

Abstract
Vagal circuit-α7 nicotinic acetylcholine receptor (α7nAChR, coded by Chrna7) signaling can modulate lung proinflammatory responses. Arginase 1 (ARG1) plays a crucial role in the resolution of lung inflammation. However, whether vagal-α7nAChR signaling can regulate lung inflammation and ARG1 expression during an influenza infection is elusive. Here, we found that lung and spleen IL-4+ cells and lung ARG1 expression were reduced; however, bronchoalveolar lavage (BAL) protein and leukocytes and lung inflammatory cytokines were increased in PR8 (A/Puerto Rico/8/1934, H1N1)-infected vagotomized mice when compared to the control. In PR8-infected α7nAChR-deficient mice, lung Arg1, Il10, and Socs3 expression and BAL Ly6C+CD206+ cells were reduced. PR8-infected Chrna7+/+ recipient mice reconstituted with Chrna7−/− bone marrow had a lower survival as compared to PR8-infected Chrna7+/+ recipient mice reconstituted with Chrna7+/+ bone marrow. Mechanistically, the activation of α7nAChR by its agonist GTS-21 could enhance IL-4-induced Arg1 expression, reduced Nos2, and TNF-α expression in PR8-infected bone marrow-derived macrophages (BMDM). Stimulation with IL-4 increased phosphorylation of STAT6 and activation of α7nAChR increased STAT6 binding with the ARG1 promoter and relieved IL-4-induced H3K27me3 methylation by increasing JMJD3 expression in PR8-infected BMDM. Inhibition of JMJD3 increased H3K27me3 methylation and abolished α7nAChR activation and IL-4 induced ARG1 expression. Activation of α7nAChR also reduced phosphorylation of AKT1 and contained FOXO1 in the nucleus. Knockdown of Foxo1a reduced α7nAChR activation and IL-4 induced Arg1 expression in PR8-infected BMDM. Therefore, vagal-α7nAChR signaling is a novel therapeutic target for treating lung inflammatory responses during an influenza infection.
Keywords: vagus circuits, α7nAChR, lung inflammation, arginase 1, influenza A virus
Introduction
Despite vaccination, influenza viral infection remains a major threat to public health, especially among children and the elderly. Influenza viruses replicate in the bronchial and alveolar epithelial cells and induce inflammatory responses in the neighboring pulmonary macrophages (MΦ). MΦ activation can be classified into ‘classical activation’ (M1 phenotype) and ‘alternative activation’ (M2 phenotype). LPS and IFN-γ can induce MΦ to be polarized into M1 MΦ. M1 MΦ could upregulate the expression of NOS2 and proinflammatory cytokines (such as TNF-α, IL-6, and CXCL2). In addition, interleukin-4 (IL-4) and IL-13 are capable of inducing the differentiation of MΦ into M2 MΦ. M2 MΦ could express high levels of IL-10, SOCS3, and arginase 1 (ARG1), which promote anti-inflammatory responses related to tissue healing and repair [1]. As a result, M2 MΦ could inhibit influenza virus-mediated lethal lung inflammatory responses and damage [2].
In the past decades, Tracey and his colleagues have found that the cholinergic anti-inflammatory pathway via the regulatory hub-spleen plays an important role in regulating systemic proinflammatory responses [3–5]. But, another study has shown that vagal efferent neurons in a rat model neither synapse with splenic sympathetic neurons nor drive their ongoing activity [6]. Besides innervating the spleen, the vagus nerve can innervate the distal airway and even the alveoli [7] and airway sensors [8]. The vagal circuits between the lung and nervous centers in the nodose ganglion and the nucleus of solitary tract in the brain stem form a unique machinery named the pulmonary parasympathetic inflammatory reflex [9, 10]. Studies have shown that vagotomy and the knockout of α7nAChR can aggravate lipopolysaccharide (LPS) or Escherichia coli-induced acute lung injury (ALI) [11, 12]. The α7nAChR+CD11b+ cells are important players in mediating the protective effects of vagal-α7nAChR signaling [10].
Some reports have shown that prestimulation of human MΦ with acetylcholine could attenuate influenza virus-mediated release of proinflammatory cytokines [13] as the activation of α7nAChR promotes LPS-induced conversion of M1 MΦ to M2 [14]. PNU-282987 (an α7nAChR agonist) treatment reduced lung mRNA levels and the frequency of M1 MΦ, whereas cells expressing the M2-related markers, ARG1 (arginase 1), CD206 [15], and IL-10, were increased in the LPS-induced lung injury model [16]. However, whether vagal-α7nAChR signaling can regulate lung inflammation and ARG1 expression (as a key indicator of M2 MΦ activation) during an influenza infection is unknown.
It has been reported that STAT6 phosphorylation enhances the expression of ARG1 in IL-4-stimulated MΦ [17]. In addition, that FOXO1 and H3K27me3 epigenetic modification might regulate the transcription of ARG1 in MΦ [18, 19]. Another study has shown that the Jmjd3-Irf4 axis regulates M2 MΦ polarization and host responses against helminth infection [20]. Also, AKT signaling has been reported to be an important factor in MΦ activation and M1/M2 polarization [21] as AKT could phosphorylate FOXO and lead to the exclusion of FOXO from the nucleus to cytoplasm, hence contributing to M2 MΦ activation [22]. Therefore, whether these factors would affect vagal-α7nAChR signaling-regulated ARG1 expression during an influenza infection is worthy of investigation.
Here, we hypothesized that the disruption of vagal-α7nAChR signaling would worsen influenza-induced lung inflammation and that the activation of α7nAChR would boost IL-4-induced ARG1 expression in PR8-infected MΦ. Therefore, we studied the following: (1) whether or not vagotomy and deletion of Chrna7 would affect influenza-induced lung inflammation and ARG1 expression; (2) whether or not the activation of α7nAChR would affect IL-4-induced ARG1 expression in PR8 or Poly I:C-challenged BMDM; (3) whether or not the activation of α7nAChR would influence the binding of STAT6 with the ARG1 promoter in influenza-infected MΦ; and (4) whether or not the activation of α7nAChR would affect AKT1-FOXO1 signaling or JMJD3-H3K27me3 methylation during IL-4-induced ARG1 expression
In this study, we have demonstrated that vagotomy reduced lung IL-4 and ARG1 expression, increased bronchoalveolar lavage (BAL) protein and leukocytes and lung inflammatory cytokines during a PR8 infection. In PR8-α7nAChR-deficient mice, lung Arg1, Il10, and Socs3 expression and BAL Ly6C+CD206+ cells were reduced. PR8-infected Chrna7+/+ mice reconstituted with Chrna7−/− bone marrow had a lower survival rate. In the in vitro study, the activation of α7nAChR could enhance IL-4-induced Arg1 expression, reduced Nos2 and TNF-α expression in PR8-infected MΦ. Mechanistically, activation of α7nAChR increased STAT6 binding with the ARG1 promoter, increased FOXO1 retention in the nucleus, reduced H3K27me3 methylation, and therefore boosted IL-4 induced ARG1 expression in the PR8-infected MΦ. These finding suggest that vagal-α7nAChR signaling is a novel therapeutic target for treating influenza lung inflammation.
Materials and methods
Animals
C57BL/6 mice were housed in groups with 12-h dark–light cycles and with free access to food. Chrna7-deficient (B6.129S7-Chrna7tm1Bay) and LysMCre mice from a C57BL/6 background were purchased from the Jackson Laboratory (Bar Harbor, ME). Akt1fl/fl mice were obtained from Professor Zhong-zhou Yang, Model Animal Research Center, Nanjing University. The operations were performed under anesthesia which was induced with an intraperitoneal (IP) injection of pentobarbital sodium (50 mg/kg). The Committees on Animal Research of the Institut Pasteur of Shanghai, Chinese Academy of Sciences, approved of the protocols (Permit Number: A2014009).
