

Abstract
Fingolimod has beneficial effects on multiple diseases, including type 1 diabetes (T1D) and numerous preclinical models of colitis. Intestinal dysbiosis and intestinal immune dysfunction contribute to disease pathogenesis of T1D. Thus, the beneficial effect of fingolimod on T1D may occur via the maintenance of intestinal homeostasis to some extent. Herein, we investigated the role of fingolimod in intestinal dysfunction in non-obese diabetic (NOD) mice and possible mechanisms. NOD mice were treated with fingolimod (1 mg · kg−1 per day, i.g.) from weaning (3-week-old) to 31 weeks of age. We found that fingolimod administration significantly enhanced the gut barrier (evidenced by enhanced expression of tight junction proteins and reduced intestinal permeability), attenuated intestinal microbial dysbiosis (evidenced by the reduction of enteric pathogenic Proteobacteria clusters), as well as intestinal immune dysfunction (evidenced by inhibition of CD4+ cells activation, reduction of T helper type 1 cells and macrophages, and the expansion of regulatory T cells). We further revealed that fingolimod administration suppressed the activation of CD4+ cells and the differentiation of T helper type 1 cells, promoted the expansion of regulatory T cells in the pancreas, which might contribute to the maintenance of pancreatic immune tolerance and the reduction of T1D incidence. The protection might be due to fingolimod inhibiting the toll-like receptor 2/4/nuclear factor-κB/NOD-like receptor protein 3 inflammasome pathway in the colon. Collectively, early-life fingolimod treatment attenuates intestinal microbial dysbiosis and intestinal immune dysfunction in the T1D setting, which might contribute to its anti-diabetic effect.
Keywords: fingolimod, type 1 diabetes, gut barrier, enteric pathogens, TLR2-NF-κB-NLRP3
Introduction
Fingolimod is commonly used to treat multiple sclerosis [1]. Notably, fingolimod also exerts therapeutic benefits against type 1 diabetes (T1D) and has been a subject of recent investigations related to the treatment of T1D and its associated complications. However, its clinical efficacy has not been determined, suggesting that the antidiabetes mechanism of fingolimod is complex and limited and requires further study [2–4]. Compared to healthy controls, T1D subjects exhibit increased intestinal inflammation and impaired intestinal barrier function [5–7], which promotes the translocation of gut luminal bacteria and initiates systemic inflammation, including pancreatic inflammation [6, 8], thus contributing to the onset and progression of T1D [9–11]. The shift of intestinal homeostasis toward a proinflammatory state predates increased susceptibility to T1D [7, 12]. T1D is caused by expansion of CD4+ interferon-gamma γ (IFN-γ) + cells (Th1 cells) or a decrease in the number or function of regulatory T cells (Tregs) [13–15]. Interestingly, fingolimod ameliorates colitis in numerous preclinical models [1, 16] due to its roles in inducing Tregs and inhibiting lymphocyte proliferation and IFN-γ secretion [1, 17, 18]. Furthermore, fingolimod prevents the loss of tight junction proteins (TJPs) in a variety of barrier-related diseases [19, 20]. These studies suggest that fingolimod may enhance the gut barrier and maintain intestinal homeostasis in the setting of T1D, which may contribute to preventing T1D. However, the effects of fingolimod on intestinal homeostasis in the setting of T1D are poorly understood. The present study aimed to elucidate the microbial and immune mechanisms of fingolimod in the gastrointestinal system in T1D and to investigate the influence of fingolimod on the pancreatic immune system in the setting of T1D.
Materials and methods
Animals and T1D mouse model
The NOD mouse model is a classical model that recapitulates the hallmarks of T1D in patients [21]. Three-week-old female NOD/LtJ mice (Su Pu Si Biotechnology, Co., Ltd., Suzhou, China) were housed in the animal facility of Jiangnan University (Wuxi, China). All mice were housed in individual ventilated caging systems (TECNIPLAST, Italy) under specific pathogen-free conditions at a controlled temperature (24 ± 1 °C) on a 12 h light-dark cycle. The animals were anesthetized with pentobarbitone sodium (90 mg/kg, Sigma-Aldrich, MO, USA) and euthanized. All studies were approved by the Institutional Animal Ethics Committee of Jiangnan University (JN No. 20131205) and carried out in compliance with national and international guidelines for the Care and Use of Laboratory Animals.
Fingolimod treatment
To evaluate the effect of fingolimod on the onset of T1D, mice were treated daily with 1 mg/kg fingolimod (MCE, New Jersey, USA) in 100 µL sterile water or an equal volume of sterile water by gavage from weaning (3-weeks-old) to the age of 31 weeks (n = 12/group), and their glucose levels were monitored. In the present study, “early-life fingolimod treatment” means treatment with fingolimod from weaning to the age of 31 weeks. The mice were euthanized when a diagnosis of diabetes was made or at the end of the study.
In a parallel study, NOD control and fingolimod-treated mice were euthanized at 12 weeks of age (n = 12/group) after 9 weeks of fingolimod treatment. The pancreatic lymph nodes (PaLNs), mesenteric lymph nodes (MLNs), pancreas, blood, and spleen were collected for immune cell detection by flow cytometry. The pancreas was collected for histological evaluation by hematoxylin and eosin staining.
To track the migration of immune cells adoptively transferred into NOD mice, we transferred fluorescently labeled splenocytes to the recipient mice by intraperitoneal injection (n = 5/group).
For the assessment of gut barrier function, 10-week-old female NOD mice were given either 1 mg/kg fingolimod in 100 µL sterile water or an equal volume of sterile water by gavage daily for 3 weeks (n = 5/group).
Blood glucose measurement
A glucometer (Roche Diagnostics, Almere, Netherlands) was used to measure glucose levels from tail vein samples. NOD mice with hyperglycemia (>11.1 mmol/L) on two consecutive daily readings were considered diabetic [22–27]. NOD mice with hyperglycemia (>11.1 mmol/L) likely require urgent institution of insulin therapy [28, 29]. Thus, in the present study, this diagnostic criterion was adopted.
Intestinal permeability
Intestinal permeability was assessed by measuring fluorescein isothiocyanate (FITC)-dextran (FD20-250 mg, average molecular weight: 20,000, Sigma-Aldrich, MO, USA) accumulation in serum. At the end of the experiment, the mice received 50 mg/100 g of FITC-dextran in phosphate-buffered saline (PBS) by gavage after 4 h of fasting. Four hours after gavage, serum was collected, diluted at 1:10 in PBS, and analyzed with the Thermo Scientific™ Varioskan™ LUX reader (Thermo Fisher Scientific, MA, USA).
Western blot analysis
Colon tissues were added to 2-mL centrifuge tubes, lysed with RIPA buffer containing protease inhibitors (Beyotime, Shanghai, China), and ground with a high-throughput tissue homogenizer (SCIENTZ-48, Ningbo, China). The colon homogenates were centrifuged at 8000 × g for 15 min at 4 °C. The supernatant was used for Western blotting. The total protein concentration was quantified by a BCA protein quantitation assay kit (Beyotime, Shanghai, China). Equal amounts of total proteins were loaded on a polyacrylamide SDS-PAGE gel. The proteins were transferred to polyvinylidene fluoride membranes and then blocked with blocking buffer for 1 h at room temperature. Primary antibodies, including ZO-1, ZO-2, Occludin, Toll-like receptor 2 and 4 (TLR2 and 4), pyrin domain-containing protein 3 (NLRP3), p-NF-κB p65 (p-P65), cleaved-IL-1β (IL-1β), caspase-1-p20 (p20), and β-actin antibodies (the antibody information is listed in Table S1), were added at 4 °C overnight. After being washed, the PVDF membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5000) at room temperature for 2 h. The protein bands were visualized with Western Lightning Plus-ECL (Pierce, Rockford, IL, USA). β-actin was used as an internal standard to control for unwanted sources of variation, and relative protein expression values were expressed as the fold relative to the mean of the controls by comparison to the corresponding control value, and the control value was normalized to 1.0. Protein expression levels were quantitated with ImageJ.
