

Abstract
Skeletal muscle symptoms strongly contribute to mortality of myotonic dystrophy type 1 (DM1) patients. DM1 is a neuromuscular genetic disease caused by CTG repeat expansions that, upon transcription, sequester the Muscleblind-like family of proteins and dysregulate alternative splicing of hundreds of genes. However, mis-splicing does not satisfactorily explain muscle atrophy and wasting, and several other contributing factors have been suggested, including hyperactivated autophagy leading to excessive catabolism. MicroRNA (miR)-7 has been demonstrated to be necessary and sufficient to repress the autophagy pathway in cell models of the disease, but the origin of its low levels in DM1 was unknown. We have found that the RNA-binding protein Musashi-2 (MSI2) is upregulated in patient-derived myoblasts and biopsy samples. Because it has been previously reported that MSI2 controls miR-7 biogenesis, we tested the hypothesis that excessive MSI2 was repressing miR-7 maturation. Using gene-silencing strategies (small interfering RNAs [siRNAs] and gapmers) and the small molecule MSI2-inhibitor Ro 08-2750, we demonstrate that reducing MSI2 levels or activity boosts miR-7 expression, represses excessive autophagy, and downregulates atrophy-related genes of the UPS system. We also detect a significant upregulation of MBNL1 upon MSI2 silencing. Taken together, we propose MSI2 as a new therapeutic target to treat muscle dysfunction in DM1.
Keywords: myotonic dystrophy, muscle dysfunction, MSI2, miR-7, autophagy, antisense oligonucleotides, muscle atrophy, myotubes
Graphical abstract
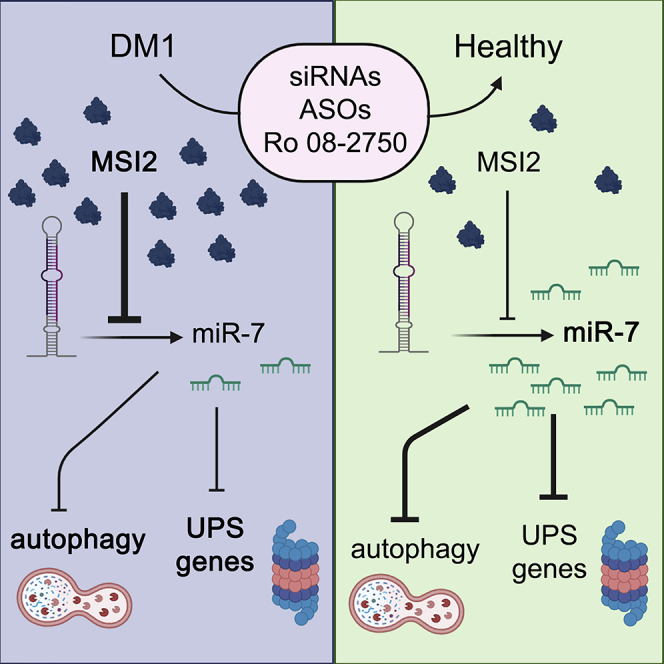
Muscle dysfunction is the strongest contributor to myotonic dystrophy type 1 (DM1) mortality. Using patient-derived muscle cells, we found that Musashi-2 (MSI2) was increased in DM1 and that their reduction by four independent approaches improved molecular and cell phenotypes. We propose MSI2 as a new therapeutic target for DM1.
Introduction
Myotonic dystrophy type 1 (DM1) is a severe and chronically debilitating disease caused by an expansion of the CTG trinucleotide repeat in the 3′ untranslated region (3′ UTR) of the DM1 protein kinase (DMPK) gene. DM1 is autosomal dominant and can affect newborns to elderly people. Adult-onset patients may become physically disabled and may have a shortened lifespan. Most symptoms are neuromuscular, including muscle weakness (myopathy), muscle stiffness and trouble relaxing muscles (myotonia), and progressive muscle wasting (atrophy), but DM1 is characteristically multisystemic and also affects the heart, the brain, and the smooth musculature. Pneumonia and respiratory distress due to muscle weakness and atrophy and heart failure are the most frequent causes of death.1
Mutant DMPK transcripts in skeletal muscle, heart, and brain tissue get retained in the cell nucleus into microscopically visible ribonuclear foci.2 CUG trinucleotide expansions fold into stable, double-stranded stem-loops that sequester the splicing factors Muscleblind-like (MBNL).3 CUGBP Elav-like family member 1 (CELF1) regulates alternative splicing antagonistically to MBNL1. In contrast to MBNL1, CELF1 is not sequestered into ribonuclear foci but is hyper-activated and stabilized in the cell nucleus.4 Like CELF1, the RNA-binding protein heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) is upregulated in DM1 and antagonizes MBNL activity inducing fetal splicing shifts.5 In the context of splicing alterations due to MBNL1/CELF1/hnRNPA1 imbalance, the post-transcriptional control of muscle genes compromises the correct skeletal muscle differentiation and function. One example is the mis-splicing of dystrophin (DMD), induced by MBNL1, that affects muscle architecture and fiber maintenance contributing to the dystrophic process characteristic of DM1 skeletal muscles.6 Additionally, the re-expression of the embryonic M2 isoform of pyruvate kinase (PKM2) in DM1 alters glucose metabolism homeostasis leading to defects in energy metabolism associated with muscle dysfunction.7
Muscle wasting is a critical symptom in DM1 patients, and multiple contributing factors have been proposed to explain muscle loss.8 Several studies support a relevant role for hyperactivated autophagy. In a Drosophila DM1 model and DM1 myotubes, the autophagic flux is overactivated, and genetic or chemical blockade of this pathway, by mechanistic target of rapamycin kinase (mTOR) upregulation or chloroquine treatment, an autophagy blocker, improves muscle function and molecular markers of muscle atrophy.9,10 In the same Drosophila model, overexpression of Muscleblind is sufficient to recover muscle area and expression of autophagy-related genes.9 Muscle wasting and histological defects are also found in an inducible mouse model that expresses 960 CUG repeats, in which proteins involved in the autophagic pathway are upregulated, thus suggesting an imbalance between anabolic and catabolic pathways that normally regulate muscle mass.11 In the widely used human skeletal actin long repeats (HSALR) mouse model, expressing 250 CUG repeats in the skeletal musculature, treatment with chloroquine also rescues muscle dysfunction defects, possibly through increased levels of MBNL1 and MBNL2.10 Importantly, primary myoblasts derived from DM1 patients show normal myogenesis but impaired apoptosis and autophagy.12
Other studies have shown that some microRNAs (miRNAs) are mis-expressed in skeletal muscle, heart, and blood samples from DM1 patients and DM1 muscle cells and mouse models.13, 14, 15, 16, 17 Specifically, miRNA (miR)-7 levels are reduced in human skeletal muscle biopsies and DM1 muscle cells. Through complementary miR-7 modulation strategies, it was demonstrated that downregulation of miR-7 in control myotubes originates DM1-like phenotypes, whereas restoration of normal miRNA levels in DM1 cells downregulates excessive autophagy and ubiquitin proteasome system (UPS). Thus, miR-7 is necessary and sufficient to inhibit major catabolic processes contributing to muscle wasting, placing miR-7 as a new target to treat DM1.13,17 Furthermore, independent studies place miR-7 as a critical inhibitor of the autophagy pathway through upregulation of the mTOR signaling and direct translational repression of critical autophagy genes, namely ATG7, ULK2, and ATG4A.18 miR-7 biogenesis is regulated by Musashi homolog 2 (MSI2) and Hu antigen R (HuR). MSI2 binds through HuR to the conserved terminal loop of pri-mir-7-1, resulting in the stabilization of the immature miRNA reducing the levels of the mature form.19 In mammals, the two family members MSI1 and MSI2 are highly expressed in stem cells but almost absent from differentiated tissues.20 Moreover, it was suggested in mouse models that Msi proteins are splicing regulators, as numerous alternative exons were altered after Msi1 or -2 knockout or overexpression.21 However, as Msi was mainly located in the cytoplasm, effects in pre-mRNA splicing might be indirect. Here, we report that MSI2 is upregulated in DM1, which explains the reduced levels of miR-7 in disease samples, and propose MSI2 as a new contributing factor to explain the muscle atrophy of the disease.
