

Abstract
NAFLD is one of the leading causes of abnormal liver function worldwide. NAFLD refers to a group of liver conditions ranging from nonalcoholic fatty liver to NASH, which involves inflammation, hepatocellular damage, and fibrosis. Triggering of inflammation in NASH is a key event in the progression of the disease, and identifying the factors that initiate or dysregulate this process is needed to develop strategies for its prevention or treatment. B cells have been implicated in several autoimmune and inflammatory diseases. However, their role in the pathogenesis of NAFLD and NASH is less clear. This review discusses the emerging evidence implicating intrahepatic B cells in the progression of NAFLD. We highlight the potential mechanisms of B‐cell activation during NAFLD, such as increased hepatic expression of B‐cell–activating factor, augmented oxidative stress, and translocation of gut‐derived microbial products. We discuss the possible effector functions by which B cells promote NAFLD, including the production of proinflammatory cytokines and regulation of intrahepatic T cells and macrophages. Finally, we highlight the role of regulatory and IgA+ B cells in the pathogenesis of NASH‐associated HCC. In this review, we make the case that future research is needed to investigate the potential of B‐cell–targeting strategies for the treatment of NAFLD.
Abbreviations
- BAFF
B‐cell–activating factor
- BCR
B‐cell receptor
- Bregs
regulatory B cells
- cDCs
classical dendritic cells
- DCs
dendritic cells
- HFD
high‐fat diet
- IFN
interferon
- IR
insulin resistance
- KCs
Kupffer cells
- LPS
lipopolysaccharide
- MOMFs
monocyte‐derived macrophages
- MYD88
myeloid differentiation primary response 88
- NAFL
nonalcoholic fatty liver
- OSEs
oxidative‐stress–derived epitopes
- sc‐RNAseq
single‐cell RNA sequencing
- Th
T helper
- TLRs
Toll‐like receptors
NAFLD is estimated to affect 30% of the population and is one of the leading causes of abnormal liver function.( 1 ) NAFLD covers a wide spectrum of liver pathology ranging from simple lipid accumulation to NASH, characterized by steatosis, inflammation, hepatocellular injury, and fibrosis.( 2 ) Hepatic inflammation, driven by liver immune cells, is a critical component in the initiation and progression of NASH.( 3 ) Notably, the liver is a unique immunological site where immune cells encounter antigen‐rich blood from the gastrointestinal tract.( 4 ) NASH is characterized by a robust recruitment of immune cells into the liver where they become activated and release molecules that cause inflammation.( 5 ) Innate immune mechanisms are a major contributing factor to the inflammatory process in NASH. However, recent evidence indicates that adaptive immunity plays an essential role in the progression of NASH, fibrosis, and HCC.( 6 ) B lymphocytes are emerging as central mediators of NAFLD because of their ability to secrete cytokines and antibodies and promote inflammation. In this review, we discuss the evidence for a role of B cells in the progression of NAFLD, including the mechanisms of their activation, their contribution to NASH‐associated HCC, and the potential for B‐cell–specific therapeutic strategies.
The NAFLD Inflammatory Environment at a Glance
Role of Innate immune cells
The liver receives a constant influx of gut‐derived products and acts as a firewall for the antigen‐rich mesenteric circulation when the mucosal–blood barrier is compromised.( 7 ) NAFLD is associated with increased intestinal permeability that facilitates the translocation of bacterial products and intact bacterium into the liver that triggers inflammation in the liver.( 8 ) Innate immune responses, particularly those initiated by macrophages, are a major contributing factor to disease progression. Hepatic macrophages are a heterogeneous population of cells that can be categorized into tissue‐resident Kupffer cells (KCs) and monocyte‐derived macrophages (MoMFs). KCs originate primarily from the yolk sac and are maintained in the liver through self‐renewal.( 9 ) During NASH, activation of KCs by lipopolysaccharide (LPS) and fatty acids promotes hepatic inflammation and fibrosis through the production of TNFα( 10 ) and IL‐1β.( 11 ) The prevailing dogma is that KC‐derived factors promote the infiltration and expansion of MoMFs, which further aggravate the disease through their inflammatory functions.( 12 ) Recent single‐cell RNA sequencing (sc‐RNAseq) of intrahepatic immune cells shows that NASH is characterized by a loss of embryonically derived KCs and increased infiltration of MoMFs.( 13 ) In addition, a remarkable subset of macrophages expressing the lipid receptor, triggering receptor expressed on myeloid cells 2, that expands in the NASH liver to coordinate hepatocyte energy supply and mitochondrial function, has been identified.( 14 ) The liver contains a reservoir of classical (cDCs) and plasmacytoid (pDCs) dendritic cells (DCs) strategically located in the subcapsular space, between hepatocytes, and in the vasculature.( 15 ) In human NASH, the ratio of cDC subsets shifts toward increased type 2 cDCs that could activate pathogenic T cells.( 16 ) However, the function of DCs in NASH is less clear given that animal studies suggest that cDCs, particularly classical type 1 DCs, prevent fibrosis and inflammation.( 17 , 18 ) Furthermore, interferon (IFN)α‐producing pDCs expand in the livers of high‐fat diet (HFD)‐fed mice and contribute to hepatic insulin resistance (IR).( 19 ) Neutrophils promote NASH through the secretion of elastase( 20 ) and myeloperoxidase.( 21 ) Accordingly, antibody‐mediated depletion of neutrophils in mice ameliorated liver steatosis and inflammation in mice fed an HFD.( 22 ) Given that neutrophils remove dead vasculature and create new channels for vascular regrowth in sterile liver injury,( 23 ) a better understanding of their reparative function in NASH is needed.
