

Abstract
Dravet syndrome is a genetic encephalopathy characterized by severe epilepsy combined with motor, cognitive, and behavioral abnormalities. Current antiepileptic drugs achieve only partial control of seizures and provide little benefit on the patient’s neurological development. In >80% of cases, the disease is caused by haploinsufficiency of the SCN1A gene, which encodes the alpha subunit of the Nav1.1 voltage-gated sodium channel. Novel therapies aim to restore SCN1A expression in order to address all disease manifestations. We provide evidence that a high-capacity adenoviral vector harboring the 6-kb SCN1A cDNA is feasible and able to express functional Nav1.1 in neurons. In vivo, the best biodistribution was observed after intracerebral injection in basal ganglia, cerebellum, and prefrontal cortex. SCN1A A1783V knockin mice received the vector at 5 weeks of age, when most neurological alterations were present. Animals were protected from sudden death, and the epileptic phenotype was attenuated. Improvement of motor performance and interaction with the environment was observed. In contrast, hyperactivity persisted, and the impact on cognitive tests was variable (success in novel object recognition and failure in Morris water maze tests). These results provide proof of concept for gene supplementation in Dravet syndrome and indicate new directions for improvement.
Keywords: SCN1A, Nav1.1., Dravet syndrome, epilepsy, encephalopathy, adenovirus, gene therapy
Graphical abstract
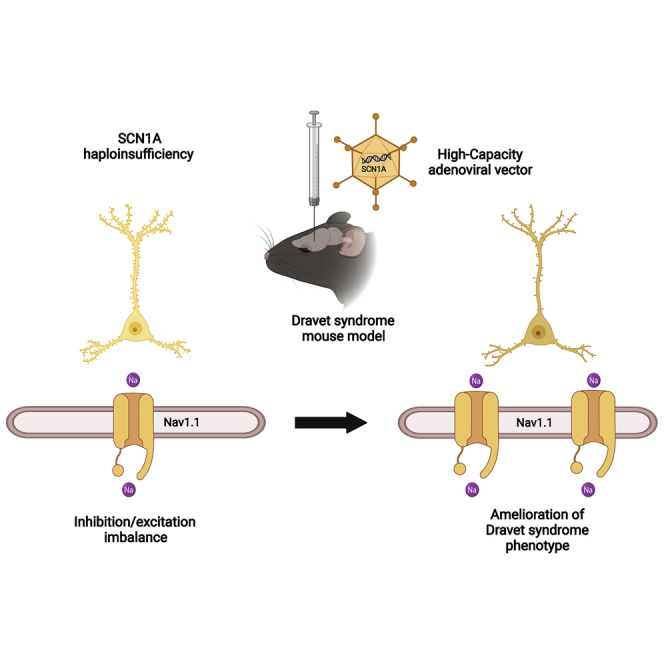
Haploinsufficiency of the SCN1A gene is the most frequent etiology of Dravet syndrome, a severe genetic encephalopathy. Delivery of the 6-kb cDNA into the brain of a mouse model with a high-capacity adenoviral vector achieves improvement of survival, brain electrophysiology, and motor function, despite limited biodistribution of the vector.
Introduction
Dravet syndrome (DS; also known as severe myoclonic epilepsy of infancy, SMEI) is a genetic encephalopathy caused in nearly 90% of cases by de novo mutations in one of the two SCN1A alleles (OMIM 607208).1 Studies in large populations estimate an incidence as high as 1:15,000 births.2 SCN1A encodes the α subunit of a voltage-gated sodium channel (Nav1.1) that is essential for the function of inhibitory interneurons in the brain.3,4 Whereas complete SCN1A loss is supposed to be embryonically lethal in humans, haploinsufficiency alters the excitatory/inhibitory equilibrium in the brain, causing a wide variety of clinical manifestations. The first symptoms appear typically at 4–8 months of age and consist of febrile seizures, which are usually refractory to antiepileptic drugs and can lead to status epilepticus (SE). During the next years a variety of febrile and afebrile seizures, myoclonic seizures, and absences increase in frequency, and a delay in psychomotor development becomes evident.5 Seizures become less frequent in late childhood and adolescence, but the risk of sudden unexpected death in epilepsy (SUDEP) remains elevated throughout the patient’s life. Mortality rate of DS is estimated as 10%–15% in industrialized countries.6 Despite intense pharmacological and support therapies, motor, intellectual, and behavioral alterations cause a dramatic deterioration in the quality of life of patients and their families. Apart from the development of improved antiepileptic drugs, intense efforts are now focused on the stimulation of Nav1.1 expression and function in order to address all disease comorbidities. Considering that DS patients harbor one healthy copy of the SCN1A gene and therefore produce a reduced amount of the Nav1.1 channel, screenings of small-molecule activators are ongoing. The main challenge is to find drugs with enough potency and selectivity to avoid unwanted activation of other channels controlling vital functions such as heart beat. Other approaches try to stimulate the function of the healthy allele by means of antisense oligonucleotides7 or stable expression of synthetic transcriptional activators delivered by adeno-associated vectors (AAVs).8 Gene supplementation with a correct copy of the SCN1A coding sequence offers the possibility of obtaining long-lasting Nav1.1 restoration without the need of expressing exogenous proteins. However, this is an important challenge because SCN1A is a large gene, with a 6-kb-long cDNA, and it is prone to rearrangements in the context of plasmids.9 This size exceeds the cloning capacity of AAV vectors, which are nowadays the most advanced vector platform for in vivo gene therapy in humans. Therefore, delivery of SCN1A requires alternative vectors with enhanced cloning capacity, high genome stability, and the ability to transduce neurons in vivo. We work on the hypothesis that high-capacity adenoviral vectors (HC-AdVs), a different viral platform with no close structural or antigenic similarities with AAVs, can meet these requirements. In contrast with early versions, HC-AdVs (also known as Helper-Dependent or “gutless”) are devoid of all viral coding genes.10,11 This feature is critical for the properties of these vectors. On one hand, it allows a cloning capacity of up to 37 kb, sufficient to accommodate most human cDNAs. On the other hand, it avoids the possibility of residual expression of viral genes in the transduced cells. As a result, cellular immune responses against these cells are blunted,12 and the transgenes remain in an episomal state for years after a single vector administration in tissues with slow cellular turnover such as brain or liver.13 Of note, HC-AdVs contain a very stable double-stranded DNA genome that is transcriptionally active soon after infection.
In this work we provide proof of concept that HC-AdVs are a feasible platform for supplementation of SCN1A in the brain. The cDNA sequence was codon-optimized to guarantee plasmid stability, and it was incorporated in a prototypic HC-AdV vector under the control of a ubiquitous promoter. Intracerebral administration in adolescent mice carrying a pathogenic SCN1A mutation was well tolerated, obtained elevation of functional Nav1.1, and showed partial restoration of the DS phenotype. This information can guide further improvements in vector specificity and biodistribution in order to maximize the therapeutic effect.
Results
Codon optimization increases the stability of the SCN1A cDNA and allows expression of a functional Nav1.1 channel
Plasmids containing the SCN1A cDNA are known to be prone to rearrangements upon amplification in E. coli. The use of recombination-free bacterial strains and growth at low temperature (30°C) has been proposed to overcome this problem.9 However, when the synthesis of this sequence was ordered from a commercial source, they could only provide a small amount of plasmid, which already contained a substantial fraction of copies with altered restriction pattern (not shown). In contrast, a codon-optimized version of the sequence could be successfully amplified using the recommended conditions, and it allowed subcloning following standard molecular biology techniques. For initial verification of expression and function, SCN1A expression was placed under the control of three ubiquitous regulatory sequences: the citomegalovirus (CMV) early enhancer and promoter; the hybrid CAG promoter (early CMV enhancer, chicken β-actin promoter with first intron and rabbit β-globin splice acceptor); and the human Elongation Factor 1α (EF1α). In the latter plasmid, SCN1A and green fluorescent protein (GFP) are co-expressed by virtue of an internal ribosomal entry site (IRES) (Figure 1A). Transfection of plasmids in cells with low endogenous Nav1.1 levels (HEK-293) confirmed efficient production of the SCN1A mRNA by qRT-PCR using primers specific for the codon-optimized sequence (Figure 1B). A robust increase of the Nav1.1 channel content was also detected by western blot in membrane-enriched cell fractions (Figure 1C) and immunofluorescence (IF) (Figure 1D). The function of the transgene was confirmed by patch clamp in GFP+ HEK-293 cells transfected with the bicistronic plasmid pEF-SCN1A-GFP (not shown).
Figure 1.

Plasmids encoding the codon-optimized SCN1A cDNA achieve efficient expression of Nav1.1. in HEK-293 cells
(A) Schematic representation of the plasmids expressing SCN1A (not drawn to scale). The plasmids were transfected in HEK-293 cells. The pCDNA3 empty plasmid was used as a negative control. Transgene expression was analyzed 48 h after transfection. (B). Quantification of SCN1A mRNA was performed by qRT-PCR. The values correspond to copies of SCN1A mRNA per cell. (C). Detection of Nav1.1 protein by western blot in membrane-enriched protein extracts. GAPDH in total extracts is shown as a housekeeping control. (D). Detection of Nav1.1 by IF. Bars indicate averages for each group, and individual values are represented by small circles. IRES, internal ribosomal entry site; pA, polyadenylation signal. ∗∗p < 0.01, Kruskal-Wallis with Dunn’s multiple comparison test. Scale bar, 25 μm.
HC-AdVs vectors expressing SCN1A are feasible
As a proof of concept for vector feasibility, we incorporated the CAG-SCN1A or EF-SCN1A-GFP expression cassette into plasmids containing the genome of a HC-AdV vector. Rescue of vector particles was performed by transfection of the pro-viral plasmids in packaging cells, followed by infection with a helper virus (HV). Subsequent steps of amplification are based on co-infection of vectors and HVs in a growing number of cells, as described previously.14 During this process, genome stability is crucial to withstand multiple rounds of replication, and the influence of transgene expression can be detrimental for the cells. Despite these potential drawbacks, we found that both prototypic vectors (HCA-CAG-SCN1A and HCA-EF-SCN1A-GFP, genomes depicted in Figure 2A) could be produced using the standard procedure. The yields were moderate (1.05 × 1012 ± 0.57 × 1012 viral particles [vp]/mL; 6.25 × 1011 ± 8.4 × 1011 viral genomes [vg]/mL; 2.27 × 1011 ± 3.08 × 1011 infectious units [iu]/mL), possibly because of over-production of Nav1.1 channels in the packaging cells. To further validate the function of the transgene product, sodium currents were analyzed by patch clamp in HEK-293 cells infected with the HCA-EF-SCN1A-GFP vector. Figures 2B and 2C show robust sodium currents in infected cells. Importantly, these currents were modulated by the selective Nav1.1 opener Hm1a, resulting in marked reduction in channel inactivation (Figure 2B).15,16 Furthermore, voltage dependence of activation (Half-activation voltage [V1/2)] of 27.7 ± 2 mV) and fast inactivation (V1/2 of −59.6 ± 2.2 mV) (Figure 2D) were similar to previous reports in HEK-293 cells transfected with human Nav1.1 expression plasmids.17 These vectors were used to infect the neuroblastoma-derived cell line SH-SY5Y, as well as primary neuronal cultures from mouse. In both cases, we found a dose-dependent increase of SCN1A mRNA (Figure 3A) and Nav1.1 protein (Figure 3B). The vector containing the CAG promoter obtained higher expression levels, consistent with its reported suitability for transgene expression in the brain.18, 19, 20, 21 This concept is supported by our in vitro data using luciferase reporter plasmids, showing higher promoter activity of CAG versus EF1α in neuronal cultures (Figure S1). As expected, the HCA-CAG-SCN1A vector was able to transduce all cell phenotypes in the primary culture, including those presenting astroglial and inhibitory and excitatory neuronal markers (Figure S2).