Reagents
The Phospho-AKT1 (Ser473) (D7F10) XP Rabbit mAb was obtained from Cell Signaling (Danvers, MA). Arginase I (H-52), STAT6 (S-20), AKT1 (C-20), p-FKHR (Ser 256), and FKHR (H-128) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). GTS-21 dihydrochloride (DMBX-A), anti-STAT6 (phospho Y641), anti-KDM6B/JMJD3, anti-Histone H3 (tri-methyl K27), and anti-Choline Acetyltransferase [EPR13024(B)] were from ABCAM (Cambridge, MA). Mouse TNF alpha ELISA Ready-SET-Go, mouse IL-4 Recombinant Protein, anti-mouse antigen F4/80 eFlour 450® antibody, anti-mouse Ly-6G (Gr-1), anti-mouse Ly-6C, anti-mouse CD11b, anti-mouse IL-4, anti-Mouse CD11c FITC, and anti-human/mouse CD45R (B220) were from eBioscince (San Diego, CA). The Poly(I:C) (LMW) was from InvivoGen (San Diego, CA). The APC anti-mouse CD206 (MMR) was from Biolegend (San Diego, CA). Rabbit (DA1E) mAb IgG XP Isotype Controls were from Cell Signaling (Danvers, MA). The Mouse/Rat CCR2 Phycoerythrin MAb (Clone 475301) was from R&D Systems (Minneapolis, MN) and the CF633-α-Bungarotoxin was from Biotium (Hayward, CA).
Bone marrow chimera experiments
To test the role of α7nAChR of bone marrow cells in mediating IAV-induced lung inflammation, the Chrna7+/+ recipient mice were lethally irradiated (6 Gy), to destroy their bone marrow, and randomly divided into two groups. Then, these mice received either Chrna7+/+ or Chrna7−/− bone marrow donor cells (5 × 106 in each mouse) by a retro-orbital injection to reconstitute their bone marrow. The mice were administered antibiotic-treated water for 3 weeks and then used for experiments at ~8 weeks after bone marrow transplantation. Using this protocol, at 8 weeks after bone marrow transplantation, more than 90% of the circulating hematopoietic cells were of donor origin.
Unilateral vagotomy
As we previously reported [10], right or sham cervical vagotomy was performed with the animals under anesthesia (IP injection of pentobarbital sodium, 50 mg/kg). The procedure involved a longitudinal midline incision in the ventral region of the neck. Using blunt dissection, the overlying muscles and fascia were separated until the right vagus and carotid artery were visible. The vagus was carefully stripped away from the carotid artery and lightly cut off from the vagotomy group. The vagus was kept intact in the sham group. The incision was sutured with a 7–0 silk thread. The respiration rhythm was not affected by the unilateral vagotomy. It was reported that the right nerve controls cardiac function and thus this side was not chosen for vagus nerve stimulation. In our study, upon comparing sham to vagotomy mice receiving an intratracheal instillation of saline, there was no difference in BAL cytokines and inflammatory cell profiles within 24 h. No mice died of heart failure after operating on the right side for the vagotomy.
Influenza viral propagation
The influenza virus, mouse adapted A/Puerto Rico/8/1934(H1N1), was kindly provided by Dr. Ertl HC [23] (Wistar Institute, Philadelphia, PA, USA). PR8 was propagated in a 10-day-old chicken embryo from specific-pathogen-free flocks (Beijing Merial, China). Each egg was injected with 0.1 mL of phosphate-buffered saline (PBS) containing ~103 infectious particles, incubated at 37 °C with forced air circulation and egg rotation, and maintained at 4 °C for 12 h before harvesting. The allantoic fluid was harvested separately from each egg. Those with high hemagglutinin activity were pooled, and aliquots were prepared and stored at −80 °C. The hemagglutinin titer of the above subtype of influenza virus was measured with 1% chicken blood cell in a V-shaped plate.
Isolation and culture of BMDM
BMDMs were obtained by in vitro differentiation of the primary femur and tibia BM cells as previously described [24]. Briefly, femurs and tibiae from wild type, Chrna7−/− and LysMcre+Akt1fl/fl mice were dissected, cleaned, disinfected in 70% ethanol, and washed with fully supplemented RPMI-1640 medium. After lysing erythrocytes, the BM cells were then cultured in a Petri dish (at a density of 4 × 106 cells/10 cm dish) supplemented with 30% L929 cell-conditioned medium. This medium contained high levels of M-CSF, because murine bone marrow MΦ, or human monocytes treated with M-CSF, differentiate into MΦ with an M2-like phenotype, while those treated with GM-CSF differentiate into a mixed population of dendritic cells and M1-like MΦ [25]. Cells were matured to phenotypic MΦ over 6 days. Adherent cells were recovered and infected with PR8 for the experiments.
Treatment strategy of BMDM
As Fig. 4b shows, we set up eight treatment strategies: the BMDMs (5 × 105/well) received α7nAChR activation with GTS-21 (25 μM in PBS) for 30 min, IL-4 stimulation (10 ng/mL in PBS) for 6 h, and then PR8 (2 MOI in PBS) influenza challenge for 18 h. The specific treatments were listed in each figure. The cells were collected after a 24-h culture was created to extract RNA and protein for analysis.
Fig. 4. Activation of α7nAChR enhances M2 MΦ activation in IL-4-treated PR8-infected BMDM.

a Histogram of F4/80 expression in BMDM. BMDM were collected after 6 days of culture and stained with fluorescent isotype or anti-F4/80 antibodies. The freshly isolated bone marrow cells were used as control. Experiments were repeated 2 times. b The experimental conditions for inducing M2 MΦ activation. Eight treatment strategies were selected, while the 8th strategy was as follows: BMDM (5 × 105/well) received α7nAChR activation with GTS-21 (25 μM in PBS) for 30 min, IL-4 stimulation (10 ng/mL in PBS) for 6 h, and then PR8 (2 MOI in PBS) influenza challenge for 18 h. The other strategies were used as controls (the same concentration of cell, GTS-21, or IL-4 as the 8th was added as required). The cells were collected after 24 h treatment to extract RNA and protein for analysis. c–e Relative expression of Arg1, Nos2, and TNF-α expressions in-PR8-infected BMDM treated with C8 condition for 18 h. The cells were collected to extract RNA for measuring Arg1 and Nos2 expression. Supernatants of media were collected to measure TNF-α by ELISA (e). n = 3 in each group. **P < 0.01, ***P < 0.001 in the indicated groups, One-way ANOVA. Cells were collected to measure p-STAT6 and ARG1 in cell lysates by Western blot (f). g Relative expression of Arg1, Fizz, and Ym1 mRNAs in α7nAChR-activated IL-4-induced PR8-infected BMDM. n = 3 in each group, *P < 0.05, unpaired t test. GTS-21 concentration was used from 0, 1, and 25 μM. Using strategies listed in (b). Data in (c–g) were representatives of two independent experiments. “-” condition indicated PBS treatment.
Animal viral infection
Male 8–10-week-old wild type, Chrna7−/−, or vagotomized mice (with a C57BL/6J background) were used in the different experimental conditions. Mice were anesthetized and then intranasally given either a PR8 1.4 × 104 FFU (in 20 µL chorioallantoic fluid) or vehicle.
Bronchoalveolar lavage
As described previously [12], BAL was done after euthanizing the mice and then placing a 20-gauge catheter into the trachea through which 1 mL of cold PBS was flushed back and forth three times. A BAL supernatant was used for measuring protein concentration. The BAL cells were quantified and lysed for Western blotting analysis.
Preparing single cells from the digested lungs
Lungs were removed and washed twice in RPMI-1640 supplemented with 1% penicillin or streptomycin. The excised lungs were minced, ground, and then transferred into a conical tube containing 3 mL of digestive solution of collagenase type 1 A 1 mg/mL plus type IV bovine pancreatic DNase 20 μg/mL in HBSS containing 5% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin). The samples were incubated for 30 min at 37 °C in a water bath shaker. The cells were collected once separated by a centrifuge for 10 min at 335 × g at 4 °C. The red cells were lysed by adding ACK lysing buffer for 1 min at room temperature.
Flow cytometric analysis
As described [26], after unspecific staining was minimized through pre-incubation for 15 min with anti-mouse CD16/32, the BMDM were labeled with fluorescent primary or isotype antibodies. Fluorescent cells were analyzed after the exclusion of debris and aggregates with LSRFortessa (BD Biosciences, San Jose, CA). Data were analyzed by FlowJo 7.6 (Tree Star Inc. Ashland, OR). All the experimental groups regarding flow cytometry and appropriate isotype controls were used to determine the gate selection. For simplicity, we did not show all the controls in the corresponding figures.
