

Abstract
Self-amplifying mRNA (saRNA) represents a promising platform for nucleic acid delivery of vaccine immunogens. Unlike plasmid DNA, saRNA does not require entry into the nucleus of target cells for expression, having the capacity to drive higher protein expression compared to mRNA as it replicates within the cytoplasm. In this study, we examined the potential of stabilized native-like HIV-1 Envelope glycoprotein (Env) trimers to elicit immune responses when delivered by saRNA polyplexes (PLXs), assembled with linear polyethylenimine. We showed that Venezuelan equine encephalitis virus (VEEV) saRNA induces a stronger humoral immune response to the encoded transgene compared to Semliki Forest virus saRNA. Moreover, we characterized the immunogenicity of the soluble and membrane-bound ConSOSL.UFO Env design in mice and showed a faster humoral kinetic and an immunoglobulin G (IgG)2a skew using a membrane-bound design. The immune response generated by PLX VEEV saRNA encoding the membrane-bound Env was then evaluated in larger animal models including macaques, in which low doses induced high IgG responses. Our data demonstrated that the VEEV saRNA PLX nanoparticle formulation represents a suitable platform for the delivery of stabilized HIV-1 Env and has the potential to be used in a variety of vaccine regimens.
Keywords: saRNA, HIV, Env, vaccine, polyplexes, PEI, mouse, macaque
Graphical abstract
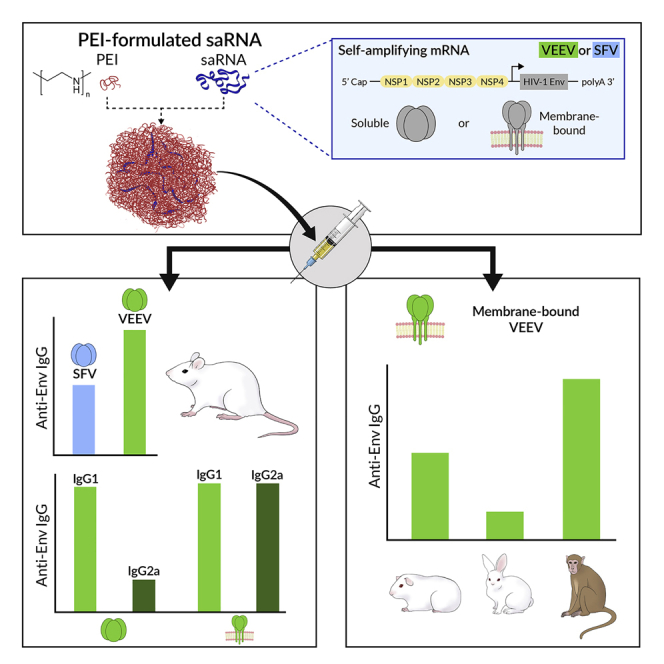
Aldon, McKay, and colleagues evaluated the potential of formulated VEEV and SFV self-amplifying mRNA (saRNA) for the delivery of HIV-1 Env stabilized trimers. The authors demonstrate great potential for PEI-polyplex formulated VEEV-saRNA platform, which elicits strong humoral responses at low doses in non-human primates.
Introduction
To date, many commercially available vaccines rely on products grown in cell culture or are from animal origin (e.g., chicken eggs for flu vaccines).1,2 The production methods involved are often complex, requiring numerous purification and control steps to ensure the safety, quality, and stability of the vaccine products.3,4 These diverse production and control processes impede rapid manufacture and testing of new vaccine immunogens. In contrast to classic vaccines, nucleic acid-based vaccines represent promising modalities, as DNA and RNA can be quickly manufactured and are versatile platforms that can be quickly and easily adapted to emerging pathogens by substitution of the encoded gene(s) of interest (GOI). However, to date DNA vaccines have only induced modest humoral immunity in humans, mostly due to limited delivery efficiency into the nucleus.5, 6, 7 In this regard, messenger RNA (mRNA) represents a promising alternative to DNA vaccines, with the major advantage that mRNA only needs to be delivered to the cytosol of target cells, where it will be directly translated into protein. In addition, recent developments in mRNA molecular engineering have greatly improved mRNA stability in vitro and in vivo, with various products now in early-stage human clinical trials and two products recently approved for SARS-CoV-2 vaccination.8, 9, 10 Self-amplifying mRNA (saRNA or replicon) represents a promising mRNA vaccine strategy, developed to support high and longer-term expression of the GOI in contrast to non-replicating mRNA platforms, which cannot self-replicate to amplify the mRNA pool available for translation.11 saRNAs are derived from single-stranded RNA viruses such as alphaviruses from which the structural genes have been removed from the genome and replaced by a GOI.12 The non-structural protein genes coding for the replication machinery (replicase) are retained and amplify the number of saRNA copies within the cytoplasm, which are then translated by the cellular machinery.
Importantly, RNA vaccines such as mRNA or saRNA are synthetic products manufactured through enzymatic reactions where cell culture is not required, thereby increasing safety and ease of manufacture.13 Because of this efficient enzymatic production methodology, high numbers of RNA vaccine doses can be produced quickly.14 Like the parental alphaviruses, saRNA vaccines generate single- and double-stranded RNA molecules within the cytoplasm during the replication cycle, each of which possesses self-adjuvanting properties by stimulating the innate immune system, a significant advantage over plasmid DNA vaccine delivery and molecular adjuvantation.15, 16, 17, 18, 19, 20, 21, 22
Early saRNA delivery strategies used virus-like replicon particles (VRPs) that resemble alphaviruses and are produced by co-transfecting cells with saRNA and helper RNA molecules expressing structural proteins, allowing budding of VRPs containing saRNA and displaying surface viral glycoproteins.23, 24, 25 These VRPs are single-cycle alphaviruses that are replication defective because of exclusion of the structural protein genes from the packaged saRNA. A number of studies previously evaluated VRPs as well as DNA-launched saRNA for the delivery of Semliki Forest virus (SFV)- or Venezuelan equine encephalitis virus (VEEV)-based saRNA expressing gp160, truncated gp160, or soluble gp140 HIV-1 Envelope glycoprotein (Env) immunogens derived from various isolates (e.g., HXB2, BH10, CN54, SF162).26, 27, 28, 29, 30 Although these studies showed that VRPs induced potent immune responses against the encoded immunogens, anti-vector neutralizing immunity was also observed against VRPs.23,31,32 Therefore, efforts to create delivery systems that bypass VRP packaging while ensuring saRNA integrity led to the development of delivery vehicles such as lipid nanoparticles (LNPs) and cationic nanoemulsions.11,33, 34, 35, 36, 37 More recently, an saRNA polyplex (PLX) nanoparticle formulation on the basis of the cationic linear polymer polyethylenimine (PEI) was developed by BioNTech, which proved to be effective at delivering saRNA. Using a similar approach, recent reports showed that robust immune responses were elicited against HIV-1 T cell and Influenza hemagglutinin (HA) immunogens.38,39 In the present work, we evaluated the potential of VEEV saRNA encoding HIV-1 Env trimers in PLX formulations to induce potent immune responses in BALB/c mice and compared it to unformulated saRNA and SFV saRNA.
Although nucleic acid platforms are attractive vaccine strategies, the expression of the GOI relies on the target cell machinery. Thus, in vivo expression of the GOI prevents downstream selection for appropriately processed and folded immunogens. Therefore, immunogens such as HIV-1 Env require engineering to stabilize their structure and limit the need for specific cellular processes such as furin cleavage.40, 41, 42 In order to bring Env trimers to nucleic acid platforms, we and others have developed native-like Env trimers that do not require furin processing and are properly folded and highly stable.40,41,43,44 In this study, we evaluated saRNA PLX nanoparticles for delivery of our HIV-1 Env ConSOSL.UFO design that produces non-cleavable native-like Env trimers.43 We compared the immune response induced by two forms of this design, one producing soluble Env trimers (ConSOSL.UFO.664) and one producing membrane-bound Env trimers (ConSOSL.UFO.750). The impact of the time interval between the prime and boost immunizations on the immune response was also investigated. Finally, as guinea pigs, rabbits, and macaques are critical pre-clinical models for the evaluation of vaccine strategies against HIV-1, we assessed the response induced in these animals using polymer formulated VEEV saRNA expressing ConSOSL.UFO.750 and demonstrated the potential of this delivery platform to induce strong humoral responses.
