

Abstract
Rheumatoid arthritis (RA) is characterized by joint leukocyte infiltration, synovial inflammation and bone damage result from osteoclastogenesis. Bruton’s tyrosine kinase (BTK) is a key regulator of B cell receptor (BCR) and Fc gamma receptor (FcγR) signaling involved in the pathobiology of RA and other autoimmune disorders. SOMCL-17-016 is a potent and selective tricyclic BTK inhibitor, structurally distinct from other known BTK inhibitors. In present study we investigated the therapeutic efficacy of SOMCL-17-016 in a mouse collagen-induced arthritis (CIA) model and underlying mechanisms. CIA mice were administered SOMCL-17-016 (6.25, 12.5, 25 mg·kg−1·d−1, ig), or ibrutinib (25 mg·kg−1·d−1, ig) or acalabrutinib (25 mg·kg−1·d−1, ig) for 15 days. We showed that oral administration of SOMCL-17-016 dose-dependently ameliorated arthritis severity and bone damage in CIA mice; it displayed a higher in vivo efficacy than ibrutinib and acalabrutinib at the corresponding dosage. We found that SOMCL-17-016 administration dose-dependently inhibited anti-IgM-induced proliferation and activation of B cells from CIA mice, and significantly decreased anti-IgM/anti-CD40-stimulated RANKL expression in memory B cells from RA patients. In RANKL/M-CSF-stimulated RAW264.7 cells, SOMCL-17-016 prevented osteoclast differentiation and abolished RANK-BTK-PLCγ2-NFATc1 signaling. In summary, this study demonstrates that SOMCL-17-016 presents distinguished therapeutic effects in the CIA model. SOMCL-17-016 exerts a dual inhibition of B cell function and osteoclastogenesis, suggesting that it to be a promising drug candidate for RA treatment.
Keywords: rheumatoid arthritis, BTK inhibitors, SOMCL-17-016, B cells, macrophages, RANKL, osteoclastogenesis
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease affecting 0.5%–1% of adults worldwide and is associated with progressive disability and socioeconomic burdens [1, 2]. It is characterized by synovial membrane inflammation and hyperplasia, the presence of particular autoantibodies and progressive cartilage and bone destruction [3]. RA is a heterogeneous disease in which different pathophysiological pathways lead to a similar clinical presentation of arthritis through a final common inflammatory pathway [4]. Although the treatment of RA has improved dramatically in the past decade, a significant proportion of RA patients do not respond effectively to the current conventional and biologic disease-modifying therapies [5–7]. Thus, new pharmacological interventions for optimizing RA treatment are urgently required.
Although the pathogenesis of RA is complicated and not completely understood, disease-specific autoantibodies, in addition to inflammation, are pivotal triggers. Recent evidence suggests that autoantibodies in RA patients potentiate bone destruction during the course of the disease in both direct and indirect ways [8]. It has been reported that activation of the Fc gamma receptor (FcγR) by cross-linked antibodies enhances osteoclastogenesis in murine bone marrow cells [9]. Furthermore, mice immunized with citrullinated protein develop greater bone loss than mice immunized with methylated bovine serum albumin (mBSA), and this effect is independent of inflammation, as the latter exhibit more severe synovitis [10]. These studies indicate that autoantibodies have a direct effect on osteoclastogenesis and bone destruction. In addition, autoantibodies and their immune complexes can induce the robust production of the cytokines TNF-α, IL-1β, IL-6 and IL-8 by macrophages via FcγR signaling, leading to chronic inflammation and joint damage. TNF-α is the strongest inducer of osteoclast differentiation and elicits this process either by activating the NF-κB/NFATc1 pathway or by upregulating RANK expression on osteoclast precursors independently or in cooperation with receptor activator of nuclear factor-κB ligand (RANKL) [11, 12].
RANKL mediates the differentiation, survival and activation of osteoclasts. Lee et al. [13] and Yuko Ariza et al. [14] proposed that RANKL induces phosphorylation of Bruton’s tyrosine kinase (BTK) and subsequent recruitment of downstream effectors, including PLCγ2, and that PLCγ2 in turn stimulates calcium signaling, which is required for activation of NFATc1, a transcription factor essential for osteoclast differentiation. Moreover, BTK and Tec double-deficient mice develop severe osteopetrosis due to cell-autonomous blockade of osteoclast differentiation [15], proving that BTK and Tec are required for osteoclastogenesis. Moreover, previous studies have demonstrated that the BTK inhibitor ibrutinib suppresses osteoclastogenesis in a murine model of collagen-induced arthritis (CIA) [16].
BTK is a member of the Tec family of kinases and is broadly expressed in cells of several hemopoietic lineages but not in T cells or natural killer (NK) cells [17, 18]. BTK is involved in multiple hematopoietic signaling pathways, including the B-cell receptor (BCR), cytokine receptor and FcγR pathways, which are related to B-cell and myeloid cell function [19]. B cell-mediated pathology in RA includes the production of RA-associated autoantibodies and the generation of RANKL in the inflamed synovium by B cells [20]. Autoantibodies react with autoantigens within the joint, leading to IgG immune complex formation and subsequent inflammatory cell recruitment. Furthermore, IgG immune complexes activate FcγR, which is mainly expressed on myeloid lineage cells, triggering inflammatory cytokine production by myeloid lineage cells, particularly macrophages [21].
The present study describes the effects of SOMCL-17-016, a highly potent and unique tricyclic inhibitor of BTK that represents a new small-molecule BTK inhibitor chemotype [22–26]. We demonstrated that SOMCL-17-016 inhibited B cell activation and the production of pathogenic autoantibodies against type II collagen. SOMCL-17-016 ameliorated arthritic manifestations and bone damage in the CIA model and decreased the levels of inflammatory cytokines in the joints and serum. Importantly, we showed that SOMCL-17-016 impeded the synovium accumulation of RANKL+ B cells and retarded osteoclast differentiation via the interaction between BTK-mediated BCR signaling and RANKL-RANK signaling. Collectively, our research highlighted a dual mechanism of action of SOMCL-17-016 as a BTK inhibitor in RA that includes suppression of pathogenic B cell responses upon BCR stimulation and inhibition of osteoclast-mediated bone loss.
