

Abstract
Although both infections and vaccines induce memory B cell (MBC) populations that participate in secondary immune responses, the MBCs generated in each case can differ. Here, we compare SARS-CoV-2 spike receptor binding domain (S1-RBD)-specific primary MBCs that form in response to infection or a single mRNA vaccination. Both primary MBC populations have similar frequencies in the blood and respond to a second S1-RBD exposure by rapidly producing plasmablasts with an abundant immunoglobulin (Ig)A+ subset and secondary MBCs that are mostly IgG+ and cross-react with the B.1.351 variant. However, infection-induced primary MBCs have better antigen-binding capacity and generate more plasmablasts and secondary MBCs of the classical and atypical subsets than do vaccine-induced primary MBCs. Our results suggest that infection-induced primary MBCs have undergone more affinity maturation than vaccine-induced primary MBCs and produce more robust secondary responses.
Keywords: SARS-CoV-2, mRNA vaccine, memory B cell, plasmablast, atypical memory B cell
Graphical abstract
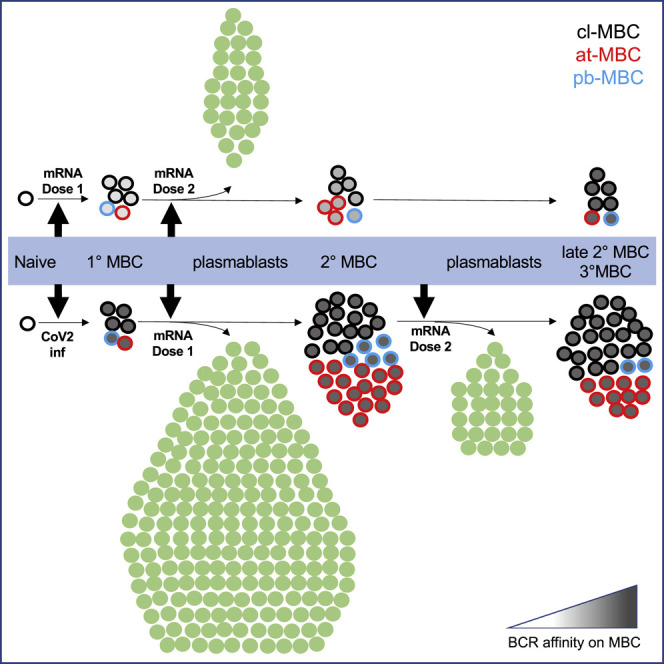
Memory B cells (MBCs) generated by different types of primary immune responses may have functional differences. Pape et al. find that SARS-CoV-2 spike-specific MBCs induced by infection had better antigen-binding capacity than did vaccine-induced primary MBCs and were more efficient at generating plasmablasts and secondary MBCs.
Introduction
mRNA vaccines encoding the SARS-CoV-2 spike protein potently induce antibodies, some capable of neutralizing the virus, and afford protective immunity from infection (Goel et al., 2021; Pascolo, 2021; Polack et al., 2020; Widge et al., 2021). The antigenic simplicity of these vaccines and their administration to millions of immunologically naive people provides an unprecedented opportunity to study the dynamics, activation, and differentiation of antigen-specific B cells in humans.
Studies in mice indicate that the B cell response to protein antigens in naive individuals is initiated when rare, naive B cells, expressing surface immunoglobulin (Ig), bind the antigen in secondary lymphoid organs, receive signals from helper T cells, and proliferate (McHeyzer-Williams and McHeyzer-Williams, 2005). This proliferation produces short-lived, Ig-secreting plasmablasts and non-plasmablasts that either become germinal center cells or germinal-center-independent memory cells, which mostly express IgM. The germinal center cells switch their Ig constant region from IgM to IgG, IgA, or IgE and acquire somatic mutations in the variable region, some of which improve antigen binding and allow the cells to survive the germinal center reaction as long-lived, switched Ig (swIg)+ memory cells or surface Ig‒ plasma cells that maintain serum Ig levels (Mesin et al., 2016; Nutt et al., 2015; Tarlinton, 2008). After subsequent exposure to antigen, the memory cells proliferate rapidly and generate plasmablasts, which boost the amount of antigen-specific Ig in the serum to aid in antigen clearance or, to a lesser extent, become germinal center cells to generate new memory cells with additional Ig mutations (Inoue et al., 2018; Pape et al., 2011; Suan et al., 2017).
In humans, the non-plasmablast population contains CD21+ classical memory B cells (cl-MBCs), and two poorly understood subsets—a CD21‒ CD27+ population, which contains a plasmablast-like subset and is sometimes referred to as the activated B cell population (Ellebedy et al., 2016; Louis et al., 2021) but will be referred to here as pb-MBCs, and a CD21‒ CD27‒ population, which is referred to as atypical memory B cells (at-MBCs) and contains a CD11c+ subset that shares features with an atypical B cell population in aged mice (Hao et al., 2011; Rubtsova et al., 2013; Wang et al., 2018). These at-MBCs, which express T-bet and may be recent products of germinal centers (Lau et al., 2017), are prominent in autoimmunity and chronic infection in humans (Isnardi et al., 2010; Jenks et al., 2018; Moir et al., 2008; Wei et al., 2007; Weiss et al., 2009) but can also be induced by vaccines (Andrews et al., 2019; Johnson et al., 2020; Kim et al., 2019; Sutton et al., 2021). The mechanisms that generate the different MBC subsets are unclear. at-MBCs are particularly enigmatic, as evidenced by conflicting studies on their lifespan, capacity for antibody production, and sensitivity to Toll-like receptor signaling (Muellenbeck et al., 2013; Obeng-Adjei et al., 2017; Pérez-Mazliah et al., 2018; Portugal et al., 2015; Rubtsov et al., 2011; Traore et al., 2009; Wong and Bhattacharya, 2019).
Although germinal center cells are mainly restricted to secondary lymphoid organs (Blink et al., 2005), plasmablasts and MBCs enter the blood and are, therefore, accessible for study in humans (Ellebedy et al., 2016). Here, we took advantage of SARS-CoV-2 mRNA vaccines to study the potential of human memory B cells generated by vaccination or infection. Our results indicate that primary MBCs formed in response to SARS-CoV-2 infection have a higher antigen-binding capacity per cell and are more efficient at producing plasmablasts and MBCs during secondary immune responses than are primary MBCs formed in response to mRNA vaccination.
Results
Tracking S1-RBD-specific B cells
We assessed B cells specific for the SARS-CoV-2 spike protein encoded by the Pfizer (Polack et al., 2020) and Moderna (Widge et al., 2021) mRNA vaccines using an antigen-based cell-enrichment flow-cytometry method. This approach was taken because it increases the sensitivity of detection of antigen-specific B cells by 100-fold over conventional methods (Pape et al., 2011; Schittek and Rajewsky, 1990; Taylor et al., 2012), thereby allowing assessment of naive B cells and MBCs at late times after antigen exposure. The N-terminal S1 receptor binding domain (S1-RBD) of the SARS-CoV-2 spike protein (Shang et al., 2020) with a single C-terminal BirA ligase sequence was expressed in insect cells, purified, biotinylated, and formed into a tetramer with Alexa Fluor 647-conjugated streptavidin (SA-AF647). This strategy was used to orient each S1-RBD molecule in the tetramer in the same configuration.
The capacity of the S1-RBD tetramer to detect S1-RBD-specific B cells was first tested in C57BL/6 mice. Spleen and lymph node cells from naive mice or mice that were immunized 14 days earlier with S1-RBD were mixed with S1-RBD/SA-AF647 tetramer and a decoy conjugate of SA-phycoerythrin (PE) plus AF647 and, then, magnetic beads conjugated with AF647 antibodies. The cell suspensions were passed over magnetized columns to enrich B cells that bound to S1-RBD, PE, SA, or AF647. The B cells were identified by flow cytometry based on expression of Ig heavy and light chains and the mouse B-cell-specific protein B220 (Figure S1A). The enriched B cell population from naive mice contained S1-RBD-specific cells that bound to S1-RBD/SA-AF647 but not to the SA-PE-AF647 decoy and a larger number of SA-, PE-, or AF647-specific cells that bound to SA-PE-AF647 but not to S1-RBD/SA-AF647 (Figure S1B). The S1-RBD-specific B cells expressed IgM and IgD but lacked swIg as expected for naive cells from the pre-immune repertoire (Cyster and Allen, 2019). In contrast, mice immunized with S1-RBD in complete Freund’s adjuvant contained more S1-RBD-specific B cells, and many were swIg+ (Figure S1C). The fact that a small population of naive-phenotype B cells was detected in unimmunized mice and a larger population of activated B cells in immunized mice is evidence that the B cells that bind the S1-RBD tetramer, but not the decoy, do so via S1-RBD-specific surface Ig receptors (BCRs).
