

Abstract
After angiogenesis-activated embryonic and early postnatal vascularization, endothelial cells (ECs) in most tissues enter a quiescent state necessary for proper tissue perfusion and EC functions. Notch signaling is essential for maintaining EC quiescence, but the mechanisms of action remain elusive. Here, we show that microRNA-218 (miR-218) is a downstream effector of Notch in quiescent ECs. Notch activation upregulated, while Notch blockade downregulated, miR-218 and its host gene Slit2, likely via transactivation of the Slit2 promoter. Overexpressing miR-218 in human umbilical vein ECs (HUVECs) significantly repressed cell proliferation and sprouting in vitro. Transcriptomics showed that miR-218 overexpression attenuated the MYC proto-oncogene, bHLH transcription factor (MYC, also known as c-myc) signature. MYC overexpression rescued miR-218-mediated proliferation and sprouting defects in HUVECs. MYC was repressed by miR-218 via multiple mechanisms, including reduction of MYC mRNA, repression of MYC translation by targeting heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), and promoting MYC degradation by targeting EYA3. Inhibition of miR-218 partially reversed Notch-induced repression of HUVEC proliferation and sprouting. In vivo, intravitreal injection of miR-218 reduced retinal EC proliferation accompanied by MYC repression, attenuated pathological choroidal neovascularization, and rescued retinal EC hyper-sprouting induced by Notch blockade. In summary, miR-218 mediates the effect of Notch activation of EC quiescence via MYC and is a potential treatment for angiogenesis-related diseases.
Keywords: miR-218, endothelial cells, Notch, MYC, angiogenesis, quiescence
Graphical abstract
Endothelial cell (EC) quiescence is important for vessel homeostasis and function, but the mechanism remains elusive. Han and colleagues report that miR-218 mediates the effect of Notch activation on promoting EC quiescence via repressing MYC, which holds potential in the treatment of angiogenesis-related diseases.
Introduction
Vascularization, the formation of organized and functional blood vessels, takes place throughout embryonic and early postnatal development. It occurs mainly through angiogenesis, which involves coordinated proliferation and differentiation of endothelial cells (ECs).1 Most ECs become quiescent after active angiogenesis, which is essential for physiological tissue perfusion and other EC functions.2 Abnormal activation of ECs is involved in many human diseases;3 therefore, it is important to understand the molecular mechanisms underlying EC quiescence in adults.
Multiple environmental and cell-intrinsic signals regulate EC quiescence.4 Notch signaling, in collaboration with vascular endothelial growth factor receptor 2 (VEGFR2) signaling, regulates angiogenesis by controlling tip-stalk cell differentiation, among other processes.5,6 Recently, single-cell RNA sequencing (RNA-seq) showed that Notch signaling is upregulated in mature capillary and arterial ECs.7 Disrupting the Notch integrating transcription factor recombination signal binding protein Jκ (RBPj) results in autonomous vessel sprouting in adult tissues, suggesting that Notch signaling is required for EC quiescence.8 Although cyclin dependent kinase inhibitor 1A (CDKN1A, also known as p21Cip1), cyclin dependent kinase inhibitor 1B (CDKN1B, also known as p27KIP1), phosphatase and tensin homolog (PTEN), and certain metabolic enzymes have been implicated in Notch-mediated proliferation arrest in ECs,9, 10, 11 and Notch signaling may also influence EC quiescence by regulating proliferation,3,12 the exact mechanism for Notch-induced EC quiescence is not completely known.
MYC proto-oncogene, bHLH transcription factor (MYC, also known as c-myc) plays a critical role in mediating EC proliferation by influencing glycolysis, mitochondrial respiration, and cell-cycle progression.7,13, 14, 15 In ECs, MYC deficiency has been shown to impair cell growth and vascular expansion.13 As a critical proto-oncogene, MYC expression is tightly controlled at multiple levels in normal cells. MYC transcription is regulated by enhancers, which can harbor oncogenic mutations.16 MYC mRNA is inherently unstable and requires cap-dependent and internal ribosome entry site (IRES)-mediated translation.17,18 IRES-dependent MYC translation is regulated by AKT serine/threonine kinase (AKT) and enhanced by rapamycin through a mitogen-activated protein kinase (MAPK)-dependent pathway.19 Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) is involved in malignant transformation20 and regulates cyclin D1 (CCND1) and MYC IRES function through AKT.21 MYC protein is rapidly degraded, mainly via the ubiquitin-proteasome system (UPS).22 Protein phosphatase 2A (PP2A) is a ubiquitously expressed serine/threonine phosphatase and dephosphorylates many critical proteins23 including MYC.24 The tyrosine phosphatase eyes absent 3 (EYA3) partners with PP2A to stabilize MYC;25 however, a relationship between Notch activation and MYC in ECs has not been characterized.26
MicroRNAs (miRNAs) are essential regulators of angiogenesis under physiological and pathological conditions.27 To identify potential Notch downstream miRNAs in ECs, we screened miRNAs upregulated after forced Notch activation in liver sinusoidal ECs (LSECs).28 In this study, we identified miR-218-5p (hereafter referred to as miR-218), hosted in the slit guidance ligand 2 (Slit2) gene intron, as a downstream molecule activated by Notch signaling in ECs.29 An anti-angiogenic function30,31 is among the several characterized activities of miR-218.32, 33, 34 Here, we show that miR-218 attenuates MYC activity via several targets, including hnRNPA1 and EYA3, and represses EC proliferation. Our results indicate Notch signaling promotes endothelial quiescence via miR-218 regulation of MYC.
Results
miR-218 is a downstream target of Notch signaling in ECs
Previously, we used small RNA-seq to show that miR-218 was upregulated in Notch-activated ECs.28 To confirm that Notch activation upregulates miR-218, we infected human umbilical vein ECs (HUVECs) with adenoviruses expressing either the Notch-intracellular domain (AdNIC) or control (AdCtrl; Figure S1A). Quantitative real-time polymerase chain reaction (PCR) showed that NIC overexpression upregulated hes related family bHLH transcription factor with YRPW motif 1 (Hey1), a downstream Notch effector, as well as miR-218 (Figure 1A). There are two human miR-218 genes, miR-218-1 and miR-218-2, accommodated in the Slit2 and Slit3 genes, respectively.29 Both quantitative real-time PCR and western blotting showed that Slit2 mRNA and protein were respectively upregulated in NIC-overexpressing ECs (Figures 1A and S1B). In contrast, Slit3 mRNA levels were comparable between HUVECs infected with AdNIC and AdCtrl (Figure S1C). When HUVECs were treated with the γ-secretase inhibitor DAPT to inhibit spontaneous ligand-dependent Notch activation (Table S1), Hey1, miR-218, and Slit2 were all downregulated (Figure 1B), whereas Slit3 showed no change (Figure S1D). Slit2 mRNA levels were much higher than Slit3 in HUVECs (Figure S1E). HUVECs cultured in culture dishes coated with soluble delta like canonical Notch ligand 4 (Dll4) protein to stimulate Notch receptors showed upregulation of Hey1, Slit2, and miR-218, which could be inhibited by DAPT (Figures 1C and 1D). These results suggest that Notch activation upregulates miR-218 in ECs, likely via the Slit2 promoter.
