

Abstract
Purpose:
The human chromosome 19q13.11 deletion syndrome is associated with a variable phenotype that includes aplasia cutis congenita (ACC) and ectrodactyly as specific features. UBA2 (ubiquitin-like modifier-activating enzyme 2) lies adjacent to the minimal deletion overlap region. We aim to define the UBA2-related phenotypic spectrum in humans and zebrafish due to sequence variants and to establish the mechanism of disease.
Methods:
Exome Sequencing was used to detect UBA2 sequence variants in 16 subjects in 7 unrelated families. uba2 loss-of-function was modeled in zebrafish. Effects of human missense variants were assessed in zebrafish rescue experiments.
Results:
7 human UBA2 loss-of-function and missense sequence variants were detected. UBA2-phenotypes included ACC, ectrodactyly, neurodevelopmental abnormalities, ectodermal, skeletal, craniofacial, cardiac, renal, and genital anomalies. uba2 was expressed in zebrafish eye, brain, and pectoral fins; uba2-null fish showed deficient growth, microcephaly, microphthalmia, mandibular hypoplasia, and abnormal fins. uba2-mRNAs with human missense variants failed to rescue nullizygous zebrafish phenotypes.
Conclusion:
UBA2 variants cause a recognizable syndrome with a wide phenotypic spectrum. Our data suggest that loss of UBA2 function underlies the human UBA2 monogenic disorder and highlights the importance of SUMOylation in the development of affected tissues.
Introduction
Features of the chromosome 19q13.11 deletion syndrome include early growth deficiencies, developmental delay, distinctive facial features, aplasia cutis congenita (ACC), hip dysplasia, digital and limb anomalies including ectrodactyly, and other malformations1–8. Deletions range in size from 1.37–11 Mb with a minimum overlapping region (MOR) of 324 kb, without clear genotype-phenotype correlation3,4,6. UBA2 lies adjacent to the MOR and has been proposed to underlie key aspects of the deletion phenotype including ACC and ectrodactyly1,2,3,5,6. Limited patient data and lack of an animal model have prevented establishing UBA2 as the causative gene.
UBA2 plays a key role in the posttranslational modification of protein (SUMOylation) by the addition of SUMO1 (small ubiquitin-like modifier) protein. UBA2 forms a heterodimer with SAE1 (SUMO-Activating Enzyme Subunit 1) and binds with SUMO1 in an ATP-dependent manner9–11. Unlike ubiquitination, SUMOylation does not only target proteins for degradation, but is involved in cell cycle regulation, subcellular trafficking, signal transduction, stress responses and chromatin structure dynamics. SUMOylation alters protein kinases and transcription factors to maintain transcriptional regulation of tissue-specific gene expression12.
In this study, we report 16 additional individuals from seven unrelated families with de novo and familial UBA2 sequence variants who have highly variable but overlapping clinical presentations. In silico modeling and a zebrafish uba2 nullizygous phenotype provides further functional evidence for the pathogenicity of UBA2 as the key gene underlying the chromosome19q13.11 microdeletion syndrome.
Material and methods
Subject enrollment and clinical evaluations
Each described patient was evaluated by a clinical geneticist. Written informed consent was obtained for exome sequencing either on a clinical or research basis. A written informed consent was also obtained from subjects to publish their photos. Genomic DNA was extracted from whole blood from affected probands and their biological parents for exome sequencing. See supplement for details.
Zebrafish modeling of the phenotypic effects of uba2 variants
All animal experiments were conducted in accordance with recommendations of the Guide for the Care and Use of Laboratory animals of the National Institutes of Health (Protocol # NEI-679). Adult AB (Tubingen) and ABTL (Tubingen long fin) zebrafish strains were raised and maintained according to standard protocols as described13.
Whole mount in situ hybridization
Wild type (WT) zebrafish embryos at different developmental stages (5 somite, 24, 35, 48, 72hpf (hours post fertilization), 5 and 7 dpf (days post fertilization) were fixed in preparation for performing in situ hybridization. See supplement method section for details.
CRISPR/Cas9 uba2 knock out line generation
CRISPR/Cas9 method was used to generate uba2 knockout zebrafish lines. See supplement method section for details.
mRNA rescue
To evaluate the impact of human UBA2 variants on encoded protein products, we utilized uba2-mutant fish to perform rescue studies with capped full-length human WT and missense alleles in mRNA transcribed with the T7 mMESSAGE mMACHINE kit (Ambion).
Please see supplement for other methodology details.
Results
Clinical studies
The cohort was gathered through GeneDx, a clinical molecular laboratory, and GeneMatcher. Investigators independently ascertained families with related phenotypes and rare candidate variants. Table 1 and the supplement contain additional clinical details.
Table 1:
Clinical features in study subjects and previous publications
| Family ID | Subject ID | Gender | UBA2 variant, Transcript | Current age or age at exam (years) | Developmental delay/ neurodevelopmental details | Height percentile (most recent or at stated age) | Weight percentile (most recent or at stated age) | Head circumference percentile (most recent / stated age) | Early growth problems | Craniofacial features | Aplasia cutis congenita | Other ectodermal variations | Ectrodactyly/ Oligodactyly | Other skeletal anomalies | Other anomalies/features: cardiac, renal, genital, ocular, miscellaneous | Other genetic/chromosomal results |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | III-2 | F | c.816_817delAT: p.Trp273Alafs*13 NM_005499 | 37 | Normal development, but had behavior problems as child, history of seizures, mini strokes. | <5 | increased | 2 | Tall forehead/ high hairline, hypertelorism, broad nasal root, facial asymmetry, cleft chin, ptosis, simple low set ears | no | Peg teeth, yellow teeth, thin hair, xerosis | no | Hypoplastic distal flexion creases, clinodactyly, syndactyly, camptodactyly, hip abnormality | Atrial fibrillation, mitral regurgitation by history but recent echo was normal. Hydronephrosis. Wears glasses. Focal nodular hyperplasia of the liver, hypofibrinogenemia. | Heterozygous for FGG LPATH variant: D344N. WNT10B heterozygous VUS: I285T. | |
| 1 | IV-1 | F | c.816_817delAT: p.Trp273Alafs*13 maternal NM_005499 |
15 | Mild delays, hypotonia, has individualized educational plan, but she's academically on target. | 25 | 67 | 2 | Yes | Tall forehead/ high hairline, downslanted palpebral fissures, hypertelorism, broad nasal root, facial asymmetry, gap between incisors, slightly bifid uvula | no | Natal tooth, peg teeth, thin hair, eczema, keratosis pilaris, dental problems | no | Hypoplastic distal flexion creases, clinodactyly, syndactyly, camptodactyly, hip abnormality, scoliosis, pectus excavatum | ASD, aberrant right subclavian. Hydronephrosis/ pelviectasis, GU reflux, urinary tract infections. High myopia. Hypofibrinogenemia. | FGG heterozygous LPATH variant: p.D344N. Microarray: dup22q11.2. |
| 1 | IV-2 | M | c.816_817delAT: p.Trp273Alafs*13 maternal NM_005499 |
13 | Autism Spectrum disorder, behavior problems, encopresis, stereotypies, mood swings, hypotonia, normal MRI. | 26 | 66 | 20 | yes | Tall forehead/ high hairline, downslanted palpebral fissures, hypertelorism, broad nasal root, facial asymmetry, triangular face, mild synophrys, telecanthus, cleft chin, micrognathia | yes, single area | Xerosis, thin hair, gaps between teeth, irregular enamel, supernumerary nipple, hyperhidrosis, hyperlinearity of palms, cutis marmorata, nail ridging, keratosis pilaris | no | Clinodactyly, syndactyly, camptodactyly, hip abnormality. Wore helmet for torticollis and plagiocephaly. | PFO (resolved). Astigmatism. | Normal CGH. Normal MID1 sequencing. |
| 1 | IV-3 | M | c.816_817delAT: p.Trp273Alafs*13 maternal NM_005499 |
8 | Autism Spectrum disorder, hypotonia, possible processing delay, poor coordination, MRI essentially normal | 18 | 54 | ~30 | yes | Tall forehead/ high hairline, orbital asymmetry, square uvula, ankyloglossia, cleft chin | no | Xerosis, keratosis pilaris, unruly hair, atopic dermatitis, history of heat exhaustion | no | Clinodactyly, syndactyly, camptodactyly, hip abnormality, wormian bones, mild pectus excavatum | PFO (resolved). Astigmatism. Hypofibrinogenemia. | FGG heterozygous LPATH variant: p.D344N. |
| 1 | IV-4 | F | c.816_817delAT: p.Trp273Alafs*13 maternal NM_005499 |
