

Abstract
Aberrant CXCR4/CXCL12 signaling is involved in many pathophysiological processes such as cancer and inflammatory diseases. A natural fragment of serum albumin, named EPI-X4, has previously been identified as endogenous peptide antagonist and inverse agonist of CXCR4 and is a promising compound for the development of improved analogues for the therapy of CXCR4-associated diseases. To generate optimized EPI-X4 derivatives we here performed molecular docking analysis to identify key interaction motifs of EPI-X4/CXCR4. Subsequent rational drug design allowed to increase the anti-CXCR4 activity of EPI-X4. The EPI-X4 derivative JM#21 bound CXCR4 and suppressed CXCR4-tropic HIV-1 infection more efficiently than the clinically approved small molecule CXCR4 antagonist AMD3100. EPI-X4 JM#21 did not exert toxic effects in zebrafish embryos and suppressed allergen-induced infiltration of eosinophils and other immune cells into the airways of animals in an asthma mouse model. Moreover, topical administration of the optimized EPI-X4 derivative efficiently prevented inflammation of the skin in a mouse model of atopic dermatitis. Thus, rationally designed EPI-X4 JM#21 is a novel potent antagonist of CXCR4 and the first CXCR4 inhibitor with therapeutic efficacy in atopic dermatitis. Further clinical development of this new class of CXCR4 antagonists for the therapy of atopic dermatitis, asthma and other CXCR4-associated diseases is highly warranted.
KEY WORDS: CXCR4 antagonist, Asthma, Allergy, Inflammation, Atopic dermatitis
Graphical abstract
Dysregulation of CXCR4/CXCL12 signaling is associated with many inflammatory diseases. EPI-X4 is an endogenous peptide antagonist of CXCR4. Molecular docking and mutational analysis allowed to increase its anti-CXCR4 activity. The optimized EPI-X4 JM#21 derivative prevented inflammatory responses in mouse models of atopic dermatitis and allergen-induced asthma.

1. Introduction
The CXCR4/CXCL12 signaling axis plays a pivotal role in numerous biologic processes, i.e., organ development, hematopoiesis, vascularization, cell and tissue renewal, hematopoietic stem cell retention, inflammation, and immune control1, 2, 3, 4, 5. In addition, deregulation of this axis, e.g., by overexpression of CXCR4, is associated with a variety of diseases, such as tumor development, progression and metastasis, neurodegenerative and autoimmune disorders, as well as immunodeficiencies or inflammatory diseases, including atopic dermatitis and several forms of asthma6, 7, 8, 9, 10, 11. CXCR4 is a long-known drug target and several small molecules, peptide or antibody-based CXCR4 antagonists are currently in preclinical and clinical development12. However, the only clinically used CXCR4 antagonist is Plerixafor (Mozobil, AMD3100), which is approved as stem cell mobilizing agent in autologous transplantation of bone marrow cells in patients with non-Hodgkin's lymphoma or multiple myeloma, but not suitable for the treatment of chronic disease because of significant adverse effects13,14.
One CXCR4/CXCL12-linked disease is asthma, a pathologic overresponse (airway hyper-responsiveness) to a wide variety of stimuli. Inhaled allergens, for example, are detected by pattern recognition receptors on the surface of airway epithelial cells. This triggers secretion of a variety of inflammatory cytokines which stimulate, e.g., lung-associated dendritic cells, that migrate into draining lymph nodes and induce a type 2 allergic response by activation of effector cells. Eosinophilia and increased influx of TH2 cells can then be measured in broncho-alveolar lavage (BAL) or bronchial biopsies. Continuous stimuli may lead to chronic inflammation resulting in severe asthma symptoms including increased mucus secretion, thickening of the airway wall and subsequent narrowing of the airway lumen, inducing shortness of breath, wheeze, cough, and chest tightness15, 16, 17. Asthma requires treatment with inhaled corticosteroids plus a second controller. Severe forms are treated with systemic corticosteroids and nowadays also monoclonal antibodies that target proinflammatory cytokines18. As in other inflammatory diseases, the CXCR4/CXCL12 axes is involved in asthma pathology: CXCL12 is released in high concentrations into BAL of patients with asthma and correlates with leukocyte infiltration suggesting that it contributes to allergic cell recruitment in asthma19. In addition, CXCR4 expression was shown to be upregulated on eosinophils in the BAL of patients with lung eosinophilia11,20 and CXCR4 overexpression on leukocytes is associated with stronger inflammatory processes19,21. In mouse models of asthma, targeting CXCR4 with an anti-CXCR4 antibody, or AMD3100, or by neutralizing CXCL12 with inhibiting ligands, reduced eosinophilia and migration of immune cells into the airways, and subsequent release of inflammatory cytokines21, 22, 23, 24, 25.
CXCR4/CXCL12 also plays a role in inflammatory diseases of the skin, in particular psoriasis, as both molecules are upregulated in inflamed areas of the skin in transgenic mice, as well as human biopsies9,26,27. Moreover, treatment with AMD3100 and CXCL12 neutralizing antibodies inhibited skin inflammation in a psoriasis mouse model26. However, little is known about the involvement of CXCR4/CXCL12 in atopic dermatitis, a chronically relapsing inflammatory skin disorder with a prevalence of about 1%–3% in adults, and 15%–20% in children, often accompanied by allergic rhinitis, asthma, food allergy, and conjunctivitis28. Inflammation is caused by an array of chemokines and adhesion factors that recruit leukocytes to the skin, where they differentiate and become activated by, e.g., dendritic cells, allergens or yet unknown factors29. These processes result in disturbed skin barrier function, an elevated and more frequent allergic response, as well as defects in antimicrobial immune defense30. Currently, there is no cure for atopic dermatitis. The inflamed skin is treated with topical immunosuppressive corticosteroids and calcineurin inhibitors or, in more severe cases, with oral corticosteroids and dupilumab, a monoclonal antibody that inhibits action of proinflammatory cytokines IL-4 and IL-1331. However, prolonged therapy with corticosteroids and calcineurin is associated with side effects and antibody therapy is relatively expensive, necessitating the development of new treatment options32.
Collectively, these data show that antagonizing CXCR4 is a seminal approach for the treatment of asthma and chronic inflammatory diseases of the skin. One promising novel CXCR4 antagonist is EPI-X4 (endogenous peptide inhibitor of CXCR4)33, 34, 35. EPI-X4 is a 16-mer peptide derived from human serum albumin (HSA) originally identified as inhibitor of CXCR4-tropic HIV-134,37. It binds to CXCR4 thereby abrogating binding of CXCL12 and the HIV-1 gp120. Interestingly, EPI-X4 not only antagonized CXCL12-induced CXCR4 signaling but also reduced the basal signaling activity of the receptor in the absence of the chemokine, rendering EPI-X4 also an inverse agonist of CXCR434. The peptide is released from albumin by ubiquitous aspartic proteases, but its physiological function is still unclear36. Binding of EPI-X4 to CXCR4 is highly specific as the peptide does not interfere with signaling of other chemokine/chemokine receptor pairs34. Furthermore, EPI-X4 was shown to block CXCL12-mediated receptor internalization and suppressed the migration and invasion of cancer and immune cells towards a CXCL12 gradient. Studies in mice provided proof-of concept evidence that EPI-X4 has therapeutic potential since the peptide mobilized hematopoietic stem/progenitor cells and inhibited recruitment of inflammatory immune cells into the lung. However, high concentrations of the endogenous peptide were administered (∼2 mg EPI-X4 per mouse i.p.)33. A preliminary structure–activity relationship (SAR) study allowed to increase the CXCR4 blocking activity of EPI-X4 by about 1.5–2 orders of magnitude33. We here set out to further optimize the anti-CXCR4 activity of EPI-X4 using rational drug design and to evaluate the therapeutic potential of the lead candidate in mouse models of asthma and atopic dermatitis. We show that the optimized EPI-X4 analog JM#21 binds CXCR4 with high affinity and antagonizes CXCL12-elicited CXCR4 responses in vitro. Most notably, topical application of EPI-X4 JM#21 prevented inflammation of the skin and lungs in mouse models of dermatitis and asthma.
2. Results
2.1. Molecular docking of EPI-X4 binding to CXCR4
Our previous SAR analysis allowed to increase the CXCR4 antagonizing activity of EPI-X4 (IC50 in the HIV-1 inhibition assay of ∼5.9 μmol/L by approximately 23-fold, resulting in the first-generation lead WSC02 with an IC50 of 254 nmol/L (Table 133). Compared to the parental peptide, the WSC02 analog is C-terminally truncated by four residues and contains four amino acid substitutions (L1I, Y4W, T5S and Q10C). The cysteine at position 10 was introduced to enable dimerization or coupling reactions to linker, fatty acid or other moieties. To further improve the CXCR4 antagonizing activity, we first aimed at obtaining an overview of the interactions of EPI-X4 with the receptor at the molecular level. For this purpose, we modeled the structure of EPI-X4 bound to CXCR4 using molecular docking. The resulting protein‒peptide complex shows that EPI-X4 binds in the extracellular pocket of CXCR4 (Fig. 1A). EPI-X4 establishes salt-bridge interactions with negatively charged residues of CXCR4 via the positively charged amino acids L1, K6 and K7. Thus, the positively charged N-terminus (L1) of EPI-X4 interacts with E31 of CXCR4 (Fig. 1B). K6 and K7 form salt-bridges with D187 and D262 of CXCR4, respectively, and the nonpolar amino acid V2 of EPI-X4 establishes hydrophobic contacts with F29 and A180 of CXCR4.