Role of Adaptive Immune Cells
Although the innate immune response in NASH has been the focus of research, growing evidence indicates that T and B cells play essential roles.( 6 ) Upon activation by type 1 IFN signals and intestinal antigens, CD8+ T cells promote nonalcoholic fatty liver (NAFL)( 24 ) and NASH( 25 ) through the secretion of IFNγ and TNFα.( 24 ) Indeed, depletion of CD8+ T cells can restore hepatic insulin sensitivity,( 24 ) decrease liver damage,( 25 ) and reduce fibrosis.( 26 ) During NASH, elevated levels of hepatic acetate and extracellular ATP stimulate resident CD8+ T cells that show an exhausted phenotype and enhanced hepatocyte killing activity.( 27 ) NASH promotes increased intrahepatic CD4+ T‐cell polarization toward IFNγ secreting T helper (Th)1 and IL‐17 secreting Th17 subsets.( 28 ) Elevated levels of intrahepatic IL‐17 accelerate the pathogenesis of NASH and HCC.( 29 ) As a result, an increased peripheral and intrahepatic Th17/regulatory T cell ratio has been proposed as a biomarker of progression from NAFL to NASH.( 28 ) Furthermore, obesity‐associated endotoxemia can also result in the expansion of IL‐17‐secreting γδ T cells in the liver, sustained by microbial antigens that enter through the portal vein.( 30 ) Natural killer T cells, a subclass of T cells that express an invariant T‐cell receptor, infiltrate the NASH liver and secrete proinflammatory cytokines, such as LT‐related inducible ligand that competes for glycoprotein D binding to herpesvirus entry mediator on T cells( 24 ) and IFNγ,( 31 ) and contribute to hepatic fibrosis through the activation of HSCs.( 32 ) Despite the substantial evidence showing the diverse functions of adaptive immune cells, such as T cells in NAFLD, the role of B cells in the pathogenesis of the disease is less understood.
B Cells and Their Subsets
B cells can be categorized primarily into B1 and B2 subsets according to the expression of cell‐surface markers (Table 1). B2 cells arise from hematopoietic progenitors in the bone marrow.( 33 ) The development of B2 cells is antigen independent and involves rearrangements of the V‐D‐J regions of the heavy‐ and light‐chain Ig gene to form an immature B cell.( 34 ) Immature B cells expressing IgM exit the bone marrow to migrate into the spleen where they mature into the marginal zone and follicular B cells in a process that requires the survival factor B‐cell–activating factor (BAFF).( 33 ) Splenic marginal zone B cells are involved in thymus‐dependent and ‐independent responses to blood‐borne pathogens and lipid antigens.( 35 ) Mature follicular B cells make up most of the B‐cell populations in the periphery and secondary lymphoid organs where they survey for antigens.( 35 ) B1 cells arise from the fetal liver and primarily reside in peritoneal and pleural cavities.( 36 ) B1 cells are classified into B1a cells that produce natural antibodies against self‐antigens and B1b cells, which can class switch to IgG3 or IgA after antigen binding.( 37 ) A functionally distinct subpopulation of B cells, known as regulatory B cells (Bregs), are strong producers of IL‐10 and IL‐35 that suppress inflammation.( 38 ) Following antigen recognition, B cells undergo thymus‐dependent or ‐independent activation, affinity maturation, and class switching.( 39 ) Activated B cells can subsequently differentiate into antibody‐secreting plasma cells and memory B cells.( 40 ) During thymus‐dependent activation, the antigen is bound by the B‐cell receptor (BCR), internalized, and presented to follicular Th cells, which become activated and provide signals to the B cell through CD40/CD40 ligand interactions and cytokines.( 41 ) In contrast, thymus‐independent B‐cell activation does not require a second signal from T cells, but instead involves signaling through Toll‐like receptors (TLRs) or cross‐linking of the BCR.( 42 ) Although their precise function at steady state is less clear, most B‐cell subsets, such as mature, immature, B2, B1, and plasma cells, have been detected in the human liver.( 43 , 44 )
TABLE 1.
Murine B‐Cell Subsets and Their Markers
| B‐Cell Subset | Markers | Refs. |
|---|---|---|
| B1 B cells | ||
| B1a | CD19+ CD5+ CD11b+/− B220lo IgMhi IgDlo CD23− CD43+ | ( 35 ) |
| B1b | CD19+ CD5− B220lo IgMhi IgDlo CD23− CD43+ | ( 35 ) |
| B2 B cells | ||
| Follicular | CD19+ B220+ IgMlow IgDhi CD21med CD1dlow CD23+ | ( 35 ) |
| Marginal zone | CD19+ B220+ IgMhi IgDlow CD1dhi CD21hi CD23‐ | ( 35 ) |
| Memory | CD19+ B220low/+ CD21+ CD95+ CD40+ CD273+ CD73+ CD80+ | ( 83 ) |
| Plasma cells | CD138+ CD93+ CXCR4+ CD44+ VLA‐4+ BCMA+ IL6‐R+ | ( 84 ) |
| Regulatory | CD19+ PD‐L1+/− Tim‐1+/− FasL+/− LAG‐3+/− | ( 38 ) |
Abbreviations: BCMA, B‐cell maturation antigen; FasL, Fas ligand; LAG‐3, lymphocyte activation gene‐3; Tim‐1, T‐cell immunoglobulin and mucin domain 1; VLA‐4, very late antigen‐4.