Preparation of single-cell suspensions
When the mice were euthanized, the PaLNs, MLNs, and pancreas were collected and immediately placed in cold PBS.
For macrophage detection, fresh pancreatic tissues were cut into small pieces, digested with 0.75 mg/mL collagenase P (Roche Basel, Switzerland) at 37 °C for 15 min, homogenized with gentle MACS™ Dissociators (MiltenyiBiotec, BergischGladbach, Germany) and filtered with a 70-μm filter [22, 30]. For T-cell detection, fresh pancreatic tissues were harvested, filtered with 70-μm polypropylene mesh, ground using a 2.5-mL syringe plunger and washed with PBS (containing 10% fetal bovine serum) [22, 30].
MLNs were harvested and filtered with 70-μm polypropylene mesh, ground using a 2.5 mL syringe plunger and washed with PBS [22, 30].
After the mice were euthanized, their spleens were harvested and dissociated into single-cell suspensions with gentle MACS™ Dissociators (MiltenyiBiotec, BergischGladbach, Germany). After centrifugation at 300 × g for 5 min, red blood cell lysis buffer (Fcmacs, Nanjing, China) was added to lyse the red blood cells. After the cells were rested for 15 min at room temperature and away from light, they were centrifuged and resuspended in PBS.
After the mice were euthanized, blood was collected in an anticoagulant tube. After centrifugation at 300 × g for 5 min, red blood cell lysis buffer was added to lyse the red blood cells. After the cells were rested for 15 min at room temperature and away from light, they were centrifuged and resuspended in PBS.
Flow cytometry
Mice were sacrificed, and single-cell suspensions were prepared from pancreatic, PaLNs, blood, spleen, and MLN tissues as described in previous studies [22, 31] and stained for 30 min at 4 °C after FcγRII/III was blocked with an anti-CD16/CD32 monoclonal antibody. The antibody information is listed in Table S2. According to the manufacturer’s protocol, for the detection of intracellular (cytoplasmic) IFN-γ, cell suspensions were incubated at 37 °C for 6 h with Cell Stimulation Cocktail (eBioscience, San Diego, CA, USA), stained and fixed. For Treg staining, cells were first surface stained, fixed and stained for intracellular (nuclear) forkhead box P3 (Foxp3). Stained cells were analyzed on an Attune NxT flow cytometer (Thermo Fisher Scientific, Massachusetts, USA). The gating strategy for flow cytometry is listed in Fig. S1–15.
Assessment of donor cell distribution
Donor cell distribution was assessed as previously described [32]. Spleens were harvested from adult NOD donors and immediately dissociated into single-cell suspensions with gentle MACS™ Dissociators (MiltenyiBiotec, BergischGladbach, Germany). After centrifugation at 300 × g for 5 min, red blood cell lysis buffer was added to lyse the red blood cells. After the cells were rested for 15 min at room temperature and away from light, they were centrifuged and resuspended in PBS. The splenocytes were labeled with the cell proliferation dye eFluor 450 (CPD450) (eBioscience, San Diego, CA, USA). CPD450-labeled cells were then injected into the peritoneal cavity. The recipients were killed 20 h after donor cell injection, and the pancreas, PaLNs, MLNs, blood, and spleen were harvested and processed into single-cell suspensions. After counting, the single-cell suspensions were subjected to cytofluorimetric analysis with an Attune NxT flow cytometer (Thermo Fisher Scientific, Massachusetts, USA).
Histopathological examination of the pancreas
Freshly harvested pancreatic tissues were collected, fixed in NEG-50 (Thermo Scientific, Massachusetts, USA), and stored at −80 °C until cut into frozen sections. Eight-micrometer sections were stained with hematoxylin and eosin following a standard procedure [33]. Then, they were scanned with a digital slice scanner (3DHISTCH, Hungary). Morphological changes in islets were evaluated based on the percentage of cell infiltration. Islets that exhibited no insulitis, few infiltrated mononuclear cells, peri-insulitis, 50% islet infiltration, and >50% islet infiltration were scored as 1, 2, 3, 4, and 5, respectively.
Collection of feces, DNA extraction, PCR amplification, and Illumina MiSeq sequencing
Fresh feces were collected from mice at 12 weeks of age and immediately stored at −80 °C until used for the extraction of fecal microbial genomic DNA with the Fast DNA Spin Kit for Soil (MP Biomedicals, cat. # 6560-200, California, USA) following the manufacturer’s instructions. In detail, 50 mg of frozen stool was added to a Lysing Matrix A tube, and then 1.0 mL CLS-TC was added to the sample tube. The mixture was homogenized in the FastPrep Instrument for 40 s at a speed setting of 6.0 and then centrifuged at 14,000 × g for 5–10 min to pellet debris. Next, the supernatant was transferred to a 2.0-mL microcentrifuge tube, an equal volume of Binding Matrix was added, and the samples were mixed and incubated with gentle agitation for 5 min at room temperature on a rotator. Then, the suspension was transferred to a SPIN™ filter, filtered and centrifuged (14,000 × g, 1 min) twice. Subsequently, the pellet was resuspended gently with 500 µL prepared SEWS-M and centrifuged (14,000 × g, 1 min). The contents of the Catch Tube were discarded, and the Catch Tube was replaced and centrifuged (14,000 × g, 1 min) without any addition of liquid. The collection tubes were replaced with new, clean tubes, the binding matrix above the SPIN™ filter was resuspended in 100 µL DES to elute DNA, and the samples were centrifuged at 14,000 × g for 1 min to collect the eluted DNA into the clean Catch Tube after the tubes were incubated at 55 °C for 5 min. The DNA was used for downstream applications and stored at −80 °C until use. The V3 and V4 regions of 16S rRNA were PCR-amplified using specific primers. The reaction conditions were as follows: 95 °C for 5 min; 95 °C for 30 s, 64 °C for 30 s, 72 °C for 30 s for 40 cycles, and a final incubation at 72 °C for 10 min. The PCR products were excised from a 1.5% agarose gel, purified by Gene Clean Turbo (MP Biomedicals, cat. # 111102400) and quantified by the Quant-iTPicoGreen dsDNA Assay Kit (Life Technologies, cat. # P7589) following the manufacturer’s instructions. Libraries were prepared using the TruSeq DNA LT Sample Preparation Kit (Illumina, cat. # FC-121-2001, San Diego, USA) and sequenced for 500 + 7 cycles on the Illumina MiSeq platform using the MiSeqReagent Kit (500 cycles-PE, cat. # MS-102-2003) [30].
Statistics
The data are presented as the mean ± standard error of the mean (SEM) and were analyzed using GraphPad Prism 7 software (San Diego, CA, USA). Two-tailed Student’s t-test was used for two independent groups. A P-value ≤ 0.05 was considered statistically significant. Principal component analysis (PCoA) figure was generated and analyzed by the Vegan 2.0 package (2.5.4) and ade4 package (1.7.13). Significantly different clusters of bacterial communities (PCoA) were explained by ANOSIM. Cumulative diabetes incidence was calculated using Kaplan–Meier estimation. Statistical significance was evaluated by the log-rank test.
