

Abstract
Proteomics has emerged as a rapidly expanding field dealing with large-scale protein analyses. It is anticipated that proteomics data will be increasingly submitted to the U.S. Food and Drug Administration (FDA) for biomarker qualification or in conjunction with applications for the approval of drugs, medical devices, and other FDA-regulated consumer products. To date, however, no established guideline has been available regarding the generation, submission and assessment of the quality of proteomics data that will be reviewed by regulatory agencies for decision making. Therefore, this commentary is aimed at provoking some thoughts and debates towards developing a framework which can guide future proteomics data submission. The ultimate goal is to establish quality control standards for proteomics data generation and evaluation, and to prepare government agencies such as the FDA to meet future obligations utilizing proteomics data to support regulatory decision.
Keywords: proteomics, mass spectrometry, quality control, standards, biomarker qualification, regulatory decision
Proteomics has emerged as a rapidly expanding field dealing with large-scale qualitative and quantitative protein analyses. The number of published research articles related to proteomics has shown a steady increase according to a search in Thompson Reuters Web of Knowledge Science Citation Index (Fig. 1). The annual increase rate is over 100% before 2002, and about 25% on average during the past decade. Although the recent relative increase rate becomes smaller because of the larger base of published papers, the absolute number of publications is still steadily increased. Proteomics has the promise to gain insights into the overall picture of proteome-wide alterations of protein abundance, interactions, activities, post-translational modifications and so forth under various physiological and pathological conditions. It is anticipated that a growing number of proteomic data will be submitted to regulatory agencies for biomarker qualification and/or in conjunction with drug applications to support, at the molecular level, drug efficacy and toxicity levels. The increasing submission of proteomic data and biomarkers will bring regulatory agencies great challenges in evaluating this type of data. It is not practical to validate a large number of proteins individually; therefore, it becomes of paramount importance that objective parameters and reliable procedures can be utilized to reflect the overall quality of a given proteomics dataset. Hence, establishing quality control standards for proteomics data generation and evaluation will help regulatory agencies meet obligations to utilize proteomics data in conjunction with drug review and biomarker qualification processes [1, 2].
Figure 1.
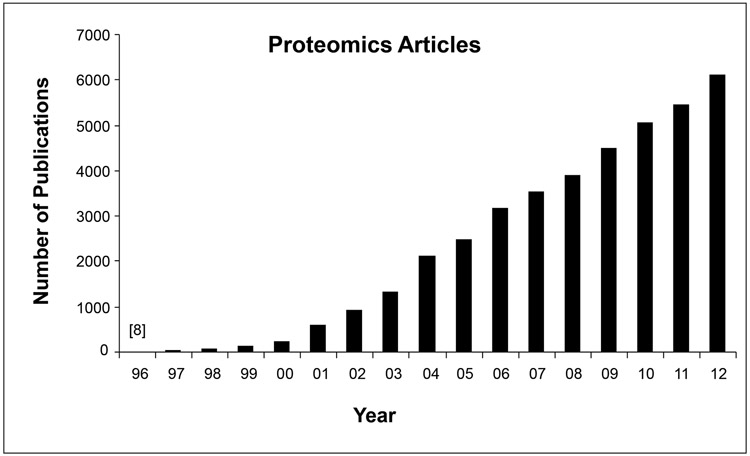
Annually published research articles (excluding reviews, abstracts, editorials, or commentaries) containing the keyword “proteomics”, “proteomic”, or “proteome” based on a search in Thompson Reuters Web of Knowledge Science Citation Index.
Compared to genomics, proteomics is a much more complicated proposition because of not only the huge number of protein species generated by variant splicing and post-translational modification, but also additional dynamic characteristics such as protein-protein interactions, interactions with other non-protein-type molecules, proteolysis, subcellular localization, high dynamic range of concentrations (particularly in plasma as a highly accessible sample type for biomarker discovery), etc. Therefore, proteomics could face more complex, challenging, and unique issues. To accurately evaluate proteomics data, simple and relevant questions have to be addressed before more specific questions relevant to a particular case are asked. For instance, how efficient is a protein extraction method? Will different methods for protein concentration measurements generate different results? These questions retain much less attention in the proteomics community than other issues such as the performance of liquid chromatography-mass spectrometry (LC-MS) platforms. Since proteins have to be extracted from membrane, cytoplasm, nucleus, and other organelles for proteomic analyses, diverse methods have been developed and utilized in protein extraction and purification processes. While in theory all proteins should be included in the analysis, it is often not the case in real experiments when diverse extraction methods with different yield efficiencies were utilized even for the same type of tissue or cell samples. It is conceivable that different protein extraction methods may yield variable amounts of protein for subsequent proteomics analyses. Thus, would it be appropriate to determine and state protein yield (e.g., mg protein/g tissue) for a particular protein extraction method and a particular type of sample? In addition, different methods to determine protein concentrations have been employed in practice. While it is not practical to endorse one method and abandon others, would it be necessary to set up a kind of conversion standard among different methods for protein concentration measurement, so that inter-methodological results could be appropriately compared and assessed?