Figure 1.

Notch activation upregulates miR-218 and its host gene Slit2 in ECs
(A) HUVECs were transfected with AdNIC or AdCtrl adenovirus for 48 h. Hey1, Slit2, and miR-218 RNA levels were determined by quantitative real-time PCR (n = 5). (B) HUVECs were cultured in the presence of DAPT (25 μM) or DMSO for 48 h, and Hey1, Slit2, and miR-218 RNA levels were determined by quantitative real-time PCR (n = 4). (C) HUVECs were cultured with dishes coated with Dll4 or PBS for 48 h. Hey1, Slit2, and miR-218 RNA levels were evaluated by quantitative real-time PCR (n = 3). (D) HUVECs were cultured as in (C) in the presence or absence of DAPT for 48 h. Hey1, Slit2, and miR-218 RNA levels were determined by quantitative real-time PCR (n = 4). (E) Reporter assay. A representative illustration of the structure of Slit2 gene and the reporter is shown on the left. Luciferase activity was determined and calibrated in HEK293T cells transfected with pGL3-Slit2 (100 ng), phRL-TK (5 ng), and increasing amount of pEFBOS-NIC (0, 50, 100 ng) for 48 h (n = 8). Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We then constructed a reporter (pGL-Slit2) using the Slit2 promoter, which harbors two consensus RBPj binding sites (Figure 1E). HEK293T cells transfected with pGL-Slit2 and increasing amounts of pEFBOS-NIC showed that pGL-Slit2 could be transactivated by transfection with NIC (Figure 1E). These results suggest that miR-218 is activated downstream of Notch signaling activation.
miR-218 inhibits EC proliferation and sprouting in vitro
Slit2 expression in both developing retinal vasculature and in vitro EC angiogenic assays was negatively correlated with proliferation-associated genes, but not with tip cell markers, suggesting that Slit2 and/or miR-218 inhibit EC proliferation (Figures S1F–S1H).35, 36, 37 We tested the function of miR-218 in HUVECs by adenovirus-mediated transfection (Figure S1I). Cell number quantification and 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays showed that overexpression of miR-218 significantly suppressed EC proliferation (Figures 2A and 2B). Cell-cycle analysis confirmed that miR-218 overexpression increased the number of cells in G0/G1 and reduced the number in S phase (Figure 2C). A sprouting assay showed that miR-218 remarkably reduced the number and length of sprouts in HUVECs (Figure 2D). Moreover, miR-218 overexpression slightly inhibited lumen formation by HUVECs (Figure 2E), but apoptosis was not affected (Figures S2A and S2B). Accordingly, an anti-miR-218 lentivirus successfully inhibited function of miR-218 and further confirmed its negative effect on EC growth, tube formation, and sprouting (Figures S3A–S3E). Taken together, these data indicate that miR-218 mainly inhibits EC proliferation and sprouting.
Figure 2.

miR-218 inhibits EC proliferation and sprouting in vitro
(A) HUVECs were transfected with miR-218 or NC by adenovirus. Cell number was evaluated using a counting chamber (n = 3). (B) HUVECs were transfected as in (A). Cell proliferation was determined by EdU assay 24, 48, and 72 h after the transfection (n = 5). (C) HUVECs were transfected as in (A). Cell-cycle progression was determined by FACS 48 h after the transfection. Cells in G0/G1, S, and G2/M were quantitatively compared between the two groups (n = 3). (D) HUVECs were transfected as in (A) and subjected to the fibrin beads assay 48 h after the transfection. Nearly all sprouts were GFP positive, which confirmed effective transfection and gene expression. The number of sprouts per bead and the average sprout length were compared between the two groups (n = 6). (E) HUVECs were transfected as in (A) and subjected to the lumen formation assay 48 h after the transfection. Lumen formation was quantitatively compared between the two groups by the number of branches, the number of loops, and cell cord length per field (n = 8). Scale bars, 100 μm. Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-218 overexpression attenuates MYC activity in ECs
To gain insight into the underlying mechanisms of miR-218-mediated inhibition of EC proliferation, we used RNA-seq to compare transcriptomes of HUVECs infected with adenovirus expressing miR-218 (AdmiR-218) or AdCtrl (Figures S4A and S4B). Bioinformatic analyses suggested that pathways associated with DNA replication, cell cycle, and metabolism were downregulated when miR-218 was overexpressed in HUVECs (Figure S4C). Gene set enrichment analysis (GSEA) also revealed that transcripts involved in cell-cycle progression were enriched in the control compared to miR-218-transfected HUVECs (Figure 3A). This analysis also indicated that MYC target genes were downregulated in miR-218-overexpressed HUVECs (Figure 3B). A heatmap comparison showed that miR-218 overexpression reduced proliferation and increased downstream proliferation-inhibiting MYC genes (Figure 3C). The effects of miR-218 overexpression in downregulating proliferation-promoting genes and upregulating proliferation-inhibiting MYC target genes were confirmed by quantitative real-time PCR and western blotting (Figures 3D and 3E); inhibiting miR-218 reversed these effects (Figures S5A and S5B). These findings suggest that miR-218 represses EC proliferation, likely by inhibiting MYC.
Figure 3.

miR-218 represses MYC signaling in ECs
(A) GSEA of gene sets related to cell-cycle progression and E2F targets in HUVECs transfected with miR-218 or NC adenovirus. (B) GSEA of MYC target genes in HUVECs transfected as in (A). (C) Heatmap of MYC target genes shown by fragments kilobase million (FPKM) of RNA-seq data from HUVECs transfected as in (A). The p value of each comparison is represented with colors (n = 3). (D) HUVECs were transfected as in (A). The expression of MYC target genes related with cell proliferation and metabolism was determined by quantitative real-time PCR 48 h after transfection (n = 4). (E) HUVECs were transfected as in (A). CCNE1, RRM2, CDK2, and CDKN1A levels were determined by western blotting (n = 3). Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
MYC rescues miR-218-induced proliferation arrest and sprouting in ECs
To confirm that miR-218 inhibits EC proliferation by attenuating MYC, we simultaneously infected HUVECs with AdmiR-218 and an adenovirus expressing MYC (AdMYC). The results showed that MYC overexpression increased cell number, possibly by decreasing the number of cells in G0/G1, and improved EC sprouting (Figures 4A–4C). Notably, miR-218-induced reduction in cell number could be significantly rescued by MYC overexpression (Figure 4A). Cell-cycle analysis further revealed that MYC overexpression overcame G1 arrest induced by miR-218 overexpression (Figure 4B). Moreover, MYC overexpression abrogated miR-218-mediated inhibition of EC sprouting, as shown by the fibrin bead sprouting assay (Figure 4C). These data further verify that miR-218 represses EC proliferation by inhibiting MYC.
Figure 4.