Results
Early-life fingolimod treatment attenuates intestinal microbial dysbiosis in diabetes-prone NOD mice
The NOD mouse model is a classical model that recapitulates the hallmarks of T1D in patients [21]. Gut microbial dysbiosis contributes to the disease pathogenesis of T1D [9, 11, 34]. 16S rRNA gene sequencing was used to determine how fingolimod modulates the gut microbiome in the setting of T1D. As shown in Fig. 1a, PCoA demonstrated that overall, the gut microbiota was significantly modified by fingolimod (ANOSIM, r = 0.3733, P = 0.002). Proteobacteria, particularly those belonging to the class γ-Proteobacteria and the family Enterobacteriaceae within this class, are associated with intestinal or systemic inflammation [35, 36]. In the healthy intestine, the abundance of the phylum Proteobacteria is low [35, 37]. In the present study, the abundances of taxonomic groups from the same phylogenetic lineage (Proteobacteria, γ-Proteobacteria, Enterobacteriales, and Enterobacteriaceae) decreased consistently in response to fingolimod treatment (Fig. 1b, c, d, e). Infection with the pathogen Helicobacter pylori contributes to T1D development [38, 39]. Our data suggested that fingolimod treatment effectively reduced the abundance of Helicobacter (also a pathogen of Proteobacteria) (Fig. 1f). Allobaculum is an important functional phylotype in metabolic dysbiosis [36]. In the present study, the abundance of the genus Allobaculum (Firmicutes phylum; Erysipelotrichi class; Erysipelotrichales order; Erysipelotrichaceae family) in fingolimod-treated mice was significantly lower than that in NOD controls (Fig. 1b–f). Furthermore, fingolimod treatment significantly increased the abundance of Prevotella (a short-chain fatty acid producer that is beneficial in T1D) (Fig. 1f) [37, 40]. Collectively, the data suggest that in diabetes-prone NOD mice, early-life fingolimod treatment attenuates intestinal microbial dysbiosis, specifically reducing the abundances of enteric pathogenic Proteobacteria clusters.
Fig. 1. Early-life fingolimod treatment attenuates intestinal microbial dysbiosis in diabetes-prone NOD mice.

Mice were treated daily with 1 mg/kg fingolimod in 100 µL sterile water or an equal volume of sterile water by gavage from weaning. Fresh feces were collected when the mice were 12 weeks of age. (a) Principal coordinate analysis (PCoA) plot of weighted UniFrac distances, with each dot representing a colonic community; the percentage of variation explained by each principal coordinate is shown in parentheses. The relative abundances of the main altered phyla (b), classes (c), orders (d), families (e), and genera (f) in control and fingolimod-treated mice. The data are the mean±SEM (n = 8–9 mice/group). *P < 0.05 versus Con, **P < 0.01 versus Con, ***P < 0.001 versus Con.
Early-life fingolimod treatment enhances the gut barrier in diabetes-prone NOD mice
Impaired intestinal barrier function occurs before T1D onset, and colonic barrier dysfunction further aggravates T1D [6, 7, 41]. TJPs, including ZO-2, Occludin, and ZO-1, participate in the regulation of intestinal barrier function. In the present study, fingolimod treatment significantly enhanced the colonic barrier in the setting of T1D, evidenced by increased expression of TJPs (Fig. 2a) and decreased passage of FITC-dextran from the gut lumen to the blood (Fig. 2b).
Fig. 2. Early-life fingolimod treatment enhances the intestinal barrier in diabetes-prone NOD mice.

(a) The expression of colonic TJPs (ZO-2, Occludin, and ZO-1) was measured by Western blotting, and gray value analysis was performed with ImageJ. Colonic tissue was collected from 12-week-old mice. (b) Assessment of intestinal barrier integrity by oral administration of FITC-dextran. The data shown are the mean ± SEM (n = 4 experiments with two pooled mice in each experiment/group in a; n = 5 experiments/group in b). *P < 0.05 versus Con, **P < 0.01 versus Con.
Early-life fingolimod treatment inhibits the activation of the colonic TLR2/4-NF-κB-NLRP3 pathway in diabetes-prone NOD mice
Activation of the TLR2/4, NF-κB, and NLRP3 inflammasome pathways are critical for the maintenance of homeostasis against pathogenic infections [42, 43]. TLRs are critically involved in the initiation of innate cellular immune responses and subsequent adaptive immune responses to microbial pathogens that may contribute to T1D development [44]. Commensal bacteria modulate intestinal inflammation, presumably through the TLR2/4 pathway [45, 46]. In the present study, colonic TLR2 and TLR4 expression was significantly inhibited in the fingolimod-treated mice compared with the NOD control mice (Fig. 3). In addition, the NLRP3 (the downstream signaling protein of TLRs) inflammasome is required for the secretion of IL-1β and IFN-γ and the differentiation of IFN-γ+ T cells [47, 48]. IL-1β induces beta-cell apoptosis under the control of the transcription factor NF-κB, subsequently contributing to insulitis lesions in T1D [48]. Furthermore, the inhibition of NLRP3 not only suppresses the activation of T cells and the differentiation of IFN-γ+ Th1 cells but also impairs the migration of diabetogenic T cells to the pancreas [49]. In the present study, early-life fingolimod treatment inhibited the activation of NF-κB (downstream of TLR2/4) and the downstream NLRP3/p20/IL-1β pathway in diabetes-prone NOD mice (Fig. 3).
Fig. 3. Early-life fingolimod treatment inhibits the activation of colonic TLR2/4-NF-κB-NLRP3 in diabetes-prone NOD mice.

Mice were treated daily with 1 mg/kg fingolimod in 100 µL sterile water or an equal volume of sterile water by gavage from weaning. Colonic tissue was collected from 12-week-old mice. Western blot analysis of colonic TLR2, TLR4, p-P65, NLRP3, p20, and IL-1β expression and gray value analysis by ImageJ. The data are the mean ± SEM (n = 4 experiments with two pooled mice in each experiment/group). *P < 0.05 versus Con, **P < 0.01 versus Con.
Early-life fingolimod treatment attenuates intestinal immune dysfunction in diabetes-prone NOD mice
In diabetes-prone NOD mice, macrophages play a major role in protecting against translocation of microbes from the gut, and the number of macrophages increases rapidly 1–2 weeks after weaning [5]. In the present study, early-life fingolimod treatment significantly inhibited the recruitment of macrophages (Fig. 4a). Furthermore, fingolimod effectively reduced CD69 expression on CD4+ cells in the MLNs (Fig. 4b). We then detected T1D-associated T-cells in the MLNs and found that fingolimod significantly reduced the frequency of pathogenic Th1 cells (Fig. 4c) and enhanced the frequency of protective Tregs (Fig. 4d). Collectively, these results suggest that early-life fingolimod treatment attenuates intestinal immune dysfunction in the setting of T1D.
Fig. 4. Early-life fingolimod treatment attenuates intestinal immune dysfunction in diabetes-prone NOD mice.
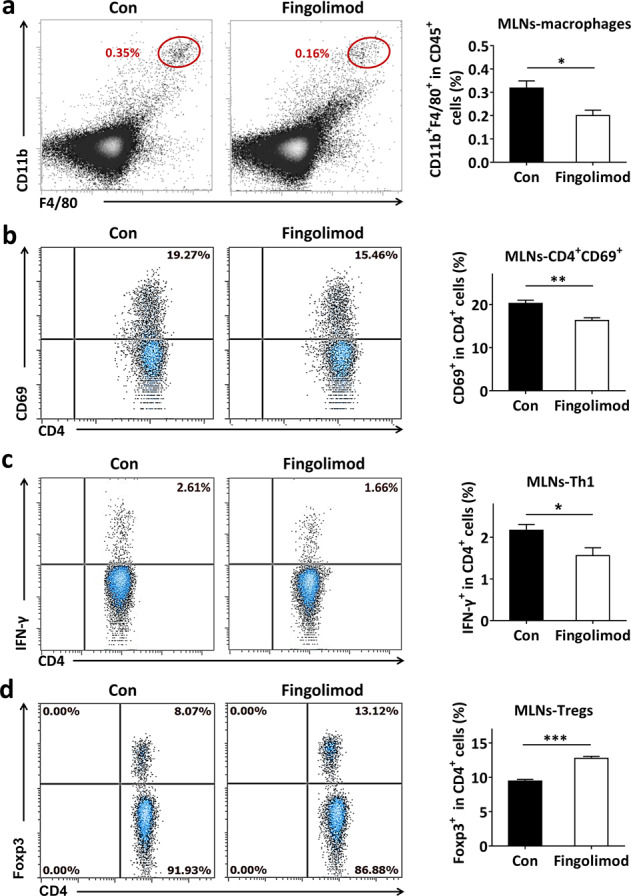
Mice were treated daily with 1 mg/kg fingolimod in 100 µL sterile water or an equal volume of sterile water by gavage from weaning. The MLNs were collected from 12-week-old mice. The frequency of stained cells was detected by flow cytometry. (a) The frequency of CD45+F4/80+CD11b+ cells among CD45+ cells (macrophages) in the MLNs. (b) The frequency of CD69+CD4+ T-cells among CD4+ cells in the MLNs. (c) The frequency of CD4+IFN-γ+ cells among CD4+ cells (Th1 cells) in the MLNs. (d) The frequency of CD4+Foxp3+ among CD4+ cells (Tregs) in the MLNs. The data are the mean ± SEM (n = 5–6 experiments with two pooled mice in each experiment/group). *P < 0.05 versus Con, **P < 0.01 versus Con, ***P < 0.001 versus Con.