Figure 2.

Functional validation of the SCN1A transgene
(A) Schematic representation of the HCA-CAG-SCN1A and HCA-EF-SCN1A-GFP genomes. (B) Representative sodium current traces from HEK-293 cells infected with HCA-EF-SCN1A-GFP before (left) and after addition of the selective Nav1.1 opener Hm1a (50 nM, right). (C) Average current densities (±SD) of cells expressing HCA-EF-SCN1A-GFP (filled symbols, n = 10) and uninfected cells (empty symbols, n = 4). (D) Voltage dependence of activation (right curves: V1/2 of −27.7 ± 2 mV) and the voltage dependence for steady-state fast inactivation (left curves, V1/2 of −59.6 ± 2.2 mV).
Figure 3.

Nav1.1 expression from HC-AdV vectors
SH-SY5Y cells and primary mouse neurons were infected with the vectors at the indicated multiplicities of infection (MOIs), and Nav1.1 was detected by qRT-PCR (A) and IF (B). Scale bars: 50 μm. Bars indicate averages for each group, and individual values are represented by small circles. ITR, inverted terminal repeats; pA, polyadenylation signal; Ѱ, packaging signal. ∗p < 0.05 Kruskal-Walliswith Dunn’s multiple comparisons test.
CAG and EF1α promoters obtain similar kinetics of transgene expression in mouse brain
Since the CAG promoter is a hybrid consisting of viral and eukaryotic sequences, some concerns about in vivo silencing have been raised in organs such as the liver.22 To compare the stability of transgene expression controlled by the CAG and EF1α promoters in the brain, we used HC-AdV vectors encoding the reporter gene luciferase (HCA-CAG-Luc and HCA-EF-Luc, respectively). The vectors were administered by stereotaxic injection in the brain of in wild-type (WT) C57BL/6 mice (basal ganglia (BG) and cerebellum (Cb), bilaterally), and luciferase expression was monitored by bioluminescence imaging (BLI) over >3 months. In both cases, we observed a biphasic pattern of light emission, with a rapid decay during the first 4 weeks and then a prolonged stabilization stage (Figure 4).
Figure 4.

CAG and EF1α promoters obtain similar kinetics of transgene expression in mouse brain
The HCA-CAG-Luc and HCA-EF-Luc vectors were administered by stereotaxic injection in basal ganglia and cerebellum of C57BL/6 mice (2 μL/injection at 6.5 × 108 vg/μL, bilateral). Luciferase activity was quantified at the indicated times by BLI after intraperitoneal administration of the substrate D-luciferin. The graph represents average light emission (±SEM) from the head.
This phenomenon has been previously observed after intracerebral12 and systemic23, 24, 25 administration of HC-AdVs. It may reflect the initial transduction of heterogeneous cell populations with different lifespans or the influence of transient, vector-related inflammation. In our case, the result is compatible with long-term expression from neurons, using both promoters.
Basal ganglia and cerebellum are favorable locations for AdV injection
Based on the previous data, the HCA-CAG-SCN1A vector was selected to evaluate the therapeutic potential of SCN1A supplementation in a DS mouse model. Considering the multifocal epileptogenic origin of seizures in DS26,27 and the global imbalance of inhibition/excitation accounting for other comorbidities,28,29 the need of widespread transgene expression is predicted. To determine the optimal route of administration, biodistribution studies were carried out in WT mice. To this end, we produced an E1/E3-deleted AdV vector expressing the fusion protein GFP-Luciferase under the control of the CAG promoter (Ad-CAG-GFPLuc). Since E1/E3-deleted and HC-AdV vectors share the same capsid structure, this is an efficient way to study the access of the vectors to different tissues. The GFP-Luciferase reporter combines the sensitivity and high dynamic range of luciferase quantification with the possibility of identifying individual transduced cells upon histological examination using epifluorescence or immunohistochemistry/IF techniques. The vector was administered by stereotaxic injection in the brain of 5-week-old mice, using bilateral coordinates specific for the following structures: cortex (Ctx: prefrontal, somatosensory, and visual); hippocampus (HC); bBG (audate-putamen, in the interface with globus pallidum); Cb; a combination of the latter two regions (BG/Cb); and the combination of them with the pre-frontal cortex (BG/Cb/pCtx). See Materials and methods for details. Stereotaxic coordinates are displayed in Table S1. Two days later, light emission from the head was visualized by BLI. However, in vivo imaging and quantification of luciferase activity from the brain gives poor spatial resolution because the light is heavily quenched and distorted by the skull (Figure S3). Therefore, mice were sacrificed for dissection of different brain structures from one hemisphere. Luciferase activity was measured in protein extracts and expressed as relative luciferase units (RLU)/μg protein (Figure 5). The other hemisphere was processed for detection of GFP by IF. Note that the global luciferase activity from each region depends on its total protein content (Figure S3).
Figure 5.

Basal ganglia and cerebellum are favorable locations for AdV injection
The Ad-CAG-GL vector was administered by stereotaxic injection in different locations of C57BL/6 mice (1.5 μL/injection at 3.5 × 107 vg/μL, bilateral): (A) cortex (Ctx, prefrontal, parietal, and occipital); (B) hippocampus (HC); (C) basal ganglia (BG); (D) cerebellum (Cb); (E) basal ganglia and cerebellum (BG/Cb); (F) basal ganglia, cerebellum, and prefrontal cortex (BG/Cb/pCtx). 48 h later, luciferase activity was quantified by BLI after intraperitoneal administration of the substrate D-luciferin. One h later, mice were sacrificed and brains were collected. One hemisphere was processed for detection of GFP, and the other was dissected for separation of the following brain structures: pCtx; ptCtx (parieto-temporo-occipital cortex); HC; Cb; brain stem (BS); BG; and hypothalamus (HT). The liver (Li) was collected to analyze the expression in extracerebral organs. Luciferase activity was measured in protein extracts from these structures and represented as RLU/μg protein. (G) representative images of GFP detection (green), with nuclei stained by DAPI (blue). For each injection location, two sections with different lateralization are shown. Note that in the case of the cortex the vector was injected in three different coordinates. Parietal and occipital are shown in the left image and prefrontal in the right image. Bars indicate averages for each group, and individual values are represented by small circles. Scale bar, 2 mm.
Our results indicate that among all single injection locations, the one achieving wider distribution of expression was BG. We detected intense luciferase activity not only in these structures (including the thalamus) but also in the hypothalamus, and to a lesser extent in the HC and the Ctx (Figure 5C). Injection in Cb achieved efficient expression in this organ, but only some spread was observed in the BS, as expected because of its anatomy (Figure 5D). More surprisingly, direct injection in HC showed a very restricted expression in this area. In addition, IF analysis revealed that most transduced cells were in the corpus callosum, leaving some critical areas such as the dentate gyrus relatively untargeted (Figures 5B and 5G). The combination of injections in BG and Cb allowed expression in most areas, with only the pCtx being relatively excluded (Figure 5E). Of note, systemic exposure to the vector was marginal, as indicated by the low luciferase activity in Li. As expected, luciferase activity and GFP expression showed good correlation in most regions. Regarding the identity of transduced cells, a combination of neurons and glia was detected, as previously described12 (Figure S4). On the basis of these results, we evaluated simultaneous injections in pCtx, BG, and Cb (Figure 5F). Although this is a more invasive procedure, it was well tolerated and allowed an improvement of brain coverage using a clinically compatible technique.
Intracerebral injection of HCA-CAG-SCN1A in adolescent DS mice is well tolerated and improves most disease manifestations
Expression of functional Nav1.1 in the brain and reduction of interictal epileptiform discharges (IED´s)
For therapeutic evaluation, the HCA-CAG-SCN1A vector was injected in the brain of mice harboring a pathogenic SCN1A missense mutation (Scn1aWT/A1783V mice, hereafter referred to as DS mice). These mice show a severe DS phenotype with a full spectrum of epileptic, motor, cognitive, and behavioral manifestations.30 In order to validate the functionality of the transgene, some animals received the vector in BG (4 × 106 or 2 × 107 vg/injection) and others were injected with the same volume of saline solution as a control. During the same surgical procedure, deep electrodes were placed in BG, close to the site of injection. In addition, a superficial electrode (screw attached to the skull) was placed over the pCtx. After recovery, this approach allows chronic recordings of behaving mice, as described in Materials and methods. One week after surgery, electrophysiological recordings were performed in both groups of mice. A significant reduction of IEDs was observed in animals receiving the HCA-CAG-SCN1A vector at the highest dose, whereas the lower dose had only a partial effect (Figure 6A). This is in agreement with partial restoration of the Nav1.1 function. Additional mice were treated with the vector to identify transduced cells overexpressing Nav1.1 by IF (Figure 6B). Note that WT and DS mice show equivalent endogenous Nav1.1 content, as previously described,30 which means that transduced cells expressing low levels cannot be distinguished from untransduced cells. Co-staining of Nav1.1 with neuronal (neuronal nuclei, NeuN) or astroglial (glial fibrillary acidic protein, GFAP) markers was performed to determine the identity of transduced cells. The number of neurons and astrocytes overexpressing Nav1.1 in the injected area was 18.65 ± 2.91 and 29.23 ± 15.66 cells/mm2, respectively, in mice injected with the highest dose (mean ± SEM, Figure 6C). Taking into account the predominance of neurons versus astroglia in the analyzed region, the estimated rate of transduction was 1.65% of neurons and 9.71% of astrocytes. Among cells overexpressing Nav1.1, 43% were neurons and 57% were astrocytes. In animals treated with the lowest vector dose, the nature of infected cells was maintained, but the percentage of transduced neurons dropped to 0.51%. Quantification of transgenic SCN1A mRNA was performed by qRT-PCR using primers specific for the codon-optimized sequence in mice injected with the highest dose. We detected an average of 7.44 × 105 ± 4.28 × 105 SCN1A copies/μg mRNA in treated mice (Figure 6D). To analyze the influence on endogenous Scn1a expression, murine mRNA was quantified with a different pair of primers (see Materials and methods) and compared with WT mice. No significant differences in endogenous Scn1a expression were observed among groups (WT: 1.08 × 106 ± 7.35 × 105, n = 3; DS: 1.18 × 106 ± 1.41 × 105, n = 3; DS+SCN1A: 1.35 × 106 ± 6.65 × 104, n = 4, p = 0.58, Kruskal-Wallis with Dunn’s multiple comparisons test). In agreement with this result, no major changes in the global content of Nav1.1 were detected in the BG of mice by western blotting (Figure 6E). These results indicate that a significant reduction of epileptogenic activity can be achieved by supplementation of SCN1A in a relatively low percentage of neurons in BG.