Results
MSI2 overexpression correlates with decreased levels of miR-7 in DM1
We previously reported that miR-7 is reduced in DM1 and that its normalization rescues muscle wasting phenotypes.17 However, the reasons leading to its reduction remained unknown. To shed light on this problem, we first measured miR-7 levels in deltoid muscle biopsies from 17 healthy controls (37 years ± 3; median ± SEM) and 16 DM1 donors (45 years ± 3), in six primary myoblast lines obtained from the above biopsies (3 control and 3 DM1), and DM1 fibroblasts expressing 1,300 CTG repeats22 transdifferentiated into multinucleated myotubes (TDMs) for 7 days (Figure 1A). In all cases, we confirmed significantly lower levels of miR-7 in DM1 samples. Considering previous work demonstrating that the RNA-binding protein MSI2 blocks miR-7 biogenesis,19 we hypothesized that MSI2 could be upregulated in DM1 cells (Figures 1B−1E). Indeed, we found an excess of MSI2 in muscle biopsies and TDMs (near 60 times at the protein level compared to control TDMs) but not in primary myoblasts. Furthermore, we immunodetected MSI2 in healthy control and DM1 TDMs, where we confirmed the increased expression in DM1 muscle cells and found that MSI2 localization was predominantly cytoplasmic (Figures 1C and 1D). Consistent with our hypothesis that increased MSI2 in DM1 might contribute to low levels of mature miR-7, the relative expressions of miR-7 and MSI2 protein correlated negatively and significantly in biopsies (r = −0.54, p = 0.045) (Figure 1F). To further validate MSI2 overexpression in DM1 and that this alteration was relevant in the disease, we resorted to publicly available RNA sequencing (RNA-seq) and ankle dorsiflexion strength data from 40 patients and ten controls.23 Importantly, MSI2 was significantly overexpressed (over 35%) in DM1 compared to controls, in full agreement with results obtained in our muscle sample cohort (Figure 1G). The analysis also supported that MSI2 levels negatively correlated with ankle dorsiflexion strength (Figure 1H; r = −0.48, p = 0.0017).
Figure 1.

MSI2 is upregulated in DM1 samples
Quantification by qRT-PCR of the relative expression of (A) miR-7 and (B) MSI2 in human biopsies (gray; n = 17 controls; n = 16 DM1), cultured primary myoblasts (blue; n = 3), and transdifferentiated myoblasts (TDMs; orange; n = 1). miR-7 quantification is relative to endogenous U1 and U6 levels, and MSI2 is referenced to GADPH and GPI expression. Sample sizes indicate biological replicates. At least three independent experiments were carried out for primary myoblasts and TDMs, and three technical replicates were performed from each biological sample in all cases. Representative confocal images of MSI2 immunostained (green; C) healthy controls and (D) DM1 TDMs. Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm. (E) Analysis of relative protein levels of MSI2 by western blot and representative blots of protein extracts obtained from human biopsies, cultured primary myoblasts, and TDMs. β-actin and GAPDH were used as endogenous controls to normalize protein levels of TDMs or human biopsies and primary myoblasts, respectively. Samples sizes as in (A) except n = 3 for primary myoblasts and TDMs. The scatterplots show the median with interquartile range for human biopsies and mean ± SEM for cultured cells. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 according to Student’s t test. (F) Pearson’s correlations between the relative miR-7 and MSI2 protein expression levels in human biopsies (gray). (G) The scatterplot represents MSI2 transcripts per million (TPM) in biopsies from 40 DM1 patients and 10 controls (according to Wang et al.23) with the median and interquartile range. ∗∗∗∗p < 0.0001 according to Student’s t test. (H) Pearson’s correlation between MSI2 TPM in DM1 biopsies and ankle dorsiflexion strength (ADS) measured in biopsy donors according to Wang et al.23
Silencing of MSI2 rescues cell parameters related to muscle disease in TDMs
To confirm that excessive MSI2 was involved in muscle-wasting mechanisms, we targeted MSI2 transcripts by small interfering RNAs (siRNAs) and gapmers, triggering RNAi and RNase-H degradation, respectively. We tested a combination of two siRNAs and a control with a random sequence for 48 h in DM1 TDMs. MSI2 levels were quantified finding a reduction of transcripts of almost 70% compared to the same cells treated with the control (Figure 2A). Importantly, this reduction had functional consequences in the cell since the direct target miR-7 increased (more than three times). Additionally, we quantified TGFBR1 levels, as it was demonstrated to be directly regulated by MSI224 and also P21, which, in contrast to TGFBR1, is inhibited by MSI2.25 TGFBR1 levels dropped (near 40%), and P21 levels, however, did not significantly change (Figure 2B). MSI2 silencing was also assessed at the level of the strong myotubes fusion phenotype described for TDMs, which we previously showed that miR-7 overexpression was sufficient to improve.17 After 7 days of differentiation, TDMs treated with siRNAs were immunodetected for Desmin, a class-III intermediate filament protein widely used as a marker of myogenic cells (Figures 2C and 2D). The reduction in MSI2 levels was sufficient to improve the fusion capacity of the cells to more than 70% compared to a mere 40% in the same cells treated with the scrambled siRNA (Figure 2E). Myotube diameter trended higher than controls upon MSI2 silencing but did not reach statistical significance (Figure 2F).
Figure 2.

MSI2 is reduced after siRNA treatment in DM1 TDMs
Quantification by qRT-PCR of (A) MSI2 and (B) miR-7, P21, and TGFBR1 in DM1 TDMs treated with 100 nM scrambled (sc) siRNA or a combination of two siRNAs targeting MSI2 at a final concentration of 100 nM. Levels of MSI2, P21, and TGFBR1 are relative to the mean of GAPDH and GPI, whereas miR-7 was normalized to U1 and U6. Data come from three biological replicates. (C and D) Representative confocal images of Desmin-immunostained (green) human TDMs for 7 days after 100 nM siRNA transfection into DM1 cells. Nuclei were counterstained with DAPI (blue). Scale bar, 200 μm. Quantification of (E) myogenic fusion index and (F) myotube diameter of DM1 myoblasts transfected with the indicated siRNA (between 10 and 15 images analyzed, totaling some 400−500 nuclei). The statistical analysis was performed comparing values from treated cells with their corresponding scramble (black dashed line in B). The bar graphs show mean ± SEM. ∗p < 0.05 and ∗∗∗p < 0.001 according to Student’s t test.
As alternatives to siRNAs, we designed three antisense oligonucleotides (ASOs) with RNase-H triggering activity (also known as gapmers) against different exons within the MSI2 transcript isoform 1 and a scrambled control oligonucleotide with the same chemistry. Initially, we assayed their toxicity profile in control TDMs and observed low toxicity, as at near 1,000 nM, the cell viability was still higher than 50%. Two different concentrations were used in subsequent experiments: 30 nM, at which around 90% of cells were viable, and 150 nM, at which between 70% and 80% of the cells were viable when transfected with any of the three ASOs (Figure 3A). To confirm the activity of the ASOs, we measured MSI2 relative expression upon transfection in DM1 TDMs. We observed a significant reduction with ASO1 and -3, but ASO1 was better at both tested concentrations. Similar results were observed quantifying MSI2 protein levels (Figures 3B and 3C). The scrambled sequence ASO did not significantly reduce MSI2 transcripts at 150 nM in transfected control TDMs (Figure S1). Overall, the level of silencing achieved with ASOs was higher than that obtained with siRNAs and, as expected, led to significant changes in direct MSI2 targets. miR-7 was derepressed in ASO-treated DM1 cells, except for ASO2, which did not significantly alter miR-7 levels compared to scrambled ASO (Figure 3D). Given the lack of activity on MSI2 transcripts and the miR-7 target, ASO2 was discarded as a potential molecule to silence the gene. The effect of reduced MSI2 levels on P21 and TGFBR1 targets was also studied (Figure 3E). P21 was strongly derepressed in muscle cells treated with ASO3 at the lowest concentration, whereas TGFBR1 behaved the opposite at 150 nM of ASO1. After 7 days of differentiation, TDMs were stained with an antibody against Desmin. Quantification of fusion capacity and myotube diameter showed that reducing MSI2 levels in DM1 TDMs restored the fusion index dramatically, in a dose-dependent manner in the case of ASO3, and the size of the myotubes (Figures 3F−3J).
Figure 3.