Early-life fingolimod treatment inhibits the migration of peritoneal CD45+ leukocytes and CD4+ cells to the pancreas in diabetes-prone NOD mice
The recruitment of autoreactive T cells to islets is a critical step in beta-cell destruction during the pathophysiological process of T1D [50]. To track the migration of immune cells adoptively transferred into NOD mice, we transferred fluorescently labeled CPD450+ splenocytes to recipient mice by intraperitoneal injection. Twenty hours later, the pancreas, PaLNs, blood, MLNs, and spleen were harvested from the recipient mice and examined by flow cytometry. The donor cells did not proliferate within this short period of time after transfer. We observed fewer CPD450+ cells (donor cells) in the pancreas (Fig. 5a), PaLNs (Fig. 5b), and blood (Fig. 5c) of fingolimod-treated mice than in those of NOD control mice. However, fingolimod treatment did not affect the number of CPD450+ cells (donor cells) in the MLNs or spleen (Fig. 5d, e), suggesting that the impacts of fingolimod on the pancreas and its draining lymph nodes may be specific to some extent. More importantly, significantly lower densities of CPD450+CD45+ leukocytes (Fig. 5f) and CPD450+CD4+ cells (Fig. 5g) were found in the PaLNs of fingolimod-treated mice than in those of NOD control mice. These data demonstrate that fingolimod may exert its immunosuppressive activity at least in part by inhibiting the migration of intraperitoneal CD45+ leukocytes, as well as CD4+ cells to the pancreas and its draining lymph nodes.
Fig. 5. Early-life fingolimod treatment inhibits the migration of peritoneal CD45+ leukocytes and CD4+ cells to the pancreas in diabetes-prone NOD mice.

Recipient mice were treated daily with 1 mg/kg fingolimod in 100 µL sterile water or an equal volume of sterile water by gavage from weaning to 12 weeks of age. CPD450-labeled cells were then injected into the peritoneal cavity of the recipient mice. The recipients were killed 20 h after donor cell injection. The pancreas, PaLNs, blood, MLNs, and spleen were harvested and subjected to flow cytometry analysis. (a) The frequency of CPD450+ cells among total pancreatic cells. (b) The frequency of CPD450+ cells among total PaLNs cells. (c) The frequency of CPD450+ cells among total blood cells. (d) The frequency of CPD450+ cells among total MLNs cells. (e) The frequency of CPD450+ cells among total spleen cells. (f) The frequency of CD45+ cells among total CPD450+ cells in the PaLNs. (g) The number of CD4+ cells among total CPD450+ cells in the PaLNs. The data are the mean ± SEM (n = 5 mice per group/group). *P < 0.05 versus Con, ***P < 0.001 versus Con.
Early-life fingolimod treatment contributes to the maintenance of pancreatic immune tolerance in diabetes-prone NOD mice
Impairment of the gut barrier leads to bacterial leakage into the PaLNs, which enhances the activation of autoreactive T-cells and promotes the differentiation of Th1 cells in the PaLNs [5, 6]. In the pancreas, fingolimod treatment significantly suppressed the recruitment of macrophages (Fig. 6a) and inhibited the activation of CD4+ cells (evidenced by reduced expression of the activation marker CD69) (Fig. 6b). T1D-associated T cells were detected in the pancreas to verify the effects of fingolimod-induced attenuation of intestinal dysfunction on pancreas-directed autoimmunity. The results showed that fingolimod treatment effectively inhibited the recruitment of pathogenic Th1 cells (Fig. 6c) and enhanced the frequency of protective Tregs (Fig. 6d). These results demonstrate that early-life fingolimod treatment contributes to the maintenance of pancreatic immune tolerance in the setting of T1D.
Fig. 6. Early-life fingolimod treatment contributes to pancreatic immune tolerance in diabetes-prone NOD mice.
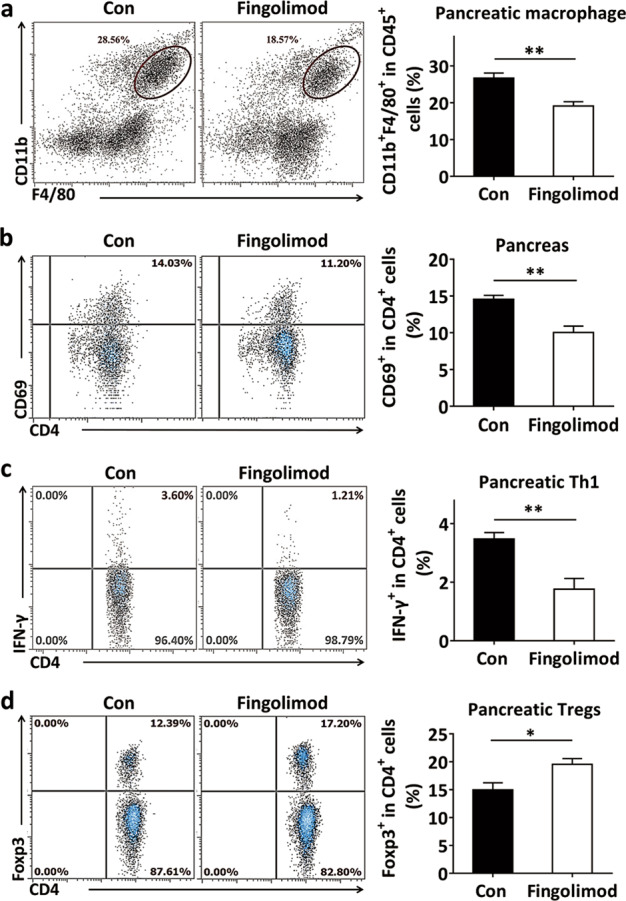
Mice were treated daily with 1 mg/kg fingolimod in 100 µL sterile water or an equal volume of sterile water by gavage from weaning. The pancreas was collected from 12-week-old mice. The number of stained cells was detected by flow cytometry. (a) The frequency of CD45+F4/80+CD11b+ cells among CD45+ cells (macrophages) in the pancreas. (b) The frequency of CD69+CD4+ T-cells among CD4+ cells in the pancreas. (c) The frequency of CD4+IFN-γ+ cells among pancreatic CD4+ cells (Th1 cells). (d) The frequency of CD4+Foxp3+ cells among pancreatic CD4+ cells (Tregs). The data are the mean ± SEM (n = 5–6 experiments with two pooled mice in each experiment/group). *P < 0.05 versus Con, **P < 0.01 versus Con.