7 | Mild delays, intermittent intention tremor, brisk patellar reflexes, poor balance, hypotonia, normal cognitive skills, MRI normal. | 11 | 11 | ~2 | yes | Tall forehead/ high hairline, downslanted palpebral fissures, broad nasal root, facial asymmetry, low set ears, simple cartilage, wide uvula, cleft chin, micrognathia | yes, multiple areas | Xerosis, mild ichthyosis, keratoderma follicular prominence, thin dry hair, frayed toenails, hyperlinear palms, hypohidrosis. | yes, unilateral hand | Hypoplastic distal flexion creases, brachydactyly of toes, clinodactyly, syndactyly, camptodactyly, hip abnormality, mild pectus excavatum and hyperextensibility, wormian bones | PFO (resolved). Early myopia, -4 diopters, improved. History of “twisted optic nerves.” Hypofibrinogenemia, reduced IgA, IgM. | Normal SNP microarray. Normal TP63 gene sequencing. FGG heterozygous LPATH variant: p.D344N. WNT10B heterozygous VUS: p.I285T. |
| 2 | II-1 | M | c.1376_1377insT: p.Thr460Aspfs*24 "de novo" NM_005499.2 |
21 | Delays. Hypotonia, sensory integration problems, normal cognitive development. | 10 | <3 | ~5 | no | Tall forehead/ high hairline, downslanted palpebral fissures, broad nasal root, facial asymmetry, epicanthal folds, long and smooth philtrum, high arched palate, dental crowding | no | no | no | Long thin fingers, foot anomalies, clinodactyly, pectus excavatum, plagiocephaly | Cryptorchidism, hydrocele | |
| 2 | II-2 | M | c.1376_1377insT: p.Thr460Aspfs*24 "de novo" NM_005499.2 |
12 | Delayed motor skills, hypotonia, sensory integration problems. | "low" | <5 | yes | Downslanted palpebral fissures, broad nasal root, zygomatic arch hypoplasia, simple, low set posteriorly rotated ears, preauricular tag, long smooth philtrum, high arched palate, micrognathia | no | no | no | Dysplastic metatarsals, toes point outward, kyphoscoliosis | Hypospadias, inguinal hernia. | ||
| 2 | II-3 | M | c.1376_1377insT: p.Thr460Aspfs*24 "de novo" NM_005499.2 |
6 | Delays, unstable gait, poor fine motor skills, sensory integration problems, poor balance, hypotonia, normal cognitive skills. | <3 | <3 | <3 | yes | Normally set ears | not noted | no | yes, unilateral partial central cleft of hand, polydactyly of third finger | Syndactyly, camptodactyly | Cryptorchidism, hydrocele | 46, XY and normal microarray |
| 3 | I-2 | M | c.364C>T: p.Arg122* NM_005499 |
45 | Delays, learning difficulties in school, depression in adulthood. | <5 | >95 | ~5–10 | Tall forehead/ high hairline, hypertelorism, broad nasal root, low set prominent ears, thin vermilion border, mild micrognathia | yes, multiple areas | Supernumerary nipples | no | none reported | Recurrent urinary tract infections; asymmetric renal sizes with reduced function of smaller kidney. Hypothyroidism, s/p cholecystectomy. Inguinal herniorrhaphy. History of pseudotumor cerebri. | ||
| 3 | II-1 | F | c.364C>T: p.Arg122* maternal NM_005499 |
24.5 | Delays (walked at 17 months, first words at 22 months), special education, depression and anxiety as an adult. | ~15 | ~93 | <3rd | Tall forehead/ high hairline, hypertelorism, broad nasal root, thin upper lip, smooth philtrum, everted lower lip, thick, lowset, and laterally protruding ears, medial eyebrow flare, micrognathia | yes, 3 areas | Supernumerary nipple | no | none reported | Bicuspid aortic valve. Astigmatism. S/p cholecystectomy, migraines. Low back pain. | ||
| 3 | II-2 | M | c.364C>T: p.Arg122* maternal NM_005499 |
21.5 | Delays recognized at 16 months, learning difficulties and special education, bipolar disease, panic attacks and social phobias as an adult. | ~20 | ~15 | ~60–70 | Tall forehead/ high hairline, downslanted palpebral fissures, hypertelorism, broad nasal root, prominent columella, bulbous tip of the nose, micrognathia | yes, single area | Supernumerary nipple | no | none reported | Cryptorchidism. Astigmatism. Asthma. | ||
| 3 | III-1 | F | c.364C>T: p.Arg122* maternal NM_005499 |
2.75 | Delays (at 16 months, cognitive function was at the 8 month old level, motor skills were at the 9 month level; at 30 months: still no sentences) | 50 | 75–90 | 3–10 | no | Tall forehead/ high hairline, downslanted palpebral fissures, hypertelorism, broad nasal root, lowset ears, micrognathia | yes, multiple areas | Supernumerary nipple | no | none reported | Frequent otitis. Constipation. | |
| 4 | II-1 | F | c.167A>C: p.Asn56Thr de novo NM_005499.2 |
21 | Delayed motor skills, attention deficit disorder, sat independently at 12 months, walked at 22 months, first word at 18 months, sentences after 2 years. | <3 | <3 | <3 | yes | High hairline, broad forehead, hypertelorism, broad nasal root, delayed dentition, mild facial dysmorphism | yes | Thin, sparse hair, coarse skin, poor sweating, cries with tears. | no | Clinodactyly, overlapping toes on right foot (3,4), delayed bone age, kyphoscoliosis treated with bracing | Renal hypoplasia, chronic kidney disease, stable. Bilateral optic nerve hypoplasia with normal vision. Postural orthostatic hypotension. Hypothyroidism, growth hormone treatment, headaches, no breast development (budding only), menarche at 14 years, pubic hair, no axillary hair. | BAZ1B:heterozygous VUS, de novo fs c.3317delA; SOS1: heterozygous variant c.281T>C; COX1: homoplasmic p.I97V; NRXN1: heterozygous VUS, p.G744R (maternal) |
| 5 | II-1 | F | c.1447G>A: p.Glu483Lys de novo NM_005499.2 |
4.75 | Global delay (gross motor and speech), nonverbal, refractory seizures, infantile spasms, hyperactivity, hypotonia. | ~75 | ~10 | ~25 | no | Epicanthal folds | yes | Normal hair and nails. | Pes planus | Hemangiomas (left ear, back). Anteriorly placed anus. | Normal microarray, normal Prader Willi/Angelman methylation, epilepsy panel: heterozygous VUS in GABRB3, paternal: p.R409Q | |
| 6 | II-1 | M | c.800T>A: p.Leu267* de novo NM_005499.2 |
1.5 | Normal development | 10–25 | 10–25 | 5–10 | no | Tall forehead/ high hairline, hypertelorism, epicanthal folds, pseudostrabismus | no | yes, bilateral ectrodactyly of the feet | Clinodactyly, complete bilateral 2–3 finger syndactyly camptodactyly |
Cryptorchidism. Bilateral inguinal hernias. | Normal SNP microarray | |
| 7 | II-1 | M | c.364C>G: p.Arg122Gly de novo NM_005499.2 |
3.9 | Gross, fine motor and speech delays, persistent. | 75–90 | ~75 | 25 | no | Tall forehead/ high hairline, broad nasal root, left preauricular ear tag, narrow palate, vertical cleft/groove in chin | no | Diffuse patches of hypopigmentation. | 4 limb ectrodactyly, oligodactyly of both feet | Syndactyly as part of ectrodactyly | VSD, not clinically significant | Normal prenatal microarray; normal WES at another clinical lab |
| Marble et al. 201718 | F | c.71G>T: p.Gly24Val de novo NM_005499 |
2.5 | Delayed motor development, normal cognitive ability. | 25–50 | 3rd | 25–50 | yes | Tall forehead/ high hairline, downslanted palpebral fissures, suspected hypertelorism and broad nasal root | yes, single large area | Thin hair in photos. | no | Clinodactyly, hip abnormality | Duane anomaly, strabismus. Recurrent otitis media, croup, tonsillitis. | ||
| Yamoto et al. 201919 | II-1 | M | c.1324dupT: p.Tyr442Leufs*17 de novo NM_005499.2 |
Bilateral ectrodactyly, oligodactyly, hands and feet | Clinodactyly, long bone deficiency of tibias | Undermasculinized external genitalia | ||||||||||
| Wang et al. 201921 | M | c.327delT: p.Phe109Leufs*3 maternal NM_005499.3 | 4 | Normal development | 10–25 (birth) | <3 (birth) | <10 (birth) | Yes, two areas | Bilateral ectrodactyly, oligodactyly | Syndactyly as part of ectrodactyly, low lying conus medullaris | Horseshoe kidney, tracheo-esophageal fistula | Normal karyotype, microarray | ||||
| Wang et al. 201921 | F | c.327delT: p.Phe109Leufs*3 NM_005499.3 | 35 | Normal development | yes | no | none reported | |||||||||
| Aerden et al. 202020 | M | c.612delA: p.Glu205Lysfs*63 de novo NM_005499.2 |
8 | Speech delay, normal motor milestones, learning difficulties, autism diagnosed at 8 years, intelligence quotient 76. | ~25–50 (3.6 years) | ~25 (3.6 years) | ~20 (3.6 years) | yes | Retrognathia, low set and prominent ears, fullness of upper eyelids. | Supernumerary nipple, increased hair on back, dry, sparse scalp hair. | yes | Polydactyly with six metatarsals on right foot, multiple bony anomalies in feet, syndactyly of toes, normal hands, transient hip instability. Normal hands. | Strabismus, hypermetropia |
Family 1:
Family 1 (Fig. 1 and 2) is comprised of an affected mother and her four offspring. Two children have ACC. By report, the maternal grandmother and great grandmother also have histories of ACC. Other ectodermal changes are variable including thin scalp hair, xerosis and dental anomalies. The index case (IV-4, Fig. 1a and 1b) has unilateral ectrodactyly of hand. All of the other affected examined individuals have more subtle digital variations including camptodactyly, syndactyly, clinodactyly and diminished distal flexion creases of the fingers. All affected individuals share a high anterior hairline and mild frontal bossing, and several, including the proband (IV-4), have slightly down-slanted palpebral fissures. All have had highly variable neurodevelopmental problems, ranging from hypotonia to autism spectrum disorder in two of the brothers. Hypotonia generally persisted throughout childhood. Affected individuals had early growth deficiencies that improved with age. See supplement for other details. All affected individuals studied are heterozygous for a UBA2 frameshift variant: c.816_817delAT, p.Trp273Alafs*13.