Materials and methods
Mice
Male DBA/1J mice were obtained from Beijing Charles River Laboratories (Certificate No SCXK 2016-0011, Beijing, China). The mice were housed under specific pathogen-free conditions and kept on a 12 h light/dark cycle at a controlled humidity (55% ± 5%) and temperature (22 ± 1 °C). All experiments were performed according to the institutional ethical guidelines on animal care and approved by the Institute Animal Care and Use Committee at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences (IACUC protocol # 2018-06-ZJP-82). All mice had free access to standard laboratory water and food and were allowed to acclimatize to our facility for 1 week before the initiation of any experiments.
Experimental design
Male DBA/1J mice were immunized with 100 μg bovine type II collagen (CII, Tokyo, Japan) in 0.1 M acetic acid emulsified in an equal volume of complete Freund’s adjuvant (CFA) containing Mycobacterium tuberculosis strain H37Rv (Wako Pure Chemical Industries Ltd., Osaka, Japan) at the base of the tail on day 0. A boost injection of 100 μg collagen-incomplete Freund’s adjuvant (IFA) emulsion was given in the same manner 21 days later. Ten days after booster immunization, the immunized mice were randomly divided into the following six groups according to clinical scores: the vehicle (ddH2O)-treated group (n = 9); the 25 mg·kg−1·d−1 ibrutinib (PCI-32765)-treated group (n = 8); the 25 mg·kg−1·d−1 acalabrutinib (ACP-196)-treated group (n = 8); the 25 mg·kg−1·d−1 SOMCL-17-016-treated group (n = 9); the 12.5 mg·kg−1·d−1 SOMCL-17-016-treated group (n = 8); and the 6.25 mg·kg−1·d−1 SOMCL-17-016-treated group (n = 8). Both ibrutinib and acalabrutinib were purchased from Selleck Company. SOMCL-17-016 was obtained through a medicinal chemistry campaign featuring a novel tricyclic pyrimido [5,4-b] pyrrolizine skeleton. Mice were treated intragastrically for 15 days. An additional 8 DBA/1J mice were used as normal controls. Clinical scores were determined for each paw of each mouse daily. Clinical score assessment was performed as previously described [27]. Briefly, the mice were assigned arthritis scores on a scale of 0–4 based on the degree and extent of paw swelling according to the following scale: 0 = normal; 1 = detectable arthritis with erythema of one or several digits; 2 = erythema and moderate swelling extending from the ankle to the midfoot; 3 = severe swelling and redness from the joint to digit; and 4 = maximal swelling with ankyloses. The clinical arthritis score was the cumulative score of the four limbs (the maximum score for each mouse was 16).
Histological examination and TRAP staining
The hind limbs of each mouse were fixed in 10% phosphate-buffered saline (PBS)-buffered formalin at room temperature, decalcified in 5% formic acid and embedded in paraffin. Each limb was sectioned at a thickness of 5 μm, and the sections were stained with hematoxylin and eosin (H&E). Histopathological features were examined microscopically (magnification of ×100). Pannus formation, cellular infiltration, synovial proliferation, and cartilage erosion were scored in a blinded manner on a severity scale ranging from 1 to 3 (0: absent; 1: weak; 2: moderate; and 3: severe) [28, 29].
For TRAP staining, each limb section was treated with an acid phosphatase leucocyte kit (Sigma-Aldrich, St. Louis, MO, USA). TRAP-positive multinucleated cells containing three or more nuclei in a field of the whole section (magnification of ×100) were counted.
Immunohistochemistry and immunofluorescence
Formalin-fixed paraffin-embedded and decalcified tissues were sectioned at a thickness of 5 μm and collected on coherent glass slides. The tissue sections were dewaxed in xylene and rehydrated through graded alcohols and water. The masked antigens were retrieved in 0.01 M citrate buffer solution by heat. For immunohistochemistry, the tissues were stained with an anti-CD11b antibody (Abcam, Cambridge, UK). For immunofluorescence, the tissues were stained using a PE-conjugated anti-CD19 antibody (BD Biosciences, Franklin Lakes, NJ, USA) and an APC-conjugated anti-CD254 antibody (RANKL, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and then counterstained with DAPI (Thermo Fisher Scientific, Waltham, MA, USA). All images were collected with a Leica TCS SPS microscope (Leica Biosystems, Wetzlar, Germany).
Micro-CT scans
The hind limbs of each mouse were fixed in 10% PBS-buffered formalin at room temperature and were scanned by micro-CT (Inveon MM system, Siemens Preclinical Solutions) at a resolution of 8.5 μm as previously described [30]. The following parameters were calculated using the Inveon Research Workplace (Siemens Medical Solutions) and manufacturer-supplied software: bone volume/total volume (bone volume fraction, BV/TV), trabecular number (Tb. N), trabecular thickness (Tb. Th), and trabecular spacing (Tb. Sp).
Flow cytometry assay
Single suspensions of mouse splenic cells or peripheral blood mononuclear cells (PBMCs) were prepared. Surface staining was conducted and analyzed as previously reported [31, 32]. Briefly, cells were incubated with PE-conjugated anti-CD19, PerCP-Cy5.5-conjugated anti-CD69, PE-conjugated anti-CD11b, BV510-conjugated anti-F4/80, FITC-conjugated anti-CD3, BV421-conjugated anti-B220, BUV395-conjugated anti-CD138, APC-conjugated anti-CD27, BV510-conjugated anti-CD19, and PE-conjugated anti-CD254 antibodies (BD Biosciences, NJ, US) after being collected and blocked with anti-CD16/CD32 mAb (eBioscience, San Diego, CA, USA) or anti-CD16/CD32 hAb (eBioscience, San Diego, CA, USA). Flow cytometry was performed on a 4-laser/13-color BD LSR II Fortessa instrument (BD Biosciences, San Diego, CA, USA), and the data were analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
Antibody assay
The levels of specific anti-CII antibodies in the serum were measured by enzyme-linked immunosorbent assay (ELISA) as previously described [30]. Briefly, a 96-well ELISA plate was coated with bovine CII (50 μg/mL) overnight at 4 °C. The serum samples were added and incubated. Horseradish peroxidase-conjugated goat anti-mouse IgG (H + L), IgG1, IgG2a and IgG2b Abs (Invitrogen, San Diego, CA, USA) were used. The optical density was measured with a spectrophotometer at 450 nm.