Figure 6.

Intracerebral administration of HCA-CAG-SCN1A increases the expression of functional Nav1.1 in DS mice
Two groups of 5-week-old DS mice received the vector by stereotaxic injection in basal ganglia at 4.6 × 106 or 2 × 107 vg/injection (DS+SCN1A (LD) and (HD), respectively). Control mice were injected with saline solution in the same location. Electrodes were placed close to the injection site and in the prefrontal area during the same surgical session. (A) One week later, electrophysiological signals were recorded from awake, freely moving animals. Representative signals from deep and superficial electrodes are shown at top and bottom, respectively. The number of IEDs per min was quantified and is represented at right. ∗p < 0.05, Kruskal-Wallis with Dunn’s multiple comparisons test. Note the presence/absence of IEDs in the DS and DS+SCN1A (HD) traces, respectively, and the partial effect in the DS+SCN1A (LD). (B) The basal ganglia of additional mice receiving the same treatment were processed for identification of cells overexpressing Nav1.1 by IF, in green, counterstained with DAPI in blue. (C) Co-staining of Nav1.1 (green) with NeuN and GFAP (in red) was performed to distinguish transduced neurons and astrocytes, respectively. The graph on the right indicates the percentage of cells overexpressing Nav1.1 in the injected region. (D) Quantification of endogenous scn1a and codon-optimized SCN1A mRNA expression in DS+SCN1A (HD), together with DS and WT controls. (E) Detection of Nav1.1 protein in the same groups of mice by western blot. In (A) and (D), bars indicate averages for each group, and individual values are represented by small circles. In (C), bars indicate average ± SEM. Scale bars, 250 μm in (B) and 50 μm in (C).
Improvement of survival and attenuation of sensitivity to hyperthermia-induced seizures
For evaluation of safety and therapeutic potential, the HCA-CAG-SCN1A vector was administered to adolescent DS mice (5-week-old) following two methods selected from our biodistribution study: injection in BG/Cb and triple injection in BG/Cb/pCtx, in order to obtain the maximal extension of gene supplementation. Control groups included untreated DS mice (ut) or DS mice treated with the vector expressing luciferase. All vectors and procedures were well tolerated, and mice rapidly recovered from surgery. Although the mortality rate in DS mice is moderate after the first 5 weeks of life,30 a significant increase of survival was still observed in mice treated with HCA-CAG-SCN1A by both administration methods (ut n = 17; BG/Cb n = 16, p < 0.04; BG/Cb/pCtx n = 12, p < 0.02, log rank test, Figure 7A). This result is consistent with protection from SUDEP. One month after treatment, mice were subjected to controlled hyperthermia to determine their seizure threshold temperature. As shown in Figure 7B, only mice that received the triple bilateral injection of HCA-CAG-SCN1A showed a significant increase in the threshold, compared with control mice or their own pre-treatment values (WT: >42°C, n = 11; ut: 39.87°C ± 0.17°C, n = 15; Luc: 39.36°C ± 0.19°C, n = 14; BG/Cb: 39.56°C ± 0.13°C, n = 14; BG/Cb/pCtx: 40.72°C ± 0.19°C, n = 14; p < 0.01, one-way ANOVA with Sidak’s multiple comparison test). In contrast, no change could be demonstrated when the vector was administered only in BG/Cb (39.5 ± 0.5°C, n = 14). The effect on spontaneous seizures is difficult to quantify in adolescent and young DS mice, but our observations during animal husbandry suggest that the BG/Cb route is able to reduce the lethality but not the frequency of crisis (Figure S6). These results suggest that reducing the predisposition to thermal-induced tonic-clonic seizures requires wider distribution of SNC1A expression compared with protection from early death.
Figure 7.

Intracerebral administration of HCA-CAG-SCN1A improves survival and attenuates sensitivity to hyperthermia-induced seizures in DS mice
Five-week-old DS mice received bilateral stereotaxic injections of the vector (2 × 107 vg/injection) in basal ganglia and cerebellum (BG/Cb) or basal ganglia, cerebellum, and prefrontal cortex (BG/Cb/pCtx). Control DS mice were left untreated (ut) or were injected with the reporter vector HCA-CAG-Luc. (A) Survival after treatment. p = 0.02 in BG/Cb/pCtx versus ut; p = 0.04 in BG/Cb versus ut, log-rank test. (B) Mice were subjected to controlled hyperthermia 1 month after treatment. The graphs represent the average seizure threshold temperature with indication of individual values (left). Right: the cumulative fraction of mice suffering a generalized tonic-clonic seizure for each body temperature before (pre) and 1 month after treatment (post). More than 80% WT mice experienced no seizures at the maximal temperature tested (42.5°C). ∗∗p < 0.01, ∗∗∗p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test.
Amelioration of motor and some behavioral manifestations but no correction of hyperactivity and learning delay
After demonstration that HCA-CAG-SCN1A attenuates the most life-threatening consequences of SCN1A deficiency, we started the evaluation of other important comorbidities. To this end, mice treated with a triple bilateral injection of the therapeutic vector or the corresponding controls were subjected to a series of motor, cognitive, and behavioral tests. Evaluation started 2 months after treatment and spanned 1 additional month. Of note, cognitive and behavioral manifestations were already present in adolescent DS mice (Figure S5), whereas motor defects were evident only after the third month of life.30 Quantification of cognitive functions started with the novel object recognition (NOR) test (Figure 8A). Mice treated with HCA-CAG-SCN1A (SCN1A group) showed an improvement in the discrimination index (DI) between novel and known objects (WT: 63.5 ± 2.45 s, n = 21; ut: 47.4 ± 4.56 s, n = 20; Luc: 47.7 ± 7.69 s, n = 14; SCN1A: 64.6 ± 2.71%, n = 12; p < 0.05, one-way ANOVA with Sidak’s multiple comparison test), suggesting amelioration of long-term visuospatial recognition memory. However, the Morris water maze test (MWM) failed to demonstrate any improvement in task learning (Figure 8B). The visible platform (VP) phase of the test (Figure 8B, left) confirmed that all DS mice can learn a task because the time needed to reach the platform is reduced by training (p < 0.0001, Friedman test), but this time is consistently higher than in WT mice (p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test), irrespective of the treatment. The invisible platform (IP) phase of the MWM test (Figure 8B, center) evidenced the inability of DS mice to use visual cues (mean ± SEM, p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test; p > 0.05, Friedman test), and the treatment with HCA-CAG-SCN1A was unable to improve it (p > 0.05, Friedman test). Consistent with the persistence of visuospatial memory retention defects in treated mice, the probe test (P) showed no amelioration in the time spent in the right quadrant, as shown in Figure 8B, right (mean percentage of time in the last probe, WT: 52.53 ± 2.92 s, n = 20; ut: 22.59 ± 2.24 s, n = 17; Luc: 27.04 ± 3.29 s, n = 15; SCN1A: 29.37 ± 3.74 s, n = 11, all DS groups p < 0.0001 versus WT, p > 0.05 SCN1A versus ut and Luc, one-way ANOVA with Sidak’s multiple comparison test). Overall, a global improvement in cognitive performance could not be demonstrated when the HCA-CAG-SCN1A vector was administered in adolescent DS mice. This result is consistent with the failure to correct the hyperactive behavior, analyzed in the open-field (OF) test (Figure 8C). In Figure 8C, left we show that the increase in movement velocity observed in DS versus WT mice (WT: 6.27 ± 0.28 cm/s, n = 23; ut: 9.87 ± 0.51 cm/s, n = 22; Luc: 11.5 ± 0.60 cm/s, n = 17, p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test) was unchanged upon administration of the HCA-CAG-SCN1A vector (SCN1A: 9.8 ± 0.5 cm/s, n = 10, p > 0.05). The same outcome was observed when we analyzed the avoidance to explore the center of the arena (Figure 8C, center), which is indicative of anxiety behavior. Mice treated with HCA-CAG-SCN1A showed no difference in the time spent in the center of the arena compared with untreated or luciferase vector-treated groups (ut: 48.80 ± 6.04 s, n = 21; Luc: 60.9 ± 7.49 s, n = 18; SCN1A: 40.1 ± 8.73 s, n = 11, p > 0.05, one-way ANOVA with Sidak’s multiple comparisons test), which is lower than WT mice (WT: 95.2 ± 10.8 s, n = 23, p < 0.001). The hyperactive behavior of DS mice was also apparent when we measured the number of stereotypies (Figure 8C, right) (WT: 3.18 ± 0.26 stereotypies/min, n = 21; ut: 8.34 ± 0.54 stereotypies/min, n = 24; Luc: 7.55 ± 0.30 stereotypies/min, n = 12, p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test), which was unchanged in mice treated with the therapeutic vector (SCN1A: 8.04 ± 0.70 stereotypies/min, n = 11, p > 0.05). In contrast, species-adapted motivation-related tests indicated improvement of behavioral traits related to interaction with the environment, which are a good indicator of the individual’s performance in daily life activities.31 On one hand, the marble burying test (Figure 8D) revealed an improvement in exploratory behavior, with a reduction of uncovered objects compared with the control groups (WT: 2.05 ± 0.32 marbles, n = 23, ut: 7.28 ± 0.73 marbles, n = 28; Luc: 8.48 ± 0.64 marbles, n = 14; SCN1A: 3.98 ± 0.59 marbles, n = 11, p < 0.05 SCN1A versus ut; p < 0.01 SCN1A versus Luc, Kruskal-Wallis with Dunn’s multiple comparisons test). On the other hand, the nest building test, which is considered an indicator of the animal’s welfare,31 showed better nest assembly skills in DS mice treated with the HCA-CAG-SCN1A vector (Figure 8E), although the amelioration was only partial.
Figure 8.

Intracerebral administration of HCA-CAG-SCN1A in adolescent DS mice improves motor skills and ameliorates some behavioral manifestations but not hyperactivity and learning
Five-week-old DS mice received bilateral stereotaxic injections of the HCA-CAG-SCN1A vector (2 × 107 vg/injection) in basal ganglia, cerebellum, and prefrontal cortex (BG/Cb/pCtx). Control mice were left untreated (ut) or were injected with the reporter vector HCA-CAG-Luc (Luc). Wild-type littermates (WT) are included as a reference of normal score for each test. Mice were subjected to the following tests: NOR (A); MWM (B); OF (C); marble burying (D); nest building (E); rotarod (F); and clasping (G). Bars represent the average value of each group, and individual scores are indicated by symbols (WT, gray inverted triangles; ut, empty orange diamonds; Luc, red circles; SCN1A, green triangles). In the MWM (B), the visible and invisible platform phases of the test (VP and IP, respectively, mean ±SEM) and the 60-s probe tests (P60) are represented at left, center, and right, respectively. (C) In the OF the mean velocity, time spent in the center of the arena, and number of stereotypies/min are represented at left, center, and right, respectively. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test (A–D and F) and Kruskal-Wallis with Dunn’s test (G).
