

Abstract
Prime editing enables efficient introduction of targeted transversions, insertions, and deletions in mammalian cells and several organisms. However, genetic disease models with base deletions by prime editing have not yet been reported in mice. Here, we successfully generate a mouse model with a cataract disorder through microinjection of prime editor 3 (PE3) plasmids to efficiently induce targeted single-base deletion. Notably, a generated mouse with a high G-deletion rate (38.2%) displays a nuclear cataract phenotype; the PE3-induced deletions in mutant mice achieve high rates of germline transmission to their progenies, with phenotypic inheritance of cataract. Our data propose that modeling a genetic disease with a single nucleotide deletion in mice can be achieved with prime genome editing in vivo.
Keywords: prime editing, cataract disorder, PE3, base deletion, germline transmission
Graphical abstract
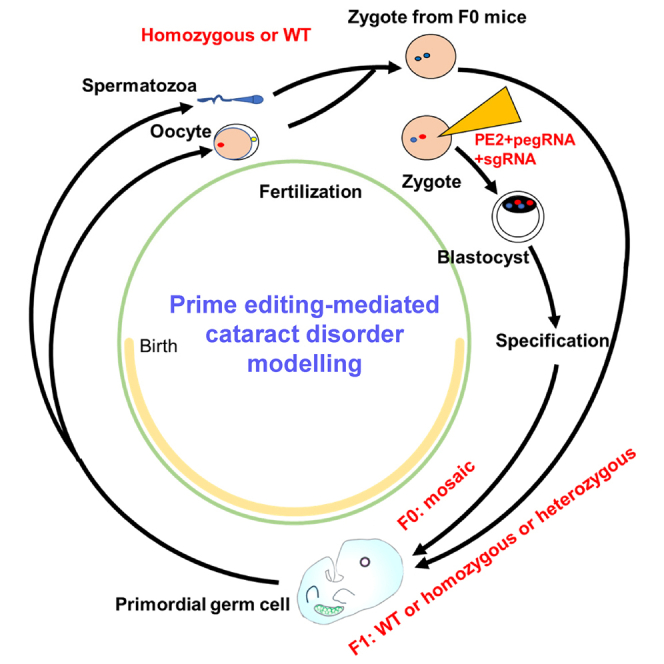
Lin et al. use the prime editing system to introduce a G-deletion into Crygc and successfully establish a mouse model with a cataract disorder, which can be transmitted to the next generation.
Introduction
Single nucleotide mutations in the human genome, including inversions, insertions, and deletions, raised from endogenous error-prone processes or exposure to exogenous factors, are well-characterized causes of various human diseases.1, 2, 3 CRISPR-Cas9-based genome editing enables correction of disease-causing mutations.4 Previously, the CRISPR-Cas9 system has been employed to correct a cataract disorder with a base deletion via homology-directed repair (HDR) based on an exogenously supplied oligonucleotide or the endogenous wild-type allele.5 In particular, the development of cytosine base editor (CBE)6 and adenine base editor (ABE)7 holds great promise for genetic correction of C-to-T and A-to-G substitutions in some genetic and pathogenic diseases in our and others’ studies.8, 9, 10 However, genetic installation of mutations, such as base transversion, insertions, and deletions, which collectively account for the most known pathogenic causes,11 is still a great challenge for gene therapy.
Another CRISPR-Cas9-based genome-editing tool, the prime editing system, applies a “search and replace” strategy to achieve all types of base conversions, small fragment insertions, and deletions.12 The final optimized version of prime editor (prime editor 2 [PE2]) consists of a fusion of a Cas9 nickase (nCas9) and an engineered reverse transcriptase (Moloney murine leukemia virus reverse transcriptase [M-MLV RT]). Under the guidance of a prime editing guide RNA (pegRNA), nCas9 searches and nicks the target DNA, and genetic mutations are installed by reverse transcription and subsequent DNA repair. To further improve the priming editing efficiency in the prime editor 3 (PE3) system, an additional single guide RNA (sgRNA) near the target site is introduced to induce a nick on the non-edited strand to promote wild-type strand excision in the PE3 system.12 The PE3 device enables efficient installation of targeted transversions, insertions, and deletions, with fewer by-products and lower off-target editing, compared to conventional CRISPR-Cas9 genome-editing systems.13 With the versatility and accuracy of this technology, PE3 can be used for targeted mutagenesis and genetic correction in mammalian cells, organoids, plants,12,14, 15, 16 as well as in mouse embryos in our recent report.17 Nevertheless, the editing efficiency of transversions in embryos or adult mice remains very low;17 successful modeling of a genetic mutation-caused disease in mice has not been reported yet.