Early-life fingolimod treatment prevents T1D in diabetes-prone NOD mice
The effect of fingolimod on T1D development was studied by evaluating the time of onset and the incidence of diabetes in NOD mice. As shown in Fig. 7a, early-life fingolimod treatment delayed the onset of T1D and reduced the incidence of T1D. Compared with no treatment, early-life fingolimod treatment resulted in a delay in T1D onset (Fig. 7a). At 31 weeks of age, 41.67% (5/12) of NOD control mice were diabetes-free, whereas 75% (9/12) of fingolimod-treated mice were diabetes-free (Fig. 7a). In addition, the average insulitis score of fingolimod-treated mice was significantly lower than that of NOD controls (Fig. 7b–c and Fig. S16).
Fig. 7. Early-life fingolimod treatment prevents T1D in diabetes-prone NOD mice.
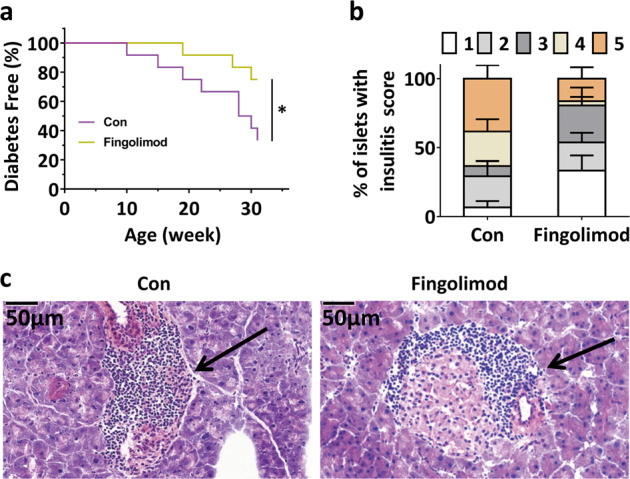
To evaluate the effect of fingolimod on the onset of T1D, NOD mice were randomly assigned to two groups and received fingolimod or an equal volume of sterile water by gavage from weaning to 31 weeks of age, and glucose levels were monitored. (a) Delayed onset and reduced incidence of diabetes in fingolimod-treated mice. Mice were diagnosed with diabetes when blood glucose levels exceeded 11.1 mmol/L on 2 consecutive days. (b) Insulitis scores for NOD controls and fingolimod-treated mice. Percentage of islets with a given score in 12-week-old NOD controls and fingolimod-treated mice (1: no infiltration; 2: few mononuclear cells infiltrated; 3: peri-insulitis; 4: <50% islet infiltration; 5: >50% islet infiltration.). (c) Histological examination of pancreatic islet infiltration by immune cells in female NOD mice at 12 weeks of age. The data are the mean ± SEM (n = 12 mice/group in a; n = 8 mice/group in b, c). *P < 0.05 versus Con.
Discussion
In the present study, early-life fingolimod treatment attenuated intestinal microbial dysbiosis and intestinal immune dysfunction, subsequently contributing to pancreatic immune tolerance in NOD mice, a model of T1D. This protective effect may be due to enhancement of the gut barrier and inhibition of the activation of the colonic TLR2/4-NF-κB-NLRP3 pathway by early-life fingolimod treatment, which may be attributed to reductions in the abundances of pathogenic Proteobacteria clusters. This study demonstrates that early-life fingolimod treatment may affect diabetes progression via modulation of gut microbiota composition and provides us with alternative treatment options for T1D patients.
T1D is an autoimmune disease, and the gut microbiota and gut immune system appear to contribute to its pathogenesis. Expansion of CD4+IFN-γ+ Th1 cells or a decrease in the number or function of Tregs leads to beta-cell destruction and T1D [13–15]. Intriguingly, increased expression of IFN-γ, reduced expression of Foxp3 (a transcription factor of Tregs), and reduced or impaired Tregs are found in the intestine and its draining lymph nodes in addition to the pancreas in T1D patients [51–53]. Previous studies have shown that relieving intestinal inflammation or improving the intestinal microbiota may contribute to the treatment of T1D [7, 54]. Fingolimod has beneficial effects on both T1D and colitis. In addition, in patients with multiple sclerosis, oral administration of fingolimod is effective and more commonly used than intravenous injection of fingolimod, suggesting that restoring intestinal homeostasis may be partly responsible for fingolimod’s therapeutic effects. Furthermore, fingolimod ameliorates colitis in numerous preclinical models, including oxazolone [55] /2,4,6-trinitrobenzene sulfonic acid/dextran sulfate sodium [56]/colitogenic T-cell transfer-induced colitis model, models of memory T-cell-mediated autoimmune inflammatory bowel diseases [57], Th1-cell-mediated colitis model [17] and interleukin-10 gene-deficiency-mediated colitis model [18]. Moreover, fingolimod treatment bolsters the integrity of the blood–brain barrier and enhances the expression of TJPs in diabetic retinopathy and a variety of barrier-related diseases [19, 20, 58]. Based on the present study in conjunction with these previous studies, the inhibitory effect of fingolimod on anti-islet cell autoimmunity may also depend on its modulation of intestinal homeostasis during early life to some extent.
Normally, the intestinal epithelium provides a protective barrier that prevents both pathogenic and commensal bacteria from escaping from the intestinal lumen and activating the systemic immune system. Infection with enteric bacterial pathogens results in a loss of gut barrier integrity [59]. Macrophages play a significant role in guarding parenteral tissue against microbial threats from the gut [5]. Compared to healthy control mice, T1D mice display an altered gut microbiota, increased intestinal inflammation, and increased intestinal permeability [5–7], as well as increased translocation of intestinal bacteria to the PaLNs [5–7]. Subsequently, increased gut bacterial leakage leads to an increase in the number of macrophages in the peritoneal cavity [5–7] and promotes the activation of CD4+ cells (evidenced by increased expression of CD69) and excessive differentiation of pathogenic Th1 cells in both the intestine and pancreas, which may contribute to the pathogenesis of T1D [5, 6, 10, 59]. These studies reveal that changes in the intestinal environment may initiate an innate immune response extraintestinally in the setting of T1D. Fingolimod, as a sphingosine-1-phosphate receptor agonist, prevents the loss of TJPs [1, 19, 20] and inhibits the proliferation of activated lymphocytes and the expression of CD69 on T-cells [60–62]. Furthermore, fingolimod increases the anti-inflammatory properties of macrophages, thus enhancing the immune suppression function of Tregs and maintaining the gut tolerogenic milieu [63]. Based on the present study in conjunction with these previous studies, fingolimod may enhance the gut barrier, thus reducing the recruitment of macrophages and inhibiting the activation of CD4+ cells and the proliferation of pathogenic Th1 cells.
Continuous TLR2 and TLR4 signaling occurs in the microenvironment of the large intestine, where commensal bacteria are extremely abundant [46]. TLR2 and TLR4 are located on gut epithelial cells, as well as on cells of the immune system (especially macrophages and dendritic cells in the lamina propria) [64–66]. Compared to those in the healthy colon, the expression levels of TLR2 and TLR4 are increased in intestinal macrophages in inflamed mucosa [67, 68]. In both diabetes-prone NOD mice and T1D patients, the TLR (particularly TLR2/4)-NF-κB-NLRP3/IL-1β pathway is critically involved in the initiation of host cellular immune responses against enteric pathogens (especially Enterobacteriaceae) and the development of T1D [42, 44, 47, 69]. Interestingly, several studies have demonstrated that fingolimod effectively inhibits the TLR2/4-NF-κB signaling pathway [70] and blocks the activation of NLRP3 and the production of IL-1β [71, 72]. In infection with Enterobacteriaceae (a cluster of enteric pathogens, such as Escherichia coli, Citrobacter rodentium, Salmonella enterica ssp., Enterica serovar Typhimurium, and Salmonella typhimurium), the activation of mammalian TLR2/4 on the cell surface and within endosomes triggers NF-κB activation and is critical for the activation of their downstream noncanonical NLRP3 inflammasome pathway in macrophages [42, 69]. Furthermore, the TLR2-NLRP3 pathway is critical to specific immune regulation in infection with the bacterial pathogen Helicobacter pylori [73, 74]. In the setting of T1D, NLRP3 deficiency not only reduces the number of macrophages and suppresses T-cell activation and Th1-cell differentiation but also impairs the migration of pathogenic T-cells to islets [49], which are thought to be beneficial in reducing the risk of T1D. Based on the present study in conjunction with these previous reports, fingolimod may reduce the abundances of enteric pathogenic Proteobacteria clusters (especially Enterobacteriaceae), subsequently inhibiting the activation of the colonic TLR2/4-NF-κB-NLRP3 pathway and the development of T1D.