Finally, the vector obtained a clear improvement of motor function, as demonstrated in the rotarod test (Figure 8F), which evaluates coordination and balance. The latency to fall from the rotating surface was increased in the therapeutic group compared with DS controls (SCN1A: 101 ± 10.1 s, n = 12; ut: 63.9 ± 6.21 s, n = 17; Luc: 52.8 ± 7.18 s, n = 17, p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test), and it was equivalent to WT mice (WT: 84 ± 7.02 s, n = 19; p > 0.05). In agreement with the improvement of cerebellar ataxia, a reduction in the clasping index was observed in mice treated with HCA-CAG-SCN1A (WT: 0.04 ± 0.04, n = 21; ut: 2.29 ± 0.16, n = 14; Luc: 2.09 ± 0.31, n = 11; SCN1A: 1.33 ± 0.31, n = 12, p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test) (Figure 8G).
Discussion
Gene transfer of the SCN1A coding sequence is a straightforward mechanism of action for the treatment of DS from a conceptual standpoint. It offers the opportunity of permanent disease control upon a single vector administration, without the need of expressing exogenous genes. However, this approach needs to overcome important technical barriers. In this work we have dissipated some uncertainties by demonstrating that a codon-optimized version of the SCN1A cDNA can be efficiently amplified and manipulated to produce HC-AdVs. The production of these vectors is more complicated than AAVs, but recent improvements are increasing their clinical feasibility.32 DS-treated animals were protected from SUDEP, their epileptic features were attenuated, and a consistent amelioration of motor function and some behavioral traits was observed. Our results are in line with a recent work using an AAV vector to deliver guide RNAs for a transgenically expressed Cas9-based transcriptional activator specific for the SCN1A promoter.33 This elegant proof-of-principle study limits Nav1.1 expression to inhibitory neurons and leverages the efficacy of the AAV-PHP.eB serotype in mouse brain, obtaining a partial therapeutic effect when administered to adolescent (P30) mice.
As expected, generalized transgene expression cannot be obtained after a single intracerebral administration of adenoviral vectors, and the coverage obtained is different depending on the injection site. The CAG promoter was chosen because it guarantees transgene expression in all cell types, it ensures long-term ubiquitous expression in the brain, and it facilitates the characterization of different routes of administration. Using this tool, we have found that the need for homogeneous and widespread transgene expression is not as stringent as predicted in DS. The hippocampus is considered an important target for this disease,34,35 but we found that HAdV5 vectors administered in this location reached a relatively low number of neurons and did not transduce neighboring structures. Injection in the cortex obtained efficient but locally restricted transgene expression, while cerebellum and especially basal ganglia were more permissive for vector transduction, in agreement with previous reports.36 Further optimization of vector dissemination in the brain could improve not only seizure control but also other DS manifestations, as discussed below. However, until efficient methods to deliver large particles across the blood-brain or ependymal-brain barrier are developed, selective stereotaxic injections are the most realistic option. We believe the method described here is clinically feasible and allows the transduction of brain areas with relevance for DS. The therapeutic benefit of focal transgene expression cannot be generalized for other genetic encephalopathies, and it should be investigated in specific preclinical models in order to evaluate the scope of this approach.
Interestingly, injection of HCA-CAG-SCN1A in basal ganglia and cerebellum improved survival of DS mice. As shown in our biodistribution experiments, transduction of cerebellum produced a concomitant expression of the transgene in brain stem (pons). Both structures play a central role in autonomic and respiratory regulation and, therefore, in SUDEP.37 In fact, the pons is connected with the thalamus, and a marked reduction in the resting-state functional connectivity between them has been described for patients at high risk of suffering SUDEP38. Moreover, an extensive cerebellar volume loss has been described in these patients.39 Tissue loss may be derived from excitotoxicity in Purkinje cells due to a hyperactivation of pontine or long climbing fibers of olivary projections. Damaged Purkinje cells would affect the function of the deep nuclei, impairing the recovery from hypotension and apnea following a generalized tonic-clonic seizure.40 Altogether, this evidence suggests that restoration of SCN1A expression in cerebellum and brain stem is a key factor for protection from SUDEP in DS mice, probably improving autonomic and respiratory regulation. We cannot rule out the possibility that SCN1A expression in the thalamus could also contribute to control the excitatory stimuli in distant brain regions not reached by the vector.41,42 In addition, the efficient transduction of the hypothalamus could exert a positive effect by normalization of circadian rhythms,43,44 although this possibility has not been directly investigated. The potential role of this region in the therapeutic effect of HCA-CAG-SCN1A requires further study to identify the GFP+ cells by IF, since this small structure was entirely collected for luciferase analysis.
Despite the effect observed after administration of the vector in basal ganglia and cerebellum, the addition of a prefrontal cortex injection was needed to provide protection from febrile seizures in the DS mouse model, suggesting the requirement of wider distribution in order to control the generalization of crisis. This concept is supported by a recent study carried out in patients suffering from Lennox-Gastaut syndrome,45 showing the importance of the prefrontal cortex in the initiation of epileptic activity, followed by propagation to the brain stem and then to the thalamus. This region is also well positioned to promote rapid communication with spinal motor neurons involved in tonic seizures. In addition, the crucial role of the prefrontal cortex in cognitive functions46 is in agreement with the improvement observed in the NOR test in treated mice. Some reports suggest the implication of this region in spatial memory.47,48 However, DS mice receiving the HCA-CAG-SCN1A vector after the triple bilateral injection (BG/Cb/pCtx) showed no improvement in the MWM test, which is the gold standard for evaluation of spatial learning and retention. This result supports the importance of the hippocampus in these brain functions. In the same vein, although HCA-CAG-SCN1A improved the general welfare of DS mice—evaluated with the marble burying and nest building tests—it failed to avoid the hyperactive behavior, suggesting that wider vector distribution is required, especially in cortical and hippocampal areas. This could be accomplished by advanced delivery methods (convection-enhanced infusion36,49 or sonoporation50) or by using vectors derived from alternative adenoviral members with superior neuronal tropism. In addition, better control of SCN1A expression could improve the therapeutic efficacy. Even if the SCN1A gene is normally expressed in different cell populations (including inhibitory interneurons, excitatory neurons, and glia), overexpression in some of these cells may be detrimental for the restoration of the excitatory/inhibitory balance in the brain. The use of the CAG promoter, which ensures long-term expression without cell specificity, has demonstrated the safety of this approach. Still, obtaining physiological expression of SCN1A is one of our current priorities, aided by the ability of HC-AdVs to accommodate large regulatory regions. The use of the endogenous promoter could modulate expression without assuming the identity of the optimal target cell/s, which are still not fully defined. While these are relevant options for treatment optimization, our results demonstrate that significant amelioration of disease manifestations can be achieved upon SCN1A transfer in a fraction of neurons, highlighting the feasibility of this approach. Importantly, optimization of gene transfer should occur in parallel with the improvement in transgene regulation in order to avoid a generalized increase in Nav1.1 function. Although it is not clear whether SCN1A overexpression and gain-of-function mutants are functionally equivalent, the latter have been associated with serious diseases.51, 52, 53
In this work we took the challenge of treating adolescent DS mice, in which the full spectrum of disease comorbidities is already established, with the only exception of motor alterations.30 Although treatment at earlier ages could render better results, we believe this group of patients is clearly in need of new therapies. Therefore, it was important to determine to what extent pre-existing defects can be reverted by disease-modifying therapies. Apart from mortality and sensitivity to hyperthermia-induced seizures, we demonstrated improvement of some behavioral tests (marble burying, nest building) and the NOR test, as well as prevention of motor deterioration. One limitation of our study is the rudimentary evaluation of spontaneous seizures. Proper assessment requires specific equipment for long-term video-electroencephalogram (EEG) recording, taking into account the relatively low frequency of seizures in adolescent/adult DS mice. Therefore, we cannot reach strong conclusions on this disease manifestation. On the other hand, hyperactivity and spatial learning delay were not significantly ameliorated. At this moment it is not possible to determine whether this is due to the suboptimal vector distribution and selectivity or is a consequence of irreversible functional/structural alterations. Of note, seizures can provoke pathogenic remodeling that is promoted by—but not specific to—SCN1A mutations.54 In addition, alterations of the neurogenic niche have been recently described in the hippocampus of DS mice.55 This issue deserves special attention in order to define the scope of novel SCN1A-targeted therapies.
Despite all the current limitations, our results demonstrate that the transfer of the full SCN1A cDNA using HC-AdVs is feasible and potentially therapeutic for DS. The next challenge is to increase the efficacy and selectivity of transgene expression into the brain, depending on vector biodistribution, infectivity, and transcriptional regulation. Each one of these parameters may require specific optimization, which goes beyond the scope of the present work. Here we have defined a basic approach with therapeutic potential and indicate potential ways of improvement.
Materials and methods
Plasmids and construction of vector genomes
pcDNA3.1 was purchased from Invitrogen (Carlsbad, CA, USA). The pCMV-SCN1A plasmid encoding the codon-optimized human SCN1A cDNA was obtained from Invitrogen (GeneArt). The CAG promoter was obtained from the pCAGGS plasmid.56 The EF1α regulatory sequence was obtained from the pTGC1100 plasmid. The IRES and eGFPLuc sequences were derived from the pIRES and pEGFPLuc plasmids, respectively (Clontech, Mountain View, CA, USA). All subclonings were performed by standard molecular biology techniques using restriction endonucleases from New England Biolabs (Ipswich, MA, USA). Ligase was from Promega (Madison, WI, USA). Oligonucleotides were purchased from Sigma-Aldrich (St. Louis, MO, USA). For the construction of the Ad-CAG-GFPLuc vector, the CAG-GFPLuc expression cassette was inserted into the ClaI (blunted)-XbaI sites of pSAdBst. This plasmid contains the sequence of HAdV5 with a small poly-cloning site (ClaI-XbaI-BstBI) substituting the E1 region and a deletion of E3. For the construction of the HC-AdV vectors, the CAG-SCN1A and EF-SCN1A-GFP expression cassettes were introduced into the AscI sites of pD23E422 or pD20-E4 plasmids, respectively. pD20-E4 was obtained from pDelta28E457 by deletion of the stuffer region comprising SwaI-BstEII sites and insertion of NotI and MluI sites.