Results
Highly efficient installation of a G-deletion (G-del) mutation into Crygc with the prime editing system in a mouse cell line
A single nucleotide indel-associated event is a general mutation mechanism18 and a major cause of genetic diseases.19 To test the editing versatility of the prime editing system to introduce a single base deletion for genetic disease modeling, we chose to generate a mouse model of dominant cataract disorder that is caused by a defined mutation in the Crygc gene,20 with 1 bp (G) deletion in exon 3 of Crygc. This mutation leads to a premature stop codon downstream of the deletion site and thus produces a truncated γC-crystallin protein and nuclear cataracts (Figure 1A) in both homozygous and heterozygous mutant mice.20 We first designed two pegRNAs starting with a primer binding site (PBS) length of 13 nucleotide (nt) and a RT template length of 10 nt (pegRNA1) or 13 nt (pegRNA2) (Table S1), according to the optimized principles for pegRNA design,12 to produce an expected deletion at +1 position. The PE3 machinery for Crygc editing was composed of a PE2 protein and a transcribed pegRNA (Figure 1B); an additional nicking sgRNA targeting the non-edited strand, 25 bp downstream of the 3′ of the pegRNA-induced nick, was constructed (Figure 1C).
Figure 1.

Highly efficient installation of a G-deletion (G-del) mutation into Crygc with a prime editing system in N2a cells
(A) Schematic diagram showing the target site in mouse Crygc locus. The PAM sequence is underlined in black, and the spacer sequence of pegRNA is underlined in blue. The cataract disorder-associated G nucleotide, the deletion of which results in a premature stop codon (highlighted in the lower panel), is highlighted by a red arrow in the upper panel. (B) The expected structure of PE2 and pegRNA1 used for inducing G-deletion in Crygc. (C) Schematic diagram for the pegRNA spacer (for prime editing) and sgRNA spacer (for nicking) design in the PE3 system for mouse Crygc editing. (D) Editing frequency of PE3 (pegRNA1 and pegRNA2)-induced G-deletion in N2a cells analyzed from Sanger sequencing results. (E) Analysis of editing efficiency from Sanger sequencing results with EditR. Two replicates were independently performed and analyzed. (F) Targeted deep-sequencing analysis of amplicons from two replicates of N2a cells transfected with PE2 and pegRNAs. The proportions of G-deletion reads were presented. (G) The proportions of sequencing reads with indels were presented.
We first tested the feasibility of PE3-mediated base deletion in mouse neuro-2a (N2a) cells. After co-transfection with PE2, pegRNA, and nicking sgRNA-expressing plasmids for 72 h, successfully transfected cells were collected for PCR amplification and detection by Sanger sequencing. It showed that both pegRNAs induced highly efficient mutations (about 80%) with a single G-deletion at the expected site (Figures 1D and 1E), which was further validated by targeted deep-sequencing data (Figure 1F). Further analysis demonstrated that PE3 devices induced 2.5%–3.2% indels within the amplicons spanning pegRNA and nicking sgRNA target sites, and the indel rates were much higher than that in wild-type cells, in which the indels should be elicited by sequencing errors (Figure 1G). These data suggest that both pegRNAs can specifically introduce a G-deletion mutation within Crygc with low by-products.
Modeling a cataract disorder in mice with prime genome editing by injection of PE3 machinery plasmids
Considering the low editing efficiency in mouse embryos or mice with transcribed PE3 mRNA micro-injection,17 we tested the editing versatility of PE3 in mouse preimplantation embryo by direct injection of PE3 plasmids (PE2, pegRNA, and nicking sgRNA), which may extend the expression period to prolong its editing time. Comparably, pegRNA1 but not pegRNA2 (Figure S1A) successfully induced apparent G-deletion mutation in four embryonic day (E)3.5 embryos (n = 4/30 in total; Figures S1B and 2A) with editing efficiency ranging from 13.8% to 100% (n = 4), whereas most obtained blastocysts were lowly or not edited, with no obvious 1 bp shifting starting from the target site in Sanger sequencing results, which were not counted as edited blastocysts. In addition, 82.5% of injected zygotes, in total, developed into blastocysts, indicating a low toxicity of the injected PE3 plasmids (Figure 2A). To further improve the editing efficiency, we measured the effect of PBS length, showing that consistent with previous reports,12,17 13 bp length of PBS was most efficient for targeted prime editing of Crygc here (Figure S1C).