Results
Polymer formulated VEEV-based saRNA induces high HIV-Env-specific humoral responses in mice
As the intramuscular route is the preferred route used to administer vaccines, we evaluated the saRNA vaccine platform referred to as Amplified Immune Response (A.I.R), allowing rapid production and effective delivery of saRNA, in in vivo studies. The VEEV-based saRNA was formulated as PLX nanoparticles from linear PEI with an optimized protocol.38,39 Hereafter, the complexed saRNA is referred to as PLX A.I.R saRNA. To evaluate the performance of PLX A.I.R saRNA for in vivo expression of Env immunogens, we analyzed the immune responses induced by PLX A.I.R/VEEV saRNA in mice through intramuscular injections and compared these to animals immunized with unformulated A.I.R/VEEV saRNA, all expressing the soluble Env ConSOSL.UFO.664 immunogen. A control group was included with mice immunized with a SFV-based saRNA expressing the HA antigen derived from Influenza virus H1N1 A/California/7/2009 strain (Cf7-HA), as this saRNA previously demonstrated its ability to induce high responses against HA in BALB/c mice.39
We immunized groups of BALB/c mice 3 times at 3 week intervals, collected serum samples at each immunization time point, and proceeded to euthanasia at week 8 to assess the cellular response from the systemic compartment (Figure 1A). Four groups received PLX A.I.R/VEEV-gp140—expressing ConSOSL.UFO.664—with incremental saRNA doses: 0.01, 0.1, 1, or 10 μg in the same injection volume. One group was injected with unformulated A.I.R/VEEV-gp140, and an additional group received a 1 μg saRNA dose of PLX A.I.R/SFV-H1N1/Cf7-HA. Analysis of the serum antigen-specific immunoglobulin G (IgG) response by ELISA showed that a minimum of 1 μg of PLX A.I.R/VEEV-gp140 was necessary to induce consistent anti-Env IgG responses after 2 injections, whereas doses < 1 μg failed to induce relevant IgG responses (Figure 1B). In addition, the 10 μg dose of PLX A.I.R/VEEV-gp140 presented significantly higher IgG titers by week 6 compared to the 1 μg dose, with a mean anti-Env IgG titer of 91 and 12 μg/mL, respectively (p = 0.0159). This enhanced titer compared to the 1 μg dose was maintained after the third injection (p = 0.0079). Interestingly, the second injection only marginally boosted antigen-specific IgG titer for either Env or H1N1/Cf7-HA. Importantly, the unformulated A.I.R/VEEV-gp140 induced significantly lower humoral responses compared to PLX A.I.R/VEEV-gp140, with on average <1 μg/mL anti-Env IgG titer after 3 immunizations. In addition, we observed that PLX A.I.R/SFV-H1N1/Cf7-HA induced quicker and higher responses than PLX A.I.R/VEEV-gp140 for the same dose, and therefore the SFV platform should be evaluated for HIV-1 Env immunogens. We also measured the Env-specific IgG content in the vaginal fluid at the final time point as a marker of mucosal response. Anti-Env IgG was detected in the highest dose group for PLX A.I.R/VEEV-gp140, likely reflecting the serum IgG titer, as similar levels of vaginal antigen-specific IgG were measured for the PLX A.I.R/SFV-H1N1/Cf7-HA group (Figure 1C). Altogether, these results demonstrate that the linear PEI formulated saRNA was more immunogenic than unformulated saRNA and that anti-Env humoral responses were induced by PLX A.I.R/VEEV-gp140 in a dose-dependent manner.
Figure 1.

Polymer formulated VEEV-based saRNA induces a high antigen-specific humoral response
Groups of n = 5 BALB/c mice were immunized 3 times 3 weeks apart with escalating doses of polymer formulated (PLX) VEEV-based saRNA encoding the ConSOSL.UFO.664 gp140 protein (PLX A.I.R/VEEV-gp140) and compared to unformulated VEEV-gp140. A PLX SFV-based saRNA encoding H1N1/Cf7-HA (PLX A.I.R/SFV-H1N1/Cf7-HA) was included as a positive control group. (A) The immunization schedule is depicted. (B) The specific IgG titers against H1N1/Cf7-HA and ConSOSL.UFO.664 were determined by ELISA. The RNA dose used for each group of animals is indicated under the x axis. For clarity, only p values corresponding to the last time point are indicated. (C) HA- and gp140-specific IFN-γ ELISpots are reported as spleen forming units (SFU)/106 splenocytes. ELISpots were performed at week 8 after euthanasia. (D) The antigen-specific vaginal IgG was determined by ELISA. (C and D) Box and whiskers, with median, min-max, and 25th–75th percentile. Mann-Whitney unpaired t test, p values: ∗<0.05, ∗∗<0.01.
Although there were no or very low anti-Env IgG titers for PLX A.I.R/VEEV-gp140 in the 0.01 and 0.1 μg groups, detectable cellular responses were generated (Figure 1C). Interestingly, the antigen-specific spot forming units (SFU) for these two groups were comparable to the 10 μg group. In contrast, the 1 μg dose group appeared to induce the highest cellular response, with an average of 366 SFU/106 splenocytes compared to 26 for the 0.01 μg group (p = 0.0159), 53 for the 0.1 μg group (p = 0.0952), and 65 for the 10 μg group (p = 0.0952). These data suggest a different dose relationship for the induction of cellular and humoral responses. Importantly, unformulated A.I.R/VEEV-gp140 failed to induce cellular interferon (IFN)-γ responses, and this was strongly significant compared to the PLX A.I.R/VEEV-gp140 1 μg and 10 μg groups (p < 0.01). These results further support the conclusion that the polymer vehicle enhances the delivery and expression of saRNA into target cells and fosters high antigen-specific humoral and cellular responses in mice.
VEEV-based saRNA induces higher anti-HIV Env responses than SFV-based saRNA
As the SFV saRNA expressing HA induced higher IgG titers at a lower dose in comparison to anti-HIV Env IgG titers using VEEV saRNA, we assessed whether the SFV-based saRNA could induce higher anti-HIV-Env immune responses. To this aim, we immunized groups of BALB/c mice 3 times 3 weeks apart with either PLX A.I.R/VEEV-gp140 or PLX A.I.R/SFV-gp140 with increasing doses of saRNA: 0.1 μg, 1 μg, or 10 μg (Figure 2A). For the 3 doses tested, the PLX A.I.R/VEEV-gp140 saRNA induced significantly higher levels of Env-specific IgG compared to the SFV-based saRNA (Figure 2B). Although PLX A.I.R/VEEV-gp140 efficiently mounted an anti-Env IgG response after 2 injections for the lowest doses, PLX A.I.R/SFV-gp140 did not induce consistent responses with the 0.1 μg or 1 μg dose. For the PLX A.I.R/SFV-gp140 1 μg group, a fair level of anti-Env response was only observed after the third immunization, with a median titer of 7.2 μg/mL, and this was significantly lower than the equivalent dose of VEEV-based saRNA (p = 0.0159). In addition, by week 9 the PLX A.I.R/VEEV-gp140 10 μg group reached a median Env-specific IgG titer of 100 μg/mL, which was ∼15-fold higher than the corresponding SFV group with a median titer of 6.1 μg/mL (p = 0.0079).
Figure 2.

The VEEV-based saRNA induces higher immune responses against gp140 than SFV-based saRNA
Groups of n = 5 BALB/c mice were immunized 3 times 3 weeks apart with escalating doses of polymer formulated (PLX) VEEV-based saRNA encoding the ConSOSL.UFO.664 gp140 protein (PLX A.I.R/VEEV-gp140) and compared to a SFV-based saRNA platform (PLX A.I.R/SFV-gp140). A PLX A.I.R/SFV-H1N1/Cf7-HA was included as a positive control group. (A) The immunization schedule is depicted. (B) Antigen-specific IgG titers as determined by ELISA. (C) The HA-specific and gp140-specific IgG1 and IgG2a were measured by ELISA, and the IgG2a-to-IgG1 ratios at week 9 are reported here. For each animal, when both specific IgG2a and IgG1 titers were <500 ng/mL, the ratio was not determined. (D) Week 9 HA- and gp140-specific IFN-γ ELISpots reported as spleen forming units (SFU)/106 splenocytes. For (B), the indicated p values compare VEEV to SFV for each week and each specific RNA dose. In (D), p values indicated by arrows compare VEEV to SFV for each week and each specific RNA dose. (B–D) Box and whiskers with median, min-max, and 25th–75th percentile. Mann-Whitney unpaired t test, p values: ∗<0.05, ∗∗<0.01, and ∝<0.05. Also see Figure S1.