Vector production
The E1/E3-deleted Ad-CAG-GFPLuc vector was obtained by transfection of the Ad-CAG-GFPLuc plasmid in HEK-293 cells, and the culture was maintained until cytopathic effect (CPE) was apparent. The virus was cloned by end-limiting dilution and amplified in HEK-293 cells. Purification was performed by double CsCl density-gradient ultracentrifugation and desalting in Sepharose size-exclusion columns. Production of HC-AdVs was performed as previously described.14 Quantification in vg was performed by qPCR of purified virus using the primers 5′ AGCATCCGTTACTCTGAGTTGG 3′ (forward); GCATGTTGGTATGCAGGATGG (reverse) for E1/E3-deleted vectors and 5′ TAGTGTGGCGGAAGTGTGATGTTG 3′ (forward); 5′ ACGCCACTTTGACCCGGAACG 3′ (reverse) for HC-AdVs. The same primers were used for quantification of iiu, as previously described.58
Cell culture
The cell lines HEK-293 (ATCC CRL-1573, American Type Culture Collection), A549 (ATCC CRL-185, American Type Culture Collection), and 293Cre4 (courtesy of Stefan Kochanek, University of Ulm, Germany) were maintained in DMEM. G-418 (400 μg/mL) was added to 293Cre4 cells. SH-SY5Y cells (ATCC CRL-2266, American Type Culture Collection) were maintained in a 1:1 mixture of Eagle’s minimum essential medium and F12 medium supplemented with 1% non-essential amino acids. All culture media were supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine. Reagents were obtained from Gibco (Gaithersburg, MD, USA). All cells were maintained at 37°C with 5% CO2 in a humidified incubator. Cells were routinely tested for mycoplasma contamination. HEK-293 cells were seeded in 24-well, 6-well, or P100 plates at a density of 2.5 × 105, 1 × 106, or 5 × 105, respectively; SH-SY5Y cells were seeded at 2.5 × 105 or 2.5 × 106 in 24-well or 6- well plates.
Brains obtained from C57BL/6 mice at the 14.5th day of embryogenesis (E14.5) were used to prepare primary neuronal cultures. Pregnant mice were sacrificed, and embryos were extracted. Embryo brains were removed from the skull; after dissection of the cortex and hippocampus, the tissues were collected in Hank’s balanced salt solution (HBSS) (Life Technologies) with 10 mM HEPES (Gibco). To dissociate cells, 60 μL of DNase I at 10 mg/mL (Roche) and 120 μL of 2.5% trypsin solution (Gibco) were added, followed by a 15-min incubation at 37°C. Enzymatic digestion was stopped by adding 5 mL of plating medium (PM: Neurobasal medium [Gibco] supplemented with 10% FBS [Sigma], 2% B-27 [Thermo], 1% GlutaMAX [Gibco], and 100 U/mL penicillin and 100 μg/mL streptomycin [Gibco]). After a centrifugation of 5 min at 90 × g, supernatant was removed and 3 mL of maintenance medium (MM: Neurobasal medium [Gibco] supplemented with 2% B-27 [Thermo], 1% GlutaMAX [Gibco], and 100 U/mL penicillin and 100 μg/mL streptomycin [Gibco]) was added to carry out the mechanical dissociation. Neurons were seeded at a density of 4 × 105 neurons per well on 24-well or 4 × 106 neurons per well on 6-well (Eppendorf) pre-treated plates (coated with 100 μg/mL poly-l-lysine [Sigma] diluted in sterilized double-distilled water and 2.5 mg/mL Matrigel [Corning, 374234] diluted in Leibovitz’s L-15 medium [Gibco]) and grown in 800 μL of MM; 12 h later cell culture medium was complemented with 50 ng/mL brain-derived neurotrophic factor (BDNF, PeproTech).
Transfections and luciferase assay
Cells were seeded in 24-well plates at 80%–90% confluence. Transfection was performed 24 h or 4 days later—for stable cell lines or primary cultures, respectively—by Lipofectamine 2000 (Invitrogen) using 0.5 μg of each firefly luciferase reporter plasmid. The transfection mixture was removed 5 h later and replaced by fresh culture medium. Cells were lysed 48 h later, and 10 μL of each sample was analyzed for luciferase activity with the Luciferase Reporter Assay System (Promega) on a Lumat LB 9507 Luminometer (Berthold Technologies). Results were normalized by μg of protein loaded, determined by Bradford assay (Bio-Rad).
Patch clamp
HEK-293 cells (ATCC CRL-1573) were infected with 400 vp/cell. 24 h after infection, recordings were performed as described previously.17 Briefly, Sutter IPA amplifier was used (Sutter Instrument, Novato, CA, USA). The pipette solutions contained 140 mM CsF, 10 mM NaCl, 1 mM EGTA, 10 mM HEPES, and 10 mM glucose, adjusted to pH 7.3 with NaOH. The external solution contained 140 mM NaCl, 20 mM glucose, 10 mM HEPES, 1 mM MgCl2, 3 mM KCl, and 1 mM CaCl2, adjusted to pH 7.35 with NaOH. Chemicals were purchased from Sigma-Aldrich or Fisher Chemical. The voltage dependence of activation was measured from a holding potential of −120 mV. Cells were depolarized for 20 ms to potentials ranging from −70 to +40 mV in 10-mV increments. The selective Nav1.1 opener Hm1a (50 nM, Alomone lab, Jerusalem, Israel; Cat# STH-601) was added to a subset of cells.
Infections in cell cultures
SH-SY5Y cells and primary neurons were seeded at a density of 2.5 × 105 or 4 × 106 in 24-well or 6- well plates, respectively. Infections were performed 24 h or 4 days later, respectively, preparing the infection mix in 100 or 500 μL of growing medium, respectively. 48 h later, cells were processed for qRT-PCR or IF analyses.
Animals and stereotaxic injections
The conditional Scn1a-A1783V mice (B6(Cg)-Scn1atm1.1Dsf/J, The Jackson Laboratory, stock no. 026133) were bred to mice expressing Cre recombinase under the control of the CMV promoter (B6.C-Tg(CMV-Cre)1Cgn/J, The Jackson Laboratory, stock no. 00605459). Breeding pairs consisted of heterozygous male Scn1a-A1783V and homozygous female CMV-Cre mice. See https://www.jax.org/strain/026133 for details about allele modification and genotyping. Offspring carrying one mutated allele (genotype hereafter referred to as DS) express the A1783V mutation in the Scn1a gene in all body tissues, mimicking what happens in DS. Animals were housed 4–6 per cage with free access to food and water, weighed weekly, and maintained in a temperature- and light-controlled (12 h/12 h light/dark cycle) environment. Breeding and experimental protocols were approved by the Ethical Committee of the University of Navarra (in accord with Spanish Royal Decree 53/2013).
For stereotaxic administration 5-week-old animals were anesthetized with ketamine-xylazine (80:10 mg/kg i.p.) and placed in a stereotactic frame. After the scalp was shaved and disinfected, a longitudinal incision was made along the midline to expose the skull, which was cleaned with iodine and hydrogen peroxide to remove the periosteum and prevent infection. Next, 2–6 burr holes were drilled at the defined positions. Coordinates (Table S1) were determined according to the Paxinos and Watson mouse brain atlas (1998). A 10-μL Hamilton syringe (Hamilton) was used for vector delivery. Viral suspensions (1.0–1.5 μL) were administered per injection point following a 0.4 μL/min infusion rate. The needle remained in place for 3 min before and after vector infusion to favor the absorption of the vector. The incision was stitched, and the animals were observed until full recovery from anesthesia and daily during the following days to avoid surgical complications.
Quantitative PCR
In order to minimize the number of animals employed in this part of the study (n = 3 or 4), one brain hemisphere of each animal was dissected and employed to determine mRNA levels of Scn1a or SCN1Aco genes by quantitative PCR, and the other was fixed for the identification of Nav1.1 protein by IF. Brain tissues or cell pellets from the in vitro experiments were processed with the Maxwell 16 LEV simplyRNA Cells/Tissue Kit (Promega) for total RNA isolation according to manufacturer’s indications. Two micrograms of RNA were then treated with DNase I and retro-transcribed into cDNA with Moleney Murine Leukemia Virus (M-MLV) retro-trasncriptase enzyme (Invitrogen) and random primers (Life Technologies). These procedures were performed in a GeneAmp PCR System 2400 (Applied Biosystems). Quantitative analysis was performed by real-time PCR using iQTM SYBR Green Supermix reagent (Bio-Rad) in the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Mouse Scn1a and human SCN1Aco expression levels were determined with specific primers (mScn1a: FP 5′-CATGTATGCTGCAGTTGATTCCA-3′ and RP 5′-AACAGGTTCAGGGTAAAGAAGG-3′; hSCN1Aco: FP 5′- TCAACATGTACATTGCCGTC-3′ and RP 5′-ATCAGCTGCAGTTTGTTGG-3′),60 and mouse/human 36b4 was used as housekeeping gene (FP 5′-AACAATCTCCCCCTTCTCCTT-3′ and RP 5′-GAAGGCCTTGACCTTTTCAG-3′). Primers were purchased from Sigma. All samples were tested in triplicate, and at least two independent qPCR experiments were performed. The relative quantification was carried out using the 2-Delta threshold cycle (2-ΔCt) corrected with primer efficiencies.61
Preparation of membrane-enriched extracts and western blotting
Membrane-enriched protein fractions (P2) were obtained from cells and brain tissues as previously described.62 Protein concentration was determined by Bradford assay (Bio-Rad), and part of the preparation was solubilized in denaturing conditions as described previously.11,63 Protein samples were mixed with 4× urea-EDTA buffer, resolved onto 7.5% SDS-polyacrylamide gels, and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% BSA and 0.05% Tween 20 in Tris-buffered saline (TBS), followed by overnight incubation at 4°C with the following primary antibodies: rabbit polyclonal anti-Nav1.1 (Alomone Labs Cat# ASC-001, 1:500) and rabbit monoclonal (14C12) anti-GAPDH (Cell Signaling Technology, Cat# 2118S, 1:5,000) diluted in 2.5% BSA, 0.05% Tween 20, and 0.01% azide in TBS. Immunolabeled protein bands were detected by using an anti-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP) conjugate (GE Healthcare, Cat# NA934V, 1:10,000) and an enhanced chemiluminescence system (Lumigen ECL Ultra TMA-6, Lumigen, Cat# TLA-100). Images were acquired with a ChemiDoc system (Bio-Rad).