Table 1.
Structure–activity relationship (SAR) study using WSC02 as a template.
| Compd. | Sequence | IC50 (nmol/L)a |
|---|---|---|
| EPI-X4 | LVRYTKKVPQVSTPTL | 5924 ± 2490 |
| WSC02 | IVRWSKKVPCVS | 254 ± 34 |
| JM#1 | ILRWSKKVPCVS | 27 ± 10 |
| JM#2 | IFRWSKKVPCVS | 123 ± 37 |
| JM#3 | IVRWSRKVPCVS | 166 ± 26 |
| JM#4 | IVRWSHKVPCVS | 202 ± 67 |
| JM#5 | IVRWSKKLPCVS | 241 ± 68 |
| JM#6 | IVRWSKKIPCVS | 204 ± 22 |
| JM#7 | IVRWSKKFPCVS | 119 ± 68 |
| JM#8 | ILRWSRKVPCVS | 36 ± 18 |
| JM#9 | ILRWSHKVPCVS | 42 ± 34 |
| JM#10 | IFRWSRKVPCVS | 50 ± 39 |
| JM#11 | IFRWSHKVPCVS | 69 ± 64 |
| JM#13 | ILRWSRKMPCVS | 16 ± 9 |
| JM#18 | ILRWSRKMPCLS | 51 ± 20 |
| JM#19 | ILRWSRKMPCFS | 55 ± 12 |
| JM#20 | ILRWSRKMPCMS | 121 ± 23 |
| JM#21 | ILRWSRKLPCVS | 7 ± 1 |
| JM#23 | ILRWSRKVPSVS | 42 ± 8 |
IC50 values shown are means derived from one representative X4-HIV-1 inhibition experiment±SEM. IC50 values were calculated in GraphPad Prism by non-linear regression; amino acid single letter code is used; amino acid residues labeled in red indicate changes as compared to WSC02.
Figure 1.

Binding mode of EPI-X4 with CXCR4 obtained by molecular docking. (A) Position of EPI-X4 (cyan) in the binding pocket of CXCR4 (cartoon). (B) Important interactions that stabilize the protein‒peptide complex: L1 (N-terminal amine), K6 and K7 of EPI-X4 are involved in salt-bridge interactions (indicated by green lines) with E31, D187 and D262, respectively. Hydrophobic interactions (indicated by yellow lines) are established between V2 of EPI-X4 and F29 and A180 of CXCR4.
2.2. Generation of EPI-X4 WSC02 analogs with increased anti-CXCR4 activity
The docking model suggests that mutation at position 2 to a slightly longer aliphatic chain (such as V2L) or to a relatively large non-polar sidechain (such as V2F) might improve the hydrophobic contacts between the peptide and the protein. As mentioned above, K6 was also found to be involved in salt-bridge interactions with CXCR4. This supports the hypothesis that changing this amino acid by arginine would increase the binding affinity, since the guanidinium group of arginine can interact strongly with carboxylate groups (via bidentate H-bonds), as compared to the ammonium group. Thus, these and other amino acid substitutions at position 2 and 6 were introduced in WSC02 in order to enhance anti-CXCR4 activity (Fig. 2A and Table 1). The CXCR4 antagonizing activity of this new set of EPI-X4 peptides (JM#1–JM#7) was then determined in a CXCR4-tropic HIV-1 infection assay, as described33. We used this assay as primary screening test because i) it allows the accurate and fast determination of IC50 values, and ii) the anti-HIV-1 activity correlates well with the activity in T-cell migration assays, which are more elaborate33. We found, that replacement of the aliphatic and hydrophobic valine (V) at position 2 by leucine (L), harboring a longer functional side-chain (JM#1), increased the antiviral activity of WSC02 by 9-fold (IC50 = 27 nmol/L). Substitution by the aromatic phenylalanine (F) had a smaller beneficial effect (JM#2, IC50 = 123 nmol/L). Also, the replacement of the positively charged lysine (K) at position 6 by the positively charged arginine (R) increased the activity of WSC02 (JM#3, IC50 = 166 nmol/L). Replacement of K6 with the weakly basic amino acid histidine (H), however, had no enhancing effect (JM#4, IC50 = 202 nmol/L). Substitution of V at position 8 by either L (JM#5) or isoleucine (I, JM#6) did not significantly alter antiviral activity (IC50 values of 241 and 204 nmol/L, respectively). However, substitution with F increased the anti-HIV-1 activity about 2-fold (JM#7, IC50 = 119 nmol/L, Fig. 2A and Table 1). Combining beneficial substitutions (JM#8‒JM#11) resulted in analogs with antiviral activities in the two-digit nanomolar range, with JM#8 (V2L and K6R) as the most active analog (IC50 = 36 nmol/L, Fig. 2B and Table 1). Subsequently, based on those findings, additional amino acid substitutions were introduced (JM#13–JM#23, Fig. 2C). First, the V at position 8 in JM#8 was replaced by methionine (M, JM#13) or L (JM#21). Those two derivatives were even more active with IC50 values of 16 and 7 nmol/L, respectively. Based on the sequence of JM#13, V at position 11 was further replaced by either L, F, or M (JM#18–JM#20) with no beneficial effects. Finally, cysteine (C) 10 in JM#8 was substituted with non-reactive serine. The resulting JM#23 analog was about as active as JM#8 (42 vs. 36 nmol/L) suggesting that C10 is not involved in receptor binding (Fig. 2C and Table 1). EPI-X4 analog JM#21 consistently blocked HIV-1 infection with the lowest IC50 of 7 ± 1 nmol/L. Thus, it was selected as lead candidate for further characterization.
Figure 2.

EPI-X4 WSC02 analogues inhibit X4-tropic HIV-1 infection with increased potency. WSC02 derivatives with either a single amino acid substitution (A), two distinct amino acid substitutions (B) or a combination of amino acid substitutions (C) were tested against X4-tropic HIV-1. TZM-bl reporter cells were preincubated with serially diluted peptide and afterwards inoculated with the virus. Infection rates were determined by β-galactosidase assay 3 days later. Data were represented as mean ± SEM (n = 3).
2.3. 3D NMR-derived solution structure of JM#21 and WSC02
We next analyzed the secondary structures of WSC02 and JM#21 by NMR. The ten best fitting calculated structures of WSC02 (Fig. 3A) and JM#21 (Fig. 3B) based on NMR experimental data, have a backbone root-mean-square deviation (RMSD) of 1.16 ± 0.33 and 0.83 ± 0.21, respectively. WSC02 (Fig. 3C) shows a ring formed by the complete peptide-chain. This ring involves, in the best fitting state, a hydrogen bond interaction between the sidechains of S12 and W4 (Fig. 3C). Additionally, the carboxylate group of S12 can form a salt-bridge with the amino group of I1. In contrast, JM#21 shows a C-terminal loop, which is, again in the best fitting structural state, based on an interaction between the backbone carboxylate of S12 and the sidechains of R6, K7, L8 and R3, leaving the N-terminus free (Fig. 3E). The fragment W4-S5-R6 exhibits a turn which can be classified as an α-turn with four peptide-bonds being involved (Fig. 3D). The superimposition of the two peptides shows that WSC02 and JM#21 are slightly similar to each other (backbone RMSD = 2.78 Å, Fig. 3E), with the main difference that the optimized EPI-X4 JM#21 contains a flexible N-terminus which might facilitate receptor binding to the extracellular pocket (Fig. 1).
Figure 3.

Solution structure of JM#21 and WSC02. Superimposition of the WSC02 (A) and JM#21 (B) conformers with the lowest final target function values calculated with ARIA. The visualization was performed with the Molecular Graphics Viewer of Visual Molecular Dynamics (VMD)37. (C) and (D) Intramolecular H-bonds in the WSC02 (C) and JM#21 (D) peptides color code: C = pink/blue, H = white, N = blue, and O = red. H-bonds are shown as dotted lines in orange. (E) Superimposed structures of WSC02 (red) and JM#21 (blue).
Next, to compare the putative interactions of EPI-X4 and the optimized derivative with CXCR4, we also performed docking calculations of JM#21 with CXCR4. We found that residues L2, R6 and L8 of JM#21 establish favorable interactions with the pocket of CXCR4. L2 of JM#21 features hydrophobic contacts with F29 at a minimum distance of 3.7 Å (Fig. 4). Notably, the distance between V2 of EPI-X4 and the F29 sidechain (Fig. 1) was larger at 4.3 Å. This agrees with our finding that large non-polar amino acids (L and F) are favored at position 2 (Table 1). Another remarkable difference between JM#21 and EPI-X4 is given by the introduction of R6 in JM#21. R6 forms a strong bi-dentate H-bond with the carboxylate group of D262, compared to the K6EPI-X4/D187CXCR4 interaction in the case of EPI-X4. We also note that the presence of large non-polar amino acids (L, I and F) at position 8 favors the interaction with the receptor, which is exemplified by the hydrophobic contact between L8 of JM#21 and I185 of CXCR4. All these interactions may collectively explain the increased activity of JM#21 with respect to EPI-X4. In addition, we also observed the H-bonding contacts R3JM#21/D187CXCR4 and K7JM#21/E288CXCR4.