Western blotting analysis
As previously described [11, 12], the cells were lysed in an ice-cold lysis buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and a protease inhibitor cocktail (for cell signaling) as well as a phosphatase inhibitor cocktail (Santa Cruz) for detecting phosphorylation. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce). Denatured proteins (10–20 μg) were loaded and run on a 10% Bis-Tris gel (Invitrogen, Carlsbad, CA). The proteins were then transferred to a PVDF membrane and incubated with primary antibodies. Membranes were exposed to corresponding HRP-labeled secondary antibodies and developed with an ECL kit (Amersham, Piscataway, NJ). Images taken were analyzed using Tanon Gel Image System 4200 (Shanghai Tanon Technology Co., Ltd., Shanghai, China).
Quantitative real-time PCR
The total RNA from the lungs or cultured cells was homogenized and extracted using Trizol (Invitrogen, CA), as described by the manufacturer. The total RNA obtained was suspended in RNase-free water and stocked at −80 °C. Real-time PCR was performed on an ABI PRISM Step-One sequence-detection system (TIANGEN, Bejing) by using a SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA) after a reverse transcription reaction of 2 µg of RNA by using M-MLV reverse transcriptase (TIANGEN, Beijing). The relative expression levels of the corresponding genes were determined by the 2−ΔΔCT method [27], normalized by GAPDH. The mouse primer pairs were as follows:
Arg 1 F 5′-AGACCACAGTCTGGCAGTTG-3′; R 5′-CCACCCAAATGACACATAGG-3′.
Fizz1 F 5′‐TTTTGGGAGATCCAGAGTGG‐3′; R 5′‐CAAGAAGCAGGGTAAATGGG‐3′.
Ym1 F 5′‐GACAGGCCAATAGAAGGGAGTTTCA‐3′; R 5′‐GACGGTTCTGAGGAGTAGAGACCAT‐3′.
Chrna7 F 5′‐TGGAAACCAGACATTCTCCTC‐3′; R 5′‐AGGGAGATACTGGCAATGCC‐3′.
Nos2 F 5′-GGTCTTTGACGCTCGGAACTGTAG-3′; R 5′-CACAACTGGGTGAACTCCAAGGTG-3′.
Cxcl2 F 5′-GCTGTCAATGCCTGAAG-3′; R 5′-GGCGTCACACTCAAGCTCT-3′.
Il6 F 5′-GGCCTTCCCTACTTCACAAG-3′; R 5′-ATTTCCACGATTTCCCAGAG-3′.
Il10 F 5′-CCCTTTGCTATGGTGTCCTT-3′; R 5′-TGGTTTCTCTTCCCAAGACC-3′.
Socs3 F 5′-CCTTCAGCTCCAAAAGCGAG-3′; R 5′-GCTCTCCTGCAGCTTGCG-3′.
Foxo1a F 5′-CCCAGGCCGGAGTTTAACC-3′; R 5′-GTTGCTCATAAAGTCGGTGCT-3′.
Gpadh F 5′‐AGCCCAGGATGCCCTTTAGT‐3′; R 5′‐GACATGCCGCCTGGAGAAAC‐3′.
ChIP assay
Chromatin immunoprecipitation (ChIP) was performed using a ChIP assay kit (Millipore, Billerica, MA) according to the manufacturer’s instruction, with minor modifications. Briefly, 5 × 106 BMDM cells were spread in a six-well cell culture cluster (Corning) and stimulated with and without GTS-21 (25 μM) and then challenged with either PBS or PR8 (2 MOI). After stimulation, the cells were cross-linked and then washed and resuspended in an SDS lysis buffer. Nuclei were fragmented by sonication and chromatin fractions were precleared with protein A-agarose beads followed by immunoprecipitation overnight with an anti-STAT6 Ab or a control IgG. Cross-linking was reversed and proteinase K digestion was performed. The input and immunoprecipitated DNA were amplified by real-time PCR. The ratio of the amount of DNA recovered relative to the input control was calculated. The primers for detecting Agr1 were as follows:
Arg43–192 F 5′-GATGGGGAGGTTCTGTTGAC-3′, R 5′-GGGCTGTCCAGCTCCTCT-3′.
Arg532–655 F 5′-CCTGACAACCCAAAGCAGTT-3′, R 5′-GGCACACGACAAAGACAACT-3′.
Arg796–907 F 5′-AATCGAAACGGAGCAATGGG-3′, R 5′-GCCTCTCTCATCTGCCCTAG-3′.
Knockdown of Foxo1a
The oligos for shRNA- Foxo1a were as follows: forward 5′ CCGGAGCGTGCCCTACTTCAAGGATAACTCGAGTTATCCTTGAAGTAGGGCACGCTTTTTTG 3′ and reverse 5′ AATTCAAAAAAGCGTGCCCTACTTCAAGGATAACTCGAGTTATCCTTGAAGTAGGGCACGCT 3′, the sequence was used as [28]. The oligos were mixed with 10× NEB buffer 2, and annealed at 95 °C for 4 min and 70 °C for 10 min then slowly cooled to room temperature. The annealed oligos were cloned into lentiviral vector pLKO.1 (digested with AgeI and EcoRI). Two micrograms shRNA PLKO.1, 1.5 µg psPAX2, and 0.5 µg pMD2.G plasmids were co-transfected into 293T cells in one well of a six-well plate. The virus-containing medium was harvested at 48 h and added to the BMDM presented with polybrene (1 μg/mL). After incubation for 12 h, the virus-containing medium was removed. The cells were replaced in a fresh medium, cultured overnight, and then the treatment procedure listed in Fig. 4b was followed in both the experimental and control groups.
Statistical analysis
For results regarding the qPCR, Western blot, and ChIP assay, the presentation in each figure is a representative result of two or three independent experiments. Statistics were calculated using SPSS software (SPSS Inc., Chicago, IL) or GraphPad Prism software (GraphPad, San Diego, CA). An unpaired t-test was used unless there were multiple comparisons, in which case we used one-way ANOVA with a post hoc Bonferroni test (with a significance level of P < 0.05). The results are shown as mean ± SD.
Results
Vagotomy reduces lung anti-inflammatory responses during an influenza infection
We collected lung and spleen cells from saline-treated sham and vagotomized mice and PR8-infected sham and vagotomized mice to detect IL-4 (an inducer of M2 MΦ activation for anti-inflammatory responses) expression by flow cytometry 6 dpi. We found that the IL-4 expression in PR8-infected sham lungs and spleens was increased as compared to that in the saline-treated mice; however, the expression was reduced in the lungs and spleens from PR8-infected vagotomized mice as compared to PR8-infected sham ones (Fig. 1a–d). Considering that ChAT (choline acetyltransferase) expressing CD4+ T [3] and α7nAChR-expressing CD11b+ cells [10] might contribute to the development of lung inflammation during influenza infection, we also analyzed these cells. Lung CD4+CHAT+ and α7nAChR+CD11b+ cells were increased in PR8-infected sham mice but did not differ between PR8-infected sham and PR8-infected vagotomized mice (Supplementary Fig. 1a, b). Blood Ly6hiCD206+ (CD206, another M2 MΦ marker) cells were increased in PR8-infected sham mice; however, these cells were reduced in PR8-infected vagotomized mice as compared to PR8-infected sham mice (Fig. 1e, f). Western blotting demonstrated that lung ARG1 (an important anti-inflammatory factor and M2 MΦ activation) expression was increased in PR8-infected mice as compared to saline-treated sham mice, but was reduced in PR8-infected vagotomized mice when compared with PR8-infected sham mice (Fig. 1g, h). These findings suggested that vagal circuits facilitate lung anti-inflammatory responses during an influenza infection.
Fig. 1. Vagotomy reduces IL-4 production in influenza-infected lung and spleen, blood Ly6ChiCD206+ monocytes, and lung ARG1 expression.

Saline-treated sham and vagotomized (VX), PR8-challenged sham and VX mice (i.n. 1.4 × 104 FFU/mouse) were sacrificed 6 dpi. Lung and spleen cells were collected for flow cytometry analysis of IL-4 expressing cells. a, b Analysis of lung IL-4+ cells by flow cytometry. c, d Analysis of spleen IL-4+ cells by flow cytometry. Data were pooled from two independent experiments. n = 6–11 in each group, *P < 0.05, ***P < 0.001, between the indicated groups, one-way ANOVA. e, f Flow cytometry analysis of blood Ly6ChiCD206+ monocytes 6 dpi. Blood cells were isolated from saline-treated sham and VX, PR8-challenged sham and VX mice (i.n. 1.4 × 104 FFU/mouse) sacrificed 6 dpi. *P < 0.05, between the indicated groups, one-way ANOVA. g, h Western blot analysis of ARG1 in lung cell lysates in saline-treated sham, PR8-challenged sham, and PR8-infected VX mice (i.n. 1.4 ×104 FFU/mouse) were sacrificed 6 dpi. Each lane represents one individual mouse. n = 3–4 in each group. *P < 0.05.