Figure 1: a. Clinical phenotypes and b. Pedigrees associated with UBA2-related syndrome.
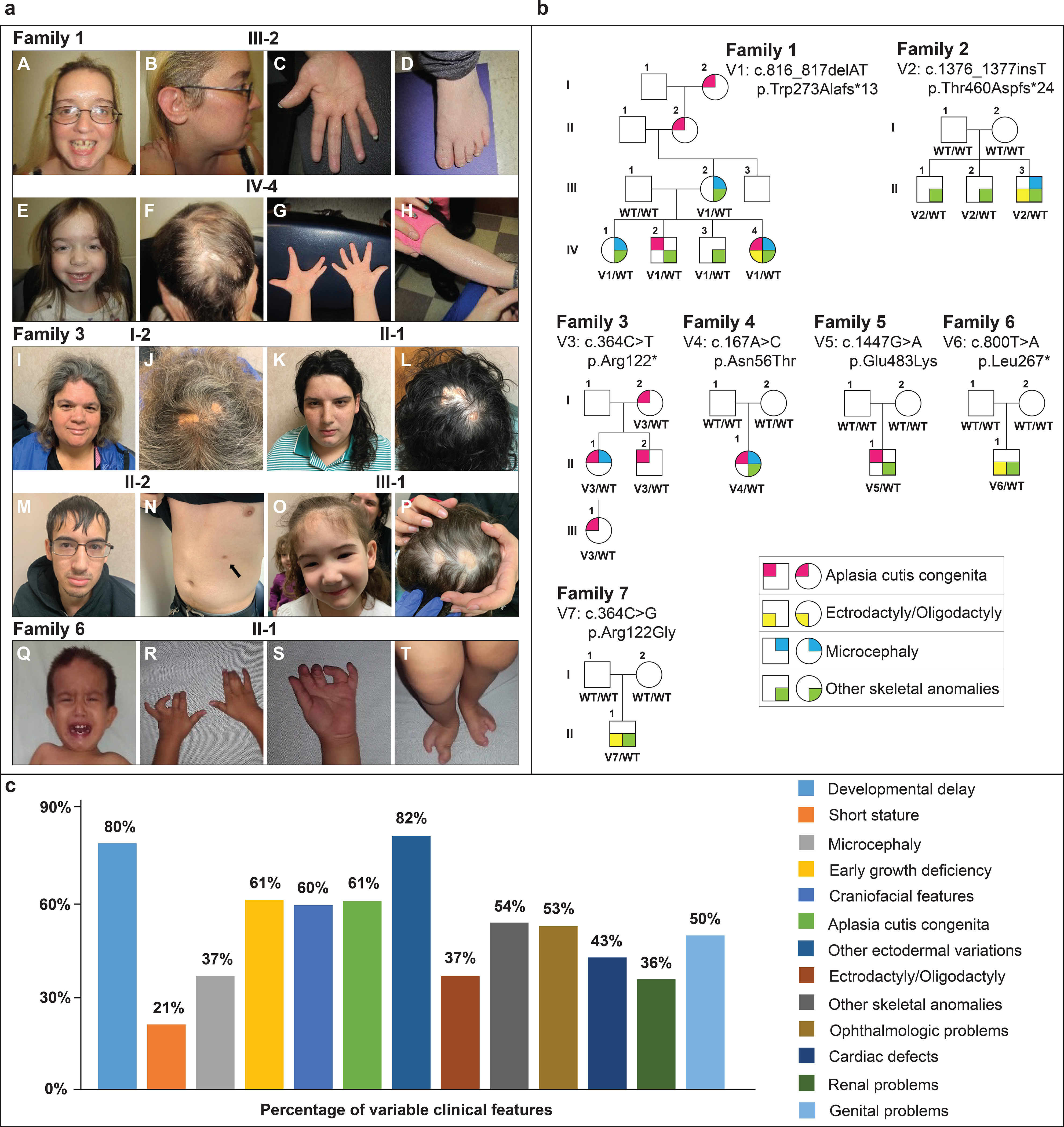
a. Family 1, III-2: A. prominent forehead, high hairline, discolored peg-shaped teeth with gap between upper incisors, cleft chin B. low set ear with simple cartilaginous pattern C. diminished distal flexion creases D. brachydactyly, mild 2–3 syndactyly, clinodactyly of the 4th toe.
Family 1, IV-4: E. prominent forehead, high hairline, cleft chin, mildly down-slanted palpebral fissures F. ACC G. repaired ectrodactyly, hypoplastic distal flexion creases H. ichthyosis
Family 3, I-1: I. tall forehead, hypertelorism, broad nasal root, mild micrognathia J. ACC
Family 3, II-1: K. tall forehead, hypertelorism, broad nasal root, thin upper lip, medial eyebrow flare L. ACC
Family 3, II-2: M. facial features N. supernumerary nipple (arrow)
Family 3, III-1: O. tall forehead, low-set ears, micrognathia P. ACC
Family 6, II-1 Q. high forehead, hypertelorism, bilateral epicanthal folds R, S. bilateral 2–3 finger syndactyly, camptodactyly T. bilateral ectrodactyly of the feet
b. Affected individuals are shown as filled symbols. Genotypes are shown below each individual who was genotyped.
c. Percentages of different clinical features variably expressed in UBA2-affected individuals based on available data. Previously reported UBA2 patients are also included in the percentages.
Figure 2: UBA2 syndrome-associated variants and molecular modeling.