Figure 4.
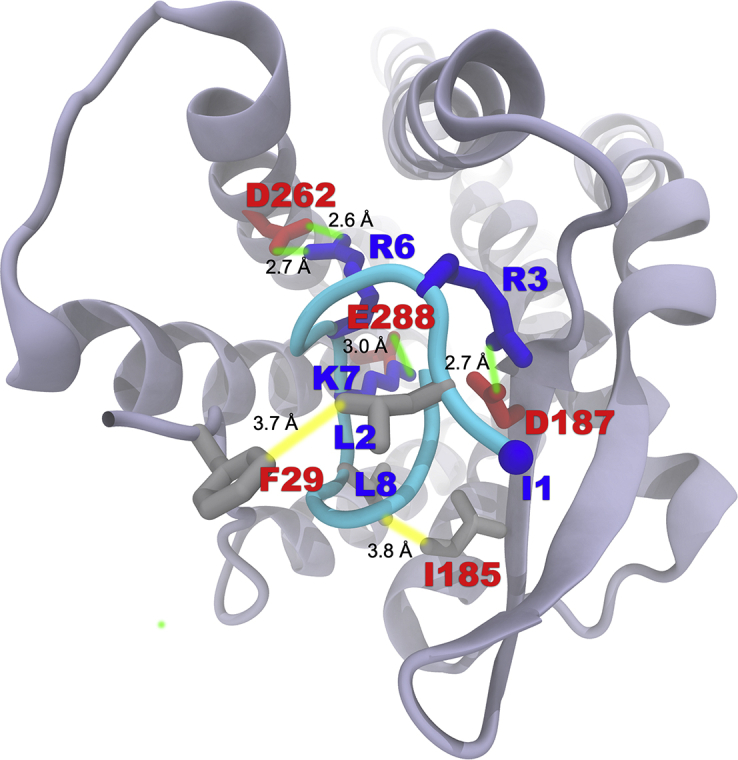
Binding mode of EPI-X4 JM#21 (cyan) with CXCR4 obtained by molecular docking. The color scheme is the same as in Fig. 1.
2.4. EPI-X4 JM#21 is a potent CXCR4 antagonist
We next determined binding affinities of EPI-X4 and analogs to CXCR4 using a recently described CXCR4 antibody competition assay38. This assay is based on the observation that most if not all CXCR4 antagonists interact with CXCR4 in a way that binding of the CXCR4 12G5 antibody raised against the extracellular loop 2 of the receptor is prevented. As shown in Fig. 5, the first- (WSC02) and second-generation (JM#21) EPI-X4 analogs competed with 12G5 binding even more efficiently than AMD3100. JM#21 (IC50 = 223 nmol/L) was ∼2-fold more active than WSC02 (IC50 = 424 nmol/L) and about 3-fold more active than AMD3100 (IC50 = 687 nmol/L, Table 2). For EPI-X4 an IC50 value of 3.24 μmol/L was determined (Table 2).
Figure 5.

Competition of EPI-X4, WSC02, JM#21 and AMD3100 with the CXCR4 ECL-2 antibody 12G5. CXCR4 expressing SupT1 cells were precooled and assay buffer was removed. EPI-X4 and the optimized derivatives WSC02 and JM#21, and the small molecule AMD3100 were diluted in cold PBS and added to the cells. Immediately afterwards, a constant concentration of the APC-labelled CXCR4 antibody (clone 12G5) was added. After 2 h incubation at 4 °C unbound antibody was removed and cells analyzed in flow cytometry. Data were represented as mean ± SEM (n = 3). IC50 values were calculated by non-linear regression.
Table 2.
Comparison between JM#21, WSC02, EPI-X4 and AMD3100.
| Compd. | IC50±SEM (μmol/L) |
||||
|---|---|---|---|---|---|
| 12G5a | X4-HIV-1b | pERKc | pAKTc | Migrationd | |
| EPI-X4 | 3.24 ± 0.62 | 5.924 ± 2.49 | >100 | 67.60 ± 30.63 | 681.63 ± 305.54 |
| WSC02 | 0.42 ± 0.07 | 0.25 ± 0.03 | 15.25 ± 3.97 | 37.97 ± 26.94 | 15.83 ± 3.50 |
| JM#21 | 0.22 ± 0.02 | 0.007 ± 0.001 | 1.54 ± 0.51 | 2.62 ± 1.40 | 0.48 ± 0.08 |
| AMD3100 | 0.69 ± 0.05 | 0.012 ± 0.002 | 3.28 ± 0.45 | 35.02 ± 27.22 | 0.06 ± 0.01 |
Competition with the CXCR4 specific 12G5 antibody.
Inhibition of CXCR4-tropic HIV-1 on TZM-bl cells.
Inhibition of CXCL12 induced ERK or AKT phosphorylation.
Inhibition of SupT1 migration towards a CXCL12 gradient. IC50 values are represented as mean ± SEM (n = 3).
Calcium ions (Ca2+) are important intracellular messengers since they exert regulatory effects on cytosolic enzymes and proteins and are often induced after CXCR4 activation39. Thus, in the next set of experiments, the effect of EPI-X4, its analogs and AMD3100 on CXCL12-induced intracellular Ca2+ mobilization was analyzed in BCR-ABL transformed bone marrow cells via flow cytometry40. As shown in Fig. 6A and B, CXCL12 treatment induced a transient Ca2+ response which was entirely disrupted by 100 μmol/L of the JM#21 analog. At the same concentration, WSC02 only suppressed Ca2+ release by ∼80%, and EPI-X4 was almost inactive at the tested concentrations (Fig. 6A and B). Here, AMD3100 was more active and reduced Ca2+ mobilization already at a concentration of 1 μmol/L, and completely abrogated it at a concentration of 10 μmol/L.
Figure 6.

Effect of EPI-X4 derivatives and AMD3100 on CXCL12-evoked Ca2+ mobilization and ERK and AKT signaling. (A) Inhibition of CXCL12-induced calcium release. BCR-ABL expressing murine pro/pre B cells were loaded with Indo-1 AM and incubated with inhibitors for 10 min. Baseline fluorescence signal was recorded for 30 s and calcium flux induced by stimulation with 100 ng/mL mCXCL12 (black arrow). Signal was recorded for additional 260 s. (B) Areas under the curves (AUC) was calculated after baseline subtraction. Data were represented as mean ± SEM (n = 3). (C) and (D) Inhibition of CXCL12-induced ERK and AKT phosphorylation. SupT1 cells were preincubated with indicated concentrations of compounds for 15 min in starvation medium and afterwards stimulated with 100 ng/mL CXCL12 for 2 min. Cells were then permeabilized and stained for phosphorylated ERK (C) or phosphorylated AKT (D) and analyzed in flow cytometry. Data were represented as mean ± SEM (n = 3). IC50 values were determined by non-linear regression. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P > 0.0001; ns = not significant (one-way ANOVA in comparison to PBS control).
CXCL12 binding to CXCR4 also activates the protein kinase B (PKB, AKT) and extracellular-regulated kinase (ERK) signaling pathways, which are both aberrantly activated in a wide range of human cancers and inflammatory diseases41, 42, 43. We determined the effect of the CXCR4 antagonists on CXCL12-induced phosphorylation of ERK and AKT in T lymphoblasts by phospho-flow cytometry. JM#21 potently suppressed CXCL12-induced ERK and AKT phosphorylation with IC50 values of 1.5 μmol/L (Fig. 6C) and 2.6 μmol/L (Fig. 6D), respectively (Table 2). AMD3100 was slightly less active compared to JM#21 for inhibition of ERK phosphorylation (Fig. 6C) and markedly less potent for AKT phosphorylation with IC50 values of 3.3 and 35.0 μmol/L, respectively (Fig. 6D and Table 2). EPI-X4 was least active for inhibition of pERK and pAKT (Table 2).
Finally, the four compounds were analyzed for their ability to block migration of the T lymphoblastic lymphoma cell line SupT1 along a CXCL12 gradient. JM#21 efficiently blocked CXCL12-induced cell migration with IC50 values of 0.48 μmol/L (Fig. 7), which was 33-fold more efficient as compared to WSC02 (IC50 = 15.8 μmol/L) and more than 1400-fold more efficient than the wild type peptide EPI-X4 (IC50 = 681.6 μmol/L). In this assay, AMD3100 was most potent with an IC50 of 0.06 μmol/L (Fig. 7 and Table 2).
Figure 7.

Migration of SupT1 cells in the presence of CXCR4 ligands towards a CXCL12 gradient. The assay was performed in a transwell plate with 5 μm pore size with 100 ng/mL CXCL12 in the lower chamber. The number of migrated cells was determined by CellTiterGlo assay. Data were represented as mean ± SEM (n = 3).