Adipose Tissue B Cells in Metabolic Disease
B2 cells accumulate in the obese adipose tissue where they contribute to local and systemic IR through pathogenic IgG antibodies( 45 ) and activation of T cells and macrophages.( 46 ) In humans, adipocyte‐specific IgG against antigens released by dying cells are found locally and in the plasma, suggesting that obesity promotes B‐cell responses against “self.”( 47 ) During obesity, IgG undergoes hyposialylation and can promote IR through activation of its receptor, functionally impaired Fcγ receptor IIB.( 48 ) Although the mechanisms underlying the expansion or infiltration of B cells into obese adipose tissue are poorly defined, signaling through the chemokine, leukotriene B4, and its receptor is required.( 46 ) B cells are among the first immune populations to infiltrate the mesenteric adipose tissue of mice fed an HFD from where they migrate to the liver and promote hepatic inflammation.( 49 ) In contrast to B2 cells, Bregs maintain adipose tissue homeostasis through IL‐10, and their loss during obesity leads to adipose tissue inflammation and IR.( 50 ) In mice fed an HFD, adipose tissue B1 cells ameliorate disease attributable to their production of IL‐10 and polyclonal IgM( 51 ); however, a role for B1 cells in adipose tissue is yet to be confirmed in humans. Furthermore, whether B cells travel between adipose tissue and the liver as a strategy to coordinate whole‐body metabolism during obesity or NAFLD is still being uncovered.
Intrahepatic B Cells in the Progression of NAFLD
Although limited, increasing evidence suggests that intrahepatic B cells participate in the progression of NAFLD (Table 2; Fig. 1). Patients with NAFL or NASH show increased intrahepatic B cells in association with higher levels of circulatory IgG against oxidative‐stress–derived epitopes.( 52 ) In patients with NASH, intrahepatic B cells are more abundant in the portal tract than in the lobules, suggesting that they participate in the portal inflammatory process.( 53 ) Studies using rodent models suggest a pathogenic function for these intrahepatic B cells in the pathogenesis of NAFLD. Mice fed an HFD that develop liver steatosis show an increased accumulation of B cells that express increased amounts of proinflammatory cytokines and have a higher capacity to activate T cells.( 54 ) Whether this accumulation and activation of B cells are causal or consequential to the triggering of NASH, or are merely bystander effects of the inflammatory environment, has been less clear. Our laboratory recently showed that intrahepatic B cells, primarily B2 cells, accumulate in the liver of NASH mice where they express a proinflammatory gene expression profile and secrete increased amounts of IL‐6 and TNFα, thereby promoting inflammation and fibrogenesis.( 55 ) Although we show that B‐cell–deficient mice have improved glucose tolerance during NASH, whether intrahepatic B cells can directly promote local IR, similar to their adipose tissue counterparts, remains to be determined. Recent transcriptome and surface proteome assessments of immune cells in mice fed a NASH‐inducing Western diet showed a distinct cluster of CD38+ B cells whose proportions were altered with NASH progression.( 56 ) Using sc‐RNAseq with superloading and multiplexing, we were able to identify three distinct clusters of B cells, including a predominant mature B‐cell population as well as two less‐abundant subsets of immature and metabolically active cells enriched with mitochondrial genes.( 55 ) Supporting a fibrogenic function of B cells, sc‐RNAseq and ligand‐receptor network analysis of NASH livers shows that activated HSCs express increased C‐X‐C motif chemokine ligand 12 with C‐X‐C motif chemokine receptor 4 (Cxcr4) in B cells as the potential target.( 57 ) Mature and plasma B‐cell clusters have been also detected by sc‐RNAseq within areas of scarring in fibrotic livers of patients with cirrhosis.( 58 ) Consistently, B cells have been shown to directly contribute to the evolution of inflammation and fibrosis in mouse models of NASH and hepatotoxicity. In mice fed a methionine‐choline–deficient diet, depletion of B2 cells reduces Th1 responses and partially prevents NASH‐related inflammation.( 52 ) B cells also seem to promote hepatic fibrosis in the CCl4‐induced mouse model of hepatotoxicity.( 59 , 60 ) B‐cell–deficient mice treated with CCl4 have substantially attenuated liver fibrosis with reduced collagen deposition, infiltration of immune cells, and HSC activation, suggesting that B cells are required for liver fibrogenesis.( 60 ) In turn, HSC‐derived retinoid acid induces B‐cell survival and innate‐like proinflammatory function in a feed‐forward mechanism that drives liver fibrosis.( 60 ) The profibrotic role of B cells during CCl4‐induced liver injury depends on the adaptor protein, myeloid differentiation primary response 88 (MYD88), which is indispensable for innate immunity and involved in the retinoid acid signaling pathway.( 60 )
TABLE 2.
