

Summary
Here, we describe a detailed step-by-step protocol to detect SARS-CoV-2 RNA using RT-PCR-mediated amplification and CRISPR/Cas-based visualization. The optimized assay uses basic molecular biology equipment such as conventional thermocyclers and transilluminators for qualitative detection. Alternatively, a fluorescence plate reader can be used for quantitative measurements. The protocol detects two regions of the SARS-CoV-2 genome in addition to the human RNaseP sample control. Aiming to reach remote regions, this work was developed to use the portable molecular workstation from BentoLab.
For complete details on the use and execution of this protocol, please refer to Alcántara et al., 2021.
Subject areas: Clinical Protocol, Immunology, Microbiology, Molecular Biology, CRISPR, Biotechnology and bioengineering
Graphical Abstract
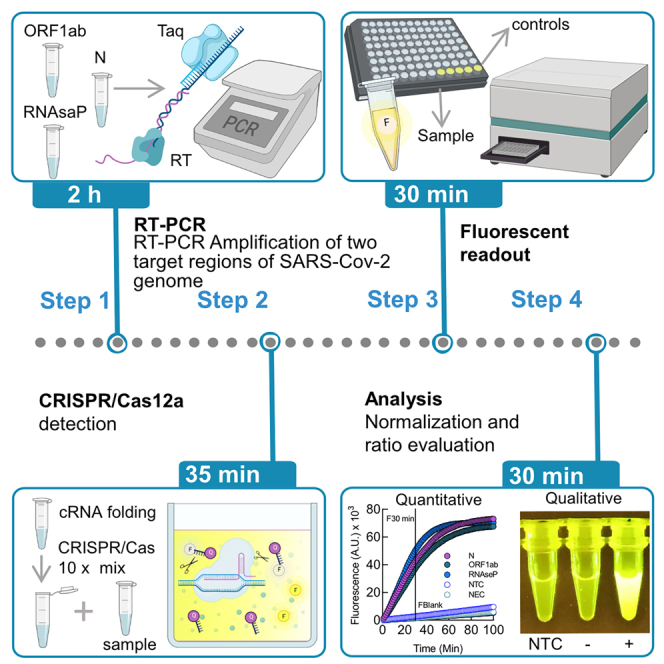
Here, we describe a detailed step-by-step protocol to detect SARS-CoV-2 RNA using RT-PCR-mediated amplification and Crispr/Cas-based visualization. The optimized assay uses basic molecular biology equipment such as conventional thermocyclers and transilluminators for qualitative detection. Alternatively, a fluorescence plate reader can be used for quantitative measurements. The protocol detects two regions of the SARS-CoV-2 genome in addition to the human RNaseP sample control. Aiming to reach remote regions, this work was developed to use the portable molecular workstation from BentoLab.
Highlights
-
•
Molecular detection of SARS-CoV-2 using RT-PCR coupled to CRISPR/Cas technology
-
•
Compatibility for applying UnCovid as a Laboratory-developed test (LDT)
-
•
Protocol validated with clinical samples
-
•
Versatility for developing molecular detection of other RNA viruses
Before you begin
The UnCovid protocol couples nucleic acid amplification using RT-PCR with detection of the amplified DNA mediated by CRISPR/Cas12a (Chen et al., 2018). Cas12a is a DNA nuclease that uses an adaptor RNA (CRISPR RNA) to identify the target DNA to be cleaved (Zetsche et al., 2015). Upon cleavage of the target double strand DNA (dsDNA), Cas12a activates a promiscuous off target activity that can cut single stranded DNA (ssDNA) (Jinek et al., 2012). This off target activity is used to cleave a dually labeled ssDNA. i.e., a ssDNA labeled with a donor fluorophore and a silent acceptor (quencher) results in measurable increase of fluorescence upon Cas12a-mediated cleavage. The Föster Resonance Energy Transfer (FRET) between the dyes is released due to a distance increase between the dyes. The UnCovid protocol is intended to be used with locally produced enzymes (M-MLV reverse transcriptase, Taq DNA polymerase, and LbCas12a) or their commercial counterparts of your preference. Albeit the crRNA can be chemically synthetized and purchased, in vitro transcription is most cost efficient. This segment of the protocol describes the production, purification, quantification, and storage of crRNA for CRISPR/Cas reaction. For details about enzyme production refer to (Mendoza-Rojas et al., 2021).
General experimental considerations
-
1.
This protocol is intended to be executed by experienced personnel. Users require to have a basic molecular biology training and experience, be accustomed with proper pipetting technique and trained for good clinical laboratory practice (GCLP).
-
2.
Good clinical laboratory practices (GCLP) are recommended for use of this protocol. A 90 min online training in GCLP is available at the following link: https://www.cdc.gov/labtraining/training-courses/good-lab-practices-molecular-genetics-testing.html.
-
3.
Correct use of basic equipment is necessary to avoid cross contamination. Avoid using the same gloves across all areas, amplicon contamination must be strongly contained (Davidi et al., 2021).
-
4.
It is recommended to work with a unidirectional flow of samples considering “clean” (preparing reactions, sample manipulation) and “dirty” lab areas (where the thermal cyclers are located, and for post-processing steps (electrophoresis)). A Basic Molecular Biology Moduls for Laboratory Practice is available at the following link: https://www.cdc.gov/labtraining/training-courses/basic-molecular-biology/laboratory-practice.html
crRNA purification
Timing: 4 h
-
5.
In vitro transcription reaction
In vitro transcription (IVT) requires a double-stranded DNA (dsDNA) template containing the T7 promoter sequence. Here, we used the following dsDNA templates to transcribe crRNAs, complemental to two SARS-CoV-2 genes and for the human RNaseP as sample quality control: ∗The T7 promoter sequence is highlighted in red.Note: Use the in vitro transcription kit of your choice to prepare crRNA. Here, the TranscriptAid T7 High Yield Transcription Kit (Thermo Fisher) was used.
∗The T7 promoter sequence is highlighted in red.Note: Use the in vitro transcription kit of your choice to prepare crRNA. Here, the TranscriptAid T7 High Yield Transcription Kit (Thermo Fisher) was used. CRITICAL: Pay attention to the following general recommendations to avoid RNase contamination (Green and Sambrook, 2019): (1) Maintain a separate pipette set, pipette tips, and any other material required for RNA lab work. (2) Use gloves at all times. Change them frequently. (3) Preferably use RNase-free water (i.e., nuclease-free water aliquots) to prepare reactions or buffers. (4) Clean working surfaces, pipettes, and equipment with 20% bleach or alternative commercial solutions. To avoid accelerated deterioration of any plastics and metals, wipe down with 70% ethanol after using 20% bleach.
CRITICAL: Pay attention to the following general recommendations to avoid RNase contamination (Green and Sambrook, 2019): (1) Maintain a separate pipette set, pipette tips, and any other material required for RNA lab work. (2) Use gloves at all times. Change them frequently. (3) Preferably use RNase-free water (i.e., nuclease-free water aliquots) to prepare reactions or buffers. (4) Clean working surfaces, pipettes, and equipment with 20% bleach or alternative commercial solutions. To avoid accelerated deterioration of any plastics and metals, wipe down with 70% ethanol after using 20% bleach.-
a.Dissolve the dsDNA templates using nuclease-free water to 100 μM concentration.