2.5. Stability of EPI-X4 JM#21 in human plasma
EPI-X4 and EPI-X4 WSC02 have a relatively low proteolytic stability in human and mouse plasma with half-lives of ∼17 and 9 min, respectively33,38. Compared to that, AMD3100 was resistant to plasma proteases38. To follow up on those results, we determined stability of EPI-X4 JM#21 in human plasma under identical conditions. JM#21 exhibits an even lower stability compared to the parental peptides with a half-life of ∼6 min (Fig. 8) arguing for topical or intranasal administration.
Figure 8.
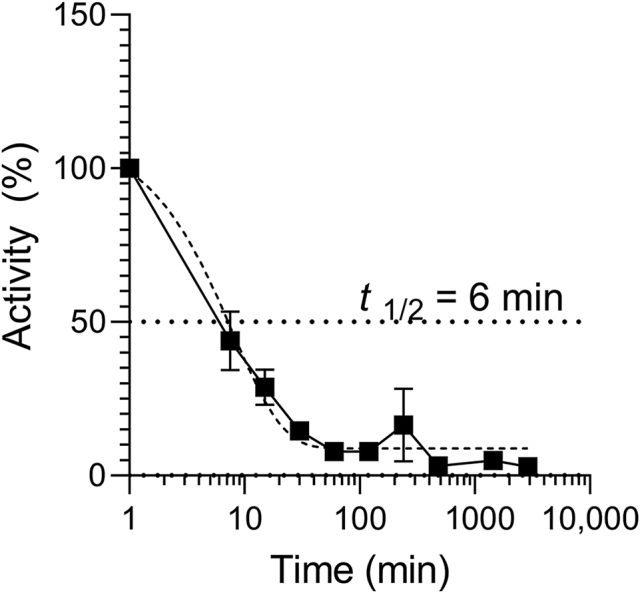
Stability of EPI-X4 JM#21 in human plasma. 20 μmol/L of EPI-X4 JM#21 was diluted in 100% human plasma. The mixture was incubated at 37 °C and samples taken at indicated time points and stored at −80 °C. Samples were thawed in parallel, serially diluted in PBS and activity determined by 12G5-antibody competition. IC50 values were determined by non-linear regression and remaining activity calculated by IC50 (t = 0)/IC50 (t) × 100. Data were represented as mean ± SEM (n = 3).
2.6. EPI-X4 JM#21 shows no toxicity in zebrafish
Zebrafish embryos provide a useful in vivo system for toxicity assessments of chemicals, since their organs and tissues are similar to those of mammals on the molecular, physiological, and basic anatomical level44. To evaluate toxicity, zebrafish embryos were exposed for 24 h to a maximum of 300 μmol/L of the peptides or controls, and then analyzed in a stereomicroscope for acute toxicity (lethality, necrosis and lysis), developmental toxicity (developmental delay and malformations) and cardiotoxicity (heart edema, reduced or absent circulation). None of the EPI-X4 analogs induced signs of toxicity whereas the membrane damaging antimicrobial peptide pleurodicin NRC0345 was acutely toxic (Fig. 9).
Figure 9.

JM#21 is not toxic in an embryonic zebrafish model. At 24 h post fertilization, dechorionated zebrafish embryos were exposed to EPI-X4 (A), WSC02 (B) or JM#21 (C) for 24 h. As negative control PBS was used, and as positive control the antimicrobial peptide NRC-03. Shown are data derived from 60 embryos per group.
2.7. EPI-X4 JM#21 suppresses airway inflammation in a mouse model of allergic airway hypereosinophilia
It has previously been shown that high doses of EPI-X4 reduce inflammatory cell infiltration in a mouse model of allergic airway hypereosinophilia33. To examine whether the EPI-X4 derivative JM#21 may be more potent in preventing airway inflammation, parental EPI-X4 and analogues analogs (16 μmol/kg body weight) and AMD3100 (12.6 μmol/kg body weight) were administered intranasally prior to allergen (OVA) challenge. As controls, mice were not challenged with OVA, and solvent only was administered. At day 8, bronchoalveolar lavage was collected and the number of infiltrated cells determined by flow cytometry (Fig. 10A). Allergen challenge resulted in significant infiltration of eosinophils (Fig. 10C), T cells (Fig. 10D), B cells (Fig. 10E) and neutrophils (Fig. 10F), also demonstrated by the total increase in cell count (Fig. 8B), compared to unchallenged control mice. The parental peptide EPI-X4 inhibited the total cell count by 55 ± 7% (P < 0.01), the recruitment of eosinophils by 76 ± 7% (P < 0.01), neutrophils by 63 ± 7% (P < 0.01), T cells by 61 ± 8% (P < 0.05) and B cells by 54 ± 9% (P < 0.05, Fig. 10B‒E). The N-terminally truncated version of EPI-X4 (ALB409-423), which does not antagonize CXCR434, did not interfere with most immune cells, but showed a significant activity in blocking neutrophil infiltration, for unknown reasons. Interestingly, the lead candidate JM#21 was most effective in suppressing migration of immune cells into the airways. Specifically, JM#21 inhibited the recruitment of eosinophils by 80 ± 1% (P < 0.01), T cells by 73 ± 13% (P < 0.01), B cells by 71 ± 5% (P < 0.05), and neutrophils by 42 ± 15% (P < 0.05), as well as the total inflammatory cell influx by 61 ± 12% (P < 0.01) (Fig. 8B‒F). The control CXCR4 antagonist, AMD3100, also inhibited recruitment of total cells (41 ± 13%, P < 0.05), eosinophils (56 ± 13%, P < 0.05), T cells (47 ± 15%, P < 0.05), B cells (47 ± 17%, P < 0.05) and neutrophils (57 ± 14%, P < 0.05), but was on average less active than JM#21. None of the compounds analysed resulted in infiltration of cells in unchallenged mice. Thus, EPI-X4 JM#21 is an effective inhibitor of lung inflammation and particularly potent in preventing infiltration of eosinophils, which are believed to play a key role in asthma exacerbations46.
Figure 10.

EPI-X4 JM#21 prevents immune cell infiltration into lungs in a mouse model of airway hypereosinophilia. (A) BALB/c mice were sensitized intraperitoneal (i.p.) with a mixture containing 50 μg OVA and 2 mg alum in 0.2 mL saline on day 0, 1 and 2. Mice were treated by intranasal (i.n.) administration with JM#21 (16 μmol/kg), EPI-X4 (16 μmol/kg), ALB409-423 (16 μmol/kg) or AMD3100 (12.6 μmol/kg), 2 h before each OVA or saline challenge on Day 5, 6 and 7. Mice were used for OVA-challenged groups (n = 4–6) and unchallenged controls (n = 2). Number of cells infiltrated into BAL were measured by flow cytometry, (B) all cells; (C) eosinophils; (D) T cells; (E) B cells; (F) neutrophils and (G) macrophages. Data were represented as means ± SEM. #P ≤ 0.05, ##P ≤ 0.01 compared to unchallenged control group, and ∗P ≤ 0.05, ∗∗P ≤ 0.01 compared to CTR + OVA group.
2.8. EPI-X4 JM#21 prevents skin inflammation in a murine model of atopic dermatitis
To evaluate the therapeutic potential of EPI-X4 JM#21 in an inflammatory skin disorder, we tested the peptide in a mouse model of atopic dermatitis. For this, ears of BALB/c mice were treated every 2 days with the vitamin D analog MC903 that induces changes in skin morphology and inflammation resembling immune perturbations observed in acute lesions of patients with AD47. EPI-X4 JM#21 and reference compounds AMD3100 and dexamethasone were applied topically daily (Fig. 11A). Thickness of ears was measured, and severity of the disease scored daily as described in Table 3. None of the compounds induced skin inflammation when administered in the absence of MC903 (Fig. 11B‒D). At day 11, MC903 induced a progressive and significant 3.1 ± 0.1-fold increase of the ear thickness (Fig. 11B and C), and a high disease score of 9.4 ± 0.1 (Fig. 11D) as compared to control (P < 0.0001). Treatment of the ears with JM#21 significantly inhibited ear thickening by 88 ± 4% (P < 0.0001), and the disease score by 88 ± 4% (P < 0.0001). Thus, it was almost as effective as dexamethasone (96 ± 2% and 97 ± 2%, respectively, P < 0.0001, Fig. 11 B‒D). The CXCR4 antagonist AMD3100 had only a minor effect on MC903 induced ear thickness (25 ± 7% inhibition, P < 0.001) and disease score (18 ± 6% inhibition, P < 0.001, Fig. 11 B‒D). These results demonstrate that the optimized second-generation CXCR4 antagonist EPI-X4 JM#21 efficiently inhibits skin inflammation in a mouse model of dermatitis without causing any obvious side effects.
Figure 11.