Vagotomy worsens lung inflammatory responses during an influenza infection
We intranasally challenged sham and vagotomized mice with either saline or PR8 and followed the body weight loss in these saline or PR8-challenged sham or vagotomized mice. The body weight loss during 6 days post PR8 infection was greater in PR8-infected vagotomized mice than PR8-infected sham mice (Fig. 2a). Using the same mode, BAL was collected at 6 dpi. It was found that BAL protein levels and leukocyte counts were higher in PR8-infected vagotomized mice than PR8-infected sham mice at 6 dpi (Fig. 2b, c). Lungs were collected at 6 dpi to extract RNA for qPCR. The lung Cxcl2 and Il6 expressions were higher in PR8-infected vagotomized mice than PR8-infected sham mice (Fig. 2d, e). Lung cells were isolated for flow cytometry. Considering CCR2+ inflammatory cells can be recruited into the PR8-infected lungs in response to CXCL2, we analyzed CCR2+CD11b+ cells. We found that lung CCR2+CD11b+ cell levels were increased in PR8-infected vagotomized mice as compared to PR8-infected sham mice (Fig. 2f, g). Lung inflammatory MΦ (B220−CD11c+CD11b+) were increased in PR8-infected vagotomized mice as compared to PR8-infected sham mice at 6 dpi (Fig. 2h, i). These findings support the hypothesis that vagal circuits suppress lung inflammatory responses during an influenza infection.
Fig. 2. Disruption of vagal circuits worsens influenza-induced lung inflammation.

a The change of body weight loss in saline-treated sham and VX, PR8-challenged sham and VX mice. The mice received PR8 (i.n. 1.4 × 104 FFU/mouse) and body weight changes were followed up for 6 days. *P < 0.05, between PR8-challenged sham and PR8-challenged-VX mice, repeated measures, two-way ANOVA. b, c Saline-treated sham and VX, PR8-challenged sham and VX mice (i.n. 1.4 × 104 FFU/mouse) were sacrificed 6 dpi. BAL cells were collected to measure protein concentration and leukocyte numbers. n = 4–5 in each group, *P < 0.05, **P < 0.01, between the indicated groups, one-way ANOVA. d, e Lungs were collected from the above groups and homogenized to extract RNA and detect Cxcl2 and Il6 expression. n = 4–5 in each group, *P < 0.05, between the indicated groups, one-way ANOVA. f–g Lung CCR2+CD11b+ cells analyzed by flow cytometry 6 dpi n = 3–5 in each group, *P < 0.05, between the indicated groups, one-way ANOVA. h–i Lung cells were collected for flow cytometry to analyze lung B220−CD11C+LCD11b+ inflammatory MΦ 6 dpi. n = 3–5 in each group, *P < 0.05, between the indicated groups, one-way ANOVA.
Deletion of Chrna7 reduces lung anti-inflammatory responses and survival in PR8-infected mice
We intranasally challenged Chrna7+/+ and Chrna7−/− mice with either saline or PR8 (1.4 × 104 FFU/mouse). We removed the lungs to measure Chrna7 and other anti-inflammatory (Arg1, Il10, and Socs3) genes by qPCR at 6 dpi. We found that lung Chrna7, Arg1, Il10, and Socs3 expressions were upregulated during the influenza infection; but these genes were reduced in PR8-infected Chrna7−/− mice as compared to PR8-infected Chrna7+/+ mice at 6 dpi (Fig. 3a–d). We isolated BAL cells for flow cytometry and found that BAL Ly6ChiCD206+ cells were increased in PR8-infected Chrna7+/+ mice; however, these cells were reduced in PR8-infected Chrna7−/− mice as compared to PR8-infected Chrna7+/+ mice at 6 dpi (Fig. 3e, f). These findings suggested that α7nAChR signaling favored M2 MΦ activation during an influenza infection. We intranasally challenged Chrna7+/+ recipients receiving Chrna7+/+ donor bone marrow and Chrna7+/+ recipients receiving Chrna7−/− donor bone marrow with PR8 (1.4 × 104 FFU/mouse). The mice were observed for 3 weeks and we found that PR8-infected Chrna7+/+ recipients receiving Chrna7−/− donor bone marrow had a reduced survival as compared to PR8-infected Chrna7+/+ recipients receiving Chrna7+/+ donor bone marrow (Fig. 3g), further supporting that α7nAChR expressed by bone marrow cells facilitates anti-inflammatory responses during an influenza infection.
Fig. 3. Deletion of Chrna7 in bone marrow cells reduces lung anti-inflammatory responses.

a–d Saline-treated Chrna7+/+, PR8-challenged Chrna7+/+ and Chrna7−/− mice (i.n. 1.4 × 104 FFU/mouse) were sacrificed 6 dpi. The lungs were removed and homogenized for extracting RNA to detect Chrna7 (a), Arg1 (b), Il10 (c), and Socs3 (d) by qPCR. n = 3–8 in each group, *P < 0.05, **P < 0.01, between the indicated groups, one-way ANOVA. e–f Flow cytometry analysis of BAL Ly6ChiCD206+ cells. As stated in (a–d), BAL was performed, and cells were isolated for fluorescent antibodies staining. n = 3–5 in each group, *P < 0.05, **P < 0.01, between the indicated groups, one-way ANOVA. g Effect of deletion of Chrna7 in bone marrow cells on survival in influenza-induced lung infection. Chrna7+/+ recipient mice were separately reconstituted with either Chrna7+/+ or Chrna7−/− donor bone marrow. The two groups of mice were challenged with PR8 (i.n. 1.4 × 104 FFU/mouse) and followed up for 21 days. The survival was analyzed by log rank test. n = 10 in each group, *P < 0.05.
Activation of α7nAChR increases IL-4-induced Arg1 expression but reduces Nos2 and TNF-α expression in PR8-infected BMDM
We developed BMDM using L929 cell-secreted M-CSF stimulation in the isolated bone marrow cells. By the flow cytometric analysis, we confirmed that BMDM were 96.6% F4/80+ cells (Fig. 4a). To test whether the activation of α7nAChR could regulate M2/M1 MΦ activation genes Arg1/Nos2 in PR8-infected MΦ, we pretreated the BMDM with eight different treating strategies (Fig. 4b). At 24 h, Arg1 expression was increased, however, Nos2 expression was reduced in the GTS-21 (α7nAChR agonist) treated PR8-infected IL-4-induced BMDM as compared to the PBS-treated PR8-infected IL-4-stimulated BMDM (Fig. 4c, d). TNF-α in the culture media, as an M1 phenotype signature molecule, was found to be reduced in the GTS-21 treated PR8-infected IL-4-induced BMDM as compared to the PBS-treated PR8-infected IL-4-stimulated BMDM (Fig. 4e). By Western blotting, we found that GTS-21 did not affect p-STAT6 levels in PR8-infected IL-4-stimulated BMDM, but significantly increased ARG1 expression at the protein level as compared to PBS-treated PR8-infected IL-4-stimulated BMDM (Fig. 4f). The other M2 MΦ activation markers, Fizz and Ym1, were also found to be increased in the GTS-21 treated PR8-infected IL-4-induced BMDM as compared to the other group (Fig. 4g). These findings suggest that activation of α7nAChR enhances M2 and suppresses M1 MΦ activation in PR8-infected IL-4-induced BMDM.