Overexpression of MYC rescues miR-218-mediated suppression of EC proliferation and sprouting
(A) HUVECs were transfected with NC, MYC, miR-218, or miR-218 plus MYC using adenovirus. Cell number was determined by a counting chamber (n = 4). (B) HUVECs were transfected as in (A). Cells in G0/G1, S, or G2/M phase were evaluated with FACS and quantitatively compared (n = 3). (C) HUVECs were transfected as in (A) and subjected to the fibrin beads sprouting assay 48 h after transfection. The number of sprouts per bead and the average sprout length were compared between the four groups (n = 4). Scale bars, 100 μm. Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-218 likely downregulates MYC via hnRNPA1 and EYA3
MYC expression and activity are regulated at multiple levels. We first examined MYC mRNA and protein levels in AdmiR-218-infected HUVECs. The results showed that miR-218 overexpression reduced, while inhibiting miR-218 increased, MYC expression at both the mRNA and protein levels (Figures 5A and 5B). This could not be attributed to forkhead box O1 (FOXO1), an upstream transcription factor of MYC (Figure S6A).13 Notably, MYC protein levels decreased to a much larger extent than mRNA (45.8% ± 10.8% versus 15.0% ± 7.3%), suggesting that translational and/or post-translational mechanisms might be involved. MG-132, a proteasome inhibitor, partially but significantly rescued miR-218-mediated MYC downregulation at the protein level (percentage of reduction from 45.8% ± 10.8% to 26.1% ± 14.7%, p < 0.01; Figure 5B). Consistently, blocking transcription with actinomycin D (ActD) or translation with cycloheximide (CHX) showed that miR-218 did not influence MYC mRNA decay but slightly accelerated MYC protein decay (Figures 5C and 5D). Glycogen synthase kinase 3 beta (GSK3β) is a critical inducer of MYC degradation.38 The levels of phosphorylated GSK3β at Ser9, which is catalytically impaired, increased after miR-218 upregulation, inconsistent with increased MYC degradation (Figure S6B). Using the DIANA microT-CDS tools,39 we found that EYA3, which inhibits MYC degradation,25 is a potential miR-218 target (Figure S7A). Downregulation of EYA3 expression by miR-218 was demonstrated by quantitative real-time PCR, western blotting, and reporter assays (Figures 5E–5G). These results suggest that miR-218 inhibits EYA3 to promote MYC degradation.
Figure 5.

miR-218 downregulates MYC expression via multiple targets at different levels
(A) HUVECs were transfected with miR-218 or NC adenovirus or anti-miR-218 (Anti-218) or control (Anti-Ctrl) lentivirus. The expression of MYC mRNA was determined by quantitative real-time PCR 48 h after transfection (n = 9 for miR-218 and NC groups; n = 4 for Anti-218 and Anti-Ctrl groups). (B) HUVECs were transfected as in (A) in the absence or presence of MG-132. The expression of MYC protein was determined by western blotting 48 h after transfection (n = 9 for miR-218 and NC groups; n = 3 for Anti-218 and Anti-Ctrl groups). (C) HUVECs were transfected with miR-218 or NC adenovirus and cultured in the presence of ActD. MYC mRNA levels were determined by quantitative real-time PCR at different time points after the addition of ActD (n = 5). (D) HUVECs were transfected as in (C) and cultured in the presence of CHX. MYC protein level was determined by western blotting at different time points after the addition of CHX (n = 5). (E and F) HUVECs were transfected with as in (A). The expression of hnRNPA1 and EYA3 were determined by (E) quantitative real-time PCR and (F) western blotting 48 h after transfection (n = 6). (G) Reporter assay. HEK293T cells were transfected with either pGL-hnRNPAUTR3 or pGL-EYAUTR3 and increasing amount of pGV317-miR-218 plasmid. Luciferase activity in cell lysates was determined 48 h after transfection (n = 6). (H) Reporter assay. HEK293T cells were transfected with pGL-Ctrl, pGL-MycUTR5, or pGL-MycUTR3 as reporters, and increasing amount of pGV317-miR-218 plasmid. Luciferase activity in cell lysates was determined 48 h after transfection (n = 6). Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, we examined the translational regulation of MYC. Consistent with previous reports, GSEA showed impaired mammalian target of rapamycin complex 1 (mTORC1) signaling,31 but we failed to detect any significant change in the phosphorylation of eukaryotic translation initiation factor 4E (EIF4E) and eukaryotic translation initiation factor 4E binding protein 1 (EIF4EBP1) in AdmiR-218-transfected HUVECs (Figures S6C and S6D). MYC might not be a predicted target of miR-218, as supported by a reporter assay using a MYC mRNA 3′ untranslated region (UTR) reporter (Figure 5H), however, the activity of a reporter containing the MYC mRNA 5′ UTR, which hosts an IRES that promotes MYC mRNA translation,17, 18, 19 was inhibited by miR-218 (Figure 5H). We also found that hnRNPA1, which promotes MYC translation by binding to IRES,20,21 is likely targeted by miR-218 (Figure S7B). Using quantitative real-time PCR, western blotting, and reporter assays, we confirmed that miR-218 significantly downregulated hnRNPA1 expression (Figures 5E–5G). We also identified several genes of the nuclear factor κB (NF-κB) pathway,38 including NFKB1, IKBKB, and VOPP1, as predicted miR-218 targets. However, nuclear localization of p65 remained unchanged, although the mRNA levels of these molecules decreased slightly after miR-218 transfection (Figures S6E and S6F). Together, these data indicate that miR-218 attenuates MYC protein levels through multiple mechanisms, including mRNA translation and protein degradation via hnRNPA1 and EYA3, respectively.
Notch activation inhibits EC proliferation and sprouting by upregulating miR-218
In HUVECs infected with NIC-overexpressing lentivirus, cell-cycle-related genes and MYC target genes were downregulated, as shown by GSEA (GSE45750; Figures S8A and S8B),12 coincident with the miR-218 upregulation signature (Figures 3A and 3B). We therefore infected HUVECs with AdNIC to activate Notch signaling, and with a miR-218-inhibiting lentivirus to downregulate miR-218 (Figure 6A). We found that reduction of miR-218 partially rescued Notch-induced MYC reduction at the protein level (Figure 6B). Moreover, NIC overexpression resulted in decreases in cell number, cell-cycle arrest, and compromised sprouting, which were also partially rescued by miR-218 knockdown (Figures 6C–6E). These results indicate that miR-218 mediates the inhibition of EC proliferation and sprouting upon Notch activation.
Figure 6.