-
b.Thaw all reagents and maintain the enzyme mix on ice.
-
c.Prepare the 25 mM NTPs (ATP, GTP, CTP, and UTP) mix by adding an equal volume of each nucleotide (100 mM stock) in a single tube.
-
d.Prepare the master mix for 40 μL reactions as follows:
Reagent Initial concentration Final concentration Volume (μL) per reaction Volume (μL) for 3 reactions 5× Reaction buffer 5× 1× 8 24 NTP mix 25 mM 10 mM 16 48 Enzyme mix - - 4 12 Nuclease-free water a - - 11 33 Final volume 39 117 aWater volume can be adjusted depending on template volume -
e.Dispense 39 μL of master mix in a 1.5 mL tube.
-
f.Add 4 μg of the respective dsDNA template to the reaction tube. Here, one microliter of a 100 μM dsDNA stock (100 pmoles) was used.
-
g.Incubate the reaction at 37°C for 3 h.
-
a.
-
6.crRNA purificationNote: Use the RNA purification kit of your choice to purify the transcribed crRNA. Here, the Direct-zol RNA Miniprep purification Kit from Zymo Research was used.
-
a.Add 3 volumes (i.e., 120 μL for a 40 μL reaction) of TRI-Reagent® (Zymo Research) or other similar product (TRIzol®, RNAzol®, TriPureTM, or TriSureTM) to each volume of the in vitro transcription reaction. Mix thoroughly and spin briefly.
-
b.Add 1 volume (i.e., 160 μL) of 95%–100% cold ethanol. Mix thoroughly and spin briefly.
-
c.Transfer the mixture to the spin column and centrifuge at 10000 × g for 30 s. Discard the flow-through (FT).
-
d.Add 400 μL of RNA wash buffer and centrifuge at 10000 × g for 30 s. Discard the FT.CRITICAL: DNase I treatment is recommended for DNA template removal. Deoxyribonuclease I (DNase I) cuts both single-stranded and double-stranded DNA. Recombinant DNase I is typically used for selectively degrading DNA in the presence of RNA. One application of the DNase I treatment is removal of DNA template after in vitro transcription prior to downstream applications. Here, the DNase I and digestion buffer are provided with the Direct-zol RNA Miniprep kit (Zymo Research, Cat # E1010). The lyophilized enzyme can be reconstituted with 275 μL of MilliQ water. DNase I digestion step is performed on the spin column. Prepare the working stock of DNase I by mixing 75 μL of DNA digestion buffer (provided in the kit) with 5 μL of DNase I (6 U/μL stock concentration, 0.38 U/μL final concentration) (Zymo Research, Cat # E1010). Alternatively, use DNase I in 1× DNase I buffer (10 mM Tris-HCl, 2.5 mM MgCl2, 0.5 mM CaCl2, pH 7.6 25°C).
-
e.Add 80 μL of DNase I working stock to the column and incubate at 25°C for 15 min.
-
f.Add 400 μL of RNA pre-wash buffer and centrifuge at 10000 × g for 30 s. Discard the FT.
-
g.Add 700 μL of RNA wash buffer and centrifuge at 10000 × g for 30 s. Discard the FT.
-
h.Transfer the column to a clean collection 1.5 mL tube
-
i.Add 80 μL of nuclease-free water. Incubate for 2–5 s, and centrifuge at 10000 × g for one minute. Collect the tube and place it on ice.
-
j.Quantify the RNA by measuring the absorbance at 260 nm (A260) in a spectrophotometer. Here, the NanoDrop One microvolume UV-Vis spectrophotometer was used (Thermo Fisher Scientific). An A260 nm absorbance of 1 is equivalent to 40 μg/mL single-stranded RNA.
-
k.Calculate the molar concentration using the corresponding molecular weight (ORF1ab = 13.5 kDa, N = 13 kDa, RNaseP = 13 kDa).
-
l.Dilute your RNA stocks to 10–20 μM using pre-cooled nuclease-free water, mix briefly and spin down.
-
m.Dispense 20 μL aliquots in 0.5–1.5 mL tubes and store at −80°C.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Nasopharyngeal swabs | Instituto de Investigación Nutricional, Lima, Peru | N/A |
| Quantitative PCR (qPCR) control RNA from heat-inactivated SARS-CoV-2, Isolate USA-WA1/2020 | Centers for Disease Control and Prevention | BEI Resources NR-52347 |
| Chemicals, peptides, and recombinant proteins | ||
| Potassium chloride | Supelco | Cat # 104936 |
| Magnesium chloride hexahydrate | Supelco | Cat # 105833 |
| Triton X-100 | Sigma-Aldrich | Cat # 10789704001 |
| Trehalose | Sigma-Aldrich | Cat # T9449-25G |
| Sodium chloride | Supelco | Cat # 106404 |
| Glycerol 85% | Supelco | Cat # 104094 |
| EDTA | Merck Millipore | Cat # 324503 |
| Tris, Hydrochloride, Molecular Biology Grade | Merck Millipore | Cat # US1648317 |
| Tris base ULTROL grade | Calbiochem | Cat # 643811 |
| Dithiothreitol (DTT) | Thermo Fisher | Cat # R0682 |
| dNTP Mix (10 mM each) | Thermo Fisher | Cat # R0194 |
| SafeGreen | abm | Cat # G108-G |
| TriTrack DNA loading dye 6× | Thermo Fisher | Cat # R1161 |
| Agarose powder | Cleaver Scientific | Cat # CSL-AG500 |
| OmniPur Water DEPC treated, sterile, nuclease-free | Sigma-Aldrich | Cat # 9610-1L |
| DNaseI | PanReac Applichem | Cat # A3778 |
| GeneRuler 100 bp DNA Ladder | Thermo Scientific | Cat # SM0242 |
| Bovine Serum Albumin, lyophilized powder | Sigma-Aldrich | Cat # A9418-100G |
| Critical commercial assays | ||
| TranscriptAid T7 High Yield Transcription Kit | Thermo Fisher | Cat # K0441 |
| Direct-zol RNA Miniprep | Zymo Research | Cat # R2051 |
| 2× One-Step RT-PCR Master Mix | Norgen BioTek | Cat # 28113 |
| QIAamp viral RNA Mini Kit | Qiagen | Cat # 52906 |
| Deposited data | ||
| Data S1 and S2 | This paper | https://doi.org/10.17632/8m8z37v8xz.1 |
| Oligonucleotides | ||
| dsDNA oligo: crRNA sequence N target SARS-CoV-2 TAATACGACTCACTATAGGTAATTTCTACTAAGTGT AGATCCCCCAGCGCTTCAGCGTTC |