Immunofluorescence procedures
Animals were perfused transcardially with 0.9% saline under an overdose of ketamine-xylazine anesthesia (240:30 mg/kg body weight; Ketamidor 100 mg/mL injectable solution, Richter Pharma, Wels, Austria, and Rompun 20 mg/mL injectable solution, Bayer Animal Health, Leverkusen, Germany; respectively). After perfusion, brains were removed and one hemisphere was fixed in 4% paraformaldehyde (PFA) (PanReac) for 48 h at room temperature (RT) and then cryopreserved in 30% sucrose solution in phosphate saline buffer 0.125 M (PBS, Gibco) at 4°C until they sank. Microtome sections (thickness: 30 μm) were cut sagittally with a freezing microtome and stored in cryopreserving solution (30% ethylene glycol, 30% glycerol in phosphate buffer 0.1 M, Sigma) at −20°C until processed. To carry out the IF, two to four free-floating tissue sections per animal were processed (n = 3 or 4 in each group). Brain sections were washed 3 times with PBS at RT, and then a blocking step was performed (2% donkey or goat normal serum [Jackson ImmunoResearch], 0.5% Triton X-100 [Sigma], and 1% BSA [Sigma] in PBS [Gibco]), followed by overnight incubation at 4°C with the primary antibodies (rabbit polyclonal anti-Nav1.1, Alomone Labs Cat# ASC-001, 1:400; rabbit anti-GFP, Abcam, Cat# MAB377, 1:200; mouse anti-NeuN clone A60, Merck Millipore, Cat# MAB377, 1:400; mouse anti-GFAP [GA5], Cell Signaling Technology, Cat# 3670, 1:500) diluted in blocking solution (simple IF anti-GFP) or Chemmate antibody diluent (Dako, for double IF or simple IF anti-Nav1.1). After being washed 3 times with PBS, slices were incubated with the secondary antibodies (Donkey anti-Rabbit IgG [H+L] Alexa Fluor 488 Cat# A-21206, 1:400; Goat anti-Mouse IgG [H+L] Alexa Fluor 568 Cat# A-11031, 1:400) for 90 min at RT and protected from light. To enable the visualization of nuclei, sections were incubated for 5 min with the DNA marker 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific Cat# D1306, 300 nM) protected from light and washed twice with PBS. For Nav1.1 immunostaining, slices were also incubated for 3 min at RT with the autofluorescence eliminator reagent (Millipore). Finally, slices were mounted on Superfrost plus slides (Thermo), air-dried for 24 h protected from light, and rinsed in toluene (5 min). Coverslips (Thermo) were placed with Immu-Mount mounting medium (Thermo Fisher Scientific Cat# 9990402). To ensure comparable immunostaining, sections were processed together under identical conditions. For the assessment of non-specific primary and secondary immunostaining, some sections from each experimental group were incubated without primary or secondary antibody, and no immunostaining was observed in any case. Fluorescence signals were acquired with the Axio Imager M1 microscope (Zeiss) using the EC Plan Neofluor 20×/0.50 M27 objective. Images were acquired with the AxioCam MR R3 camera and the program ZEN 2 blue edition (Zeiss). The employed excitation and emission wavelengths were 488 and 509 nm for Nav1.1 and GFP, 558 and 575 nm for NeuN and GFAP, and 353 and 465 nm for DAPI. Emissions were color-coded in green, red, and blue, respectively. Acquired fluorescence images were adjusted in parallel for brightness and contrast in ZEN 2 blue edition (Zeiss).
For IF detection of Nav1.1, GFAP, VGAT, and VGlut1 in culture, cells were fixed for 5 min in 4% PFA (PanReac) and 4% sucrose (Sigma) diluted in PBS with calcium and magnesium (PBS+/+, Gibco). Cells were then permeabilized with 0.1% Triton X-100 (Sigma) in PBS+/+, blocked for 30 min at RT in blocking solution (2% BSA [Sigma] and 3% Normal Goat Serum [Jackson ImmunoResearch] in PBS+/+), and incubated overnight with the rabbit anti-Nav1.1 (1:400, Alomone Labs Cat# ASC-001), mouse anti-GFAP (GA5), (1:500, Cell Signaling Technology, Cat#3670), mouse anti-VGAT CL2-793 (1:500, Thermo Fisher, Cat# MA5-24643), and mouse anti-VGlut1 (1:1.000, Sigma, Cat# AMAb91041) diluted in blocking solution. After washes, cells were incubated with the secondary antibodies diluted 1:400 for 2 h at RT and protected from light (Goat anti-Rabbit IgG [H+L] Alexa Fluor 546 Cat# A-11010; Goat anti-mouse IgG [H+L] Alexa Fluor 488 Cat# A-11019). The GFP expression was detected by epifluorescence. Fluorescence signals were acquired with the DM IL LED inverted fluorescence microscope (Leica) equipped with a fluorescence cube slider (Filter 1, cube GFP for Alexa 488 staining: EF BP 470/40 nm, SP BP 525/50 nm, and DM 500 nm and filter 2, cube N2.1 for Alexa 546 staining: EF 515-560 nm, SP 590 nm, DM 580 nm), coupled to a mercury metal halide lamp (EL6000, Leica) using the HL Plan I 10×/0.22 PH1 or the HCX PL Fluotar L 20×/0.44 CORR objectives. Images were acquired with the DFC345FC camera (Leica Capture quality: 2.048 × 1.536 pixels [3 megapixels]) and the program Leica Application Suite (Leica Image information: 8 bits, dimensions 1,600 × 1,200, calibration 1.14 μm/px for 10× objective and 2.27 μm/px for 20× objective). Emissions were color-coded in red for Nav1.1 and green for GFP, GFAP, VGAT and VGlut1. For each panel, acquired fluorescence images were adjusted in parallel for brightness and contrast in ImageJ 1.52p (NIH, Bethesda, MD, USA). Sharpness was improved by employing an unsharp mask filter (radius [sigma]: 5.0 px, and mask weight; 0.50).
Quantifications of labeled cells and colocalizations were performed by two independent blinded investigators using ImageJ software.
Analysis of in vivo or ex vivo bioluminescence in mice
For in vivo BLI, mice were anesthetized with intraperitoneal injection of a ketamine-xylazine mixture (80:10 mg/kg i.p.). The substrate D-luciferin (Regis Technologies) was administered intraperitoneally (200 μL of a 30 μg/μL solution in PBS). Light emission was detected 5, 10, 15, and 30 min later with a PhotonImager Optima apparatus (Biospace) for identification of the peak values. Data were analyzed with M3Vision software (Biospace), representing the maximal value obtained for each animal. In Figure 5, mice were sacrificed 1 h later for collection of brain and liver. Brain structures (prefrontal cortex, parieto-temporo-occipital cortex, basal ganglia, brain stem, hypothalamus, hippocampus and cerebellum) were dissected before freezing in liquid nitrogen. Luciferase activity was measured in these samples as described for cell lines.
Electrophysiological recordings
Electrophysiological studies were carried out in 6-week-old animals (n = 5 per group). For the vector/saline administration the same protocol described above was followed. Three miniature stainless steel screws were placed, one of them over the prefrontal cortex, pCTX (AP: +2.58 mm and ML: ±1.5 mm) and the other two over the cerebellum contralateral and ipsilateral to the registration point, being used as ground and reference, respectively. Animals were left to recover from surgery, and 1 week after implantation they were connected to an acquisition system for electrophysiological recordings (Intan RHD2000 system, IntanTech). Recording sessions were simultaneously video recorded to evaluate the behavioral state of the animals and the appearance of clinical seizures. Animals were recorded for 30 min freely moving at RT on an OF box (60 cm × 60 cm × 60 cm). Electrophysiological data were converted from Intan format into Spike2 format (Cambridge Electronic Design) with custom-made routines in MATLAB (MathWorks, Natick, MA, USA). Reviewed features of the Spike2 software were used to analyze the recordings in order to assess the presence of IEDs, which were semi-automatically annotated by performing an initial detection following previously described methods64 and further validated by visual inspection. To do this, signals were loaded with MATLAB scripts, resampled to 1,000 Hz, band-pass filtered in the 60–100 Hz range, and rectified. IED events were detected when the filtered envelope was >3 times above standard deviation of baseline. Envelope was computed by estimating the root mean square (RMS) value within a 200-ms window. Then, candidate events were uploaded into the Spike2 file as a marker channel that was further reviewed and curated by two specialists.
Thermal induction of seizures
Epileptogenic thermal threshold of mice was tested by induction of hyperthermia seizures. The animals were body restrained into a methacrylate cylinder coupled to an infrared lamp that gradually increased ambient temperature inside the chamber. Mouse body temperature was measured before initiation of heating with a RET-4 rectal probe (Physitemp Instruments) connected to a TCAT-2LV controller (Physitemp Instruments). Animals were introduced into the cylinder prewarmed at 25°C, and the temperature was gradually increased 0.5°C every 2 min to a maximum of 42.5°C or until a generalized seizure was observed. Body temperature was measured again just after removal of the animals from the cylinder, and they were placed in contact with a rubber bag filled with cold water to accelerate the recovery. Animals were tested twice: at least 2 days before administration of vectors (pre-surgery) and ∼30 days after vector administration (post-surgery). Both pre- and post-surgery heat-induced seizure periods were also emulated in control animals (WT mice and DS mice untreated or treated with the HCA-CAG-Luc vector).
Behavioral assessment
Morris water maze test
The MWM is a hippocampus-dependent learning task that serves to test the working and reference memory function. The test was carried out as described previously.30 The water maze consisted of a circular pool of 1.2-m diameter and 0.6-m height (LE820120, Panlab Harvard Apparatus) filled with water tinted with non-toxic white paint and maintained at 20°C. During the first part of the test, visible platform (VP), mice were trained to find the platform in order to escape from the water. During this phase, the platform was raised above the water surface and mice were trained for 5 consecutive days (four trials per day). In the second part of the test, invisible platform (IP), the platform was placed in the opposite quadrant and hidden below water level, and visible cues were placed in each of the four quadrants of the maze to allow spatial learning. Mice were trained for 8 consecutive days (four trials per day). In both phases, mice were placed into the maze facing toward the wall of the pool in selected locations pseudo-randomly established. Each trial was finished when the mouse reached the platform (escape latency) or after 60 s; when an animal was unable to find the platform, it was gently guided onto it. After each trial, animals remained on the platform for 15 s. On the 4th, 7th and 9th days of the IP phase all mice were subjected to a probe trial in order to evaluate their retention. The platform was removed from the pool, and mice were allowed to swim for 60 s in the pool, measuring the time spent in the quadrant where the platform was placed during the IP. All trials were recorded and analyzed with the program WaterMaze3 (Actimetrics, Evanston, IL, USA).
Evaluation of spontaneous seizures
Mice were placed in the OF box and video recorded for 1 h/day for 12 weeks after treatment. In all cases, recordings took place in the morning. Identification of tonic-clonic seizures was performed by trained personnel in a blinded fashion.
Novel object recognition test
The NOR test was conducted for the assessment of visuospatial memory. The test was carried out as described previously.30 A standard squared four-compartment mildly illuminated OF box was employed (LE800SC, Panlab Harvard Apparatus, 90 × 90 × 40 cm). Before performing the test, animals were habituated to the box over 15 min; these data were employed to evaluate their motor spontaneous activity and stereotypes as described below. The test consists of two different stages; during the first one (habituation phase) two identical objects were placed into the box, symmetrically separated from each other, and each mouse was allowed to explore them for 5 min. After a delay of 24 h, the mouse was placed again in the cage and exposed for 5 min to one familiar object in the same position and to a novel object placed in a new location (NOR 24 h phase). All trials were video recorded, and the total time spent exploring each object was measured manually with a stopwatch. To avoid the presence of olfactory trails, the apparatus and the objects were thoroughly cleaned after each trial. The DI was calculated as a percentage according to this equation: (Exploration time of the novel object/Total exploration time) × 100. Consequently, a ratio of 50% reflects equal exploration of the familiar and the novel object, indicating no learning retention, and ratios above 50% are indicative of visuospatial learning retention.