Cytokine assay
The levels of cytokines in the serum and joints were quantified using Luminex x-MAP technology (Luminex Corp, Austen TX, USA). Serum and joint homogenates from CIA mice were analyzed using a Milliplex multi-analyte magnetic bead panel obtained from Thermo Fisher Scientific (Waltham, MA, USA), and all data were collected on a Luminex 200 instrument. All standard curves with four-parameter logistic fitting generated from the known cytokine concentrations supplied by the manufacturer had calculated R2 values of equal to or close to 1, and quality controls provided the expected results.
Gene expression analysis
For the in vitro experiment, RAW264.7 cells were seeded in 24-well plates (5 × 105 cells/well), pretreated with ibrutinib (1000 nM), acalabrutinib (1000 nM) or tenfold serial dilutions of SOMCL-17-016 from 1 nM to 1000 nM for 2 h and then stimulated with 50 ng/mL of mouse recombinant RANKL (Sigma-Aldrich, St. Louis, MO, USA) and 40 ng/mL recombinant mouse M-CSF (R&D, Minneapolis, MN, USA) for an additional 24 h. Total RNA was extracted from the ankles of the tested mice or cells with a total RNA kit (Tiangen, Beijing, China) and reverse transcribed into cDNA with the RT Master Mix kit (Takara, Dalian, China). Real-time PCR was performed with SYBR® Green Real-time PCR Master Mix (TOYOBO, Osaka, Japan) on an Applied Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Relative mRNA expression levels were normalized to the level of β-actin using the ΔΔCt method. The primer sequences used are listed in Table 1.
Table 1.
Sequences of primers used for quantitative real-time PCR.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| mouse TRAP | CTGGAGTGCACGATGCCAGCGACA | TCCGTGCTCGGCGATGGACCAGA |
| mouse CTSK | AATACCTCCCTCTCGATCCTACA | TGGTTCTTGACTGGAGTAACGTA |
| mouse DC-STAMP | GGGGACTTATGTGTTTCCACG | ACAAAGCAACAGACTCCCAAAT |
| mouse OPG | ACCCAGAAACTGGTCATCAGC | CTGCAATACACACACTCATCACT |
| mouse RANKL | CAGCATCGCTCTGTTCCTGTA | CTGCGTTTTCATGGAGTCTCA |
| mouse NFATc1 | CCGTTGCTTCCAGAAAATAACA | TGTGGGATGTGAACTCGGAA |
| mouse TRAF6 | AAAGCGAGAGATTCTTTCCCTG | ACTGGGGACAATTCACTAGAGC |
| mouse c-Fos | CGGGTTTCAACGCCGACTA | TTGGCACTAGAGACGGACAGA |
| mouse c-Jun | ACTCGGACCTTCTCACGTCG | TAGACCGGAGGCTCACTGTG |
| mouse β-actin | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
TRAP tartrate-resistant acid phosphatase, CTSK Cathepsin K; DC-STAMP, dendritic cell-specific transmembrane protein, OPG osteoprotegerin, RANKL receptor activator of nuclear factor κB ligand, NFATc1 nuclear factor for activated T cells 1, TRAF6 tumor necrosis factor receptor-associated factor 6.
Western blotting
RAW264.7 cells were allowed to rest at 37 °C for 12 h. The cells were then pretreated with ibrutinib (1000 nM), acalabrutinib (1000 nM) or tenfold serial dilutions of SOMCL-17-016 from 1 nM to 1000 nM for 2 h and then stimulated with RANKL (50 ng/mL) and M-CSF (40 ng/mL) for 24 h. The cultured cells were lysed with sodium dodecyl sulfate (SDS) sample buffer containing protease inhibitor cocktail (Roche Life Science, Mannheim, Germany), and the protein concentration was measured by the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein were separated by 10% SDS-PAGE and blotted with antibodies against BTK, PLCγ2, phosphorylated BTK (Y223), phosphorylated-PLCγ2 (Y1217), NFAT2 (Cell Signaling Technology, Beverly, MA, USA) and GAPDH (KangChen, Biotechnology, China). The densities of the bands were quantified with a computerized densitometer (ImageJ Launcher, Broken Symmetry Software).
Ex vivo proliferation
Splenocytes from different groups were prepared and cultured with medium alone or with anti-IgM antibodies (10 μg/mL) for 72 h. After incubation, the cultures were pulsed with 0.5 μCi/well of [3H-TdR] thymidine to evaluate cell proliferation activity.
Culture of PBMCs
PBMCs from treatment-naive RA patients and age- and gender-matched healthy controls were collected and studied in this research. All RA patients met the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism collaborative RA classification criteria (Ref: 10.1002/art.27584). Informed consent was obtained from all the participants before the study. The study was approved by the Ethics Committee of Renji Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, China. PBMCs were isolated from heparinized blood by Ficoll-Hypaque density gradient centrifugation (Pharmacia Biotech, Sweden) and then pretreated with ibrutinib (1000 nM) and acalabrutinib (1000 nM) or SOMCL-17-016 (1000 nM) for 2 h. Then, the cells were stimulated with 2.5 μg/mL anti-CD40 (eBioscience, San Diego, CA, USA) and 10 μg/mL anti-IgM (Southern Biotech, Birmingham, AL, USA) at 37 °C in an atmosphere of 5% CO2 for 24 h. The expression of RANKL on memory B cells was examined by flow cytometry.
Cell viability assay
To evaluate the cytotoxicity of ibrutinib, acalabrutinib and SOMCL-17-016 on splenocytes and RAW264.7 cells, the MTT assay for cell viability was performed as previously reported [33]. Briefly, fresh splenocytes (8 × 105 cells) were cultured in triplicate for 48 h in the presence or absence of the compounds. MTT (5 mg/mL) reagent was added 4 h before the end of culture, and then the cells were lysed with DMSO. RAW264.7 cells were seeded in 96-well plates at a density of 2 × 103 cells/well. After overnight incubation at 37 °C, the medium was replaced with 200 μL of fresh media containing the compounds at various concentrations for 4 days. Then, MTT (5 mg/mL) reagent was added 2 h before the end of culture, and the formazan was dissolved in DMSO. Absorbance was measured at 570 nm with a microplate reader (Spectramax 190, Molecular Devices Corporation, Sunnyvale, CA, USA).