Atrophic muscle markers ameliorate in DM1 TDMs upon MSI2 reduction
(A) Cell growth inhibition assay by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) method. Human control (CNT) myoblasts were transfected with a range of concentrations of ASO1, -2, and -3 (n = 4). TC10 (33.1 nM, 158.3 nM, and 40.6 nM for ASO1, -2 and -3, respectively) was obtained using the least-squares non-linear regression model. Vertical dashed lines indicate 30 and 150 nM concentrations used in the subsequent experiments. MSI2 quantification by (B) qRT-PCR relative to GAPDH and GPI and (C) by western blot relative to β-actin expression. (D) Quantification of miR-7 relative to U1 and U6 in DM1 TDMs transfected with the indicated ASOs. (E) Quantification by qRT-PCR of P21 and TGFBR1 expression relative to the mean of GAPDH and GPI levels after treatment of the muscle cells with the indicated concentrations of ASOs. Relative expression of control myotubes 1.412 ± 0.068 and 0.643 ± 0.042 for P21 and TGFBR1, respectively. (D and E) The sample size was three biological replicates. (F) Analysis of myogenic fusion index and (G) myotube diameter of DM1 myoblasts transfected with the indicated concentrations of ASOs or scramble (n = 10−15 images analyzed). (H−J) Representative confocal images of Desmin-immunostained (green) human TDMs for 7 days after ASO or scrambled control transfection at 150 nM. Nuclei were counterstained with DAPI (blue). Scale bar, 40 μm. The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, according to Student’s t test. In all cases, the statistical analysis compares the values of ASO and scrambled-treated cells (black dashed line or gray bar).
Reduction of MSI2 inhibits hyperactivated autophagy and muscle atrophy-related genes in DM1 TDMs
Given the involvement of MSI2 in miR-7 biogenesis,19 we investigated the expression of critical autophagy pathway genes in DM1 and control TDMs, namely ATG3, ATG4A, ATG5, ATG7, and mTOR (Figure 4A). Their expression was rescued significantly, approaching normal levels in all cases, after treatment with ASOs at one or both concentrations tested, except ATG5, which only significantly reduced its expression at the highest concentration of ASO1 (Figure 4B). Autophagy status in DM1 TDMs in response to MSI2 levels was also assessed at the protein level. We evaluated the expression of ATG4A and ATG7, direct miR-7 targets, and observed that ATG4A was dramatically reduced (around 50% compared to scrambled ASO) at the highest concentration of ASOs, whereas ATG7 was also repressed in response to low MSI2 upon ASO transfection (Figures 4C and 4D). Additionally, we tested the activation of the AKT pathway by calculating the phospho(Ser473)-AKT (AKT-P)-to-total-AKT ratio. This is a relevant parameter, as AKT controls both protein synthesis via mTOR and protein degradation (including autophagy) via the FoxO family of transcription factors.26,27 Our data show a robust increase in AKT-P/AKT ratio in cells treated with 150 nM ASO1 or ASO3 (Figure 4E). The soluble-to-autophagosome-associated-LC3 (LC3-I and LC3-II, respectively) ratio was also quantified since the transformation of LC3-I into LC3-II is an indicator of increased autophagic flux in cells.28 Notably, autophagic flux dropped to about one-half when MSI2 was silenced with the highest concentration of ASOs (Figure 4F). Finally, we quantified P62 scaffold protein levels that deliver proteins committed for lysosomal degradation to the autophagosome. Low levels of autophagic activity lead to the accumulation of P62, as autophagy itself degrades the protein.29,30 We did not detect any significant increase in P62 levels upon ASO treatment (Figure S2).
Figure 4.

Inhibition of MSI2 expression in DM1 TDMs restores impaired autophagic markers
(A) qRT-PCR quantification of the relative expression of the indicated genes involved in different autophagy pathway steps in DM1 and control (green dashed lines) myotubes. Statistical analyses were performed comparing DM1 versus controls. (B) Analyses of the expression of genes quantified in (A) in DM1 myotubes treated with ASOs targeting MSI2 transcripts. All comparisons for the statistical analyses were performed using their corresponding scrambled control (dashed lines). The mean of GAPDH and GPI expression was used as reference (n = 3). Quantification and representative western blots of (C) ATG4A, (D) ATG7, (E) AKT-P/AKT total protein levels, and (F) LC3-II/LC3-I ratio in DM1 TDMs treated with MSI2 targeting or scrambled ASOs at the indicated concentrations. Green dashed lines indicate protein expression levels in control myotubes. β-actin was used as an endogenous control to normalize protein levels (n = 3). The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 according to Student’s t test.
To further evaluate the autophagy status in DM1 TDMs treated with ASOs, we performed immunofluorescence staining to detect LC3. In cells transfected with the scrambled ASO, a heavily punctated pattern was observed. These spots correspond to LC3 conjugated to membrane-bound phosphatidylethanolamine (LC3-II) in autophagosomes.31 However, upon reduction in MSI2 levels, these spots disappear, and the signal becomes mainly cytoplasmic and diffuse, indicating an increase in soluble LC3-I and, therefore, lower autophagic flux (Figures 5A−5F). We analyzed LC3 immunofluorescence results by quantifying LC3 dots per unit of area (Figure 5G). Analyzed dots correspond to the LC3-II isoform involved in the autophagosome formation, an essential step in the mechanism underlying autophagy. Our results confirmed increased puncta formation in DM1 TDMs (around four times) and also that this parameter can be modulated upon MSI2 targeting with ASOs. Both ASO1 and -3 caused a significant decrease in LC3 dots when added to DM1 TDMs compared to cells treated with the scrambled ASO at the same concentration. The acidotropic dye LysoTracker marks the acidic cellular compartments, including lysosomes and autophagolysosomes, which are excellent markers of autophagy flux levels. When the muscle cells were treated with the ASOs at the lowest concentration, no noticeable change was observed in the signal; however, treatment with high ASO concentration strongly lowered the signal, thus confirming the reduction in autophagic activity when MSI2 gets silenced (Figures 5H−5M).
Figure 5.

Blocking MSI2 expression improves muscle wasting markers in DM1 TDMs
(A−F) Fluorescence confocal images of LC3 immunostaining (red) or (H−M) LysoTracker staining (red) in 7-day TDMs treated for 48 h with (A, D, H, and K) scramble, (B, E, I, and L) ASO1, or (C, F, J, and M) ASO3 at the indicated concentrations. Nuclei were counterstained with DAPI. Scale bar, 20 μm. (G) Quantification of LC3 puncta per square micrometer. Each condition was compared to cells treated with its corresponding scrambled oligonucleotides. The green dashed line shows the mean value obtained for control muscle cells. Quantification by qRT-PCR of the relative expression of genes involved in catabolic pathways leading to muscle degradation in DM1 and control myotubes (N) or DM1 cells treated with the indicated concentration of ASOs (O). Statistical analyses were carried out comparing DM1 with the control condition (N) or comparing ASOs with their corresponding scramble (O). GAPDH and GPI were used as endogenous controls (n = 3). The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 according to Student’s t test.
Since we have previously reported that increased miR-7 levels rescued expression of muscle atrophy-related genes,17 we quantified the transcripts of a set of genes involved in protein degradation belonging to the UPS system, namely FBXO32, MuRF1, FOXO1, FOXO3, AKT1, IGF1, IL-1β, and MSTN. We observed that all of them were misregulated in DM1 myotubes demonstrating pathological upregulation of this pathway (Figure 5N). Consistent with the observations upon miR-7 modulation, MSI2 silencing rescued the expression of most of the genes studied, at least at one of the two concentrations tested (Figure 5O). IGF1, FBXO32, and MSTN levels were significantly rescued by at least one ASO concentration. Taken together, these results indicate that ASO-mediated reduction in MSI2 transcript level mimic gene expression rescues in autophagy and UPS systems brought about by miR-7 agomiRs in DM1 TDMs.
Reducing MSI2 boosts MBNL1 levels in DM1 TDMs
Considering the key role of MBNL and CELF1 proteins in the pathogenesis of DM1 and previous results where inhibition of autophagy in DM1 promoted MBNL1 activity,17 we quantified MBNL1 and MBNL2 and CELF1 levels after ASO treatments. Our data show that when DM1 muscle cells were treated with ASOs at 150 nM, MBNL1 protein was enhanced, achieving levels 50% higher than those obtained in cells treated with scrambled ASO (Figure 6A). No effect was observed on MBNL1 transcript levels upon ASO treatment (Figure S3). In contrast, MBNL2 and CELF1 levels remained unchanged after ASO treatment (Figures 6B and 6C; Figure S3). These results were confirmed by immunofluorescence staining to detect MBNL1 and MBNL2 in ASO-treated DM1 TDMs. For MBNL1, a low concentration of ASOs did not change the intensity or the pattern of the signal compared to scrambled-treated controls, but at the high ASO concentration, a boost in the green signal became evident both in the cytoplasm and in the cell nucleus (Figures 6D−6I). An antibody against MBNL2 gave results consistent with the western blot quantification, except for a slight increase with 150 nM ASO1, although the signal was very weak given the low expression of MBNL2 (Figures 6J−6O). To confirm that the additional protein detected was functional, we studied splicing defects regulated by MBNL proteins (Figures 6P−6R). ASO-induced MBNL1 increase was sufficient to reduce the inclusion of pyruvate kinase (PK) exon 10 (fetal PKM2 isoform)7 in all of the experimental conditions evaluated. The inclusion of SERCA1 exon 22 (ASO3) or NFIX exon 7 (both tested ASOs) further confirms the relevance of the extra MBNL1 detected.