Activation of α7nAChR promotes IL-4-induced ARG1 expression in PR8-infected BMDM by reducing AKT1 phosphorylation and enhancing binding of STAT6 with the ARG1 promoter
To test the specificity of GTS-21 on α7nAChR and the dependent role of α7nAChR in mediating ARG1 expression in MΦ, we treated the Chrna7+/+ and Chrna7−/− BMDM with either PBS or GTS-21, followed with by PR8-challenge and IL-4 stimulation as stated in Fig. 4b. We found that ARG1 expression in the GTS-21-treated (25 μM) PR8-infected IL-4-stimulated-Chrna7+/+ BMDM was increased as compared to the PBS-treated PR8-infected IL-4-stimulated-Chrna7+/+ BMDM. The ARG1 expression in the GTS-21-treated (25 μM) PR8-infected IL-4-stimulated-Chrna7−/− BMDM was lower than the ARG1 expression in the GTS-21-treated (25 μM) PR8-infected IL-4-stimulated-Chrna7+/+ BMDM (Fig. 5a), suggesting that GTS-21 could specifically act on α7nAChR. To test whether the activation of α7nAChR would affect p-AKT1, p-STAT6, and ARG1 levels in PR8-infected IL-4-stimulated BMDM in a dose-dependent way, we pretreated the BMDM with different doses of GTS-21 followed by PR8 challenge and IL-4 stimulation. The Western blot demonstrated that the activation of α7nAChR dose-dependently increased ARG1, but reduced phosphorylation of AKT1 in the PR8-infected IL-4-stimulated BMDM. The p-STAT6 expression did not change with the GTS-21 pretreatment (Fig. 5b). GTS-21 at 50 μM did not further increase the expression of ARG1 in PR8-infected IL-4-stimulated BMDM (Fig. 5b). We replaced PR8 with Poly I:C to observe the dose-dependent effects of GTS-21 on p-AKT1, p-STAT6, and ARG1 expression in IL-4-stimulated BMDM. We found that GTS-21 at 25 μM also reduced p-AKT1, increased ARG1, but did not change p-STAT6 in Poly I:C-challenged IL-4-stimulated BMDM as compared to PBS-treated Poly I:C-challenged IL-4-stimulated BMDM (Fig. 5c). GTS-21 at 50 μM did not further increase the expression of ARG1 in Poly I:C-infected IL-4-stimulated BMDM (Fig. 5c). To test the dose-dependent effects of IL-4 in triggering the expression of ARG1 in the GTS-21-treated PR8-infected BMDM, we used IL-4 (at concentration of 10, 25, and 50 ng/mL) to stimulate the GTS-21-treated PR8-infected BMDM. We found that IL-4 at a concentration of 10 ng/mL was the optimal concentration for inducing higher levels of ARG1 expression in the GTS-21-treated PR8-infected BMDM (Fig. 5d). IL-4 stimulation increased p-STAT6 expression, but this change was not in an IL-4-dose-dependent manner (Fig. 5d). To examine whether the activation of α7nAChR would increase STAT6 binding with the ARG1 promoter, we approached the ChIP assay and found that Arg1 relative expression (% input) was increased in the anti-STAT6 antibody (Ab) immunoprecipitated group as compared to the isotype Ab immunoprecipitated group in GTS-21-pretreated PR8-infected BMDM at 7 h (Fig. 5e), indicating that α7nAChR activation could enhance STAT6 binding with the ARG1 promoter in PR8-infected BMDM.
Fig. 5. Activation of α7nAChR reduces AKT1 phosphorylation and increases p-STAT6 binding with ARG1 promoter.

a Changes of ARG1, and p-STAT6 in GTS-21-PR8-IL-4 treated Chrna7+/+ or Chrna7−/− BMDM. The Chrna7+/+ or Chrna7−/− BMDM were treated according to the procedures listed in Fig. 4b. Two concentrations of GT-21 (25 and 50 μM) were used. b Using Western blot to detect dose-dependent effects of GTS-21 on ARG1, p-STAT6, and p-AKT1 in the GTS-21-PR8-IL-4 treated BMDM following procedure of Fig. 4b. GTS-21 concentration was chosen at 0, 10, 25, and 50 μM. c Using Western blot to detect dose-dependent effects of GTS-21 on ARG1, p-STAT6, and p-AKT1 in the GTS-21-Poly I:C-IL-4 treated BMDM following procedure of Fig. 4b. GTS-21 concentration was chosen at 0, 10, 25, and 50 μM. The Poly I:C concentration was 100 μg/mL. d Dose-dependent effect of IL-4 on p-STAT6 and ARG1 detected by Western blot. Experiments followed the procedures listed in Fig. 4b, but IL-4 concentration was chosen at 0, 10, 20, and 50 ng/mL. Data presented in Fig. 2a–d were repeated at least in two independent experiments. e Detecting STAT6 binding with Arg1 promoter by ChIP assay. The BMDM were treated GTS-21 (25 μM) for 30 min followed by PR8 (2 MOI) challenge for 6 h. The control groups received PBS. The data were pooled from three independent experiments, “-” condition indicated PBS treatment, **P < 0.01 in the indicated groups, unpaired t test.
Activation of α7nAChR promotes IL-4-induced ARG1 expression in PR8-infected BMDM by regulating JMJD3-H3K27me3 methylation
Since H3K27me3 is a transcription repressor, we compared H3K27me3 expressions using strategies listed in Fig. 4b. By flow cytometric analysis, we found that H3K27me3 expression was lower in the GTS-21-treated PR8-infected IL-4 stimulated BMDM when compared with the PBS-treated PR8-infected IL-4 stimulated BMDM. The GTS-21 treatment also reduced H3K27me3 expression in PR8-infected BMDM as compared to PBS-treated PR8-infected BMDM (Fig. 6a). These findings suggested that the activation of α7nAChR mitigated the suppressive effects of H3K27me3 on transcription. Considering JMJD3 (coded by Kdm6b) is a key regulator of H3K27me3, we measured Kdm6b expression and found that Kdm6b was increased in the GTS-21-treated PR8-infected IL-4 stimulated BMDM when compared with the PBS-treated PR8-infected IL-4 stimulated BMDM (Fig. 6b). By Western blotting, we confirmed that the expression of p-AKT1 was reduced, JMJD3 was increased, and H3K27me3 was decreased in the GTS-21-treated PR8-infected IL-4 stimulated BMDM when compared with the PBS-treated PR8-infected IL-4 stimulated BMDM (Fig. 6c). We also found that the GTS-21 pretreatment particularly reduced H3K27me3 expression when BMDM were stimulated with an IL-4 no matter challenge with or without PR8 (Fig. 6d). The above results suggested that the activation of α7nAChR might reduce the phosphorylation of AKT1 and increased JMJD3 expression under IL-4 stimulation which modifies H3K27me3 methylation. To test whether JMJD3 would regulate ARG1 expression, we pretreated the GTS-21-treated PR8-infected IL-4 stimulated BMDM with JMJD3 inhibitor-GSK-J1 and found that the inhibition of JMJD3 reduced the ARG1 expression in the GTS-21-treated PR8-infected IL-4 stimulated BMDM as compared to the vehicle-pretreated GTS-21-treated PR8-infected IL-4 stimulated BMDM (Fig. 6e). We further studied whether the deletion of Akt1 would affect JMJD3 and ARG1 expressions and H3K27me3 methylation in GTS-21-treated PR8-infected IL-4 stimulated BMDM. By Western blotting, we found that JMJD3 expression was reduced, H3K27me3 methylation was elevated, and consequently ARG1 expression was reduced in the GTS-21-treated PR8-infected IL-4 stimulated LysMCre+Akt1fl/fl BMDM as compared to the GTS-21-treated PR8-infected IL-4 stimulated Akt1fl/fl BMDM (Fig. 6f). These findings indicated that the phosphorylation of AKT1 and AKT1 itself have deferential roles in regulating ARG1 expression in GTS-21-treated PR8-infected IL-4 stimulated BMDM.
Fig. 6. Activation of α7nAChR increases IL-4-induced ARG1 expression depending on JMJD3-H3K27me3 methylation signaling.

a Flow cytometric analysis of H3K27me3 expression in GTS-21-PR8-IL4-treated BMDM following the procedure in Fig. 4b. The percentage of positive cells was listed. The results were repeated in two independent experiments, *P < 0.05 in the indicated groups, unpaired t test. b qPCR analysis of Jmjd3 in the GTS-21-PR8-IL4 treated BMDM. Cells were treated with GTS-21 and PR8 according to Fig. 4b procedure except IL-4 treatment for 1 h. The RNA was extracted for qPCR. **P < 0.01, One-way ANOWA. c Using the procedure as listed in Fig. 4b, the cells collected for Western blot for detecting p-STAT6, p-AKT1, ARG1, JMJD3 and H3K27me3. The histone 3 was used as a control. d Effect of activation of α7nAChR on H3K27me3 in PR8, IL-4, or PR8 + IL-4-stimulated MΦ. “-” condition indicated PBS treatment. e Effect of GSK-J1 on ARG1 expression in GTS-21-PR8-IL-4-treated BMDM. The BMDM were pretreated with either vehicle or GSK-J1 (30 nM) for 15 min, then went through the protocol listed in Fig. 4b. “-” condition indicated PBS treatment. f Effect of deletion of Akt1 in myeloid cells on expression of JMJD3, H3K27me3, and ARG1 in GTS-21-PR8-IL-4-treated BMDM. The results in Fig. 6c–f were repeated in at least two independent experiments followed the experimental setting in Fig. 4b.