Generation of S1-RBD-specific primary MBCs from their rare, naive precursors after mRNA vaccination
A similar strategy was used to identify S1-RBD-specific B cells in the blood of human volunteers. An example of the gating strategy is shown in Figure S2 for a subject who received an mRNA vaccine 14 days earlier. The S1-RBD/SA-AF647 tetramer and the decoy fluorochrome SA-PE-AF647 were mixed with peripheral blood mononuclear cells (PBMCs) before enrichment using magnetic beads conjugated with AF647 antibodies. The bound and unbound fractions from the magnetized columns were then stained with fluorochrome-labeled antibodies specific for markers of interest and gated for viable single cells with the light-scatter properties of lymphocytes that did not express CD3, CD14, or CD16 (Figures S2A–S2C). B cells were identified in this population based on expression of CD19 (Figure S2D), and S1-RBD-specific B cells were identified as cells that bound the S1-RBD but not the decoy tetramer (Figure S2E). The S1-RBD-specific B cells were further divided into CD19lo CD20– plasmablasts (Sanz et al., 2019) that also expressed CD27 and CD38, and CD19+ CD20+ non-plasmablasts that were CD38lo (Figure S2F). S1-RBD tetramer-binding cells were only present in the unbound fraction from samples that contained many plasmablasts (Figures 1 D, S2E, and S2F). These cells bound few S1-RBD tetramers, likely because of reduced expression of surface Ig (Ellebedy et al., 2016).
Figure 1.

Primary response after mRNA vaccination
(A) S1-RBD total Ig titers for SARS-CoV-2-naive (n = 18–22) and SARS-CoV-2-infected (n = 8–11) individuals at the indicated times after the first or second dose of SARS-CoV-2 spike mRNA vaccine. The mean titers are indicated over each group.
(B) Flow cytometry plots of the indicated cell types and molecules for samples from a SARS-CoV-2-naive subject before vaccination, pre-gated as described in Figure S2. Percentages of cells in each gate are indicated.
(C) Total log10 number of S1-RBD tetramer-binding B cells per million total B cells in the peripheral blood of SARS-CoV-2-naive individuals (n = 15–26) at the indicated times after the first vaccination. The values for each sample are shown as open circles, the horizontal bar in each box represents the median value, the values at the top and bottom error bar are the maximum and minimum values, and the means are indicated above each box. Values were compared with one-way ANOVA. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns, not significant.
(D) Flow cytometry plots of the indicated cell types and molecules for samples from a SARS-CoV-2-naive subject 13 days after the first vaccination. Percentages of cells in each gate are indicated.
(E and F) Total log10 number of S1-RBD tetramer-binding plasmablasts (E) or non-plasmablasts (F) per million total B cells in the peripheral blood of SARS-CoV-2-naive individuals (n = 15–26) at the indicated times after the first vaccination. Box and whisker plots are formatted as in (C). Values were compared with one-way ANOVA. ∗∗∗∗p < 0.0001, ns = not significant. Any cell per million value that was ≤1 was set to 0 on the log10 cell per million plots. Such values were at or below the limit of detection.
We first used this approach to detect S1-RBD-specific B cells in the blood of unvaccinated subjects ages 21–65 years who had not had a previous SARS-CoV-2 infection (SARS-CoV-2-naive), as evidenced by the lack of S1-RBD antibodies (Figure 1A). The magnetized, column-bound fraction, but not the unbound fraction, from these individuals contained a few S1-RBD-specific B cells (Figure 1B), which were present at an average frequency of 10 per million total B cells (Figure 1C). The S1-RBD tetramer-binding population contained 92% ± 10% (mean ± SD, n = 11) non-plasmablasts, 80% ± 15% expressing IgM and IgD, and most expressing CD21 but not the classical MBC marker CD27 (Figure 1B). Thus, S1-RBD tetramer-binding cells were rare in pre-vaccination SARS-CoV-2-naive subjects, and most had the phenotype of naive follicular B cells.
We then measured the response of the S1-RBD-specific naive B cell population after SARS-CoV-2-naive subjects received their first dose of the Pfizer or Moderna mRNA vaccines. The S1-RBD tetramer-binding cells retained the naive phenotype and remained around 10 per million on post-vaccination days 6–10 but increased to 385 per million on days 12–16 (Figure 1C). Plasmablasts accounted for much of the expanded population (Figures 1D and 1E) and, on average, were 17% ± 16% (mean ± SD, n = 23) IgM+, 31% ± 21% IgA+, and 48% ± 20% IgG+. The increase in plasmablasts preceded an increase in S1-RBD antibodies on day 21–28 (Figure 1A). The plasmablasts declined significantly to 5 cells per million by days 21–28, as expected for this short-lived cell type (Sze et al., 2000) (Figure 1E). S1-RBD tetramer-binding non-plasmablasts increased to a frequency of about 50 per million on days 12–16 and to 62 per million at the time of the second vaccination on days 21–28 (Figures 1D and 1F). For the purposes of this study, the memory phase is considered to begin on day 21 because that is when MBCs achieve the same frequency in the blood as that in the secondary lymphoid organs in which they are generated (Blink et al., 2005). Non-plasmablasts in SARS-CoV-2-naive subjects on days 21–28 after their first vaccination will, therefore, be referred to as vaccine-induced primary MBCs. Many of these cells expressed CD27, a common marker of human MBCs (Sanz et al., 2019), and tended to bind a bit more of the decoy reagent than did naive S1-RBD-specific B cells (Figure 2 A).
Figure 2.

Secondary response by mRNA-vaccine- and SARS-CoV-2-induced primary MBCs
(A) Flow cytometry plots of the indicated cell types and molecules, pre-gated as described in Figure S2, for samples from SARS-CoV-2-naive subjects on day 28 after the first vaccination or on days 7, 14, or 33 after the second vaccination. Percentages of cells in each gate are indicated. CD19hi CD11c+ at-MBCs are shown in the gate in the fourth plot in each row.
(B–F) Total log10 number of S1-RBD tetramer-binding non-plasmablasts (nPB) (B), plasmablasts (C), CD21+ cl-MBCs (D), CD21‒ CD27+ pb-MBCs, (E), or CD21‒ CD27‒ CD11c+ at-MBCs (F) per million total B cells in the peripheral blood of SARS-CoV-2-naive subjects (n = 8–29) at indicated times after the first (primary) or second (secondary) vaccination (black circles) or SARS-CoV-2-infected individuals at the time of (primary) or the indicated times after (secondary) the first vaccination (red circles). CD21‒ CD27‒ CD11c‒ MBCs were not included in the analysis because the nature of these cells is unknown. Box and whisker plots are formatted as in Figure 1C. Values were compared with one-way ANOVA. ∗p < 0.05, ∗∗p < 0. 01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns = not significant. Any cell per million value that was ≤1 was set to 0 on the log10 cell per million plots. Such values were at or below the limit of detection.
(G) Flow cytometry plots of the indicated cell types and molecules as in (A) for samples from SARS-CoV-2-infected subjects before vaccination or on days 7, 14, or 28 after the first vaccination.
In summary, rare S1-RBD-specific, naive B cells proliferated after mRNA vaccination to form a large population of transient plasmablasts and a smaller population of primary MBCs, which persisted in the blood until the time of the booster vaccination 3–4 weeks later.
Booster vaccination induces a rapid secondary response
We then assessed the secondary immune response to the booster dose of the mRNA vaccine in SARS-CoV-2-naive subjects. In contrast to the primary response, which peaked on days 12–16 and generated 271 S1-RBD-specific plasmablasts per million (Figure 1E), the secondary S1-RBD-specific plasmablast response peaked at 828 cells per million on days 5–10 after booster vaccination, before declining by days 11–18 (Figures 2C and 2D). As expected, the secondary plasmablast response was accompanied by a boost in S1-RBD-specific antibodies (Figure 1A). Non-plasmablasts peaked at 266 cells per million on days 5–10, then fell to about 100 per million on days 11–18 and remained near that number on days 21–38 (Figure 2B). The non-plasmablasts present on day 21 and thereafter were defined as secondary MBCs. In summary, the booster vaccination generated a larger and more rapid plasmablast response than the primary, leaving subjects with 10-fold more S1-RBD-specific antibodies and 1.6 times more secondary MBCs than they had at the time of the boost.
Both primary and secondary vaccine-induced MBC populations contain cl-MBC, pb-MBC, and at-MBC subsets
The composition of the MBC populations changed during course of the booster vaccination. Before the second vaccination, the S1-RBD-specific B cell population consisted of 62 non-plasmablasts (Figures 2A and 2B) and 5 plasmablasts (Figures 2A and 2C) per million, and the non-plasmablast population consisted of 34 CD21+ cl-MBC, 15 CD21‒ CD27+ pb-MBC, and 3 CD21‒ CD27‒ CD11c+ at-MBC per million (Figures 2A and 2D–2F). On days 11–18 after the boost, there were 50 cl-MBC, 21 pb-MBC, and 25 at-MBC per million (Figures 2A and 2D–2F). The 8.3-fold increase in at-MBCs over the numbers of this subset in the primary response was statistically significant (Figure 2F). The cl-MBCs and pb-MBCs did not change on days 21–38 (Figures 2D and 2E) from the numbers on day 11–18. Although the at-MBCs appeared to decline from 25 cells per million on days 11–18 to 13 cells per million on days 21–38, this drop was not statistically significant (Figure 2F). Thus, a month after the booster vaccine, SARS-CoV-2-naive subjects had about the same number of secondary cl-MBCs and pb-MBCs as they had after the primary response and a 4-fold larger population of at-MBCs.