We further characterized the antibody response and determined anti-Env IgG1 and IgG2a titers, as these can be used as a surrogate to assess the type of T helper (Th1 or Th2) response induced by the vaccine using the IgG2a-to-IgG1 ratio.45 By the end of the vaccination schedule, both SFV- and VEEV-based saRNA appeared to prompt IgG1 and IgG2a responses similar to Env as shown by the IgG2a-to-IgG1 ratios (Figure 2C; Figures S1A and S1B). We observed a trend toward an IgG1 bias for the 0.1 and 1 μg doses and a more balanced response for the 10 μg dose. In marked contrast, the anti-HA response appeared biased toward IgG2a, consistent with Vogel et al.39 We then assessed the Env-specific IgG in the vaginal fluid and detected the highest levels in the PLX A.I.R/VEEV-gp140 10 μg group, whereas only limited amounts of specific IgG were detected in the SFV saRNA groups (Figure S1C). Altogether, these results demonstrated that the VEEV-based saRNA outperformed the SFV-based saRNA at inducing high levels of anti-Env specific IgG. Furthermore, our results seemed to indicate that the polarization of the antigen-specific IgG isotype response is primarily antigen dependent rather than driven by the genetics of the alphavirus from which the saRNA is derived.
Next, we assessed the antigen-specific cellular IFN-γ response by enzyme-linked immunosorbent spot (ELISpot). The response proved to be significantly higher (p < 0.05) in animals immunized with 0.1 and 1 μg PLX A.I.R/VEEV-gp140 compared to PLX A.I.R/SFV-gp140 0.1 and 1 μg groups, respectively (Figure 2D). Although the PLX A.I.R/SFV-gp140 1 and 10 μg groups induced similar levels of cellular responses, the 0.1 μg dose struggled to produce detectable responses. In contrast, the 0.1 and 10 μg PLX A.I.R/VEEV-gp140 groups induced equivalent IFN-γ responses that were lower than the 1 μg regimen (p = 0.0079). Hence, these results indicate that the VEEV saRNA was more effective at mounting an IFN-γ cellular response against the encoded Env immunogen.
Finally, comparison of this experiment with PLX A.I.R/VEEV-gp140 immunizations from the first experiment showed that IgG titers induced with 1 and 10 μg doses were consistent across the two experiments, with some variability in the lower 0.1 μg dose (Figures 1B and 2B). Furthermore, IFN-γ ELISpot results were analogous in both experiments, with the 1 μg dose reproducibly producing the strongest response, which reached statistical significance in the second experiment compared to the other two dose regimens (p < 0.01) (Figures 1C and 2D).
Increasing the interval between prime and boost induces similar responses
Having determined that the VEEV saRNA induced higher antigen-specific responses to our Env immunogen, we determined whether the interval between prime and boost immunizations modulates the induced response. We immunized groups of BALB/c mice, following two schedules (Figure 3A). Mice were immunized at 0 and 3 weeks (schedule 1) or at 0 and 6 weeks (schedule 2), with 1 μg PLX A.I.R/VEEV-gp140 and IFN-γ cellular responses assessed after each immunization at weeks 3 and 5 for schedule 1 and weeks 6 and 8 for schedule 2. We first looked at the Env-specific serum IgG titer and observed that whether the boost was given at week 3 or week 6, titers were comparable at the final time point, with a mean IgG titer of 39 and 54 μg/mL, respectively (Figure 3B). This demonstrated that the response could be efficiently recalled with a delayed boost immunization. This also demonstrated that high IgG titers could quickly be achieved with a boost injection closer to the initial one, whereas the longer schedule 2 only marginally increased the titer by week 6 prior to a second injection with a mean titer of 0.374 μg/mL compared to the combined schedule 1/schedule 2 week 3 mean titer of 0.134 μg/mL (p < 0.01) and to schedule 1 week 5 mean titer of 39 μg/mL (p < 0.01). In addition, IFN-γ ELISpot results showed that the response was similar for schedules 1 and 2 by week 3 and week 6, respectively, as well as for the last time point (Figure 3C). Altogether, these results show that the time interval between prime and boost could be increased with no impact on the magnitude of the humoral and cellular IFN-γ responses.
Figure 3.

Impact of the time interval between prime and boosting
(A) Two groups of n = 10 BALB/c mice were primed with 1 μg RNA dose of PLX A.I.R/VEEV-gp140 and assigned to immunization schedule 1 (Sch. 1) or schedule 2 (Sch. 2). In schedule 1, 5 mice were euthanized at week 3 (cull 1) and 5 mice were boosted at week 3 and then euthanized at week 5 (cull 2). In schedule 2, 5 mice were euthanized at week 6 (cull 1) and 5 mice were boosted at week 6 and then euthanized at week 5 (cull 2). (B) Antigen-specific IgG titers as determined by ELISA. (C) gp140-specific IFN-γ ELISpots were performed after each euthanasia time point and are reported as spleen forming units (SFU)/106 splenocytes. Box and whiskers, with median, min-max, and 25th–75th percentile. Mann-Whitney unpaired t test, p values: ∗<0.05, ∗∗<0.01, ∗∗∗<0.001.
Membrane-bound Env elicits a faster humoral kinetic and higher antigen-specific IgG2a compared to soluble Env
We previously established that the membrane-bound ConSOSL.UFO.750 Env triggered a faster humoral kinetic and a different IgG2a:IgG1 profile compared to its soluble counterpart ConSOSL.UFO.664 when delivered as DNA.43 Therefore we decided to examine the response elicited by both immunogens using PLX A.I.R/VEEV saRNA. Mice were immunized following schedule 2 with either PLX A.I.R/VEEV-UFO.750 (UFO.750) or PLX A.I.R/VEEV-gp140 (UFO.664) (Figure 4A). After one injection, the membrane-tethered UFO.750 design induced 17-fold higher Env-specific IgG titer compared to the soluble UFO.664 design (p < 0.01) (Figure 4B). By week 8, both groups reached comparable IgG levels, with a median titer at 21.2 μg/mL for UFO.664 and 33.3 μg/mL for UFO.750. Although the IgG levels were similar at week 8, we observed that Env-specific IgG2a titers for the UFO.750 group were on average >10-fold higher (p < 0.05) compared to UFO.664, whereas the IgG1 titers were equivalent in both groups (Figures 4C and 4D). In addition, we noted that the UFO.664 IgG response tended toward a marked IgG1 skew, with 4 animals having an IgG2a:IgG1 ≤ 0.01 whereas animals in the UFO.750 group ranged from 0.1 to 18 and presented a range of IgG1- or IgG2a-skewed responses (Figure 4E). However, with one animal in the UFO.664 group showing an IgG2a:IgG1 of 40, the trends between the two groups were not statistically different. Finally, IFN-γ ELISpot analysis showed that UFO.664 induced higher levels of cellular responses by week 8 compared to UFO.750, albeit not significant (p = 0.0556), and these levels were consistent with the results displayed in Figure 3C. Of note, the ELISpots were performed using ConSOSL.UFO.664 soluble protein as stimulating antigen, as peptide pools were not available. Thus, potential responses against the membrane proximal external region (MPER) and cytoplasmic tail domains of Env, which are included in ConSOSL.UFO.750 but not ConSOSL.UFO.664, were not assessed. Overall, these results confirmed our previous observations that the membrane-bound ConSOSL.UFO.750 Env induced a quicker humoral response with a different IgG isotype signature compared to ConSOSL.UFO.664. This phenomenon does not appear to be dependent on the delivery platform, whether it is plasmid DNA or saRNA delivered intramuscularly.
Figure 4.