Activation of α7nAChR promotes IL-4-induced ARG1 expression in PR8-infected BMDM via suppressing phosphorylation of AKT1-FOXO1 signaling
Considering that the phosphorylation of FOXO1 by AKT1 could lead to exclusion of FOXO1 from the nucleus to cytoplasm [22], we first studied the corresponding changes of p-FOXO1, p-AKT1, and ARG1 expression in cell lysates by Western blotting. We found that p-FOXO1 and p-AKT1 were reduced, while in contrast, ARG1 was increased in the GTS-21 treated PR8-infected IL-4-stimulated BMDM as compared to the PBS-treated PR8-infected IL-4-stimulated BMDM (Fig. 7a). We then isolated the nucleus portion in the cultured cells to measure the ratio of FOXO1 to p-FOXO1. We found that ratio of FOXO1/p-FOXO1 in the nucleus was markedly increased in the GTS-21 treated PR8-infected IL-4-stimulated BMDM as compared to the PBS-treated PR8-infected IL-4-stimulated BMDM (Fig. 7b). Considering that FOXO1 is a positive regulator of M2 MΦ activation genes (Arg1, Fizz1, and IL-13ra1) [18], we built scrambled shRNA and Foxo1a shRNA constructs in Lentivirus. After the BMDM was infected with scrambled shRNA and Foxo1a shRNA Lentivirus constructs, we found that the knockdown of Foxo1a (Fig. 7c) markedly reduced Arg1 expression in the GTS-21 treated PR8-infected IL-4-stimulated BMDM as compared to the scrambled shRNA infected GTS-21 treated PR8-infected IL-4-stimulated BMDM (Fig. 7d). To test whether the deletion of Akt1 from BMDM would affect p-FOXO1 and ARG1 expression, we compared GTS-21-treated PR8-infected IL-4-stimulated Akt1fl/fl BMDM and LysMCre+Akt1fl/fl BMDM. We found that p-FOXO1 and ARG1 expression was reduced in the GTS-21 treated PR8-infected IL-4-stimulated LysMCre+Akt1fl/fl BMDM as compared to the GTS-21 treated PR8-infected IL-4-stimulated Akt1fl/fl BMDM (Fig. 7e). These findings also support the hypothesis that AKT1 existence is required for ARG1 expression in MΦ, and its role is different from the phosphorylation of AKT1.
Fig. 7. Activation of α7nAChR augments IL-4-induced ARG1 expression relying on AKT1-FOXO1 signaling.

a Effect of GTS-21 on FOXO1/p-FOXO1, AKT1/p-AKT1, and ARG1 in IL-4 stimulated PR8-infected BMDM. The BMDM were treated with strategies listed in Fig. 4b. Experiments were repeated two times. b Analysis of nuclear p-FOXO1/FOXO1 ratio in GTS-21 treated IL-4-stimulated PR8-infected BMDM. The BMDM were treated using treating conditions in the Fig. 4b except IL-4 stimulation was given for 1 h instead of 18 h. The results were repeated two times. c, d The efficiency and effect of knockdown of Foxo1a on Arg1 expression in GTS-21 treated IL-4-stimulated PR8-infected BMDM. The BMDM were first given Scrambled ShRNA and Foxo1a ShRNA treatment and then received treatment using strategies listed in Fig. 4b. Data were pooled from three experiments. *P < 0.05, **P < 0.01, unpaired t test or One-way ANOVA. e Effect of deletion of Akt1 in myeloid cells on p-STAT6, p-AKT1, p-FOXO1, and ARG1 in α7nAChR-activated IL-4-stimulated PR8-infected BMDM. The Akt1fl/fl and LysMCre Akt1fl/fl bone marrow cells were isolated and BMDM cells were developed. The cells were treated following the procedure listed in Fig. 4b. The results were repeated at least two times.
In summary, vagal circuit-α7nAChR signaling boosts lung anti-inflammatory responses and increases ARG1 expression during an influenza infection. In PR8-infected IL-4-stimulated MΦ, activation of α7nAChR could augment IL-4-induced ARG1 expression via STAT6, JMJD3-H3K27me3, and AKT1-FOXO1 signaling (Fig. 8).
Fig. 8. Hypothetic model: activation of α7nAChR boosts ARG1 expression in the influenza-infected macrophages.
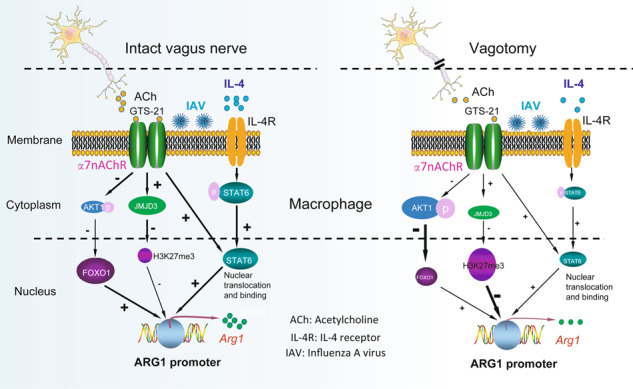
When vagus nerve is intact, acetylcholine release is normal. Influenza infection can increase lung α7nAChR, CHAT, and IL-4 expression, which facilitates the development of anti-inflammatory responses. Activation of α7nAChR could increase STAT6 binding with ARG1 promoter. In PR8-infected IL-4-stimulated MΦ, activation of α7nAChR can reduce phosphorylation of AKT1 and exclusion of FOXO1 from nucleus, and increase JMJD3 expression by which suppressing H3K27me3 methylation. These changes augment ARG1 expression in M2 macrophages. In contrast, vagotomy compromises acetylcholine release and expression of IL-4 during influenza infection. The reduction of activation of α7nAChR and IL-4 mitigates STAT6 binding with ARG1 promoter, promotes exclusion of FOXO1 from nucleus, and increases H3K27me3 methylation. These changes impair lung anti-inflammatory responses and ARG1 expression.
Discussion
In this study, we have found that the disruption of vagal circuits could reduce lung and spleen IL-4 expression during an influenza infection. ARG1, an M2 MΦ activation marker, was reduced in PR8-infected vagotomized mice. BAL protein and leukocytes and lung M1 MΦ activation genes (Cxcl2 and Il6) were increased in PR8-infected vagotomized mice. In PR8-α7nAChR-deficient mice, lung M2 MΦ activation genes: Arg1, Il10, and Socs3 and BAL Ly6C+CD206+ cells were reduced. The PR8-infected bone marrow α7nAChR−/− mice had a lower survival. In the in vitro study, activation of α7nAChR could enhance IL-4-induced Arg1 expression and reduce Nos2 and TNF-α expression in PR8-infected MΦ. Mechanistically, activation of α7nAChR boosted IL-4 induced ARG1 expression in the PR8-infected MΦ via STAT6, JMJD3-H3K27me3, and AKT1-FOXO1 signaling.