Con A and LPS-induced proliferation assay
Splenocytes (5 × 105 cells) were cultured in triplicate for 48 h in the presence or absence of the compounds and were stimulated with 5 μg/mL concanavalin A (Con A) to induce T cell proliferation and 10 μg/mL lipopolysaccharide (LPS) to induce B cell proliferation. The cells were pulsed with 0.5 μCi/well of [3H] thymidine for 8 h and harvested on glass fiber filters. The incorporated radioactivity was counted using a Beta Scintillation Counter (MicroBeta TriLux, PerkinElmer Life Sciences, Boston, MA, USA). The IC50 values were calculated using the log(inhibitor) versus normalized response nonlinear fit (GraphPad Prism 6.0).
In vitro osteoclastogenesis assay
For differentiation of osteoclasts, RAW264.7 cells were seeded at a density of 2 × 103 cells/well in 96-well plates. After overnight incubation, the medium was replaced with 10% FBS DMEM containing 50 ng/mL recombinant mouse RANKL and 40 ng/mL recombinant mouse M-CSF in the absence or presence of the compound at the indicated concentrations (tenfold serial dilutions of ibrutinib, acalabrutinib or SOMCL-17-016 from 1 nM to 1000 nM). The cultures were incubated at 37 °C for another 4 days. TRAP staining was performed with the aid of a commercial kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. TRAP-positive multinucleated cells containing three or more nuclei were counted.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA) software. Statistically significant differences were determined using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. P values < 0.05 were considered significant.
Results
SOMCL-17-016 dose-dependently prevented arthritis progression in CIA mice
SOMCL-17-016 is a BTK inhibitor with a novel chemical structure, as shown in Fig. 1. The in vitro immunosuppressive activity of SOMCL-17-016 on Con A- and LPS-induced proliferation of murine splenocytes was evaluated. SOMCL-17-016 exhibited a higher selective index (SI) than ibrutinib and acalabrutinib for LPS-induced B cell proliferation but exerted a much milder effect on Con A-induced T cell proliferation (Table 2). DBA1/J mice immunized with CII were assigned to five treatment groups (6.25, 12.5 or 25 mg·kg−1·d−1 SOMCL-17-016 or 25 mg·kg−1·d−1 ibrutinib or acalabrutinib) and administered the indicated compounds orally after disease was established (Fig. 2a). As Fig. 2b shows, SOMCL-17-016 application significantly retarded the progression of arthritis in CIA mice until the end of the experiment, and 25 mg·kg−1·d−1 SOMCL-17-016 had more potent therapeutic effects than ibrutinib and acalabrutinib at the corresponding dosages (Fig. 2b). In addition, SOMCL-17-016 administration markedly improved paw swelling in CIA mice (Fig. 2c–e) and ameliorated joint pathology, including pannus formation, cellular infiltration, synovial proliferation, and cartilage destruction (Fig. 2f, g).
Fig. 1. The chemical structure of the tricyclic BTK inhibitor SOMCL-17-016.
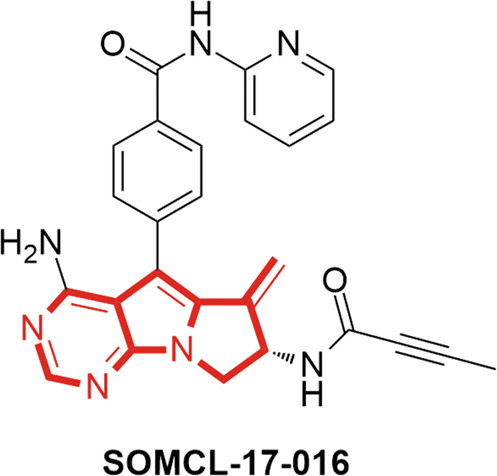
SOMCL-17-016 is a BTK inhibitor with a novel tricyclic chemical structure.
Table 2.
Summary of cytotoxicity and suppressive activities.
| Compounds | CC50 | Con A IC50 | SI | LPS IC50 | SI |
|---|---|---|---|---|---|
| (μM) | (μM) | (μM) | |||
| SOMCL-17-016 | 9.44 ± 0.25 | 0.59 ± 0.17 | 17.44 ± 4.5 | 0.02 ± 0.01 | 601.88 ± 202.29 |
| Ibrutinib | 1.91 ± 0.16 | 0.46 ± 0.22 | 5.19 ± 2.16 | 0.07 ± 0.02 | 28.20 ± 5.45 |
| Acalabrutinib | 30.87 ± 2.17 | 13.87 ± 3.42 | 2.41 ± 0.75 | 0.66 ± 0.11 | 47.81 ± 4.56 |
CC50, The cytotoxic concentration of the compound that reduces cell viability by 50%; IC50, The inhibitory concentration of the compound that reduces cell proliferation by 50%; Selective Index (SI) = CC50/IC50.
Fig. 2. SOMCL-17-016 dose-dependently prevented arthritis progression in CIA mice.

a Experimental design of CIA induction and the treatment strategy. b Mice were orally treated with ibrutinib, acalabrutinib or different doses of SOMCL-17-016 for 15 days. Arthritis clinical scores were recorded every day after administration. c, d The thickness of right and left paw swelling was measured at the end of the study (n = 8 or 9). e Representative photographs displaying the gross features of the left hind paws. f Representative ankle joint sections stained with H&E (magnification of ×100). g Histopathological evaluation of the ankle joint for the presence of pannus formation, cellular infiltration, synovial proliferation, and cartilage erosion (n = 4). The data are shown as the means ± SEMs; *P < 0.05, **P < 0.01 and ***P < 0.001, #P < 0.05, ##P < 0.01, and ###P < 0.001, ∆P < 0.05, ∆∆P < 0.01, and ∆∆∆P < 0.001; SOMCL-17-016, acalabrutinib and ibrutinib respectively, significantly different from the vehicle-treated group.