(A) EPI-X4 JM#21 is active in a mouse model of atopic dermatitis. Ears of BALB/c mice were applied every two days with mock or MC903 (2.5 nmol/ear). (B) EPI-X4 JM#21 (400 nmol/ear), AMD3100 (315 nmol/ear), dexamethasone (Dexa, 20 nmol/ear) or ethanol (EtOH) were applied topically daily. Representative photographs of ears are taken at day 12. Ear thickness (C) and the inflammatory score (D) were determined daily. Mice were used for MC903 (n = 8–16) and controls (n = 4–7). Data shown are mean ± SEM. ####P ≤ 0.0001 compared to CTR + EtOH group, and ∗∗∗P ≤ 0.01, ∗∗∗∗P ≤ 0.0001 compared to MC903 + EtOH group.
Table 3.
Scoring system for atopic dermatitis model.
| Severity | Clinical sign |
||||
|---|---|---|---|---|---|
| Redness | Blooding | Eruption | Crusting | Sum | |
| Absent | 0 | 0 | 0 | 0 | 0/Score min |
| Mild | 1 | 1 | 1 | 1 | 4 |
| Moderate | 2 | 2 | 2 | 2 | 8 |
| Severe | 3 | 3 | 3 | 3 | 12/Score max |
3. Discussion
In this study we show that rational drug design allowed to develop an optimized derivative of an endogenous CXCR4 antagonist that effectively prevents inflammatory reactions in murine models of atopic dermatitis and asthma. EPI-X4 JM#21 differs from WSC02, a first-generation derivative33, by three amino acid substitutions. Two exchanges were introduced because molecular docking studies suggested enhanced contacts of the peptide with the extracellular pocket in CXCR4. A third exchange was thought to achieve extra-flexibility of the mid part of EPI-X4 JM#21. Interestingly, NMR analyses revealed structural differences between JM#21 and WSC02, most notably the free N-terminal end in JM#21. This could explain the superior activity of JM#21 over WSC02 in cell-based assays, because binding with CXCR4 occurs in a random coil state of the peptide and as the peptide enters the CXCR4 core with its N-terminus (Fig. 1), where a free and accessible N-terminal end could be of advantage. Thus, a combination of empiric and rational drug design allowed to increase the anti-CXCR4 activity of EPI-X4 by nearly three orders of magnitude, as measured in the X4-HIV inhibition assay. EPI-X4 JM#21 represents the lead of the 2nd generation EPI-X4 derivatives. It inhibited CXCR4 antibody binding and X4-HIV-1 infection, as well as CXCL12-mediated ERK and AKT activation, Ca2+ mobilization and cell migration more efficiently than WSC02. EPI-X4 JM#21 was also more active than the clinically approved AMD3100, as determined in antibody competition, antiviral and ERK/AKT signaling assays. However, JM#21 was less active than AMD3100 in cell migration and Ca2+ mobilization assays. The reason(s) for these different activities in antagonizing CXCR4 remain to be determined and may reflect different binding modes of the small molecule AMD3100 versus the JM#21 peptide to CXCR4. Notably, none of the EPI-X4 derivatives induced acute, cardio- or developmental toxicity in zebrafish embryos, a widely accepted vertebrate model for toxicity assessment. Similarly, we did not observe visible or behavioral adverse effects of EPI-X4, WSC02 or JM#21 when administered topically onto the ears or intranasally into lungs of mice.
Intranasal application of EPI-X4 and the JM#21 derivative efficiently reduced influx of various immune and inflammatory cells but not macrophages into BAL in a mouse asthma model. Both peptides were more potent that AMD3100, which has previously been shown to attenuate allergen-induced infiltration of inflammatory cells into the airways25,48,49. The mechanism(s) underlying CXCR4-mediated inhibition of asthma are not fully understood, but may involve CXCR4-mediated suppression of inflammatory cell (mainly eosinophil) migration21,23,24, reduced expression of matrix metalloproteinase MMP-9 that drives airway remodeling48, or specific inhibition of CXCR4-expressing neutrophils or eosinophils releasing extracellular traps involved in asthma pathology21,50. Eosinophils are main contributors to the development of bronchial asthma and its exacerbations46,50. EPI-X4 and the optimized JM#21 derivative reduced the number of eosinophils in lungs by 76 ± 7% and 80 ± 1%, respectively, suggesting a preferential application of the peptides in eosinophilic asthma. On average, application of 16 μmol/kg EPI-X4 and the optimized JM#21 analog suppressed inflammatory cell infiltration to a similar extent, which is not reflecting the different activities in antagonizing CXCR4. Perhaps both peptides have been applied at concentrations reaching maximum effects, suggesting that lower doses or less frequent application of JM#21 may result in similar outcomes, which will be analyzed in advanced preclinical development.
Notably, topical application of EPI-X4 JM#21 onto the ears effectively reduced skin inflammation in an atopic dermatitis mouse model. To our best knowledge, this is the first report demonstrating that a CXCR4-antagonizing agent shows therapeutic efficacy in an in vivo model of this inflammatory skin disease. Daily treatment with JM#21 reduced ear thickness and the disease score significantly more efficiently than AMD3100, and almost as efficiently as dexamethasone, an approved corticosteroid. The reason(s) for the differential efficacy of JM#21 and AMD3100 remain to be addressed. Differences in CXCR4 receptor binding and antagonization, or impaired penetration of AMD3100 into the skin, may be an explanation. It will be interesting to see whether a further increase of the EPI-X4 JM#21 dose will result in a complete suppression of disease, as shown for dexamethasone. Thus, these findings warrant further development of EPI-X4 JM#21 as therapeutic agent for atopic dermatitis, especially because the actual continuous treatment of inflamed skin with standard corticosteroids used nowadays is associated with side-effects and novel treatment options are urgently required32.
Our findings demonstrate that EPI-X4 and improved derivatives impair inflammatory reactions in mouse models of atopic dermatitis and asthma more efficiently than the FDA-approved small molecule AMD3100. It will be interesting to see how EPI-X4 derivatives will perform in comparison to other classes of CXCR4 inhibitors, since several different molecules are currently under pre-clinical development for several different diseases with CXCR4/CXCL12 axis involvement. For EPI-X4 JM#21, topical treatment of inflamed skin, or intranasal administration in the airway might be an optimal application strategy, since it circumvents problems associated with peptide drugs, e.g., enzymatic instability and rapid excretion. Since, the CXCR4/CXCL12 axis is also involved in other inflammatory diseases of skin and lungs (e.g., psoriasis26, and pulmonary fibrosis51,52), EPI-X4 JM#21 should also be evaluated as therapeutic agent for these disorders. In contrast to AMD3100, that is known for severe side effects after continued treatment and is therefore not approved for chronic diseases14, a peptide drug based on the body-own EPI-X4 will also most likely be well tolerated. Thus, further preclinical and clinical development of JM#21 as a novel class of CXCR4 inhibitors is highly warranted.
4. Experimental
4.1. Molecular modeling
The reported crystal structure of CXCR4 (PDB ID: 3ODU) was used for the modelling studies53. The missing N-terminal loop was added using the NMR structure of the N-terminus of CXCR4 (PDB ID: 2K04)54. The NMR structures of EPI-X4 (PDB ID: 2N0X)33 and of JM#21 (reported here) were used for the docking studies with CXCR4. The molecular docking calculations were carried out using the HADDOCK v2.2 webserver available at http://www.bonvinlab.org/software/haddock2.2/55,56.
4.2. Reagents and peptide synthesis
All chemicals were used as provided by the manufacturers. Amino acids were purchased from Novabiochem (Merck KGaA, Darmstadt, Germany). N,N-dimethylformamide (DMF), 20% (v/v) piperidine in DMF, O-benzotriazole-N,N,N′,N′-tetramethyluronium-hexafluoro-phosphate (HBTU) and trifluoroacetic acid (TFA) were purchased from Merck Millipore (Merck KGaA). Triisopropylsilane (TIS), diisopropylethylamine (DIEA) were purchased from Sigma–Aldrich (Sigma–Aldrich Chemie GmbH Munich, Germany). Acetonitrile was purchased from JT.Baker (Avantor Performance Materials B.V. 7418 AM Deventer Netherlands). The peptides were synthesized automatically on a 0.10 mmol scale using standard Fmoc solid phase peptide synthesis techniques with the microwave synthesizer (Liberty blue; CEM). A preloaded resin was used and provided in the reactor. The resin was washed with DMF. The Fmoc protecting group was removed with 20% (v/v) piperidine in DMF and initialized with microwave followed by washing with DMF. Amino acids were added in 0.2 mol/L equivalent to the reactor, then HBTU in a 0.5 mol/L equivalent was dosed to the amino acid solution. After that, 2 mol/L equivalent of DIEA was added to the resin. The coupling reaction was proceeded with microwaves for a few minutes then the resin was washed in DMF. These steps were repeated for all amino acids in the sequence. The last amino acid was deprotected. Once the synthesis was completed, the peptide was cleaved in 95% (v/v) trifluoroacetic acid (TFA), 2.5% (v/v) triisopropylsilane (TIS), and 2.5% (v/v) H2O for 1 h. The peptide residue was precipitated and washed with cold diethyl ether (DEE) by centrifugation. The peptide precipitate was then allowed to dry under air flow to remove residual ether. The peptide was purified using reversed phase preparative high-performance liquid chromatography (HPLC; Waters) in an acetonitrile/water gradient under acidic conditions on a Phenomenex C18 Luna column (5 mm pore size, 100 Å particle size, 250 mm × 21.2 mm). Following purification, the peptide was lyophilized on a freeze dryer (Labconco) for storage prior to use. The purified peptide mass was verified by liquid chromatography mass spectroscopy (LCMS; Waters). AMD3100 octahydrochloride hydrate was purchased from Sigma–Aldrich (#A5602) and dissolved in H2O. CXCL12 was purchased from Peprotech (#300-28A) and dissolved at a concentration of 100 μg/mL in H2O. Information about purities, yields and structural identification spectra are provided in the supplement.