Notch activation represses endothelial proliferation and sprouting via miR-218
(A) HUVECs were transfected with AdCtrl, AdNIC, or AdNIC plus anti-miR-218 lentivirus. The level of miR-218 was tested by quantitative real-time PCR 48 h after transfection (n = 8). (B) HUVECs were transfected as in (A), and MYC expression was determined by western blotting 48 h after the transfection (n = 4). (C and D) HUVECs were transfected as in (A). (C) Cell number was evaluated using counting chamber 48 h after transfection (n = 5). (D) Cell-cycle progression in each group was analyzed 48 h after transfection (n = 4). (E) HUVECs were transfected as in (A) and subjected to the fibrin beads sprouting assay 48 h after transfection. The number of sprouts per bead and the average sprout length are shown (n = 4). Scale bars, 100 μm. Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-218 compromises physiological and pathological angiogenesis
Next, we examined the effect of miR-218 on angiogenesis in vivo. Retinal vasculature develops after birth in mice through angiogenesis, and EC proliferation decreases after postnatal day 10 (P10).40 Using quantitative real-time PCR, miR-218 levels decreased in retinal ECs at P5, and then increased up until P11 (Figure 7A). Coincidentally, levels of MYC mRNA also increased at P5 and decreased at P11 (Figure 7A). P3 pups were intravitreally injected with either miR-218 agomiR or antagomir and whole retinal flat-mounts were immunostained at P5. The number of proliferating (bromodeoxyuridine, BrdU positive) ECs, as well as vessel density at the growth edge, were significantly reduced in the agomiR-injected group, and slightly increased in the antagomiR-injected group (Figures 7B and S9A). Meanwhile, western blotting demonstrated that MYC, EYA3, hnRNPA1, and ribonucleotide reductase regulatory subunit M2 (RRM2, a MYC target) protein levels decreased significantly in miR-218 upregulated retinas, while the levels of CDKN1A protein increased significantly (Figure 7C). In the laser-induced choroidal neovascularization (CNV) model, miR-218 in ECs decreased on day 7, then increased thereafter, as shown by quantitative real-time PCR (Figure 7D). We also intravitreally injected mice with miR-218 agomiR on day 0.5 after laser photocoagulation (Figure S9B). Compared with the control group, mice treated with miR-218 agomiR showed a significant decrease in CNV area (Figure 7E). To confirm the relationship between miR-218 and Notch in vivo, we intravitreously injected miR-218 agomiR into P3 pups, which were pre-treated with DAPT (Figure S9C). Immunostaining of whole retinal flat-mounts at P5 showed that miR-218 could significantly rescue EC hypersprouting and hyperproliferation caused by Notch blockade (Figure 7F). These results suggest that the expression pattern of miR-218 is closely related to angiogenesis, and miR-218 overexpression could suppress physiological, as well as pathological, neovascularization in vivo.
Figure 7.

miR-218 overexpression compromises physiological and pathological angiogenesis
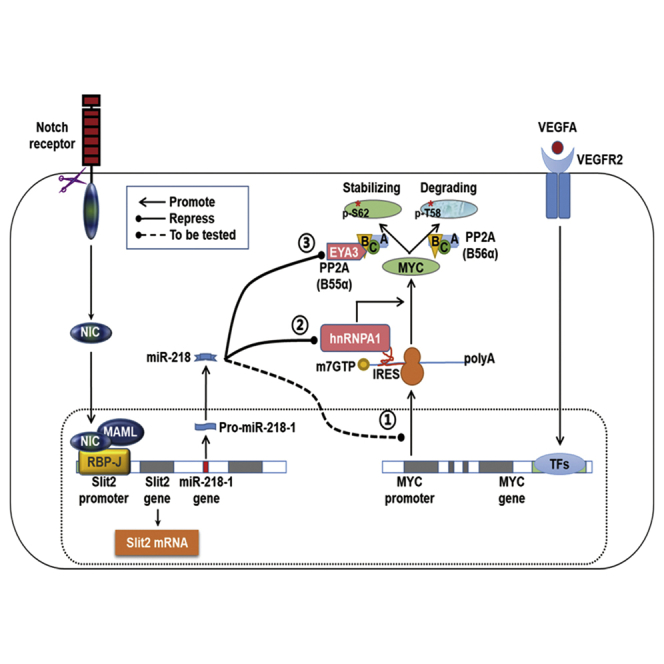
(A) Expression levels of miR-218 and MYC mRNA from retinal vasculature isolated from pups at P3, P5, P7, and P11 (n = 3). (B) Immunofluorescence staining with IB4 (green), BrdU (red), and Erg (white) of P5 retina injected with miR-218 agomiR and NC. BrdU+Erg+ cells and the number of loops were quantitatively compared in the leading growth edge between the two groups (n = 5). (C) P3 pups were injected intravitreally with miR-218 agomiR or NC. Retinas were collected on P5 and MYC, EYA3, hnRNPA1, RRM2, and CDKN1A levels were determined by western blotting (n = 3 or 4). (D) Mice were subjected to laser-induced CNV. Choroidal tissues were collected on the indicated days, and miR-218 level was determined by quantitative real-time PCR (n = 3). (E) Immunofluorescence staining with IB4 (green) of flat mounted RPE-choroid-sclera complexes on day 7. The CNV volumes (marked by circles with dashed lines) were compared between the miR-218 and NC groups (n = 4 for each group, 5 laser burns per sample). (F) Pups were injected subcutaneously with DAPT or DMSO at P1 and P3, followed by intravitreous injection with miR-218 agomiR or NC at P3, and injected i.p. with EdU at P5. Retinas were collected 4 h later, followed by immunofluorescence staining with IB4 (green), EdU (red), and Erg (white). The number of loops and EdU+Erg+ cells were quantitatively compared in the leading growth edge among the three groups (n = 6). (G) A summary of miR-218-mediated repression of MYC expression in ECs. Notch activation in ECs upregulates miR-218 by transactivating the Slit2 promoter. miR-218 represses MYC via at least three mechanisms, namely, (1) reducing MYC mRNA level, (2) inhibiting MYC translation via hnRNPA1, and (3) promoting MYC degradation via EYA3. MYC could be activation by various pro-angiogenic pathways such as VEGFR2 signaling (as an example). Scale bars, 50, 100, or 200 μm as indicated. Error bars, means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
Notch signaling plays a critical role in blood vessel development by regulating EC functions.41 In quiescent phalanx ECs, Notch signaling is active in maintaining quiescence, because disruption of RBPj results in autonomous angiogenic sprouting.7,8 Several downstream effectors mediating EC quiescence by Notch signaling have been implicated.9, 10, 11 Moreover, Notch signaling may interact with other morphogen and cytokine pathways to repress EC proliferation. VEGFR2 signaling is typically repressed by Notch activation.5,6 Notch activation also represses Wnt signaling at different levels, which inhibits EC proliferation.42 In the current study, we demonstrate that Notch activation upregulates miR-218. There are two miR-218 genes in the human genome, miR-218-1 and miR-218-2, harbored by Slit2 and Slit3, respectively.29 Our data revealed that Notch activation upregulates miR-218-1, likely by transactivating the Slit2 promoter, in ECs. It has been well established that characteristic metabolic patterns determine EC quiescence,3,12,43,44 and that MYC critically regulates EC metabolism in different contexts.13, 14, 15 Our results demonstrate that miR-218 mediates the effects of Notch activation on EC quiescence by repressing MYC (Figure 7G). The role of Slit2, which is a roundabout guidance receptor (Robo) ligand and regulates sprouting and vessel stability via Robo receptors,45 requires further investigation.