SOMCL-17-016 hampered bone erosion and diminished excessive osteoclasts in CIA mice
A micro-CT scan was subsequently utilized to verify that SOMCL-17-016 has direct protective effects against bone destruction. Three-dimensional reconstruction of the right hind paws revealed severe articular bone loss in vehicle-treated CIA mice and that SOMCL-17-016 had an equal ameliorative effect on bone erosion as ibrutinib and acalabrutinib (Fig. 3a). Specifically, compared with control mice, vehicle-treated CIA mice showed a marked decrease in bone volume/total volume (bone volume fraction, BV/TV), trabecular number (Tb. N) and trabecular thickness (Tb. Th) but increased trabecular spacing (Tb. Sp). Treatment with 25 mg·kg−1·d−1 SOMCL-17-016 significantly improved all these bone parameters (Fig. 3b). It is well accepted that bone erosion in RA is attributed to the abnormal differentiation of osteoclasts, which results in an imbalance between bone resorption and bone formation [34]. Thus, we identified osteoclasts in the inflamed joint by tartrate-resistant acid phosphatase (TRAP) staining. As expected, SOMCL-17-016 abrogated the accumulation of TRAP-positive multinucleated osteoclasts in the joints of CIA mice in a dose-dependent manner (Fig. 3c, d).
Fig. 3. SOMCL-17-016 hampered bone erosion and diminished excessive osteoclasts in CIA mice.

a Representative three-dimensional reconstructions of the right hind paws of normal control mice and CIA mice. b Histomorphometric analysis of the distal tibia for each treatment group and the bone volume/total volume (BV/TV), trabecular thickness (Tb. Th), trabecular number (Tb. N), and trabecular spacing (Tb. Sp) values of mice from each group. c TRAP staining (magnification of ×100) of representative inflamed joints in the hind paws of mice from the different groups. d Numbers of TRAP-positive osteoclast cells in a field of joint sections of mice from each group. The data are shown as the means ± SEMs (n = 3); *P < 0.05, **P < 0.01, and ***P < 0.001, significantly different from the vehicle-treated group.
SOMCL-17-016 impeded B cell activation and suppressed autoantibody production in CIA mice
As B cell activation may contribute to autoimmunity in CIA mice, we determined the impact of SOMCL-17-016 on anti-IgM-induced B cell reactions. Functionally, SOMCL-17-016 dose-dependently inhibited the proliferation (Fig. 4a) and activation (Fig. 4b, c) of splenic B cells from CIA mice. To evaluate the critical role of plasma cells in autoantibody production, we analyzed the proportion of plasma cells in the spleen. As shown in Fig. 4d, the CD3-CD138+B220med population was markedly augmented in the peripheral blood of vehicle-treated CIA mice compared to control mice, and SOMCL-17-016 treatment significantly inhibited the expansion of this subset of cells. Consistent with the decrease in the number of plasma cells, SOMCL-17-016-treated CIA mice exhibited lower serum levels of anti-CII IgG antibody, as well as the IgG1, IgG2a and IgG2b isotypes, than vehicle-treated CIA mice, although the decrease in the level of the IgG2a subtype was not statistically significant (Fig. 4e).
Fig. 4. SOMCL-17-016 impeded B cell activation and suppressed autoantibody production in CIA mice.

a Splenocytes isolated from CIA mice were stimulated with anti-IgM (10 μg/mL) and cultured for 72 h. Cell proliferation was measured by the [3H]-thymidine incorporation assay. Splenocytes isolated from CIA and control mice were stimulated with anti-IgM (10 μg/mL) and cultured for 24 h. Flow cytometric analysis of B cell activation by CD19 and CD69 staining (b) or CD19 and CD86 staining (c). Representative (left) and statistical results (right) are shown. d Flow cytometric analysis of plasma cells by B220 and CD138 staining. Each plot shows data for an individual mouse (n = 5). e Serum levels of total anti-CII IgG, IgG1, IgG2a and IgG2b in CIA mice and normal control mice. Each plot shows data for an individual mouse (n = 8 or 9). The data are shown as the means ± SEMs; *P < 0.05, **P < 0.01 and ***P < 0.001, significantly different from the vehicle group.
SOMCL-17-016 decreased osteoclastogenesis and reduced pathogenic B cell-derived RANKL expression in RA
The mast osteoclastogenic genes TRAP and cathepsin K (CTSK) and fusion-mediating molecule dendritic cell-specific transmembrane protein (DC-STAMP) orchestrate the process of osteoclastogenesis. SOMCL-17-016 restrained the expression of these genes in the joint tissues of CIA mice and promoted the transcription of osteoprotegerin (OPG), the decoy receptor for RANKL that functions as a negative regulator of osteoclastogenesis (Fig. 5a). Furthermore, the level of RANKL was extremely high in the joints of CIA mice, as displayed in Fig. 5b, c. Intriguingly, administration of a BTK inhibitor markedly inhibited local accumulation of RANKL while moderately affecting the RANKL content in the peripheral blood (Fig. 5d). Specifically, we identified a distinct CD19+RANKL+ B cell cluster in the synovium of CIA mice, the number of which decreased obviously after BTK inhibitor treatment (Fig. 5e). Due to this phenomenon, we speculated that BTK inhibitors might alter the phenotype of a B cell subset with RANKL-expressing potential when they encounter autoantigens as memory B cells migrate from lymphoid tissue, which is similar to the events that occur in the inflamed joint. Thus, we conducted an in vitro assay using PBMCs collected from RA patients and healthy volunteers and found that BCR and CD40 costimulation provoked obvious expression of RANKL on CD19+CD27+ memory B cells but not on healthy control cells. This upregulation of RANKL expression was markedly reversed in memory B cells from RA patients cultured with SOMCL-17-016, ibrutinib or acalabrutinib (Fig. 5f).
Fig. 5. SOMCL-17-016 decreased osteoclastogenesis and reduced pathogenic B cell-derived RANKL expression in RA.

a The mRNA expression of TRAP, CTSK, DC-STAMP and OPG in joint tissue. Each plot shows data for an individual mouse (n = 3). b Soluble receptor activator of nuclear factor-κB ligand (sRANKL) production in joint tissue. Each plot shows data for an individual mouse (n = 6). c The mRNA expression of RANKL in joint tissue. Each plot shows data for an individual mouse (n = 3). d Levels of sRANKL in the serum. Each plot shows data for an individual mouse (n = 6). e Joint tissues were subjected to immunofluorescence staining for CD19 and RANKL, and the nuclei were stained with DAPI. The data are shown as the means ± SEMs; **P < 0.01 and ***P < 0.001, significantly different from the vehicle-treated group. f Representative histograms showing the MFI of RANKL on the surface of CD19+CD27+ memory B cells from RA patients following stimulation, incubation with medium alone or incubation with compounds. The dot plots show the MFI of RANKL induction under different conditions (n = 4). The data are shown as the means ± SEMs; *P < 0.05 and **P < 0.01, significantly different from cells stimulated with anti-IgM/anti-CD40 antibodies only.