Open-field test
Motor activity was tested for 15 min in a standardized squared four-compartment mildly illuminated OF box (LE800SC, Panlab Harvard Apparatus, 90 × 90 × 40 cm). Trials were video recorded and automatically analyzed with a video tracking system (Ethovision XT 5.0, Noldus Information Technology). Parameters of mean speed (cm/s) and time spent in the central zone (15 cm apart from the walls) were measured and represented. Of note, mice showing very low exploratory activity were excluded from the analysis, since they tend to remain in one corner of the cage and would introduce a bias in the evaluation of anxiety. The presence of stereotypies, indicative of anxiety and hyperactivity, was also evaluated by visual inspection and manually counted.
Marble burying test
Normal exploratory behavior was assessed with the marble burying test.30 Twelve glass marbles were put uniformly in a cage, three marbles per line, and mice were placed in the center of each cage and allowed to interact with them for 30 min. After this period of time, two blind experimenters quantified the number of unburied marbles.
Nest building test
This test is useful for identifying abnormal behavior in mice.65 Animals were placed in a box provided with a piece of tightly packed cotton material (Nestlets Nesting Material, Ancare) and allowed to interact with it overnight. The next day, the nesting index was evaluated and scored as no nest, partial nest, or complete nest covering the mouse.
Rotarod test
Motor coordination, balance, and physical condition were tested in a rotarod apparatus (LE8200 Panlab, Harvard Apparatus). The test was carried out as described previously.30 The day before the test, animals were trained to walk over the rotating rod for 5 min at a constant speed of 12 rpm. For the test, animals were positioned on a rod programmed to rotate with lineal increasing speed, going from 4 to 40 rpm in 5 min. Animals underwent three trials with a resting time of 60 min between them on 2 consecutive days. The time spent in the accelerating cylinder was recorded, representing the mean value.
Hindlimb clasping test
This test can be used as a marker of disease progression in several mouse models of neurodegeneration.66 Animals are grasped by the tail near its base and lifted. Hindlimb position is evaluated for 30 s. The animal receives a score of 0 when hindlimbs are splayed outward, 1 when one of them is retracted toward the abdomen for >50% of the duration of the test, 2 if both hindlimbs are partially retracted for >50% of the time, and 3 if they are completely retracted.
Statistical analysis
GraphPad Prism software was used for analysis. Datasets following normal distribution (D’Agostino and Pearson normality test) were compared by one-way ANOVA with Sidak’s multiple comparisons tests. Otherwise, groups were compared by Kruskal-Wallis with Dunn’s multiple comparison test. The Friedman test was applied to identify intra-group improvement over trials in the MWM test. The log-rank test was used for analysis of survival.
Acknowledgments
This research was funded by projects RTI2018-097730-B-I00/MCI/AEI/FEDER, ERA-NeT E-Rare, AC17/00029 (ISCIII)/FEDER, and Inocente-inocente Foundation. L.M.-J. is a recipient of a Pedro Lopez Berastegui fellowship. We thank the fundraising department of the University of Navarra for its contribution to the sustainability of this project and the animal facility at CIMA for the maintenance of mice. We thank Dr. Julio Artieda for his valuable advice. We would like to express our deepest gratitude to the families of DS patients for their unconditional support and inspiration.
Author contributions
A.R., R.S.-C., M.V., G.G.-A., and R.H.-A. conceived the experiments. A.R., L.M.-J., J.T., M.B., M.G.-A., S.L., E.F.-P., M.J.N., M.R, S.F., C.M., P.G.-M., and E.P. conducted the experiments. A.R., M.V., J.T., M.R., C.M., R.S.-C., and R.H.-A. analyzed the results. A.R., M.V., M.R., R.S.-C., and R.H.-A. wrote the manuscript. All authors reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.08.003.
Contributor Information
Ana Ricobaraza, Email: aricobaraza@unav.es.
Ruben Hernandez-Alcoceba, Email: rubenh@unav.es.
Supplemental information
References
- 1.Claes L., Del-Favero J., Ceulemans B., Lagae L., Van Broeckhoven C., De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y.W., Sullivan J., McDaniel S.S., Meisler M.H., Walsh E.M., Li S.X., Kuzniewicz M.W. Incidence of Dravet Syndrome in a US Population. Pediatrics. 2015;136:e1310–e1315. doi: 10.1542/peds.2015-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu F.H., Mantegazza M., Westenbroek R.E., Robbins C.A., Kalume F., Burton K.A., Spain W.J., McKnight G.S., Scheuer T., Catterall W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 4.Yamakawa K. Molecular and cellular basis: insights from experimental models of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):70–71. doi: 10.1111/j.1528-1167.2011.03006.x. [DOI] [PubMed] [Google Scholar]
- 5.Dravet C. Dravet syndrome history. Dev. Med. Child Neurol. 2011;53(Suppl 2):1–6. doi: 10.1111/j.1469-8749.2011.03964.x. [DOI] [PubMed] [Google Scholar]
- 6.Sakauchi M., Oguni H., Kato I., Osawa M., Hirose S., Kaneko S., Takahashi Y., Takayama R., Fujiwara T. Retrospective multiinstitutional study of the prevalence of early death in Dravet syndrome. Epilepsia. 2011;52:1144–1149. doi: 10.1111/j.1528-1167.2011.03053.x. [DOI] [PubMed] [Google Scholar]
- 7.Hsiao J., Yuan T.Y., Tsai M.S., Lu C.Y., Lin Y.C., Lee M.L., Lin S.W., Chang F.C., Liu Pimentel H., Olive C. Upregulation of Haploinsufficient Gene Expression in the Brain by Targeting a Long Non-coding RNA Improves Seizure Phenotype in a Model of Dravet Syndrome. EBioMedicine. 2016;9:257–277. doi: 10.1016/j.ebiom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colasante G., Lignani G., Brusco S., Di Berardino C., Carpenter J., Giannelli S., Valassina N., Bido S., Ricci R., Castoldi V. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol. Ther. 2020;28:235–253. doi: 10.1016/j.ymthe.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldman D.H., Lossin C. The Nav channel bench series: Plasmid preparation. MethodsX. 2014;1:6–11. doi: 10.1016/j.mex.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cots D., Bosch A., Chillón M. Helper dependent adenovirus vectors: progress and future prospects. Curr. Gene Ther. 2013;13:370–381. doi: 10.2174/156652321305131212125338. [DOI] [PubMed] [Google Scholar]
- 11.Ricobaraza A., Gonzalez-Aparicio M., Mora-Jimenez L., Lumbreras S., Hernandez-Alcoceba R. High-capacity adenoviral vectors: Expanding the scope of gene therapy. Int. J. Mol. Sci. 2020;21:3643. doi: 10.3390/ijms21103643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barcia C., Jimenez-Dalmaroni M., Kroeger K.M., Puntel M., Rapaport A.J., Larocque D., King G.D., Johnson S.A., Liu C., Xiong W. One-year expression from high-capacity adenoviral vectors in the brains of animals with pre-existing anti-adenoviral immunity: clinical implications. Mol. Ther. 2007;15:2154–2163. doi: 10.1038/sj.mt.6300305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunetti-Pierri N., Ng P. Gene therapy with helper-dependent adenoviral vectors: lessons from studies in large animal models. Virus Genes. 2017;53:684–691. doi: 10.1007/s11262-017-1471-x. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez-Aparicio M., Mauleon I., Alzuguren P., Bunuales M., Gonzalez-Aseguinolaza G., San Martín C., Prieto J., Hernandez-Alcoceba R. Self-inactivating helper virus for the production of high-capacity adenoviral vectors. Gene Ther. 2011;18:1025–1033. doi: 10.1038/gt.2011.58. [DOI] [PubMed] [Google Scholar]
- 15.Osteen J.D., Herzig V., Gilchrist J., Emrick J.J., Zhang C., Wang X., Castro J., Garcia-Caraballo S., Grundy L., Rychkov G.Y. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature. 2016;534:494–499. doi: 10.1038/nature17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards K.L., Milligan C.J., Richardson R.J., Jancovski N., Grunnet M., Jacobson L.H., Undheim E.A.B., Mobli M., Chow C.Y., Herzig V. Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc. Natl. Acad. Sci. USA. 2018;115:E8077–E8085. doi: 10.1073/pnas.1804764115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nissenkorn A., Almog Y., Adler I., Safrin M., Brusel M., Marom M., Bercovich S., Yakubovich D., Tzadok M., Ben-Zeev B., Rubinstein M. In vivo, in vitro and in silico correlations of four de novo SCN1A missense mutations. PLoS ONE. 2019;14:e0211901. doi: 10.1371/journal.pone.0211901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosch M.K., Nerbonne J.M., Ornitz D.M. Dual transgene expression in murine cerebellar Purkinje neurons by viral transduction in vivo. PLoS ONE. 2014;9:e104062. doi: 10.1371/journal.pone.0104062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yaguchi M., Ohashi Y., Tsubota T., Sato A., Koyano K.W., Wang N., Miyashita Y. Characterization of the properties of seven promoters in the motor cortex of rats and monkeys after lentiviral vector-mediated gene transfer. Hum. Gene Ther. Methods. 2013;24:333–344. doi: 10.1089/hgtb.2012.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobsson J., Ericson C., Jansson M., Björk E., Lundberg C. Targeted transgene expression in rat brain using lentiviral vectors. J. Neurosci. Res. 2003;73:876–885. doi: 10.1002/jnr.10719. [DOI] [PubMed] [Google Scholar]
- 21.Sehara Y., Shimazaki K., Kurosaki F., Kaneko N., Uchibori R., Urabe M., Kawai K., Mizukami H. Efficient transduction of adeno-associated virus vectors into gerbil hippocampus with an appropriate combination of viral capsids and promoters. Neurosci. Lett. 2018;682:27–31. doi: 10.1016/j.neulet.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Poutou J., Bunuales M., Gonzalez-Aparicio M., German B., Zugasti I., Hernandez-Alcoceba R. Adaptation of vectors and drug-inducible systems for controlled expression of transgenes in the tumor microenvironment. J. Control. Release. 2017;268:247–258. doi: 10.1016/j.jconrel.2017.10.032. [DOI] [PubMed] [Google Scholar]
- 23.Morral N., O’Neal W., Rice K., Leland M., Kaplan J., Piedra P.A., Zhou H., Parks R.J., Velji R., Aguilar-Córdova E. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. USA. 1999;96:12816–12821. doi: 10.1073/pnas.96.22.12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrhardt A., Xu H., Kay M.A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 2003;77:7689–7695. doi: 10.1128/JVI.77.13.7689-7695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuah M.K.L., Schiedner G., Thorrez L., Brown B., Johnston M., Gillijns V., Hertel S., Van Rooijen N., Lillicrap D., Collen D. Therapeutic factor VIII levels and negligible toxicity in mouse and dog models of hemophilia A following gene therapy with high-capacity adenoviral vectors. Blood. 2003;101:1734–1743. doi: 10.1182/blood-2002-03-0823. [DOI] [PubMed] [Google Scholar]