Multiple immunosuppressive drugs cause islet dysfunction, limiting their use for T1D therapy [75–77]. The conservation of the insulin secretory function of islets after high-dose fingolimod treatment indicates that this drug is not overtly toxic to islets [75–77], demonstrating that fingolimod can serve as a potential therapeutic for T1D. Nevertheless, there are still many problems associated with the use of fingolimod, such as the high-dose required, its apparent side effects on the entire body, and the low concentration of the drug in target organs [1, 78], suggesting that the development of new therapeutic strategies based on fingolimod should be pursued further. In recent years, many efforts have been made to address these issues. The graphene-fingolimod complex has a drug-releasing effect [79]. Interestingly, compared with oral fingolimod application, local application of the graphene-fingolimod releasing system can alleviate the harmful effects of fingolimod on the systemic immune system and achieve better blood glucose control [79]. Furthermore, either delayed or abrogated efficacy is observed when >0.5% w/w fingolimod is packaged in a fingolimod-three‐dimensional polydimethylsiloxane scaffold (which modulates local delivery of fingolimod in a sustained‐release manner), despite the fact that drug release rates are estimated to be ~80-fold less than published systemic delivery reports where no detrimental effects were noted [80]. Moreover, a preconditioning implant scheme with fingolimod nanofibers effectively prevents exposure of islets to harmful concentrations of fingolimod while simultaneously preserving the therapeutic effects of fingolimod on diabetes [81]. Previous studies and our data suggest that the selection of an appropriate administration method and dose can mitigate the adverse effects of fingolimod and allow better blood glucose control, suggesting the need for further basic medical studies and prospective randomized clinical trials to determine the proper dose and administration route of fingolimod for T1D therapy and bringing a new understanding to the field of the potential for fingolimod as a therapeutic for T1D.
Furthermore, multiple immunoregulatory drugs, including fingolimod, are most effective when applied very early in the clinical course of multiple sclerosis, an autoimmune disease [82]. In the present study, our data indicated that oral administration of fingolimod from early life has beneficial effects in the NOD mouse model of T1D, suggesting that it may represent an effective new therapeutic strategy for T1D. This is consistent with previous reports that fingolimod can prevent T1D if administered before or during stimulation and expansion of autoreactive T cells or in the early stages of insulitis [3, 4]. The effectiveness of fingolimod is diminished in each successive stage of diabetes [3, 4]. Thus, it is possible that an early window of therapeutic opportunity may exist in which the biology of the disease can be modified for a long-term benefit. Furthermore, the number of peritoneal macrophages increases rapidly 1–2 weeks after weaning in NOD mice [5], suggesting that the gut barrier changes around weaning (3-weeks-old) in the setting of T1D. During early life, the intestinal environment undergoes significant developmental changes, and until the entire ecosystem is established, minor intestinal modifications in this critical stage can have significant consequences for intestinal and systemic immune homeostasis [12, 83, 84]. Therefore, effective fingolimod intervention may have to begin in early life. It is also possible that fingolimod intervention in early life is sufficient to protect against T1D.
Collectively, early-life fingolimod treatment maintains intestinal homeostasis (attenuates intestinal microbial dysbiosis and intestinal immune dysfunction) and enhances the gut barrier, thus suppressing the activation of CD4+ cells and the differentiation of Th1 cells by inhibiting the activation of the colonic TLR2/4-NF-κB-NLRP3 signaling pathway (Fig. 8). These results indicate that intestinal microbiota and intestinal immunity may play an essential role in the anti-inflammatory effect of fingolimod in the setting of T1D, providing novel insights into the antidiabetes mechanisms of fingolimod and complementing the conclusions of previous studies, which deserve clinical investigation in the future.
Fig. 8. Graphical abstract.

In diabetes-prone NOD mice, early-life fingolimod treatment enhances the gut barrier and attenuates intestinal microbial dysbiosis and intestinal immune dysfunction, thus suppressing CD4+ T-cell activation and Th1-cell differentiation in the pancreas and promoting the expansion of Tregs. This protective effect of fingolimod may be due to its inhibition of the activation of the colonic TLR2/4-NF-κB-NLRP3 pathway to some extent, which may be mediated by decreases in pathogenic Proteobacteria clusters.
Supplementary information
Acknowledgements
This work was supported by funds from the National Natural Science Foundation of China (Grant Nos. 80270666, 81870439, 81901670, 81973322, and National Youth 1000 Talents Plan), the Natural Science Foundation for Distinguished Young Scholars of Jiangsu Province (Grants No.: BK20200026), the Recruitment Plan for High-level, Innovative and Entrepreneurial Talents (Innovative Research Team) of Jiangsu Province, the Jiangsu Province Qing Lan Project, Jiangsu Province “Six Summit Talents” program (Grant No. 2019-YY-038), the collaborative innovation center of food safety and quality control in Jiangsu Province, National First-Class Discipline Program of Food Science and Technology (Grant No. JUFSTR20180103), Wuxi Social Development Funds for International Science & Technology Cooperation (Grant No. WX0303B010518180007PB), the Fundamental Research Funds for the Central Universities (Grant Nos.: 81870439 and JUSRP22007), Chinese Postdoctoral Science Foundation (Grant No. 2018M642169), Jiangsu Postdoctoral Research Foundation (Grant No. 2018K237C) and Wuxi Taihu Talent Project.
Author contributions
LLP, JS, and LLJ designed the study. LLJ, MZ, and HL conducted the studies. LLJ, MZ, and HL analyzed the data. LLJ wrote the paper. All authors read and approved the final paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Jia Sun, Email: jiasun@jiangnan.edu.cn.
Li-long Pan, Email: llpan@jiangnan.edu.cn.
Supplementary information
The online version of this article (10.1038/s41401-020-00590-4) contains supplementary material, which is available to authorized users.