Figure 6.

Degradation of MSI2 transcripts enhances MBNL1 levels in 7-day differentiated TDMs
(A) Quantification and representative blots of MBNL1 levels in protein extracts obtained from DM1 TDMs treated with the indicated concentrations of MSI2-targeting or scrambled ASOs. (B and C) The western blots shown are representative of the amounts of MBNL2 and CELF1. β-actin was used as an endogenous control (n = 3). Representative confocal images of (D−I) MBNL1 (green) or (J−O) MBNL2 (red) immunodetection in DM1 TDMs treated for 48 h with scramble (D, J, G, and M), ASO1 (E, K, H, and N), or ASO3 (F, L, I, and O) at the indicated concentrations. Nuclei were counterstained with DAPI (scale bar, 50 μm). (P−R) Semiquantitative RT-PCR analyses and representative agarose gels of splicing events altered in DM1 cells: (P) PKM isoforms represented as the percent of PKM2 and (Q) SERCA1 (exon 22) and (R) NFIX (exon 7) represented as the percent of exon inclusion. GAPDH was used as an internal control (n = 3). In all cases, the statistical analyses were performed comparing values from treated cells with those obtained for cells treated with the scrambled ASO at the same concentration. Green dashed lines indicate values obtained for healthy control TDMs. The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, according to Student’s t test.
MSI2 activity inhibitor Ro 08-2750 improves phenotypes in DM1 muscle cells
MSI2 contains two highly conserved RNA-binding domains. The first is crucial for the binding specificity, whereas the second adds affinity for its target sequences.32 It was recently demonstrated that Ro 08-2750 binds to the first RNA-binding site, thus blocking MSI2 ability to bind to its target RNAs and consequently, the regulation that MSI2 exerts over them.33 Considering this, we treated DM1 TDMs differentiated for 7 days with 10 and 15 μM Ro 08-2750, which were concentrations below TC10 (11.44 μM) and below TC50 (20.06 μM; Figure S4A), respectively. MSI2 levels were analyzed in cells treated with the small molecule, and according to the described mechanism of action of Ro 08-2750,33 a mild effect on MSI2 levels was observed upon the treatment (Figures 7A and 7B; Figure S4). Consistent with its role in miR-7 biogenesis, DM1 cells treated with the compound at 10 μM showed a significant 2.5-fold increase in miR-7 levels compared to control DM1 TDMs (DMSO; Figure 7C; Figure S4). TGFBR1 expression, a direct target of MSI2, was also significantly reduced by 10 μM of Ro 08-2750, whereas for P21, no change was detected (Figure 7D; Figure S4). Since the strongest effect of the molecule was detected at 10 μM, we decided to test the dose response on evaluated phenotypes of lower concentrations. For this purpose, we treated DM1 TDMs with 1 and 3 μM of Ro 08-2750. No effect was detected on MSI2 levels or its targets P21 and TGFBR1 at 1 μM. However, 3 μM was sufficient to significantly de-repress miR-7 biogenesis leading to levels similar to those obtained with the 10 μM treatment (Figure S4)
Figure 7.

Downregulation of MSI2 activity by treatment with the small molecule Ro 08-2750 improves DM1-related phenotypes
MSI2 quantification by (A) qRT-PCR, relative to the mean of GAPDH, GPI, and HPRT1 levels, and (B) western blot in 7-day differentiated DM1 TDMs treated for 48 h with 10 or 15 μM Ro 08-2750 or with 0.8% DMSO as control. Bar graph and representative blot images of MSI2 immunodetection are shown. β-actin expression was used as an endogenous control. (A and B) n = 6 for myotubes treated with DMSO and 10 μM Ro 08-2750. (C) Relative expression levels of miR-7 were measured by qRT-PCR. Data were normalized to the mean of U1 and U6 levels. (D) Quantification by qRT-PCR of TGFBR1 after small-molecule treatment relative to the mean of GAPDH, GPI, and HPRT1 expression levels used as an endogenous reference. In all qRT-PCR and western blots, the sample size was n = 3, unless otherwise specifically indicated. Representative confocal images of Desmin-immunostained (green) human DM1 TDMs for 7 days after treatment with (E) DMSO as control or with Ro 08-2750 compound at (F) 10 μM or (G) 15 μM. Quantification of the percentage of myogenic fusion index (H) and myotube diameter (I) of DM1 TDMs with the indicated concentrations of the compound (n = 10−15 images). Quantification by western blot of (J) ATG4A, (K) ATG7, and (L) P62. Representative blots used for quantification are shown below the bar graphs. β-actin was used as an endogenous control to normalize protein levels (n = 3). Representative confocal images of LysoTracker staining (red) of 7-day DM1 TDMs treated with (M) DMSO as control or with the Ro 08-2750 compound at (N) 10 μM or (O) 15 μM. (E−G and M−O). Scale bars correspond to 20 μm, and the nuclei were counterstained with DAPI. The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 according to Student’s t test.
The fusion index and myotube diameter were obtained in Desmin-immunostained cells to check whether the observed reduction in MSI2 levels and activity inhibition had any impact on the DM1 cell model (Figures 7E−7G). The fusion index and diameter of the cells improved significantly at both concentrations of Ro 08-2750, but the rescue was more robust at 10 μM (Figures 7H and 7I). Additionally, we evaluated the fusion index after treating the cells with 1 or 3 μM of Ro 08-2750, and we confirmed that the recovery was in a dose-dependent manner reaching a peak at 10 μM (Figure S4). Specifically, the recovery percentage was 48%, 77%, 98%, and 63% when cells were treated at 1, 3, 10, and 15 μM of Ro 08-2750. Considering the results obtained at the different concentrations of Ro 08-2750 tested, we found that 10 μM was the optimum concentration to work with the compound, since when treating DM1 TDMs with concentrations below or above, the rescues disappeared or worsened, respectively. Given the regulation of the autophagy pathway by MSI2 and miR-7, three proteins involved in the process (ATG4A, ATG7, P62) were quantified by western blot (Figures 7J−7L). The results confirmed the contribution of MSI2 to the hyperactivation of the autophagy pathway, since DM1 TDMs treated with 10 μM of the MSI2 inhibitor significantly rescued the levels of the autophagy-related proteins and qualitatively decreased the formation of autophagolysosomes according to LysoTracker stainings (Figures 7M−7O). Overall, these results demonstrate that similar molecular and cell rescues can be obtained by MSI2 silencing and protein activity inhibition.
To validate results, we treated independent DM1 cell lines with the MSI2 inhibitor Ro 08-2750. Specifically, we treated immortalized myoblasts differentiated for 7 days from the same donor as for TDMs (Figure 8) and two primary myoblast lines obtained from additional patient biopsies (Table S1; Figure 9). We confirmed that MSI2 levels were significantly increased compared to controls, whereas miR-7 behaved inversely (Figures 8A and 8B; Figure 9A). Notably, Ro 08-2750 at 10 μM rescued the expression of the direct MSI2 targets miR-7 and P21 in all cell lines and TGFBR1 in the DM1−14 (Figure 8B; Figure 9A). Similarly, UPS system and muscle degradation genes showing impaired expression in DM1 muscle cells were significantly rescued upon treatment (Figure 8C; Figure 9B). Additionally, we evaluated by anti-Desmin immunofluorescence the fusion capacity of immortalized myoblasts at 7, 10, and 14 days of differentiation (Figures 8D−8G; Figure S5). In all cases, the capacity of DM1 muscle cells was much lower when compared to control counterparts. Notably, the fusion index was significantly improved after treatment with the compound, namely 160%, 77%, and 95% at days 7, 10, and 14, respectively, compared to DM1 cells treated with vehicle only. In the case of primary myoblasts cultured for 7 days, it was also confirmed that the fusion capacities of both DM1−14 and DM1−16 lines were significantly impaired compared to healthy controls (Figures 9C−9H) and that this phenotype was dramatically reversed after the addition of 10 μM Ro 08-2750. Finally, DM1 cells showed increased LC3 puncta formation (Figures 8H−8K; Figures 9I−9N), confirming the hyperactivation of the autophagic pathway. In immortalized myotubes, this was reinforced by the detection of increased ATG4A and ATG7 protein levels (Figures 8L and 8M). Treatment with the small molecule was sufficient to reverse autophagic markers to a non-DM1 state dramatically.