Direct Evidence of B‐Cell Involvement in the Pathogenesis of NAFLD
| Key Finding | Refs. | |
|---|---|---|
| Human studies | ||
| NAFL‐NASH patients | B cells found in T‐cell–rich aggregates in the liver of NAFLD patients. B‐cell infiltration was associated with elevated levels of IFNγ, IgG against OSE antigens, and lobular inflammation and fibrosis. | ( 52 ) |
| NAFL‐NASH patients | Elevated serum BAFF levels correlated with hepatocyte ballooning and fibrosis. | ( 61 ) |
| NAFL‐NASH patients | Elevated serum IgA was associated with and predictive of liver fibrosis. | ( 67 ) |
| Mouse models | ||
| High‐fat, high‐carbohydrate diet‐induced NASH, muMT (B‐cell–deficient) mice, anti‐CD20 B‐cell depletion, B‐cell–specific MYD88‐deficient mice | Intrahepatic B cells drive NASH progression through activation by microbial triggers in a MYD88‐dependent mechanism and through modulation of T‐cell–mediated inflammation. | ( 55 ) |
| HFD‐induced NAFL, muMT (B‐cell–deficient mice) | Mesenteric adipose tissue (MAT) B cells promote MAT inflammation by stimulating proinflammatory macrophages. MAT B cells also migrate into the liver where they induce decreased IL‐10 and increased TNFα and MCP‐1 expression in hepatocytes. | ( 49 ) |
| HFD‐induced NAFL | Intrahepatic B cells promote inflammation by producing TNFα, IL‐6, and IgG2a and stimulating Th1 responses. | ( 54 ) |
| Methionine‐choline–deficient diet‐induced NASH, TACI‐Ig (B2‐cell deficient), BAFF neutralizing monoclonal antibody | B2 cells mature into plasma cells. Increased circulating anti‐OSE IgG. B2‐cell deficiency ameliorates NASH progression and reduces IFN‐γ+ CD4 T cells in the liver. | ( 52 ) |
Abbreviations: MCP‐1, monocyte chemoattractant protein‐1; TACI, transmembrane activator and CAML interactor.
FIG. 1.
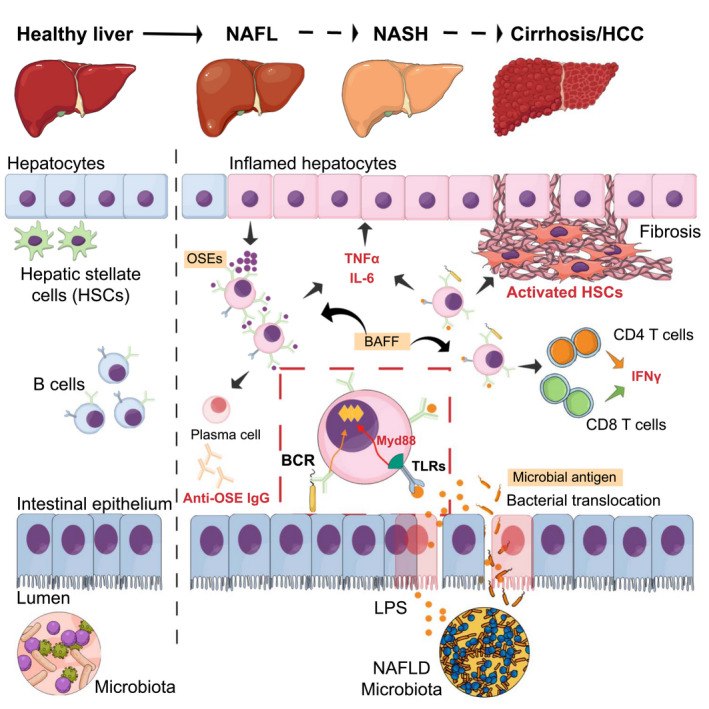
Role of B cells in the progression of NAFLD. Intrahepatic B cells promote disease through cytokine and antibody production. BAFF and OSEs are involved in B‐cell activation. Intestinal‐derived microbial factors resulting from dysbiosis activate intrahepatic B cells in a Myd88‐dependent manner. B‐cell activation may also occur in an antigen‐specific manner through BCR signaling. Activated B cells promote NASH progression through modulation of CD4 and CD8 T‐cell activation and IFNγ responses. The mechanisms of B‐cell–mediated activation of HSCs leading to fibrosis in NASH are unknown.
Potential Mechanisms for B‐Cell Activation
A central objective in the field is the identification of the factors that trigger inflammation in the transition from NAFLD to NASH.( 3 ) In this regard, limited research has been conducted to determine the intrinsic and extrinsic mechanisms of B‐cell activation. In a mouse model of NASH without obesity, B‐cell activation preceded T‐cell responses and was correlated with increased hepatic expression of BAFF, suggesting that this cytokine is involved in B‐cell survival and maturation.( 52 ) Compared with subjects with simple steatosis, NASH patients have higher BAFF serum levels, which are positively correlated with hepatocyte ballooning and liver fibrosis.( 61 ) Despite increased weight gain and adiposity, BAFF‐deficient mice fed an HFD have improved insulin sensitivity, decreased adipose tissue inflammation, and reduced hepatic steatosis in an adipokine‐like role.( 62 ) In mice fed a methionine‐choline–deficient diet to induce NASH, antibody‐mediated BAFF neutralization prevented liver plasma maturation and ameliorated parenchymal damage and lobular inflammation.( 52 ) These findings suggest either direct effects of BAFF on hepatic and adipose tissue metabolism or indirect effects mediated by B‐cell survival and maturation.