miR-218 is a tumor suppressor that targets a broad range of signaling pathways.46 In ECs, miR-218 has been shown to restrain angiogenesis by downregulating Robo1 or RPTOR independent companion of MTOR, complex 2 (Rictor).30,31 Our transcriptomic analysis showed that miR-218 overexpression in HUVECs exclusively repressed genes involved in cell proliferation and biomass synthesis. These transcriptomic changes pointed to attenuated MYC activity. Indeed, a panel of MYC downstream molecules was downregulated in miR-218-overexpressing HUVECs, and ectopic expression of MYC rescued the miR-218-overexpressing phenotype, confirming that miR-218 targets MYC to repress EC proliferation and metabolism. Our data also confirmed repression of the mTORC1 signature in miR-218-overexpressing HUVECs, consistent with previous findings.31 Reduction in mTORC1 activity may further disfavor MYC activity and reinforce EC quiescence via metabolic control after Notch activation and miR-218 upregulation.47
As the central regulator of cell proliferation, MYC is tightly regulated at multiple levels in cells, including miRNA-mediated regulation.48,49 Interestingly, miR-218 does not attenuate MYC levels by directly targeting MYC mRNA, but by targeting at least three MYC regulators as documented in our experiments. First, miR-218 overexpression slightly reduced the MYC mRNA levels. Several pathways, including Wnt, FOXO1, and NF-κB,12,48 regulate MYC transcription, but we failed to find significant transcriptomic changes in these pathways in miR-218-overexpressing HUVECs, leaving the mechanism for miR-218-mediated MYC transcriptional regulation an open question. The second mechanism of miR-218-mediated MYC downregulation may be a retarded translation. We identified hnRNPA1 as a potential miR-218 target in ECs. hnRNPA1 regulates alternative pre-mRNA splicing, nuclear export of mature mRNAs, and both cap-dependent and IRES-mediated translation initiation.17, 18, 19 The 5′ UTR of the MYC mRNA contains an IRES, which recruits the 40S ribosomal subunit to initiate translation.17,19 Importantly, it has been reported that hnRNPA1 acts as an IRES trans-acting factor (ITAF) of MYC mRNA, and therefore maintains MYC translation when cap-dependent translation is inhibited.21 miR-218 downregulates hnRNPA1 via its 3′ UTR, leading to compromised MYC translation. The third mechanism underlying miR-218-mediated MYC repression is protein degradation. This involves miR-218-mediated repression of EYA3, which promotes MYC degradation. EYA proteins serve as transcriptional co-activators and haloacid dehalogenase-family tyrosine phosphatases. EYA proteins have an unexpected connection with PP2A, the major serine/threonine phosphatase that dephosphorylates many critical cellular molecules, such as AKT, tumor protein p53 (TP53), and catenin beta 1 (CTNNB1).22,23 PP2A consists of a catalytic (C), structural (A), and regulatory/variable B-type subunit, which can be grouped into four subfamilies (B [B55/PR55], B′ [B56/PR61], B″ [B72/PR72], and BΔ [PR93/PR110]) and determines PP2A function. The B56α subunit directs PP2A to MYC, leading to dephosphorylation of Ser62 and proteasome-dependent degradation of MYC. EYA3 binds to B55α, leading to dephosphorylation of MYC Thr58 instead of Ser62, which increases MYC stability.24,25 Therefore, miR-218 downregulation of EYA3 leads to decreased MYC stability. In summary, our data suggest that miR-218 downregulates MYC in a multi-layered manner, which may achieve the regulatory purpose without causing catastrophic alterations in cell signaling (Figure 7G).
Previously, we reported that Notch activation upregulates a group of miRNAs, and one of them, miR-342-5p, represses multiple angiogenic pathways in ECS including VEGFR2 and transforming growth factor beta (TGF-β) signaling.28 In the current study, we showed that miR-218 is also upregulated by Notch activation and represses angiogenesis by inhibiting EC proliferation via MYC. Many miRNAs have been implicated in regulating angiogenesis.29,50 Our studies suggest that Notch activation could upregulate a group of miRNAs that coordinate the effects of Notch signaling to repress angiogenesis via multiple mechanisms. These anti-angiogenic miRNAs could be used in combination for the treatment of angiogenic diseases, such as CNV and cancer.
Materials and methods
Animals
C57BL/6 mice were maintained under specific pathogen-free conditions. P3 pups were intravitreally injected with 0.2 nmol synthetic miR-218 agomiR or antagomiR in 0.5 μL RNA-free phosphate-buffered saline (PBS) in one eye and with nonsense control (NC; RiboBio, Guangzhou, China) in the other. On P5, pups were intraperitoneally (i.p.) injected with BrdU (Sigma-Aldrich, St Louis, MO, USA) at 100 μg/g of body mass or EdU (RiboBio) at 50 μg/g of body mass for 4 h before sacrifice. For Notch signal blockade in vivo, 30 g/kg DAPT (Selleck Chemicals, Houston, TX, USA) was subcutaneously injected on P1 and P3. The laser-induced CNV model was established in 4-week-old mice as previously described.8 Mice received an intravitreal injection of miR-218 agomiR or Cy3-labeled NC on day 0.5 after photocoagulation and were sacrificed on day 7. Retinal pigment epithelia (RPE)-choroid-sclera complexes were dissected after perfusion with PBS. All animal experiments followed the guidelines issued by the Animal Experiment Administration Committee of the Fourth Military Medical University.
Cell culture and transfection
HUVECs were cultured in EC medium (ECM; ScienCell, San Diego, CA, USA) supplemented with 5% fetal bovine serum (FBS), EC growth supplements (ECGS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells between passages two and six were used. MRC5 (human embryonic lung fibroblasts) and HEK293T cell lines (American Type Culture Collection [ATCC], Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA) containing 2 mM L-glutamine and 10% FBS.
Adenoviruses expressing miR-218 (AdmiR-218, from pGV317-miR-218) was purchased from Genechem (Shanghai, China), and AdMYC or AdNIC were purchased from HanBio (Shanghai, China). All adenovirus-encapsulated plasmids contained a green fluorescent protein (GFP) sequence to monitor infection efficiency. HUVECs were infected with adenovirus at a multiplicity of infection (MOI) of 30 according to standard procedures. To knockdown miR-218, we infected HUVECs with a lentivirus expressing miR-218 inhibitor (Anti-218; Genechem) at an MOI of 30. DAPT and the proteasome inhibitor MG-132 (Selleck Chemicals) were used at concentrations of 25 μmol/L and 50 μmol/L, respectively, with dimethyl sulfoxide (DMSO) as a control. For RNA and protein decay assays, HUVECs were treated with ActD (Selleck Chemicals) at 5 μmol/mL or CHX (Selleck Chemicals) at 50 μg/mL, respectively, for the stated period of time. To coat culture dishes with soluble Dll4, we added recombinant Dll4 (Sino-Biological, Beijing, China) solution or PBS to the culture wells (0.5 μg) and incubated them for 12 h at 4°C. The solutions were discarded, and HUVECs (6 × 104 cells/well) were seeded after washing with PBS. In some experiments, DAPT or DMSO was added after cell adherence and cultured for 48 h.