Figure 8.

MSI2 inhibition in DM1 myoblasts improves pathological phenotypes
(A) MSI2 quantification by western blot and representative blots in control (orange) and DM1 myoblasts treated with vehicle 0.8% DMSO (gray) or 10 μM Ro-08-2750 (red) during the last 48 h of 7-day differentiated myotubes; GAPDH expression was used as an endogenous control. (B) Quantification by qRT-PCR of MSI2 and its direct targets miR-7, P21, and TGFBR1 in the same conditions as those described in (A). (C) Quantification by qRT-PCR of the relative expression of genes involved in catabolic pathways leading to muscle degradation in DM1 treated with vehicle (gray dashed line) and control myoblasts (orange) or DM1 cells treated with compound (red). Statistical analyses were carried out comparing control and DM1 treated with Ro-08-2750 with DM1 myoblasts treated with vehicle. mRNA levels shown in (A) and (B) are relative to the mean of GAPDH, GPI, and HPRT1; miR-7 was normalized to the mean of U1 and U6 (n = 3). Representative confocal images of Desmin immunostaining (green; D−F) and LC3 staining (red; H−J) of 7-day control and DM1 myoblasts treated with DMSO (D, H, and E−I) as control or with 10 μM Ro 08-2750. (F and J) Scale bars correspond to 40 μm, and the nuclei were counterstained with DAPI. (G) Quantification of myogenic fusion index of the conditions represented in (D)−(F) at 7, 10, and 14 days of differentiation (n = 10−15 images). (K) Quantification of the number of LC3 puncta per square micrometer. (G and K) Each condition was compared to DM1 cells treated with DMSO. Quantification by western blot of (L) ATG4A and (M) ATG7. Representative blots used for quantification are shown on the side of the bar graphs. GAPDH was used as an endogenous control to normalize protein levels (n = 3). The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 according to Student’s t test.
Figure 9.

Ro 08-2750 treatment of immortalized DM1 myoblasts differentiated for 7 days validates MSI2 as a new therapeutic target
(A and B) Quantification by qRT-PCR of the indicated genes in control primary myoblasts (line CNT-10, orange dashed line) and two DM1 lines, DM1−14 and DM1−16, treated with vehicle (0.8% DMSO; gray) or 10 μM Ro-08-2750 (red) for 48 h in 7-day differentiation cultures. Asterisks above gray bars indicate statistical differences between CNT-10 and the DM1 lines. Asterisks over the black lines indicate the significant differences between both treatments (DMSO or Ro 08-2750). Gene expression is relative to the mean of GAPDH and GPI, and miR-7 was normalized to the mean of U1 and U6 (n = 3). (C) Quantification of the myogenic fusion index and (D−H) representative confocal images of Desmin immunostaining (green) of control primary myoblasts (D) and DM1−14 and DM1−16 lines treated with vehicle (E and G) or with 10 μM Ro-08-2750 (F and H). (I−M) Representative confocal micrographs of LC3 immunostaining (red) and (N) quantification of the number of LC3 puncta per square micrometer in cell cultures identical to (D)−(H). Scale bar corresponds to 40 μm, and the nuclei were counterstained with DAPI (blue). The bar graphs show mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 according to Student’s t test.
Taken together, all results support that MSI2 represents a valid new therapeutic target to treat muscle dysfunction in DM1.
Discussion
A key aspect of DM1 pathogenesis is progressive muscle wasting. DM1 muscle atrophy research has focused on the RNA toxicity concept and the increased activity and stability of glycogen synthase kinase 3 beta (GSK3b).34 However, this paradigm has changed in the last years, as strong evidence supports alterations in various signaling pathways contributing to DM1 muscle dysfunction.8 miR-7 downregulation originates, at least in part, autophagy overactivation, which is one of the most critical contributors to muscle atrophy in DM1 together with other atrophy-related pathways such as the UPS.9,12,17,35 However, the mechanisms promoting miR-7 dysregulation remain unclear. Here, we report that MSI2 is pathologically overexpressed in DM1 samples, and its depletion by different gene-silencing strategies (siRNA, ASOs) or small-molecule inhibition increases miR-7 levels and ameliorates disease-related muscle markers. The rescues detected were qualitatively similar to those observed in previous work, where miR-7 was replenished with a miRNA mimic in DM1 TDMs.17 Nevertheless, a quantitative comparison between the different strategies to silence MSI2 indicates that the lower the MSI2 levels and the higher miR-7, the better recovery parameters in DM1 TDMs. This association was apparent between inhibition of MSI2 and muscle growth rescue, showing the highest increase in diameter when the levels of MSI2 were the lowest. The previously reported treatment with a miR-7 mimic or chloroquine in DM1 muscle cells achieved a milder recovery of the cells’ diameter than the one observed in the present work.10,17 This suggests that together with the MSI2-miR-7-atrophy axis, MSI2 overexpression may have additional consequences independent of repression of miR-7 biogenesis. However, based on the obtained results, it cannot be ruled out that there are other alterations in the disease that are contributing, additionally to MSI2, to the deregulation of miR-7. Specifically, it has been described that miR-7 biogenesis is also regulated by the quaking protein, which, similarly to MSI2, binds to immature miR-7.37 Moreover, it was reported that Cyrano, a long non-coding RNA (lncRNA), binds to miR-7 and effectively promotes its degradation via target-directed miRNA degradation.38 In the context of DM1, of particular interest is the regulation of miR-7 by the NF90-NF45 complex (also known as ILF2 and ILF3, respectively), which binds to pri-miR-7-1, inhibiting the synthesis of mature miRNA.41 Interestingly, upregulation of ILF2 and ILF3 was demonstrated in biopsies from DM1 skeletal muscles.17 Globally, it is possible that other new targets exist in a DM1 context to modulate miR-7 levels.
Besides using strategies to inhibit MSI2, we treated four different DM1 cell models with the small molecule Ro 08-2750 that binds directly and selectively to MSI2 and competes for its RNA binding, thus leading to inhibition of MSI2 RNA-binding activity and the regulation of downstream targets.33 Globally, we fully validated data obtained in the TDMs, provide evidence in multiple systems, and build more confidence in the robustness of modulating this pathway as a potential therapeutic strategy against DM1. Finally, our data are consistent with previous observations demonstrating that the axis miR-7-autophagy is involved in myotubes differentiation.39
Skeletal muscle cells regulate their size by a balance of protein synthesis and degradation pathways.40 Noteworthy, the reduction of MSI2 levels showed effects on both sides of this complex balance, because in addition to dampening down catabolism via autophagy repression, it also acted on muscle anabolism. MSTN, a member of the transforming growth factor-β (TGF-β) superfamily, negatively regulates muscle mass by inhibiting Akt phosphorylation, resulting in decreased protein synthesis and reduced cell size.42 Importantly, we observed that treatment of DM1 TDMs with MSI2-targeting ASOs significantly reduced MSTN expression levels and increased AKT1 and its active form (p-AKT), thus derepressing muscle mass anabolism. Complementary, upon ASO treatment, IGF-1 was significantly upregulated. IGF-1 promotes muscle protein synthesis via activating the Akt/mTOR pathway,43 and consistently, in MSI2 silenced cells, we also detected increased mTOR expression together with AKT activation. These results and the fact that MSTN and IGF-1 have opposing roles in regulating the growth and size of skeletal muscle42 indicate that MSTN and IGF-1 rescue may ameliorate ASO-treated DM1 TDMs via the Akt/mTOR pathways. Remarkably, reduction of MSI2 expression in DM1 TDMs with ASOs was sufficient to rescue the expression of MuRF1, FBXO32, FoxO1, and FoxO3 genes involved in protein degradation via the AKT/mTOR pathway, thus contributing to improved fusion index and diameter of treated TDMs. Finally, it has been reported that increased expression of PKM2 in type 1 myofibers leads to type 1 myofiber atrophy.7 In our experiments, the percentage of the PKM2 isoform was significantly reduced after MSI2 silencing with the ASOs. This suggests that re-establishing normal MSI2 levels promotes a PKM2-to-PKM1 switch characteristic from differentiated myotubes, contributing to the improved fusion index and myotube diameter observed in treated DM1 TDMs.