Major standardization efforts for certain proteomic approaches and for setting scientific publication guidelines have been made by proteomics communities. Several attempts have been initialized such as the Proteomics Standards Initiative by the Human Proteome Organization (HUPO-PSI) [3] and the Clinical Proteomic Technology Assessment for the Cancer by National Cancer Institute [4]. A series of proposals have been recommended and published [5-17]. More recently, the ProteomeXchange (www.proteomexchange.org) consortium has been set up to provide a single point of submission of MS proteomics data to the main existing proteomics repositories such as PRIDE and PeptideAtlas. The primary goals of these initiatives were aimed at facilitating data comparisons from different laboratories and for overall data quality evaluation. Although these frameworks recommend the minimal reporting information regarding experimental procedures, unfortunately they do not address all parameters that can have major influences on the accuracy of experimental data. To date, there is no established guideline and standard available concerning the generation of proteomics data for regulatory evaluation. A recent report by the HUPO Test Sample Working Group revealed that out of the 27 laboratories which examined the same sample that consisted of 20 highly purified human proteins, only 7 laboratories reported all 20 proteins correctly [18]. Notably, this was only at the qualitative level, i.e., identification of the correct proteins rather than determination of the relative abundance of each protein in the sample. Many MS-based quantitative proteomic approaches were developed in the past decade for global measurement of proteome changes or targeted analysis of biomarkers of disease or drug response [19-23]. These technologies were further improved more recently to achieve ultra high resolution, selectivity, sensitivity, and speed of peptide measurement [24-27], which resulted in higher throughput and nearly complete analysis of proteomes [28-31]. Although recent efforts were made to initiate the evaluation of the intra- and inter-laboratory performance of targeted proteomics approaches [32, 33], the performance of majority of global quantitative proteomic approaches has not been evaluated thoroughly. What should be considered as the “gold-standard” for proteomics data evaluation? In our opinion, similar as already proposed by others [34], a high quality proteomics dataset should fulfill at least two criteria: 1) repeatability of data generated within the same laboratory and 2) consistency of data generated from different laboratories. However, without further quantitative definition of consistence, questions will remain such as what is the minimum level of quality required to accept or reject proteomics data?
In order to begin addressing these questions, we reviewed research articles published in leading proteomics journals including Molecular & Cellular Proteomics, Journal of Proteome Research, Journal of Proteomics, Proteomics, Proteome Science, and Proteomics - Clinical Applications. Our objective was to use the published papers in a calendar year as an example to provide a snapshot of the diversity of subjects and methodologies in the proteomics field. We began our effort in 2009 when the full-year literature of 2008 became available to us. Although the quantitation numbers in such a snapshot might change from year to year, the diversity of the proteome analyzing techniques, protein concentration determination methods, and the proteome study subjects will remain unchanged in the foreseeable future. The analysis determined there were 903 papers published in those journals in that year. Excluding method/technology/software development papers, short/brief/rapid communications, and papers focused on sub-proteomes (e.g., membrane proteomics, mitochondria proteomics, and nuclear proteomics), 521 received further in-depth review (Supplemental Table 1). Based on these publications, we were able to draw the following conclusions. First, diverse protein extraction methods have been used, not only for samples of different origins (e.g., tissues, cell lines, microorganisms, and plants) but also for the same type of sample. It is unknown how different the extraction efficiencies are for these variable protein extraction methods and in what degree the variety of methods affect the experimental outcomes. As an indicator for protein extraction efficiency, one could simply state that X gram protein was yielded from Y gram of tissue or Z mL of body fluid. However, only a small number of papers (< 4%) reported protein yield from original samples. While the extracted protein amount from a sample might not be a major concern for some research laboratories that study differential protein levels in a relative sense, it is highly relevant from the regulatory perspective, especially concerning data acquisition following Good Laboratory Practice, and when data from different sources need to be compared and reviewed by the regulatory agencies. Next, different methods such as Bradford, BCA, and Lowry assays are employed for protein concentration measurements. Although these are routine laboratory procedures, different assays may or may not lead to different conclusions depending on what specific protein targets are analyzed by the subsequent analytical procedure. Also, a large number of the papers (~40%) did not mention what method was used in determining protein concentrations in their experimental procedures at all, which make data comparisons even more difficult. Finally, mass spectrometry (MS) is by far the most-widely used tool for proteomic analyses, employed in ~ 95% of the studies. While our literature review may not provide the most comprehensive and up-to-date statistics, since a huge number of changes have occurred since 2009 in the proteomics field including the increased usage of labeled quantitative MS analyses, it nevertheless demonstrated the diversity of the assessed parameters in the proteomics field. Since the trend may not change anytime soon, current efforts towards developing guidelines and standardizations in the proteomics field should focus on MS approaches.