4.3. Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance spectra were collected on a Bruker 700 MHz AvanceIII spectrometer with a 5 mm BBI 1H/BB probe equipped with a z-gradient and on a 850 MHz AVANCE III system equipped with a 5 mm triple resonance TXI 1H/13C/15N probe with a z-gradient at 298 K. NMR samples with JM#21 or WSC02 were prepared with 5 mg peptide each in 500 μL MilliQ-water containing 10% (v/v) D2O. Several homo- and hetero-nuclear 2D NMR spectra (HSQC, HMBC, COSY, TOCSY, and NOESY) were recorded for the assignment of the 1H and 13C resonances at natural isotope abundance. NMR spectra were processed using Topspin 3.6 and were analyzed using NMRFAM-SPARKY55. After manual peak assignment and extraction of the peak intensities, the data was incorporated into the program ARIA for structure calculations57.
4.4. Cell culture
TZM-bl HIV-1 reporter cells stably expressing CD4, CXCR4 and CCR5 and harboring the lacZ reporter genes under the control of the HIV LTR promoter were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl cells (Cat#8129) from Dr. John C. Kappes, and Dr. Xiaoyun Wu. TZM-bl cells and HEK293T cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS), 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine (Gibco). SupT1 cells were cultured in RPMI supplemented with 10% FCS, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mmol/L l-glutamine and 1 mmol/L HEPES (Gibco). The BCR-ABL cell line was generated introducing the BCR-ABL expression vector into bone marrow-derived wild type pro/pre B cells. Cells were cultured in Iscove's basal medium supplemented with 10% FCS, 100 U/mL penicillin, 100 U/mL streptomycin, 2 mmol/L l-glutamine and 50 μmol/L β-mercaptoethanol.
4.5. HIV-1 inhibition assay
Viral stocks of CXCR4-tropic NL4-3 were generated by transient transfection of HEK293T cells with proviral DNA as described before33. Inhibition of viral infection was performed in TZM-bl reporter cells. For this 70 μL of 1 × 105 cells (in growth medium supplemented with 2.5% FCS) were pretreated with 10 μL of inhibitors for 15 min at 37 °C. Cells were then inoculated with 20 μL of diluted virus. Infection rates were determined after 3 days using Gal-Screen system (Applied Biosystems).
4.6. Ca2+ mobilization assay
A well-established murine cell line expressing the fusion protein BCR-ABL was used for the experiments. Measurement of intracellular calcium flux was performed as described previously58. 1 × 106 cells were incubated with 5 μg/mL of Indo-1 AM (Invitrogen) and 0.5 μg/mL of pluronic F-127 (Invitrogen) in Iscove's medium supplemented with 1% FCS (Pan Biotech) at 37 °C for 45 min. Cells were then pelleted by centrifugation and resuspended in Iscove's medium with 1% FCS and treated with the EPI-X4 derivatives for 10 min at 37 °C. Prior measurement cells were pre-warmed for 5 min at 37 °C. Calcium flux was assessed by FACS measurement at BD LSR Fortessa. After 30 s of baseline recording, CXCR4-dependent calcium signaling was determined by stimulating with 100 ng/mL of mouse SDF-1α (PeproTech). To quantify CXCL12-induced Ca2+ mobilization, the kinetics analysis platform of FlowJo analysis software (BD Biosciences) was used to determine the area under the curve (AUC) of each calcium flux plot. In order to allow for inter-experimental comparison, the AUC was calculated after baseline subtraction for each single plot. AUC values were exported from FlowJo into MS–Excel where calculations were performed. Statistical analysis was performed using GraphPad Prism 8. One-way ANOVA with Tukey's multiple comparisons test was performed. Degree of significance is indicated as follows: ∗P < 0.05; **P < 0.01; ***P < 0.001; ns = not significant.
4.7. ERK and AKT signaling
CXCL12 induced phosphorylation of ERK and AKT was monitored by phosphoflow cytometry as described before38. For this 100,000 SupT1 cells were starved for 2 h at 37 °C (growth medium supplemented with 1% FCS). Inhibitors were then added for 10 min and cells subsequently stimulated with 100 ng/mL CXCL12 for 2 min. The reaction was stopped by adding 1% PFA and shifting to 4 °C for at least 10 min. Cells were then permeabilized with ice cold methanol and stained with phospho-p44/42 MAPK (ERK1) (Tyr204)/(ERK2) (Tyr187) (D1H6G) mouse mAb (Cell Signaling, #5726) and phospho-AKT (Ser473) (193H12) rabbit mAb (Cell Signaling, #4058) and adequate secondary antibodies for flow cytometry. Signal in the unstimulated control without inhibitor was defined as background and set to 0%. CXCL12 induced signal without inhibitor was set 100%.
4.8. Cell migration assay
Migration assays towards a 100 ng/mL CXCL12 gradient were performed with SupT1 cells using 96-well transwell assay plates (Corning Incorporated, Kennebunk, ME, USA) with 5 μm polycarbonate filters. First, 50 μL (0.75 × 105) SupT1 cells resuspended in assay buffer (RPMI supplemented with 0.1% BSA) were seeded into the upper chamber in the presence or absence of compounds and allowed to settle down for around 15 min. In the meantime, 200 μL assay buffer supplemented with or without 100 ng/mL CXCL12 as well as compounds were filled into a 96 well-V plate. Cells were allowed to migrate towards CXCL12 by putting upper chamber onto the 96 well-V plate. After a migration time of 4 h at 37 °C (5% CO2) lower compartment were analyzed for cell content by Cell-Titer-Glo® assay (Promega, Madison, WI, USA). Percentages of migrated cells were calculated as described before59 and normalized to the CXCL12-only control.
4.9. Antibody competition assay
Competition with the ECL-2 specific CXCR4 antibody was performed as described before38. Compounds were serially diluted in cold PBS and afterwards added to 50,000 SupT1 cells. APC-conjugated anti-human CXCR4 antibody (clone 12G5, #555976, BD) was diluted in PBS containing 1% FCS and added immediately afterwards at a final concentration of 0.245 nmol/L. After 2 h incubation at 4 °C, unbound antibody was removed and cells analyzed in flow cytometry.
4.10. Toxicity in zebrafish
Wild-type zebrafish embryos were dechorionated at 24 h post fertilization (hpf) using digestion with 1 mg/mL pronase (Sigma) in E3 medium (83 μmol/L NaCl, 2.8 μmol/L KCl, 5.5 μmol/L CaCl2, 5.5 μmol/L MgSO4). Embryos were exposed for 24 h in groups of 3–100 μL of E3 containing the test substances. Peptides were tested at 3, 30 and 300 μmol/L. Each concentration was tested in two independent assays, each of which was performed on 10 × 3 embryos. The peptide solvent (PBS), diluted in E3, was used as negative control at the same amount as introduced by the highest peptide concentration. As positive control for acute toxicity the pleurocidin antimicrobial peptide NRC-03 (GRRKRKWLRRIGKGVKIIGGAALDHL-NH2) was used at a concentration of 6 μmol/L45. At 48 hpf (after 24 h of incubation) embryos were scored in a stereomicroscope for signs of acute toxicity (lysis and/or necrosis), developmental toxicity (delay and/or malformations), or cardiotoxicity (heart edema and/or reduced or absent circulation). Chi-Square test was used to calculate whether the distribution of embryos into toxicity classes differed significantly between the PBS negative control and the test substances.
4.11. Mice
Nine-week-old male BALB/c mice (Janvier, France) were used. The animals were maintained on a 12/12-h light/dark cycle, with food and water ad libitum. All animals received humane care in compliance with the guidelines formulated by the French Ministry of Agriculture and of Higher Education and Research, who authorized the procedures (APAFiS #10067 and #1341).
4.12. Effect of EPI-X4 JM#21 in a mouse model of atopic dermatitis
Mice were anesthetized by isoflurane 5% (Dechra) before each topical application. MC903 (calcipotriol hydrate, Sigma) was dissolved in EtOH 100% and topically applied at 2.5 nmol/ear every two days from day 0 to day 12 on both sides of the mouse ears. EtOH 100% was applied to control mice every two days. All peptides were solubilized in EtOH 80% and applied topically at 400 nmol/ear (=16 μmol/kg/ear) in a volume of 25 μL/ear every day from day 0 to day 12, 1 h after MC903 application. Reference compound dexamethasone (Dexa, Sigma) was also used topically and daily at 20 nmol/ear (=0.8 μmol/kg/ear) and dissolved in EtOH 80%. The reference CXCR4 non peptidic antagonist AMD3100 (Euromedex) was used daily, topically at 315 nmol/ear (=12.6 μmol/kg/ear) and dissolved in EtOH 80%. Ear thickness was measured with a digital micrometer every day before the next topical application, and a mean value of the measurements of both ears was calculated. Scores were registered daily to evaluate the severity of the atopic dermatitis, following the scoring system on Table 3. Photographs of the ears were taken at day 12.