The pulmonary parasympathetic inflammatory reflex has afferent and efferent arcs, which could modulate lung infection and immunity [9, 10, 29, 30] as the distal airways and alveoli are innervated by vagus nerve endings [7]. A study has shown that peritoneal ACh levels were reduced at days 1, 3, and 7 post vagotomy and that this change altered resting peritoneal lipid mediator profiles and increased peritoneal leukocyte recruitment [31]. Previously, we measured AChE activity in alveolar proinflammatory cells and BAL choline concentrations in E. coli pneumonia and found that the AChE activity and choline levels in the BAL cells were increased [12]. The ACh concentration, however, did not increase in the BAL in E. coli pneumonia, which is consistent with the rapid hydrolysis of ACh (half-life, 2 min) occurring after ACh release from nerve endings [32]. During an influenza infection, the ChAT-expressing immune cells, especially the CD4+ T cells, might synthesize acetylcholine [3, 33]. We found that CD4+ChAT+ cells were increased in PR8-infected sham mice, but did not differ between PR8-infected sham and vagotomized mice. This finding indicated that CD4+CHAT+ cells might contribute to ACh production in PR8-infected sham mice’s lungs. Thus, we speculate that the disruption of vagal circuits might reduce lung ACh, which impairs the activation of α7nAChR expressed by the adjacent MΦ. As reported, the resident MΦ and recruited monocytic cells expressed α7nAChR in the lung [10–12]. The α7nAChR+CD11b+ cells were increased in PR8-infected lungs but did not differ between PR8-infected sham and vagotomized mouse lungs. The finding also suggested that the reduced activation of α7nAChR resulting from vagotomy might compromise M2 MΦ activation. More importantly, lung IL-4 expression was significantly reduced in the PR8-infected vagotomized lungs versus in the PR8-infected sham lungs. This change might majorly account for the reduced M2 MΦ activation under vagotomy conditions.
M2 MΦ activation can express a variety of signature genes, such as Arg1, Fizz, Ym1, and Il10. In this study, we aimed to study whether and how the activation of α7nAChR would affect M2 MΦ activation. We knew that it is impossible to examine all the signature gene changes; we simply selected ARG1 as our target. It is reported that IL-4 stimulation could alter arginine metabolism by inducing ARG1 expression and inhibiting nitric oxide production in MΦ. The induction of Arg1 transcription by IL-4 stimulation requires a composite DNA response element for STAT6 [34]. In our study, the ChIP assay demonstrated that the activation of α7nAChR by GTS-21 could increase STAT6 binding with the ARG1 promoter in PR8-infected BMDM, which provides a molecular basis for synergizing with IL-4 stimulation to boost M2 activation in MΦ under an influenza challenge.
The PI3K/AKT pathway is an important signaling pathway that regulates inflammation. PI3K plays a key role in phosphorylating AKT1, and the latter can then affect alternative MΦ activation [35, 36]. In LPS-stimulated MΦ, AKT1 ablation gives rise to an M1 and AKT2 ablation results in an M2 phenotype [37]. In this study, we found that the activation of α7nAChR reduced p-AKT1S473 and upregulated the IL-4-induced ARG1 in PR8-stimulated MΦ. However, in PR8-infected LysMCre+Akt1fl/fl BMDM, GTS-21 pretreated IL-4 induced ARG1 expression was suppressed. Therefore, we speculate that AKT1 itself and phosphorylation of AKT1S473 might regulate ARG1 expression in IL-4 induced PR8-infected BMDM differently.
In the GTS-21-treated PR8-infected IL-4-induced BMDM, the activation of α7nAChR reduces p-AKT1. Lower p-AKT1 coincided with higher JMJD3 expression at both mRNA and protein levels. Meanwhile, the GTS-21-treated PR8-infected IL-4-induced BMDM had a reduced H3K27me3 expression as compared to the PBS-treated PR8-infected IL-4-stimulated BMDM. It is reported that IL-4 treatment of human monocytes could increase the transcription of Kdm6b [38]. Thus, the reduction of p-AKT1 and IL-4 stimulation might cooperatively boost Kdm6b expression in the GTS-21-treated PR8-infected IL-4-induced BMDM. Furthermore, the activation of α7nAChR could reduce p-AKT1 and p-FOXO1, which retains FOXO1 in the nucleus to enhance IL-4-induced Arg1 transcription [18, 39]. The knockdown of Foxo1a could abolish the upregulated Arg1 transcription in GTS-21-treated PR8-infected IL-4-induced BMDM, suggesting that FOXO1 is a key regulator in boosting Arg1 transcription in GTS-21-treated PR8-infected IL-4-induced BMDM. There is an evidence showing that FOXO1 overexpression promotes H3 acetylation and phosphorylation, and reduces H3 methylation [40]. Therefore, α7nAChR boosting Arg1 transcription in PR8-infected IL-4-induced BMDM might allow for synergism between JMJD3-H3K27me3 and FOXO1 signaling.
Different from the effect of the reduction of phosphorylation of AKT1 on ARG1 transcription, the knockout of AKT1 reduces Arg1 transcription in GTS-21-treated PR8-infected IL-4-induced BMDM by suppressing JMJD3 expression and increasing H3K27me3 expression. Thus, we consider that AKT1 itself is a positive regulator of ARG1 expression in monocytic cells during influenza-induced lung inflammation. It needs to be noted that we cannot exclude the other AKT isoforms play a role in mediating Arg1 expression, for example, AKT2 could also regulate pulmonary inflammation and fibrosis via modulating macrophage activation [41].
A study has demonstrated that GTS-21 has cell-specific anti-inflammatory effects, which are independent of α7nAChR [42]. In the present study, GTS-21 could not boost ARG1 expression in PR8-infected IL-4-induced BMDM from Chrna7−/− macrophages. Our result indicates that GTS-21 specifically acts on α7nAChR. To exclude the role of α7nAChR in the other type of cells, we approached the bone marrow transplantation mouse model to figure out the role of α7nAChR in bone marrow cells in regulating influenza-induced lung inflammation. As shown in Fig. 3g, we have demonstrated that wild-type mice receiving Chrna7−/− bone marrow-derived cells had higher mortality, supporting the hypothesis that activation of α7nAChR in bone marrow cells might favor M2 MΦ activation and dampen M1 type proinflammatory responses in lung inflammation due to an influenza infection. One study has shown that α7nAChR knockout mice had more body weight loss than wild-type mice after cigarette smoke exposure and H9N2 infection [13], which supports our findings from an inflammatory point of view.
Meanwhile, we have extensively studied whether activation of α7nAChR would affect virus replication in the influenza-infected lungs. We found that activation of α7nAChR could promote influenza replication in the lung epithelial cells. Mice with vagotomy or knockout of α7nAChR had lower viral titers in influenza-infected lungs. We have also elucidated the possible mechanisms by which activation of α7nAChR promotes influenza replication in lung epithelial cells (unpublished data). The findings in Fig. 3g supported that α7nAChR expressed by bone marrow-derived cells (rather than lung epithelial cells) contributes to reduction of influenza lung inflammation.
Many experiments have demonstrated stimulation of vagus nerve could suppress inflammatory responses via activation of α7nAChR in macrophages [3, 5, 30]. Activation of α7nAChR in macrophages inhibits LPS or Poly I:C induced NF-κB activation or triggers similar anti-inflammatory responses. It is reported that in LPS-challenged microglia, activation of α7nAChR inhibits the transformation of M1 microglia and promotes the M2 phenotype [14]. TLR3 agonist Poly I:C also induced M2 macrophage activation [43]. In our study, we found that Poly I:C did not increase ARG1 expression; however, activation of α7nAChR could significantly increase IL-4-triggered ARG1 expression (Fig. 5c) in Poly I:C-challenged macrophages. Therefore, vagotomy or knockout of α7nAChR could mitigate LPS or Poly I:C induced M2 macrophage activation.
In summary, vagal-α7nAChR signaling is required for lung anti-inflammatory responses during an influenza infection. The activation of α7nAChR boosts IL-4-induced ARG1 expression in MΦ via STAT6, JMJD3-H3K27me3 methylation, and AKT1-FOXO1 signaling.
Supplementary information
Acknowledgements
This work is supported by NSFC programs 81730001, 91942305, and 81970075, the Strategic Leading Project (B) of CAS XDPB0303, the International Collaboration project of CAS 153831KYSB20170043, and Innovative Research Team of High-level Local Universities in Shanghai. The authors appreciate Yi-yi Jiang, Ya-qiong Cui, and Lian-ping Cheng for breeding and genotyping animals.
Author contributions
XS and JFX designed the experiments and analyzed the data; ZWG, YYH, LL, CQZ, SJX, and JC performed the experiments; ZZY provided us Akt1 flox mice; XS and JFX provided funds and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Ethical approval
The animal study was reviewed and approved by the Committees on Animal Research of the Institut Pasteur of Shanghai; Chinese Academy of Sciences approved the protocols.
Footnotes
These authors contributed equally: Zhao-wei Gao, Ling Li, Yuan-yuan Huang
Contributor Information
Jin-fu Xu, Email: jfxucn@gmail.com.
Xiao Su, Email: xsu@ips.ac.cn.