The study of MBNL protein levels upon MSI2 silencing demonstrated increased MBNL1 after treatment with the highest concentration of ASO1. Moreover, extra MBNL1 protein was functional as splicing events regulated by MBNL1 were significantly improved. Previous results have shown that miR-7 levels were MBNL1 independent.17 MBNL1 levels may increase because of autophagy repression via MSI2/miR-7, resulting in reduced MBNL1 disposal as similarly observed when autophagy was blocked with chloroquine in DM1 muscle cells.10 Another possibility would be for MSI2 to act independently of the miR-7/autophagy axis since MSI2 regulates many targets involved in varied processes, both at the level of mRNA stability or post-transcriptionally.44,45
Maintenance of fetal-splicing patterns of certain transcripts is a key phenotype in DM1.46 A potential direct role for MSI2 on alternative splicing misregulation in DM1, combined with MBNL1, CELF1, and HNRNPA1 dysregulations, cannot be discarded, but so far, the involvement of this protein in splicing has been suggested to be indirect, through the control of other regulators. Indeed, when we quantified PKM2 levels after ASO treatments, we observed a significant reduction at all tested conditions. However, MBNL1 levels were only increased when cells were treated at the highest concentration of ASO1. These results suggest that MSI2 could indirectly contribute to PKM regulation, as it was reported that c-MYC,33 a direct target of MSI2, upregulates transcription of PTB, hnRNPA1, and hnRNPA2, which, in turn, promotes PKM2 levels.47
Upon MSI2 modulation, we observed a surprisingly modest effect on the MSI2 mRNA-binding targets’ levels TGFBR1 and P21.24,48 However, these results do not exclude the possibility that other molecular alterations in DM1, in addition to MSI2 overexpression, may also contribute to TGFBR1 and P21 misregulation and, consequently, counteract the effect of MSI2 silencing. Specifically, CELF1 was reported to regulate P21 expression in DM1.49,50 Consistent with these observations, CELF1 remained unchanged after MSI2 blocking, so this fact could also be contributing to the mild effect over P21 levels after MSI2 reduction.
In summary, this study sheds new light on the molecular mechanisms leading to muscle atrophy in DM1 and, more specifically, on miR-7 downregulation and subsequent autophagy regulation. We demonstrate that the RNA-binding protein MSI2 is strongly upregulated in DM1 and, for the first time, propose that this protein is a new therapeutic target to treat DM1 muscle dysfunction. Remarkably, we observed a significant negative correlation between MSI2 levels and ankle dorsiflexion force in biopsies from 40 DM1 patients, highlighting the relevance of MSI2 dysregulation as a contributor to the muscle phenotype. Moreover, we demonstrate that the reduction of MSI2 levels by three different strategies was enough to significantly reduce miR-7 levels, constituting essential proof-of-concept studies to develop new therapeutic strategies.
Materials and methods
Chemically modified oligonucleotides
siRNAs targeting MSI2 transcripts (ID S42755 and S42757; catalog number 4392421) and negative control (catalog number 4390843) were from the Invitrogen (Madrid, Spain) Silencer Select siRNA collection. ASO and scrambled control were synthesized by biomers.net. The ASO sequences were as follows: ASO1, 5′ +T+G+A∗C∗T∗T∗C∗T∗T∗T∗C∗G∗G+C+T+G 3′; ASO2, 5′ +C+A+C∗G∗A∗A∗G∗T∗T∗G∗G∗G∗A+T+A+T 3′; ASO3, 5′ +T+T+G∗G∗A∗T∗T∗A∗A∗G∗G∗T∗T+G+C+C 3′; and scrambled ASO, 5′ +T+T+C∗C∗C∗T∗G∗A∗A∗G∗G∗T∗T+C+C+T 3′, which are complementary to sequences from exon 9 (ASO1), exon 10 (ASO2), and exon 14 (ASO3). A + sign denotes locked nucleic acid nucleotides, and ∗ indicates phosphorothioate linkages.
Cell culture and treatment with oligonucleotides and small molecule
Unaffected (control) and patient-derived (DM1) cells were kindly provided by Dr. Furling (Institute of Myology, Paris, France).22 Briefly, DM1 cells were obtained from skin or skeletal muscle biopsy of an 11-year-old female donor expressing 1,300 CTG repeats, and counterpart control cells were obtained from a 25-year-old non-DM1 donor. Both fibroblasts and myoblasts have been immortalized. Additionally, fibroblasts were transduced to express MyoD (doxycycline) inducibly. Forced expression of MyoD transdifferentiates fibroblasts into myotubes.22 TDMs were cultured as previously described.17 After 4 days in differentiation medium (MDM), cells were transfected with a combination of the two siRNAs targeting MSI2 transcripts (final concentration 100 nM) using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Madrid, Spain) following the manufacturer’s recommendations. After 48 h, the medium containing the siRNAs was replaced by fresh MDM. 24 h later, cells were collected. As a control, the same protocol was performed using 100 nM of a siRNA with a scrambled sequence. Gapmers were transfected following the same protocol at 30 and 150 nM. To treat cells with the small molecule Ro 08-2750 (Tocris catalog number 2272; Bristol, UK), cells were cultured as before. Briefly, after 5 days in MDM, the medium was supplemented with Ro 08-2750 to a final concentration of 1, 3, 10, or 15 μM (0.8% DMSO) or with 0.8% DMSO (vehicle) for 48 h. Immortalized myoblasts were cultured as previously described.22 Immortalized myoblasts cultured for 5 days in MDM were treated with Ro 08-2750 to a final concentration of 10 μM for 48 h. In the case of fusion index determinations at 7, 10, and 14 days, the compound was added for the last 48 h.
Primary human skeletal muscle cultures
Primary human myoblasts from muscle biopsies from 3 DM1 patients and three controls were obtained as previously described.51 A positive selection for the CD56 surface marker was performed to enrich for myoblasts, using CD56-coated microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions. Purified myoblasts were seeded in six-well plates (2.5 × 105 cells/well), and the following day, the medium was replaced with MDM (DMEM + 2% horse serum + 1% penicillin-streptomycin). The medium was changed every 2 days. To treat primary myoblasts, CNT-10, DM1−14, and DM1−16 with the small molecule Ro 08-2750 cells were cultured as before. After 5 days in differentiation conditions, the medium was supplemented with Ro 08-2750 to a final concentration of 10 μM (0.8% DMSO) or with 0.8% DMSO (vehicle) for 48 h. Information about biopsy donors is compiled in Table S1.
DM1 patients and skeletal muscle biopsies
All muscle biopsies were taken after informed consent by patients and approval by the Experimentation Ethics Committee of the University Hospital La Fe (Valencia, Spain; authorization 2014/0799). Methods for the determination of the size of the CTG repeat expansion were as reported in Fernandez-Costa et al.13 A detailed description of muscle type, sex, age, and repeats length is provided in Table S1.
Immunofluorescence methods
Desmin immunostaining and fusion index and diameter determination were performed as previously described.10 For MSI2 immunodetection, 3.5 × 104 fibroblasts/well were seeded in 24-well plates. After 7 days of differentiation into myotubes, cells were fixed with 4% paraformaldehyde (PFA) for 15 min. After three washes with 0.1% Triton X-100 in PBS 1× (PBS-T), cells were blocked (PBS-T, 1% BSA, 5% normal goat serum) for 1 h and incubated with rabbit anti-MSI2 (1:100, EP1305Y; Abcam) in blocking buffer for 48 h at 4°C. After three washes with PBS-T, cells were incubated for 1 h with goat anti-rabbit fluorescein isothiocyanate (FITC) immunoglobulin G (IgG; 1:200; Sigma-Aldrich, St. Louis, MO, USA) in blocking buffer. Finally, cells were washed thrice with PBS 1× and were counterstained and mounted with VECTASHIELD mounting medium containing 40,6-dia-midino-2-phenylindole (DAPI; Vector Laboratories, London, UK) to detect the nuclei.