4.13. Effect of EPI-X4 JM#21 in a mouse model of allergic airway hypereosinophilia
Mice were sensitized by intraperitoneal (i.p.) injection of a mixture containing 50 μg ovalbumin (OVA, grade V, Sigma) and 2 mg Al(OH)3 (alum, Sigma) in saline on day 0, 1 and 2. On day 5, 6 and 7, mice were treated with peptides (16 μmol/kg), AMD3100 (12.6 μmol/kg, Euromedex) or solvent (NaCl 0.9%) by intranasal administration (i.n., 12.5 μL/nostril). Two hours later, mice were challenged i.n. with 10 μg of OVA in saline. The unchallenged controls were administered i.n. with saline. Before each intranasal administration, mice were anesthetized with ketamine (75 mg/kg, Merial) and xylazine (5 mg/kg, Bayer). Mice were kept on a hot plate (37 °C) until complete awakening. On day 8, mice were anesthetized with ketamine (300 mg/kg, Merial) and xylazine (20 mg/kg, Bayer) and bronchoalveolar lavage performed with PBS-EDTA 2 mmol/L23. Briefly, a catheter was inserted into the trachea, and lungs were washed 10 times with 500 μL of PBS-EDTA 2 mmol/L. BAL fluid was centrifuged (300×g for 5 min, 4 °C), cell pellet suspended in 500 μL of PBS-EDTA and total cells counted on a Muse Cell Analyser (Millipore) using anti-CD45-PE antibody (eBioscience, 1:500). Differential cell counts were assessed by flow cytometry (LSRII® cytometer, BD Bioscience). BAL cells were added with Fc Block (BD Bioscience) for 20 min and then marker antibodies were incubated for 30 min: CD11c-FITC (BD Bioscience, 1:250), GR-1-PE-eFluor610 (eBioscience, 1:500), CD11b-APC-Cy7 (BD Bioscience, 1:200), CD45-AlexaFluor700 BioLegend, 1:200), CD3-BV650 (BD Bioscience, 1:250), CD19-BV605 (BD Bioscience, 1:250), DAPI (BD Bioscience, 1:500). Live leukocytes were identified as CD45+DAPI− cells then differentiated into T cells (CD11blowCD19−CD3+), B cells (CD11blowCD19+CD3−), eosinophils (CD11b+CD11c−GR1−), neutrophils (CD11b+CD11c−GR1+) and macrophages (CD11b+GR1−CD11c+).
4.14. Calculations and statistical analysis
Statistical analysis was performed in GraphPad Prism (version 8.3.0). IC50 curves were determined by nonlinear regression. Differences between groups were tested for statistical significance using a one-way ANOVA followed by Bonferroni's post-test. Data were considered significantly different when P ≤ 0.05.
Acknowledgments
This work was supported by the German Research Foundation (DFG) through the CRC1279 to Ludger Ständker, Gönül Kizilsavas, Elsa Sanchez-Garcia, Manfred Wagner, Gilbert Weidinger, Frank Kirchhoff and Jan Münch, the DFG by individual grants (MU 3115/11-1 and MU 3115(8-1)) to Jan Münch, the Baden-Württemberg Stiftung to Jan Münch, and an ERC-PoC grant “EPI-X4 Health” to Frank Kirchhoff and Jan Münch. Elsa Sanchez-Garcia was also supported by the DFG under Germany's Excellence Strategy-EXC 2033-390677874-RESOLV and by the Boehringer Ingelheim Foundation (Plus-3 Program).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2020.12.005.
Author contributions
Pandian Sokkar, Sara Carmignani and Elsa Sanchez-Garcia did the computational docking analysis. Mirja Harms, Monica Habib and Ashraf H. Abadi did the rational drug design and peptides were synthesized by Nico Preising and Ludger Ständker, Monica Habib contributed in the peptides' synthesis, purification and identification, Mirja Harms did most of the in vitro experiments, except for the Ca2+ release experiments that were done by Antonella Nicolò and Hassan Jumaa, and the migration experiment that was performed by Andrea Gilg. Gönül Kizilsavas and Manfred Wagner determined all NRM structures. Martina Raasholm. and Gilbert Weidinger performed the zebrafish toxicity assay, and Simona Nemska and Nelly Frossard performed all mice experiments. Mirja Harms and Jan Münch coordinated the whole study and wrote the manuscript with assistance of Frank Kirchhoff.
Conflicts of interest
Ludger Ständker, Mirja Harms, Ashraf H. Abadi, Monica M.W. Habib, Frank Kirchhoff, Elsa Sanchez-Garcia, Pandian Sokkar and Jan Münch are coinventors of patents claiming the use of EPI-X4 and derivatives for the therapy of CXCR4 associated diseases.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Cavallero S., Shen H., Yi C., Lien C.L., Kumar-Ram S., Sucov H. CXCL12 signaling is essential for maturation of the ventricular coronary endothelial plexus and establishment of functional coronary circulation. Dev Cell. 2015;33:469–477. doi: 10.1016/j.devcel.2015.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ara T., Tokoyoda K., Okamoto R., Koni P.A., Nagasawa T. The role of CXCL12 in the organ-specific process of artery formation. Blood. 2005;105:3155–3161. doi: 10.1182/blood-2004-07-2563. [DOI] [PubMed] [Google Scholar]
- 3.Salcedo R., Oppenheim J.J. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation. 2003;10:359–370. doi: 10.1038/sj.mn.7800200. [DOI] [PubMed] [Google Scholar]
- 4.Nie Y., Waite J., Brewer F., Sunshine M.J., Littman D., Zou Y.R. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med. 2004;200:1145–1156. doi: 10.1084/jem.20041185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petit I., Ponomaryov T., Zipori D., Tsvee L. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S., Behnam-Azad B., Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiang Z., Zou Z.J., Zhang X.H., Wei Z.W., Zhu J.T., Yu J., Chen W., He Y., Schwarz R.E., Brekken R.A., Awasthi N., Zhang C.H. A positive crosstalk between CXCR4 and CXCR2 promotes gastric cancer metastasis. Oncogene. 2017;36:5122–5133. doi: 10.1038/onc.2017.108. [DOI] [PubMed] [Google Scholar]
- 8.Bonham L.W., Karch C.M., Fan C.C., Tan C., Geier G.E., Wang Y. CXCR4 involvement in neurodegenerative diseases. Transl Psychiatry. 2018;8:73. doi: 10.1038/s41398-017-0049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García-Cuesta E.M., Santiago C.A., Vallejo-Díaz J., Juarranz Y., Redríguez-Frade J.M., Mellado M. The role of the CXCL12/CXCR4/ACKR3 axis in autoimmune diseases. Front Endocrinol. 2019;10:585. doi: 10.3389/fendo.2019.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abboud D., Hanson J. Chemokine neutralization as an innovative therapeutic strategy for atopic dermatitis. Drug Discov Today. 2017;22:702–711. doi: 10.1016/j.drudis.2016.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Nagase H., Kudo K., Izumi S., Ohta K., Kobayashi N., Yamaguchi M. Chemokine receptor expression profile of eosinophils at inflamed tissue sites: decreased CCR3 and increased CXCR4 expression by lung eosinophils. J Allergy Clin Immunol. 2001;108:563–569. doi: 10.1067/mai.2001.118292. [DOI] [PubMed] [Google Scholar]
- 12.Tahirovic Y.A., Pelly S., Jecs E., Miller E., Sharma S., Liotta D. Small molecule and peptide-based CXCR4 modulators as therapeutic agents. A patent review for the period from 2010 to 2018. Expert Opin Ther Pat. 2020;30:87–101. doi: 10.1080/13543776.2020.1707186. [DOI] [PubMed] [Google Scholar]
- 13.Micallef I., Stiff P., DiPersio J., Maziarz R.T., McCarty J.M., Angell J. Successful stem cell mobilization rescue by AMD3100 (Plerixafor) + G-CSF for patients who failed primary mobilization: Results from the phase III (3101-NHL) study. Blood. 2007;110:602. [Google Scholar]
- 14.de Clercq E. Mozobil® (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir Chem Chemother. 2019;27 doi: 10.1177/2040206619829382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holgate S.T. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. [DOI] [PubMed] [Google Scholar]
- 16.Brightling C.E., Symon F., Birring S., Bradding P., Wardlaw A.J., Pavord I.D. Comparison of airway immunopathology of eosinophilic bronchitis and asthma. Thorax. 2003;58:528–532. doi: 10.1136/thorax.58.6.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett N.A., Austen K.F. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity. 2009;31:425–437. doi: 10.1016/j.immuni.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brussino L., Heffler E., Bucca C., Nicola S., Rolla G. Eosinophils target therapy for severe asthma: critical points. BioMed Res Int. 2018;2018:7582057. doi: 10.1155/2018/7582057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Negrete-García M.C., Velazquez A., Popoca-Coyotl A., Montes-Vizuet A.R., Juarez-Carvajal E., Teran L.M. Chemokine (C-X-C motif) ligand 12/stromal cell-derived factor-1 is associated with leukocyte recruitment in asthma. Chest. 2010;138:100–106. doi: 10.1378/chest.09-2104. [DOI] [PubMed] [Google Scholar]