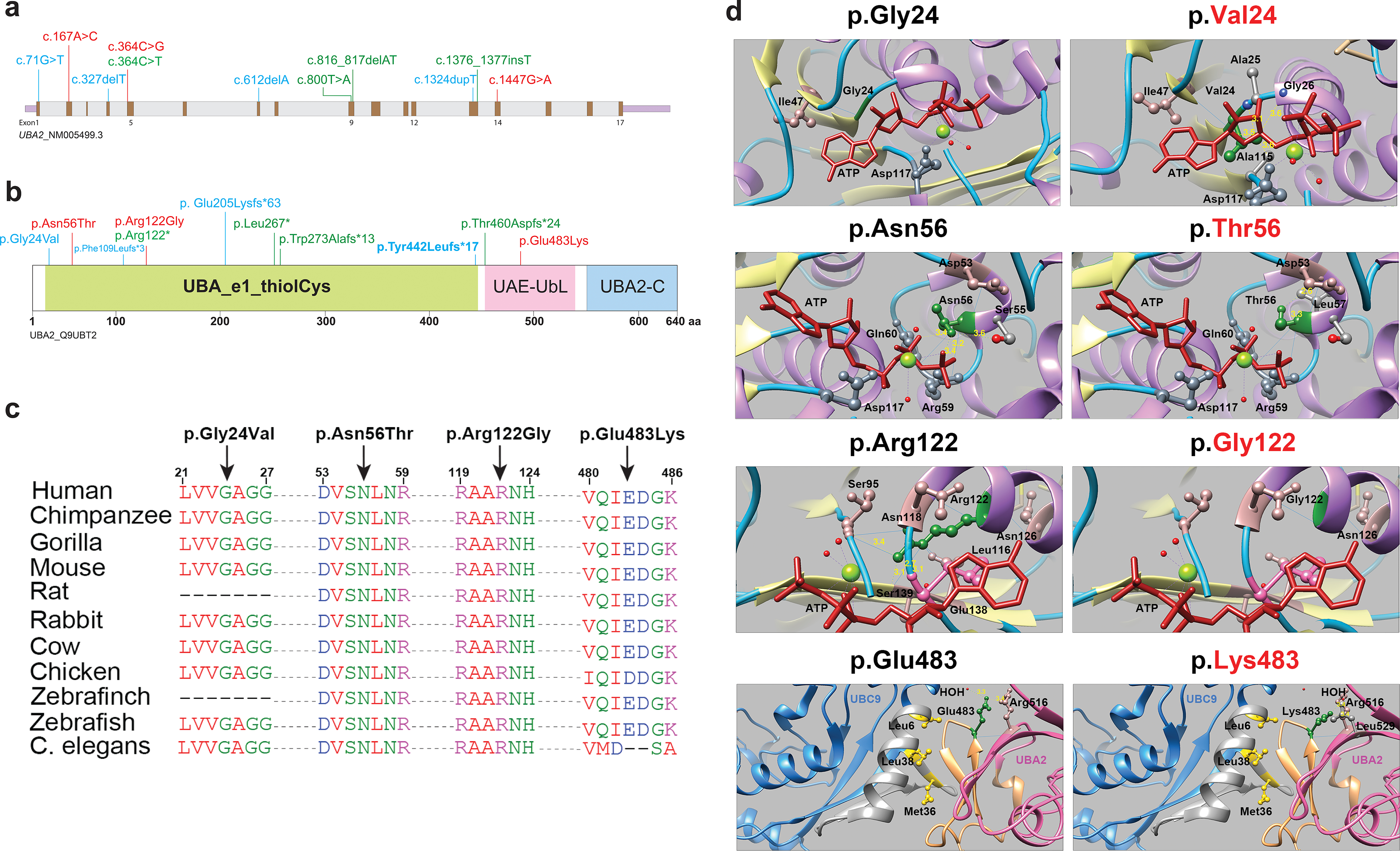
a. Schematic representation of the UBA2 gene. Exons are shown in brown color boxes; introns and 3’ and 5’ UTRs are in light grey and purple, respectively. Newly reported UBA2 missense and loss-of-function variants are shown in red and green, respectively, while blue is used to represent previously reported UBA2 variants. b. Schematic representation of UBA2 protein domains. The UBA2 protein domain carrying catalytically active sites of ubiquitin-activating enzyme is shown in light green. This domain has putative active sites to bind ATP, substrate, and zinc with the last of five conserved cysteine residues playing an important role in ubiquitin thioester complex formation. UBA2-C (C-terminus) and UAE-UbL (ubiquitin-like) domains are shown in pink and blue, representing the C-terminus of UBA2 protein. UAE-UbL is structurally similar to ubiquitin and is involved in E1-SUMO-thioester transfer to E2 conjugation protein. The amino acid changes for the aforementioned variants are shown in the same color scheme as Figure 2B. c. Amino acid sequence alignments of the human UBA2 protein across different species at each of the residues reported with missense variants. d. Molecular modeling of human UBA2 protein. Secondary structure helix, strand and coil regions are shown in purple, yellow and cyan blue shades, respectively. Forest green color is used to show residues of interest in proteins with WT and missense changes and the ATP molecule is shown in brick red color. Blue color shows regions of hydrogen bonding and light pink shows residues involved in hydrogen bond formation with residues of interest. The distances to nearby residues are shown by dashed yellow lines. Last panel: UBA2 and UBC9 are shown in hot pink and cyan blue color, respectively. As per molecular modeling predictions, p.Gly24Val: Glycine is flexible enough to maintain torsion angles and is buried in the protein core to maintain local secondary structure. p.Asn56Thr. Introducing a smaller but more hydrophobic residue at Asparagine 56 results in an empty space in the protein core and subsequent loss of hydrogen bonding with Asp53. p.Arg122Gly: The typical Arginine 122 residue is involved in hydrogen bonding with Asn118, Gly138 and Ser139. Replacement with Glycine is predicted to disrupt this array.
Family 2:
This family consists of three affected brothers (Fig. 1b: II-1, II-2, II-3); neither parent is affected. Parentage was genetically confirmed prior to exome sequencing. All affected individuals have histories of hypotonia through childhood that impeded motor development and even feeding ability in early infancy, and sensory integration problems, but normal cognitive abilities. Neither ACC nor other ectodermal changes are noted, but the youngest brother (II-3) has unilateral cleft hand and polydactyly. More subtle foot, toe, and other minor digital anomalies vary among the three affected males. All three also have histories of cryptorchidism and/or hypospadias. Each is heterozygous for a “de novo” frameshift UBA2 variant, c.1376_1377insT, p.Thr460Aspfs*24, not detected in blood of either parent with either NextGen (130X coverage at 10X depth) or Sanger sequencing.
Family 3:
Clinical details about part of this family were reported previously14 but are now updated and expanded along with results of exome analysis. The male proband (II-2, Fig. 1a and 1b) has a single area of ACC, supernumerary nipple, cryptorchidism, early developmental delay, astigmatism, learning disability, depression, bipolar disorder, and social phobia. His mother (I-2) has multiple areas of healed ACC, supernumerary nipples, small head circumference, and asymmetric kidneys with reduced renal function. Neither have documented hand or foot anomalies. They are both heterozygous for a nonsense variant in UBA2: c.364C>T, p.Arg122*. Two other affected individuals (II-1 and III-1) have similar facial features, ACC, and supernumerary nipples and were each confirmed to harbor the familial UBA2 variant.
Family 4:
The female proband (II-1, Fig. 1b), 21 years old at examination, has a history of delayed motor skills and attention deficit disorder. Height, weight, and head circumference are all currently less than the third percentile; she also had early growth deficiency, delayed dentition and bone age. Features include ACC, thin scalp hair, clinodactyly, and overlapping toes. See Table 1 and supplement for additional endocrine, renal, and ophthalmologic concerns. She is heterozygous for a de novo missense variant in UBA2, c.167A>C: p.Asn56Thr.
Family 5:
The female proband (Fig. 1b, II-1), 4 years and 9-months-old at exam, has developmental delay, absent speech, hemangiomas, ACC, and seizures. She has relative macrocephaly, epicanthal folds, anteriorly placed anus, and pes planus. She carries a de novo missense UBA2 variant: c.1447G>A, p.Glu483Lys.
Family 6:
The proband is a male toddler (Fig. 1a and 1b, II-1) with cryptorchidism, bilateral inguinal hernias, and multiple limb deformities including bilateral ectrodactyly of the feet, complete 2–3 finger syndactyly, clinodactyly and camptodactyly. He has low- normal growth and normal developmental milestones. Facial features include hypertelorism, bilateral epicanthal folds and pseudostrabismus. He does not have ACC or other ectodermal abnormalities. He is heterozygous for a de novo UBA2 nonsense variant c.800T>A, p.Leu267*.
Family 7:
The proband (Fig. 1b, II-1) is a 3 year 11-month-old Caribbean male born at 35 weeks gestational age. At two weeks, height and weight (corrected for prematurity) were normal, but head circumference measured at the 2nd centile. He had global developmental delay and four limb ectrodactyly, tall and prominent forehead, deep-set eyes, broad nasal root, left preauricular tag, narrow palate, and a vertical cleft chin. Pre-surgery, he had left 2–3 finger syndactyly with a nodule adjacent to the medial aspect of the PIP joint of the 4th finger. The right 3rd digit is missing; other digits are relatively normal. On the left foot, 2 malformed digits are divided by a deep central cleft; the right foot also has a deep central cleft with 3 malformed digits, and 4–5 toe syndactyly. He does not have ACC but has large areas of faint hypopigmentation over his torso and limbs. He is heterozygous for a de novo missense variant in UBA2: c.364C>G, p.Arg122Gly.