SOMCL-17-016 inhibited systemic inflammatory macrophages and tissue inflammation in joints
Autoantibodies and immune complexes exert their pathogenic effects in RA by activating FcγR expressed on myeloid lineage cells, which results in subsequent inflammatory cytokine production and phagocytic events. Indeed, joint macrophages accumulate during RA progression, and the number of macrophages is correlated with the severity of cartilage damage [21]. Analysis of local inflammatory cytokine production in the joints showed that SOMCL-17-016 treatment substantially reduced IL-6, TNF-α, IL-1β and monocyte chemoattractant protein-1 (MCP-1) expression (Fig. 6a) at the protein level and inhibited the infiltration of CD11b+ monocytes in affected articulation at the end of the study (Fig. 6b). Although SOMCL-17-016 decreased the elevated percentage of CD11bhiF4/80+ macrophages in both the peripheral blood (Fig. 6c) and spleens (Fig. 6d) of CIA mice, it did not significantly lower serum TNF-α and MCP-1 levels, different from the efficacious effect on IL-6 or IL-1β production (Fig. 6e).
Fig. 6. SOMCL-17-016 inhibited systemic inflammatory macrophages and tissue inflammation in joints.

a Cytokine production profile in tissue homogenates. Each plot shows data for an individual mouse (n = 6). b Immunohistochemical staining for CD11b in joint tissue (magnification of ×100). Representative (left) and statistical results (right) are shown. Each plot shows data for an individual mouse (n = 3). Flow cytometric analysis of CD11bhiF4/80+ macrophages in the (c) peripheral blood and (d) spleens of mice from each group. Representative (left) and statistical results (right) are shown. Each plot shows data for an individual mouse (n = 3). e Levels of cytokines in the serum. Each plot shows data for an individual mouse (n = 6). The data are shown as the means ± SEMs; *P < 0.05, **P < 0.01 and ***P < 0.001, significantly different from the vehicle-treated group.
SOMCL-17-016 inhibited osteoclastogenesis in RAW264.7 cells via RANK-BTK signaling
Given the protective effects of BTK inhibition on bone destruction and osteoclastogenesis in joints, we evaluated the direct influence of SOMCL-17-016 on osteoclastogenesis in an in vitro culture system. SOMCL-17-016, ibrutinib and acalabrutinib concentration-dependently inhibited RANKL and M-CSF-driven osteoclast differentiation at doses ranging from 1 nM to 1000 nM (Fig. 7a, b) without causing overt cytotoxicity to osteoclast precursor cells (Fig. 7c). In line with the reduction in osteoclast number, the expression of NFATc1 and TRAP was decreased after SOMCL-17-016 treatment, whereas the transcription of the tumor necrosis factor receptor-associated factor 6 (TRAF6) gene and its downstream elements AP-1 (c-Fos and c-Jun) was unaffected (Fig. 7d). Coincidently, BTK inhibition had no effect on the phosphorylation of NF-κB (Fig. 7e, f).
Fig. 7. SOMCL-17-016 inhibited osteoclastogenesis in RAW264.7 cells via RANK-BTK signaling.

a Representative TRAP staining images of RAW264.7 cells treated with or without ibrutinib, acalabrutinib or SOMCL-17-016 (1000 nM) and then stimulated with RANKL/M-CSF for 4 days. b The number of TRAP-positive multinucleated osteoclasts (≥3 nuclei) was quantified. c The MTT assay was performed to analyze the viability of RAW264.7 cells treated with the indicated concentrations of ibrutinib, acalabrutinib or SOMCL-17-016 for 4 days. d mRNA levels of the osteoclast marker genes NFATc1, TRAP, TRAF6, c-Fos and c-Jun. e, f Western blot analysis of the indicated proteins in the RANKL signaling pathway. The data are shown as the means ± SEMs; *P < 0.05, **P < 0.01 and ***P < 0.001, significantly different from cells stimulated with RANKL/M-CSF only.
Since BTK inhibitors barely affected TRAF6 expression and its downstream pathways in macrophages following the RANKL-RANK interaction, an alternative signaling pathway mediated by BTK activation other than TRAF6-dependent activation might be involved in osteoclastogenesis suppression by BTK inhibition. In addition, BTK deficiency inhibits the PLCγ2-dependent positive feedback loop governing NFATc1 expression, which is indispensable during the later stage of osteoclast maturation [13]. Figure 7e, f show the results of Western blot analysis of osteoclasts in the presence of M-CSF and RANKL for 24 h. Surprisingly, 1000 nM ibrutinib did not significantly inhibit BTK phosphorylation but markedly suppressed PLCγ2 phosphorylation and NFATc1 levels at some concentrations. In contrast, SOMCL-17-016 prevented BTK activation and the downstream PLCγ2 phosphorylation and NFATc1 expression. Since ibrutinib exhibited poor selectivity for BTK and Tec kinase, the inhibition of PLCγ2 and NFATc1 by ibrutinib might be due to Tec inhibition instead. These data suggested that SOMCL-17-016 impeded RANKL signaling via the inhibition of BTK and that SOMCL-17-016 has better selectivity for BTK than ibrutinib.
Discussion
BTK has attracted considerable interest as a therapeutic target for multiple autoimmune diseases. Notably, inhibition of BTK is a prospective strategy for the prevention of bone loss in osteoclast-associated bone disorders [14, 16]. In the current study, we described the pharmacological properties and unique mechanisms of a novel, irreversible and selective tricyclic BTK inhibitor, SOMCL-17-016, that was obtained by cyclizing the sidechain of the bicyclic inhibitor ibrutinib.