Figure 2.

PE3-mediated efficient base deletion to model a cataract disorder in the mouse
(A) The editing efficiency in blastocysts induced by PE2, pegRNA1, and a nicking sgRNA (three components are all plasmids). Three independent experiments were performed, and the number of blastocysts, as well as edited embryos (with apparent editing efficiency; >5% as estimated by EditR for Sanger sequencing results), was presented. Three embryos were not successfully amplified for Sanger sequencing. (B) The editing frequencies in mice induced by the PE3 (pegRNA1) system. After microinjection, 80 embryos were transferred, and 19 mice were born. The editing frequencies of two mice with apparent G-deletion were analyzed, and Sanger sequencing results were presented. (C) Two female mice with apparent G-deletion were pictured, and a nuclear cataract in the lens of the mouse with high editing frequency (#3; 41%) was highlighted by red arrows. (D) Editing frequencies in mouse tails (#3 and #15) by targeted deep sequencing. The fragments flanking the editing sites were PCR amplified, and the PCR products were subjected to deep sequencing and analysis. The proportions of reads with “Other” (not including wild-type [WT] and G-deletion reads) and G-deletion were presented. (E) Summary of whole genome sequencing (WGS) analysis. The genomic DNA from tails of the two mutant mice (#3 and #15) was sequenced with high coverage. Off-target (OT) indels induced by pegRNAs or nicking sgRNAs were analyzed. Potential off-target sites were predicted by an online tool Cas-OFFinder3 (http://www.rgenome.net/cas-offinder/). Spacer sequences with ≤5 mismatches and “NGG” PAM were considered as potential off-target sites.
Intriguingly, one embryo was completely mutated with G-deletion in Sanger sequencing results (Figure 2A), which proved the high feasibility of prime editing for disease modeling in vivo. Thus, we utilized pegRNA1 to generate an adult mouse model of cataract disorder. After transferring 80 injected embryos into pseudopregnant mice, 19 mice were born, among which two mice with apparent G-deletion were obtained with mutation rates of 41% (#3) and 17% (#15) (Figure 2B). 4 weeks after birth, the cataract disorder was observed in the lenses of the mouse with 41% mutation efficiency, whereas another mouse (17% mutation efficiency) exhibited normal lenses (Figure 2C). To further validate the efficiency of prime editing, targeted deep sequencing was performed for analyzing the genome sequences in two mice. We demonstrated that 38.2% (#3) and 26.6% (#15) of sequenced reads were detected with expected G-deletion (Figure 2D), which was close to Sanger sequencing results, with very low levels of by-products and indels (Figure 2D).
Subsequently, we performed whole genome sequencing (WGS) using the genomic DNA from mouse tails to comprehensively investigate off-targeting throughout the genome induced by the pegRNA or nicking sgRNA. After filtering out naturally occurring variants in the wild-type mouse with the same genetic background,17,21 potential off-targeting sites for used pegRNA or sgRNA were examined, and no base deletions or insertions possibly induced by the PE3 device were uniquely found in mice #3 and #15 (Figure 2E). We also performed targeted deep-sequencing analysis to assess putative off-target sites that were predicted by Cas-OFFinder3 induced by pegRNA or nicking sgRNA with mismatch ≤3, and no parent off-targeting activity was observed (<0.1%) (Figures S2A and 2B). Considering the usage of plasmid DNA in the PE3 device, we analyzed the integration of PE3 plasmids in the genome, and no PE2 or pegRNA coding DNA sequences were detected in both mice. Considering the possibility of scaffold sequence insertion that contributes to indels at the target locus,12 we analyzed the PE3 editing experiments in obtained mice (#3 and #15). As shown in Figure S2C, about 0.15% average total insertion of any number of pegRNA scaffold nucleotides was observed, which was much lower than that in PE3-edited N2a cells. These data suggest that PE3-mediated base deletion is feasible and specific in our adult mouse model with a cataract disorder.