None of the detected UBA2 variants was found in the GnomAD database15. Results of in silico predictor analyses for missense variants and variant classification is provided in Supplementary Tables 1 and 2. All would be classified as pathogenic or likely pathogenic using American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines (classification criteria)16 in Supplemental Table 2.
Modeling effects of missense variants on UBA2 function
UBA2 in complex with SAE1 plays a key role in the SUMOylation pathway. Observed human UBA2 variants are distributed across the gene (Fig. 2a and b). All truncating variants are expected to undergo nonsense-mediated decay based on their position within the mRNA. Missense variants occur at residues that are strongly conserved across vertebrates (Fig. 2c). Given the similarities in phenotypes between individuals with truncating and missense alleles, we hypothesized that missense alleles also lead to loss-of-function.
To understand how missense alleles might disrupt UBA2 function, molecular modeling using published crystal structures17 and simulated substitutions were performed for each detected human missense variant. In the UBA2 protein, p.Gly2418 is directly involved in ATP binding; its substitution with valine results in altered protein conformation and is predicted to result in loss of ATP binding and ectopic interactions with nearby residues (Fig. 2d)17. Similarly, asparagine replacement with threonine at position 56 putatively abolishes ATP-dependent activation. The p.Arg122Gly substitution is predicted to result in loss of interaction with ATP. Human UBA2 protein interacts with a conjugating enzyme called UBC9 (amino acids 6–38) via amino acid residues 478–509, which include Glutamate 483. UBA2 forms a hydrophobic bond with Leu6, Met36 and Leu38 of UBC9; replacing Glutamate 483 with Lysine is predicted to disrupt UBA2-UBC9 binding. In summary, missense alleles observed in patients with UBA2-associated syndrome are observed to occur at functionally critical residues and potentially disrupt ATP-binding, protein folding, or protein-protein interactions.
Zebrafish uba2 expression in affected tissues
By whole mount in situ hybridization, uba2 transcript was detected on the dorsoventral axis of 5-somite stage embryos (Fig. S1a and b). At later stages, uba2 is expressed in developing brain, eye, craniofacial structures, and fins. At 24 hpf, uba2 expression was restricted to the head region, including the eye and nervous system (Fig. S1c). At 35 hpf, prominent signal was observed in pectoral fins (arrows, Fig. S1d). At all other examined stages (48 and 72 hpf, 5 and 7 dpf), uba2 mRNA signal localized to the head region, specifically brain, neural retina, and lens (Fig. S1e–h). Therefore, zebrafish uba2 is expressed in some structures that are analogous to those affected in humans harboring deleterious UBA2 variants.
Variable expressivity observed with uba2 loss-of-function
uba2 knockout zebrafish lines were generated by CRISPR/Cas9-targeted deletion. The phenotype of homozygous fish was notable for failure to inflate swim bladders. At 5–8 dpf, we observed severe gross morphological defects in uba2−/− zebrafish (Fig. 3) including small eyes, hydrocephalus and craniofacial edema, ventrally-curved body axis, and uninflated swim bladder. Faint heartbeat and severe pericardial edema were observed in 41% of embryos (Fig. 3a and b). Edema became generalized at 8 dpf when most lethality was noted. To further examine the effect of uba2 on zebrafish development, we calculated the survival rate of uba2−/− zebrafish which was significantly lower than control (WT) and heterozygous fish. uba2−/− zebrafish showed a mortality rate of approximately 50% at 8 dpf; however, 100% of mutant fish were dead by day 12 (Fig. 3d).
Figure 3: Severe dysmorphic features in embryonic uba2−/− zebrafish.
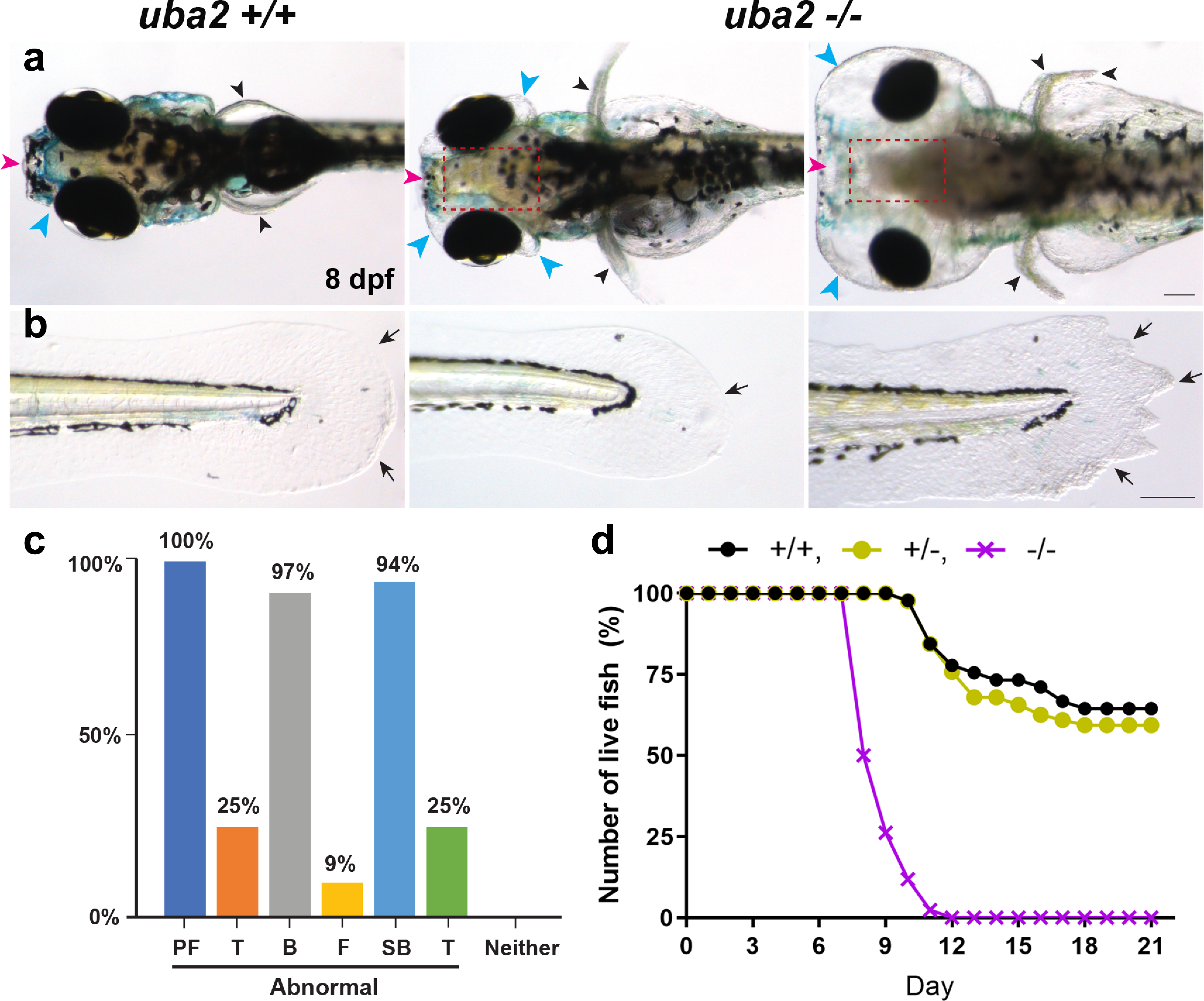
a-b. Dorsal and lateral views of uba2 zebrafish at 8 dpf. Compared to WT controls, mutant lines showed aberrant head development with small eyes and hydrocephalus. c. Bar graph representing the percentage of uba2−/− zebrafish with gross morphological defects. d. Survival curve showing the number of live fish over the course of 21 days. WT and heterozygous fish showed similar death curves, but homozygous fish had steeper death curves with 100% mortality by day 12. PF (pectoral fins), T (tail), B (brain), F (craniofacial) and SB (swim bladder).
Nullizygous fish exhibited a wide phenotypic range. We observed a pair of normal extended pectoral fins in WT zebrafish versus uba2−/− fish, where pectoral fins were found to be short and upright-oriented (Fig. 3a) confirming uba2 function in fish extremity development. WT zebrafish had thin lines originating from base to fin tips showing normal actinotrichia. In contrast, uba2−/− fish displayed collapsed (Fig. 3b, middle image) and irregular fin fold edges (Fig. 3b, last image).