- 20.Nagase H., Miyamasu M., Yamaguchi M., Fujisawa T., Ohta K., Yamamoto K., Morita Y., Hirai K. Expression of CXCR4 in eosinophils: Functional analyses and cytokine-mediated regulation. J Immunol. 2000;164:5935–5943. doi: 10.4049/jimmunol.164.11.5935. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalo J.A., Lloyd C.M., Peled A., Delaney T., Coyle A.J., Gutierrez-Ramos J.C. Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1α in the inflammatory component of allergic airway disease. J Immunol. 2000;165:499–508. doi: 10.4049/jimmunol.165.1.499. [DOI] [PubMed] [Google Scholar]
- 22.Hachet-Haas M., Balabanian K., Rohmer F., Pons F., Franchet C., Lecat S. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J Biol Chem. 2008;283:23189–23199. doi: 10.1074/jbc.M803947200. [DOI] [PubMed] [Google Scholar]
- 23.Gasparik V., Daubeuf F., Hachet-Haas M., Rohmer F., Gizzi P., Haiech J. Prodrugs of a CXC chemokine-12 (CXCL12) neutraligand prevent inflammatory reactions in an asthma model in vivo. ACS Med Chem Lett. 2012;3:10–14. doi: 10.1021/ml200017d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daubeuf F., Hachet-Haas M., Gizzi P., Gasparik V., Bonnet D., Utard V., Hibert M., Frossard N., Galzi J.L. An antedrug of the CXCL12 neutraligand blocks experimental allergic asthma without systemic effect in mice. J Biol Chem. 2013;288:11865–11876. doi: 10.1074/jbc.M112.449348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukacs N.W., Berlin A., Schols D., Skerlj R.T., Bridger G.J. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol. 2002;160:1353–1360. doi: 10.1016/S0002-9440(10)62562-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zgraggen S., Huggenberger R., Kerl K., Detmar M. An important role of the SDF-1/CXCR4 axis in chronic skin inflammation. PLoS One. 2014;9 doi: 10.1371/journal.pone.0093665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takekoshi T., Wu X., Mitsui H., Tada Y., Kao M.C., Sato S., Dwinell Hwang S.T. CXCR4 negatively regulates keratinocyte proliferation in IL-23-mediated psoriasiform dermatitis. J Invest Dermatol. 2013;133:2530–2537. doi: 10.1038/jid.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths C.E.M., van de Kerkhof P., Czarnecka-Operacz M. Psoriasis and atopic dermatitis. Dermatol Ther. 2017;7:31–41. doi: 10.1007/s13555-016-0167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Homey B., Steinhoff M., Ruzicka T., Leung D.Y.M. Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol. 2006;118:178–189. doi: 10.1016/j.jaci.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 30.Weidinger S., Novak N. Atopic dermatitis. Lancet. 2016;387:1109–1122. doi: 10.1016/S0140-6736(15)00149-X. [DOI] [PubMed] [Google Scholar]
- 31.Guttman-Yassky E., Bissonnette R., Ungar B., Suarez-Farinas M., Ardeleanu M., Esaki H., Suprun M. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143:155–172. doi: 10.1016/j.jaci.2018.08.022. [DOI] [PubMed] [Google Scholar]
- 32.Coondoo A., Phiske M., Verma S., Lahiri K. Side-effects of topical steroids: a long overdue revisit. Indian Dermatol Online J. 2014;5:416. doi: 10.4103/2229-5178.142483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zirafi O., Kim K.A., Ständker L., Mohr K.B., Sauter D., Heigele A., Kluge S.F. Discovery and characterization of an endogenous CXCR4 antagonist. Cell Rep. 2015;11:737–747. doi: 10.1016/j.celrep.2015.03.061. [DOI] [PubMed] [Google Scholar]
- 34.Buske C., Kirchhoff F., Münch J. EPI-X4, a novel endogenous antagonist of CXCR4. Oncotarget. 2015;6:35137–35138. doi: 10.18632/oncotarget.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller J.A., Zirafi O., Roan N.R., Lee S.J., Münch J. Evaluation of EPI-X4 as a urinary peptide biomarker for diagnosis and prognosis of late acute GvHD. Bone Marrow Transplant. 2016;51:1137–1139. doi: 10.1038/bmt.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zirafi O., Hermann P.C., Münch J. Proteolytic processing of human serum albumin generates EPI-X4, an endogenous antagonist of CXCR4. J Leukoc Biol. 2016;99:863–868. doi: 10.1189/jlb.2MR1115-521RR. [DOI] [PubMed] [Google Scholar]
- 37.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 38.Harms M., Gilg A., Ständker L., Beer A., Mayer B., Gruber C., Münch J. Microtiter plate-based antibody-competition assay to determine binding affinities and plasma/blood stability of CXCR4 ligands. Sci Rep. 2020;10:16036. doi: 10.1038/s41598-020-73012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pozzobon T., Goldoni G., Viola A., Molon B. CXCR4 signaling in health and disease. Immunol Lett. 2016;177:6–15. doi: 10.1016/j.imlet.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Abdelrasoul H., Vadakumchery A., Werner M., Lenk L., Khadour A., Young M. Synergism between IL7R and CXCR4 drives BCR-ABL induced transformation in Philadelphia chromosome-positive acute lymphoblastic leukemia. Nat Commun. 2020;11:3194. doi: 10.1038/s41467-020-16927-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thalhamer T., McGrath M.A., Harnett M.M. MAPKs and their relevance to arthritis and inflammation. Rheumatology. 2007;47:409–414. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 42.Hers I., Vincent E.E., Tavaré J.M. Akt signalling in health and disease. Cell Signal. 2011;23:1515–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 44.Raldúa D., Piña B. In vivo zebrafish assays for analyzing drug toxicity. Expet Opin Drug Metabol Toxicol. 2014;10:685–697. doi: 10.1517/17425255.2014.896339. [DOI] [PubMed] [Google Scholar]
- 45.Morash M.G., Douglas S.E., Robotham A., Ridley C.M., Gallant J.W., Soanes K.H. The zebrafish embryo as a tool for screening and characterizing pleurocidin host-defense peptides as anti-cancer agents. Dis Model Mech. 2011;4:622–633. doi: 10.1242/dmm.007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Travers J., Rothenberg M.E. Eosinophils in mucosal immune responses. Mucosal Immunol. 2015;8:464–475. doi: 10.1038/mi.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li M., Herner P., Zhang Z., Kato S., Metzger D., Chambon P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A. 2006;103:11736–11741. doi: 10.1073/pnas.0604575103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen H., Xu X., Teng J., Chen S., Bunjhoo H., Cao Y., Liu J. CXCR4 inhibitor attenuates ovalbumin-induced airway inflammation and hyperresponsiveness by inhibiting Th17 and Tc17 cell immune response. Exp Ther Med. 2016;11:1865–1870. doi: 10.3892/etm.2016.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doyle T.M., Ellis R., Park H.J., Inman M.D., Sehmi R. Modulating progenitor accumulation attenuates lung angiogenesis in a mouse model of asthma. Eur Respir J. 2011;38:679–687. doi: 10.1183/09031936.00133210. [DOI] [PubMed] [Google Scholar]
- 50.Choi Y., Pham D.L., Lee D.H., Lee S.H., Kim S.H., Park H.S. Biological function of eosinophil extracellular traps in patients with severe eosinophilic asthma. Exp Mol Med. 2018;50:104. doi: 10.1038/s12276-018-0136-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu J., Mora A., Shim H., Stecenko A., Brigham K.L., Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol. 2007;37:291–299. doi: 10.1165/rcmb.2006-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li F., Xu X., Geng J., Wan X., Dai H. The autocrine CXCR4/CXCL12 axis contributes to lung fibrosis through modulation of lung fibroblast activity. Exp Ther Med. 2020;19:1844–1854. doi: 10.3892/etm.2020.8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu B., Chien E., Mol C., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veldkamp C.T., Seibert C., Peterson F., De la Cruz N.B., Haugner J.C., Basnet H. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dominguez C., Boelens R., Bonvin A.M.J.J. HADDOCK: A protein−protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 56.van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E. The HADDOCK2.2 webserver: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 57.Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T.E., Nilges M. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 58.Storch B., Meixlsperger S., Jumaa H. The Ig-α ITAM is required for efficient differentiation but not proliferation of pre-B cells. Eur J Immunol. 2007;37:252–260. doi: 10.1002/eji.200636667. [DOI] [PubMed] [Google Scholar]
- 59.Balabanian K., Lagane B., Infantino S., Chow K., Harriague J., Moepps B. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.