Immunostaining
Retinas were dissected and post-fixed in 4% paraformaldehyde (PFA) overnight at 4°C, blocked, and permeabilized in PBS containing 1% bovine serum albumin (BSA) and 0.5% Triton X-100 overnight. Samples were incubated with isolectin B4 (IB4) (1:100) or anti-Erg (1:200) in PBS containing 1% BSA and 0.5% Triton X-100 overnight at 4°C, followed by incubation with Alexa Fluor 647-conjugated goat anti-rabbit immunoglobulin G (IgG; H+L) secondary antibody (1:400) in PBS. Retinas were then treated with 2 mol/L HCl for 30 min at 37°C, washed with 0.1 mol/L sodium tetraborate solution, and blocked in PBS containing 1% BSA for 2 h at room temperature (RT). Anti-BrdU (1:400) was incubated in PBS containing 1% BSA and 0.5% Triton X-100, followed by incubation with Alexa Fluor 488-conjugated goat anti-rat IgG (H+L) secondary antibody (1:400). EdU staining was performed according to the manufacturer’s instructions. Each step was washed three times with PBS for 10 min. Retinas were flat-mounted under a dissecting microscope (Olympus, Tokyo, Japan), examined, and photographed under a confocal laser scanning microscope (FV1000, Olympus). Antibody reagents are listed in Table S2.
Reporter assay
The human Slit2 promoter (−2,000 to +1 bp of human Slit2 gene, NC_000004.12:20251905-20620561) was amplified by PCR with HUVEC DNA as a template, and subcloned into the pGL3-basic plasmid (Promega, Madison, WI, USA) to construct pGL-Slit2. The 5′ and 3′ human MYC UTRs (NM_002467.5), hnRNPA1 3′ UTR (NM_002136.3), and human EYA3 3′ UTR (NM_001990.3) were amplified from DNA or cDNA derived from HUVECs using the primers listed in Table S3. Amplified fragments were cloned into the pGL3-promoter plasmid (Promega) to construct pGL-MycUTR5, pGL-MycUTR3, pGL-hnRNPAUTR3, and pGL-EYAUTR3, respectively.
HEK293T cells were co-transfected with pGL-Slit2 (100 ng), phRL-TK (5 ng), and pEFBOS-NIC (0, 50, 100 ng, balanced by pEFBOS-neo).51 In other cases, HUVECs were co-transfected with 100 ng pGL-MycUTR5, pGL-MycUTR3, pGL-hnRNPAUTR3, pGL-EYAUTR3, together with phRL-TK (5 ng) and pGV317-miR-218 (0, 50, 100 ng, Genechem). Cells were lysed 48 h after transfection, and firefly and Renilla luciferase activities were analyzed using a Dual-Luciferase Reporter Assay System (Promega).
Cell proliferation assay
EdU incorporation was performed after infection with AdmiR-218 or AdCtrl for 24, 48, or 72 h as previously described.28 For cell-cycle analysis, HUVECs were cultured in ECM containing 0.5% FBS for 24 h, then cultured in complete ECM for another 24 h. Cells were trypsinized and fixed in 75% ethanol at 4°C for 2 h, then incubated with PBS containing 0.2% Triton X-100, 3,000 U/mL RNase A (Sigma-Aldrich), and 50 μg/mL propidium iodide (PI; P4170, Sigma-Aldrich) for 30 min at 37°C. Cells were analyzed by fluorescence-activated cell sorting (FACS) using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Data were analyzed using ModFitLT software (Verity Software House, Topsham, ME, USA). For cell counting, HUVECs were digested and resuspended in complete ECM. The number of cells per well was quantified using a counting chamber.
Cell apoptosis assay
Cell apoptosis was detected using a DNA fragmentation imaging kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. DNA fragmentation was measured using terminal deoxynucleotidyl transferase and fluorescein-labeled dUTP (TUNEL assay). Briefly, HUVECs were fixed in 4% PFA for 10 min at RT, and then incubated with the reaction solution at 37°C for 1 h following the nuclei dye mixture at RT for 5 min. For the positive control, HUVECs were pre-treated with DNase I at RT for 10 min before incubation with the reaction solution. The cells were analyzed using a fluorescence microscope (BX51, Olympus).
Lumen formation assay
HUVECs (1 × 105 cells/well) were seeded in 96-well plates precoated with 200 μL Matrigel (1:1, BD Biosciences) and incubated at 37°C for 4 h. The number of branches, loops, and total length of the cell cords were measured.
Fibrin beads sprouting assay
HUVECs were incubated with Cytodex 3 microcarrier beads (Sigma-Aldrich) in EGM-2 medium (Lonza, Basel, Switzerland) at 37°C for 4 h, then cultured overnight. The beads were embedded in fibrinogen (Sigma-Aldrich) containing 0.625 U/mL thrombin (Sigma-Aldrich) the next day at a density of 100 beads/mL in a 48-well plate, and 0.5 mL EGM-2 medium was added with MRC5 (5 × 103 cells/well). HUVECs were allowed to sprout for 4 days. Sprouting was quantified by measuring the number and length of the sprouts.
RNA-seq analysis
HUVECs infected with AdmiR-218 or AdCtrl for 48 h were analyzed by RNA-seq. RNA extraction and sequencing were performed using a custom service provided by Megagenomics (Beijing, China) using the Illumina HiSeq3000 platform. Raw data reported in this paper have been deposited in the Genome Sequence Archive of the BIG Data Center (Beijing, China; accession number CRA002986; http://bigd.big.ac.cn/gsa). Bioinformatics analysis was performed using OmicShare (https://www.omicshare.com/tools).
Quantitative real-time PCR
Total RNA from HUVECs or retinal vasculature was extracted using TRIzol reagent (Invitrogen), and cDNA was synthesized using a reverse transcription kit (Takara Dalian, Dalian, China). Real-time PCR was performed using a TB Green Premix Ex Taq kit (Takara) and a QuantStudio 3 real-time PCR system (Life Technologies, Waltham, MA, USA). Gene expression levels are reported as relative fold-change, with RPS18 as an internal control.52 The miRNA levels were determined using a miRNA quantitative real-time PCR kit (Takara), with U6 RNA as an internal control. The primer sequences are listed in Table S3.
Western blotting
HUVECs or retinal vasculature were lysed in radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China) containing phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich). To detect the cytoplasmic and nuclear p65 protein levels, we performed cytoplasmic and nuclear extraction using an extraction kit (Beyotime) according to the manufacturer’s instructions. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto polyvinylidene fluoride (PVDF) membranes. Membranes were incubated with primary antibodies, then horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG or horse anti-mouse IgG secondary antibody (1:2,000, Cell Signaling Technology, Boston, MA, USA). Each step was followed by three washes in PBS containing 0.1% Tween 20 for 10 min. Bands were visualized using an enhanced chemiluminescence (ECL) system (Clinx Science Instruments, Shanghai, China). Quantification was performed using ImageJ2x software (Rawak Software, Stuttgart, Germany). The antibodies used are listed in Table S2.
Retinal vasculature assessment
Assessment of the retinal vessels was based on a published protocol.53 For quantitation of vascular density and proliferative ECs, the equivalent area of the retina with adequate capillary plexus (to avoid vessel-free zone) at the leading edge was selected as a “field” between different samples. Vascular density was calculated as the number of capillary loops, and proliferative ECs were calculated as the number of BrdU+Erg+ or EdU+Erg+ cells per field.
Statistical analysis
Statistical analysis was performed using Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA) and GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). All quantitative data are presented as the mean ± standard deviation (SD). Statistical significance was calculated using Student’s t test or one-way analysis of variance (ANOVA). Statistical significance was set at p < 0.05.
