is associated with insulin resistance,13,34 and STAT3-deficient mice spontaneously develop a form of dilated cardiomyopathy similar to that occurring in diabetic mice.14 Although pharmacologic or ischaemic conditioning enhance the STAT3 signalling, however, due to fundamental alteration of JAK/STAT activity in the presence of hyperinsulinaemia and hyperglycaemia, this pathway is considerably depressed and harder to stimulate in the diabetic myocardium.35 Multiple studies established the reduction of post-ischaemic STAT3 phosphorylation and/or activation due to diabetes in heart, which may contribute to increased susceptibility to myocardial I/R injury in diabetes.16,36–38 Interestingly, diabetic patients demonstrated increased apoptosis and oxidative stress with attenuation of the JAK2/STAT3 pathway in right atrial tissue compared with non-diabetic patients following cardiopulmonary bypass.35 Earlier, we demonstrated that treatment with RAPA at the onset of reperfusion protects diabetic heart by restoring phosphorylation of STAT3 and AKT (target of mTORC2), with reduction of ribosomal protein S6 phosphorylation (target of mTORC1).16 Administration of RAPA at reperfusion reduced infarct size in diabetic WT mice, but not in diabetic tamoxifen-inducible cardiomyocyte-specific STAT3-deficient mice following I/R. In the present study, our results show that chronic RAPA treatment increased STAT3 (Tyr-705) phosphorylation in the heart of C57 WT mice, which was even more pronounced in diabetic hearts. By taking advantage of our novel inducible cardiomyocyte-specific STAT3-deficient mouse strain placed on HFD to induce obesity and T2D, we interrogated the essential role of STAT3 in RAPA-induced cardioprotection against I/R injury. The results show that RAPA exerted a robust infarct-sparing effect in HFD-fed WT littermates following global I/R injury which paralleled with the improvement of post-ischaemic cardiac functional recovery. The cardioprotective effect of RAPA against I/R was abolished in HFD-fed tamoxifen-inducible cardiomyocyte-specific STAT3-deficient mice. At the cellular level, RAPA treatment protects cardiomyocytes of diabetic mice against SI/RO injury, which was blunted in cardiomyocytes of HFD-fed STAT3-deficient mice. These results further corroborate the essential role of STAT3 in mediating the protection of diabetic heart with chronic RAPA treatment.
A cardiomyocyte-restricted deletion of STAT3 leads to spontaneous heart failure associated with disturbed sarcomere organization and a reduced myocardial capillary density in male mice.14,39 In STAT3-KO hearts, the ubiquitin-conjugating enzymes Ube2i and Ube2g1 expression were markedly reduced by induction of miR-199a-5p (a microRNA that is transcriptionally suppressed by STAT3).39 Human terminal failing hearts harbouring low STAT3 protein levels displayed increased miR-199a levels and decreased Ube2g1 expression.39 miR-199a targets the Ube2i and Ube2g1, thereby disturbing the protein turnover by the ubiquitin-proteasome system (UPS), which leads to the reduced expression of α- and β-sarcomeric myosin heavy-chain (MHC) proteins.39 We recently demonstrated reduced expression of MHCα6 in diabetic heart, which was restored upon chronic RAPA treatment with improvement of cardiac contractile function.12 Interestingly, the present result may explain the potential role of reduced STAT3 activation in suppression of MHCα6 expression in diabetic heart mice by impairment of UPS. Conversely, STAT3 activation following chronic RAPA treatment may restore MHCα6 resulting in improved contractile function in diabetic mice.12
4.2 Critical role of STAT3 in regulation of mTOR signalling
Prior studies established the cross-talk between STAT3 and AKT signalling, which are impaired in diabetic conditions and render the hearts susceptible to I/R injury.40,41 In diabetic myocardium, the efficacy of ischaemic pre- or post-conditioning is significantly impaired through inhibition of STAT3 phosphorylation as well as STAT3-regulated AKT activity.13,34 Our results show that the post-I/R repression of phosphorylation of STAT3 (Tyr-705) and AKT (Ser473) were restored in hearts of RAPA-treated diabetic mice, but not in hearts of STAT3-deficient mice. However, the inhibition of phosphorylation of S6 with RAPA treatment remained unchanged in diabetic STAT3-deficient mice. These results confirm the regulatory role of STAT3 in activation of mTORC2 in RAPA-induced protection against I/R injury, without having impact on mRORC1 activity in diabetic mice.