PE3-induced base deletion is transmitted to the next generation
Next, we asked whether PE3-induced base deletion can be transmitted to the next generation. The two mutated mice (Figure 2C), one with cataract disorder and the other with normal lenses, were crossed with wild-type male mice. Among the 8 and 9 progenies, two and three mice with a heterozygous genotype were obtained, respectively, and the other 12 mice displayed a wild-type genotype (Figures 3A and 3B). Interestingly, two mice that were born by mice #3 and #15, respectively, were observed with cataract disorders as expected, whereas the other three mice carrying heterozygous G-deletion displayed normal lenses (Figure 3A), which was also observed in a previous study.5 Moreover, the heterozygous genotype from Sanger sequencing was further confirmed by targeted deep-sequencing data (Figure 3C), with ∼50% wild-type reads and ∼50% reads with G-deletion, as well as neglectable levels of by-products in F1 mice (Figure 3C).
Figure 3.

PE3-induced base deletion is transmitted to the next generation
(A) The two mutant female mice (#3 and #15) were crossed with WT male mice, and 8 and 9 mice were born, respectively. Two (from #3) and three (from #15) mice with heterozygous genotype were pictured, and the lens with nuclear cataract was highlighted by red arrows. (B) Sanger sequencing results for F1 mice with heterozygous genotypes. (C) Targeted deep-sequencing analysis of genotypes in F1 mice with heterozygous genotypes. (D) The frequency of indels in mouse tails (#3 and #15) induced by the PE3 system (pegRNA1) was analyzed in deep-sequencing data. (E) The frequency of indels containing reads flanking the nicking sgRNA spacer was presented in mutant mice (#3 and #15). (F) The frequency of indels in tails of F1 mice (born by #3 and #15). (G) A model for PE3-mediated genome editing and genetic transmission in vivo.
We also analyzed the frequency of indels in F0 or F1 mice in targeted deep-sequencing data. Among the minimal levels of by-products in F0 mice, there were only 0.18% and 0.02% of sequencing reads with indels around the pegRNA targeting site (Figure 3D). We also validated that only less than 1% reads with indels were not matched with the wild-type sequences around the nicking site (Figure 3E). It indicates that rare indels are induced by the PE3 system with a pegRNA/sgRNA combination. Similarly, there were only about 0.02% indels among sequencing reads in F1 mice (Figure 3F), which was close to the indel rate in wild-type N2a cells (Figure 1G), indicating that PE3-induced minimal indels in F0 mice are not readily transmitted to F1 mice.
In total, 29.4% of offspring (n = 5/17; 2/8 from #3 and 3/9 from #15) was heterozygous, which was close to the average percentage (32.2%) of targeted editing efficiency in two generated mice (#3 and #15). We postulate that a total of ∼29.4% of alleles in somatic cells or ∼29.4% of germ cells in F0 mice was successfully mutated by the PE3 system. It indicates that somatic mosaicism is induced by PE3 through zygotic injection and that even a low rate of mutations can be transmitted to the next generation through crossing, particularly facilitating disease modeling (Figure 3G).
PE3-mediated repair of G-deletion in Crygc in N2a cells
To test whether the PE3 device can be used for correction of disease-related genetic mutations, N2a cells with G-deletion induced by PE3 were subjected to establish single cell clones with homozygous genotypes. Among established 8 cell lines, expected mutations were observed in 5 clones, without other unexpected mutations or indels (Figure 4A), which can be used as a cellular model for cataract disorder. We then constructed PE3 plasmids with a specific pegRNA3 and the same nicking sgRNA to introduce a G-insertion into clones 6 and 8, to repair the genetic mutation in cataract disorder (Figure 4B). As expected, G-deletion was successfully repaired by our designed PE3 editing strategy with pegRNA3, with 33.3% and 26.7% editing efficiency in the two clones, respectively, as demonstrated by Sanger sequencing and targeted deep-sequencing results, with very low levels of by-products (Figures 4B and 4C). Thus, we proved the conceptional feasibility to repair a genetic cataract disorder in a cellular model.
Figure 4.