To better characterize variable expression and the relationship between the zebrafish knockout and the human disorders, we quantified craniofacial (F), brain (B), pectoral fin (PF), tail fin (TF) and swim bladder (SB) defects. Defects at later stages of development were studied in uba2−/− fish bred from the same parent at 8 dpf, when approximately half the fish survive (n=32; Fig. 3c). Tissue-level malformations were observed in craniofacial structures (9.38%), brain size (90.6%), tail fin (25%), pectoral fin (100%) and swim bladder (93.75%) (Fig. 3c and as described below). Thus, across individual fish with similar genetic backgrounds, total uba2 function loss recapitulates some tissue-level phenotypes and the variable expression observed in human UBA2-related phenotypes.
Neuronal reduction in uba2 zebrafish
Tissue-level analysis was performed in zebrafish to elucidate abnormalities resulting from uba2 loss-of-function. First, we conducted immunohistochemistry studies on 8 dpf zebrafish cryosections through eye and brain. Compared to WT controls, uba2-null fish showed small heads, reduced midbrain size, low nuclei cell count with high accumulation of actin signal (orange, Fig. S2), implying a decreased proportion of gray to white matter. In addition, uba2−/− fish had smaller eyes, reduced retinal thickness, retinal laminations, and lens defects (see supplement).
Skeletal and extremity phenotypes in the uba2 zebrafish model
To investigate the impact of uba2 on zebrafish skeletal development, we stained uba2 WT (+/+), heterozygous (+/−) and homozygous (−/−) fish with alcian blue dye at 5 dpf. In both uba2 WT (Fig. 4a) and heterozygous zebrafish (data not shown), alcian staining demonstrated a normal pattern of cartilage element development including typical ceratohyoid, Meckel’s cartilage, ceratobranchials arches and pectoral fin cartilage. However, complete loss of uba2 in homozygous fish resulted in abnormal craniofacial development. In addition to jaw malformations, other craniofacial malformations included malformed and hypoplastic ventral and dorsal cartilage structures with lack of basihyal and hypohyal development. We also noted an apparently abnormal fusion of Meckel’s cartilage with the palatoquadrate, resulting in a small, narrow mandible (Fig. 4b). Moreover, Meckel’s cartilage was flattened at the midline fusion point with completely absent ceratohyal cartilage and ceratobranchials arches, the equivalent of micrognathia in these fish.
Figure 4: Cranial cartilage patterns observed in uba2−/− zebrafish and rescue of uba2 mutant phenotype with human UBA2 mRNA.
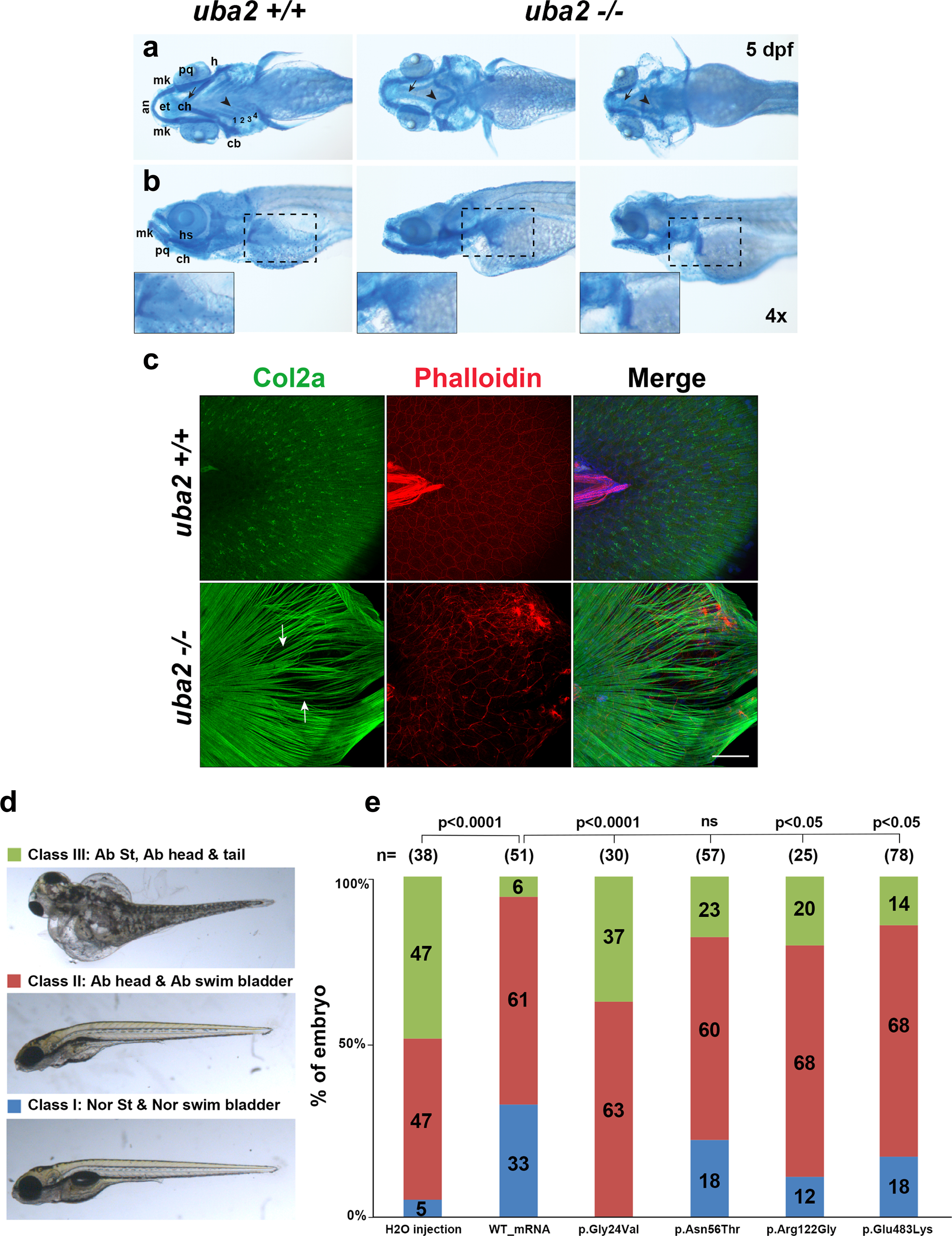
a-b. Brightfield ventral and lateral views of cartilage stained uba2 in wild type and homozygous mutant fish are shown in top and bottom panels, respectively. Closeups of pectoral fin cartilage phenotype are shown in inserts in the bottom panel highlighted by black dashed boxes on lateral views. an (anterior), mk (Meckel’s cartilage), pq (palatoquadrate), ep (ethmoid plate), ch (ceratohyal), h (hypohyal), cb (ceratobranchials 1–4), hs (hyosymplectic). c. Z-stack images of uba2 zebrafish median fins stained with Col2a (green), Rhodamine-Phalloidin (red) and Dapi (blue). Arrows are used to show the gaps between actinotrichia fibers. Scale bar: 50 μm. d. Suppression of uba2 in zebrafish produces an abnormal phenotype which is classified into three categories. e. Proportions of uba2−/− zebrafish embryos representing each phenotype category after injecting with WT or mutation harboring human UBA2 mRNA. Landmark abbreviations: Nor St (normal structure), Ab (abnormal) and ns (not significant). Chi Square test p-values are shown above the phenotypes for each rescue experiment.
To explore whether uba2 mutation causes skeletal phenotypes in adult fish, we performed micro-computed tomography (CT) comparing WT (n=3) and uba2+/− (n=3) fish, as nullizygous fish did not survive to this stage. We noted abnormal, wavy ribs and dysmorphic fin girdles in uba2+/− fish (Fig. S3).
In teleosts, finfolds are typically made of type II collagen matrix structures called actinotrichia that line the epidermis. Brightfield microscopy of uba2−/− fish revealed structural defects in median fins (Fig. 3b). To examine the effect of uba2 truncation on zebrafish median fin structure development, we stained uba2 zebrafish (+/+, +/− and −/−) larvae with type II collagen (Col2a) and Phalloidin (F-actin) antibodies to label actinotrichia (Fig. 4c).
Actinotrichia fibrils initiate fin development and become the future fin connective tissue. At 5 dpf, both WT (Fig. 4c, top panel) and heterozygous (data not shown) larvae develop median fins showed normally arrayed Col2a-labeled actinotrichia fibers; however, we observed non-rigid, non-parallel and bent actinotrichia in uba2−/− (Fig. 4c, arrows) fish. Phalloidin staining in uba2−/− fish revealed disorganized and disrupted organization, corresponding to areas of this abnormal collagen pattern (Fig. 4c).