4.3 Anti-hypertrophic effect of RAPA in diabetic mice
Earlier we established that RAPA improves metabolic status and cardiac function in db/db mice.12 Our present results also show that RAPA treatment significantly reduced body weight, liver weight, and plasma glucose levels in HFD-fed WT mice, but not in STAT3-deficient mice. Although LV fractional shortening was not different between HFD-fed WT and STAT3-deficient mice, AWDT and the ratio of heart weight to tibia length were significantly higher in these mice as compared to WT mice, indicating LV hypertrophy in diabetic STAT3-deficient mice. These results suggest the potential role of STAT3 in mitigating hypertrophy or other underlying adverse remodelling that occurs in the diabetic heart. Moreover, there was marked decline in the development of cardiac hypertrophy with chronic RAPA treatment in HFD-fed STAT3-deficient mice as demonstrated by reduction in heart weight, AWDT and AWST. These results suggest that the anti-hypertrophic effects of the RAPA involve alternate pathways that remain functional in the absence of STAT3. To this context, inhibition of mTORC1 with PRAS40 has been reported to mitigate pathologic myocardial hypertrophy.42
4.4 Interplay between STAT3 and miR-17-92 in diabetic heart
There is a close interaction between STAT3 and miR-17/20a due to the highly conserved STAT3-binding in the promoter region of the miR-17-92 gene (C13orf25)17,18 A persistent activation of STAT3 leads to strong up-regulation of miR-17/20a in human pulmonary arterial endothelial cells.17 miR-17 and 20a are part of the miR-17-92 cluster, which comprises seven microRNAs (miRs-17-3p, 17-5p, 18a, 19a, 19b, 20a, and 92a) and transcribed as a common primary transcript.43 Knockout of the miR-17-92 cluster in mice caused severe developmental defects in the cardiovascular system by triggering lung hypoplasia and ventricular septal defects.19 Transgenic overexpression of miR-17-92 in cardiomyocytes induces cardiomyocyte proliferation in embryonic, postnatal, and adult hearts.20 In addition, overexpression of miR-17-92 protected the adult heart from MI-induced injury.20 Specifically, intra-cardiac injection of miR-19a/19b mimics enhanced cardiomyocyte proliferation and protected heart against ischaemic injury.44 A series of studies have indicated that miR-17-92 is involved in inhibiting apoptosis, and promoting angiogenesis through interacting with c-Myc and E2F1 transcription factors.45,46 Due to anti-apoptotic properties of miR-17-92, the transgenic mice with miR-17-92 over-expression in lymphocytes developed lymphoproliferative disease and autoimmunity, and died prematurely due to suppression of c-Myc-induced apoptosis by targeting Bcl-2-2-like 11 (BIM) and PTEN, thereby increasing the level of anti-apoptotic BCL2.47
Frank et al.48 demonstrated that overexpression of miR-20a significantly inhibited hypoxia-induced apoptosis, while its targeted knockdown exacerbated cardiomyocyte death by inducing apoptosis. Adenoviral overexpression of miR-20a caused suppression of the pro-apoptotic transcription factor E2F1 expression in cardiomyocytes. Interestingly, miR-20a is up-regulated after hypoxia/reoxygenation in cardiomyocytes, while chronic pressure overload leads to its down-regulation.48 Moreover, the anti-apoptotic effect of miR-20a is mediated through down-regulation of the pro-apoptotic factor Egln3/PHD3, a prolyl hydroxylase domain protein.48
Our results also show significant induction of miR-17 and -20a, in the hearts and cardiomyocytes isolated from RAPA-treated db/db mice and in the hearts of RAPA-treated HFD-fed WT mice (Figure 4). Such induction of miR-17 and -20a with RAPA was abolished in HFD-fed STAT3-deficient mice (Figure 4E), which indicates the increases of miR-17 and miR-20 expression in diabetic hearts via a STAT3-dependent mechanism. We also noticed that HFD-fed miR-17-92-deficient mice had higher infarct size as compared to WT following I/R injury (Figure 5). The infarct-limiting effect of RAPA was lost in HFD-fed miR-17-92-deficient mice. In addition, RAPA-induced post-ischaemic cardiac function recovery was abolished in HFD-fed miR-17-92-deficient mice.