Oxidative stress may also trigger B‐cell immune responses as shown in a cohort of NAFLD patients with increased levels of IgG antibodies against oxidatively damaged molecules known as oxidative‐stress–derived epitopes (OSEs).( 63 ) In mice, inducing NASH with a methionine‐deficient diet leads to the expansion of liver plasma cells and increased circulatory anti‐OSE IgG, whereas B2 cell deficiency blunts this response.( 52 ) Another possibility is that gut‐derived antigens are involved in the activation and subsequent proinflammatory function of B cells. Indeed, fecal microbiota transfer from NAFLD patients into lean mice results in increased B‐cell accumulation in the liver accompanied by increased liver steatosis and fibrosis.( 55 ) LPS is the main bacterial product implicated in NAFLD pathogenesis upon translocation across the intestinal epithelium in a TLR4‐dependent mechanism.( 64 ) Although TLR4 activation in NAFLD has been mainly attributed to KCs,( 65 ) TLR4 signaling can also activate B cells through the MYD88 pathway.( 55 ) In addition, we have shown that NASH promotes a strong BCR activation, which suggests a direct antigen stimulation of the BCR independent from inflammatory signals.( 55 ) Whether B‐cell activation in the liver during NAFLD is antigen dependent, the identity of the antigenic stimuli, and the potential T‐dependent or ‐independent mechanisms involved in B‐cell responses are unknown. Alternatively, changes in the microbiota can influence B cells through mechanisms that are independent of antigen translocation. For instance, gut‐microbiota–derived metabolites, including tryptophan, short‐chain fatty acids, and bile acids, are potent immunomodulatory molecules that can affect host metabolism.( 66 )
Effector Functions of Intrahepatic B Cells
B cells are important effector cells in immune‐mediated inflammatory diseases because of their ability to differentiate into antibody‐producing cells and modulate immune responses through antigen presentation and secretion of cytokines. Although the cellular sources and mechanisms leading to their production are unclear, some patients with NAFL and NASH show increased serum IgA, IgM, and IgG.( 67 ) Indeed, patients with NASH have elevated anti‐OSE IgG antibodies, which are positively correlated with hepatic inflammation, suggesting that antibody‐mediated responses against oxidative stress are involved in disease progression.( 63 , 68 ) In mice fed a methionine‐choline–deficient diet to induce experimental NASH, the production of anti‐OSE IgG correlates with the severity of liver injury and hepatic inflammation.( 69 ) Furthermore, inducing an immune response against OSEs worsens hepatic inflammation and parenchymal injury and activates Th1 cells and macrophages, suggesting a causative role for anti‐OSE IgGs in NASH progression.( 69 ) In addition to anti‐OSE IgGs, patients with NAFLD and NASH have elevated IgA, which is an independent predictor of advanced fibrosis.( 67 ) In mice and humans with NASH, liver‐resident IgA+ cells suppress the protective role of CD8+ cytotoxic T cells leading to HCC.( 70 )
Beyond their traditional role as antibody‐producing cells, intrahepatic B cells use cytokine‐mediated responses to promote inflammation and regulate the function of neighboring T cells. In NASH patients, liver biopsies show evidence of B‐cell aggregation within T‐cell–rich regions, suggesting crosstalk between these cell types.( 52 ) Intrahepatic B cells have been shown to secrete greater amounts of IL‐6 and TNFα and promote the activation of CD4 Th cells and their differentiation into Th1 cells in steatotic( 54 ) and NASH( 55 ) livers. These data suggest a proinflammatory, innate‐like function of intrahepatic B cells in the progression of NAFLD. Whether this secretion of proinflammatory cytokines by B cells promotes the transition from NAFLD to NASH, is the consequence of earlier inflammatory cascades, or simply a bystander response are unknown.
A key event in liver fibrogenesis is the activation of quiescent HSCs, which differentiate into myofibroblasts and secrete large amounts of collagen and profibrosis factors.( 71 ) Multiple mechanisms have been implicated in HSC profibrotic functions, but cytokine‐mediated activation is indispensable.( 72 ) Intrahepatic B cells from CCl4‐treated fibrotic mice show increased activation and production of proinflammatory cytokines whereas their genetic ablation results in suppressed fibrosis.( 59 , 60 ) Mice with a normal number of B cells, but impaired Ig production, are not protected from CCl4‐induced liver fibrosis, suggesting an antibody‐independent mechanism.( 59 ) Importantly, TNF signaling has a prominent role in promoting fibrosis, given that TNFα and its receptor 1 are key regulators of HSC activation.( 73 ) Our findings show that B cells are strong producers of TNFα and that their activation promotes fibrosis in NASH, independent from lipid accumulation.( 55 ) Together, this evidence suggests that B cells may be strong inducers of HSC activation through the release of proinflammatory cytokines. However, the precise mechanisms through which B cells contribute to NASH‐related fibrosis have not been investigated.