- 26.Wirrell E.C., Laux L., Donner E., Jette N., Knupp K., Meskis M.A., Miller I., Sullivan J., Welborn M., Berg A.T. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations From a North American Consensus Panel. Pediatr. Neurol. 2017;68:18–34.e3. doi: 10.1016/j.pediatrneurol.2017.01.025. [DOI] [PubMed] [Google Scholar]
- 27.Tran C.H., Vaiana M., Nakuci J., Somarowthu A., Goff K.M., Goldstein N., Murthy P., Muldoon S.F., Goldberg E.M. Interneuron Desynchronization Precedes Seizures in a Mouse Model of Dravet Syndrome. J. Neurosci. 2020;40:2764–2775. doi: 10.1523/JNEUROSCI.2370-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tai C., Abe Y., Westenbroek R.E., Scheuer T., Catterall W.A. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA. 2014;111:E3139–E3148. doi: 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruffolo G., Cifelli P., Roseti C., Thom M., van Vliet E.A., Limatola C., Aronica E., Palma E. A novel GABAergic dysfunction in human Dravet syndrome. Epilepsia. 2018;59:2106–2117. doi: 10.1111/epi.14574. [DOI] [PubMed] [Google Scholar]
- 30.Ricobaraza A., Mora-Jimenez L., Puerta E., Sanchez-Carpintero R., Mingorance A., Artieda J., Nicolas M.J., Besne G., Bunuales M., Gonzalez-Aparicio M. Epilepsy and neuropsychiatric comorbidities in mice carrying a recurrent Dravet syndrome SCN1A missense mutation. Sci. Rep. 2019;9:14172. doi: 10.1038/s41598-019-50627-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rock M.L., Karas A.Z., Rodriguez K.B., Gallo M.S., Pritchett-Corning K., Karas R.H., Aronovitz M., Gaskill B.N. The time-to-integrate-to-nest test as an indicator of wellbeing in laboratory mice. J. Am. Assoc. Lab. Anim. Sci. 2014;53:24–28. [PMC free article] [PubMed] [Google Scholar]
- 32.Brücher D., Kirchhammer N., Smith S.N., Schumacher J., Schumacher N., Kolibius J., Freitag P.C., Schmid M., Weiss F., Keller C. iMATCH: an integrated modular assembly system for therapeutic combination high-capacity adenovirus gene therapy. Mol. Ther. Methods Clin. Dev. 2021;20:572–586. doi: 10.1016/j.omtm.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamagata T., Raveau M., Kobayashi K., Miyamoto H., Tatsukawa T., Ogiwara I., Itohara S., Hensch T.K., Yamakawa K. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol. Dis. 2020;141:104954. doi: 10.1016/j.nbd.2020.104954. [DOI] [PubMed] [Google Scholar]
- 34.Stein R.E., Kaplan J.S., Li J., Catterall W.A. Hippocampal deletion of NaV1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet Syndrome. Proc. Natl. Acad. Sci. USA. 2019;116:16571–16576. doi: 10.1073/pnas.1906833116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansen N.A., Dehghani A., Breukel C., Tolner E.A., van den Maagdenberg A.M.J.M. Focal and generalized seizure activity after local hippocampal or cortical ablation of NaV 1.1 channels in mice. Epilepsia. 2020;61:e30–e36. doi: 10.1111/epi.16482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Idema S., Caretti V., Lamfers M.L.M., van Beusechem V.W., Noske D.P., Vandertop W.P., Dirven C.M.F. Anatomical differences determine distribution of adenovirus after convection-enhanced delivery to the rat brain. PLoS ONE. 2011;6:e24396. doi: 10.1371/journal.pone.0024396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen L.A., Harper R.M., Kumar R., Guye M., Ogren J.A., Lhatoo S.D., Lemieux L., Scott C.A., Vos S.B., Rani S., Diehl B. Dysfunctional brain networking among autonomic regulatory structures in temporal lobe epilepsy patients at High Risk of sudden unexpected death in epilepsy. Front. Neurol. 2017;8:544. doi: 10.3389/fneur.2017.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang Y., Chen Q., Yu X., Xia W., Luo C., Huang X., Tang H., Gong Q., Zhou D. A resting-state functional connectivity study in patients at high risk for sudden unexpected death in epilepsy. Epilepsy Behav. 2014;41:33–38. doi: 10.1016/j.yebeh.2014.08.140. [DOI] [PubMed] [Google Scholar]
- 39.Allen L.A., Vos S.B., Kumar R., Ogren J.A., Harper R.K., Winston G.P., Balestrini S., Wandschneider B., Scott C.A., Ourselin S. Cerebellar, limbic, and midbrain volume alterations in sudden unexpected death in epilepsy. Epilepsia. 2019;60:718–729. doi: 10.1111/epi.14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryvlin P., Nashef L., Lhatoo S.D., Bateman L.M., Bird J., Bleasel A., Boon P., Crespel A., Dworetzky B.A., Høgenhaven H. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- 41.Ritter-Makinson S., Clemente-Perez A., Higashikubo B., Cho F.S., Holden S.S., Bennett E., Chkhaidze A., Eelkman Rooda O.H.J., Cornet M.C., Hoebeek F.E. Augmented Reticular Thalamic Bursting and Seizures in Scn1a-Dravet Syndrome. Cell Rep. 2019;26:54–64.e6. doi: 10.1016/j.celrep.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalume F., Oakley J.C., Westenbroek R.E., Gile J., de la Iglesia H.O., Scheuer T., Catterall W.A. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol. Dis. 2015;77:141–154. doi: 10.1016/j.nbd.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han S., Yu F.H., Schwartz M.D., Linton J.D., Bosma M.M., Hurley J.B., Catterall W.A., De La Iglesia H.O. Na V1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc. Natl. Acad. Sci. USA. 2012;109:E368–E377. doi: 10.1073/pnas.1115729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papale L.A., Makinson C.D., Christopher Ehlen J., Tufik S., Decker M.J., Paul K.N., Escayg A. Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+) Epilepsia. 2013;54:625–634. doi: 10.1111/epi.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warren A.E.L., Harvey A.S., Vogrin S.J., Bailey C., Davidson A., Jackson G.D., Abbott D.F., Archer J.S. The epileptic network of Lennox-Gastaut syndrome: Cortically driven and reproducible across age. Neurology. 2019;93:e215–e226. doi: 10.1212/WNL.0000000000007775. [DOI] [PubMed] [Google Scholar]
- 46.Barker G.R.I., Bird F., Alexander V., Warburton E.C. Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J. Neurosci. 2007;27:2948–2957. doi: 10.1523/JNEUROSCI.5289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Bruin J.P.C., Sànchez-Santed F., Heinsbroek R.P.W., Donker A., Postmes P. A behavioural analysis of rats with damage to the medial prefrontal cortex using the Morris water maze: evidence for behavioural flexibility, but not for impaired spatial navigation. Brain Res. 1994;652:323–333. doi: 10.1016/0006-8993(94)90243-7. [DOI] [PubMed] [Google Scholar]
- 48.Jo Y.S., Park E.H., Kim I.H., Park S.K., Kim H., Kim H.T., Choi J.S. The medial prefrontal cortex is involved in spatial memory retrieval under partial-cue conditions. J. Neurosci. 2007;27:13567–13578. doi: 10.1523/JNEUROSCI.3589-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carty N., Lee D., Dickey C., Ceballos-Diaz C., Jansen-West K., Golde T.E., Gordon M.N., Morgan D., Nash K. Convection-enhanced delivery and systemic mannitol increase gene product distribution of AAV vectors 5, 8, and 9 and increase gene product in the adult mouse brain. J. Neurosci. Methods. 2010;194:144–153. doi: 10.1016/j.jneumeth.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorick C.M., Sheybani N.D., Curley C.T., Price R.J. Listening in on the Microbubble Crowd: Advanced Acoustic Monitoring for Improved Control of Blood-Brain Barrier Opening with Focused Ultrasound. Theranostics. 2018;8:2988–2991. doi: 10.7150/thno.26025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cestèle S., Schiavon E., Rusconi R., Franceschetti S., Mantegazza M. Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc. Natl. Acad. Sci. USA. 2013;110:17546–17551. doi: 10.1073/pnas.1309827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhifallah S., Lancaster E., Merrill S., Leroudier N., Mantegazza M., Cestèle S. Gain of function for the SCN1A/HNAv1.1-L1670W mutation responsible for familial hemiplegic migraine. Front. Mol. Neurosci. 2018;11:232. doi: 10.3389/fnmol.2018.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berecki G., Bryson A., Terhag J., Maljevic S., Gazina E.V., Hill S.L., Petrou S. SCN1A gain of function in early infantile encephalopathy. Ann. Neurol. 2019;85:514–525. doi: 10.1002/ana.25438. [DOI] [PubMed] [Google Scholar]
- 54.Salgueiro-Pereira A.R., Duprat F., Pousinha P.A., Loucif A., Douchamps V., Regondi C., Ayrault M., Eugie M., Stunault M.I., Escayg A. A two-hit story: Seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol. Dis. 2019;125:31–44. doi: 10.1016/j.nbd.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 55.Martín-Suárez S., Abiega O., Ricobaraza A., Hernandez-Alcoceba R., Encinas J.M. Alterations of the Hippocampal Neurogenic Niche in a Mouse Model of Dravet Syndrome. Front. Cell Dev. Biol. 2020;8:654. doi: 10.3389/fcell.2020.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 57.Toietta G., Pastore L., Cerullo V., Finegold M., Beaudet A.L., Lee B. Generation of helper-dependent adenoviral vectors by homologous recombination. Mol. Ther. 2002;5:204–210. doi: 10.1006/mthe.2002.0532. [DOI] [PubMed] [Google Scholar]
- 58.Jager L., Ehrhardt A. Persistence of high-capacity adenoviral vectors as replication-defective monomeric genomes in vitro and in murine liver. Hum. Gene Ther. 2009;20:883–896. doi: 10.1089/hum.2009.020. [DOI] [PubMed] [Google Scholar]
- 59.Schwenk F., Baron U., Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W., Sheng J., Guo J., Gao F., Zhao X., Dai J., Wang G., Li K. Tumor necrosis factor-α enhances voltage-gated Na+ currents in primary culture of mouse cortical neurons. J. Neuroinflammation. 2015;12:126. doi: 10.1186/s12974-015-0349-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 62.Dunah A.W., Wyszynski M., Martin D.M., Sheng M., Standaert D.G. α-actinin-2 in rat striatum: localization and interaction with NMDA glutamate receptor subunits. Brain Res. Mol. Brain Res. 2000;79:77–87. doi: 10.1016/s0169-328x(00)00102-9. [DOI] [PubMed] [Google Scholar]
- 63.Ricobaraza A., Cuadrado-Tejedor M., Pérez-Mediavilla A., Frechilla D., Del Río J., García-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 64.Gelinas J.N., Khodagholy D., Thesen T., Devinsky O., Buzsáki G. Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nat. Med. 2016;22:641–648. doi: 10.1038/nm.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deacon R.M. Assessing nest building in mice. Nat. Protoc. 2006;1:1117–1119. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- 66.Guyenet S.J., Furrer S.A., Damian V.M., Baughan T.D., La Spada A.R., Garden G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010;39:1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.