Membrane-bound Env elicits faster humoral kinetic and higher specific IgG2a compared to soluble Env
Groups of n = 5 BALB/c mice were immunized twice 6 weeks apart with 1 μg RNA dose of PLX A.I.R/VEEV-Env expressing ConSOSL.UFO.664 (UFO.664) or ConSOSL.UFO.750 (UFO.750). (A) The immunization schedule is depicted. (B) The specific IgG titers were determined by ELISA using ConSOSL.UFO.664 as the coating antigen. (C) and (D) depict the Env-specific IgG1 and IgG2, respectively. (E) The IgG2a-to-IgG1 ratios for weeks 6 and 8 are plotted. For each animal, when both specific IgG2a and IgG1 titers were <500 ng/mL, the ratio was not determined. (F) Env-specific IFN-γ ELISpots are reported as spleen forming units (SFU)/106 splenocytes. ELISpots were performed using ConSOSL.UFO.664 protein on splenocytes from week 8. Box and whiskers, with median, min-max, and 25th–75th percentile. Mann-Whitney unpaired t test, p values: ∗< 0.05, ∗∗< 0.01. ns, Non-significant.
Env-specific IgG response elicited by saRNA UFO.750 in guinea pig, rabbit, and macaque models
To determine whether the immune response observed in BALB/c mice would be reproduced in other animal models, we tested PLX A.I.R/VEEV-UFO.750 in larger animal models and measured the Env-specific serum IgG. First, we tested PLX A.I.R/VEEV-UFO.750 in guinea pigs. Four groups of 4 animals were immunized with a 0.5, 2, 8, or 16 μg saRNA dose of PLX A.I.R/VEEV-UFO.750 following a schedule previously used for induction of neutralizing antibodies against HIV-1 Env.43,46 Animals were immunized 3 times at weeks 0, 4, and 8. Doses of 8 and 16 μg induced detectable but low antibody binding responses 3 weeks after the first injection, whereas the 0.5 and 2 μg doses did not induce detectable responses (Figure 5A). Although IgG titers were increased after the boost immunizations, IgG levels only reached a mean titer of ∼5 μg/mL for the 8 and 16 μg groups and <1 μg/mL for the lower doses. Next, we immunized groups of rabbits with the same incremental saRNA doses, including an additional group injected with a 32 μg dose (Figure 5B). These animals were injected twice 4 weeks apart, and the last serum sample was taken at week 6. Groups that received 0.5, 2, and 8 μg of saRNA did not appear to respond, and, in contrast to guinea pigs, we observed that a second boost was necessary to induce a detectable response for the 16 μg dose. However, both 16 and 32 μg groups produced titers by week 6 that were comparable to that induced in guinea pigs by week 6 for the 8 and 16 μg doses. Thus, these data show that there was no advantage in doubling the saRNA dose past a threshold in guinea pigs (8 μg) and rabbits (16 μg) for the induction of humoral responses.
Figure 5.

Env-specific humoral response elicited by saRNA encoding membrane-bound Env in guinea pig, rabbit, and macaque
Animals were immunized with escalating saRNA doses of PLX A.I.R/VEEV-ConSOSL.UFO.750. The Env-specific serum IgG titers were assessed at the indicated time points by capture ELISA using ConSOSL.UFO.664 MycHIS. (A) Guinea pigs were immunized at weeks 0, 4, and 8 (n = 4 animals per group). (B) Rabbits were immunized at weeks 0 and 4 (n = 4 animals per group). (C) Cynomolgus macaques were immunized at weeks 0, 4, and 8 (black arrows). 1 macaque received 5 μg 3 times, 2 macaques received 20 μg 3 times, 2 other macaques received 80 μg 3 times, and 1 macaque received an alternative regimen with 5 then 20 and 5 μg. (A and B) Box and whiskers, with median, min-max, and 25th–75th percentile. Mann-Whitney unpaired t test performed, p values: ∗<0.05. ns, Non-significant.
Next, we performed a pilot study to assess the efficiency of the PLX A.I.R/VEEV saRNA platform for expression in cynomolgus macaques, a non-human primate (NHP) model relevant to pre-clinical evaluation of HIV-1 vaccines. We injected 5 cynomolgus macaques with increasing doses of saRNA: 5 μg (n = 1), 20 μg (n = 2), and 80 μg (n = 2) doses. Animals received 3 injections of PLX A.I.R/VEEV-UFO.750 at weeks 0, 4, and 8, and serum samples were collected throughout the immunization schedule (Figure 5C). An additional animal received an alternative regimen with a low dose for prime (5 μg) followed by a 20 μg boost and a low-dose (5 μg) last boost. After the prime, low responses (<0.1 μg/mL) could be measured by week 4 in 3 animals. By week 8, 4 weeks after the first boost, 5/6 animals showed high anti-Env IgG titers, including 4 that had titers ranging from 16 to 47 μg/mL. However, a sixth animal in the highest dose group displayed a limited response, with only 0.64 μg/mL IgG titer at week 12. Interestingly, after the second boost, titers appeared to peak or plateau by week 8/10 and then started declining. These data suggest that PLX A.I.R/VEEV-UFO.750 is capable of inducing higher IgG titers in cynomolgus macaques than in guinea pigs or rabbits. It appeared that the alternative regimen induced similar IgG titer compared to the 4 animals that showed high titers by week 10. Interestingly, the 5 μg dose performed similarly to higher doses, suggesting a potential plateau effect with no advantage in increasing the dose of delivered saRNA. In addition, there appeared to be no advantage from inclusion of a second boost 4 weeks after the first boost, likely reflecting that there was no contraction of response initiated by the first boost. Although these data are preliminary, they provide some important insight for the planning of future studies.
Discussion
Although nucleic acid-based vaccines present production advantages, to date the immunogenicity of DNA-based vaccines has induced limited immunity in humans and usually required physical methods of delivery such as electroporation to efficiently deliver DNA in vivo.5,7,15 In contrast to DNA vaccines, which need to cross the nuclear envelope for expression, RNA vaccines only require entry into the cytoplasm by crossing the plasma membrane, without any risk of spontaneous integration into the host’s genome. Delivering RNA into target cells in sufficient quantities while maintaining its stability and integrity remains challenging, but the development of delivery vehicles such as LNPs, cationic nanoemulsions, and PLXs have greatly improved the delivery efficiency of RNA molecules.10 In addition to these improvements, saRNA itself appears to have advantages over non-replicating mRNA, as it potentially requires lower amounts of material while supporting high and longer-term expression of the GOI.36,39,47,48 In our study, we investigated a polymer formulated VEEV saRNA for the delivery of soluble native-like Env trimer immunogens. Importantly, we showed in mice that this platform induced higher immune responses than unformulated saRNA expressing the same soluble HIV-1 Env immunogen. This PLX A.I.R/VEEV-gp140 also induced >100-fold higher IgG titers compared to a previous LNP formulation for the same immunogen.35 In addition, we observed that the alphavirus species used to design the saRNA can impact the level of the immune response against Env. Although the SFV saRNA proved to be an excellent platform for HA antigen, the response to Env using SFV saRNA was markedly lower compared to HA and compared to the VEEV saRNA. Although studies suggest that VEEV saRNA might replicate more efficiently and potentially be less cytopathic than SFV saRNA, more studies comparing these two saRNAs are required to characterize fundamental properties that may explain the observed immune responses.49,50 Therefore, we suggest that immunogenicity of selected antigens should be evaluated using SFV and VEEV saRNA in order to determine which saRNA genetic background suits a particular antigen.
In the BALB/c mouse model, our data indicate that 2 immunizations with 10 μg of PLX A.I.R/VEEV-gp140 induce high anti-Env IgG titers (111.5 ± 21.0 μg/mL, mean ± SEM, n = 10) that were comparable to DNA + electroporation using 20 μg of ConSOSL.UFO.664 expression plasmid (76.3 ± 19.9 μg/mL, mean ± SEM, n = 5).43 In addition, we demonstrated that the interval between prime and boost immunizations (i.e., 3 versus 6 weeks) could be increased without impacting the level of the humoral and IFN-γ cellular responses for saRNA expressing soluble Env. In contrast, Hekele et al. showed that a saRNA expressing full-length HA and delivered through LNP encapsulation could induce higher IgG responses when the time between prime and boost was increased from 3 to 8 weeks.51 These differences could be intrinsic to the nature of the antigen—HA versus HIV-1 Env—but we hypothesized that the presentation of HA in the membrane context plays a key role and induces distinct responses to the soluble antigen. We also investigated the immunogenicity of a membrane-bound version of the ConSOSL.UFO design using the VEEV saRNA platform. We observed that the ConSOSL.UFO.750 design delivered through the VEEV saRNA platform induced a faster humoral response kinetic as well as a different IgG subtype signature compared to ConSOSL.UFO.664. Furthermore, the membrane-bound Env induced higher IgG2a responses than its soluble counterpart, in line with our previous report evaluating DNA delivery of these immunogens.43 Thus, our data support the conclusion that the membrane-tethered context of ConSOSL.UFO.750 is a key feature leading to IgG2a class switch.