Our results also show that the restoration of post-I/R phosphorylation of STAT3 was abolished in hearts of HFD-fed miR-17-92-deficient mice (Figure 7), suggesting a potential reciprocal regulatory role of miR-17-92 with STAT3 activation. Previous studies demonstrated one of the members of miR-17/92 cluster, miR-18a, enhanced the transcriptional activity of STAT3 not only by targeting STAT3 directly but also through indirect mechanisms,49,50 which support our present observations in ischaemic diabetic hearts. Further in depth studies are warranted to clarify this interplay between STAT3 and miR-17-92.
4.5 miR-17-92 suppresses PHD3 in RAPA-treated diabetic hearts
Prolyl hydroxylases (PHDs) are attractive therapeutic targets in a wide range of diseases including cardiovascular and MI.51 Three PHDs (named PHD1, PHD2, and PHD3) effect the proteasome-mediated degradation of HIF by catalyzing the hydroxylation of key proline residues in the HIF-1α subunit under normoxic conditions.52 When oxygen tension is reduced, PHD-mediated hydroxylation cannot occur and HIF-1α accumulates in the nucleus, resulting in HIF-mediated gene transcription. PHD3 is up-regulated following MI as well as ischaemic and diabetic cardiomyopathy, which are associated with increased apoptosis.52–54 Myocardial infarct size was significantly increased in cardiomyocyte-specific transgenic mice with overexpression of PHD3.55 Depletion of PHD3 protected the heart from myocardial injury and inhibited cardiomyocyte apoptosis.56 Also, a chemotherapeutic drug, doxorubicin, increased apoptosis through induction of Egln3/PHD3.57 Moreover, overexpression of Egln3/PHD3 increased apoptosis, while its inhibition/silencing of Egln3 substantially ameliorated doxorubicin-induced apoptosis. The pro-apoptotic mechanism in cardiomyocytes involved association of PHD3 with Bcl-2, which prevented the association of Bcl-2 with Bax, resulting in an acceleration of apoptosis.57 In the present study, RAPA suppressed the transcript and expression level of Egln3/PHD3 in diabetic hearts. Post-I/R induction of myocardial pro-apoptotic proteins Egln3/PHD3 and Bax were significantly repressed by RAPA treatment in diabetic WT mice, but not in miR-17-92-deficient mice. These studies suggest that miR-17-92 is critical in the cardioprotective effect of RAPA against I/R injury in diabetic mice.
4.6 miR-17-92 regulates mTORC2 activity after rapamycin treatment
miR-17-92 contributes to the homeostatic regulation of proliferation, cell cycle, cell survival, apoptosis, and angiogenesis by regulating AKT-mTOR pathways.19,46,58,59 The conditional deletion of pancreatic β-cell specific miR-17-92 in mouse has higher phosphatase and tensin homologue (PTEN) and lower phosphorylated AKT in islets.60 In the present study, we revealed that cardiac-specific miR-17-92-deficiency abolished RAPA-induced restoration of AKT phosphorylation following I/R injury in diabetic hearts, without interfering the RAPA-induced inhibition of S6 phosphorylation (target of mTORC1) (Figure 7). Interestingly, a cross-talk between hypoxia and insulin signalling through PHD3-regulated glucose and lipid metabolism has been established in HFD-induced diabetic mice with hepatic PHD3 deletion.61 The deletion of hepatic PHD3 improved insulin sensitivity and ameliorated diabetes by specifically stabilizing HIF-2α and enhancing IRS2 expression, which led to phosphorylation of AKT and improved glucose homeostasis.61 In the present study, we identified that treatment with RAPA repressed post-I/R stimulation of PHD3 expression with restoration of phosphorylation of AKT in diabetic mouse heart, but such regulations were abolished in miR-17-92-deficient diabetic mice. These results suggest a possible interplay between miR-17-92 with PHD3-AKT signalling in post-I/R diabetic mice.