Primary MBC populations from SARS-CoV-2-infected subjects and single-vaccinated SARS-CoV-2-naive subjects are similar in size and composition
We then assessed the primary MBCs induced by SARS-CoV-2 infection. People aged 20–76 years who had SARS-CoV-2 infections 2–9 months earlier were recruited to the study, and their prior infection was confirmed by assessment of S1-RBD antibodies. All of these individuals had antibodies, although the titers were slightly lower than those observed in SARS-CoV-2-naive subjects after the first vaccination (Figure 1A). The S1-RBD-specific B population in SARS-CoV-2-infected subjects consisted mostly of non-plasmablasts (Figure 2G), which averaged 118 cells per million (Figure 2B) and was not significantly different than the 62 vaccine-induced primary MBCs per million observed from SARS-CoV-2-naive subjects. The memory subsets in these SARS-CoV-2-induced primary MBC populations averaged 77 cl-MBCs, 16 pb-MBCs, and 2 at-MBCs per million (Figures 2D–2F), again similar to the vaccine-induced primary MBCs.
The secondary response in SARS-CoV-2-infected individuals is more robust
We then analyzed the secondary responses of people with prior SARS-CoV-2 infection. SARS-CoV-2-infected subjects generated a rapid secondary response on days 5–10 after their first vaccination, similar to the response of the SARS-CoV-2-naive subjects after their second vaccination. However, S1-RBD-specific secondary plasmablasts peaked at 5,117 cells per million on days 5–10 (Figures 2C and 2G), 6-fold higher than that observed in SARS-CoV-2-naive subjects, before declining to 9 cells per million on days 11–18 (Figures 2C and 2G). The burst of plasmablasts coincided with a 30-fold increase in S1-RBD-specific antibodies (Figure 1A), which was also greater than the 10-fold increase in SARS-CoV-2-naive subjects after their second vaccination.
Non-plasmablasts increased from 118 cells per million at the time of vaccination to 512 cells per million on days 5–10, which was not significantly different from the 266 cells per million generated during the secondary response in SARS-CoV-2-naive subjects (Figure 2B). However, the secondary non-plasmablasts in SARS-CoV-2-infected subjects remained stable at 625 and 614 cells per million, respectively, on days 11–18 and 21–38 (Figures 2B and 2G), in contrast to a 2–3-fold decline in SARS-CoV-2-naive subjects. Thus, the secondary response to mRNA vaccination in SARS-CoV-2-infected subjects generated 6-fold more secondary MBCs than the secondary response did in SARS-CoV-2-naive subjects.
We also examined the composition of the secondary MBCs in SARS-CoV-2-infected subjects to determine which subsets were elevated over those in SARS-CoV-2-naive subjects. On days 21–38 after vaccination, the secondary MBCs in SARS-CoV-2-infected subjects contained 230 cl-MBCs, 70 pb-MBCs, and 164 at-MBCs per million, compared with 47 cl-MBCs, 23 pb-MBCs, and 13 at-MBCs per million in SARS-CoV-2-naive subjects after their second dose of the vaccine (Figures 2D–2F). Thus, the secondary response to single mRNA vaccination in SARS-CoV-2-infected subjects generated 5-, 3-, and 12-fold more cl-MBCs, pb-MBCs, and at-MBCs, respectively, than the secondary response did in SARS-CoV-2-naive subjects. Given that unvaccinated SARS-CoV-2-infected and singly vaccinated SARS-CoV-2-naive subjects had similar numbers of primary MBCs (Figures 2D–2F), these results suggest that SARS-CoV-2-induced primary MBCs are more efficient than vaccine-induced primary MBCs at generating plasmablasts and secondary MBCs.
The IgA isotype is over-represented in the plasmablast population compared with the MBC population
The Ig isotype determines the effector functions of antibodies, with IgA having a critical role at mucosal surfaces (Seifert and Küppers, 2016). We, therefore, examined isotype switching in vaccine- and SARS-CoV-2-induced primary MBCs. At the time of the second dose, 29%, 4%, and 65% of the cells in the vaccine-induced primary MBC population expressed IgM, IgA, or IgG, respectively (Figures 3 A and 3B), whereas 4%, 29%, or 64% of the plasmablasts generated on days 5–10 after the second vaccination expressed those isotypes (Figures 3B and 3C). Thus, the secondary plasmablast population had a much higher fraction of IgA+ cells and much lower fraction of IgM+ cells than the vaccine-induced primary MBC population. The secondary MBC population that followed on days 21–38 after the plasmablasts disappeared contained 22%, 2%, and 74% IgM-, IgA-, and IgG-expressing cells, respectively, which was similar to the primary MBC population (Figures 3B and 3D). SARS-CoV-2-induced primary MBCs and their secondary plasmablasts and MBCs showed a similar pattern (Figure 3E). Thus, although the isotype profile of secondary MBCs reflected that of primary MBC precursors, MBC populations with very few IgA+ cells generated a disproportionately large fraction of IgA+ plasmablasts.
Figure 3.

Isotype expression by S1-RBD-specific MBCs and plasmablasts
(A, C, and D) Flow cytometry plots of the indicated cell types and molecules, pre-gated as described in Figure S2, for samples from SARS-CoV-2-naive subjects on day 28 after the first vaccination (A) or on days 7 (C) or 33 (D) after the second vaccination. Percentages of cells in each gate are indicated. IgG+ plasmablasts (PB) were identified as swIg+ IgA‒ IgM‒ cells because plasmablasts express less surface IgG than do other isotypes.
(B and E) Percentages of IgM+, IgA+, or IgG+ S1-RBD-specific vaccine- (B) (n = 13–30 subjects) or SARS-CoV-2-induced primary MBCs (E) (n = 11–14 subjects), and their PB or MBC progeny. Box and whisker plots are formatted as in Figure 1C.
mRNA vaccination induces progressive affinity maturation
Ig affinity is thought to be critical for antigen neutralization (Kepler et al., 2014; Scheid et al., 2009). We, therefore, measured the relative affinities of the S1-RBD-specific B cell populations. The mean fluorescence intensity of S1-RBD tetramer bound per cell was divided by that of CD79b, a component of the BCR complex that is proportional to the amount of Ig on the cell surface, to normalize antigen binding to the levels of surface BCR. As illustrated by examples in Figures 4 A and 4B, this assay revealed that IgM+ cells early in the primary response (Figure 4A) bind less S1-RBD than do IgG+ MBCs (Figure 4B). Our previous studies of antigen-specific B cells in mice showed that this difference is due to increased BCR affinity for antigen in the latter population and correlates with numerous somatic mutations (Pape et al., 2018).
Figure 4.

Affinity maturation by S1-RBD-specific MBCs
(A and B) Flow cytometry plots of the indicated cell types and molecules, pre-gated as described in Figure S2, for samples from a SARS-CoV-2-naive subject on day 6 after their first vaccination (A) or a SARS-CoV-2-infected subject before vaccination (B). The contour plot shows an overlay of S1 RBD-specific IgM+ IgD+ cells (red) from the sample shown in (A), and IgG+ cells (blue) from the sample show in (B).
(C) S1-RBD tetramer mean fluorescence intensity (MFI)/CD79b ratios for IgM+ cells from SARS-CoV-2-naive individuals before vaccination (n = 8, gray) or IgG+ cells from SARS-CoV-2-naive (black) (n = 7–18) or SARS-CoV-2-infected (red) individuals after the primary (1°) (n = 10), secondary (2°) (n = 22 samples from 11 individuals) or tertiary (3°) (n = 20 samples from 10 individuals) exposure to SARS-CoV-2 S1-RBD via the infection or the vaccine. Box and whisker plots are formatted as in Figure 1C. For a sample to be used in this analysis, it had to contain at least 10 S1-RBD-specific cells. For the naive IgM time point, there was one outlier that had an MFI/CD79b ratio of eight, which was excluded from the analysis because the subsequent day 11–14 and 21–28 IgG time points from this individual had an MFI/CD79b ratio of less than four. Values were compared with one-way ANOVA. ∗p < 0.05, ∗∗p < 0. 01, ns, not significant.
The IgM+ IgD+ S1-RBD tetramer-binding naive cells from pre-vaccination SARS-CoV-2-naive subjects, which presumably expressed un-mutated germline-encoded Igs (Cyster and Allen, 2019), had a mean S1-RBD tetramer/CD79b ratio of 2.1 (Figure 4C). This ratio increased to 3.1 for the S1-RBD tetramer-binding swIg+ non-plasmablast population present on days 11–18 and to 3.8 for the primary MBC population on days 21–28 at the time of the second vaccination (Figure 4C). The secondary MBCs present on days 21–38 after the second vaccination had a ratio of 4.7. Thus, there was a progressive increase in antigen-binding capacity with time after vaccination from the precursor IgM+ and IgD+ naive cells to switched primary and then secondary MBCs, consistent with progressive affinity maturation. Furthermore, the detection of precursor IgM+ and IgD+ naive S1-RBD-specific B cells, which likely do not have somatic mutations/affinity maturation, validates the sensitivity of the antigen-enrichment method used in this study.