Broughton et al.,2020 | N/A |
| dsDNA oligo: crRNA sequence ORF1ab target SARS-CoV-2 TAATACGACTCACTATAGGTAATTTCTACTA AGTGTAGATTTAGAGACGGTTGGGAAATTG |
This paper and Alcántara et al., 2021 | N/A |
| dsDNA oligo: crRNA sequence RNaseP target human sample control TAATACGACTCACTATAGGTAATTTC TACTAAGTGTAGATAATTACTTGGGTGTGACCCT |
Broughton et al.,2020 | N/A |
| ssDNA oligo: primer forward N target SARS-CoV-2 TACAAACATTGGCCGCAAATTGC | This paper and Alcántara et al., 2021 | N/A |
| ssDNA oligo: primer reverse N target SARS-CoV-2 CCAATGCGCGACATTCCG | This paper and Alcántara et al., 2021 | N/A |
| ssDNA oligo: primer forward ORF1ab target SARS-CoV-2 GTTGTTCAGTTGACTTCGC | This paper and Alcántara et al., 2021 | N/A |
| ssDNA oligo: primer reverse ORF1ab target SARS-CoV-2 GACAATTTCACAAGCACAGG | This paper and Alcántara et al., 2021 | N/A |
| ssDNA oligo: primer forward RNaseP target human sample control ACTCAGCCATCCACATCC | This paper and Alcántara et al., 2021 | N/A |
| ssDNA oligo: primer reverse RNaseP target human sample control CACCCTCAATGCAGAGTC | Broughton et al.,2020 | N/A |
| reporter_FQ: /56-FAM/TTATT/3IABkFQ/ | Chen et al. (2018) | N/A |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism version 9 | GraphPad Software, La Jolla, CA | www.graphpad.com |
| Stata v14.0 | StataCorp, College Station, TX | https://www.stata.com/ |
| Other | ||
| 96-well flat clear bottom black polystyrene TC-treated microplates | Corning® | Cat # 3603 |
| NanoDrop One microvolume UV-Vis spectrophotometer | Thermo Fisher | Cat # ND-ONE-W |
| safeVIEW: LED/Blue Light Transilluminator | Cleaver Scientific | Cat # SAFEVIEW |
| CytationTM 5 Cell Imaging Multi-Mode Reader | BioTek Instruments | N/A |
| SynergyTM H1 Hybrid Multi-Mode Reader | BioTek Instruments | N/A |
Materials and equipment (optional)
-
•
Buffers:
10× CrB Buffer
| Stocks | Final concentration | Amount |
|---|---|---|
| Tris-HCl (1 M) (pH 7.9, 25°C) | 100 mM | 2.5 mL |
| NaCl (1 M) | 100 mM | 2.5 mL |
| Nuclease-free water | n/a | 20 mL |
| Total | n/a | 25 mL |
∗Store at −20°C
1× CrB1 Buffer
| Stocks | Final concentration | Amount |
|---|---|---|
| 10× CrB Buffer | 1× | 3.9 mL |
| Nuclease-free water | n/a | 100 μL |
| Total | n/a | 4 mL |
∗Always use fresh working stocks.
1× CrB2 Buffer
| Stocks | Final concentration | Amount |
|---|---|---|
| 1× CrB1 Buffer | 1× | 3340 L |
| MgCl2 (1 M) | 17.65 mM | 60 μL |
| Total | n/a | 3.4 mL |
∗Always use fresh stocks.
Note: If using locally produced enzymes, prepare the following buffers
5× RPB Buffer
| Stocks | Final concentration | Amount |
|---|---|---|
| Tris-HCl (1 M) (pH 8.4, 25°C) | 250 mM | 6.25 mL |
| KCl (1 M) | 375 mM | 9.375 mL |
| MgCl2 (1 M) | 15 mM | 375 μL |
| Trehalose dihydrate | 50% | 12.5 g |
| DTT (1 M) | 50 mM | 1.25 mL |
| EDTA (0.5 M) | 0.5 mM | 25 μL |
| Nuclease-free water | n/a | to 25 mL |
| Total | n/a | 25 mL |
∗Storage at −20°C.
4× RPB Buffer
| Stocks | Final concentration | Amount |
|---|---|---|
| 5× RPB Buffer | 4× | 200 μL |
| 10 mM dNTPs mixture | 1.6 mM | 40 μL |
| Nuclease-free water | n/a | 10 μL |
| Total | n/a | 250 μL |
∗Storage at – 20°C. Each 250 μL aliquot yields 50 RT-PCR reactions.
MB6 buffer
| Stocks | Final concentration | Amount |
|---|---|---|
| Tris-HCl (1 M) (pH 8.0, 25°C) | 50 mM | 1.25 mL |
| NaCl (1 M) | 100 mM | 2.5 mL |
| DTT (1 M) | 5 mM | 125 μL |
| EDTA (0.5 M) | 0.1 mM | 5 μL |
| Triton X-100 | 0.1% | 25 μL |
| Glycerol | 50% | 12.5 mL |
| Nuclease-free water | n/a | 8.5 mL |
| Total | n/a | 25 mL |
∗Storage at −20°C
-
•
Oligonucleotides: The following primers were used at 5 μM stocks:
Note: Dissolve the primers using nuclease-free water to 100 μM concentration. Then dilute to 5 μM in an appropriate volume.
| Gene | Orientation | Name | Sequence (5’ – 3′) | Amplicon Size (bp) |
|---|---|---|---|---|
| ORF1ab | Forward | FCb3 | GTTGTTCAGTTGACTTCGC | 168 |
| Reverse | RCb2 | GACAATTTCACAAGCACAGG | ||
| N | Forward | FN1 | TACAAACATTGGCCGCAAATTGC | 131 |
| Reverse | RN2 | TCATCCAATTTGATGGCACC | ||
| RNAseP | Forward | FR1 | ACTCAGCCATCCACATCC | 175 |
| Reverse | RR2 | CACCCTCAATGCAGAGTC |
-
•Additional reagents:
-
•Working stock 2 mg/mL BSA, dissolved in Nuclease-free water
-
•Working stock 1 M MgCl2
-
•Nuclease-free water
-
•56-FAM/TTATT/3IABkFQ dually-labeled probe should be dissolved at 100 μM concentration in nuclease-free water. HPLC-purified probe is recommended and can be purchased from any oligo synthesis supplier.
-
•
Note: If using locally produced enzymes, then prepare a working 20× stock of enzymes as follows:
-
•Enzymes:
-
•20× working stock M-MLV reverse transcriptase: Dilute your enzyme stock in MB6 buffer to a final concentration of 34 ng/μL.
-
•20× working stock Taq DNA polymerase: Dilute your enzyme stock to a final concentration of 32 ng/μL.
-
•
-
•Equipment:
-
•The protocol described here for SARS-CoV-2 detection can be used in any conventional thermal cycler. Here, we used the T-100 thermal cycler (Bio-Rad) (Cat. No. 1861096) and the portable BentoLab (https://www.bento.bio/).
-
•Fluorescence signal visualization used the microplate reader Synergy H1 (BioTek Instruments) for a quantitative readout, or a transilluminator safeVIEW: LED/Blue Light (Cleaver Scientific) for a qualitative readout.