4.7 Limitation of the study
In the present study, we used HFD-fed cardiac-specific miR-17-92 deficient diabetic mice to identify the critical role of miR-17 and miR-20 in RAPA-induced cardioprotection against I/R injury. However, such an effect could be due to the cluster of miR-17-92, instead of individual miRNAs. Future studies by suing specific miRNA-deficient mice may help to address this issue. We also used isolated perfused heart that were subjected to global I/R injury as opposed to the in vivo model of MI. One of the concerns is the occlusion of left coronary artery to induce ischaemia in the db/db or HFD-fed diabetic mice is fraught with high mortality as compared to non-diabetic WT mice. In addition, the perfusion buffer contained glucose as a metabolic substrate, and lacked long-chain fatty acids. Despite the importance of fatty acids as the preferred metabolic fuel of the hearts, fatty acids are generally omitted from the perfusion buffer due to their poor solubility in aqueous solutions and the formation of frothing during vigorous oxygenation of the buffer.62 Furthermore, the hearts were isolated from mice fed with HFD (containing 60% fat calories, for 16 weeks) to induce obesity and diabetes, therefore, the lack of long chain fatty acid in perfusion medium may not largely impact the outcome of the present study.
5. Conclusion
In conclusion, our results show a novel mechanism by which mTOR inhibition with chronic RAPA treatment induces protective effects against myocardial I/R injury in diabetic mice. Specifically, the essential role of STAT3-miR-17-92 signalling axis was found to be critical in this protection. These studies may have potential clinical relevance in preventing I/R injury in diabetics because RAPA has relatively mild side effects at low dose. No negative impact on the immune system nor changes in blood glucose, insulin secretion, or insulin sensitivity, in healthy old individuals given RAPA were reported for at least 8 weeks.63 This indicates that RAPA alleviates the adverse conditions that compromise the diabetic heart when encountered with MI and possibly may provide better outcomes in diabetic patients following acute MI. Therefore, chronic treatment with low dose of RAPA may be a promising pharmacological intervention for attenuating MI and improving prognosis in diabetic patients.
Authors’ contributions
A.S., S.K.R., M.P., and D.D. contributed to the acquisition, analysis, and interpretation of data. F.N.S. was responsible for the study concept and acquisition and interpretation of data. A.D. contributed to the study concept and design the experiments, to the acquisition and interpretation of data and to preparing the manuscript. R.C.K. contributed to the study concept and critically editing the manuscript. All authors had editorial oversight of the final manuscript version, approved the final version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Yaoliang Tang, Augusta University, for providing us the breeding pair of miR-17-92-deficient mice.
Conflict of interest: none declared.
Funding
This work was supported by the National Institute of Health [RO1HL134366 (to A.D. and R.C.K.), R01HL133167 and R01HL142281 (to F.N.S.)], and VETAR [Value and Efficiency Teaching and Research Project sponsored by Virginia Commonwealth University School of Medicine to A.D.].
Translational perspective
The prevalence of heart disease and associated cardiovascular complications are a major cause of morbidity and mortality in diabetics. This study provides a novel mechanism by which mammalian target of rapamycin (mTOR) inhibition with chronic low-dose rapamycin (RAPA) treatment induces protective effect against acute myocardial infarction in diabetic mice. Specifically, the results show an essential role of a highly innovative STAT3-miR-17-92-mTOR signalling axis in cardioprotection in diabetes, which may have potential clinical relevance in preventing ischaemia/reperfusion (I/R) injury in diabetics because RAPA has relatively mild side effects at low dose. Future studies using cardiac tissue from diabetic and non-diabetic patients undergoing elective cardiac surgery may further confirm the role of this signalling pathway in the context of myocardial I/R injury.