Further investigating these extremity defects at a cellular level, we performed ultrastructural analysis of the uba2 zebrafish body wall near the median finfold at 5 dpf. Detailed examination by TEM revealed a typical dynamically-assembled dense striated pattern of actinotrichia in WT fish (Fig. S4). Similarly, in WT fish we observed a normal and organized distribution of skeletal muscles with normal nuclei and mitochondria. However, in uba2−/− zebrafish, we observed disorganized (or incompletely developed) and scattered actinotrichia with abnormal epidermal cells (arrow). The skeletal muscle layer in homozygous fish was also observed to be discontinuous or atrophic with degenerated nuclei and mitochondria. Therefore, absent uba2 impacts connective and epithelial tissue and skeletal muscle and causes extremity malformations in developing fish.
Conserved function of UBA2 candidate variants in zebrafish
To further confirm the specificity of the uba2 knockout phenotype, we attempted phenotypic rescue of developmental fish malformations by injecting human UBA2 mRNA. Injected fish were grouped into three phenotypic classes and genotyped at 5 dpf, and the uba2−/− subset was analyzed. Embryos were classified as Class I (grossly normal body structure), Class II (decreased head size, absent swim bladder), and Class III (small head and body, generalized edema) (Fig. 4d). As compared to H2O-injected controls, injecting human WT UBA2 mRNA grossly rescued phenotypes in a significant number of fish. The proportion of Class I fish increased from 5% to 33%, and the proportion of Class III fish decreased from 47% to 6% (p<0.0001) (Fig. 4e). Even though WT-UBA2 mRNA injection rescued gross phenotypes, most uba2−/− zebrafish still did not show inflated swim bladder (data not shown), suggesting that early uba2 deficiency permanently impacts zebrafish physiology despite substitution with human mRNA.
Human mRNAs encoding p.Gly24Val, p.Arg122Gly and p.Glu483Lys all failed to rescue the uba2−/− phenotypes in contrast to WT mRNA. The p.Asn56Thr substitution demonstrated statistically similar rescue to control mRNA; however, there were more Class III fish (23% vs 6%) and fewer Class I fish (18% vs 33%) following p.Asn56Thr injection, indicating possible partial loss-of-function for this missense substitution (Fig. 4e). Because the mRNAs containing the missense variants failed to rescue uba2-null phenotypes to a similar level as did WT UBA2 mRNA, we conclude that the most likely mechanism of disease is loss-of-function.
Discussion
In this study, we describe a cohort of patients harboring deleterious variants in the UBA2 gene. They show highly variable inter- and intra-familial expression of dermatologic, skeletal, extremity, neurologic, cardiac, and renal features, similar to those of the chromosome 19q13.11 microdeletion syndrome1–8. These observations further support UBA2 as the critical gene in the microdeletion syndrome and suggest its essential role in early human growth and development. There are only a few other reports of intragenic UBA2 variants (summarized in Table 1). Marble et al.18 reported a de novo UBA2 missense variant (c.71G>T, p.Gly24Val) in a 2.5-year-old female with ACC, thin hair, tall forehead, Duane anomaly, hip dysplasia, clinodactyly, and poor weight gain. Wang et al.18 reported an inherited UBA2 frameshift variant [c.327delT, p.Phe109Leufs*3] in a young boy and his mother. The mother had ACC but was otherwise healthy. The son had ACC, microcephaly, bilateral ectrodactyly, low‐lying conus medullaris, horseshoe kidney and tracheoesophageal fistula. A de novo UBA2 loss-of-function variant [c.1324dupT, p.Tyr442Leufs*17] was associated with four extremity split hand and foot malformation with tibial deficiency and under-masculinized external genitalia19. Aerden et al.20 reported a male proband with ectrodactyly of the feet, autism spectrum disorder, craniofacial variations, dry sparse scalp hair, strabismus and hypermetropia who was heterozygous for a de novo frameshift variant in UBA2 [c.612delA, p.Glu205Lysfs*63]; this was considered to be responsible for the phenotype20.
The four patients previously reported with intragenic UBA2 variants were added to our clinical summary table (Table 1) to compare phenotypes18–21. We’ve estimated the percentage of key traits in UBA2 subjects (Fig. 1c) based on available clinical information. The most specific aspects of the UBA2-related phenotype are ACC, seen in 61%, and ectrodactyly, which is less common (37%). Early growth deficiency and neurodevelopmental delay are reported in 61% and 80% of affected individuals, respectively. More variable digital and skeletal abnormalities are also present (56%) but are sometimes subtle and potentially overlooked (e.g., Fig. 1a, panels C, D). These include clinodactyly (62%), syndactyly (59%), camptodactyly (57%), and hip abnormality (35%).The most common craniofacial variations are tall forehead/high hairline (76%), down-slanted palpebral fissures (47%), hypertelorism (62%), broad nasal root (81%), microcephaly (37%), and micrognathia (53%). Other observed features among our subjects include other ectodermal variations (~82%), ocular abnormalities (53%), cardiac (43%), genital (50%, in males) and renal (36%) abnormalities.
In C. elegans, Uba-2 is also noted to be a critical element of the SUMOylation pathway; its ablation leads to embryonic lethality22. UBA2 acute knockdown in xenograft tumors by conditional shRNAs causes marked growth arrest, cell proliferation defects and increased apoptosis23. In mice, loss of any key component of the SUMOylation pathway can lead to severe impairment of cellular functions and lethality24,25. An in-situ hybridization study conducted in mouse embryos (8.5 to 11.5 days post-coitum) revealed Uba2 ample expression at multiple morphogenetic activity sites, e.g. neural folds, branchial arches and limb buds24, suggesting that Uba2 is essential for normal cellular function/development. Recently, SUMOylation was reported to regulate differentiation of several ocular tissues26,27.
Phenotypic features in our human UBA2-related syndrome cohort and the uba2 knockout zebrafish are reminiscent of disorders associated with pathogenic variants in DLX5/6 (split-hand/foot malformation (SHFM1, OMIM: 220600), TP63 (e.g., Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3, EEC3, OMIM: 604292, split hand/foot malformation syndrome 4, SHFM4, OMIM: 605289 and others) and FBXW4, a candidate for SHFM3 (OMIM: 246560). tp63−/− zebrafish embryos have ectodermal defects involving skin, absent pectoral fin buds and reduced size fin folds at 36hpf and embryos died between 40–50hpf28. tp63 zebrafish morphants affect skin integrity by making the skin more prone to microbial infection29. fndc3a−/−zebrafish show broken actinotrichia, aberrant collagen localization and cellular defects in epidermal cells during caudal fin development30. It is possible that these genes function downstream of the SUMOylation pathway, leading to phenotypes that overlap the UBA2-related syndrome.
In the current study, the mRNA rescue experiments showed that WT-UBA2 mRNA injection partially rescued the abnormal head/eye, tail, and uninflated swim bladder phenotype in uba2−/− zebrafish (33%). Notably, three of four human missense UBA2 mRNAs did not rescue the uba2−/−phenotype to a significant degree, suggesting a loss-of-function mechanism for these disease-associated alleles. As wide phenotypic variability is observed in both fish and human UBA2/uba2-related phenotypes, additional studies are warranted to define potential modifiers. Morpholinos (MOs) have been used in reverse genetic studies in a range of animal models31,32. However, MOs may be hard to interpret as they typically result in more severe phenotypes33. mRNA rescue in CRISPR-generated stable mutant lines are potentially useful in the interpretation of MO-related inconsistencies. Precise single nucleotide variant animal models of human diseases can help to better understand underlying molecular processes and may aid in management of UBA2-related abnormalities34.
In conclusion, we report clinical details in 16 individuals from seven unrelated families with inherited or de novo heterozygous UBA2 sequence variants, who present with highly variable phenotypes. Definition of the UBA2-related autosomal dominant phenotypic spectrum in humans, in silico modeling predictions, uba2 expression and characterization of the knock out phenotype in zebrafish support the significance of UBA2/uba2 in development, potentially by affecting post-translational modification of SHFM-associated genes. mRNA rescue experiments in zebrafish also suggest that loss of gene function is the primary mechanism of disease. The highly variable expressivity of the human UBA2 phenotype, either via sequence alteration or contiguous gene deletion, even within the same family, remains incompletely explained; there are likely other modifiers, still to be identified. However, our studies define a human disorder associated with UBA2 sequence variants with a phenotype that overlaps key aspects of the chromosome 19q13.11 microdeletion syndrome.