Acknowledgments
This study was supported by The National Natural Science Foundation of China (grant numbers 31730041, 91339115, 31671523, 81670863, 81900870) and the Natural Science Foundation of Guangdong Province (grant number 2018A030313571).
Author contributions
H.H., X.-C.Y., and Y.-S.W. designed the experiments and prepared the manuscript. J.-X.S., G.-R.D., Z.-Y.Y., and L.L. performed the experiments and collected data. J.-L.D., B.R., M.-H.L., and T.-F.C. assisted with experiments and data collection. X.-Y.X. and J.-J.C. assisted with the data collection.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.07.023.
Contributor Information
Yu-Sheng Wang, Email: wangys003@126.com.
Xian-Chun Yan, Email: yanxianchun@163.com.
Hua Han, Email: huahan_biochem8796@163.com.
Supplemental information
References
- 1.Eilken H.M., Adams R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010;22:617–625. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Godo S., Shimokawa H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017;37:e108–e114. doi: 10.1161/ATVBAHA.117.309813. [DOI] [PubMed] [Google Scholar]
- 3.Li X., Sun X., Carmeliet P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 2019;30:414–433. doi: 10.1016/j.cmet.2019.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Schlereth K., Weichenhan D., Bauer T., Heumann T., Giannakouri E., Lipka D., Jaeger S., Schlesner M., Aloy P., Eils R. The transcriptomic and epigenetic map of vascular quiescence in the continuous lung endothelium. eLife. 2018;7:e34423. doi: 10.7554/eLife.34423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siebel C., Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017;97:1235–1294. doi: 10.1152/physrev.00005.2017. [DOI] [PubMed] [Google Scholar]
- 6.Margadant C. Positive and negative feedback mechanisms controlling tip/stalk cell identity during sprouting angiogenesis. Angiogenesis. 2020;23:75–77. doi: 10.1007/s10456-020-09706-0. [DOI] [PubMed] [Google Scholar]
- 7.Rohlenova K., Goveía J., García-Caballero M., Subramanian A., Kalucka J., Treps L., Falkenberg K.D., de Rooij L.P.M.H., Zheng Y., Lin L. Single-cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. 2020;31:862–877.e14. doi: 10.1016/j.cmet.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Dou G.R., Wang Y.C., Hu X.B., Hou L.H., Wang C.M., Xu J.F., Wang Y.S., Liang Y.M., Yao L.B., Yang A.G., Han H. RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J. 2008;22:1606–1617. doi: 10.1096/fj.07-9998com. [DOI] [PubMed] [Google Scholar]
- 9.Noseda M., Chang L., McLean G., Grim J.E., Clurman B.E., Smith L.L., Karsan A. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression. Mol. Cell. Biol. 2004;24:8813–8822. doi: 10.1128/MCB.24.20.8813-8822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rostama B., Turner J.E., Seavey G.T., Norton C.R., Gridley T., Vary C.P., Liaw L. DLL4/Notch1 and BMP9 interdependent signaling induces human endothelial cell quiescence via P27KIP1 and Thrombospondin-1. Arterioscler. Thromb. Vasc. Biol. 2015;35:2626–2637. doi: 10.1161/ATVBAHA.115.306541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serra H., Chivite I., Angulo-Urarte A., Soler A., Sutherland J.D., Arruabarrena-Aristorena A., Ragab A., Lim R., Malumbres M., Fruttiger M. PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat. Commun. 2015;6:7935. doi: 10.1038/ncomms8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 13.Wilhelm K., Happel K., Eelen G., Schoors S., Oellerich M.F., Lim R., Zimmermann B., Aspalter I.M., Franco C.A., Boettger T. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Testini C., Smith R.O., Jin Y., Martinsson P., Sun Y., Hedlund M., Sáinz-Jaspeado M., Shibuya M., Hellström M., Claesson-Welsh L. Myc-dependent endothelial proliferation is controlled by phosphotyrosine 1212 in VEGF receptor-2. EMBO Rep. 2019;20:e47845. doi: 10.15252/embr.201947845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu P., Wilhelm K., Dubrac A., Tung J.K., Alves T.C., Fang J.S., Xie Y., Zhu J., Chen Z., De Smet F. FGF-dependent metabolic control of vascular development. Nature. 2017;545:224–228. doi: 10.1038/nature22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancho O., Herranz D. The MYC Enhancer-ome: long-range transcriptional regulation of MYC in cancer. Trends Cancer. 2018;4:810–822. doi: 10.1016/j.trecan.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meristoudis C., Trangas T., Lambrianidou A., Papadopoulos V., Dimitriadis E., Courtis N., Ioannidis P. Systematic analysis of the contribution of c-myc mRNA constituents upon cap and IRES mediated translation. Biol. Chem. 2015;396:1301–1313. doi: 10.1515/hsz-2015-0190. [DOI] [PubMed] [Google Scholar]
- 18.Hinnebusch A.G., Ivanov I.P., Sonenberg N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016;352:1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y., Sharma A., Wu H., Lichtenstein A., Gera J. Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J. Biol. Chem. 2005;280:10964–10973. doi: 10.1074/jbc.M407874200. [DOI] [PubMed] [Google Scholar]
- 20.Roy R., Huang Y., Seckl M.J., Pardo O.E. Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip. Rev. RNA. 2017;8:e1431. doi: 10.1002/wrna.1431. [DOI] [PubMed] [Google Scholar]
- 21.Jo O.D., Martin J., Bernath A., Masri J., Lichtenstein A., Gera J. Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J. Biol. Chem. 2008;283:23274–23287. doi: 10.1074/jbc.M801185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas L.R., Tansey W.P. Proteolytic control of the oncoprotein transcription factor Myc. Adv. Cancer Res. 2011;110:77–106. doi: 10.1016/B978-0-12-386469-7.00004-9. [DOI] [PubMed] [Google Scholar]
- 23.Seshacharyulu P., Pandey P., Datta K., Batra S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013;335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnold H.K., Sears R.C. A tumor suppressor role for PP2A-B56alpha through negative regulation of c-Myc and other key oncoproteins. Cancer Metastasis Rev. 2008;27:147–158. doi: 10.1007/s10555-008-9128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L., Zhou H., Li X., Vartuli R.L., Rowse M., Xing Y., Rudra P., Ghosh D., Zhao R., Ford H.L. Eya3 partners with PP2A to induce c-Myc stabilization and tumor progression. Nat. Commun. 2018;9:1047. doi: 10.1038/s41467-018-03327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo W., Garcia-Gonzalez I., Fernández-Chacón M., Casquero-Garcia V., Sanchez-Muñoz M.S., Mühleder S., Garcia-Ortega L., Andrade J., Potente M., Benedito R. Arterialization requires the timely suppression of cell growth. Nature. 2021;589:437–441. doi: 10.1038/s41586-020-3018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kir D., Schnettler E., Modi S., Ramakrishnan S. Regulation of angiogenesis by microRNAs in cardiovascular diseases. Angiogenesis. 2018;21:699–710. doi: 10.1007/s10456-018-9632-7. [DOI] [PubMed] [Google Scholar]