References
- 1.Plutzky J.Macrovascular effects and safety issues of therapies for type 2 diabetes. Am J Cardiol 2011;108:25B–32B. [DOI] [PubMed] [Google Scholar]
- 2.Yeung CY, Lam KS, Li SW, Lam KF, Tse HF, Siu CW.. Sudden cardiac death after myocardial infarction in type 2 diabetic patients with no residual myocardial ischemia. Diabetes Care 2012;35:2564–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah AD, Langenberg C, Rapsomaniki E, Denaxas S, Pujades-Rodriguez M, Gale CP, Deanfield J, Smeeth L, Timmis A, Hemingway H.. Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1.9 million people. Lancet Diabetes Endocrinol 2015;3:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sung MM, Koonen DP, Soltys CL, Jacobs RL, Febbraio M, Dyck JR.. Increased CD36 expression in middle-aged mice contributes to obesity-related cardiac hypertrophy in the absence of cardiac dysfunction. J Mol Med 2011;89:459–469. [DOI] [PubMed] [Google Scholar]
- 5.Turdi S, Kandadi MR, Zhao J, Huff AF, Du M, Ren J.. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol 2011;50:712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li SY, Fang CX, Aberle NS, Ren BH, Ceylan-Isik AF, Ren J.. Inhibition of PI-3 kinase/Akt/mTOR, but not calcineurin signaling, reverses insulin-like growth factor I-induced protection against glucose toxicity in cardiomyocyte contractile function. J Endocrinol 2005;186:491–503. [DOI] [PubMed] [Google Scholar]
- 7.Mariappan MM, Feliers D, Mummidi S, Choudhury GG, Kasinath BS.. High glucose, high insulin, and their combination rapidly induce laminin-beta1 synthesis by regulation of mRNA translation in renal epithelial cells. Diabetes 2007;56:476–485. [DOI] [PubMed] [Google Scholar]
- 8.Sataranatarajan K, Mariappan MM, Lee MJ, Feliers D, Choudhury GG, Barnes JL, Kasinath BS.. Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am J Pathol 2007;171:1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumgart D, Klauss V, Baer F, Hartmann F, Drexler H, Motz W, Klues H, Hofmann S, Volker W, Pfannebecker T, Stoll HP, Nickenig G.. One-year results of the SCORPIUS study: a German multicenter investigation on the effectiveness of sirolimus-eluting stents in diabetic patients. J Am Coll Cardiol 2007;50:1627–1634. [DOI] [PubMed] [Google Scholar]
- 10.de Waha A, Dibra A, Kufner S, Baumgart D, Sabate M, Maresta A, Schomig A, Kastrati A.. Long-term outcome after sirolimus-eluting stents versus bare metal stents in patients with diabetes mellitus: a patient-level meta-analysis of randomized trials. Clin Res Cardiol 2011;100:561–570. [DOI] [PubMed] [Google Scholar]
- 11.Lee SW, Park SW, Kim YH, Yun SC, Park DW, Lee CW, Kang SJ, Rhee KS, Chae JK, Ko JK, Park JH, Lee JH, Choi SW, Jeong JO, Seong IW, Cho YH, Lee NH, Kim JH, Chun KJ, Kim HS, Park SJ.. A randomized comparison of sirolimus- versus paclitaxel-eluting stent implantation in patients with diabetes mellitus: 4-year clinical outcomes of DES-DIABETES (drug-eluting stent in patients with DIABETES mellitus) trial. JACC Cardiovasc Interv 2011;4:310–316. [DOI] [PubMed] [Google Scholar]
- 12.Das A, Durrant D, Koka S, Salloum FN, Xi L, Kukreja RC.. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem 2014;289:4145–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drenger B, Ostrovsky IA, Barak M, Nechemia-Arbely Y, Ziv E, Axelrod JH.. Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology 2011;114:1364–1372. [DOI] [PubMed] [Google Scholar]
- 14.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H.. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 2004;95:187–195. [DOI] [PubMed] [Google Scholar]
- 15.Das A, Salloum FN, Durrant D, Ockaili R, Kukreja RC.. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J Mol Cell Cardiol 2012;53:858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das A, Salloum FN, Filippone SM, Durrant DE, Rokosh G, Bolli R, Kukreja RC.. Inhibition of mammalian target of rapamycin protects against reperfusion injury in diabetic heart through STAT3 signaling. Basic Res Cardiol 2015;110:31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC.. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 2009;104:1184–1191. [DOI] [PubMed] [Google Scholar]
- 18.Zhang M, Liu Q, Mi S, Liang X, Zhang Z, Su X, Liu J, Chen Y, Wang M, Zhang Y, Guo F, Zhang Z, Yang R.. Both miR-17-5p and miR-20a alleviate suppressive potential of myeloid-derived suppressor cells by modulating STAT3 expression. J Immunol 2011;186:4716–4724. [DOI] [PubMed] [Google Scholar]