-
•RT-PCR product visualization uses 5% agarose gel electrophoresis. In this protocol, the multiSUB Choice, Wide Midi Horizontal Electrophoresis System was used (Cleaver Scientific).
-
•
Step-by-step method details
The present protocol describes the procedure for a conventional RT-PCR reaction with locally produced enzymes (Mendoza-Rojas et al., 2021) coupled to CRISPR/Cas12a detection using a fluorescence microplate reader, or direct fluorescence visualization (Alcántara et al., 2021). This protocol is intended to be used for SARS-CoV-2 RNA detection. However, the described methodology could be set up as a base protocol for molecular detection of nucleic acid molecules (RNA/DNA) of any other pathogen or microorganism of interest.
Note: In this protocol, we describe RT-PCR and CRISPR/Cas recipes for 32 reactions, to fit in the BentoLab portable system. All the examples in the article correspond to a 32 reactions workflow (28 samples + controls). Scale the required volume of each reagent depending on the total number of samples to test or if a 96-well thermocycler is available.
Sample RNA extraction
This protocol has been validated with nasopharyngeal swab samples. Use your locally approved viral RNA extraction or other approved commercial alternative kits. Here, the viral RNA material was extracted using the QIAamp viral RNA Mini kit (QIAGEN).
-
1.
To perform RNA extraction, 200 μL of sample or negative extraction control (NEC) was used according to the manufacturer protocol (https://www.qiagen.com/us/resources/download.aspx?id=c80685c0-4103-49ea-aa72-8989420e3018&lang=en). Proceed with the Spin protocol or Vacuum protocol depending on vacuum pump and manifold availability.
-
2.
Elute RNA with 100 μL of nuclease-free water.
Note: A negative extraction control (NEC) should be included during the RNA extraction step to evaluate contamination during sample handling. A NEC could be a pool of verified negative patient samples, commercial alternatives (i.e., human DNA in stabilizing synthetic matrix, such as the one available in the HDPCRTM SARS-CoV-2 assay (ChromaCode)), or even molecular-grade nuclease-free water.
One-step RT-PCR
This protocol was standardized to amplify two gene SARS-CoV-2 targets, at the 5′- and 3′-end of the genome, as well as a region of the RNAseP human gene as RNA extraction control using a one-step RT-PCR reaction. Examples used M-MLV reverse transcriptase (RT) and Taq DNA polymerase produced in the laboratory. Commercial enzymes can be used as an alternative.
Alternatives: The following commercial kits have been evaluated in replacement of the enzymes mentioned here, M-MLV reverse transcriptase and/or Taq-polymerase. Commercial kits can be used as one- or two-step RT-PCR reactions without interfering with the CRISPR/Cas detection assay. For one-step reactions we tested the 2× One-Step RT-PCR Master Mix (Cat. No. 28113, Norgen Biotek). For two-step reactions we tested RevertAid First Strand cDNA Synthesis Kit (Cat. No. K1622, ThermoFisher) coupled with DreamTaq Green PCR Master Mix (2×) (Cat. No. K1081, ThermoFisher) or Taq DNA polymerase with Standard Taq Buffer (Cat. No. M0273S, NEB). Other commercial enzymes are expected to be suitable for nucleic acid amplification followed by detection with the proposed CRISPR/Cas system described in this protocol.
The one-step RT-PCR reaction produces first a DNA copy (cDNA) of the target RNA molecules extracted from nasopharyngeal swabs or saliva samples. After this reverse transcription step, the cDNA template is amplified by the Taq DNA polymerase to generate dsDNA copies of the target region of interest (Álvarez-Fernández, 2013; Chaubal et al., 2018; Graham et al., 2021; Mahony et al., 2011).
Note: Each gene target is amplified individually by RT-PCR.
-
3.Master mix preparationCRITICAL: It is strongly recommended to prepare the master mix in a different laboratory area from where clinical samples are manipulated or where the samples are analyzed. Amplicon contamination can severely compromise your results. Follow the general recommendations for molecular analysis of clinical samples (WHO. World Health Organization. (2018))
 Pause point: Thaw all reagents on ice, while pre-heating a thermoblock or thermocycler at 50°C.
Pause point: Thaw all reagents on ice, while pre-heating a thermoblock or thermocycler at 50°C.-
a.Prepare the master mix according to the following recipe (i.e., scale the required volume of each reagent depending on the total number of samples to test). Here we work with 33 reactions corresponding to a final volume of 660 μL considering a 2 μL sample to be analyzed and a total volume of 20 μL for each reaction.
Reagent Initial concentration Final concentration Volume (μL) per reaction Volume (μL) for 33 reactions RPB4× 4× 1× 5 165 M-MLV RT 20× 1× 1 33 Taq polymerase 20× 1× 1 33 Reverse primer 5 μM 0.2 μM 0.8 26.4 Forward primer 5 μM 0.2 μM 0.8 26.4 Nuclease-free water - - 9.4 310.2 Final volume 18 594 Note: It is recommended to prepare 1-2 additional reactions due to some volume loss by pipetting.CRITICAL: We strongly recommend preparing and maintaining the master mix on ice (or use an IsoFreeze® PCR rack) to avoid unspecific product amplification (i.e., primer dimerization) (Graham et al., 2021). -
b.Mix by gently vortexing and spin down for few seconds.
-
c.Dispense 18 μL in PCR tubes (0.2 mL). Individual tubes or PCR strips can be used.
-
d.Securely close all tubes and spin down the tubes for few seconds.
-
a.
Place the tubes on ice (or using an IsoFreeze® PCR rack)
-
4.
Add 2 μL of extracted RNA samples to each reaction (step 2). To evaluate potential reaction contamination and reaction performance, a no template control (NTC), a NEC (step 1), and a positive control should be included in each assay, respectively.
Note: For the NTC, nuclease-free water must be added to the respective PCR tube. For the positive control, genomic RNA, RNA standards (e.g., as the ones available in the BEI Resources catalogue (https://www.beiresources.org/)) or sample pools from certified positive samples for SARS-CoV-2 can be used.
-
5.
Insert the tubes or strips into the thermocycler from the BentoLab system (alternative thermocyclers can be used instead) (Figure 1).
-
6.
Run the following RT-PCR program:
| PCR cycling conditions | |||
|---|---|---|---|
| Step | Temperature | Time | Cycles |
| Reverse transcription | 50°C | 20 min | 1 |
| Initial denaturation | 95°C | 5 min | 1 |
| Denaturation | 95°C | 5 s | 45 cycles |
| Annealing/Extension | 55°C | 30 s | |
| Hold | 10°C | Forever | |
Note: Total run time 1:40 hours
-
7.
Store the RT-PCR products at 4°C for short term or at −20°C for mid/long term until the CRISPR/Cas detection assay is performed.
Note: Optional, the RT-PCR product can be visualized by agarose gel electrophoresis. RT-PCR products are expected to be 168 bp for the ORF1ab target, 131 bp for the N target, and 175 bp for the RNAseP target (Figure 2). If RT-PCR product bands are not visible, see troubleshooting section – problem 1.
Figure 1.

Recommended plate layout for 32 reactions to be processed in the BentoLab
Figure 2.