Next, as the membrane-bound Env form induced a faster humoral kinetic and higher IgG2a responses in mice, we selected it for evaluation in larger animal models. Guinea pigs immunized with 8 or 16 μg PLX A.I.R/VEEV-UFO.750 developed low anti-Env IgG responses, but these are similar to levels achieved with 20 μg DNA + electroporation immunizations.43 In contrast, rabbits hardly developed an anti-Env response after 2 injections with 16 or 32 μg of PLX A.I.R/VEEV-UFO.750. These results indicate that the PLX A.I.R/VEEV platform might not be well adapted for these animal models. Therefore, we sought to evaluate the potential of the PLX A.I.R/VEEV-UFO.750 to induce humoral responses in NHPs. Although previous studies have shown that linear PEI-based formulation was an efficient delivery vehicle for SFV saRNA,38,39 we demonstrated in the present study that this technology was also efficient at delivering VEEV saRNA in NHPs, with strong humoral responses being raised. Importantly, we show that PLX A.I.R/VEEV saRNA prompted high responses against Env in macaques with saRNA doses as low as 5 μg. This represents 10 times less saRNA compared to the 50 μg dose administered to NHPs by Bogers et al. for the delivery of a gp140 immunogen.33 Similarly to this report, we observed that a second immunization at week 4 was required to induce high IgG titers in NHPs with an apparently faster kinetic in our study. This difference possibly reflects a synergy of the linear PEI-based formulation and the membrane-bound Env immunogen, as ConSOSL.UFO.750 induces a faster humoral kinetic than ConSOSL.UFO.664 (Aldon et al.43 and this study).
Although two recent studies evaluated saRNA delivery of HIV immunogens, their immunogens and design strategies largely differ from the present study. Moyo et al. evaluated the delivery of immunogens engineered to elicit T cell responses using a SFV-based saRNA,38 and Melo et al. evaluated the delivery of a particulate immunogen that presents on its surface a small domain of Env focused on the CD4 binding site (eOD-GT8).34 Our work evaluated the delivery of trimeric native-like trimers (soluble and membrane bound), which greatly differ from eOD-GT8 particulate presentation and the T cell immunogens. Moreover, we compared SFV- and VEEV-based saRNA delivery of soluble trimers in mouse and evaluated the performance of the PLX VEEV saRNA platform in 4 animal models, whereas the Moyo et al. and Melo et al. studies were carried out solely using mouse models. The observation that both guinea pigs and rabbits had low responses whereas NHPs showed promising and substantial responses after two immunizations is interesting. Previously, Hubby et al. showed that HA delivered by a VEEV-based VRP induced no humoral response in rabbits when administered intramuscularly but induced antibody responses after only a single subcutaneous injection.52 In addition, they showed that NHPs injected intramuscularly with the same VRPs developed good humoral responses. This, together with our data, suggests that the immunization route and the type of target cells greatly modulate the saRNA replication and may reflect variability in IFN response/restriction factors between cell types (e.g., muscle versus skin) and species. Of note, no toxicity was observed in the four animal models, consistent with previous clinical studies showing good safety profiles for linear PEI 22 kDa formulations.53,54
In case of an outbreak for a pathogen that can spread rapidly, a vaccine platform that is able to induce high antibody titers with a limited number of injections is desirable and could warrant quick and effective protection in an emergency setting. Here, we demonstrated that the PLX A.I.R/VEEV saRNA can induce high antigen-specific serum IgG after 2 injections in mice and NHPs. Therefore, this platform represents a promising vaccine strategy for both conventional and emergency vaccination settings where the delivery platform will not require extensive and complex GMP manufacture as for other classic vaccine approaches.
Materials and methods
saRNA synthesis by in vitro transcription
T7 in vitro transcription is based on protocols provided by the MEGAscript T7 Transcription Kit (Thermo Fisher, formerly Ambion). The general procedure starting with linear DNA template containing the T7 promoter, and particularly with respect to co-transcriptional capping with the synthetic cap analog beta-S-ARCA(D1) (used in 4-to-1 ratio regarding guanosine triphosphate [GTP] concentration), is carried out similarly to previously described protocols.55 Based on previous work, e.g., Pokrovskaya and Gurevich,56 high-yielding processes qualified for our particular systems were developed; here, protocols have been modified and optimized with respect to long saRNA with up to 10,000 nt.
Linear polyethylenimine formulation of saRNA
Polyelectrolyte complexes, also known as PLXs, comprising a 22.5 kDa linear PEI and saRNA were manufactured according to an internal developed and optimized protocol (https://worldwide.espacenet.com/patent/search/family/065234527/publication/WO2019137999A1?q=WO2019137999A1). Linear PEI was obtained from Polyplus-transfection (Illkirch, France). PLXs were formulated for a final RNA concentration of 0.1 mg/mL and a N-to-P ratio (N/P) of 12 in MBG Buffer (MES-buffered-glucose, pH 6.1). The N/P was calculated as the bulk stoichiometric ratio between the total amount of nitrogen within linear PEI and the phosphates in the RNA. For the calculation of the N/P from the concentrations in mg/mL, PEI was assumed to consist of repeat units with the molar mass of 43 Da comprising one nitrogen, and for the saRNA the molar mass of the repeat units (nucleotides) comprising the phosphates was taken as 330 Da. For manufacturing the PLXs equivoluminar amounts of PEI and saRNA solutions were mixed and immediately vortexed for 6 s. The RNA solution in MBG was prepared by mixing concentrated MBG Buffer (2×) and saRNA from stock solution. Similarly, PEI solution was prepared with the required volume of linear PEI stock, and concentrated MBG Buffer (2×). Water was added for final adjustment of concentrations if required. After mixing and vortexing, the formulations were incubated for 15 min at room temperature. For in vivo experiments with multiple injection time points, PLX stock formulations with the required amount for each time point were produced prior to experiment start and stored at −80°C. For quality control, physicochemical characterization of the PLX formulations was performed, including the following parameters: particle size by dynamic light scattering (Wyatt Technology, Dernbach, Germany), RNA concentration by UV- absorption at 260 nm (NanoDrop 2000, Thermo Fisher Scientific, Darmstadt, Germany), RNA integrity by capillary electrophoresis (Fragment Analyzer System, Agilent), ζ-potential by laser Doppler electrophoresis (WALLIS Zeta Potential, Cordouan, Pessac, France), and osmolality by freezing point depression (Osmomat 010, Gonotec, Berlin, Germany). Typical particle size after formulation ranged from 70 to 80 nm with a polydispersity of ∼0.24 and a ζ-potential between +22 and 25 mV. In average, osmolality of the formulations ranged between 300 and 330 mOsm. RNA integrity was monitored after thawing PLX, with no observed degradation of RNA during formulation of PLX, storing at −80°C, or thawing process.
Animals and immunization procedure
BALB/c mice aged 6–8 weeks old were placed into groups of n = 5. Animals were handled and procedures were performed in accordance with the terms of a project license granted under the UK Home Office Animals (Scientific Procedures) Act 1986. Mice were immunized intramuscularly in the quadriceps muscle with various doses of PLX A.I.R saRNA (SFV or VEEV) in a 50 μL injection volume following the immunization schedules indicated in the figures. Blood samples were collected from the tail vein throughout the study and were centrifuged to collect serum samples and then stored at −20°C. At the end of each experiment, mice were euthanized and spleens were collected to perform ELISpot analysis. In addition, for some experiments, vaginal washes were performed to analyze the IgG content. Mouse vaginas were washed with 3 × 25 μL of 1× PBS + protease inhibitor cocktail (Merck Millipore), and the collected samples were stored at −20°C.
Four groups of n = 4 of Dunkin-Hartley guinea pigs (Charles River) aged 10 weeks old were immunized intramuscularly in the quadriceps 3 times at 4 week intervals with PLX A.I.R/VEEV-UFO.750 in a 50 μL injection volume. Blood samples were collected at weeks 0, 4, 8, and 12 from the saphenous vein with a 23G needle (BD Biosciences) to prick the vain of the pre-warmed animals. After centrifugation of the blood, the serum was collected and stored at −20°C.