In addition to the above discussion about the protein sample preparation and concentration measurement based on literature, we examined a typical procedure from sample extraction to on-line nanoflow liquid chromatography-tandem mass spectrometry (nanoflow LC-MS/MS) to illustrate additional issues, which might have previously been thought as trivial. This approach, without isotopic labeling, has been widely applied to the identification and quantification of proteins from a variety of biological matrixes, which has been termed as label-free quantitative proteome analysis [35, 36]. There are also many other label-based proteomic approaches relying on stable isotope labeling, including stable isotope labeling by amino acids in cell culture (SILAC) [37], isotope-coded affinity tags (ICAT) [38, 39], isobaric multiplexing tagging reagents for relative and absolute protein quantitation (iTRAQ) [40], tandem mass tags (TMT) [41], enzyme-catalyzed 16O/18O labeling [42-44], etc. All these approaches have been used for the discovery and quantification of biomarker candidates. An emerging tool for verification of biomarker candidates is mass spectrometry-based selected reaction monitoring (SRM) or multiple reaction monitoring (MRM) in which specific biomarker candidates are targeted and detected [20, 45]. For all the various mass spectrometry-based biomarker discovery and verification tools, some technologies may be more sophisticated than the others. Technical evaluation and discussion of each technology is beyond the scope of this commentary. However, each approach involves protein sample preparation, LC-MS/MS, and data analysis. Here we focus on these common techniques and discuss associated issues that might be concerns in regulatory evaluation.
With respect to sample preparation, we examined three commonly used protein concentration assays, Bradford, BCA, and Lowry, and which together cover about 93% of all protein concentration assays according to the literature review. We measured protein concentration of the same mammalian proteome sample using each of the three protein assays and generated standard curves for each method using the same protein standards (see Supplemental Materials for more details). The results demonstrated that different assay methods could indeed generate significantly different outcomes (Fig. 2). Since most laboratories use the same assay for all sample measurements for a specific project, this might not be an issue. However, when different laboratories use diverse assays to determine protein concentrations, it might become a critical issue. For example, one might assume one microgram of peptides was injected onto the LC column while the actual amount could be significantly more or less than one microgram depending on which protein assay method was used.
Figure 2.
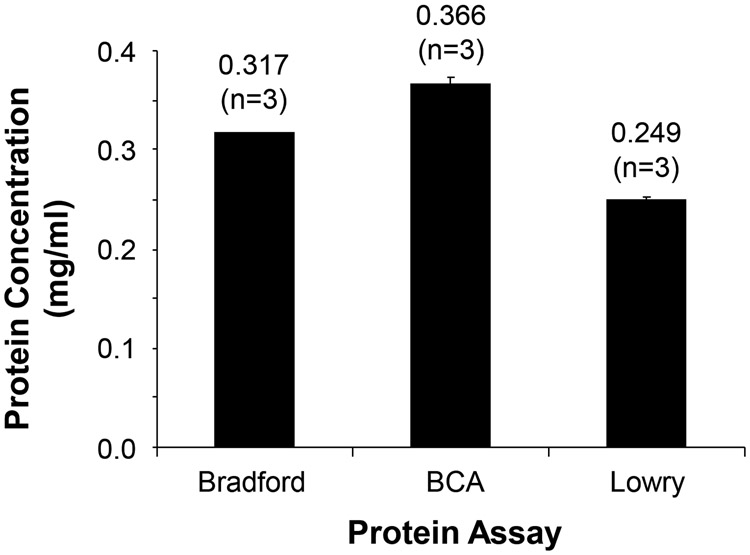
Protein concentration of the same sample was determined using three different protein assay methods (BCA, Bradford, and Lowry). The same bovine serum albumin concentration series were used for generating the standard curve for each method. Error bars indicate standard error of the mean. There is a statistically significant difference among different protein assay methods (p < 0.0001, ANOVA, n = 3 replicates).
To examine the ramifications of varying the amount of protein injected, a study was done to examine the quantitative outcomes. The rationale was that the LC-MS intensities of a peptide, defined as the area under the extracted ion chromatogram (XIC) of the peptide, should be proportional to the amounts of peptides injected onto the LC column. We not only observed that some low-abundant peptides in the sample became undetectable when the injection amount was reduced to a half, but also found that many peptides did not respond in a linear fashion in MS intensities to the sample amounts. For example, some peptides had a reduction of MS intensities of 20-30% instead of the expected 50% (Fig. 3). The inconsistency could be due to variable ionization efficiency of the same peptide in different amounts, peptide species co-elution, slight change of solvent electrospray microenvironment in which the peptide is eluted, etc. The results demonstrated that careful control of sample loading amounts is critical when comparative proteomic analysis is performed and unexpected differential injection of sample amounts could indeed generate significantly different outcomes, especially when label-free quantitative proteomics approaches are used. The results from this study and others [18] could explain some of the cross-laboratory inconsistencies due to unexpected sample loading variations.
Figure 3.