Role of B Cells in NASH‐Associated HCC
B cells, particularly IgA+ B cells, have been shown to play a critical role in the progression of NASH‐driven HCC given that the number of tumor‐infiltrating B cells correlate with tumor progression in patients with HCC.( 74 ) In humans, sc‐RNAseq shows that B cells, predominantly plasma cells, are enriched in HCC‐infiltrating immune cells (24.26%) compared to the cirrhotic (5.41%) and healthy liver (5.82%).( 75 ) In agreement, patients with HCC who have a lower proportion of tumor‐infiltrating plasma cells present a better prognosis.( 75 ) Inflammation‐induced IgA+ B cells seem to disrupt the antitumor immune response during NASH‐induced HCC, as shown in MUP‐μPA mice fed an HFD.( 70 ) Genetic ablation of B cells has been shown to strongly reduce tumor formation in a different mouse model where the animals fail to secrete phospholipid and develop portal inflammation and HCC.( 74 ) Mechanistically, IgA+ B cells inhibit the activation and cytotoxic activity of CD8+ T cells through expression of programmed death‐ligand 1 (PD‐L1) and production of the immunosuppressive cytokine, IL‐10.( 70 ) Bregs can also inhibit antitumor immunity through IL‐10 production and promote HCC growth through direct interactions with tumor cells.( 76 ) However, liver B cells might play multifaceted roles given that several studies show that patients with HCC with increased infiltration of total B cells have a favorable prognostic outcome.( 77 , 78 ) In addition, in mice transplanted with murine hepatomas or diethylnitrosamine‐induced cancer, tumor growth was enhanced when all B cells were depleted.( 78 ) These contradictory results might be attributed to the stages of human HCC studied, different mouse models, specificity for B‐cell subsets, and composition of microbiotas. Future studies are needed to clarify the precise role of liver B cells in NASH‐related HCC.
Concluding Remarks and Future Directions
NAFLD is a heterogeneous liver disease with a spectrum ranging from simple lipid accumulation to inflammation, liver injury, fibrosis, and increased risk of HCC. Activation of the immune system during NASH and the recruitment of proinflammatory cells into the liver is a key event in the pathogenesis of the disease. Although B lymphocytes are central mediators of autoimmune and inflammatory disease because of their ability to secrete cytokines and antibodies, their role in NAFL and NASH is poorly understood. Future research is needed to identify additional antigen‐dependent and ‐independent mechanisms by which intrahepatic B cells become activated and promote NASH, particularly as activators of HSCs. Advances in single‐cell omics are likely to help elucidate the functional relevance of B1 subsets in disease progression as well as the crosstalk between B cells and other intrahepatic immune and nonimmune cells. Given the role of B cells in the progression of NAFLD, it is conceivable that B‐cell–directed therapies can modulate disease development. Such strategies may include B‐cell depletion, impairment of B‐cell survival and proliferation, modulation of BCR signaling, immunization, and approaches targeting B‐cell inflammatory mediators such as TNFα. B‐cell–directed therapies, such as rituximab (anti‐CD20) and belimumab (BAFF signaling blocker), have been used for years and are effective for the treatment of several autoimmune diseases.( 79 , 80 ) Although B‐cell depletion therapies might ameliorate NASH progression, unwanted side effects may involve immunosuppression and toxicity. Therefore, more selective strategies that target the costimulatory pathways, CD80/CD86, and inhibit the interaction between T cells and B cells could be viable.( 81 ) Another possibility is to modulate BCR signaling using a monoclonal antibody that binds CD22, currently in trials for the treatment of systemic lupus erythematosus.( 82 ) However, more preclinical evidence is needed before considering the BCR signaling pathway as a therapeutic target in NAFLD.
Author Contributions
F.B., S.K., H.W., and X.S.R. contributed to the writing of the manuscript and the generation of figures. X.S.R. conceptualized the topic, acquired funding for, and supervised the writing of the article.
Supported by NIH grant DK122056 (to X.R.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11‐20. [DOI] [PubMed] [Google Scholar]
- 2. Marengo A, Jouness RI, Bugianesi E. Progression and natural history of nonalcoholic fatty liver disease in adults. Clin Liver Dis 2016;20:313‐324. [DOI] [PubMed] [Google Scholar]
- 3. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349‐364. [DOI] [PubMed] [Google Scholar]
- 4. Brandl K, Kumar V, Eckmann L. Gut‐liver axis at the frontier of host‐microbial interactions. Am J Physiol Gastrointest Liver Physiol 2017;312:G413‐G419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology 2019;70:1026‐1037. [DOI] [PubMed] [Google Scholar]
- 6. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 2020;17:81‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Balmer ML, Slack E, de Gottardi A, Lawson MAE, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med 2014;6:237ra266. [DOI] [PubMed] [Google Scholar]
- 8. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009;49:1877‐1887. [DOI] [PubMed] [Google Scholar]
- 9. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue‐resident macrophages originate from yolk‐sac‐derived erythro‐myeloid progenitors. Nature 2015;518:547‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, et al. Hyperresponsivity to low‐dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin‐mediated signaling. Cell Metab 2012;16:44‐54. [DOI] [PubMed] [Google Scholar]
- 11. Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol 2018;332:111‐120. [DOI] [PubMed] [Google Scholar]
- 12. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012;61:416‐426. [DOI] [PubMed] [Google Scholar]
- 13. Daemen S, Gainullina A, Kalugotla G, He LI, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep 2021;34:108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, et al. TREM2 sustains macrophage‐hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Investig 2021;131:e135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 2016;151:1176‐1191. [DOI] [PubMed] [Google Scholar]