For LC3B immunodetection, cells were differentiated in MDM for 7 days (3 × 105 cells/well in 24-well plates). After 48 h of gapmer treatment, cells were fixed with 4% PFA in PBS for 15 min at room temperature. After fixation, cells were permeabilized with methanol 100% for 15 min at −20°C and blocked with blocking buffer (PBS 1×, 0.5% BSA, 1% goat serum) for 1 h at room temperature. Cells were incubated overnight (O/N) with rabbit anti-LC3B (1:200, ab51520; Abcam, Cambridge, UK) diluted in blocking buffer. Then, samples were incubated with goat anti-rabbit IgG (H+L) Alexa Fluor Plus 594 (1:200; Invitrogen, Carlsbad, CA, USA) for 2 h. Between antibodies, cells were washed three times with PBS 1×. Finally, cells were washed three times with PBS and mounted in Vectashield (Vector Laboratories, London, UK) with 2 μg/mL DAPI. MBNL1 and MBNL2 were detected in cells differentiated in MDM for 7 days as previously described.10
For LysoTracker staining, 2.5 × 104 cells were seeded in a 24-well plate. After treatment with ASOs for 48 h, samples were incubated for 30 min at 37°C with 100 nM LysoTracker RED-DND99 (Invitrogen, Madrid, Spain). After two washes with warmed PBS, cells were fixed with 4% PFA for 15 min, followed by washes in 1× PBS. Then, cells were mounted using Vectashield (Vector Laboratories, London, UK) with 2 μg/mL DAPI. Images were acquired in an LSM800 confocal microscope (Zeiss) at 400× magnification.
LC3 puncta quantification
Image analyses of LC3-stained myotubes were performed using the Ifdotmeter software52 following the authors’ recommendations. Briefly, the quantification of the number of LC3 dots or LC3 puncta was performed with 15−20 images per condition. Total LC3 dots per image were normalized relative to the total area corresponding to all of the myotubes observed in each image. Myotube area was measured by using ImageJ software. Data were expressed as the number of LC3 dots/μm2.
Toxicity assay
Control cells were aliquoted in a 96-well plate with 1.0 × 105 cells per well. After 24 h, cells were transfected with different ASOs as described17 (concentrations ranging from 1 nM to 1 μM) or treated with Ro 08-2750 (Tocris Bioscience, Bristol, UK) at concentrations ranging from 0.0195 to 80 μM (in 1:2 serial dilutions). Cell viability was measured as previously described.10
RNA extraction, semiquantitative PCR, and real-time PCR
All of the procedures were performed as previously described.17 Primers not previously reported are listed in Table S2. qRT-PCR data were obtained using the delta-delta CT method. Commercial TaqMan probes (QIAGEN) were used to detect MBNL1 (6-carboxyfluorescein (FAM)-labeled probe) and reference GAPDH (NHS esterfluoro-phore (MAX)-labeled probe) genes (Integrated DNA Technologies, Leuven, Belgium). Relative quantification of the relevant genes in healthy control TDMs was previously reported.17
Western blotting
Protein extraction, quantification, and immunodetection were performed as previously described.10,17 Specifically, MSI2 detections were performed by incubating membranes O/N with a rabbit anti-MSI2 antibody (1:1,000, EP1305Y; Abcam, Cambridge, UK). Goat horseradish peroxidase (HRP)-conjugated anti-rabbit-IgG (1:3,500; Sigma-Aldrich, St. Louis, MO, USA) was used as the secondary antibody. Images were acquired with an ImageQuant LAS 4000 or Amersham ImageQuant 800 (GE Healthcare). Quantification was performed using ImageJ software (NIH). Relative quantification of the relevant proteins in healthy control TDMs was previously reported.17
Statistical analyses
In all molecular studies, for comparison on mean data, we assumed that all parameters follow a normal distribution, and the samples were compared using two-tailed t tests (α = 0.05), applying Welch’s correction when necessary. The statistical differences were estimated by Student’s t tests (p < 0.05) on normalized data. Sample sizes (n) are included in the figure legends.
Acknowledgments
The Wolfson Centre for Inherited Neuromuscular Disease provided antibody MB1a (4A8). This work was possible by research grants RTI2018-094599-B-100 from the Ministerio de Ciencia e Innovación-Agencia Estatal de Investigación, which included funds from the European Regional Development Fund (ERDF), and PROMETEO/2020/081 from the Generalitat Valenciana to R.A. Additional funding was from the “Fundación para la Innovación y la Prospectiva en Salud en España, FIPSE” to R.A. A.B. and M.S.-A. thank the support of the Conselleria d’Educació, Investigació and Cultura i Esport (Generalidad Valenciana) as postdoctoral (APOSTD2017/077) and predoctoral (ACIF/2018/071) grantees, respectively. N.M. was supported by a predoctoral fellowship (PRE2019-090622) from the Ministerio de Ciencia e Innovación-Agencia Estatal de Investigación. Part of the equipment employed in this work has been funded by Generalitat Valenciana and co-financed with ERDF funds (OP ERDF of Comunitat Valenciana 2014-2020).
Author contributions
R.A. provided the conceptual framework for the study. R.A., A.B., and M.S.-A. conceived and designed the experiments, helped in the interpretation of results, and prepared the manuscript. J.P.-G. and J.J.V. provided the muscle biopsies and protein extracts and RNA from primary myoblasts. M.S.-A., A.B., and N.M., performed the experiments and analyzed data.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.08.010.
Supplemental information
References
- 1.Smith C.A., Gutmann L. Myotonic Dystrophy Type 1 Management and Therapeutics. Curr. Treat. Options Neurol. 2016;18:52. doi: 10.1007/s11940-016-0434-1. [DOI] [PubMed] [Google Scholar]
- 2.Pettersson O.J., Aagaard L., Andrejeva D., Thomsen R., Jensen T.G., Damgaard C.K. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res. 2014;42:7186–7200. doi: 10.1093/nar/gku352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Todd P.K., Paulson H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 2010;67:291–300. doi: 10.1002/ana.21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuyumcu-Martinez N.M., Wang G.S., Cooper T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li M., Zhuang Y., Batra R., Thomas J.D., Li M., Nutter C.A., Scotti M.M., Carter H.A., Wang Z.J., Huang X.-S. HNRNPA1-induced spliceopathy in a transgenic mouse model of myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 2020;117:5472–5477. doi: 10.1073/pnas.1907297117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rau F., Lainé J., Ramanoudjame L., Ferry A., Arandel L., Delalande O., Jollet A., Dingli F., Lee K.-Y., Peccate C. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun. 2015;6:7205. doi: 10.1038/ncomms8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Z., Cooper T.A. Reexpression of pyruvate kinase M2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 2013;110:13570–13575. doi: 10.1073/pnas.1308806110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozimski L.L., Sabater-Arcis M., Bargiela A., Artero R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol. Rev. Camb. Philos. Soc. 2021;96:716–730. doi: 10.1111/brv.12674. [DOI] [PubMed] [Google Scholar]
- 9.Bargiela A., Cerro-Herreros E., Fernandez-Costa J.M., Vilchez J.J., Llamusi B., Artero R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis. Model. Mech. 2015;8:679–690. doi: 10.1242/dmm.018127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bargiela A., Sabater-Arcis M., Espinosa-Espinosa J., Zulaica M., Lopez de Munain A., Artero R. Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. USA. 2019;116:25203–25213. doi: 10.1073/pnas.1820297116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morriss G.R., Rajapakshe K., Huang S., Coarfa C., Cooper T.A. Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1. Hum. Mol. Genet. 2018;27:2789–2804. doi: 10.1093/hmg/ddy192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loro E., Rinaldi F., Malena A., Masiero E., Novelli G., Angelini C., Romeo V., Sandri M., Botta A., Vergani L. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. 2010;17:1315–1324. doi: 10.1038/cdd.2010.33. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Costa J.M., Garcia-Lopez A., Zuñiga S., Fernandez-Pedrosa V., Felipo-Benavent A., Mata M., Jaka O., Aiastui A., Hernandez-Torres F., Aguado B. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2013;22:704–716. doi: 10.1093/hmg/dds478. [DOI] [PubMed] [Google Scholar]