RT-PCR products from positive and negative controls for ORF1ab, N and RNAseP targets
The amplification products were run on a 5% agarose gel at 70 V for 60 min. AmpliSize Molecular Ruler (Cat. No. 1708200, Bio-Rad) was used as ladder. Products were visualized using SafeGreen (Cat. No. G108-G, abm) in the loading buffer TriTrack DNA loading dye 6× (Cat. No. R1161, ThermoFisher).
CRISPR/Cas12a-mediated detection
This protocol was standardized to detect two loci at the 5′- and 3′-end of the SARS-CoV-2 genome, respectively, and a region of the RNAseP human gene. The following crRNA sequences were used for protocol standardization and validation:
| Target | Sequence (5’ – 3′) | length (bp) |
|---|---|---|
| ORF1ab | UAAUUUCUACUAAGUGUAGAUUUAGAGACGGUUGGGAAAUUG | 42 |
| N | UAAUUUCUACUAAGUGUAGAUCCCCCAGCGCUUCAGCGUUC | 41 |
| RNAseP | UAAUUUCUACUAAGUGUAGAUAAUUACUUGGGUGUGACCCU | 41 |
The LbCas12a enzyme detects a specific DNA sequence by recognizing the base pairing between crRNA and the target (Chen et al., 2018; Swarts et al., 2017). Upon recognition of the target DNA, Cas12a undergoes structural changes in its RuvC active site that result in an unspecific trans-cleavage activity on any available single-stranded DNA (ssDNA) molecule in the vicinity (Chen et al., 2018; Li et al., 2019). This has been adapted for in vitro detection of a target DNA of interest for diagnostic proofs of concept (Broughton et al., 2020; Chen et al., 2018). The CRISPR/Cas12a assay uses a double-labeled reporter probe (i.e., with a fluorophore and quencher) that is cleaved upon Cas12a trans-activity only in the presence of the specific target DNA in the reaction, thereby generating a fluorescent signal that can be detected with a fluorescence plate reader or by direct visualization using a transilluminator (Bonini et al., 2020; Kumar et al., 2020).
Note: The following example concentrations and volumes are indicated for the previous 32 RT-PCR reactions for the ORF1ab target that include 28 unknown samples, two positive controls and two no template controls (NTC). Each target RT-PCR product (ORF1ab, N, and RNaseP) is evaluated individually using the CRISPR/Cas12a assay. An Excel template (Mendeley Data: https://doi.org/10.17632/8m8z37v8xz.1 Reaction recipe template) is provided to calculate the necessary volume of the required reagents for crRNA re-folding and Cas12a master mix preparation.
Note: All buffers required in this segment must be prepared in advance.
-
8.
Thaw all reagents on ice
-
9.crRNA re-folding
-
a.Prepare the working stock by diluting the crRNA stock in nuclease-free water to get a final concentration of 1.3 μM (see before you begin, Step 6I, for crRNA production).
-
b.Mix gently by vortexing or tapping, and spin briefly.
-
c.Incubate at 65°C for 10 min.
-
d.Cool at 25°C for 10 min.
-
e.Mix gently by vortexing or tapping. Then spin briefly.
-
a.
-
10.10× Cas12a complex
-
a.Prepare the 10× Cas12a complex according to the following recipe (i.e., scale the required volume of each reagent depending on the total number of samples to test or your stocks’ concentration).
Reagent Initial concentration Final concentration Volume (μL) per reaction Volume (μL) for 33 reactionsa Cas12a 5.4 μM 0.1μM 0.184 6.1 Re-folded crRNA 1.3 μM 0.15 μM 1.164 38.4 Probe 100 μM 3 μM 0.3 9.9 1× CrB1 - - 8.351 275.6 Final volume - 330 aConsidering one additional reaction due to volume loss by pipetting. -
b.Protect from light by wrapping the tube with aluminum foil or using dark tubes, if available.
-
c.Incubate the 10× Cas12a complex at 25°C for 10 min.
-
a.
-
11.CRISPR/Cas12a reaction preparation
-
a.Each CRISPR/Cas detection reaction is prepared according to the following scheme:
Pipetting
OrderReagent Initial concentration Final concentration Final volume (μL) per assay Notes 1 CrB2 (17.65 mM MgCl2) 1× 1× 85 Mix separately 2 RT-PCR product - - 5 3 Cas12a complex 10× 1× 10 Add right before measuring Final volume - - 100 Note: CrB2 buffer contains 17.65 mM MgCl2, which upon addition of the RT-PCR product and the 10× Cas12a complex brings MgCl2 to 15 mM. -
b.Dispense 85 μL of 1× CrB2 in separate tubes, PCR strips, or a 96-well PCR plate.
-
c.Add 5 μL of the RT-PCR product (Step 7).
-
d.Mix gently by tapping or using a plate mixer if available. If working with PCR tubes or strips, spin briefly.Note: When working with a multichannel pipette consider increasing the volumes of the first two pipetting steps to avoid inconsistent pipetting. 102 μL CrB2 and 6 μL RT-PCR product for a total of 108 μL from which 90 μL are taken with the multichannel pipette for further analysis.
-
e.Dispense 10 μL of the 10× Cas12a complex in every well of the 96-well microplate (Step 10c) (Figure 3).
-
f.Add 90 μL of the diluted RT-PCR products according to the above plate layout (Step 11c).
-
a.
Figure 3.

Recommended plate layout for CRISPR/Cas reactions in a 96-well microplate
For each target 32 reactions are included (28 unknown samples + duplicates of positive controls, negative extraction, and no template controls).
Alternative for the Cas12a complex to set up CRISPR/Cas reactions: the EnGen® Lba Cas12a (Cpf1) (Cat. No. M0653S – M0653T, NEB) can be used with the NEB buffer 1×.
-
12.
Outcome readout
A quantitative and a qualitative approach have been evaluated using a microplate reader or by direct fluorescence visualization in microtubes or plates, respectively. The fluorescence is produced by the cleavage of the double-labeled probe (56-FAM/TTATT/3IABkFQ) due to target recognition by the CRISPR/Cas12a system.-
a.Quantitative readoutNote: The assay was tested using the SynergyTM H1 Hybrid Multi-Mode microplate reader (BioTek), with software Gen5 v3.10.
-
i.Set-up the instrument according to the following parameters:
General Plate type Select the respective black plate Temperature 25°C Kinetic step Runtime 35 min Interval 1 min Read methoda Detection method Fluorescence intensity Read type Endpoint/Kinetic Optics type Monochromators Excitation (nm) 491 Emission (nm) 525 Gain 85 Read height 7 mm aIf using an equipment that works with a variable bandwidth (i.e., Cytation 5 Multi-Mode reader, Biotek) consider using the following wavelengths: ex 491±9 nm, and em 525±21 nm. If your instrument uses excitation and emission filters, use the corresponding set for Fluorescein (Usually pre-set as GFP) -
ii.Transfer the microplate to the reading instrument and proceed with the measurement.
-
i.
-
b.Qualitative readoutNote: The protocol was tested using the transilluminator - safeVIEW: LED/Blue Light (470 nm blue LEDs) (Cleaver Scientific).