Supplementary Material
Acknowledgments:
We gratefully thank the individuals and families who participated in this project. We would like to express our deepest gratitude to Dr. Mary Ella Pierpont for her valuable contribution and dedicate this report to her memory. WKC received financial support from the JPB Foundation. NR’s work is supported by NHGRI grant 1U54HG006542. We would like to thank Dr. Sunit Dutta (National Eye Institute, NIH, Bethesda) for assistance in establishing uba2 zebrafish knock out lines. We thank the Zebrafish facility, Confocal, transmission electron microscopy and micro-computed tomography mouse imaging facilities at NIH for their support and technical assistance. The research work carried out at NIH was supported by funds provided by National Eye Institute, National Institutes of Health, Bethesda, MD.
Footnotes
Declaration of Interests:
RES, IMW, MJGS, LR and JJ are employees of GeneDx, Inc., Gaithersburg, MD. The other authors declare no competing interests.
Ethics declaration:
Study participants were enrolled in approved protocols as per the policies of the Institutional Review Board Committees of the institutions at which patients were identified, or via GeneDx, following the tenets of the Declaration of Helsinki. The main IRB for this study is Western Institutional Review Board, Study Number 1175206, WIRB protocol # 20171030 (GeneDx). Written informed consent for inclusion in this study was obtained as required from all subjects, including specific consent to use photographs. All zebrafish-related experiments were conducted in accordance with recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, protocol # NEI-679.
Web Resources:
ClinVar Database https://www.clinicalgenome.org/data-sharing/clinvar
gnomAD https://gnomad.broadinstitute.org/
GeneMatcher https://genematcher.org/
Pathogenicity predictions https://varsome.com/
OMIM http://www.omim.org/
Clustal omega https://www.ebi.ac.uk/Tools/msa/clustalo/
Supplemental Data
Supplemental data include a supplemental material and methods and results section, four figures and one table.
Data availability:
All data is mentioned in the main text and supplement, available to readers.
References
- 1.Abe KT, Rizzo I, Coelho ALV, Sakai N Jr., Carvalho DR, Speck-Martins CE. 19q13.11 microdeletion: Clinical features overlapping ectrodactyly ectodermal dysplasia-clefting syndrome phenotype. Clin Case Rep. 2018;6:1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chowdhury S, Bandholz AM, Parkash S, et al. Phenotypic and molecular characterization of 19q12q13.1 deletions: a report of five patients. Am J Med Genet A. 2014;164A:62–69. [DOI] [PubMed] [Google Scholar]
- 3.Gana S, Veggiotti P, Sciacca G, et al. 19q13.11 cryptic deletion: description of two new cases and indication for a role of WTIP haploinsufficiency in hypospadias. Eur J Hum Genet. 2012;20:852–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malan V, Raoul O, Firth HV, et al. 19q13.11 deletion syndrome: a novel clinically recognisable genetic condition identified by array comparative genomic hybridisation. J Med Genet. 2009;46:635–640. [DOI] [PubMed] [Google Scholar]
- 5.Melo JB, Estevinho A, Saraiva J, Ramos L, Carreira IM. Cutis Aplasia as a clinical hallmark for the syndrome associated with 19q13.11 deletion: the possible role for UBA2 gene. Mol Cytogenet. 2015;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuurs-Hoeijmakers JH, Vermeer S, van Bon BW, et al. Refining the critical region of the novel 19q13.11 microdeletion syndrome to 750 Kb. J Med Genet. 2009;46:421–423. [DOI] [PubMed] [Google Scholar]
- 7.Urquhart JE, Williams SG, Bhaskar SS, Bowers N, Clayton-Smith J, Newman WG. Deletion of 19q13 reveals clinical overlap with Dubowitz syndrome. J Hum Genet. 2015;60:781–785. [DOI] [PubMed] [Google Scholar]
- 8.Venegas-Vega C, Nieto-Martinez K, Martinez-Herrera A, et al. 19q13.11 microdeletion concomitant with ins(2;19)(p25.3;q13.1q13.4)dn in a boy: potential role of UBA2 in the associated phenotype. Mol Cytogenet. 2014;7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desterro JM, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J Biol Chem. 1999;274:10618–10624. [DOI] [PubMed] [Google Scholar]
- 10.He P, Sun X, Cheng HJ, et al. UBA2 promotes proliferation of colorectal cancer. Mol Med Rep. 2018;18:5552–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsen SK, Capili AD, Lu X, Tan DS, Lima CD. Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature. 2010;463:906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang SC, Ding JL. Ubiquitination and SUMOylation in the chronic inflammatory tumor microenvironment. Biochim Biophys Acta Rev Cancer. 2018;1870:165–175. [DOI] [PubMed] [Google Scholar]
- 13.Westerfield M The zebrafish book : a guide for the laboratory use of zebrafish (Danio rerio). [Eugene, OR]: M. Westerfield; 2007. [Google Scholar]
- 14.Marble M, Pridjian G. Scalp defects, polythelia, microcephaly, and developmental delay: a new syndrome with apparent autosomal dominant inheritance. Am J Med Genet. 2002;108:327–332. [DOI] [PubMed] [Google Scholar]
- 15.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lois LM, Lima CD. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005;24:439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marble M, Guillen Sacoto MJ, Chikarmane R, Gargiulo D, Juusola J. Missense variant in UBA2 associated with aplasia cutis congenita, duane anomaly, hip dysplasia and other anomalies: A possible new disorder involving the SUMOylation pathway. Am J Med Genet A. 2017;173:758–761. [DOI] [PubMed] [Google Scholar]
- 19.Yamoto K, Saitsu H, Nishimura G, et al. Comprehensive clinical and molecular studies in split-hand/foot malformation: identification of two plausible candidate genes (LRP6 and UBA2). Eur J Hum Genet. 2019;27:1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aerden M, Bauters M, Van Den Bogaert K, et al. Genotype-phenotype correlations of UBA2 mutations in patients with ectrodactyly. Eur J Med Genet. 2020:104009. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Dupuis L, Jobling R, Kannu P. Aplasia cutis congenita associated with a heterozygous loss-of-function UBA2 variant. Br J Dermatol. 2020;182:792–794. [DOI] [PubMed] [Google Scholar]
- 22.Jones D, Crowe E, Stevens TA, Candido EP. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin-conjugating enzymes, ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 2002;3:RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrington B, Varshney GK, Burgess SM, Sood R. CRISPR-STAT: an easy and reliable PCR-based method to evaluate target-specific sgRNA activity. Nucleic Acids Res. 2015;43:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costa MW, Lee S, Furtado MB, et al. Complex SUMO-1 regulation of cardiac transcription factor Nkx2–5. PLoS One. 2011;6:e24812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao J Sumoylation regulates diverse biological processes. Cell Mol Life Sci. 2007;64:3017–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nie Q, Xie J, Gong X, et al. Analysis of the Differential Expression Patterns of Sumoylation Enzymes E1, E2 and E3 in Ocular Cell Lines. Curr Mol Med. 2018;18:509–515. [DOI] [PubMed] [Google Scholar]
- 27.Gong X, Nie Q, Xiao Y, et al. Localization Patterns of Sumoylation Enzymes E1, E2 and E3 in Ocular Cell Lines Predict Their Functional Importance. Curr Mol Med. 2018;18:516–522. [DOI] [PubMed] [Google Scholar]
- 28.Santos-Pereira JM, Gallardo-Fuentes L, Neto A, Acemel RD, Tena JJ. Pioneer and repressive functions of p63 during zebrafish embryonic ectoderm specification. Nat Commun. 2019;10:3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H, Kimelman D. A dominant-negative form of p63 is required for epidermal proliferation in zebrafish. Dev Cell. 2002;2:607–616. [DOI] [PubMed] [Google Scholar]
- 30.Liedtke D, Orth M, Meissler M, et al. ECM alterations in Fndc3a (Fibronectin Domain Containing Protein 3A) deficient zebrafish cause temporal fin development and regeneration defects. Sci Rep. 2019;9:13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yousaf R, Ahmed ZM, Giese AP, et al. Modifier variant of METTL13 suppresses human GAB1-associated profound deafness. J Clin Invest. 2018;128:1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yousaf S, Sethna S, Chaudhary MA, Shaikh RS, Riazuddin S, Ahmed ZM. Molecular characterization of SLC24A5 variants and evaluation of Nitisinone treatment efficacy in a zebrafish model of OCA6. Pigment Cell Melanoma Res. 2020;33:556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stainier DYR, Raz E, Lawson ND, et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017;13:e1007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prykhozhij SV, Berman JN. Zebrafish knock-ins swim into the mainstream. Dis Model Mech. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is mentioned in the main text and supplement, available to readers.