SOMCL-17-016 dose-dependently reduced the serum levels of CII-specific autoantibodies, rapidly ameliorated bone damage and inhibited joint inflammation in CIA mice, and exhibited more potent effects in vivo than ibrutinib and acalabrutinib at the corresponding dosages. B cells and myeloid cells play significant contributory roles in the initiation and effector phases, respectively, of autoimmune disease, with the former driving antigen presentation, costimulation functions and antibody production and the latter orchestrating joint destruction by producing inflammatory mediators, including TNF-α, IL-1β and IL-6 [35]. The ability of SOMCL-17-016 to suppress the activation of, proliferation of, and costimulatory molecule expression by antigen-specific B cells is comparable to that of the representative first- and secondary-generation BTK inhibitors ibrutinib and acalabrutinib. The therapeutic benefits of SOMCL-17-016 in CIA mice was associated with inhibition of the production of rheumatoid-associated inflammatory cytokines within the joints and serum. Therefore, SOMCL-17-016 was likely effective in treating CIA at several levels.
Previous studies have shown that RANKL is highly expressed in synovial fibroblasts and activated T cells in RA [36–38]. In recent years, accumulating research has shown that highly activated CD80+CD86+ synovium B cells are another important source of RANKL in RA [20, 39]. We also observed more abundant accumulation of RANKL+CD19+ B cells in the affected joint synovium of CIA mice than in those of their normal counterparts in the current study. Furthermore, we demonstrated that upon activation via BCR and CD40, CD19+CD27+ memory B cells, PBMCs from RA patients but not those from healthy controls predominantly expressed high levels of RANKL. These findings emphasize a potential role of effector memory B cells in osteoclastogenesis involved in RA, and we showed that both in vivo and ex vivo application of SOMCL-17-016 decreased the number of RANKL-expressing B cells to a similar extent as ibrutinib and acalabrutinib. Nevertheless, the most puzzling issue is the discrepancy between the in vitro induction of RANKL expression in CD19+CD38+ murine memory B cells and the observed clusters of RANKL+ synovium B cells in vivo. Although increased RANKL expression was seen in the joint synovium of CIA mice, an appreciable increase was not observed in anti-IgM and anti-CD40 antibody-activated CD19+CD38+ B cells from the bone marrow, spleens or peripheral blood of normal BALB/c mice, even upon costimulation with interferon-γ (IFN-γ) (data not shown). Whether B cells originating from lymphoid tissues encounter specific antigens or yet-to-be defined stimuli that favor their differentiation toward RANKL+ effector B cells or whether synovium residential B cells acquire the propensity to express RANKL in response to the bone-devastating inflammatory milieu within a regulatory network requires further investigation.
Further biological research revealed that SOMCL-17-016 directly blocked osteoclast differentiation in a concentration-dependent manner and inhibited the transcription factor NFATc1 at the protein level in vitro without affecting TRAF6 and its downstream elements. These results confirmed that BTK inhibition disrupts the pathogenic signaling pathway transduced via BTK-PLCγ2 following the RANKL-RANK interaction independent of TRAF6, hence contributing to the downstream suppression of NFATc1 activation and osteoclastogenesis. Furthermore, SOMCL-17-016 consistently inhibited BTK, PLCγ2 and NFATc1 upon RANKL signaling in osteoclast precursor cells, probably owing to its better selectivity for BTK than Tec kinase.
Taken together, our results show that in a complex disease state, SOMCL-17-016 simultaneously disrupted several pathogenic processes, including autoantibody production, inflammatory responses and osteoclastogenesis. These findings suggest that SOMCL-17-016, a selective BTK inhibitor with a novel chemical structure, is a promising drug candidate for RA and other autoimmune disorders.
Conclusion
Collectively, our study demonstrated that oral administration of SOMCL-17-016 protected against joint destruction and inflammation in a murine RA model with better efficacy than ibrutinib and acalabrutinib. In addition, SOMCL-17-016 inhibited the activation and RANKL expression of B cells while reducing inflammatory cytokine production locally and generally in vivo. Furthermore, SOMCL-17-016 inhibited osteoclast differentiation in vitro by disrupting the transduction of RANK-BTK-PLCγ2 signals.
Acknowledgements
This work was supported by the Personalized Medicines—Molecular Signature-based “Drug Discovery and Development,” Strategic Priority Research Program of the Chinese Academy of Sciences (grant No. XDA12020369); the National Natural Science Foundation of China (NSFC) (grant No. 81903882); and the National Science and Technology Major Project “New Drug Creation and Manufacturing Program,” China (grant No. 2018ZX09711002-014-001).
Author contributions
YTL, HHD, AZ, SJH, and JPZ contributed to the conception and design of this study; YTL and ZML completed the main part of the experiment; QW, LC, XQY and FHZ participated in the in vivo experiment; HHD, SSL, YTH, SQC, FMY participated in the in vitro experiment; ZLS contributed to the synthesis of the compound; AZ, SJH, JPZ, JD, MYG and HX contributed to drafting the paper and revising it critically for important intellectual content; and all authors contributed to this paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Yu-ting Liu, Hui-hua Ding
Contributor Information
Ao Zhang, Email: aozhang@simm.ac.cn.
Shi-jun He, Email: heshijun@simm.ac.cn.
Jian-ping Zuo, Email: jpzuo@simm.ac.cn.