The SARS-CoV-2-induced primary MBC population had an S1-RBD tetramer/CD79b ratio of 7.1 before vaccination, and it did not increase in the secondary MBC population induced by the first vaccination (Figure 4C).
Most subjects with previous SARS-CoV-2 infection make poor tertiary responses
We also assessed the tertiary response of SARS-CoV-2-infected subjects to their second vaccination. At the time of the second vaccine dose, these subjects had an average of 614 secondary MBC per million (Figure 5 A), compared with the 144 secondary MBCs that SARS-CoV-2-naive subjects had on days 56–78 after their booster shot. Surprisingly, 8 of 12 subjects generated very few plasmablasts on days 5–7 after the second vaccination, for an average of 226 plasmablasts per million (Figure 5B), which was about 20-fold fewer plasmablasts than these subjects generated during the secondary response. The weak plasmablast response corresponded with the lack of an increase in S1-RBD-specific serum antibodies 2 weeks after the second vaccination (Figure 1A) as observed in other studies (Goel et al., 2021; Stamatatos et al., 2021). Furthermore, on average, there was no increase in the number of non-plasmablasts on days 5–7 after the secondary vaccination, and the number of tertiary MBCs remained constant until days 58–96, when there were 663 cells per million (Figure 5A). The tertiary MBC subsets did not significantly change in number between days 12–15 and 58–96 (Figures 5C–5E), although the at-MBCs trended down during that interval. Finally, these tertiary MBCs had similar S1-RBD tetramer/CD79b ratios to the secondary MBCs (Figure 4C) before the booster vaccine, suggesting that no further affinity maturation occurred during the tertiary response.
Figure 5.

S1-RBD-specific MBCs in SARS-CoV-2-infected individuals make a poor tertiary response
(A–E) Total log10 number of S1-RBD tetramer-binding total MBCs (A), plasmablasts (B), CD21+ cl-MBCs (C), CD21‒ CD27+ pb-MBCs (D), or CD21‒ CD21‒ CD11c+ at-MBCs (E) per million total B cells in the peripheral blood of SARS-CoV-2-naive (black circles) or SARS-CoV-2-infected (red circles) subjects (n = 5–12) at the indicated times after the second vaccination. Box and whisker plots are formatted as in Figure 1C. Values were compared with one-way ANOVA. ∗p < 0.05, ∗∗∗∗p < 0.0001, ns, not significant.
MBCs in fully vaccinated SARS-CoV-2-infected subjects outnumbered those in SARS-CoV-2-naive subjects but had similar capacity to bind antigens and recognize the B.1.351 variant
Despite the poor tertiary response, the SARS-CoV-2-infected subjects ended up with 4.6 times greater MBCs per million than SARS-CoV-2-naive subjects had on days 56–78 after their second vaccinations (Figure 5A), and this included 5.3 times more cl-MBCs and 7.4 times more at-MBCs (Figures 5C and 5E). At this time, the S1-RBD-specific MBCs present in both the SARS-CoV-2-infected and SARS-CoV-2-naive individuals had similar S1-RBD tetramer/CD79b ratios (Figure 4C). We also determined whether the MBCs present in fully vaccinated SARS-CoV-2-infected and SARS-CoV-2-naive individuals bound to the S1-RBD from the B.1.351 variant (Khateeb et al., 2021), which contains three amino acid substitutions compared with the wild-type S1-RBD in the vaccine. PBMCs were stained simultaneously with the wild-type S1-RBD tetramer labeled with one fluorochrome and the B.1.351 S1-RBD tetramer labeled with another (Figure 6 A). An average of 74% of the secondary MBCs from SARS-CoV-2-naive subjects and 62% from SARS-CoV-2-infected subjects that bound to the wild-type S1-RBD also bound the B.1.351 S1-RBD (Figure 6B). In summary, even though both MBC populations present several months after the second vaccination had a similar degree of affinity maturation and cross-reactivity on the B.1.351 variant, there were more cl-MBC and at-MBCs in individuals that had previous SARS-CoV-2 infection.
Figure 6.

S1-RBD-specific MBCs in fully vaccinated SARS-CoV-2-infected and SARS-CoV-2-naive individuals recognize the B.1.351 variant to a similar degree
(A) Flow cytometry plots, pre-gated as described in Figure S2, for a sample from a SARS-CoV-2-naive subject on day 79 after their second vaccination, stained with both AF647-labeled wild-type (WT) S1-RBD and BV650-labeled B.1.351 S1-RBD tetramers.
(B) Percentages of WT S1-RBD-specific MBCs that also bound to the B.1.351 variant in SARS-CoV-2-naive (n = 10) or SARS-CoV-2-infected (n = 4) individuals 76–189 days after their second vaccination.
Discussion
The goal of this study was to characterize the secondary immune responses of MBC populations generated by an mRNA vaccine or SARS-CoV-2 infection. Like most studies of adaptive immune responses in humans, our analysis was limited to the peripheral blood even though the S1-RBD-specific MBCs of interest were likely generated in secondary lymphoid organs and produced plasmablasts and secondary MBCs in that location during secondary responses. Other studies, however, have shown that primary MBCs have the same frequency in secondary lymphoid organs and blood at 3 weeks after immunization (Blink et al., 2005) and that plasmablasts generated from MBCs in secondary lymphoid organs rapidly enter the blood (Ellebedy et al., 2016). Thus, our results for MBCs and their plasmablast progeny in blood are likely reflective of what occurred in the lymph nodes.
Our data indicate that SARS-CoV-2-naive people have a few phenotypically naive B cells with BCRs that have a relatively low capacity for S1-RBD binding. These features suggest that these B cells had not responded or undergone affinity maturation in response to common coronaviruses—a conclusion that has also been reached from antibody studies (Sette and Crotty, 2021). The primary MBCs that formed from these naive precursors 3–4 weeks after the first mRNA vaccination in SARS-CoV-2-naive people showed evidence of minimal affinity maturation, perhaps because not enough time had passed for the process to occur. Thus, increasing the time between the first and second injections of mRNA vaccines may be beneficial because MBCs with higher affinity BCRs may accumulate during the expanded interval. We found evidence that the secondary MBCs underwent marked affinity maturation by 3–4 and especially 8–11 weeks after the second vaccination. This result indicates that mRNA vaccines are potent drivers of secondary germinal center B cell formation in SARS-CoV-2-naive people and that this process can go on for weeks as suggested by the finding that spike-specific antibodies continue to accrue somatic mutations for 6 months after the second vaccination (Sokal et al., 2021; Wang et al., 2021). In contrast to the vaccine-induced primary MBCs, we found that SARS-CoV-2-induced primary MBCs had undergone maximal affinity maturation even before vaccination, either because the inducing infection occurred many months in the past, thereby allowing time for maximal accrual of somatic mutations, or because SARS-CoV-2 infection is a stronger stimulus for affinity maturation than a single mRNA vaccination.
The S1-RBD-specific primary MBC populations induced by mRNA vaccination or SARS-CoV-2 infection had several similarities despite their different origins, ages, and antigen-binding capacities. They both contained similar numbers of cl-MBCs, pb-MBCs, and at-MBCs. Both populations generated CD21‒ CD27+ secondary pb-MBCs that survived stably for several months. Thus, S1-RBD-specific pb-MBCs may be more than short-lived, activated precursors fated to enter the cl-MBC pool (Ellebedy et al., 2016), although it remains to be seen if they will eventually decline. Both populations, which had very few at-MBCs, generated this subset efficiently early in the secondary response. This finding adds to the growing literature showing that at-MBCs are generated after vaccination and, thus, may be unappreciated components of normal antigen-specific B cell responses in humans (Andrews et al., 2019; Johnson et al., 2020; Kim et al., 2019; Sutton et al., 2021; Weiss et al., 2009).
Another similarity involved isotype switching. Although both the primary MBC populations induced by mRNA vaccination or SARS-CoV-2 infection contained as many as 25% IgM+ cells, they rapidly generated almost exclusively swIg+ plasmablasts during the secondary response. This finding is consistent with earlier work showing that swIg+ memory B cells are predisposed to produce plasmablasts much faster than naive B cells (Pape et al., 2011; Wrammert et al., 2008) and that IgM+ memory B cells are inefficient contributors to secondary antibody responses (Pape et al., 2011). In addition, although both MBC populations contained less than 5% IgA+ cells, 30% of their plasmablast progeny expressed that isotype, consistent with a previous study of SARS-CoV-2-induced primary MBCs (Dan et al., 2021). Assuming that IgA+ memory cells do not proliferate more than other memory cells and IgM+ memory cells do not contribute much to the secondary response, the most likely explanation for the abundance of IgA+ plasmablasts is rapid switching of IgG+ MBCs to IgA. Notably, secondary MBCs generated from the primary MBC populations contained less than 5% IgA+ cells, similar to their primary MBC precursors. The mechanism that prevents the efficient formation of IgA+ MBCs is not known.