References
- 1.Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol Ther. 2018;185:34–49. doi: 10.1016/j.pharmthera.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Penaranda C, Tang Q, Ruddle NH, Bluestone JA. Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes. 2010;59:1461–8. doi: 10.2337/db09-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popovic J, Kover KL, Moore WV. The effect of immunomodulators on prevention of autoimmune diabetes is stage dependent: FTY720 prevents diabetes at three different stages in the diabetes-resistant biobreeding rat. Pediatr Diabetes. 2004;5:3–9. doi: 10.1111/j.1399-543X.2004.00036.x. [DOI] [PubMed] [Google Scholar]
- 4.Morris MA, McDuffie M, Nadler JL, Ley K. Prevention, but not cure, of autoimmune diabetes in a NOD.scid transfer model by FTY720 despite effective modulation of blood T cells. Autoimmunity. 2011;44:115–28.. doi: 10.3109/08916934.2010.499885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emani R, Alam C, Pekkala S, Zafar S, Emani MR, Hanninen A. Peritoneal cavity is a route for gut-derived microbial signals to promote autoimmunity in non-obese diabetic mice. Scand J Immunol. 2015;81:102–9. doi: 10.1111/sji.12253. [DOI] [PubMed] [Google Scholar]
- 6.Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. 2016;213:1223–39.. doi: 10.1084/jem.20150744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gavin PG, Mullaney JA, Loo D, Cao KL, Gottlieb PA, Hill MM, et al. Intestinal metaproteomics reveals host-microbiota interactions in subjects at risk for type 1 diabetes. Diabetes Care. 2018;41:2178–86.. doi: 10.2337/dc18-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endesfelder D, Engel M, Davis-Richardson AG, Ardissone AN, Achenbach P, Hummel S, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome. 2016;4:17. doi: 10.1186/s40168-016-0163-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaarala O, Atkinson MA, Neu J. The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes. 2008;57:2555–62.. doi: 10.2337/db08-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao L, Van’t Land B, van de Worp W, Stahl B, Folkerts G, Garssen J. Early-life nutritional factors and mucosal immunity in the development of autoimmune diabetes. Front Immunol. 2017;8:1219. doi: 10.3389/fimmu.2017.01219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mithieux G. Does Akkermansia muciniphila play a role in type 1 diabetes? Gut. 2018;67:1373–4. doi: 10.1136/gutjnl-2017-315732. [DOI] [PubMed] [Google Scholar]
- 12.Hansen CH, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sorensen SJ, et al. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. 2012;55:2285–94.. doi: 10.1007/s00125-012-2564-7. [DOI] [PubMed] [Google Scholar]
- 13.Van YH, Lee WH, Ortiz S, Lee MH, Qin HJ, Liu CP. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-gamma-producing T-cells without affecting Th17 cells. Diabetes. 2009;58:146–55.. doi: 10.2337/db08-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Herrath MG, Oldstone MB. Interferon-gamma is essential for destruction of beta cells and development of insulin-dependent diabetes mellitus. J Exp Med. 1997;185:531–9. doi: 10.1084/jem.185.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marwaha AK, Tan S, Dutz JP. Targeting the IL-17/IFN-gamma axis as a potential new clinical therapy for type 1 diabetes. Clin Immunol. 2014;154:84–9. doi: 10.1016/j.clim.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Peyrin-Biroulet L, Christopher R, Behan D, Lassen C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun Rev. 2017;16:495–503. doi: 10.1016/j.autrev.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007;178:2458–68.. doi: 10.4049/jimmunol.178.4.2458. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima T, Ito T, Kishi D, Kai Y, Tamagawa H, Nezu R, et al. Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin-10 gene-deficient mice with colitis. Inflamm Bowel Dis. 2004;10:182–92.. doi: 10.1097/00054725-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Fan L, Yan H. FTY720 attenuates retinal inflammation and protects blood-retinal barrier in diabetic rats. Invest Ophthalmol Vis Sci. 2016;57:1254–63. doi: 10.1167/iovs.15-18658. [DOI] [PubMed] [Google Scholar]
- 20.Rolland WB, Krafft PR, Lekic T, Klebe D, LeGrand J, Weldon AJ, et al. Fingolimod confers neuroprotection through activation of Rac1 after experimental germinal matrix hemorrhage in rat pups. J Neurochem. 2017;140:776–86.. doi: 10.1111/jnc.13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wicker LS, Miller BJ, Coker LZ, McNally SE, Scott S, Mullen Y, et al. Genetic control of diabetes and insulitis in the nonobese diabetic (NOD) mouse. J Exp Med. 1987;165:1639–54. doi: 10.1084/jem.165.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, et al. Pancreatic beta-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity. 2015;43:304–17. doi: 10.1016/j.immuni.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 23.American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris MI, Hadden WC, Knowler WC, Bennett PH. International criteria for the diagnosis of diabetes and impaired glucose tolerance. Diabetes Care. 1985;8:562–7. doi: 10.2337/diacare.8.6.562. [DOI] [PubMed] [Google Scholar]
- 25.Gabir MM, Hanson RL, Dabelea D, Imperatore G, Roumain J, Bennett PH, et al. The 1997 American Diabetes Association and 1999 World Health Organization criteria for hyperglycemia in the diagnosis and prediction of diabetes. Diabetes Care. 2000;23:1108–12. doi: 10.2337/diacare.23.8.1108. [DOI] [PubMed] [Google Scholar]
- 26.Verdoia M, Schaffer A, Barbieri L, Cassetti E, Nardin M, Bellomo G, et al. Diabetes, glucose control and mean platelet volume: a single-centre cohort study. Diabetes Res Clin Pr. 2014;104:288–94. doi: 10.1016/j.diabres.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 27.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amiel SA, Pursey N, Higgins B, Dawoud D. Guideline Development G. Diagnosis and management of type 1 diabetes in adults: summary of updated NICE guidance. BMJ. 2015;351:h4188. doi: 10.1136/bmj.h4188. [DOI] [PubMed] [Google Scholar]
- 29.Gunawardana SC, Piston DW. Reversal of type 1 diabetes in mice by brown adipose tissue transplant. Diabetes. 2012;61:674–82. doi: 10.2337/db11-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia L, Shan K, Pan LL, Feng N, Lv Z, Sun Y, et al. Clostridium butyricum CGMCC0313.1 protects against autoimmune diabetes by modulating intestinal immune homeostasis and inducing pancreatic regulatory T cells. Front Immunol. 2017;8:1345. doi: 10.3389/fimmu.2017.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. Oncogene. 2017;36:3048–58. doi: 10.1038/onc.2016.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci USA. 2005;102:17729–33. doi: 10.1073/pnas.0509006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia L, Li D, Feng N, Shamoon M, Sun Z, Ding L, et al. Anti-diabetic effects of Clostridium butyricum CGMCC0313.1 through promoting the growth of gut butyrate-producing bacteria in type 2 diabetic mice. Sci Rep. 2017;7:7046. doi: 10.1038/s41598-017-07335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Oliveira GLV, Leite AZ, Higuchi BS, Gonzaga MI, Mariano VS. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology. 2017;152:1–12. doi: 10.1111/imm.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, et al. The potential of gut commensals in reinforcing intestinal barrier function and alleviating inflammation. Nutrients. 2018;10:988. [DOI] [PMC free article] [PubMed]
- 36.Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, et al. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat Commun. 2015;6:7486. doi: 10.1038/ncomms8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeon CY, Haan MN, Cheng C, Clayton ER, Mayeda ER, Miller JW, et al. Helicobacter pylori infection is associated with an increased rate of diabetes. Diabetes Care. 2012;35:520–5. doi: 10.2337/dc11-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan Z, Song L, Hu L, Hu M, Lei X, Huang Y, et al. Helicobacter pylori infection is associated with diabetes among Chinese adults. J Diabetes Investig. 2020; 11:199–205. [DOI] [PMC free article] [PubMed]
- 40.Kalinkovich A, Livshits G. A cross talk between dysbiosis and gut-associated immune system governs the development of inflammatory arthropathies. Semin Arthritis Rheum. 2019;49:474–84. [DOI] [PubMed]
- 41.Sane F, Scuotto A, Pierrat V, Kacet N, Hober D, Romond MB. Diabetes progression and alterations in gut bacterial translocation: prevention by diet supplementation with human milk in NOD mice. J Nutr Biochem. 2018;62:108–22. doi: 10.1016/j.jnutbio.2018.08.017. [DOI] [PubMed] [Google Scholar]