References
- 1.Gibofsky A. Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis. Am J Managed Care. 2012;18:S295–302. [PubMed] [Google Scholar]
- 2.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 4.Derksen V, Huizinga TWJ, van der Woude D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol. 2017;39:437–46. doi: 10.1007/s00281-017-0627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15. doi: 10.1038/s41413-018-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia XY, Chang Y, Wei F, Dai X, Wu YJ, Sun XJ, et al. CP-25 reverses prostaglandin E4 receptor desensitization-induced fibroblast-like synoviocyte dysfunction via the G protein-coupled receptor kinase 2 in autoimmune arthritis. Acta Pharmacol Sin. 2019;40:1029–39. doi: 10.1038/s41401-018-0196-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han L, Zhang XZ, Wang C, Tang XY, Zhu Y, Cai XY, et al. IgD-Fc-Ig fusion protein, a new biological agent, inhibits T cell function in CIA rats by inhibiting IgD-IgDR-Lck-NF-kappaB signaling pathways. Acta Pharmacol Sin. 2020;41:800–12. doi: 10.1038/s41401-019-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steffen U, Schett G, Bozec A. How autoantibodies regulate osteoclast induced bone loss in rheumatoid arthritis. Front Immunol. 2019;10:1483. doi: 10.3389/fimmu.2019.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeling M, Hillenhoff U, David JP, Schett G, Tuckermann J, Lux A, et al. Inflammatory monocytes and Fcγ receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc Natl Acad Sci USA. 2013;110:10729–34. doi: 10.1073/pnas.1301001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engdahl C, Bang H, Dietel K, Lang SC, Harre U, Schett G. Periarticular bone loss in arthritis is induced by autoantibodies against citrullinated vimentin. J Bone Miner Res. 2017;32:1681–91. doi: 10.1002/jbmr.3158. [DOI] [PubMed] [Google Scholar]
- 11.Komine M, Kukita A, Kukita T, Ogata Y, Hotokebuchi T, Kohashi O. Tumor necrosis factor-alpha cooperates with receptor activator of nuclear factor kappaB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone. 2001;28:474–83. doi: 10.1016/S8756-3282(01)00420-3. [DOI] [PubMed] [Google Scholar]
- 12.Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000;275:4858–64. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- 13.Lee SH, Kim T, Jeong D, Kim N, Choi Y. The tec family tyrosine kinase Btk Regulates RANKL-induced osteoclast maturation. J Biol Chem. 2008;283:11526–34. doi: 10.1074/jbc.M708935200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ariza Y, Murata M, Ueda Y, Yoshizawa T. Bruton’s tyrosine kinase (Btk) inhibitor tirabrutinib suppresses osteoclastic bone resorption. Bone Rep. 2019;10:100201. doi: 10.1016/j.bonr.2019.100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806. doi: 10.1016/j.cell.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 16.Shinohara M, Chang BY, Buggy JJ, Nagai Y, Kodama T, Asahara H, et al. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone. 2014;60:8–15. doi: 10.1016/j.bone.2013.11.025. [DOI] [PubMed] [Google Scholar]
- 17.Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–7. doi: 10.1111/j.1600-065X.2000.imr017504.x. [DOI] [PubMed] [Google Scholar]
- 18.Tomlinson MG, Kane LP, Su J, Kadlecek TA, Mollenauer MN, Weiss A. Expression and function of Tec, Itk, and Btk in lymphocytes: evidence for a unique role for Tec. Mol Cell Biol. 2004;24:2455–66. doi: 10.1128/MCB.24.6.2455-2466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia. 2015;29:1004–17. doi: 10.1038/leu.2014.351. [DOI] [PubMed] [Google Scholar]
- 20.Ota Y, Niiro H, Ota S, Ueki N, Tsuzuki H, Nakayama T, et al. Generation mechanism of RANKL(+) effector memory B cells: relevance to the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2016;18:67. doi: 10.1186/s13075-016-0957-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamilton JA, Tak PP. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009;60:1210–21. doi: 10.1002/art.24505. [DOI] [PubMed] [Google Scholar]
- 22.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 23.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075–80. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, et al. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363:240–52. doi: 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Truong H, Mitchell SA, Liclican A, Gosink JJ, Li W, et al. Homogeneous BTK occupancy assay for pharmacodynamic assessment of tirabrutinib (GS-4059/ONO-4059) target engagement. SLAS Discov. 2018;23:919–29. doi: 10.1177/2472555218786165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346:219–28. doi: 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- 27.Zhou R, Tang W, Ren YX, He PL, Zhang F, Shi LP, et al. (5R)-5-hydroxytriptolide attenuated collagen-induced arthritis in DBA/1 mice via suppressing interferon-gamma production and its related signaling. J Pharmacol Exp Ther. 2006;318:35–44. doi: 10.1124/jpet.106.101113. [DOI] [PubMed] [Google Scholar]
- 28.Schramm C, Kriegsmann J, Protschka M, Huber S, Hansen T, Schmitt E, et al. Susceptibility to collagen-induced arthritis is modulated by TGFbeta responsiveness of T cells. Arthritis Res Ther. 2004;6:R114–9. doi: 10.1186/ar1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang Y, Wu Y, Wang D, Wei W, Qin Q, Xie G, et al. Therapeutic effects of TACI-Ig on rats with adjuvant-induced arthritis via attenuating inflammatory responses. Rheumatology (Oxford) 2011;50:862–70. doi: 10.1093/rheumatology/keq404. [DOI] [PubMed] [Google Scholar]
- 30.Lin ZM, Yang XQ, Zhu FH, He SJ, Tang W, Zuo JP. Artemisinin analogue SM934 attenuate collagen-induced arthritis by suppressing T follicular helper cells and T helper 17 cells. Sci Rep. 2016;6:38115. doi: 10.1038/srep38115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou LF, He SJ, Li X, Yang Y, He PL, Zhou Y, et al. Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum. 2011;63:2445–55. doi: 10.1002/art.30392. [DOI] [PubMed] [Google Scholar]
- 32.He SJ, Lin ZM, Wu YW, Bai BX, Yang XQ, He PL, et al. Therapeutic effects of DZ2002, a reversible SAHH inhibitor, on lupus-prone NZB×NZW F1 mice via interference with TLR-mediated APC response. Acta Pharmacol Sin. 2014;35:219–29. doi: 10.1038/aps.2013.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou LF, He SJ, Wang JX, Yang Y, Zhu FH, Zhou Y, et al. SM934, a water-soluble derivative of arteminisin, exerts immunosuppressive functions in vitro and in vivo. Int Immunopharmacol. 2009;9:1509–17. doi: 10.1016/j.intimp.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 34.Sato K, Takayanagi HOsteoclasts. rheumatoid arthritis, and osteoimmunology. Curr Opin Rheumatol. 2006;18:419–26. doi: 10.1097/01.bor.0000231912.24740.a5. [DOI] [PubMed] [Google Scholar]
- 35.Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7:41–50. doi: 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- 36.Kotake S, Udagawa N, Hakoda M, Mogi M, Yano K, Tsuda E, et al. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 2001;44:1003–12. doi: 10.1002/1529-0131(200105)44:5<1003::AID-ANR179>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 37.Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–8. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 38.Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, et al. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000;43:259–69. doi: 10.1002/1529-0131(200002)43:2<259::AID-ANR4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 39.Yeo L, Toellner KM, Salmon M, Filer A, Buckley CD, Raza K, et al. Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann Rheum Dis. 2011;70:2022–8. doi: 10.1136/ard.2011.153312. [DOI] [PMC free article] [PubMed] [Google Scholar]