PE3-mediated repair of G-deletion in Crygc in N2a cells
(A) Genotyping of 8 clones from PE3 (pegRNA1)-induced N2a cells. The Sanger sequencing results for clones 6 and 8 were presented. (B) Schematic diagram for pegRNA3-induced repair of G-deletion in clones 6 and 8. (C) Targeted deep-sequencing analysis of amplicons from WT N2a cells, clone 6, clone 8, and clone 6 or clone 8 transfected with the PE3 device for correction (PE2, pegRNA3, and nicking sgRNA). G-insertion occurred in clones 6 and 8 and makes the genome become a WT status (defined as WT/G-insertion). The proportions of reads with WT, other, and G-deletion were presented.
Discussion
The powerful ability of the PE3 device to install targeted transversions, insertions, and deletions enables introduction of targeted mutagenesis in various models.12,14, 15, 16, 17 However, very low editing efficiency can be achieved by microinjection of PE3 mRNAs in our previous study.17 We also tested the editing efficiency of PE3 mRNAs for the Crygc targeting site before testing plasmid injection, whereas very low targeting efficiency was consistently observed (data not shown). Thus, we tried to inject PE3 plasmids in the present study for disease modeling. Surprisingly, we obtained edited embryos and adult mice with very high targeting efficiency (100% and 41%). More importantly, we observed a genetic cataract disorder in an obtained F0 mouse. As far as we know, this is the first report for PE3-mediated disease modeling with a disease phenotype in mice.
Additionally, we find that PE3-induced genetic mutations can be transmitted to the next generation with high efficiency. Even for edited F0 mice with low editing efficiency, without expected disease phenotype, the introduced genetic mutations can be passaged to the F1 progenies through haploid germ cells. Therefore, here, we provide a relatively low but feasible pathway to obtain disease models with the PE3 system; even sometimes the disease-related phenotype cannot be observed in F0 mice.
In summary, we successfully generate a genetic mouse model of cataract disorder by PE3-induced targeted base deletion through microinjection of PE3 plasmids but not mRNA. Notably, the F0 mouse with a high G-deletion rate displays a nuclear cataract phenotype; the PE3-induced G-deletion in mutated mice with or without cataract disorder can achieve high rates of germline transmission to their progenies, with or without phenotypic inheritance. Lastly, our data in a cellular model propose that correction of a genetic disease with base deletions by prime genome editing can be anticipated in zygotes or even adult tissues.
Materials and methods
Animals
Animal experiments in the present study were approved by the Animal Care and Use Committee of the Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China. Mice were maintained in a specific pathogen-free (SPF) facility under a 12-h dark-light cycle. B6D2F1 (C57BL/6J × DBA/2) mouse strains were used as embryo donors, and ICR (Institute of Cancer Research) were used as pseudopregnant mothers.
Microinjection, embryo in vitro culturing, and embryo transfer
Microinjection of mouse embryo was performed referring to our previously described method.22 Briefly, superovulated B6D2F1 female mice were mated with adult B6D2F1 males, and zygotes were collected from female oviducts at 20 h post-human chorionic gonadotropin (hCG; Sansheng, China) injection. pCMV-PE2 plasmid (Addgene; 132775) (100 ng/μL), pegRNA (50 ng/μL), and nicking sgRNA expression plasmids (50 ng/μL) were mixed, and 2−4 pL mixture was injected to the cytoplasm of zygotes at 21−24 h post-hCG. Microinjection was performed in a droplet of M2 (Sigma-Aldrich, USA; M7167) containing 5 μg/mL cytochalasin B by using a Piezo-driven micromanipulator (Prime Tech, Japan; Pmm4G). Then the embryos were cultured in potassium simplex optimized medium (KSOM; Millipore, USA; MR-106-D) at 37°C in 5% CO2 atmosphere. The embryos were cultured until blastocyst stage (E4.5) and then subjected to genotyping. To generate live pumps with targeted genome editing by a PE3 device, embryos were transferred to the oviducts of pseudopregnant ICR mice immediately after microinjection, with 20 embryos for each surrogate.