Five groups of n = 4 female New Zealand white rabbits (Charles River) aged 2.5 months old were immunized intramuscularly in the right leg quadriceps with PLX A.I.R/VEEV-UFO.750 twice 4 weeks apart with 0.5, 2, 8, 16, or 32 μg dose of saRNA in 50 μL. Pre-warmed animals were bled at weeks 0, 2, 4, and 6, using the marginal ear vein. Blood samples were spun and sera collected and stored at −20°C.
Female cynomolgus macaques (Macaca fascicularis), aged 39 and 44 months old and originating from Mauritian AAALAC-certified breeding centers were housed in IDMIT infrastructure facilities (Animal facility authorization #D92-032-02, Prefecture des Hauts de Seine, France) and in compliance with European Directive 2010/63/EU, the French regulations, and the Standards for Human Care and Use of Laboratory Animals of the Office for Laboratory Animal Welfare (OLAW, assurance number #A5826-01, USA). The protocols were approved by the institutional ethical committee “Comité d’Ethique en Expérimentation Animale du Commissariat à l’Energie Atomique et aux Energies Alternatives” (CEtEA #44) under statement number A15_073. The study was authorized by the “Research, Innovation and Education Ministry” under registration number APAFIS#3132-2015121014521340. Six macaques were immunized intramuscularly in the quadriceps 3 times at 4 week intervals with 5, 20, or 80 μg of PLX A.I.R/VEEV-UFO.750. One macaque received 5 μg 3 times, two macaques received 20 μg 3 times, two other macaques received 80 μg 3 times, and one macaque was immunized with an alternative regimen: 5 μg, then 20 μg followed by 5 μg. Blood samples were collected at weeks −1, 0, 2, 4, 6, 8, 10, and 12 and spun, and sera were collected and stored at −80°C.
IFN-γ ELISpots
Mice were euthanized, and were spleens removed, placed into 5 mL of RPMI, and processed as previously described.57 The IFN-γ T cell response was evaluated with the Mouse IFN-γ ELISpotPLUS kit (Mabtech), following the manufacturer’s instructions. Briefly, anti-IFN-γ pre-coated plates were blocked with complete medium. For the negative and gp140 antigen wells, 2.5 × 105 cells/well (100 μL of the cell suspension) were added to wells containing 100 μL of complete medium (negative control) or 100 μL of 10 μg/mL ConSOSL.UFO.664 protein in complete medium—final concentration of 5 μg/mL in the 200 μL final volume in duplicate wells. The positive control wells were loaded with 5 × 105 cells/well in wells containing ConA in complete medium—final concentration of 5 μg/mL in the 200 μL final volume. After overnight incubation at 5% CO2, +37°C, plates were developed as per the manufacturer’s protocol, allowed to dried, and read with the AID ELISpot Reader ELR03 and AID ELISpot Reader software (Autoimmun Diagnostika).
Antigen-specific IgG/IgG1/IgG2a ELISAs
For mouse serum and vaginal samples, the antigen-specific IgG, IgG1, and IgG2a titers were assessed by ELISA as previously described.57 MaxiSorp high-binding ELISA plates (Nunc) were coated with 100 μL/well of 1 μg/mL Influenza A H1N1 soluble protein (JPT Peptide Technologies, Berlin, Germany) or 1 μg/mL HIV-1 Env ConSOSL.UFO.664 (in house) in 1× PBS. For the standard IgG/IgG1/IgG2a, 3 columns on each plate were coated with 1:1,000 dilution of goat anti-mouse Kappa and Lambda light chains (SouthernBiotech). For total IgG (vaginal samples), the entire plate was coated with goat anti-mouse Kappa and Lambda light chains instead of the antigen. After overnight incubation at +4°C, the plates were washed, blocked for 1 h, and washed again, and the diluted samples and standard were added. Plates were then incubated for 1 h and washed, and the secondary detection antibody was added (anti-mouse IgG/IgG1/IgG2a-HRP [SouthernBiotech]). After 1 h incubation, plates were washed and developed for 5 min. The absorbance was read on a KC4 spectrophotometer at 450 nm (BioTek Instruments). The standard IgG was purchased from SouthernBiotech (cat# 0107-01). The standard IgG1 (cat# M9269) and standard IgG2a (cat# M9144) were purchased from Sigma.
For guinea pigs, rabbits, and cynomolgus macaques, Env-specific serum IgG titers were determined by 9E10 capture ELISA. ELISA plates were coated overnight at +4°C with 9E10 mouse anti-cMyc monoclonal antibody—5 μg/mL for guinea pigs and rabbits, 2.5 μg/mL for macaques—in 1× PBS, 100 μL/well. For the standard wells, 1:2,000 dilution in 100 μL/well 1× PBS of goat anti-guinea pig IgG F(ab′)2 (Jackson ImmunoResearch) was used for guinea pigs, 1:3,000 of goat anti-rabbit IgG Fc Ab (Jackson ImmunoResearch) for rabbits, and 1,2000 of goat anti-human Kappa and goat anti-human Lambda (SouthernBiotech) for macaques. Plates were washed 4 times with 1× PBS + 0.05% Tween 20 and then blocked with 200 μL/well 1× casein buffer (Thermo Fisher Scientific) for 1 h at +37°C. After a washing step, ConSOSL.UFO.664 Myc-HIS Env trimers were loaded onto the plates at 1 μg/mL in casein buffer, 100 μL/well. Diluted serum samples were then added in triplicate wells (50 μL/well in casein buffer) as well as the diluted IgG standard for each species starting at 200 ng/mL. After 1 h at +37°C, plates were washed and the detection antibody added (100 μL/well in casein buffer): 1:25,000 donkey anti-guinea pig IgG (H+L) biotinylated antibody (Sigma) for guinea pigs, 1:25,000 mouse anti-rabbit IgG biotinylated monoclonal antibody clone RG-96 (Sigma) for rabbits, and 1:50,000 mouse anti-rhesus monkey IgG Fc biotinylated antibody (Southern Biotech) for cynomolgus macaques. After 1 h incubation at +37°C, plates were washed, and poly-HRP40 (Fitzgerald) was added at 1:10,000 in casein buffer, 100 μL/well, for another hour. After a final wash, plates were developed for 5 min and the absorbance was read on a KC4 spectrophotometer at 450 nm.
Statistical analysis
Statistical analyses were carried out with Mann-Whitney unpaired t test in order to determine statistical significance, using GraphPad Prism v.7.0h.
Acknowledgments
We warmly thank Jonathan Mottl, Jakob Erk, and André Gerts for excellent technical assistance as well as Raphaël Ho Tsong Fang, Sébastien Langlois, and all the members of the ASW core lab facility at IDMIT center. We would like to thank Cristina Sala for creating the graphical abstract and the PLX cartoon. This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 681137. The sole responsibility for the content of this project lies with the authors. It does not necessarily reflect the opinion of the European Union. The European Commission is not responsible for any use that may be made of the information contained therein. We gratefully acknowledge Dormeur Investment Services Ltd for providing funds to purchase equipment used in these studies. The Infectious Disease Models and Innovative Therapies research infrastructure (IDMIT) is supported by the “Programme Investissements d’Avenir” (PIA), managed by the ANR under reference ANR-11-INBS-0008, and received the support of the Domaine d’Intérêt Majeur (DIM, Paris, France) “One Health.”
Author contributions
R.J.S., P.F.M., and A.B.V. designed the studies; Y.A., P.F.M., J.M.H., R.L., P.M., and N.D.-B. performed the experimental work; H.H. supervised the structural characterization of the vaccine; R.J.S., R.L.G., K.F., and U.S. conceived and conceptualized the work and strategy. Y.A. and P.F.M. analyzed the data; Y.A. wrote the first draft of the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
Declaration of interests
U.S. is a management board member and employee at BioNTech SE (Mainz, Germany); J.M.H., H.H., and A.B.V. are employees at BioNTech SE. J.M.H, H.H., A.B.V., and U.S. are inventors on patents and patent applications related to RNA technology and have securities from BioNTech SE. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.06.008.