The primary MBC populations induced by mRNA vaccination or SARS-CoV-2 infection, however, had several different behaviors during secondary responses to mRNA vaccination. The SARS-CoV-2-induced primary MBCs produced 6-fold more plasmablasts than did the vaccine-induced MBCs, perhaps because their more affinity-matured BCRs transduced stronger signals. Strong BCR signaling has been shown in other contexts to favor the plasmablast fate (Pape et al., 2018; Paus et al., 2006). Receipt of stronger BCR signals could also account for the finding that the affinity matured SARS-CoV-2-induced primary MBC population produced secondary MBCs more efficiently than vaccine-induced primary MBCs because other work indicates that B cells that receive strong BCR signals proliferate more and produce more apoptosis-resistant progeny than do B cells that proliferate less (Taylor et al., 2015). Strong BCR signaling may be particularly conducive to the formation of the at-MBCs because these cells accumulate in situations of chronic antigenic stimulation (Isnardi et al., 2010, Jenks et al., 2018, Moir et al., 2008, Wei et al., 2007, Weiss et al., 2009). Curiously, Andrews et al. (2019) observed that naive B cells specific for the variable head region of influenza hemagglutinin formed at-MBCs more efficiently than MBCs specific for the conserved stem region in the same individuals. We wonder whether stem-specific antibodies from prior infections limit the available antigen for stem-specific MBCs, thereby reducing their BCR signaling and capacity to form at-MBCs.
Remarkably, the S1-RBD-specific secondary MBC population in SARS-CoV-2-infected people produced few plasmablasts or tertiary MBCs after booster vaccination as described by Goel et al. (2021), perhaps because of the exceedingly large amount of antigen-clearing antibody present at the time of the boost (Pape et al., 2011). Thus, although the first vaccination greatly increased the number of MBCs in SARS-CoV-2-infected individuals, as seen in other studies (Goel et al., 2021; Mazzoni et al., 2021), we detected very little evidence that the booster vaccination provided an additional increase. Although none of the tertiary MBC subsets declined significantly over a 7–11-week period after the second vaccination, there was a trend toward a loss of at-MBCs. This result fits with studies suggesting that at-MBCs are more short-lived than other types of MBCs are (Pérez-Mazliah et al., 2018).
Finally, our results shed light on the immunological status of SARS-CoV-2-naive and SARS-CoV-2-infected people out to 3 months after completion of the two-dose vaccination series. Although both groups end up with affinity-matured MBCs, many capable of recognizing a variant of concern, SARS-CoV-2-infected people have five times more MBCs than SARS-CoV-2-naive subjects have. The significance of this difference in terms of immunity to subsequent SARS-CoV-2 infection should remain an area of active investigation, especially because the antibodies in SARS-CoV-2-infected individuals who are then vaccinated are very good at neutralizing both the original and the variant SARS-CoV-2 viruses (Reynolds et al., 2021; Stamatatos et al., 2021; Wang et al., 2021).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD90.2 (53-2.1) | Ebioscience | Cat#47-0902-82; RRID: AB_1272187 |
| Mouse: anti-F480 (BM8) | Ebioscience | Cat#47-4801-80; RRID: AB_2637188 |
| Mouse: anti-Gr1(RB6-8C5) | Ebioscience | Cat#47-5931-80; RRID: AB_1518805 |
| Mouse: anti-B220 (RA3-6B2) | BD | Cat#563793; RRID: AB_2738427 |
| Mouse: anti-IgM (11/41) | Ebioscience | Cat# 25-5790-82; RRID: AB_469655 |
| Mouse: anti-IgD (11-26c.2a) | BD | Cat#563618; RRID: AB_2738322 |
| Human: anti-CD3 (OKT3) | Ebioscience | Cat#47-0037-42; RRID: AB_2573936 |
| Human: anti-CD14 (61D3) | Ebioscience | Cat#47-0149-42; RRID: AB_1834358 |
| Human: anti-CD16 (CB16) | Ebioscience | Cat#47-0168-42; RRID: AB_11220086 |
| Human: anti-CD19 (HIB19) | Biolegend | Cat#302242; RRID: AB_2561381 |
| Human: anti-CD20 (2H7) | BD | Cat#563782; RRID: AB_2744325 |
| Human: anti-CD21 (B-ly4) | BD | Cat#562966; RRID: AB_2737921 |
| Human: anti-CD27 (O323) | Biolegend | Cat#302834; RRID: AB_1121920 |
| Human: anti-CD38 (HB-7), | Biolegend | Cat#356620; RRID: AB_2566232 |
| Human: anti-CD11c (B-ly6) | BD | Cat#562393; RRID: AB_11153662 |
| Human: anti-CD79b (CB3-1) | Biolegend | Cat#341404; RRID: AB_1595454 |
| Human: anti-IgD (IA6-2) | Biolegend | Cat#348210; RRID: AB_10683460 |
| Human: anti-IgM (MHM-88) | Biolegend | Cat#314524; RRID: AB_2562373 |
| Human: anti-IgA | Southern Biotech | Cat#2050-02; RRID: AB_2795702 |
| Human: anti-IgG (G18-145) | BD | Cat#564230; RRID: AB_2738684 |
| Human: Fc block | BD | Cat#564219; RRID: AB_2728082 |
| Mouse: Fc block CD16/CD32 | BD | Cat# 553141; RRID: AB_394656 |
| Human: HRP-labeled anti-Ig heavy and light chain | Invitrogen | Cat#31412; RRID: AB_228265 |
| Biological samples | ||
| Human blood samples | University of Minnesota | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Ghost Red 710 viability dye | TonBo | Cat#13-0871-T100 |
| Collagenase D | Roche/Boehringer | Cat# 1088874 |
| S1-RBD/SA-AF647 tetramer | This paper | N/A |
| decoy SA-PE-AF647 | This paper | N/A |
| AccuCheck Counting Beads | ThermoFisher | Cat#PCB100 |
| KPL ABTS® Peroxidase Substrate (1-Component) | Sera Care | Cat#5120-0043 |
| anti-Cy5/AF647 MicroBeads | Miltenyi | Cat#130-091-395 |
| BD Cytofix/Cytoperm | BD Biosciences | Cat#51-2090KZ |
| Flow Cytometry Perm Buffer | TONBO | Cat#TNB-1213-L150 |
| Complete Freund’s Adjuvant (CFA) | Sigma | Cat#F5881 |
| 10K Amicon Ultra Centrifugal Filters | Millipore | Cat#UFC801008 |
| 100K Amicon Ultra Centrifugal Filters | Millipore | Cat#UFC510008 |
| SA-PE Phycolink | Prozyme | Cat#PJRS27 |
| SA-AF647 | ThermoFisher | Cat#S21374 |
| Critical commercial assays | ||
| Effectene kit | QIAGEN | Cat#301425 |
| Bir A protein ligase kit | Avidity | Cat#BirA500 |
| Experimental models: Cell lines | ||
| HEK293T cells | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6NCr (B6) | Charles River/NCI | Cat#556 |
| Oligonucleotides | ||
| T7-Promoter (5′-TAATACGACTCACTATAGGG-3′) | This paper | N/A |
| BGH-reverse (5′-TAGAAGGCACAGTCGAGG-3′) | This paper | N/A |
| Recombinant DNA | ||
| gene block encoding S1-RBD | This paper | N/A |
| pcDNA3.1/Zeo (+) Mammalian Expression Vector | ThermoFisher | Cat #: V86020 |
| gene block encoding the BirA sequence | This paper | N/A |
| Software and algorithms | ||
| FlowJo software | Tree Star | https://www.flowjo.com |
| Prism software | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| CPT Mononuclear Cell Preparation Tubes-sodium citrate | BD | Cat#362761 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marc K. Jenkins (jenki002@umn.edu).
Materials availability
Constructs used in this study such as pcDNA3.1-Zeo-S1-RBD-BirA construct and all unique materials are available from the lead contact under a materials transfer agreement (MTA).
Experimental model and subject details
Human participants
Human participants (Table S1) that were CoV2-naive or had previous CoV2 infection were followed at the time of and up to 3 months after vaccination with Pfizer or Moderna mRNA vaccines. Severity of COVID-19 illness was rated using a 1-5 Likert scale that incorporated systemic, respiratory, and gastrointestinal symptoms (1 = minimal, i.e., carry on normal activities, mild discomfort, less than 1 week; 2 = mild, i.e., slightly decreased activities, mild discomfort, less than 2 weeks; 3 = moderate, i.e., significantly decreased activities, moderate discomfort; 4 = severe, i.e., significantly decreased activities, moderate to high discomfort, longer duration, no hospitalization; 5 = hospitalization).
Severity of side effects following immunization was rated using a 0-4 Likert scale (0 = no side effects except for arm pain or discomfort; 1 = minimal, i.e., carry on normal activities, mild discomfort; 2 = mild, i.e., slightly decreased activities, mild discomfort; 3 = moderate, i.e., significantly decreased activities, moderate discomfort; 4 = severe, i.e., ER visit and/or hospitalization. One of the participants with history of COVID-19 illness had severe symptoms (fevers, chest tightness) within 24 hours of his first dose of the Pfizer vaccine and did not receive a second dose.