- 19.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T.. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008;132:875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang DZ.. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 2013;112:1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolli R, Stein AB, Guo YR, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu YQ, Xuan YT.. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol 2011;50:589–597. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Salloum FN, Xi L, Rao YJ, Kukreja RC.. ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol 2009;296:H1236–H1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das A, Xi L, Kukreja RC.. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem 2005;280:12944–12955. [DOI] [PubMed] [Google Scholar]
- 24.Das A, Xi L, Kukreja RC.. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem 2008;283:29572–29585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howell JJ, Manning BD.. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 2011;22:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laplante M, Sabatini DM.. mTOR signaling in growth control and disease. Cell 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM.. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006;22:159–168. [DOI] [PubMed] [Google Scholar]
- 28.Volkers M, Doroudgar S, Nguyen N, Konstandin MH, Quijada P, Din S, Ornelas L, Thuerauf DJ, Gude N, Friedrich K, Herzig S, Glembotski CC, Sussman MA.. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med 2014;6:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, Wu WJ, Tan W, Bolli R.. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res 2004;64:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R.. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA 2001;98:9050–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R.. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res 2008;102:131–135. [DOI] [PubMed] [Google Scholar]
- 32.Gross ER, Hsu AK, Gross GJ.. The JAK/STAT pathway is essential for opioid-induced cardioprotection: jAK2 as a mediator of STAT3, Akt, and GSK-3 beta. Am J Physiol Heart Circ Physiol 2006;291:H827–H834. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Liu Z, Wang J, Wong GT, Cheung CW, Zhang L, Chen C, Xia Z, Irwin MG.. Susceptibility to myocardial ischemia reperfusion injury at early stage of type 1 diabetes in rats. Cardiovasc Diabetol 2013;12:133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, Lee MK, Danton C, Deshmukh S, Cline GW, Wu JJ, Bennett AM, Rothermel B, Kalinowski A, Russell KS, Kim YB, Kelly DP, Kim JK.. Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes 2005;54:2514–2524. [DOI] [PubMed] [Google Scholar]
- 35.Owais K, Huang T, Mahmood F, Hubbard J, Saraf R, Bardia A, Khabbaz KR, Li Y, Bhasin M, Sabe AA, Sellke F, Matyal R.. Cardiopulmonary bypass decreases activation of the signal transducer and activator of transcription 3 (STAT3) pathway in diabetic human myocardium. Ann Thorac Surg 2015;100:1636–1645; discussion 1645. [DOI] [PubMed] [Google Scholar]
- 36.Pipicz M, Demjan V, Sarkozy M, Csont T.. Effects of cardiovascular risk factors on cardiac STAT3. Int. J. Mol. Sci 2018;19:3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C, Li H, Wang S, Mao X, Yan D, Wong SS, Xia Z, Irwin MG.. Repeated non-invasive limb ischemic preconditioning confers cardioprotection through PKC-/STAT3 signaling in diabetic rats. Cell Physiol Biochem 2018;45:2107–2121. [DOI] [PubMed] [Google Scholar]
- 38.Wang T, Mao X, Li H, Qiao S, Xu A, Wang J, Lei S, Liu Z, Ng KF, Wong GT, Vanhoutte PM, Irwin MG, Xia Z.. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med 2013;63:291–303. [DOI] [PubMed] [Google Scholar]
- 39.Haghikia A, Missol-Kolka E, Tsikas D, Venturini L, Brundiers S, Castoldi M, Muckenthaler MU, Eder M, Stapel B, Thum T, Haghikia A, Petrasch-Parwez E, Drexler H, Hilfiker-Kleiner D, Scherr M.. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J 2011;32:1287–1297. [DOI] [PubMed] [Google Scholar]
- 40.Lecour S.Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol 2009;47:32–40. [DOI] [PubMed] [Google Scholar]