Plasmid construction
pCMV-PE2 was purchased from Addgene (132775). The pegRNA plasmid was constructed according to a modified method as previously described.12,17 In brief, the pegRNA expressing vector was PCR amplified from pGL3-U6-sgRNA-EGFP (Addgene; 107721) using Phanta Max Super Fidelity DNA Polymerase (Vazyme, China). The pegRNA backbone PCR primers are the following: forward 5′-agctaggtctcctttttttaaagaattctcgacctcgagac-3′; reverse 5′-tctctcggtctcacggtgtttcgt-3′. Then purified PCR products were subjected to digestion with BsaI-HFv2 (NEB) to produce cohesive ends. Spacer oligos, pegRNA 3′ extension oligos, and sgRNA scaffold sequences with phosphorylation (T4 Polynucleotide Kinase [PNK]; NEB) at 3′ ends were synthesized, and pairs of top and bottom oligos were annealed, respectively. Then, pegRNA expression backbone and annealed oligos of spacers, sgRNA scaffold sequences, and 3′ extension oligos were ligated using with T4 DNA ligase (NEB) for plasmid construction. A similar strategy was designed for pegRNA3 used in correction of G-deletion mutation in mutated N2a clones. The sequences for synthesis are listed in Table S1. For construction of sgRNAs used for nicking, DNA oligos were synthesized, annealed, and cloned into a BsaI-digested pGL3-U6-sgRNA-mCherry expression vector with T4 DNA ligase (NEB). The sgRNA oligo sequences are the following: forward 5′-accgtatgagatgcctaactaccg-3′; reverse 5′-aaaccggtagttaggcatctcata-3′.
Cell culture and transfection
N2a cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum (Gemini) at 37°C with 5% CO2 in an incubator (Thermo Fisher Scientific). Confluent cells were seeded onto 24-well plates. 24 h after seeding, transfection was performed at approximately 70% confluence using EZ Trans (Shanghai Life iLab, China) according to the manufacturer’s instructions. Briefly, a total of 1,300 ng plasmids (900 ng PE2, 300 ng pegRNAs, and 100 ng nicking sgRNAs) were transfected per well. After transfection for 72 h, pegRNA-GFP and sgRNA-mCherry double-positive cells were sorted by fluorescence-activated cell sorting (FACS). 30,000 cells were collected for each sample.
Genomic DNA extraction and genotyping
Genomic DNA of GFP and mCherry double-positive N2a cells and E4.5 blastocysts was extracted using QuickExtract DNA Extraction Solution (Lucigen) according to the manufacturer’s instructions. Genomic DNA of mouse tails was extracted by the phenol-chloroform method as we previous described.23 Purified DNA was PCR amplified with Phanta Max Super-Fidelity DNA Polymerase (Vazyme, China). Primers used for genotyping are listed in Table S2.
Estimation of editing efficiency
An online tool EditR (https://moriaritylab.shinyapps.io/editr_v10/) was applied to estimate the editing frequency with Sanger sequencing results.
Targeted deep sequencing
The fragments covering the on-target sites were amplified from genomic DNA using Phanta Max SuperFidelity DNA Polymerase (Vazyme). The paired-end sequencing of PCR amplicons was performed by Illumina NextSeq 500 (2 × 150) platform at Novogene, China. Primers used for targeted deep sequencing are listed in Table S2. The adaptor pair of the pair-end reads was removed using AdapterRemoval version (v.)2.2.2, and pair-end read alignments of 11 bp or more bases were combined into a single consensus read. All processed reads were then mapped to the target sequences using the Burrows-Wheeler Alignment-maximal exact match (BWA-MEM) algorithm (BWA v.0.7.16). For each site, the mutation rate was calculated using bam-readcount with parameters -q 20 -b 30. Indels were calculated based on reads containing at least 1 inserted or deleted nucleotide in the protospacer. Indel frequency was calculated as the number of indel-containing reads/total mapped reads. The aligned reads were visualized by using the Integrated Genome Viewer (IGV) and tabbed using Pysamstats. For analyzing the percentage of sequencing reads with scaffold insertion of any length among total reads, we used a method as previously described,12 and all reads with scaffold sequences were considered as scaffold insertions. If the wild-type nucleotides were the same to the tail of scaffold sequences, then these reads were not considered as scaffold insertions.
Estimation of off-targeting activity
The potential off-target sites were predicted by Cas-OFFinder3 with NGG protospacer adjacent motif (PAM) (http://www.rgenome.net/cas-offinder). The information for predicted off-target sites with up to 5 nt mismatches is summarized in Table S3. Primers used for targeted deep sequencing to detect putative off-target sites of pegRNA1 and nicking sgRNA with up to 3 nt mismatches are listed in Tables S4 and S5. Because there were 27 putative off-target sites for pegRNA1 with three mismatches, we randomly selected 9 sites for off-targeting analysis.