- 16. Haas JT, Vonghia L, Mogilenko DA, Verrijken AN, Molendi‐Coste O, Fleury S, et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab 2019;1:604‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013;58:589‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heier EC, Meier A, Julich‐Haertel H, Djudjaj S, Rau M, Tschernig T, et al. Murine CD103+ dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol 2017;66:1241‐1250. [DOI] [PubMed] [Google Scholar]
- 19. Revelo X, Ghazarian M, Chng M, Luck H, Kim J, Zeng K, et al. Nucleic acid‐targeting pathways promote inflammation in obesity‐related insulin resistance. Cell Rep 2016;16:717‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J, Liang B, Bian D, Luo Y, Yang J, Li Z, et al. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun 2019;518:691‐697. [DOI] [PubMed] [Google Scholar]
- 21. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri‐Sverdlov R, et al. Neutrophil‐derived myeloperoxidase aggravates non‐alcoholic steatohepatitis in low‐density lipoprotein receptor‐deficient mice. PLoS One 2012;7:e52411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ou R, Liu J, Lv M, Wang J, Wang J, Zhu LI, et al. Neutrophil depletion improves diet‐induced non‐alcoholic fatty liver disease in mice. Endocrine 2017;57:72‐82. [DOI] [PubMed] [Google Scholar]
- 23. Peiseler M, Kubes P. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Investig 2019;129:2629‐2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2017;2:eaai7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wolf M, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross‐talk with hepatocytes. Cancer Cell 2014;26:549‐564. [DOI] [PubMed] [Google Scholar]
- 26. Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar‐Gonzalez RM, Shivakumar P, et al. Hepatic natural killer T‐cell and CD8+ T‐cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun 2017;1:299‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto‐aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 2021;592:444‐449. [DOI] [PubMed] [Google Scholar]
- 28. Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol 2016;196:97‐105. [DOI] [PubMed] [Google Scholar]
- 29. Harley ITW, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, et al. IL‐17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014;59:1830‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li F, Hao X, Chen Y, Bai LI, Gao X, Lian Z, et al. The microbiota maintain homeostasis of liver‐resident γδT‐17 cells in a lipid antigen/CD1d‐dependent manner. Nat Commun 2017;7:13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maricic I, Marrero I, Eguchi A, Nakamura R, Johnson CD, Dasgupta S, et al. Differential activation of hepatic invariant NKT cell subsets plays a key role in progression of nonalcoholic steatohepatitis. J Immunol 2018;201:3017‐3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al. NKT‐associated hedgehog and osteopontin drive fibrogenesis in non‐alcoholic fatty liver disease. Gut 2012;61:1323‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pieper K, Grimbacher B, Eibel H. B‐cell biology and development. J Allergy Clin Immunol 2013;131:959‐971. [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Liu J, Burrows PD, Wang JY. B cell development and maturation. Adv Exp Med Biol 2020;1254:1‐22. [DOI] [PubMed] [Google Scholar]
- 35. Prieto JMB, Felippe MJB. Development, phenotype, and function of non‐conventional B cells. Comp Immunol Microbiol Infect Dis 2017;54:38‐44. [DOI] [PubMed] [Google Scholar]
- 36. Ghosn E, Dorshkind K, Herzenberg LA, Holodick N, Kantor A, Montecino‐Rodriguez E, et al. B1 B cell progenitors. Science 2019;364:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baumgarth N. A hard(y) look at B‐1 cell development and function. J Immunol 2017;199:3387‐3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valizadeh A, Sanaei R, Rezaei N, Azizi G, Fekrvand S, Aghamohammadi A, et al. Potential role of regulatory B cells in immunological diseases. Immunol Lett 2019;215:48‐59. [DOI] [PubMed] [Google Scholar]
- 39. Leeman‐Neill RJ, Lim J, Basu U. The common key to class‐switch recombination and somatic hypermutation: discovery of AID and its role in antibody gene diversification. J Immunol 2018;201:2527‐2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol 2020;20:229‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 2005;5:853‐865. [DOI] [PubMed] [Google Scholar]
- 42. Lanzavecchia A, Sallusto F. Toll‐like receptors and innate immunity in B‐cell activation and antibody responses. Curr Opin Immunol 2007;19:268‐274. [DOI] [PubMed] [Google Scholar]
- 43. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest 2017;127:1019‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frasca D, Diaz A, Romero M, Garcia D, Jayram D, Thaller S, et al. Identification and characterization of adipose tissue‐derived human antibodies with “anti‐self” specificity. Front Immunol 2020;11:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanigaki K, Sacharidou A, Peng J, Chambliss KL, Yuhanna IS, Ghosh D, et al. Hyposialylated IgG activates endothelial IgG receptor FcγRIIB to promote obesity‐induced insulin resistance. J Clin Invest 2018;128:309‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu Z, Xu J, Tan J, Song Y, Liu L, Zhang F, et al. Mesenteric adipose tissue B lymphocytes promote local and hepatic inflammation in non‐alcoholic fatty liver disease mice. J Cell Mol Med 2019;23:3375‐3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nishimura S, Manabe I, Takaki S, Nagasaki M, Otsu M, Yamashita H, et al. Adipose natural regulatory B cells negatively control adipose tissue inflammation. Cell Metab 2013;18:759‐766. [DOI] [PubMed] [Google Scholar]
- 51. Shen L, Chng MH, Alonso MN, Yuan R, Winer DA, Engleman EG. B‐1a lymphocytes attenuate insulin resistance. Diabetes 2015;64:593‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bruzzì S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A, et al. B2‐Lymphocyte responses to oxidative stress‐derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med 2018;124:249‐259. [DOI] [PubMed] [Google Scholar]