Supplementary information
The online version of this article (10.1038/s41401-020-00579-z) contains supplementary material, which is available to authorized users.
References
- 1.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J, Li F, Sun R, Gao X, Wei H, Li LJ, et al. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat Commun. 2013;4:2106. doi: 10.1038/ncomms3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–62.. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 6.Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol. 2012;97:1180–5. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- 7.Hertweck MS, Hung KS. Ultrastructural evidence for the innervation of human pulmonary alveoli. Experientia. 1980;36:112–3. doi: 10.1007/BF02004006. [DOI] [PubMed] [Google Scholar]
- 8.Livermore S, Zhou Y, Pan J, Yeger H, Nurse CA, Cutz E. Pulmonary neuroepithelial bodies are polymodal airway sensors: evidence for CO2/H+ sensing. Am J Physiol Lung Cell Mol Physiol. 2015;308:L807–15. doi: 10.1152/ajplung.00208.2014. [DOI] [PubMed] [Google Scholar]
- 9.Yang X, Zhao C, Gao Z, Su X. A novel regulator of lung inflammation and immunity: pulmonary parasympathetic inflammatory reflex. QJM. 2014;107:789–92.. doi: 10.1093/qjmed/hcu005. [DOI] [PubMed] [Google Scholar]
- 10.Zhao C, Yang X, Su EM, Huang Y, Li L, Matthay MA, et al. Signals of vagal circuits engaging with AKT1 in alpha7 nAChR(+)CD11b(+) cells lessen E. coli and LPS-induced acute inflammatory injury. Cell Discov. 2017;3:17009. doi: 10.1038/celldisc.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su X, Lee JW, Matthay ZA, Mednick G, Uchida T, Fang X, et al. Activation of the alpha7 nAChR reduces acid-induced acute lung injury in mice and rats. Am J Respir Cell Mol Biol. 2007;37:186–92.. doi: 10.1165/rcmb.2006-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su X, Matthay MA, Malik AB. Requisite role of the cholinergic alpha7 nicotinic acetylcholine receptor pathway in suppressing Gram-negative sepsis-induced acute lung inflammatory injury. J Immunol. 2010;184:401–10.. doi: 10.4049/jimmunol.0901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han Y, Ling MT, Mao H, Zheng J, Liu M, Lam KT, et al. Influenza virus-induced lung inflammation was modulated by cigarette smoke exposure in mice. PLoS One. 2014;9:e86166. doi: 10.1371/journal.pone.0086166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Lu Y, Bian H, Guo L, Zhu H. Activation of the alpha7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am J Transl Res. 2017;9:971–85. [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104–8. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinheiro NM, Santana FP, Almeida RR, Guerreiro M, Martins MA, Caperuto LC, et al. Acute lung injury is reduced by the alpha7nAChR agonist PNU-282987 through changes in the macrophage profile. FASEB J. 2017;31:320–32.. doi: 10.1096/fj.201600431r. [DOI] [PubMed] [Google Scholar]
- 17.Tateyama H, Murase Y, Higuchi H, Inasaka Y, Kaneoka H, Iijima S, et al. Siglec-F is induced by granulocyte-macrophage colony-stimulating factor and enhances interleukin-4-induced expression of arginase-1 in mouse macrophages. Immunology. 2019;158:340–52.. doi: 10.1111/imm.13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung S, Ranjan R, Lee YG, Park GY, Karpurapu M, Deng J, et al. Distinct role of FoxO1 in M-CSF- and GM-CSF-differentiated macrophages contributes LPS-mediated IL-10: implication in hyperglycemia. J Leukoc Biol. 2015;97:327–39.. doi: 10.1189/jlb.3A0514-251R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–54.. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–44.. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 21.Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017;198:1006–14.. doi: 10.4049/jimmunol.1601515. [DOI] [PubMed] [Google Scholar]
- 22.Hay N. Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta. 2011;1813:1965–70.. doi: 10.1016/j.bbamcr.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parzych EM, DiMenna LJ, Latimer BP, Small JC, Kannan S, Manson B, et al. Influenza virus specific CD8+ T cells exacerbate infection following high dose influenza challenge of aged mice. Biomed Res Int. 2013;2013:876314. doi: 10.1155/2013/876314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison AR, Yarovinsky TO, Young BD, Moraes F, Ross TD, Ceneri N, et al. Chemokine-coupled beta2 integrin-induced macrophage Rac2-myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J Exp Med. 2014;211:1957–68. doi: 10.1084/jem.20132130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trus E, Basta S, Gee K. Who’s in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: competing factors in macrophage polarization. Cytokine. 2020;127:154939. doi: 10.1016/j.cyto.2019.154939. [DOI] [PubMed] [Google Scholar]
- 26.Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Xiao N, Eto D, Elly C, Peng G, Crotty S, Liu YC. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat Immunol. 2014;15:657–66.. doi: 10.1038/ni.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y, Zhao C, Su X. Neuroimmune regulation of lung infection and inflammation. QJM. 2019;112:483–7. doi: 10.1093/qjmed/hcy154. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, Li L, Su X. Vagus nerve through alpha7 nAChR modulates lung infection and inflammation: models, cells, and signals. Biomed Res Int. 2014;2014:283525. doi: 10.1155/2014/283525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalli J, Colas RA, Arnardottir H, Serhan CN. Vagal regulation of Group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity. 2017;46:92–105. doi: 10.1016/j.immuni.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada H, Otsuka M, Fujimoto K, Kawashima K, Yoshida M. Determination of acetylcholine concentration in cerebrospinal fluid of patients with neurologic diseases. Acta Neurol Scand. 1996;93:76–8. doi: 10.1111/j.1600-0404.1996.tb00175.x. [DOI] [PubMed] [Google Scholar]
- 33.Reardon C, Duncan GS, Brustle A, Brenner D, Tusche MW, Olofsson PS, et al. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc Natl Acad Sci U S A. 2013;110:1410–5. doi: 10.1073/pnas.1221655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM., Jr Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene. 2005;353:98–106. doi: 10.1016/j.gene.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Binger KJ, Gebhardt M, Heinig M, Rintisch C, Schroeder A, Neuhofer W, et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J Clin Invest. 2015;125:4223–38.. doi: 10.1172/JCI80919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruckerl D, Jenkins SJ, Laqtom NN, Gallagher IJ, Sutherland TE, Duncan S, et al. Induction of IL-4Ralpha-dependent microRNAs identifies PI3K/Akt signaling as essential for IL-4-driven murine macrophage proliferation in vivo. Blood. 2012;120:2307–16.. doi: 10.1182/blood-2012-02-408252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A. 2012;109:9517–22.. doi: 10.1073/pnas.1119038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu AT, Lupancu TJ, Lee MC, Fleetwood AJ, Cook AD, Hamilton JA, et al. Epigenetic and transcriptional regulation of IL4-induced CCL17 production in human monocytes and murine macrophages. J Biol Chem. 2018;293:11415–23.. doi: 10.1074/jbc.RA118.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung S, Kim JY, Song MA, Park GY, Lee YG, Karpurapu M, et al. FoxO1 is a critical regulator of M2-like macrophage activation in allergic asthma. Allergy. 2019;74:535–48.. doi: 10.1111/all.13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu LW, Gu LY, Lu L, Chen XF, Li CF, Mei ZC. FOXO1-mediated epigenetic modifications are involved in the insulin-mediated repression of hepatocyte aquaporin 9 expression. Mol Med Rep. 2015;11:3064–8. doi: 10.3892/mmr.2014.3085. [DOI] [PubMed] [Google Scholar]
- 41.Nie Y, Sun L, Wu Y, Yang Y, Wang J, He H, et al. AKT2 regulates pulmonary inflammation and fibrosis via modulating macrophage activation. J Immunol. 2017;198:4470–80.. doi: 10.4049/jimmunol.1601503. [DOI] [PubMed] [Google Scholar]
- 42.Garg BK, Loring RH. GTS-21 has cell-specific anti-inflammatory effects independent of alpha7 nicotinic acetylcholine receptors. PLoS One. 2019;14:e0214942. doi: 10.1371/journal.pone.0214942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Natarajan C, Yao SY, Sriram S. TLR3 agonist poly-IC induces IL-33 and promotes myelin repair. PLoS One. 2016;11:e0152163. doi: 10.1371/journal.pone.0152163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.