WGS
Two mutant F0 mice, #3 and #15, were subject to WGS. 1 μg of genomic DNA extracted from mouse tails was fragmented (∼300 bp) by ultrasonication using a Covaris S2 system. Then, the sheared DNA fragments were subjected to DNA library construction, followed by high-throughput sequencing by the HiSeq X Ten platform (Illumina) as paired-end 150 bp (Novogene, China). The WGS data of wild-type mice have the same genetic background with the founders used in our previous study,17,21 and these data were used for background subtraction. The raw data were first filtered to remove low-quality reads with the following criteria: (1) sequencing quality of <3 and (2) reads with residual length of <40 bases after the adaptor sequences were trimmed. Then the filtered sequencing reads were converted into FASTQ files, and all clean reads were mapped to the mouse genome (GRCm38/mm10) by using BWA v.0.7.13 with default parameters. The Genome Analysis Toolkit (GATK; v.3.7) HaplotypeCaller was applied for identifying variants, following the criteria as follows: (1) sequencing depth (for each individual) >1/4× and <4×; (2) root mean square (RMS) mapping quality (MQ) >40.0; (3) Phred-scaled p value using Fisher’s exact test to detect strand bias <60; (4) Z score from the Wilcoxon rank sum test of alanine transaminase (Alt) versus Ref read MQs (MQRankSum) >−12.5; and (5) Z score from the Wilcoxon rank sum test of Alt versus Ref read position bias (ReadPosRankSum) >−8. After filtering out variants that were not detected in the two wild-type mice, we picked out the variants with G-deletions among the remaining variants. These remaining variants with the putative off-target sites, which were predicted by Cas-OFFinder3 (up to 5 mismatches), were scanned.
Data accession
WGS and deep-sequencing data are deposited to the Sequence Read Archive (SRA) database (SRA: PRJNA713933). The authors declare that all used plasmids, annotated DNA sequences, and other data are all available from the author upon request.
Statistical analysis
Results were obtained from two or three independent experiments and are presented as the mean ± SD values. Data plotting was carried out using GraphPad Prism 8.0.
Acknowledgments
This work was supported, in part, by the National Key Research and Development Program (2018YFC1004700; to Y.Q.), Excellent Youth Foundation of Guangdong Scientific Committee (2020B1515020018; to Y.Q.), National Natural Science Foundation of China Grant (31825018; to Q.S.), and Shanghai Municipal Science and Technology Major Project (2018SHZDZX05; to Q.S).
Author contributions
Z. Liu, Y.Q., and Q.S. designed, conceived, and supervised the work. J.L., Z. Lu, and X.L. performed the experiments and data analysis. Y.Q., J.L., and X.L. co-wrote the manuscript. S.W., W.Y., Y.L., X.Z., and X.H. helped with the experiments, data analysis, and discussion. S.H. performed computational analysis.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.06.020.
Contributor Information
Qiang Sun, Email: qsun@ion.ac.cn.
Yunbo Qiao, Email: ybqiao@gzhu.edu.cn.
Zhen Liu, Email: zliu2010@ion.ac.cn.