The study protocol was reviewed and approved by an independent Institutional Review Board at Advarra, Inc. (Columbia, MD), with permission from the University of Minnesota.
Mice
Six- to 10-week-old, sex-matched C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD) and maintained in a specific pathogen–free facility under protocols approved by the University of Minnesota Institutional Animal Care and Use Committee and in accordance with National Institutes of Health (NIH) guidelines.
Cell lines
HEK293T cells were maintained in Dulbecco’s Modification of Eagle’s Medium (cellgro) supplemented with 10% Fetal Bovine Serum, L-Glutamine, HEPES buffer, Non Essential Amino Acids (NEAA), sodium pyruvate, penicillin/streptomycin, and gentamycin, and incubated in a humidified chamber at 10% C02 and 37°C.
Method details
Tetramer and decoy production
A wild-type S1-RBD-6xHis tagged protein was generated using a 753 base pair gene block, synthesized by GenScript, that included from 5′ to 3′, a HindIII site, a Kozak sequence, codons for native signal peptide, the S1-RBD sequence (Arg319-Phe541), a 6x Histidine tag, a stop codon and an XhoI site (aagcttgccaccatgttcgtcttcctggtcctgctgcctctggtctcctcacagtgcagggtgcagccaaccgagtctatcgtgcgctttcctaatatcacaaacctgtgcccatttggcgaggtgttcaacgcaacccgcttcgccagcgtgtacgcctggaataggaagcggatcagcaactgcgtggccgactatagcgtgctgtacaactccgcctctttcagcacctttaagtgctatggcgtgtcccccacaaagctgaatgacctgtgctttaccaacgtctacgccgattctttcgtgatcaggggcgacgaggtgcgccagatcgcccccggccagacaggcaagatcgcagactacaattataagctgccagacgatttcaccggctgcgtgatcgcctggaacagcaacaatctggattccaaagtgggcggcaactacaattatctgtaccggctgtttagaaagagcaatctgaagcccttcgagagggacatctctacagaaatctaccaggccggcagcaccccttgcaatggcgtggagggctttaactgttatttcccactccagtcctacggcttccagcccacaaacggcgtgggctatcagccttaccgcgtggtggtgctgagctttgagctgctgcacgccccagcaacagtgtgcggccccaagaagtccaccaatctggtgaagaacaagtgcgtgaacttccaccaccaccaccaccactaactcgag). The gene block was cloned into pcDNA3.1/Zeo (+) using HindIII/XhoI sites and the resulting plasmid was named pcDNA3.1-Zeo(+):S1-RBD.
To generate S1-RBD-BirA, a 210 base pair gene block was synthesized with overlapping residues on both 5′ and 3′ ends for infusion cloning, including from 5′ to 3′, codons encoding part of wild-type S1-RBD, a GGGGS linker, a 6x His tag, another GGGGS linker, the BirA sequence, and a stop codon (gcgtggtggtgctgagctttgagctgctgcacgccccagcaacagtgtgcggccccaagaagtccaccaatctggtgaagaacaagtgcgtgaacttcggaggtggaggatcccatcatcatcatcatcatggaggtggaggatccggcctgaacgatatttttgaagcgcagaaaattgaatggcatgaataactcgagtctagagggc). This gene block was cloned into the aforementioned pcDNA3.1-Zeo(+):S1-RBD construct using BlpI and XhoI sites. The final construct was sequence verified using T7-Promoter (5′-TAATACGACTCACTATAGGG-3′) and BGH-reverse(5′-TAGAAGGCACAGTCGAGG-3′) primers. A South African variant B.1.351 S1-RBD-BirA construct was produced by cloning a 460 base pair gene block from IDT with K417N, E484K and N501Y mutations (tatagcgtgctgtacaactccgcctctttcagcacctttaagtgctatggcgtgtcccccacaaagctgaatgacctgtgctttaccaacgtctacgccgattctttcgtgatcaggggcgacgaggtgcgccagatcgcccccggccagacaggcaacatcgcagactacaattataagctgccagacgatttcaccggctgcgtgatcgcctggaacagcaacaatctggattccaaagtgggcggcaactacaattatctgtaccggctgtttagaaagagcaatctgaagcccttcgagagggacatctctacagaaatctaccaggccggcagcaccccttgcaatggcgtgaagggctttaactgttatttcccactccagtcctacggcttccagcccacatacggcgtgggctatcagccttaccgcgtggtggtgctgagctttgagctgc) between BsrGI and BlpI sites in the original pcDNA3.1-Zeo(+):S1-RBD-BirA construct using in-fusion cloning. The DNA constructs were transfected into 293T cells using Effectene (QIAGEN, Cat# 301425). Fresh medium was added on the following day and the supernatant was harvested and pooled on days 3 and 5. S1-RBD-BirA proteins were purified using Ni2+ affinity chromatography using standard procedures and the elute was washed and concentrated on 10K Amicon Ultra Centrifugal Filters (Millipore # UFC801008)
Wild-type or B.1.351 S1-RBD, at a concentration of 2 mg/ml in 10 mM Tris HCl, pH 8 was biotinylated using the Bir A protein ligase kit (Avidity #BirA500). Free biotin was removed by centrifugation in a 10K Amicon Ultra Centrifugal Filter with 2 PBS washes containing 2% sodium azide. Tetramer was prepared by reacting 1,000 pmoles of S1 RBD-biotin with 250 pmoles of SA-AF647 that was added in 4 increments with 20-minute incubations between each addition. The tetramer was washed 2 times in a 100K Amicon Ultra 0.5 mL filter (Millipore #UFC510008) and to remove any unreacted SA-AF647 or protein and adjusted to a final concentration of 1 μM SA.
The SA-AF647 decoy was prepared by conjugating SA-PE to AF647 (Invitrogen) for 60 minutes at room temperature. The free AF647 was removed by centrifugation in a 100K Amicon Ultra 0.5 mL filter. The SA-PE-AF647 complex concentration was calculated by measuring the absorbance of PE at 565 nm, and the solution was adjusted to 1 μM. The SA-PE-AF647 complex was then incubated with a ten-fold molar excess of free biotin for 30 minutes at room temperature to block any free sites on SA.
Mouse immunizations and lymphocyte preparation
Animals were injected interperitoneally with 50 μL of CFA (Sigma, St. Louis, MO) emulsion containing 25 μg of S1-RBD protein. Explanted lymph nodes and spleens were minced in Collagenase D (cat# 1088874 Roche/Boehringer) and 0.1 mg/ml final concentration of DNase I, incubated for 15 minutes at 37°C and then passed through fine mesh, washed in sorter buffer (PBS containing 0.1% sodium azide and 2% fetal bovine serum), and suspended in 0.1 mL of sorter buffer containing mouse Fc block (BD #553141).
Human PBMC preparation
Blood samples were collected into several 8 mL CPT Mononuclear Cell Preparation Tubes-sodium citrate (BD #362761) and stored at 4°C for up to 24 hours until preparation. Peripheral blood mononuclear cells (PBMC) were enriched by centrifuging the tubes at 1,700xg for 20 minutes at 25°C. After removing a 1 mL aliquot of plasma, the mononuclear cells were poured into a 50 mL conical tube and washed twice with MACS buffer (PBS containing 0.5% fetal bovine serum and 2 mM EDTA). The washed cell pellet was then suspended in 50 μl MACS buffer containing human Fc block (BD #564219) prior to staining.
Cell enrichment
Mouse spleen and lymph nodes or human PBMC suspensions were mixed with 1 μL of 1 μM decoy SA-PE-AF647 and incubated for 10 minutes at room temperature, and then with 0.5 μl of 1 μM wild-type S1-RBD/SA-AF647 tetramer or 1 μM wild-type S1-RBD/SA-AF647 tetramer plus 1 μM B.1.351 S1-RBD/SA-BV650 tetramer for 45 minutes at room temperature. Samples were washed with sorter buffer and 25 μl of anti-Cy5/AF647 MicroBeads (Miltenyi #130-091-395) were added and incubated for 15 minutes at 4°C. The cell suspensions were then passed over magnetized LS columns. The columns were washed 3 times to remove unlabeled cells and the cells that flowed through the columns were saved. After the last wash, the columns were removed from the magnetic field, and bound cells were eluted in 5 mL of sorter buffer. Fluorescent beads (AccuCheck, Life Technologies #PCB100) were used to calculate the number of live lymphocytes in each cell suspension.
Flow cytometry
Mouse spleen and lymph node cells that bound to the columns were stained with fluorophore-conjugated antibodies specific for CD90.2 (53-2.1 Ebioscience #47-0902-82), F480 (BM8 Ebioscience #47-4801-80), Gr1(RB6-8C5 Ebioscience#47-5931-80), B220 (RA3-6B2 BD#563793), IgM (11/41 Ebioscience#15-5790-82), and IgD (11-26c.2a BD#563618). The cells were then fixed in 250 μl Cytofix/Cytoperm (BD #51-2090KZ), washed with permeabilization buffer (TonBo #TNB-1213-L150), and incubated with AF350-labeled antibodies specific for mouse Ig heavy and light chains. Flow cytometry was performed on a 5-laser (355 nm, 405 nm, 488 nm, 561 nm, 640 nm) 18 parameter BD Fortessa H1770 and analyzed with FlowJo software (Tree Star).