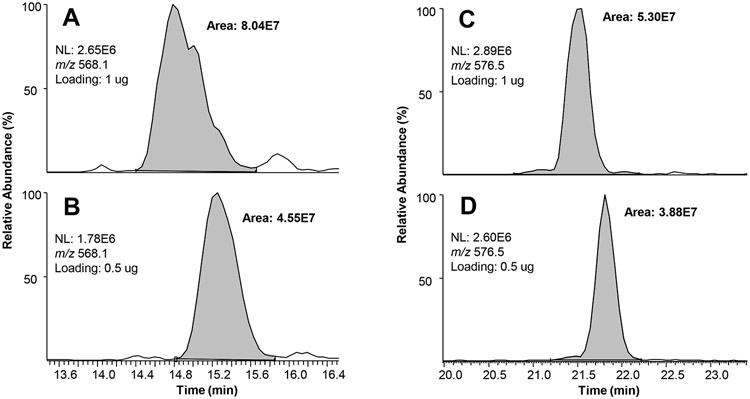
Examples of different peptide loading amounts (1 μg for A and C, and 0.5 μg for B and D, respectively) affecting peptide intensities disproportionally. For method details see Supplemental Materials. A and B show the peptide DAVTYTEHAK (m/z 568.1) had a reduction of intensities (integrated peak area) of ~43% as expected when the amount of sample loading was decreased to a half. However, C and D show an example that the peptide GESPVDYDGGR (m/z 576.5) had a reduction of intensity of ~27% instead of the expected 50%.
Shotgun proteomics is a bottom-up approach that has been increasingly utilized by the scientific community. This approach identifies proteins and characterizes their amino acid sequences and post-translational modifications by proteolytic digestion of proteins first, followed by one or more dimensions of separation of the peptides by liquid chromatography coupled to mass spectrometry for online detection. Since conventional protein extraction buffers often contain protease inhibitors to prevent protein breakdown, it would be worthwhile to know whether or not adding protease inhibitors in the protein extraction buffer would make any difference in the LC-MS/MS analysis for peptide/ protein identification. Based on the literature review mentioned above, 64 studies used the shotgun approach. Among them, 28 (44%) included protease inhibitors in the extraction buffer, while 36 (56%) did not. Thus, clearly there is no consensus of the scientific community with regard of using or omitting protease inhibitors in the shotgun approach. For this reason, a study was performed in which proteins were extracted from the same batch of cultured cells using protein extraction buffers with or without protease inhibitors, followed by trypsin digestion and subsequent 1D LC-MS/MS analyses. The results showed that protein extraction without protease inhibitors led to significantly higher numbers of identified peptides compared to those using extraction buffer with protease inhibitors (Fig. 4). Similar results were obtained for the numbers of proteins identified (data not shown). This could be due to the inhibitory effect of the remaining protease inhibitors in the protein sample on trypsin activity during the digestion process and due to an interference of the remaining protease inhibitors in the peptide sample during MS analyses. By examining the LC-MS spectra, we found that the non-small molecule protease inhibitors remained in the sample and significantly interfered with reversed-phase LC (RPLC) separation and MS/MS data acquisition for peptides. However, this effect could be minimized when the two-dimensional LC-MS/MS approach such as strong cation exchange (SCX) LC coupled with RPLC-MS/MS is employed. Therefore, addition of protease inhibitors or not during sample preparation should be carefully considered along with the subsequent analytical approaches.
Figure 4.
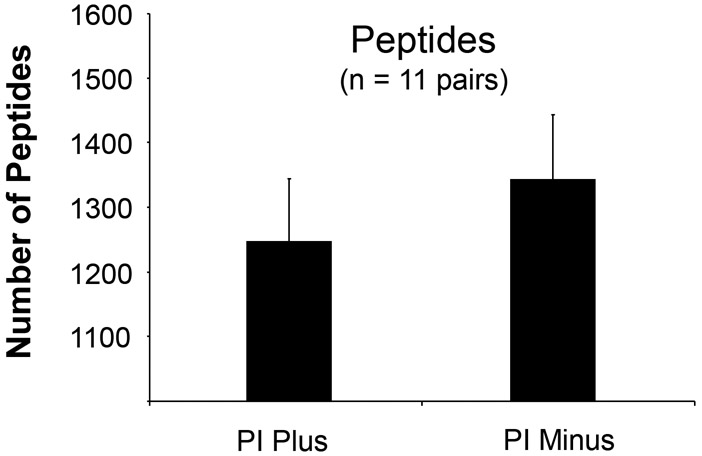
Sample preparation using the protein extraction buffer with or without protease inhibitors (PI) led to different numbers of identified peptides. Proteins were extracted from mouse lymphoma cells (L5178Y/Tk+/−3.7.2C). Error bars indicate standard error of the mean. The number of identified peptides is significantly higher for the samples without protease inhibitors than that with protease inhibitors (p < 0.002, paired t-test, n = 11 pairs of “PI Plus” versus “PI Minus”).
With the evolution of mass spectrometers towards high mass precision instruments, label-free quantification of LC-MS data has become a very appealing approach for the quantitative analysis of biological samples, as it has been demonstrated that the intensity of extracted peptide signals scales linearly in general with their molecular concentration across a dynamic range of 3 to 4 orders of magnitude [46]. Although there are potential aforementioned issues, because of its promising future, we examined this label-free approach, and evaluated consistency of experimental results by repeated mass spectrometric analyses of the same peptide mixture. Whereas variability in peptide LC retention time, sample loading, and column separation efficiency over time are potential issues as discussed in the proteomic community [47], quantitation by calculating the peak area of integrated chromatographic peak of a defined peptide with a specific mass-to-charge ratio (i.e., m/z) is also of concern. Previous reports have already identified software analysis issues [48, 49] such as differences between the two most popular search algorithms, Mascot and Sequest. In terms of accurate quantification, measurement of peak areas is a standard. However, intrinsic variability in peak integration of MS data owing to the software could be a critical factor affecting quantitation accuracy (Fig. 5). While different software may have its own issues, problems in peak integration could be common. Although proteomic scientists may be aware of this issue, the problem has not been fully resolved. While manual integration is possible, it is not practical for high-throughput analyses of hundreds and thousands of peptides. More sophisticated algorithms for MS peak integration should be developed for more accurate quantification.