Supplemental information
References
- 1.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J., Nickerson D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maki H. Origins of spontaneous mutations: specificity and directionality of base-substitution, frameshift, and sequence-substitution mutageneses. Annu. Rev. Genet. 2002;36:279–303. doi: 10.1146/annurev.genet.36.042602.094806. [DOI] [PubMed] [Google Scholar]
- 3.Cooper D.N., Krawczak M. The mutational spectrum of single base-pair substitutions causing human genetic disease: patterns and predictions. Hum. Genet. 1990;85:55–74. doi: 10.1007/BF00276326. [DOI] [PubMed] [Google Scholar]
- 4.Karimian A., Azizian K., Parsian H., Rafieian S., Shafiei-Irannejad V., Kheyrollah M., Yousefi M., Majidinia M., Yousefi B. CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell. Physiol. 2019;234:12267–12277. doi: 10.1002/jcp.27972. [DOI] [PubMed] [Google Scholar]
- 5.Wu Y., Liang D., Wang Y., Bai M., Tang W., Bao S. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molla K.A., Yang Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019;37:1121–1142. doi: 10.1016/j.tibtech.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Villiger L., Grisch-Chan H.M., Lindsay H., Ringnalda F., Pogliano C.B., Allegri G., Fingerhut R., Häberle J., Matos J., Robinson M.D. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 10.Lin X., Chen H., Lu Y.Q., Hong S., Hu X., Gao Y., Lai L.L., Li J.J., Wang Z., Ying W. Base editing-mediated splicing correction therapy for spinal muscular atrophy. Cell Res. 2020;30:548–550. doi: 10.1038/s41422-020-0304-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsoukas I.G. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front. Genet. 2020;11:528. doi: 10.3389/fgene.2020.00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampton T. DNA Prime Editing: A New CRISPR-Based Method to Correct Most Disease-Causing Mutations. JAMA. 2020;323:405–406. doi: 10.1001/jama.2019.21827. [DOI] [PubMed] [Google Scholar]
- 14.Tang X., Sretenovic S., Ren Q., Jia X., Li M., Fan T., Yin D., Xiang S., Guo Y., Liu L. Plant Prime Editors Enable Precise Gene Editing in Rice Cells. Mol. Plant. 2020;13:667–670. doi: 10.1016/j.molp.2020.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Y.Y., Chai Y.P., Lu M.H., Han X.L., Lin Q., Zhang Y., Zhang Q., Zhou Y., Wang X.C., Gao C., Chen Q.J. Prime editing efficiently generates W542L and S621I double mutations in two ALS genes in maize. Genome Biol. 2020;21:257. doi: 10.1186/s13059-020-02170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schene I.F., Joore I.P., Oka R., Mokry M., van Vugt A.H.M., van Boxtel R., van der Doef H.P.J., van der Laan L.J.W., Verstegen M.M.A., van Hasselt P.M. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020;11:5352. doi: 10.1038/s41467-020-19136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., Li X., He S., Huang S., Li C., Chen Y., Liu Z., Huang X., Wang X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020;6:27. doi: 10.1038/s41421-020-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian D., Wang Q., Zhang P., Araki H., Yang S., Kreitman M., Nagylaki T., Hudson R., Bergelson J., Chen J.Q. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature. 2008;455:105–108. doi: 10.1038/nature07175. [DOI] [PubMed] [Google Scholar]
- 19.Ku C.S., Tan E.K., Cooper D.N. From the periphery to centre stage: de novo single nucleotide variants play a key role in human genetic disease. J. Med. Genet. 2013;50:203–211. doi: 10.1136/jmedgenet-2013-101519. [DOI] [PubMed] [Google Scholar]
- 20.Zhao L., Li K., Bao S., Zhou Y., Liang Y., Zhao G., Chen Y., Xiao J. A 1-bp deletion in the gammaC-crystallin leads to dominant cataracts in mice. Mamm. Genome. 2010;21:361–369. doi: 10.1007/s00335-010-9275-5. [DOI] [PubMed] [Google Scholar]
- 21.Li J., Liu Z., Huang S., Wang X., Li G., Xu Y., Yu W., Chen S., Zhang Y., Ma H. Efficient base editing in G/C-rich regions to model androgen insensitivity syndrome. Cell Res. 2019;29:174–176. doi: 10.1038/s41422-018-0133-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y., Li J., Zhou C., Meng B., Wei Y., Yang G., Lu Z., Shen Q., Zhang Y., Yang H., Qiao Y. Allele-specific genome editing of imprinting genes by preferentially targeting non-methylated loci using Staphylococcus aureus Cas9 (SaCas9) Sci. Bull. (Beijing) 2019;64:1592–1600. doi: 10.1016/j.scib.2019.08.023. [DOI] [PubMed] [Google Scholar]
- 23.Yang G., Zhou C., Wang R., Huang S., Wei Y., Yang X., Liu Y., Li J., Lu Z., Ying W. Base-Editing-Mediated R17H Substitution in Histone H3 Reveals Methylation-Dependent Regulation of Yap Signaling and Early Mouse Embryo Development. Cell Rep. 2019;26:302–312.e4. doi: 10.1016/j.celrep.2018.12.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.