- 28.Yan X.C., Cao J., Liang L., Wang L., Gao F., Yang Z.Y., Duan J.L., Chang T.F., Deng S.M., Liu Y. miR-342-5p is a Notch downstream molecule and regulates multiple angiogenic pathways including Notch, vascular endothelial growth factor and transforming growth factor β signaling. J. Am. Heart Assoc. 2016;5:e003042. doi: 10.1161/JAHA.115.003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Small E.M., Sutherland L.B., Rajagopalan K.N., Wang S., Olson E.N. MicroRNA-218 regulates vascular patterning by modulation of Slit-Robo signaling. Circ. Res. 2010;107:1336–1344. doi: 10.1161/CIRCRESAHA.110.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X., Dong J., He Y., Zhao M., Liu Z., Wang N., Jiang M., Zhang Z., Liu G., Liu H. miR-218 inhibited tumor angiogenesis by targeting ROBO1 in gastric cancer. Gene. 2017;615:42–49. doi: 10.1016/j.gene.2017.03.022. [DOI] [PubMed] [Google Scholar]
- 31.Guan B., Wu K., Zeng J., Xu S., Mu L., Gao Y., Wang K., Ma Z., Tian J., Shi Q. Tumor-suppressive microRNA-218 inhibits tumor angiogenesis via targeting the mTOR component RICTOR in prostate cancer. Oncotarget. 2017;8:8162–8172. doi: 10.18632/oncotarget.14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fish J.E., Wythe J.D., Xiao T., Bruneau B.G., Stainier D.Y., Srivastava D., Woo S. A Slit/miR-218/Robo regulatory loop is required during heart tube formation in zebrafish. Development. 2011;138:1409–1419. doi: 10.1242/dev.060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torres-Berrío A., Nouel D., Cuesta S., Parise E.M., Restrepo-Lozano J.M., Larochelle P., Nestler E.J., Flores C. MiR-218: a molecular switch and potential biomarker of susceptibility to stress. Mol. Psychiatry. 2020;25:951–964. doi: 10.1038/s41380-019-0421-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tie J., Pan Y., Zhao L., Wu K., Liu J., Sun S., Guo X., Wang B., Gang Y., Zhang Y. MiR-218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet. 2010;6:e1000879. doi: 10.1371/journal.pgen.1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeong H.W., Hernández-Rodríguez B., Kim J., Kim K.P., Enriquez-Gasca R., Yoon J., Adams S., Schöler H.R., Vaquerizas J.M., Adams R.H. Transcriptional regulation of endothelial cell behavior during sprouting angiogenesis. Nat. Commun. 2017;8:726. doi: 10.1038/s41467-017-00738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goyal D., Goyal R. Angiogenic transformation in human brain micro endothelial cells: whole genome DNA methylation and transcriptomic analysis. Front. Physiol. 2019;10:1502. doi: 10.3389/fphys.2019.01502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao Q., Eichten A., Parveen A., Adler C., Huang Y., Wang W., Ding Y., Adler A., Nevins T., Ni M. Single-cell transcriptome analyses reveal endothelial cell heterogeneity in tumors and changes following antiangiogenic treatment. Cancer Res. 2018;78:2370–2382. doi: 10.1158/0008-5472.CAN-17-2728. [DOI] [PubMed] [Google Scholar]
- 38.Farrell A.S., Sears R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014;4:a014365. doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Vlachos I.S., Vergoulis T., Reczko M., Filippidis C., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–W173. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehling M., Adams S., Benedito R., Adams R.H. Notch controls retinal blood vessel maturation and quiescence. Development. 2013;140:3051–3061. doi: 10.1242/dev.093351. [DOI] [PubMed] [Google Scholar]
- 41.Tetzlaff F., Fischer A. Control of blood vessel formation by Notch signaling. Adv. Exp. Med. Biol. 2018;1066:319–338. doi: 10.1007/978-3-319-89512-3_16. [DOI] [PubMed] [Google Scholar]
- 42.Phng L.K., Potente M., Leslie J.D., Babbage J., Nyqvist D., Lobov I., Ondr J.K., Rao S., Lang R.A., Thurston G., Gerhardt H. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev. Cell. 2009;16:70–82. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jabs M., Rose A.J., Lehmann L.H., Taylor J., Moll I., Sijmonsma T.P., Herberich S.E., Sauer S.W., Poschet G., Federico G. Inhibition of endothelial Notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation. 2018;137:2592–2608. doi: 10.1161/CIRCULATIONAHA.117.029733. [DOI] [PubMed] [Google Scholar]
- 44.Han Y.M., Bedarida T., Ding Y., Somba B.K., Lu Q., Wang Q., Song P., Zou M.H. β-hydroxybutyrate prevents vascular senescence through hnRNP A1-mediated upregulation of Oct4. Mol. Cell. 2018;71:1064–1078. doi: 10.1016/j.molcel.2018.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai C., Gong Q., Cheng Y., Su G. Regulatory mechanisms of Robo4 and their effects on angiogenesis. Biosci. Rep. 2019;39 doi: 10.1042/BSR20190513. BSR20190513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu Y.F., Zhang L., Waye M.M., Fu W.M., Zhang J.F. MiR-218 mediates tumorigenesis and metastasis: Perspectives and implications. Exp. Cell Res. 2015;334:173–182. doi: 10.1016/j.yexcr.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 47.Allen-Petersen B.L., Risom T., Feng Z., Wang Z., Jenny Z.P., Thoma M.C., Pelz K.R., Morton J.P., Sansom O.J., Lopez C.D. Activation of PP2A and inhibition of mTOR synergistically reduce MYC signaling and decrease tumor growth in pancreatic ductal adenocarcinoma. Cancer Res. 2019;79:209–219. doi: 10.1158/0008-5472.CAN-18-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dang C.V. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackstadt R., Hermeking H. MicroRNAs as regulators and mediators of c-MYC function. Biochim. Biophys. Acta. 2015;1849:544–553. doi: 10.1016/j.bbagrm.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Leone P., Buonavoglia A., Fasano R., Solimando A.G., De Re V., Cicco S., Vacca A., Racanelli V. Insights into the regulation of tumor angiogenesis by micro-RNAs. J. Clin. Med. 2019;8:2030. doi: 10.3390/jcm8122030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu Y.Y., Fu L.A., Li S.Z., Chen Y., Li J.C., Han J., Liang L., Li L., Ji C.C., Zheng M.H., Han H. Hif-1α and Hif-2α differentially regulate Notch signaling through competitive interaction with the intracellular domain of Notch receptors in glioma stem cells. Cancer Lett. 2014;349:67–76. doi: 10.1016/j.canlet.2014.03.035. [DOI] [PubMed] [Google Scholar]
- 52.Hellemans J., Vandesompele J. Selection of reliable reference genes for RT-qPCR analysis. Methods Mol. Biol. 2014;1160:19–26. doi: 10.1007/978-1-4939-0733-5_3. [DOI] [PubMed] [Google Scholar]
- 53.Pitulescu M.E., Schmidt I., Benedito R., Adams R.H. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 2010;5:1518–1534. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.