Supplemental information
References
- 1.Manini I., Trombetta C.M., Lazzeri G., Pozzi T., Rossi S., Montomoli E. Egg-Independent Influenza Vaccines and Vaccine Candidates. Vaccines (Basel) 2017;5:18. doi: 10.3390/vaccines5030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rey-Jurado E., Tapia F., Muñoz-Durango N., Lay M.K., Carreño L.J., Riedel C.A., Bueno S.M., Genzel Y., Kalergis A.M. Assessing the Importance of Domestic Vaccine Manufacturing Centers: An Overview of Immunization Programs, Vaccine Manufacture, and Distribution. Front. Immunol. 2018;9:26. doi: 10.3389/fimmu.2018.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plotkin S., Robinson J.M., Cunningham G., Iqbal R., Larsen S. The complexity and cost of vaccine manufacturing - An overview. Vaccine. 2017;35:4064–4071. doi: 10.1016/j.vaccine.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dey A.K., Cupo A., Ozorowski G., Sharma V.K., Behrens A.J., Go E.P., Ketas T.J., Yasmeen A., Klasse P.J., Sayeed E. cGMP production and analysis of BG505 SOSIP.664, an extensively glycosylated, trimeric HIV-1 envelope glycoprotein vaccine candidate. Biotechnol. Bioeng. 2018;115:885–899. doi: 10.1002/bit.26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalams S.A., Parker S.D., Elizaga M., Metch B., Edupuganti S., Hural J., De Rosa S., Carter D.K., Rybczyk K., Frank I., NIAID HIV Vaccine Trials Network Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J. Infect. Dis. 2013;208:818–829. doi: 10.1093/infdis/jit236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim M., Badruddoza A.Z.M., Firdous J., Azad M., Mannan A., Al-Hilal T.A., Cho C.S., Islam M.A. Engineered Nanodelivery Systems to Improve DNA Vaccine Technologies. Pharmaceutics. 2020;12:30. doi: 10.3390/pharmaceutics12010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rezaei T., Khalili S., Baradaran B., Mosafer J., Rezaei S., Mokhtarzadeh A., de la Guardia M. Recent advances on HIV DNA vaccines development: Stepwise improvements to clinical trials. J. Control. Release. 2019;316:116–137. doi: 10.1016/j.jconrel.2019.10.045. [DOI] [PubMed] [Google Scholar]
- 8.Jackson N.A.C., Kester K.E., Casimiro D., Gurunathan S., DeRosa F. The promise of mRNA vaccines: a biotech and industrial perspective. NPJ Vaccines. 2020;5:11. doi: 10.1038/s41541-020-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kowalski P.S., Rudra A., Miao L., Anderson D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019;27:710–728. doi: 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geall A.J., Verma A., Otten G.R., Shaw C.A., Hekele A., Banerjee K., Cu Y., Beard C.W., Brito L.A., Krucker T. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundstrom K. Self-Replicating RNA Viruses for RNA Therapeutics. Molecules. 2018;23:3310. doi: 10.3390/molecules23123310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maruggi G., Zhang C., Li J., Ulmer J.B., Yu D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019;27:757–772. doi: 10.1016/j.ymthe.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulmer J.B., Sztein M.B. Promising cutting-edge technologies and tools to accelerate the discovery and development of new vaccines. Curr. Opin. Immunol. 2011;23:374–376. doi: 10.1016/j.coi.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flingai S., Czerwonko M., Goodman J., Kudchodkar S.B., Muthumani K., Weiner D.B. Synthetic DNA vaccines: improved vaccine potency by electroporation and co-delivered genetic adjuvants. Front. Immunol. 2013;4:354. doi: 10.3389/fimmu.2013.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villarreal D.O., Svoronos N., Wise M.C., Shedlock D.J., Morrow M.P., Conejo-Garcia J.R., Weiner D.B. Molecular adjuvant IL-33 enhances the potency of a DNA vaccine in a lethal challenge model. Vaccine. 2015;33:4313–4320. doi: 10.1016/j.vaccine.2015.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todorova B., Adam L., Culina S., Boisgard R., Martinon F., Cosma A., Ustav M., Kortulewski T., Le Grand R., Chapon C. Electroporation as a vaccine delivery system and a natural adjuvant to intradermal administration of plasmid DNA in macaques. Sci. Rep. 2017;7:4122. doi: 10.1038/s41598-017-04547-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Widera G., Austin M., Rabussay D., Goldbeck C., Barnett S.W., Chen M., Leung L., Otten G.R., Thudium K., Selby M.J., Ulmer J.B. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J. Immunol. 2000;164:4635–4640. doi: 10.4049/jimmunol.164.9.4635. [DOI] [PubMed] [Google Scholar]
- 19.Kutzler M.A., Weiner D.B. DNA vaccines: ready for prime time? Nat. Rev. Genet. 2008;9:776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deering R.P., Kommareddy S., Ulmer J.B., Brito L.A., Geall A.J. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin. Drug Deliv. 2014;11:885–899. doi: 10.1517/17425247.2014.901308. [DOI] [PubMed] [Google Scholar]
- 21.Pepini T., Pulichino A.M., Carsillo T., Carlson A.L., Sari-Sarraf F., Ramsauer K., Debasitis J.C., Maruggi G., Otten G.R., Geall A.J. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017;198:4012–4024. doi: 10.4049/jimmunol.1601877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aldon Y., Kratochvil S., Shattock R.J., McKay P.F. Chemokine-Adjuvanted Plasmid DNA Induces Homing of Antigen-Specific and Non-Antigen-Specific B and T Cells to the Intestinal and Genital Mucosae. J. Immunol. 2020;204:903–913. doi: 10.4049/jimmunol.1901184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wecker M., Gilbert P., Russell N., Hural J., Allen M., Pensiero M., Chulay J., Chiu Y.L., Abdool Karim S.S., Burke D.S., HVTN 040/059 Protocol Team. NIAID HIV Vaccine Trials Network Phase I safety and immunogenicity evaluations of an alphavirus replicon HIV-1 subtype C gag vaccine in healthy HIV-1-uninfected adults. Clin. Vaccine Immunol. 2012;19:1651–1660. doi: 10.1128/CVI.00258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pushko P., Parker M., Ludwig G.V., Davis N.L., Johnston R.E., Smith J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology. 1997;239:389–401. doi: 10.1006/viro.1997.8878. [DOI] [PubMed] [Google Scholar]
- 25.Frolov I., Frolova E., Schlesinger S. Sindbis virus replicons and Sindbis virus: assembly of chimeras and of particles deficient in virus RNA. J. Virol. 1997;71:2819–2829. doi: 10.1128/jvi.71.4.2819-2829.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brand D., Lemiale F., Turbica I., Buzelay L., Brunet S., Barin F. Comparative analysis of humoral immune responses to HIV type 1 envelope glycoproteins in mice immunized with a DNA vaccine, recombinant Semliki Forest virus RNA, or recombinant Semliki Forest virus particles. AIDS Res. Hum. Retroviruses. 1998;14:1369–1377. doi: 10.1089/aid.1998.14.1369. [DOI] [PubMed] [Google Scholar]
- 27.Ajbani S.P., Velhal S.M., Kadam R.B., Patel V.V., Lundstrom K., Bandivdekar A.H. Immunogenicity of virus-like Semliki Forest virus replicon particles expressing Indian HIV-1C gag, env and polRT genes. Immunol. Lett. 2017;190:221–232. doi: 10.1016/j.imlet.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 28.Berglund P., Quesada-Rolander M., Putkonen P., Biberfeld G., Thorstensson R., Liljeström P. Outcome of immunization of cynomolgus monkeys with recombinant Semliki Forest virus encoding human immunodeficiency virus type 1 envelope protein and challenge with a high dose of SHIV-4 virus. AIDS Res. Hum. Retroviruses. 1997;13:1487–1495. doi: 10.1089/aid.1997.13.1487. [DOI] [PubMed] [Google Scholar]