-
i.Once the CRISPR/Cas12a reaction is prepared, incubate without light exposure for 30 min.
-
ii.Transfer the microplate to the transilluminator and close the filter tap.
-
iii.Register the results of direct fluorescence visualization using a photo camera or a smartphone.Note: Using a black cabinet, box, or dark room improves the direct visualization of the results.
-
i.
-
a.
Expected outcomes
Regarding the fluorescence detection in unknown samples, the fluorescence must rapidly increase over time for positive samples. Negative samples and reaction controls do not show a marked increase. However, they can slightly increase over time. An appropriate variable to compare results is the fluorescence ratio between the analyzed sample over the non-template control (NTC) (see the quantification and statistical analysis section)(Alcántara et al., 2021; Hou et al., 2020). The fluorescence ratio should be higher for positive samples than for the negative ones. There is a positive dependence between fluorescence ratios and the viral RNA load of the sample, higher RNA loads derive in higher fluorescence ratios (Figure 4A). In the validation study with 100 clinical samples, the median fluorescence ratio for positive samples was higher for ORF1ab target (11.4) than the one observed for the N target (7.9) (Figure 4B) (Alcántara et al., 2021). For both detection targets, positive samples show a fluorescence ratio over 4-fold with respect to negative samples. For results interpretation, the fluorescence ratio of the RNaseP human gene must be considered (see below).
Figure 4.

Fluorescence ratio distribution for clinical samples in this example
Data from 50 SARS-CoV-2 positive and 50 negative samples are shown.
(A) Fluorescence distribution for SARS-CoV-2 positive samples sorted by viral RNA load for the ORF1ab and N targets.
(B) Fluorescence distribution for SARS-CoV-2 positive and negative samples for the two targets (ORF1ab, N).
(C) RNaseP fluorescence ratio distribution for SARS-CoV-2 samples, sorted by viral RNA load, positive and, negative controls. Tukey boxplots in A-C indicate the median, 25%, and 75% percentiles. Whiskers indicate minimun and maximun values excluding outliers (full circles).
The fluorescence signal of the RNaseP gene must increase over time for both SARS-CoV-2 positive and negative clinical samples. Fluorescence ratios of the RNaseP target over the NTC control are expected to be around 25 independently of the viral RNA load (Figure 4C). Fluorescence ratios for the RNaseP target are higher than the ones observed for SARS-CoV-2 targets. In the validation study, the median fluorescence ratio for the 100 tested clinical samples for RNaseP was around 25 (Alcántara et al., 2021). If the fluorescence ratio is less than two, the assay must be repeated to rule out any technical error. If the repeated test fluorescence ratio is less than two, the sample quality must be checked, or a new sample should be required (see troubleshooting section).
Fluorescence ratios for positive controls must be higher than the negative controls. In the validation study with 100 clinical samples, a mean fluorescence ratio of 13.96 ± 1.72 RFU and 0.64 ± 0.14 RFU were reported for the positive and negative controls for the ORF1ab target, respectively. Likewise, an average fluorescence ratio of 8.37 ± 0.35 RFU and 1.05 ± 0.43 RFU were calculated for the positive and negative controls for the N target, respectively (Figure 5). If fluorescence is not observed, see the troubleshooting section – problems 2 and/or 3.
Figure 5.

Fluorescence ratio for positive and negative controls
Fluorescence ratio data represent the mean of five independent experiments. Error bars show standard deviation.
Finally, the observation of fluorescence is indicative of a positive result for SARS-CoV-2. Positive samples show a high fluorescence intensity (Figure 6). If weak fluorescence signal is visualized, see the troubleshooting section – problem 4.
Figure 6.

Visual detection of the amplified N target using CRISPR/Cas12a-dependent fluorescence signal
RT-PCR products of positive and negative SARS-CoV-2 samples were observed in a blue light LED transilluminator.
Quantification and statistical analysis
In the quantitative approach, the fluorescence ratios over the NTC are evaluated to determine positivity.
Note: An Excel template (Mendeley Data: https://doi.org/10.17632/8m8z37v8xz.1. Fluorescence ratios calculation template) can be used to calculate the fluorescence ratio of each sample. Sheet 1 is the original readout report and exported as an Excel file from the Biotek instruments. The template must be modified if using a different instrument and software. Sheet 2 shows the normalized fluorescence ratios between the sample and the NTC control.
-
1.
Export the raw data for all samples as an Excel spreadsheet. Relative fluorescence unit (RFU) time courses will appear in one column for each sample/well (Table 1).
-
2.
Normalize the fluorescence signal by subtracting the first value (Time = 0) for each sample (Table 2).
-
3.
Calculate the mean for fluorescence values of the positive control.
-
4.
Calculate the fluorescence ratio for each control and sample using the following equation:
Table 1.
Example of raw data measured on a plate reader
| Time | Temp. | Raw data | |||||
|---|---|---|---|---|---|---|---|
| - | - | Sample Example | Sample Example | Positive control | Positive control | NEC | NTC |
| Plate wells in File S2 | C5 | C7 | E8 | F8 | G8 | H8 | |
| 0:00:00 | 25°C | 3255 | 2323 | 5298 | 4210 | 2388 | 2182 |
| 00:15:00 | 25°C | 11773 | 3051 | 24534 | 21618 | 3633 | 3431 |
| 00:30:00 | 25°C | 22442 | 3759 | 47499 | 43959 | 5366 | 5015 |
Table 2.
Example of normalized data measured on a plate reader
| Time | Temp. | Raw data | |||||
|---|---|---|---|---|---|---|---|
| - | - | Sample Example | Sample Example | Positive control | Positive control | NEC | NTC |
| Plate wells in File S2 | C5 | C7 | E8 | F8 | G8 | H8 | |
| 00:15:00 | 25°C | 8518 | 728 | 19236 | 17408 | 1245 | 1249 |
| 00:30:00 | 25°C | 19187 | 1436 | 42201 | 39749 | 2978 | 2833 |
Divide the fluorescence signal (RFU) of test samples (positive control or NEC) by the NTC values, both measured at the timepoint 30 min (Table 3).
-
5.
Determine positivity following the algorithm shown in Figure 7,
Note: The method using this protocol showed a Limit of Detection (LoD) of 102 ge/μL using SARS-CoV-2 genomic RNA (NR-52347, BEI Resources). During the validation process five out of 50 clinical samples showed false negative results. All discordant samples showed Cq values more than 33 (Alcántara et al., 2021). However, Cq value distribution in infected people peaked between 23 – 25 Cq values (Buchan et al., 2020). In that context, the described protocol here should be able to detect all clinical samples.
Table 3.
Example of fluorescence ratio for normalized data measured on a plate reader
| Time | Temp. | Raw data | |||||
|---|---|---|---|---|---|---|---|
| - | - | Sample Example | Sample Example | Positive control | Positive control | NEC | NTC |
| Plate wells in File S2 | C5 | C7 | E8 | F8 | G8 | H8 | |
| 00:30:00 | 25°C | 6.604 | 0.494 | 14.525 | 13.681 | 1.025 | 0.975 |
Figure 7.