Figure 5.

An example of software analysis variations in MS peak integration when the same peptide (mitochondrial Stress-70 protein) was repeatedly measured. Automatic peak integration and area calculation were performed using the PepQuan module in Bioworks (Rev. 3.3.1 SP1). The extracted ion chromatograms of the peptide were generated from 5 LC-MS/MS analyses using a linear ion trap mass spectrometer LTQ-XL for the same trypsin-digested proteome sample from mouse lymphoma cells (L5178Y/Tk+/−3.7.2C). The circle indicates an additional peak that was included in the calculation of the integrated peak area value. Since these were technical replicates, these results demonstrate another variance that is caused by the post-analytical data processing even though Good Laboratory Practice has been followed and the reproducibility was kept at an optimal level during analytical experiments.
These results warrant further debates at the next level, i.e., how a regulatory agency should evaluate proteomics data. Identifying molecular changes as early biomarkers for drug safety assessment [50] should have distinct advantages because cellular and tissue damage is often not reversible, and because often times it is already at a late stage when damage is observed by conventional methods. As high-quality data produced are fundamental to the generation of reliable biological knowledge, quality control at each step of the entire experimental and analytical processes is essential to ensure that the detected differences reflect biological but not methodological variations. From a uniquely regulatory perspective, unlike cross-laboratory data comparison for basic science research when discrepancies could be eventually explained and resolved through additional experiments in the future, regulatory agencies such as the FDA bear additional responsibilities, and have to make a timely “Yes” or “No” decision, that could affect the public health. Would there be confidence in the data generated using the same experimental procedures but from two independent laboratories? Even this is practically doable; would two datasets be sufficient to draw an accurate conclusion? Since false-positive or false-negative rates are dependent upon the pre-analytical and analytical variables, should these false-positive and false-negative rates be identified in each dataset? If so, how should they be determined and how reliable are the methods to determine such rates? While the methods and approaches for global and local false discovery rate (FDR) estimation at the peptide level are now well understood, there has been much less development in the area of protein level analysis and the field is essentially lacking a robust and statistically powerful model with a practical software implementation for the analysis of very large proteomic datasets according to a recent evaluation [51]. It is expected that improvement in tools and methods for shotgun proteomic data analysis will further increase the quality of published proteomic data. A recent study that recommended 46 performance metrics for LC-MS analyses [47] is a good start and will definitely facilitate quality control of proteomics data. A recent introduction of yeast protein extract as a proteomics quality control material to evaluate the preanalytical and analytical variables of proteomics-based experimental workflows by the National Institute of Standards and Technology (NIST) could be another helpful approach for improving the accuracy, repeatability and reproducibility [52]. In addition, the analytical instrument and software will also get further improvement over time. However, it seems that there is still a significant work ahead towards establishing a regulatory standard for proteomic data submission and evaluation. The challenge will require a joint effort from the government, academia, and private sectors.
Supplementary Material
ACKNOWLEDGMENTS
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [53] with the dataset identifier PXD000334. This work was supported in part by FDA Commissioner’s Fellowship Program and by an appointment to the Research Participation Program at the National Center for Toxicological Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration. We wish to thank Yuan Gao, Ricky Holland, Xiaoqing Guo, Nan Mei, Reagan Kelly, Joshua Xu, and Hong Fang for providing various levels of help and assistance throughout this study. The views expressed here are those of the authors and not necessarily of the U.S. Food and Drug Administration, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- [1].Goodsaid F, Papaluca M. Evolution of biomarker qualification at the health authorities. Nat Biotechnol 2010;28:441–3. [DOI] [PubMed] [Google Scholar]