- 29.Knudsen M.L., Ljungberg K., Tatoud R., Weber J., Esteban M., Liljeström P. Alphavirus replicon DNA expressing HIV antigens is an excellent prime for boosting with recombinant modified vaccinia Ankara (MVA) or with HIV gp140 protein antigen. PLoS ONE. 2015;10:e0117042. doi: 10.1371/journal.pone.0117042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ljungberg K., Whitmore A.C., Fluet M.E., Moran T.P., Shabman R.S., Collier M.L., Kraus A.A., Thompson J.M., Montefiori D.C., Beard C., Johnston R.E. Increased immunogenicity of a DNA-launched Venezuelan equine encephalitis virus-based replicon DNA vaccine. J. Virol. 2007;81:13412–13423. doi: 10.1128/JVI.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White L.J., Sariol C.A., Mattocks M.D., Wahala M P B W., Yingsiwaphat V., Collier M.L., Whitley J., Mikkelsen R., Rodriguez I.V., Martinez M.I. An alphavirus vector-based tetravalent dengue vaccine induces a rapid and protective immune response in macaques that differs qualitatively from immunity induced by live virus infection. J. Virol. 2013;87:3409–3424. doi: 10.1128/JVI.02298-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morse M.A., Hobeika A.C., Osada T., Berglund P., Hubby B., Negri S., Niedzwiecki D., Devi G.R., Burnett B.K., Clay T.M. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Invest. 2010;120:3234–3241. doi: 10.1172/JCI42672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogers W.M., Oostermeijer H., Mooij P., Koopman G., Verschoor E.J., Davis D., Ulmer J.B., Brito L.A., Cu Y., Banerjee K. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015;211:947–955. doi: 10.1093/infdis/jiu522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melo M., Porter E., Zhang Y., Silva M., Li N., Dobosh B., Liguori A., Skog P., Landais E., Menis S. Immunogenicity of RNA Replicons Encoding HIV Env Immunogens Designed for Self-Assembly into Nanoparticles. Mol. Ther. 2019;27:2080–2090. doi: 10.1016/j.ymthe.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blakney A.K., McKay P.F., Yus B.I., Aldon Y., Shattock R.J. Inside out: optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019;26:363–372. doi: 10.1038/s41434-019-0095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brito L.A., Chan M., Shaw C.A., Hekele A., Carsillo T., Schaefer M., Archer J., Seubert A., Otten G.R., Beard C.W. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014;22:2118–2129. doi: 10.1038/mt.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhuang X., Qi Y., Wang M., Yu N., Nan F., Zhang H., Tian M., Li C., Lu H., Jin N. mRNA Vaccines Encoding the HA Protein of Influenza A H1N1 Virus Delivered by Cationic Lipid Nanoparticles Induce Protective Immune Responses in Mice. Vaccines (Basel) 2020;8:123. doi: 10.3390/vaccines8010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moyo N., Vogel A.B., Buus S., Erbar S., Wee E.G., Sahin U., Hanke T. Efficient Induction of T Cells against Conserved HIV-1 Regions by Mosaic Vaccines Delivered as Self-Amplifying mRNA. Mol. Ther. Methods Clin. Dev. 2018;12:32–46. doi: 10.1016/j.omtm.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel A.B., Lambert L., Kinnear E., Busse D., Erbar S., Reuter K.C., Wicke L., Perkovic M., Beissert T., Haas H. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018;26:446–455. doi: 10.1016/j.ymthe.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma S.K., de Val N., Bale S., Guenaga J., Tran K., Feng Y., Dubrovskaya V., Ward A.B., Wyatt R.T. Cleavage-independent HIV-1 Env trimers engineered as soluble native spike mimetics for vaccine design. Cell Rep. 2015;11:539–550. doi: 10.1016/j.celrep.2015.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovacs J.M., Noeldeke E., Ha H.J., Peng H., Rits-Volloch S., Harrison S.C., Chen B. Stable, uncleaved HIV-1 envelope glycoprotein gp140 forms a tightly folded trimer with a native-like structure. Proc. Natl. Acad. Sci. USA. 2014;111:18542–18547. doi: 10.1073/pnas.1422269112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torrents de la Peña A., Sanders R.W. Stabilizing HIV-1 envelope glycoprotein trimers to induce neutralizing antibodies. Retrovirology. 2018;15:63. doi: 10.1186/s12977-018-0445-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aldon Y., McKay P.F., Allen J., Ozorowski G., Felfödiné Lévai R., Tolazzi M., Rogers P., He L., de Val N., Fábián K. Rational Design of DNA-Expressed Stabilized Native-Like HIV-1 Envelope Trimers. Cell Rep. 2018;24:3324–3338.e5. doi: 10.1016/j.celrep.2018.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong L., He L., de Val N., Vora N., Morris C.D., Azadnia P., Sok D., Zhou B., Burton D.R., Ward A.B. Uncleaved prefusion-optimized gp140 trimers derived from analysis of HIV-1 envelope metastability. Nat. Commun. 2016;7:12040. doi: 10.1038/ncomms12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng S.L., Szabo S.J., Glimcher L.H. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl. Acad. Sci. USA. 2002;99:5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chakrabarti B.K., Feng Y., Sharma S.K., McKee K., Karlsson Hedestam G.B., Labranche C.C., Montefiori D.C., Mascola J.R., Wyatt R.T. Robust neutralizing antibodies elicited by HIV-1 JRFL envelope glycoprotein trimers in nonhuman primates. J. Virol. 2013;87:13239–13251. doi: 10.1128/JVI.01247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huysmans H., Zhong Z., De Temmerman J., Mui B.L., Tam Y.K., Mc Cafferty S., Gitsels A., Vanrompay D., Sanders N.N. Expression Kinetics and Innate Immune Response after Electroporation and LNP-Mediated Delivery of a Self-Amplifying mRNA in the Skin. Mol. Ther. Nucleic Acids. 2019;17:867–878. doi: 10.1016/j.omtn.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leyman B., Huysmans H., Mc Cafferty S., Combes F., Cox E., Devriendt B., Sanders N.N. Comparison of the Expression Kinetics and Immunostimulatory Activity of Replicating mRNA, Nonreplicating mRNA, and pDNA after Intradermal Electroporation in Pigs. Mol. Pharm. 2018;15:377–384. doi: 10.1021/acs.molpharmaceut.7b00722. [DOI] [PubMed] [Google Scholar]
- 49.Garmashova N., Gorchakov R., Volkova E., Paessler S., Frolova E., Frolov I. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 2007;81:2472–2484. doi: 10.1128/JVI.02073-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrakova O., Volkova E., Gorchakov R., Paessler S., Kinney R.M., Frolov I. Noncytopathic replication of Venezuelan equine encephalitis virus and eastern equine encephalitis virus replicons in Mammalian cells. J. Virol. 2005;79:7597–7608. doi: 10.1128/JVI.79.12.7597-7608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hekele A., Bertholet S., Archer J., Gibson D.G., Palladino G., Brito L.A., Otten G.R., Brazzoli M., Buccato S., Bonci A. Rapidly produced SAM(®) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013;2:e52. doi: 10.1038/emi.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hubby B., Talarico T., Maughan M., Reap E.A., Berglund P., Kamrud K.I., Copp L., Lewis W., Cecil C., Norberg P. Development and preclinical evaluation of an alphavirus replicon vaccine for influenza. Vaccine. 2007;25:8180–8189. doi: 10.1016/j.vaccine.2007.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buscail L., Bournet B., Vernejoul F., Cambois G., Lulka H., Hanoun N., Dufresne M., Meulle A., Vignolle-Vidoni A., Ligat L. First-in-man phase 1 clinical trial of gene therapy for advanced pancreatic cancer: safety, biodistribution, and preliminary clinical findings. Mol. Ther. 2015;23:779–789. doi: 10.1038/mt.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sidi A.A., Ohana P., Benjamin S., Shalev M., Ransom J.H., Lamm D., Hochberg A., Leibovitch I. Phase I/II marker lesion study of intravesical BC-819 DNA plasmid in H19 over expressing superficial bladder cancer refractory to bacillus Calmette-Guerin. J. Urol. 2008;180:2379–2383. doi: 10.1016/j.juro.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 55.Kuhn A.N., Diken M., Kreiter S., Selmi A., Kowalska J., Jemielity J., Darzynkiewicz E., Huber C., Türeci O., Sahin U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010;17:961–971. doi: 10.1038/gt.2010.52. [DOI] [PubMed] [Google Scholar]
- 56.Pokrovskaya I.D., Gurevich V.V. In vitro transcription: preparative RNA yields in analytical scale reactions. Anal. Biochem. 1994;220:420–423. doi: 10.1006/abio.1994.1360. [DOI] [PubMed] [Google Scholar]
- 57.Mann J.F., Tregoning J.S., Aldon Y., Shattock R.J., McKay P.F. CD71 targeting boosts immunogenicity of sublingually delivered influenza haemagglutinin antigen and protects against viral challenge in mice. J. Control. Release. 2016;232:75–82. doi: 10.1016/j.jconrel.2016.04.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.