- 41.Lei S, Su W, Xia ZY, Wang Y, Zhou L, Qiao S, Zhao B, Xia Z, Irwin MG.. Hyperglycemia-induced oxidative stress abrogates remifentanil preconditioning-mediated cardioprotection in diabetic rats by impairing caveolin-3-modulated PI3K/Akt and JAK2/STAT3 signaling. Oxid Med Cell Longev 2019;2019:9836302.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, Ornelas L, Joyo E, Thuerauf DJ, Konstandin MH, Gude N, Glembotski CC, Sussman MA.. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci USA 2013;110:12661–12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mendell JT.miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008;133:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao F, Kataoka M, Liu N, Liang T, Huang ZP, Gu F, Ding J, Liu J, Zhang F, Ma Q, Wang Y, Zhang M, Hu X, Kyselovic J, Hu X, Pu WT, Wang J, Chen J, Wang DZ.. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat Commun 2019;10:1802.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB.. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc Natl Acad Sci USA 2008;105:19678–19683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT.. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005;435:839–843. [DOI] [PubMed] [Google Scholar]
- 47.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K.. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 2008;9:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frank D, Gantenberg J, Boomgaarden I, Kuhn C, Will R, Jarr KU, Eden M, Kramer K, Luedde M, Mairbaurl H, Katus HA, Frey N.. MicroRNA-20a inhibits stress-induced cardiomyocyte apoptosis involving its novel target Egln3/PHD3. J Mol Cell Cardiol 2012;52:711–717. [DOI] [PubMed] [Google Scholar]
- 49.Brock M, Trenkmann M, Gay RE, Gay S, Speich R, Huber LC.. MicroRNA-18a enhances the interleukin-6-mediated production of the acute-phase proteins fibrinogen and haptoglobin in human hepatocytes. J Biol Chem 2011;286:40142–40150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu W, Takanashi M, Borjigin N, Ohno SI, Fujita K, Hoshino S, Osaka Y, Tsuchida A, Kuroda M.. MicroRNA-18a modulates STAT3 activity through negative regulation of PIAS3 during gastric adenocarcinogenesis. Br J Cancer 2013;108:653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, Signoretti S, Liao R, Kaelin WG Jr.. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 2010;122:1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaelin WG, Ratcliffe PJ. Jr. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008;30:393–402. [DOI] [PubMed] [Google Scholar]
- 53.Willam C, Maxwell PH, Nichols L, Lygate C, Tian YM, Bernhardt W, Wiesener M, Ratcliffe PJ, Eckardt KU, Pugh CW.. HIF prolyl hydroxylases in the rat;organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol 2006;41:68–77. [DOI] [PubMed] [Google Scholar]
- 54.Xia Y, Gong L, Liu H, Luo B, Li B, Li R, Li B, Lv M, Pan J, An F.. Inhibition of prolyl hydroxylase 3 ameliorates cardiac dysfunction in diabetic cardiomyopathy. Mol Cell Endocrinol 2015;403:21–29. [DOI] [PubMed] [Google Scholar]
- 55.Zieseniss A, Hesse AR, Jatho A, Krull S, Holscher M, Vogel S, Katschinski DM.. Cardiomyocyte-specific transgenic expression of prolyl-4-hydroxylase domain 3 impairs the myocardial response to ischemia. Cell Physiol Biochem 2015;36:843–851. [DOI] [PubMed] [Google Scholar]
- 56.Xie L, Pi X, Wang Z, He J, Willis MS, Patterson C.. Depletion of PHD3 protects heart from ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis. J Mol Cell Cardiol 2015;80:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Huo Z, Yan B, Lin X, Zhou ZN, Liang X, Zhu W, Liang D, Li L, Liu Y, Zhao H, Sun Y, Chen YH.. Prolyl hydroxylase 3 interacts with Bcl-2 to regulate doxorubicin-induced apoptosis in H9c2 cells. Biochem Biophys Res Commun 2010;401:231–237. [DOI] [PubMed] [Google Scholar]
- 58.Mogilyansky E, Rigoutsos I.. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 2013;20:1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L.. miR-19 is a key oncogenic component of mir-17-92. Genes Dev 2009;23:2839–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Y, Tian L, Wan S, Xie Y, Chen X, Ji X, Zhao Q, Wang C, Zhang K, Hock JM, Tian H, Yu X.. MicroRNA-17-92 cluster regulates pancreatic beta-cell proliferation and adaptation. Mol Cell Endocrinol 2016;437:213–223. [DOI] [PubMed] [Google Scholar]
- 61.Taniguchi CM, Finger EC, Krieg AJ, Wu C, Diep AN, LaGory EL, Wei K, McGinnis LM, Yuan J, Kuo CJ, Giaccia AJ.. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med 2013;19:1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao R, Podesser BK, Lim CC.. The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am J Physiol Heart Circ Physiol 2012;303:H156–H167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kraig E, Linehan LA, Liang H, Romo TQ, Liu Q, Wu Y, Benavides AD, Curiel TJ, Javors MA, Musi N, Chiodo L, Koek W, Gelfond JAL, Kellogg DL Jr.. A randomized control trial to establish the feasibility and safety of rapamycin treatment in an older human cohort: immunological, physical performance, and cognitive effects. Exp Gerontol 2018;105:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.