- [2].Goodsaid FM, Mendrick DL. Translational medicine and the value of biomarker qualification. Sci Transl Med 2010;2:47ps4. [DOI] [PubMed] [Google Scholar]
- [3].Taylor CF, Paton NW, Lilley KS, Binz PA, Julian RK Jr., Jones AR, Zhu W, Apweiler R, Aebersold R, Deutsch EW, Dunn MJ, Heck AJ, Leitner A, Macht M, Mann M, Martens L, Neubert TA, Patterson SD, Ping P, Seymour SL, Souda P, Tsugita A, Vandekerckhove J, Vondriska TM, Whitelegge JP, Wilkins MR, Xenarios I, Yates JR 3rd, Hermjakob H The minimum information about a proteomics experiment (MIAPE). Nat Biotechnol 2007;25:887–93. [DOI] [PubMed] [Google Scholar]
- [4].Tao F 1st NCI annual meeting on Clinical Proteomic Technologies for Cancer. Expert Rev Proteomics 2008;5:17–20. [DOI] [PubMed] [Google Scholar]
- [5].Taylor CF, Binz PA, Aebersold R, Affolter M, Barkovich R, Deutsch EW, Horn DM, Huhmer A, Kussmann M, Lilley K, Macht M, Mann M, Muller D, Neubert TA, Nickson J, Patterson SD, Raso R, Resing K, Seymour SL, Tsugita A, Xenarios I, Zeng R, Julian RK Jr. Guidelines for reporting the use of mass spectrometry in proteomics. Nat Biotechnol 2008;26:860–1. [DOI] [PubMed] [Google Scholar]
- [6].Binz PA, Barkovich R, Beavis RC, Creasy D, Horn DM, Julian RK Jr., Seymour SL, Taylor CF, Vandenbrouck Y Guidelines for reporting the use of mass spectrometry informatics in proteomics. Nat Biotechnol 2008;26:862. [DOI] [PubMed] [Google Scholar]
- [7].Gibson F, Anderson L, Babnigg G, Baker M, Berth M, Binz PA, Borthwick A, Cash P, Day BW, Friedman DB, Garland D, Gutstein HB, Hoogland C, Jones NA, Khan A, Klose J, Lamond AI, Lemkin PF, Lilley KS, Minden J, Morris NJ, Paton NW, Pisano MR, Prime JE, Rabilloud T, Stead DA, Taylor CF, Voshol H, Wipat A, Jones AR. Guidelines for reporting the use of gel electrophoresis in proteomics. Nat Biotechnol 2008;26:863–4. [DOI] [PubMed] [Google Scholar]
- [8].Domann PJ, Akashi S, Barbas C, Huang L, Lau W, Legido-Quigley C, McClean S, Neususs C, Perrett D, Quaglia M, Rapp E, Smallshaw L, Smith NW, Smyth WF, Taylor CF, Minimum Information About a Proteomics E. Guidelines for reporting the use of capillary electrophoresis in proteomics. Nat Biotechnol 2010;28:654–5. [DOI] [PubMed] [Google Scholar]
- [9].Jones AR, Carroll K, Knight D, Maclellan K, Domann PJ, Legido-Quigley C, Huang L, Smallshaw L, Mirzaei H, Shofstahl J, Paton NW, Minimum Information About a Proteomics E. Guidelines for reporting the use of column chromatography in proteomics. Nat Biotechnol 2010;28:654. [DOI] [PubMed] [Google Scholar]
- [10].Hoogland C, O'Gorman M, Bogard P, Gibson F, Berth M, Cockell SJ, Ekefjard A, Forsstrom-Olsson O, Kapferer A, Nilsson M, Martinez-Bartolome S, Albar JP, Echevarria-Zomeno S, Martinez-Gomariz M, Joets J, Binz PA, Taylor CF, Dowsey A, Jones AR, Minimum Information About a Proteomics E. Guidelines for reporting the use of gel image informatics in proteomics. Nat Biotechnol 2010;28:655–6. [DOI] [PubMed] [Google Scholar]
- [11].Orchard S, Salwinski L, Kerrien S, Montecchi-Palazzi L, Oesterheld M, Stumpflen V, Ceol A, Chatr-aryamontri A, Armstrong J, Woollard P, Salama JJ, Moore S, Wojcik J, Bader GD, Vidal M, Cusick ME, Gerstein M, Gavin AC, Superti-Furga G, Greenblatt J, Bader J, Uetz P, Tyers M, Legrain P, Fields S, Mulder N, Gilson M, Niepmann M, Burgoon L, De Las Rivas J, Prieto C, Perreau VM, Hogue C, Mewes HW, Apweiler R, Xenarios I, Eisenberg D, Cesareni G, Hermjakob H. The minimum information required for reporting a molecular interaction experiment (MIMIx). Nat Biotechnol 2007;25:894–8. [DOI] [PubMed] [Google Scholar]
- [12].Bourbeillon J, Orchard S, Benhar I, Borrebaeck C, de Daruvar A, Dubel S, Frank R, Gibson F, Gloriam D, Haslam N, Hiltker T, Humphrey-Smith I, Hust M, Juncker D, Koegl M, Konthur Z, Korn B, Krobitsch S, Muyldermans S, Nygren PA, Palcy S, Polic B, Rodriguez H, Sawyer A, Schlapshy M, Snyder M, Stoevesandt O, Taussig MJ, Templin M, Uhlen M, van der Maarel S, Wingren C, Hermjakob H, Sherman D. Minimum information about a protein affinity reagent (MIAPAR). Nat Biotechnol 2010;28:650–3. [DOI] [PubMed] [Google Scholar]
- [13].Montecchi-Palazzi L, Kerrien S, Reisinger F, Aranda B, Jones AR, Martens L, Hermjakob H. The PSI semantic validator: a framework to check MIAPE compliance of proteomics data. Proteomics 2009;9:5112–9. [DOI] [PubMed] [Google Scholar]