Human PBMC that bound to the columns or flowed through were stained with a Ghost Red 710 viability dye (TonBo #13-0871-T100) and fluorophore-conjugated antibodies specific for CD3 (OKT3, Invitrogen #47-0037-42)), CD14 (61D3, Invitrogen #47-0149-42) CD16 (CB16, Invotrogen #47-0168-42, CD19 (HIB19, Biolegend #302242), CD20 (2H7, BD #563782), CD21 (B-ly4, BD #562966), CD27 (O323, Biolegend #302834), CD38 (HB-7, Biolegend #356620), CD11c (B-ly6, BD #526393), CD79b (CB3-1, Biolegend #341404), IgD (IA6-2, Biolegend #348210), IgM (MHM-88, Biolegend #314524), IgA (Southern Biotech #2050-02), and IgG (G18-145, BD-564230), washed in sorter buffer and fixed in 250 μl BD Cytofix/Cytoperm. Flow cytometry was performed on a BD Fortessa X20 and analyzed with FlowJo software (Tree Star).
Calculation of total numbers of S1-RBD tetramer binding cells per million B cells
The total number of S1-RBD specific cells per sample was obtained by multiplying the frequency of tetramer-binding B cells by the total number of single, live, lymphocytes eluted from the enrichment columns (bound fraction). At time points where plasmablasts were present (day 11-18 of the primary response and day 5-9 of the secondary response) the flow through fractions contained S1-RBD-specific plasmablasts that did not bind to the columns due to their lower levels of BCR. For these time points, the total numbers of plasmablasts in the flow through was added to the total numbers in the bound to obtain the total number of S1-RBD specific cells per sample.
To calculate the total S1-RBD specific cells per million B cells, the total number of S1-RBD specific cells in the sample was divided by the total number of single, live, CD19+ B cells in the entire sample (bound and flow through) and multiplied by 1 million. The frequency of CD20‒negative plasmablasts and CD20+ non-plasmablasts was then multiplied by the total S1 RBD binding cells per million B cells to obtain the total plasmablasts and non-plasmablasts per million B cells. For the MBC subset and isotype analysis, the total number of S1 RBD-specific non-plasmablasts per million B cells was multiplied by the frequency of the subset of interest. In order for a sample to be used for the subset analysis it had to contain at least 40 S1-RBD tetramer-binding cells.
S1-RBD ELISA
Plasma samples collected before vaccination, 21-28 days after the first vaccination, and then 14-30 days after the second vaccination from each subject were titrated in 96-well plates coated with 3 μg/ml S1-RBD in PBS and blocked with 1% BSA. Plate-bound Ig of all isotypes was detected by incubating the wells sequentially with horseradish peroxidase-labeled antibodies specific for human Ig heavy and light chains (Invitrogen/ThermoFisher #31412), and KPL ABTS Peroxidase Substrate (SeraCare #5120-0043). Each plate contained the same known positive and negative serum as a reference. The optical density (405 nm) of each well was measured in an ELISA plate reader. Titers were calculated as the reciprocal of the dilution that gave a half-maximal optical density (405) nm value.
Quantification and statistical analysis
Statistical significance between the log10 cell per million values or S1-RBD/CD79b MFI ratios for individual samples from different groups was analyzed by ordinary one-way ANOVA and Sidak’s multiple comparison tests using Prism version 9. The exact values of n are stated in the figure legends. Only samples that contained at least 40 S1-RBD tetramer-binding cells were included for analyses of MBC subsets. One value was excluded as described in the legend to Figure 4.
Acknowledgments
The authors acknowledge Jennifer Walter and Charles Elwood for technical help and maintenance of mice. We also acknowledge the contributions of the University of Minnesota Clinical Translational Science Institute (CTSI) in the start-up of this study, including Carrie McKenzie, Francoise Mercadier-Crevel, and Sydney Viel. This work was supported by a special grant from the Office of the Dean of the University of Minnesota Medical School. Partial funding for the clinical efforts was provided by the non-profit organization Achieving Cures Together.
Author contributions
Conceptualization, K.A.P. and M.K.J.; methodology, K.A.P.; formal analysis, K.A.P. and M.K.J.; investigation, all authors; writing – original draft, M.K.J. and K.A.P.; writing – review & editing, all authors; visualization, M.K.J.; funding acquisition, M.K.J. and A.K.; supervision, M.K.J.; IRB protocol development, A.K.; clinical team supervision, A.J.K., A.K., and J.R.; participant recruitment, consent, clinical metadata capture, A.J.K., D.K., R.B., C.G., C.E., A.K., B.P.V., M.M., and E.S.; sample processing, C.E. and S.L.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects.
Published: September 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109823.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andrews S.F., Chambers M.J., Schramm C.A., Plyler J., Raab J.E., Kanekiyo M., Gillespie R.A., Ransier A., Darko S., Hu J., et al. Activation dynamics and immunoglobulin evolution of pre-existing and newly generated human memory B cell responses to influenza hemagglutinin. Immunity. 2019;51:398–410.e5. doi: 10.1016/j.immuni.2019.06.024. [DOI] [PubMed] [Google Scholar]
- Blink E.J., Light A., Kallies A., Nutt S.L., Hodgkin P.D., Tarlinton D.M. Early appearance of germinal center-derived memory B cells and plasma cells in blood after primary immunization. J. Exp. Med. 2005;201:545–554. doi: 10.1084/jem.20042060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J.G., Allen C.D.C. B cell responses: Cell interaction dynamics and decisions. Cell. 2019;177:524–540. doi: 10.1016/j.cell.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy A.H., Jackson K.J., Kissick H.T., Nakaya H.I., Davis C.W., Roskin K.M., McElroy A.K., Oshansky C.M., Elbein R., Thomas S., et al. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat. Immunol. 2016;17:1226–1234. doi: 10.1038/ni.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel R.R., Apostolidis S.A., Painter M.M., Mathew D., Pattekar A., Kuthuru O., Gouma S., Hicks P., Meng W., Rosenfeld A.M., et al. Distinct antibody and memory B cell responses in SARS-CoV-2 naïve and recovered individuals following mRNA vaccination. Sci. Immunol. 2021;6:eabi6950. doi: 10.1126/sciimmunol.abi6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., O’Neill P., Naradikian M.S., Scholz J.L., Cancro M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294–1304. doi: 10.1182/blood-2011-01-330530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T., Moran I., Shinnakasu R., Phan T.G., Kurosaki T. Generation of memory B cells and their reactivation. Immunol. Rev. 2018;283:138–149. doi: 10.1111/imr.12640. [DOI] [PubMed] [Google Scholar]
- Isnardi I., Ng Y.S., Menard L., Meyers G., Saadoun D., Srdanovic I., Samuels J., Berman J., Buckner J.H., Cunningham-Rundles C., Meffre E. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115:5026–5036. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks S.A., Cashman K.S., Zumaquero E., Marigorta U.M., Patel A.V., Wang X., Tomar D., Woodruff M.C., Simon Z., Bugrovsky R., et al. Distinct effector B cells induced by unregulated Toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. 2018;49:725–739.e6. doi: 10.1016/j.immuni.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.L., Rosenthal R.L., Knox J.J., Myles A., Naradikian M.S., Madej J., Kostiv M., Rosenfeld A.M., Meng W., Christensen S.R., et al. The transcription factor T-bet resolves memory B cell subsets with distinct tissue distributions and antibody specificities in mice and humans. Immunity. 2020;52:842–855.e6. doi: 10.1016/j.immuni.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler T.B., Liao H.X., Alam S.M., Bhaskarabhatla R., Zhang R., Yandava C., Stewart S., Anasti K., Kelsoe G., Parks R., et al. Immunoglobulin gene insertions and deletions in the affinity maturation of HIV-1 broadly reactive neutralizing antibodies. Cell Host Microbe. 2014;16:304–313. doi: 10.1016/j.chom.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateeb J., Li Y., Zhang H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care. 2021;25:244. doi: 10.1186/s13054-021-03662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.C., Baccarella A.M., Bayat A., Pepper M., Fontana M.F. FCRL5+ Memory B Cells Exhibit Robust Recall Responses. Cell Rep. 2019;27:1446–1460.e4. doi: 10.1016/j.celrep.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D., Lan L.Y., Andrews S.F., Henry C., Rojas K.T., Neu K.E., Huang M., Huang Y., DeKosky B., Palm A.E., et al. Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci. Immunol. 2017;2:eaai8153. doi: 10.1126/sciimmunol.aai8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis K., Bailly E., Macedo C., Lau L., Ramaswami B., Chang A., Chandran U., Landsittel D., Gu X., Chalasani G., et al. T-bet+CD27+CD21− B cells poised for plasma cell differentiation during antibody-mediated rejection of kidney transplants. JCI Insight. 2021;6:e148881. doi: 10.1172/jci.insight.148881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni A., Di Lauria N., Maggi L., Salvati L., Vanni A., Capone M., Lamacchia G., Mantengoli E., Spinicci M., Zammarchi L., et al. COVID-19 Research Group First-dose mRNA vaccination is sufficient to reactivate immunological memory to SARS-CoV-2 in subjects who have recovered from COVID-19. J. Clin. Invest. 