Test decision scheme
Cut-off values were calculated by a Receiver Operating Characteristic (ROC) curve analysis (Hanley and McNeil, 1982). The selected cut-off values were those that reported the highest percentage of correctly classified samples according to a ROC curve analysis with 100 samples (Alcántara et al., 2021). ∗ Test again if symptoms occur. If symptoms already present RT-qPCR recommended. ∗∗ If an invalid result is obtained, see troubleshooting section – problem 5.
Limitations
This protocol has been validated only with clinical upper-respiratory samples (n = 100) and performed in one country (Peru). Although no mutations have been reported in the SARS-CoV-2 target sequences used in this protocol (ORF1ab and N), the test sensitivity could vary depending on local circulating SARS-CoV-2 strains. The detection targets, used in this protocol, have been selected considering genome regions with low variability. We cannot exclude that false negative results could arise due to future variants of SARS-CoV-2. We recommend conducting a local clinical validation before using it as a laboratory-developed test (LDT) for clinical analysis. Negative results do not exclude COVID-19 disease. Molecular tests are part of the diagnostic algorithms worldwide that include other variables such as symptoms, contact to diagnosed patients, among others. False negative results can be produced by errors during sample manipulation and processing. This protocol is not authorized by any governmental institution yet. A technical guide to validate in-house diagnostic tests can be downloaded from the FIND website (https://www.finddx.org/reports-and-landscapes/in-house-test-development-for-molecular-detection-of-sars-cov-2-en-fr-pt/). Other outcome readouts such as lateral flow assay on paper strips have not been validated with this protocol.
Troubleshooting
Problem 1
No amplification for the positive control in RT-PCR (RT_PCR_outcome)
The lack of RT-PCR products for the positive control indicates that a technical error has occurred during the preparation of the master mix or the RT-PCR running step. Also, that the quality of the reagents could be compromised. Low or null yield amplification can produce a low fluorescence signal that could be misinterpreted as a negative result.
Potential solution
Check that the used cycling conditions are correct. Storage conditions of the reagents, enzymes and aliquots of the controls must be checked. Replace the working stock of reagents, enzymes or aliquots of the controls if necessary.
Problem 2
High fluorescence signal in the no template control and/or negative extraction control (Fluorescence_outcome)
A high fluorescence signal in the NTC indicates contamination during the CRISPR/Cas reaction or more likely during the amplification step (RT-PCR). A high fluorescence signal in the NTC gives lower fluorescence ratios that could be misinterpreted as negative results in the test samples (false negative). In a five-day assay (to assess repeatability), a normalized fluorescence signal average (i.e., time zero subtracted) of 5171 ± 821 RFU (a.u.) and 2725 ± 411 RFU (a.u.) were obtained for both NTC controls of N and ORF1ab targets, respectively, at time point 30 min. Similarly, a normalized fluorescence signal average of 5350 ± 234 RFU (a.u.) and 1754 ± 418 RFU (a.u.) were observed for both NEC control of N and ORF1ab targets, respectively. However, these values may change depending on the instrument used and current setup.
Potential solution
First, it is recommended to repeat the assay in order to rule out cross contamination between samples or contamination in any reagent used in the CRISPR/Cas reaction. At the same time, it is recommended to run an electrophoresis to visualize any amplicon contamination in the NTC or NEC reaction. If amplicons are observed, contamination in reagents, equipment (i.e., pipettes or tips) or lab area used in the RT-PCR must be considered. Replace the working stock of reagents (i.e., primers, buffer, dNTP, or enzymes), and tips. Clean the pipettes and working areas with a commercial solution such as LookOut® DNA erase spray (Sigma-Aldrich) or 1–2% hypochlorite solution. Cleaning procedures of equipment and lab areas must be considered as part of the working routine.
Problem 3
Low fluorescence signal in positive controls (Fluorescence_outcome)
Low fluorescence signal in positive controls could be due to technical errors in the reaction preparation (i.e., miscalculation of reagents or enzymes concentration, improper pipetting dispense, etc.). Also, that the quality of the reagents or enzymes may be compromised. Low fluorescent signals could compromise the analysis of unknown samples, affecting the calculated ratios and the test results. This problem can arise from the RT-PCR step or the CRISPR/Cas-mediated detection. In a five-day assay (to assess repeatability), a normalized fluorescence signal average (i.e., time zero subtracted) of 43029 ± 6598.40 RFU (a.u.) and 37837.8 ± 3583.51 RFU (a.u.) were obtained for both SARS-CoV-2 N and ORF1ab targets, respectively, for a time readout of 30 min. However, the reported values may change depending on the instrument used and/or the reading setup.
Potential solution
To rule out a potential lack of RT-PCR amplification, check the reaction products by agarose gel electrophoresis. If RT-PCR products are absent see problem 1. If amplified products are observed, the problem can be solved by focusing on the CRISPR/Cas reaction. Check the reagents and enzyme calculation, equipment setup, and reagents and enzymes storage conditions. Replace the working reagents and/or enzymes stocks if necessary. If a different fluorophore is used in the double-labeled probe, equipment setup must be standardized before evaluating unknown clinical samples.
Problem 4
Weak fluorescence during direct visualization readout (Visualization_outcome)
Naked-eyed detection has been validated using a blue-light transilluminator. High fluorescence signal was reported for high- (Cq < 25) and mid-viral load samples (Cq 25–31) (Figure 6). Using other transilluminators (e.g., UV-light transilluminator) showed weaker fluorescence signal even for high- and mid- viral load samples.
Potential solution
It is recommendable to test direct visual readout for control reactions before testing clinical unknown samples, if using other than a blue-light transilluminator. If available, top filter or dark box can be used to improve direct fluorescence visualization.
Problem 5
Invalid results (Interpretation_outcome)
Invalid results are obtained when the fluorescence signal in the RNAseP target is low, producing fluorescence ratios < 2. Low fluorescence ratios can be obtained if there is a high fluorescence signal in the NTC or a low signal in the reactions with the test samples.
Potential solution
Verify the presence of RT-PCR products to rule out an amplification problem by agarose gel electrophoresis. If RT-PCR products are absent see problem 1. If amplified products are observed, the problem can be solved by focusing on the CRISPR/Cas reaction. Repeat the CRISPR/Cas reaction to rule out any technical error during the preparation of the Cas12a master mix. Verify the storage conditions of the Cas12a and crRNA stocks to assure the reagents quality. If the repeated fluorescence ratio result is < 2 or RT-PCR products are not adequately amplied, low sample quality must be considered, and a new sample must be requested.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pohl Milón (pmilon@upc.pe).
Materials availability
All unique reagents developed in this work can be purchased at any gene synthesis company (i.e., primers and crRNA templates).
Acknowledgments
We are very thankful to Dr. Marcos Milla for donating equipment that was used in this study and others. We would also like to thank all laboratory members of the Adaui and Milón groups for their help, support, and great working atmosphere. This work was supported by grants from the Fondo Nacional de Desarrollo Científico, Tecnológico y de Innovación Tecnológica [070-2020-FONDECYT] to V.A. and [036-2019-FONDECYT-BM-INC.INV] to P.M.