- 14.Perfetti A., Greco S., Cardani R., Fossati B., Cuomo G., Valaperta R., Ambrogi F., Cortese A., Botta A., Mignarri A. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016;6:38174. doi: 10.1038/srep38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koutsoulidou A., Kyriakides T.C., Papadimas G.K., Christou Y., Kararizou E., Papanicolaou E.Z., Phylactou L.A. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PLoS ONE. 2015;10:e0125341. doi: 10.1371/journal.pone.0125341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerro-Herreros E., Sabater-Arcis M., Fernandez-Costa J.M., Moreno N., Perez-Alonso M., Llamusi B., Artero R. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018;9:2482. doi: 10.1038/s41467-018-04892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabater-Arcis M., Bargiela A., Furling D., Artero R. miR-7 Restores Phenotypes in Myotonic Dystrophy Muscle Cells by Repressing Hyperactivated Autophagy. Mol. Ther. Nucleic Acids. 2020;19:278–292. doi: 10.1016/j.omtn.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu D.N., Jiang M.J., Mei Z., Dai J.J., Dai C.Y., Fang C., Huang Q., Tian L. microRNA-7 impairs autophagy-derived pools of glucose to suppress pancreatic cancer progression. Cancer Lett. 2017;400:69–78. doi: 10.1016/j.canlet.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Choudhury N.R., de Lima Alves F., de Andrés-Aguayo L., Graf T., Cáceres J.F., Rappsilber J., Michlewski G. Tissue-specific control of brain-enriched miR-7 biogenesis. Genes Dev. 2013;27:24–38. doi: 10.1101/gad.199190.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siddall N.A., McLaughlin E.A., Marriner N.L., Hime G.R. The RNA-binding protein Musashi is required intrinsically to maintain stem cell identity. Proc. Natl. Acad. Sci. USA. 2006;103:8402–8407. doi: 10.1073/pnas.0600906103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katz Y., Li F., Lambert N.J., Sokol E.S., Tam W.-L., Cheng A.W., Airoldi E.M., Lengner C.J., Gupta P.B., Yu Z. Musashi proteins are post-transcriptional regulators of the epithelial-luminal cell state. eLife. 2014;3:e03915. doi: 10.7554/eLife.03915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arandel L., Polay Espinoza M., Matloka M., Bazinet A., De Dea Diniz D., Naouar N., Rau F., Jollet A., Edom-Vovard F., Mamchaoui K. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech. 2017;10:487–497. doi: 10.1242/dmm.027367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang E.T., Treacy D., Eichinger K., Struck A., Estabrook J., Olafson H., Wang T.T., Bhatt K., Westbrook T., Sedehizadeh S. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019;28:1312–1321. doi: 10.1093/hmg/ddy432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S.M., Deering R.P., Lu Y., Tivnan P., Lianoglou S., Al-Shahrour F., Ebert B.L., Hacohen N., Leslie C., Daley G.Q. Musashi-2 controls cell fate, lineage bias, and TGF-β signaling in HSCs. J. Exp. Med. 2014;211:71–87. doi: 10.1084/jem.20130736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou L., Sheng W., Jia C., Shi X., Cao R., Wang G., Lin Y., Zhu F., Dong Q., Dong M. Musashi2 promotes the progression of pancreatic cancer through a novel ISYNA1-p21/ZEB-1 pathway. J. Cell. Mol. Med. 2020;24:10560–10572. doi: 10.1111/jcmm.15676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonaldo P., Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heras-Sandoval D., Pérez-Rojas J.M., Hernández-Damián J., Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014;26:2694–2701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 28.Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjørkøy G., Lamark T., Pankiv S., Øvervatn A., Brech A., Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 30.Rusten T.E., Stenmark H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010;12:207–209. doi: 10.1038/ncb0310-207. [DOI] [PubMed] [Google Scholar]
- 31.Tanida I., Ueno T., Kominami E. LC3 and Autophagy. Methods Mol. Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 32.Zearfoss N.R., Deveau L.M., Clingman C.C., Schmidt E., Johnson E.S., Massi F., Ryder S.P. A conserved three-nucleotide core motif defines Musashi RNA binding specificity. J. Biol. Chem. 2014;289:35530–35541. doi: 10.1074/jbc.M114.597112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minuesa G., Albanese S.K., Xie W., Kazansky Y., Worroll D., Chow A., Schurer A., Park S.-M., Rotsides C.Z., Taggart J. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat. Commun. 2019;10:2691. doi: 10.1038/s41467-019-10523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones K., Wei C., Iakova P., Bugiardini E., Schneider-Gold C., Meola G., Woodgett J., Killian J., Timchenko N.A., Timchenko L.T. GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Invest. 2012;122:4461–4472. doi: 10.1172/JCI64081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huguet A., Medja F., Nicole A., Vignaud A., Guiraud-Dogan C., Ferry A., Decostre V., Hogrel J.-Y., Metzger F., Hoeflich A. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 2012;8:e1003043. doi: 10.1371/journal.pgen.1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Vogel G., Yu Z., Richard S. The QKI-5 and QKI-6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Mol. Cell. Biol. 2013;33:1233–1243. doi: 10.1128/MCB.01604-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang A., Shao T.-J., Bofill-De Ros X., Lian C., Villanueva P., Dai L., Gu S. AGO-bound mature miRNAs are oligouridylated by TUTs and subsequently degraded by DIS3L2. Nat. Commun. 2020;11:2765. doi: 10.1038/s41467-020-16533-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L., Chen Y., Nie L., Ding X., Zhang X., Zhao W., Xu X., Kyei B., Dai D., Zhan S. MyoD-induced circular RNA CDR1as promotes myogenic differentiation of skeletal muscle satellite cells. Biochim. Biophys. Acta. Gene Regul. Mech. 2019;1862:807–821. doi: 10.1016/j.bbagrm.2019.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Cohen S., Nathan J.A., Goldberg A.L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- 41.Higuchi T., Todaka H., Sugiyama Y., Ono M., Tamaki N., Hatano E., Takezaki Y., Hanazaki K., Miwa T., Lai S. Suppression of MicroRNA-7 (miR-7) Biogenesis by Nuclear Factor 90-Nuclear Factor 45 Complex (NF90-NF45) Controls Cell Proliferation in Hepatocellular Carcinoma. J. Biol. Chem. 2016;291:21074–21084. doi: 10.1074/jbc.M116.748210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morissette M.R., Cook S.A., Buranasombati C., Rosenberg M.A., Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am. J. Physiol. Cell Physiol. 2009;297:C1124–C1132. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida T., Delafontaine P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells. 2020;9:1970. doi: 10.3390/cells9091970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett C.G., Riemondy K., Chapnick D.A., Bunker E., Liu X., Kuersten S., Yi R. Genome-wide analysis of Musashi-2 targets reveals novel functions in governing epithelial cell migration. Nucleic Acids Res. 2016;44:3788–3800. doi: 10.1093/nar/gkw207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duggimpudi S., Kloetgen A., Maney S.K., Münch P.C., Hezaveh K., Shaykhalishahi H., Hoyer W., McHardy A.C., Lang P.A., Borkhardt A., Hoell J.I. Transcriptome-wide analysis uncovers the targets of the RNA-binding protein MSI2 and effects of MSI2's RNA-binding activity on IL-6 signaling. J. Biol. Chem. 2018;293:15359–15369. doi: 10.1074/jbc.RA118.002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.López-Martínez A., Soblechero-Martín P., de-la-Puente-Ovejero L., Nogales-Gadea G., Arechavala-Gomeza V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes (Basel) 2020;11:1109. doi: 10.3390/genes11091109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.David C.J., Chen M., Assanah M., Canoll P., Manley J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H., Tan S., Wang J., Chen S., Quan J., Xian J., Zhang Ss., He J., Zhang L. Musashi2 modulates K562 leukemic cell proliferation and apoptosis involving the MAPK pathway. Exp. Cell Res. 2014;320:119–127. doi: 10.1016/j.yexcr.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Denis J.A., Gauthier M., Rachdi L., Aubert S., Giraud-Triboult K., Poydenot P., Benchoua A., Champon B., Maury Y., Baldeschi C. mTOR-dependent proliferation defect in human ES-derived neural stem cells affected by myotonic dystrophy type 1. J. Cell Sci. 2013;126:1763–1772. doi: 10.1242/jcs.116285. [DOI] [PubMed] [Google Scholar]
- 50.Picchio L., Legagneux V., Deschamps S., Renaud Y., Chauveau S., Paillard L., Jagla K. Bruno-3 regulates sarcomere component expression and contributes to muscle phenotypes of myotonic dystrophy type 1. Dis. Model. Mech. 2018;11 doi: 10.1242/dmm.031849. dmm031849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Luna N., Gallardo E., Soriano M., Dominguez-Perles R., de la Torre C., Rojas-García R., García-Verdugo J.M., Illa I. Absence of dysferlin alters myogenin expression and delays human muscle differentiation “in vitro”. J. Biol. Chem. 2006;281:17092–17098. doi: 10.1074/jbc.M601885200. [DOI] [PubMed] [Google Scholar]
- 52.Rodríguez-Arribas M., Pizarro-Estrella E., Gómez-Sánchez R., Yakhine-Diop S.M.S., Gragera-Hidalgo A., Cristo A., Bravo-San Pedro J.M., González-Polo R.A., Fuentes J.M. IFDOTMETER: A New Software Application for Automated Immunofluorescence Analysis. J. Lab. Autom. 2016;21:246–259. doi: 10.1177/2211068215600650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.