2021;131:e149150. doi: 10.1172/JCI149150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams L.J., McHeyzer-Williams M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- Mesin L., Ersching J., Victora G.D. Germinal center B cell dynamics. Immunity. 2016;45:471–482. doi: 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S., Ho J., Malaspina A., Wang W., DiPoto A.C., O’Shea M.A., Roby G., Kottilil S., Arthos J., Proschan M.A., et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J. Exp. Med. 2008;205:1797–1805. doi: 10.1084/jem.20072683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muellenbeck M.F., Ueberheide B., Amulic B., Epp A., Fenyo D., Busse C.E., Esen M., Theisen M., Mordmüller B., Wardemann H. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J. Exp. Med. 2013;210:389–399. doi: 10.1084/jem.20121970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt S.L., Hodgkin P.D., Tarlinton D.M., Corcoran L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015;15:160–171. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- Obeng-Adjei N., Portugal S., Holla P., Li S., Sohn H., Ambegaonkar A., Skinner J., Bowyer G., Doumbo O.K., Traore B., et al. Malaria-induced interferon-γ drives the expansion of Tbethi atypical memory B cells. PLoS Pathog. 2017;13:e1006576. doi: 10.1371/journal.ppat.1006576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape K.A., Taylor J.J., Maul R.W., Gearhart P.J., Jenkins M.K. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape K.A., Maul R.W., Dileepan T., Paustian A.S., Gearhart P.J., Jenkins M.K. Naive B cells with high-avidity germline-encoded antigen receptors produce persistent IgM+ and transient IgG+ memory B cells. Immunity. 2018;48:1135–1143.e4. doi: 10.1016/j.immuni.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolo S. Synthetic messenger RNA-based vaccines: From scorn to hype. Viruses. 2021;13:13. doi: 10.3390/v13020270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus D., Phan T.G., Chan T.D., Gardam S., Basten A., Brink R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006;203:1081–1091. doi: 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mazliah D., Gardner P.J., Schweighoffer E., McLaughlin S., Hosking C., Tumwine I., Davis R.S., Potocnik A.J., Tybulewicz V.L., Langhorne J. Plasmodium-specific atypical memory B cells are short-lived activated B cells. eLife. 2018;7:e39800. doi: 10.7554/eLife.39800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., et al. C4591001 Clinical Trial Group Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal S., Tipton C.M., Sohn H., Kone Y., Wang J., Li S., Skinner J., Virtaneva K., Sturdevant D.E., Porcella S.F., et al. Malaria-associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. eLife. 2015;4:e07218. doi: 10.7554/eLife.07218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C.J., Pade C., Gibbons J.M., Butler D.K., Otter A.D., Menacho K., Fontana M., Smit A., Sackville-West J.E., Cutino-Moguel T., et al. UK COVIDsortium Immune Correlates Network. UK COVIDsortium Investigators Prior SARS-CoV-2 infection rescues B and T cell responses to variants after first vaccine dose. Science. 2021 doi: 10.1126/science.abh1282. Published online April 30, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov A.V., Rubtsova K., Fischer A., Meehan R.T., Gillis J.Z., Kappler J.W., Marrack P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K., Rubtsov A.V., van Dyk L.F., Kappler J.W., Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc. Natl. Acad. Sci. USA. 2013;110:E3216–E3224. doi: 10.1073/pnas.1312348110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz I., Wei C., Jenks S.A., Cashman K.S., et al. Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.02458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid J.F., Mouquet H., Feldhahn N., Seaman M.S., Velinzon K., Pietzsch J., Ott R.G., Anthony R.M., Zebroski H., Hurley A., et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458:636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- Schittek B., Rajewsky K. Maintenance of B-cell memory by long-lived cells generated from proliferating precursors. Nature. 1990;346:749–751. doi: 10.1038/346749a0. [DOI] [PubMed] [Google Scholar]
- Seifert M., Küppers R. Human memory B cells. Leukemia. 2016;30:2283–2292. doi: 10.1038/leu.2016.226. [DOI] [PubMed] [Google Scholar]
- Sette A., Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. 2021;184:861–880. doi: 10.1016/j.cell.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Wan Y., Liu C., Yount B., Gully K., Yang Y., Auerbach A., Peng G., Baric R., Li F. Structure of mouse coronavirus spike protein complexed with receptor reveals mechanism for viral entry. PLoS Pathog. 2020;16:e1008392. doi: 10.1371/journal.ppat.1008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal A., Chappert P., Barba-Spaeth G., Roeser A., Fourati S., Azzaoui I., Vandenberghe A., Fernandez I., Meola A., Bouvier-Alias M., et al. Maturation and persistence of the anti-SARS-CoV-2 memory B cell response. Cell. 2021;184:1201–1213.e14. doi: 10.1016/j.cell.2021.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatatos L., Czartoski J., Wan Y.H., Homad L.J., Rubin V., Glantz H., Neradilek M., Seydoux E., Jennewein M.F., MacCamy A.J., et al. mRNA vaccination boosts cross-variant neutralizing antibodies elicited by SARS-CoV-2 infection. Science. 2021:eabg9175. doi: 10.1126/science.abg9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan D., Sundling C., Brink R. Plasma cell and memory B cell differentiation from the germinal center. Curr. Opin. Immunol. 2017;45:97–102. doi: 10.1016/j.coi.2017.03.006. [DOI] [PubMed] [Google Scholar]
- Sutton H.J., Aye R., Idris A.H., Vistein R., Nduati E., Kai O., Mwacharo J., Li X., Gao X., Andrews T.D., et al. Atypical B cells are part of an alternative lineage of B cells that participates in responses to vaccination and infection in humans. Cell Rep. 2021;34:108684. doi: 10.1016/j.celrep.2020.108684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze D.M., Toellner K.M., García de Vinuesa C., Taylor D.R., MacLennan I.C. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J. Exp. Med. 2000;192:813–821. doi: 10.1084/jem.192.6.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlinton D.M. Evolution in miniature: selection, survival and distribution of antigen reactive cells in the germinal centre. Immunol. Cell Biol. 2008;86:133–138. doi: 10.1038/sj.icb.7100148. [DOI] [PubMed] [Google Scholar]
- Taylor J.J., Pape K.A., Jenkins M.K. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J. Exp. Med. 2012;209:597–606. doi: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.J., Pape K.A., Steach H.R., Jenkins M.K. Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naïve B cell. Science. 2015;347:784–787. doi: 10.1126/science.aaa1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traore B., Koné Y., Doumbo S., Doumtabé D., Traoré A., Crompton P.D., Mircetic M., Huang C.Y., Kayentao K., Dicko A., et al. The TLR9 agonist CpG fails to enhance the acquisition of Plasmodium falciparum-specific memory B cells in semi-immune adults in Mali. Vaccine. 2009;27:7299–7303. doi: 10.1016/j.vaccine.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Wang J., Kumar V., Karnell J.L., Naiman B., Gross P.S., Rahman S., Zerrouki K., Hanna R., Morehouse C., et al. Autoimmunity Molecular Medicine Team IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat. Commun. 2018;9:1758. doi: 10.1038/s41467-018-03750-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Muecksch F., Schaefer-Babajew D., Finkin S., Viant C., Gaebler C., Hoffmann H.H., Barnes C.O., Cipolla M., Ramos V., et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature. 2021;595:426–431. doi: 10.1038/s41586-021-03696-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C., Anolik J., Cappione A., Zheng B., Pugh-Bernard A., Brooks J., Lee E.H., Milner E.C., Sanz I. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J. Immunol. 2007;178:6624–6633. doi: 10.4049/jimmunol.178.10.6624. [DOI] [PubMed] [Google Scholar]
- Weiss G.E., Crompton P.D., Li S., Walsh L.A., Moir S., Traore B., Kayentao K., Ongoiba A., Doumbo O.K., Pierce S.K. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J. Immunol. 2009;183:2176–2182. doi: 10.4049/jimmunol.0901297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widge A.T., Rouphael N.G., Jackson L.A., Anderson E.J., Roberts P.C., Makhene M., Chappell J.D., Denison M.R., Stevens L.J., Pruijssers A.J., et al. mRNA-1273 Study Group Durability of responses after SARS-CoV-2 mRNA-1273 vaccination. N. Engl. J. Med. 2021;384:80–82. doi: 10.1056/NEJMc2032195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R., Bhattacharya D. Basics of memory B-cell responses: lessons from and for the real world. Immunology. 2019;156:120–129. doi: 10.1111/imm.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrammert J., Smith K., Miller J., Langley W.A., Kokko K., Larsen C., Zheng N.Y., Mays I., Garman L., Helms C., et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.