Author contributions
Conceptualization and methodology, R.A., K.P., V.A., and P.M.; investigation, R.A., K.P., J.A.N., G.M.R., E.D., and D.A.; formal analysis, R.A., K.P., J.A.N., and P.M., writing – original draft, supervision, and visualization, R.A., K.P., and P.M., writing – review & editing, R.A., K.P., J.A.N., G.M.R., E.D., D.A., V.A., and P.M.; project administration, P.M.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100878.
Contributor Information
Roberto Alcántara, Email: pcmeralc@upc.edu.pe.
Pohl Milón, Email: pmilon@upc.edu.pe.
Supplemental information
Excel spreadsheet intended to be used for reagents and enzymes volume calculation for CRISPR/Cas reaction preparation. Data about number of reactions, crRNA and Cas12a concentrations must be replaced with the appropriated values. Recommended setup values for the fluorescence reader Synergy H1 (BioTek) are also indicated.
Excel spreadsheet intended to be used for fluorescence signal data analysis from the fluorescence reader Synergy H1 (BioTek) (or another similar alternative equipment). Fluorescence signal data must be replaced directly in the “Fluorescence analysis template” tab, whenever the experiment plate was setup according to the recommended plate layout for CRISPR/Cas reaction (Figure 3). for manipulating samples
Data and code availability
Supplemental data files have been deposited to Mendeley Data: https://doi.org/10.17632/8m8z37v8xz.1.
References
- Alcántara R., Peñaranda K., Mendoza G., Nakamoto J.A., Martins-Luna J., Valle J.D., Adaui V., Milón P. Unlocking SARS-CoV-2 detection in low- and middle-income. Cell Reports Methods. 2021 doi: 10.1016/j.crmeth.2021.100093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Fernández R. Chapter one explanatory chapter PCR primer design. Methods Enzymol. 2013;529:1–21. doi: 10.1016/B978-0-12-418687-3.00001-X. [DOI] [PubMed] [Google Scholar]
- Bonini A., Poma N., Vivaldi F., Kirchhain A., Salvo P., Bottai D., Tavanti A., Francesco F.D. Advances in biosensing: the CRISPR/Cas system as a new powerful tool for the detection of nucleic acids. J. Pharm. Biomed. 2020;192:113645. doi: 10.1016/j.jpba.2020.113645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton J.P., Deng X., Yu G., Fasching C.L., Servellita V., Singh J., Miao X., Streithorst J.A., Granados A., Sotomayor-Gonzalez A. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020;38:870–874. doi: 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan B.W., Hoff J.S., Gmehlin C.G., Perez A., Faron M.L., Munoz-Price L.S., Ledeboer N.A. Distribution of SARS-CoV-2 PCR cycle threshold values provide practical insight into overall and target-specific sensitivity among symptomatic patients. Am. J. Clin. Pathol. 2020;154:aqaa133. doi: 10.1093/ajcp/aqaa133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaubal G., Sarkale P., Kore P., Yadav P. Development of single step RT-PCR for detection of Kyasanur forest disease virus from clinical samples. Heliyon. 2018;4:e00549. doi: 10.1016/j.heliyon.2018.e00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.S., Ma E., Harrington L.B., Costa M.D., Tian X., Palefsky J.M., Doudna J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:eaar6245. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidi D., Fitzgerald S., Glaspell H.L., Jalbert S., Klapperich C.M., Landaverde L., Maheras S., Mattoon S.E., Britto V.M., Nguyen G.T. Amplicon residues in research laboratories masquerade as COVID-19 in surveillance tests. Cell Rep. Methods. 2021;1:100005. doi: 10.1016/j.crmeth.2021.100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T.G.W., Dugast-Darzacq C., Dailey G.M., Nguyenla X.H., Dis E.V., Esbin M.N., Abidi A., Stanley S.A., Darzacq X., Tjian R. Open-source RNA extraction and RT-qPCR methods for SARS-CoV-2 detection. PLoS One. 2021;16:e0246647. doi: 10.1371/journal.pone.0246647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M.R., Sambrook J. How to win the battle with RNase. Cold Spring Harb. Protoc. 2019;2019 doi: 10.1101/pdb.top101857. [DOI] [PubMed] [Google Scholar]
- Hanley J.A., McNeil B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- Hou T., Zeng W., Yang M., Chen W., Ren L., Ai J., Wu J., Liao Y., Gou X., Li Y. Development and evaluation of a rapid CRISPR-based diagnostic for COVID-19. PLoS Pathog. 2020;16:e1008705. doi: 10.1371/journal.ppat.1008705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Malik Y.S., Ganesh B., Rahangdale S., Saurabh S., Natesan S., Srivastava A., Sharun K., Yatoo M.I., Tiwari R. CRISPR-cas system: an approach with potentials for COVID-19 diagnosis and therapeutics. Front. Cell. Infect. Microbiol. 2020;10:576875. doi: 10.3389/fcimb.2020.576875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Li S., Wu N., Wu J., Wang G., Zhao G., Wang J. HOLMESv2: a CRISPR-cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019;8:2228–2237. doi: 10.1021/acssynbio.9b00209. [DOI] [PubMed] [Google Scholar]
- Mahony J.B., Petrich A., Smieja M. Molecular diagnosis of respiratory virus infections. Crit. Rev. Clin. Lab. Sci. 2011;48:217–249. doi: 10.3109/10408363.2011.640976. [DOI] [PubMed] [Google Scholar]
- Mendoza-Rojas G., Sarabia-Vega V., Sanchez-Castro A., Tello L., Cabrera-Sosa L., Nakamoto J.A., Peñaranda K., Adaui V., Alcántara R., Milón P. A low-cost and open-source protocol for expression of key molecular biology enzymes. Star Protoc. 2021 doi: 10.1016/j.xpro.2021.100899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts D.C., van der Oost J., Jinek M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-cas12a. Mol. Cell. 2017;66:221–233.e4. doi: 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. (2018). Dos and Dont's for molecular testing. Available at https://www.who.int/teams/global-malaria-programme/case-management/diagnosis/nucleic-acid-amplification-based-diagnostics/dos-and-don-ts-for-molecular-testing (Accessed: 19 Sep 2020).
- Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel spreadsheet intended to be used for reagents and enzymes volume calculation for CRISPR/Cas reaction preparation. Data about number of reactions, crRNA and Cas12a concentrations must be replaced with the appropriated values. Recommended setup values for the fluorescence reader Synergy H1 (BioTek) are also indicated.
Excel spreadsheet intended to be used for fluorescence signal data analysis from the fluorescence reader Synergy H1 (BioTek) (or another similar alternative equipment). Fluorescence signal data must be replaced directly in the “Fluorescence analysis template” tab, whenever the experiment plate was setup according to the recommended plate layout for CRISPR/Cas reaction (Figure 3). for manipulating samples
Data Availability Statement
Supplemental data files have been deposited to Mendeley Data: https://doi.org/10.17632/8m8z37v8xz.1.