- [14].Gloriam DE, Orchard S, Bertinetti D, Bjorling E, Bongcam-Rudloff E, Borrebaeck CA, Bourbeillon J, Bradbury AR, de Daruvar A, Dubel S, Frank R, Gibson TJ, Gold L, Haslam N, Herberg FW, Hiltke T, Hoheisel JD, Kerrien S, Koegl M, Konthur Z, Korn B, Landegren U, Montecchi-Palazzi L, Palcy S, Rodriguez H, Schweinsberg S, Sievert V, Stoevesandt O, Taussig MJ, Ueffing M, Uhlen M, van der Maarel S, Wingren C, Woollard P, Sherman DJ, Hermjakob H. A community standard format for the representation of protein affinity reagents. Mol Cell Proteomics 2010;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gibson F, Hoogland C, Martinez-Bartolome S, Medina-Aunon JA, Albar JP, Babnigg G, Wipat A, Hermjakob H, Almeida JS, Stanislaus R, Paton NW, Jones AR. The gel electrophoresis markup language (GelML) from the Proteomics Standards Initiative. Proteomics 2010;10:3073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tabb DL, Vega-Montoto L, Rudnick PA, Variyath AM, Ham AJ, Bunk DM, Kilpatrick LE, Billheimer DD, Blackman RK, Cardasis HL, Carr SA, Clauser KR, Jaffe JD, Kowalski KA, Neubert TA, Regnier FE, Schilling B, Tegeler TJ, Wang M, Wang P, Whiteaker JR, Zimmerman LJ, Fisher SJ, Gibson BW, Kinsinger CR, Mesri M, Rodriguez H, Stein SE, Tempst P, Paulovich AG, Liebler DC, Spiegelman C. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J Proteome Res 2010;9:761–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mischak H, Apweiler R, Banks RE, Conaway M, Coon J, Dominiczak A, Ehrich JH, Fliser D, Girolami M, Hermjakob H, Hochstrasser D, Jankowski J, Julian BA, Kolch W, Massy ZA, Neusuess C, Novak J, Peter K, Rossing K, Schanstra J, Semmes OJ, Theodorescu D, Thongboonkerd V, Weissinger EM, Van Eyk JE, Yamamoto T. Clinical proteomics: A need to define the field and to begin to set adequate standards. Proteomics Clin Appl 2007;1:148–56. [DOI] [PubMed] [Google Scholar]
- [18].Bell AW, Deutsch EW, Au CE, Kearney RE, Beavis R, Sechi S, Nilsson T, Bergeron JJ, Group HTSW. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat Methods 2009;6:423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sabido E, Selevsek N, Aebersold R. Mass spectrometry-based proteomics for systems biology. Curr Opin Biotechnol 2012;23:591–7. [DOI] [PubMed] [Google Scholar]
- [20].Meng Z, Veenstra TD. Targeted mass spectrometry approaches for protein biomarker verification. J Proteomics 2011;74:2650–9. [DOI] [PubMed] [Google Scholar]
- [21].Gerszten RE, Carr SA, Sabatine M. Integration of proteomic-based tools for improved biomarkers of myocardial injury. Clin Chem 2010;56:194–201. [DOI] [PubMed] [Google Scholar]
- [22].Ning MM, Lopez M, Sarracino D, Cao J, Karchin M, McMullin D, Wang X, Buonanno FS, Lo EH. Pharmaco-proteomics opportunities for individualizing neurovascular treatment. Neurol Res 2013;35:448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yu LR. Pharmacoproteomics and toxicoproteomics: the field of dreams. J Proteomics 2011;74:2549–53. [DOI] [PubMed] [Google Scholar]
- [24].Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, Viner R, Schwartz J, Remes P, Belford M, Dunyach JJ, Cox J, Horning S, Mann M, Makarov A. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics 2012;11:O111 013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Altelaar AF, Heck AJ. Trends in ultrasensitive proteomics. Curr Opin Chem Biol 2012;16:206–13. [DOI] [PubMed] [Google Scholar]
- [26].Boja ES, Rodriguez H. Mass spectrometry-based targeted quantitative proteomics: achieving sensitive and reproducible detection of proteins. Proteomics 2012;12:1093–110. [DOI] [PubMed] [Google Scholar]
- [27].Shi T, Sun X, Gao Y, Fillmore TL, Schepmoes AA, Zhao R, He J, Moore RJ, Kagan J, Rodland KD, Liu T, Liu AY, Smith RD, Tang K, Camp DG 2nd, Qian WJ. Targeted quantification of low ng/mL level proteins in human serum without immunoaffinity depletion. J Proteome Res 2013;12:3353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nagaraj N, Kulak NA, Cox J, Neuhauser N, Mayr K, Hoerning O, Vorm O, Mann M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol Cell Proteomics 2012;11:M111 013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hebert AS, Merrill AE, Stefely JA, Bailey DJ, Wenger CD, Westphall MS, Pagliarini DJ, Coon JJ. Amine-reactive Neutron-encoded Labels for Highly Plexed Proteomic Quantitation. Mol Cell Proteomics 2013;12:3360–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ding C, Jiang J, Wei J, Liu W, Zhang W, Liu M, Fu T, Lu T, Song L, Ying W, Chang C, Zhang Y, Ma J, Wei L, Malovannaya A, Jia L, Zhen B, Wang Y, He F, Qian X, Qin J. A fast workflow for identification and quantification of proteomes. Mol Cell Proteomics 2013;12:2370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. The One Hour Yeast Proteome. Mol Cell Proteomics 2013;12:M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJ, Pearson TW, Paulovich AG, Carr SA. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics 2012;11:M111 013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abbatiello SE, Mani DR, Schilling B, Maclean B, Zimmerman LJ, Feng X, Cusack MP, Sedransk N, Hall SC, Addona T, Allen S, Dodder NG, Ghosh M, Held JM, Hedrick V, Inerowicz HD, Jackson A, Keshishian H, Kim JW, Lyssand JS, Riley CP, Rudnick P, Sadowski P, Shaddox K, Smith D, Tomazela D, Wahlander A, Waldemarson S, Whitwell CA, You J, Zhang S, Kinsinger CR, Mesri M, Rodriguez H, Borchers CH, Buck C, Fisher SJ, Gibson BW, Liebler D, Maccoss M, Neubert TA, Paulovich A, Regnier F, Skates SJ, Tempst P, Wang M, Carr SA. Design, implementation and multisite evaluation of a system suitability protocol for the quantitative assessment of instrument performance in liquid chromatography-multiple reaction monitoring-MS (LC-MRM-MS). Mol Cell Proteomics 2013;12:2623–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Eisenacher M, Schnabel A, Stephan C. Quality meets quantity - quality control, data standards and repositories. Proteomics 2011;11:1031–6. [DOI] [PubMed] [Google Scholar]
- [35].Wiener MC, Sachs JR, Deyanova EG, Yates NA. Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal Chem 2004;76:6085–96. [DOI] [PubMed] [Google Scholar]
- [36].Wang M, You J, Bemis KG, Tegeler TJ, Brown DP. Label-free mass spectrometry-based protein quantification technologies in proteomic analysis. Brief Funct Genomic Proteomic 2008;7:329–39. [DOI] [PubMed] [Google Scholar]
- [37].Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002;1:376–86. [DOI] [PubMed] [Google Scholar]
- [38].Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 1999;17:994–9. [DOI] [PubMed] [Google Scholar]
- [39].Yu LR, Conrads TP, Uo T, Issaq HJ, Morrison RS, Veenstra TD. Evaluation of the acid-cleavable isotope-coded affinity tag reagents: application to camptothecin-treated cortical neurons. J Proteome Res 2004;3:469–77. [DOI] [PubMed] [Google Scholar]
- [40].Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 2004;3:1154–69. [DOI] [PubMed] [Google Scholar]
- [41].Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 2003;75:1895–904. [DOI] [PubMed] [Google Scholar]
- [42].Schnolzer M, Jedrzejewski P, Lehmann WD. Protease-catalyzed incorporation of 18O into peptide fragments and its application for protein sequencing by electrospray and matrix-assisted laser desorption/ionization mass spectrometry. Electrophoresis 1996;17:945–53. [DOI] [PubMed] [Google Scholar]
- [43].Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem 2001;73:2836–42. [DOI] [PubMed] [Google Scholar]
- [44].Gao Y, Gopee NV, Howard PC, Yu LR. Proteomic analysis of early response lymph node proteins in mice treated with titanium dioxide nanoparticles. J Proteomics 2011;74:2745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics 2006;5:573–88. [DOI] [PubMed] [Google Scholar]
- [46].Levin Y, Schwarz E, Wang L, Leweke FM, Bahn S. Label-free LC-MS/MS quantitative proteomics for large-scale biomarker discovery in complex samples. J Sep Sci 2007;30:2198–203. [DOI] [PubMed] [Google Scholar]
- [47].Rudnick PA, Clauser KR, Kilpatrick LE, Tchekhovskoi DV, Neta P, Blonder N, Billheimer DD, Blackman RK, Bunk DM, Cardasis HL, Ham AJ, Jaffe JD, Kinsinger CR, Mesri M, Neubert TA, Schilling B, Tabb DL, Tegeler TJ, Vega-Montoto L, Variyath AM, Wang M, Wang P, Whiteaker JR, Zimmerman LJ, Carr SA, Fisher SJ, Gibson BW, Paulovich AG, Regnier FE, Rodriguez H, Spiegelman C, Tempst P, Liebler DC, Stein SE. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol Cell Proteomics 2010;9:225–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Callister SJ, Barry RC, Adkins JN, Johnson ET, Qian WJ, Webb-Robertson BJ, Smith RD, Lipton MS. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J Proteome Res 2006;5:277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Balgley BM, Laudeman T, Yang L, Song T, Lee CS. Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol Cell Proteomics 2007;6:1599–608. [DOI] [PubMed] [Google Scholar]
- [50].Gao Y, Holland RD, Yu LR. Quantitative proteomics for drug toxicity. Brief Funct Genomic Proteomic 2009;8:158–66. [DOI] [PubMed] [Google Scholar]
- [51].Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics 2010;73:2092–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Beasley-Green A, Bunk D, Rudnick P, Kilpatrick L, Phinney K. A proteomics performance standard to support measurement quality in proteomics. Proteomics 2012;12:923–31. [DOI] [PubMed] [Google Scholar]
- [53].Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 2013;41:D1063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.