- 53. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393‐1405. [DOI] [PubMed] [Google Scholar]
- 54. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, Chi YJ, et al. Role of intrahepatic B cells in non‐alcoholic fatty liver disease by secreting pro‐inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis 2016;17:464‐474. [DOI] [PubMed] [Google Scholar]
- 55. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota‐driven activation of intrahepatic B cells aggravates nonalcoholic steatohepatitis through innate and adaptive signaling. Hepatology 2021. Feb 20. 10.1002/hep.31755. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from Kupffer cells in the fatty liver. Immunity 2020;53:641‐657.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell 2019;75:644‐660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramachandran P, Dobie R, Wilson‐Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single‐cell level. Nature 2019;575:512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest 2005;115:3072‐3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thapa M, Chinnadurai R, Velazquez VM, Tedesco D, Elrod E, Han JH, et al. Liver fibrosis occurs through dysregulation of MyD88‐dependent innate B‐cell activity. Hepatology 2015;61:2067‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miyake T, Abe M, Tokumoto Y, Hirooka M, Furukawa S, Kumagi T, et al. B cell‐activating factor is associated with the histological severity of nonalcoholic fatty liver disease. Hepatol Int 2013;7:539‐547. [DOI] [PubMed] [Google Scholar]
- 62. Nakamura Y, Abe M, Kawasaki K, Miyake T, Watanabe T, Yoshida O, et al. Depletion of B cell‐activating factor attenuates hepatic fat accumulation in a murine model of nonalcoholic fatty liver disease. Sci Rep 2019;9:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Albano E, Mottaran E, Vidali M, Reale E, Saksena S, Occhino G, et al. Immune response towards lipid peroxidation products as a predictor of progression of non‐alcoholic fatty liver disease to advanced fibrosis. Gut 2005;54:987‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rivera CA, Adegboyega P, van Rooijen N , Tagalicud A, Allman M, Wallace M. Toll‐like receptor‐4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non‐alcoholic steatohepatitis. J Hepatol 2007;47:571‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll‐like receptor 4 is involved in the development of fructose‐induced hepatic steatosis in mice. Hepatology 2009;50:1094‐1104. [DOI] [PubMed] [Google Scholar]
- 66. Krishnan S, Ding Y, Saedi N, Choi M, Sridharan GV, Sherr DH, et al. Gut microbiota‐derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep 2018;23:1099‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McPherson S, Henderson E, Burt AD, Day CP, Anstee QM. Serum immunoglobulin levels predict fibrosis in patients with non‐alcoholic fatty liver disease. J Hepatol 2014;60:1055‐1062. [DOI] [PubMed] [Google Scholar]
- 68. Nobili V, Parola M, Alisi A, Marra F, Piemonte F, Mombello C, et al. Oxidative stress parameters in paediatric non‐alcoholic fatty liver disease. Int J Mol Med 2010;26:471‐476. [DOI] [PubMed] [Google Scholar]
- 69. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014;59:886‐897. [DOI] [PubMed] [Google Scholar]
- 70. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation‐induced IgA+ cells dismantle anti‐liver cancer immunity. Nature 2017;551:340‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest 2017;127:55‐ 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chiu YS, Wei CC, Lin YJ, Hsu YH, Chang MS. IL‐20 and IL‐20R1 antibodies protect against liver fibrosis. Hepatology 2014;60:1003‐1014. [DOI] [PubMed] [Google Scholar]
- 73. Tarrats N, Moles A, Morales A, Garcia‐Ruiz C, Fernandez‐Checa JC, Mari M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology 2011;54:319‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, et al. B lymphocytes limit senescence‐driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018;67:1970‐1985. [DOI] [PubMed] [Google Scholar]
- 75. Zhang S, Liu Z, Wu D, Chen L, Xie L. Single‐cell RNA‐Seq analysis reveals microenvironmental infiltration of plasma cells and hepatocytic prognostic markers in HCC with cirrhosis. Front Oncol 2020;10:596318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shao Y, Lo CM, Ling CC, Liu XB, Ng KP, Chu ACY, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett 2014;355:264‐272. [DOI] [PubMed] [Google Scholar]
- 77. Zhang Z, Ma L, Goswami S, Ma J, Zheng B, Duan M, et al. Landscape of infiltrating B cells and their clinical significance in human hepatocellular carcinoma. OncoImmunology 2019;8:e1571388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour‐infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017;66:342‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hofmann K, Clauder AK, Manz RA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol 2018;9:835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Porsch F, Binder CJ. Impact of B‐cell‐targeted therapies on cardiovascular disease. Arterioscler Thromb Vasc Biol 2019;39:1705‐1714. [DOI] [PubMed] [Google Scholar]
- 81. Keating GM. Abatacept: a review of its use in the management of rheumatoid arthritis. Drugs 2013;73:1095‐1119. [DOI] [PubMed] [Google Scholar]
- 82. Clowse MEB, Wallace DJ, Furie RA, Petri MA, Pike MC, Leszczyński P, et al. Efficacy and safety of epratuzumab in moderately to severely active systemic lupus erythematosus: results from two phase III randomized, double‐blind, placebo‐controlled trials. Arthritis Rheumatol 2017;69:362‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Weisel F, Shlomchik M. Memory B cells of mice and humans. Annu Rev Immunol 2017;35:255‐284. [DOI] [PubMed] [Google Scholar]
- 84. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody secreting plasma cells. Nat Rev Immunol 2015;15:160‐171. [DOI] [PubMed] [Google Scholar]