- 42.Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem. 2012;287:34474–83. doi: 10.1074/jbc.M112.401406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JJ, Jo EK. NLRP3 inflammasome and host protection against bacterial infection. J Korean Med Sci. 2013;28:1415–23. doi: 10.3346/jkms.2013.28.10.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc Natl Acad Sci USA. 2015;112:9973–7. doi: 10.1073/pnas.1508740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villena J, Kitazawa H. Modulation of intestinal TLR4-inflammatory signaling pathways by probiotic microorganisms: lessons learned from Lactobacillus jensenii TL2937. Front Immunol. 2014;4:512. doi: 10.3389/fimmu.2013.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macpherson AJ, Smith K. Mesenteric lymph nodes at the center of immune anatomy. J Exp Med. 2006;203:497–500. doi: 10.1084/jem.20060227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tai N, Wong FS, Wen L. The role of the innate immune system in destruction of pancreatic beta cells in NOD mice and humans with type I diabetes. J Autoimmun. 2016;71:26–34. doi: 10.1016/j.jaut.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.S97. [DOI] [PubMed] [Google Scholar]
- 49.Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci USA. 2015;112:11318–23. doi: 10.1073/pnas.1513509112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savinov AY, Burn P. Interference with islet-specific homing of autoreactive T cells: an emerging therapeutic strategy for type 1 diabetes. Drug Discov Today. 2010;15:531–9. doi: 10.1016/j.drudis.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Badami E, Sorini C, Coccia M, Usuelli V, Molteni L, Bolla AM, et al. Defective differentiation of regulatory FoxP3+ T cells by small-intestinal dendritic cells in patients with type 1 diabetes. Diabetes. 2011;60:2120–4. doi: 10.2337/db10-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alam C, Bittoun E, Bhagwat D, Valkonen S, Saari A, Jaakkola U, et al. Effects of a germ-free environment on gut immune regulation and diabetes progression in non-obese diabetic (NOD) mice. Diabetologia. 2011;54:1398–406. doi: 10.1007/s00125-011-2097-5. [DOI] [PubMed] [Google Scholar]
- 53.Westerholm-Ormio M, Vaarala O, Pihkala P, Ilonen J, Savilahti E. Immunologic activity in the small intestinal mucosa of pediatric patients with type 1 diabetes. Diabetes. 2003;52:2287–95. doi: 10.2337/diabetes.52.9.2287. [DOI] [PubMed] [Google Scholar]
- 54.Yap YA, Marino E. An insight into the intestinal web of mucosal immunity, microbiota, and diet in inflammation. Front Immunol. 2018;9:2617. doi: 10.3389/fimmu.2018.02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daniel C, Sartory NA, Zahn N, Schmidt R, Geisslinger G, Radeke HH, et al. FTY720 ameliorates oxazolone colitis in mice by directly affecting T helper type 2 functions. Mol Immunol. 2007;44:3305–16. doi: 10.1016/j.molimm.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 56.Karuppuchamy T, Behrens EH, Gonzalez-Cabrera P, Sarkisyan G, Gima L, Boyer JD, et al. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017;10:162–71. doi: 10.1038/mi.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujii R, Kanai T, Nemoto Y, Makita S, Oshima S, Okamoto R, et al. FTY720 suppresses CD4+CD44highCD62L- effector memory T cell-mediated colitis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G267–74. doi: 10.1152/ajpgi.00496.2005. [DOI] [PubMed] [Google Scholar]
- 58.Cheng Y, Desse S, Martinez A, Worthen RJ, Jope RS, Beurel E. TNFalpha disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav Immun. 2018;69:556–67. doi: 10.1016/j.bbi.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP. Gut barrier disruption by an enteric bacterial pathogen accelerates insulitis in NOD mice. Diabetologia. 2010;53:741–8. doi: 10.1007/s00125-009-1626-y. [DOI] [PubMed] [Google Scholar]
- 60.Zeng X, Wang T, Zhu C, Ye Y, Song B, Lai X, et al. FTY720 mediates activation suppression and G0/G1 cell cycle arrest in a concanavalin A-induced mouse lymphocyte pan-activation model. Inflamm Res. 2012;61:623–34. doi: 10.1007/s00011-012-0454-6. [DOI] [PubMed] [Google Scholar]
- 61.Srinivasan S, Bolick DT, Lukashev D, Lappas C, Sitkovsky M, Lynch KR, et al. Sphingosine-1-phosphate reduces CD4+ T-cell activation in type 1 diabetes through regulation of hypoxia-inducible factor short isoform I.1 and CD69. Diabetes. 2008;57:484–93. doi: 10.2337/db07-0855. [DOI] [PubMed] [Google Scholar]
- 62.Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28:102–7. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 63.Dominguez-Villar M, Raddassi K, Danielsen AC, Guarnaccia J, Hafler DA. Fingolimod modulates T cell phenotype and regulatory T cell plasticity in vivo. J Autoimmun. 2019;96:40–9. doi: 10.1016/j.jaut.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hug H, Mohajeri MH, La Fata G. Toll-like receptors: regulators of the immune response in the human gut. Nutrients. 2018;10:203. [DOI] [PMC free article] [PubMed]
- 65.Santaolalla R, Fukata M, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol. 2011;27:125–31. doi: 10.1097/MOG.0b013e3283438dea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–60. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 67.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7. doi: 10.1128/IAI.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hausmann M, Kiessling S, Mestermann S, Webb G, Spottl T, Andus T, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 69.Waldhuber A, Puthia M, Wieser A, Cirl C, Durr S, Neumann-Pfeifer S, et al. Uropathogenic Escherichia coli strain CFT073 disrupts NLRP3 inflammasome activation. J Clin Invest. 2016;126:2425–36. doi: 10.1172/JCI81916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dong YF, Guo RB, Ji J, Cao LL, Zhang L, Chen ZZ, et al. S1PR3 is essential for phosphorylated fingolimod to protect astrocytes against oxygen-glucose deprivation-induced neuroinflammation via inhibiting TLR2/4-NFkappaB signalling. J Cell Mol Med. 2018;22:3159–66. doi: 10.1111/jcmm.13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doyle TM, Chen Z, Durante M, Salvemini D. Activation of sphingosine-1-phosphate receptor 1 in the spinal cord produces mechanohypersensitivity through the activation of inflammasome and IL-1beta pathway. J Pain. 2019;20:956–64. doi: 10.1016/j.jpain.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao S, Li L, Sun X, Hua J, Zhang K, Hao L, et al. FTY720 inhibits MPP+-induced microglial activation by affecting NLRP3 inflammasome activation. J Neuroimmune Pharmacol. 2019;14:478–92. doi: 10.1007/s11481-019-09843-4. [DOI] [PubMed] [Google Scholar]
- 73.Pachathundikandi SK, Muller A, Backert S. Inflammasome activation by helicobacter pylori and its implications for persistence and immunity. Curr Top Microbiol Immunol. 2016;397:117–31. doi: 10.1007/978-3-319-41171-2_6. [DOI] [PubMed] [Google Scholar]
- 74.Koch KN, Muller A. Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes. 2015;6:382–7. doi: 10.1080/19490976.2015.1105427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fu F, Hu S, Deleo J, Li S, Hopf C, Hoover J, et al. Long-term islet graft survival in streptozotocin- and autoimmune-induced diabetes models by immunosuppressive and potential insulinotropic agent FTY720. Transplantation. 2002;73:1425–30. doi: 10.1097/00007890-200205150-00011. [DOI] [PubMed] [Google Scholar]
- 76.Truong W, Emamaullee JA, Merani S, Anderson CC, James, Shapiro AM. Human islet function is not impaired by the sphingosine-1-phosphate receptor modulator FTY720. Am J Transpl. 2007;7:2031–8. doi: 10.1111/j.1600-6143.2007.01880.x. [DOI] [PubMed] [Google Scholar]
- 77.Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, et al. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. 1999;68:396–402. doi: 10.1097/00007890-199908150-00012. [DOI] [PubMed] [Google Scholar]
- 78.Comi G, Hartung HP, Bakshi R, Williams IM, Wiendl H. Benefit-risk profile of sphingosine-1-phosphate receptor modulators in relapsing and secondary progressive multiple sclerosis. Drugs. 2017;77:1755–68. doi: 10.1007/s40265-017-0814-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu B, Gao Q, Liu R, Ren M, Wu Y, Jiang Z, et al. Effect of a new drug releasing system on microencapsulated islet transplantation. Int J Clin Exp Pathol. 2015;8:12390–9. [PMC free article] [PubMed] [Google Scholar]
- 80.Frei AW, Li Y, Jiang K, Buchwald P, Stabler CL. Local delivery of fingolimod from three-dimensional scaffolds impacts islet graft efficacy and microenvironment in a murine diabetic model. J Tissue Eng Regen Med. 2018;12:393–404. doi: 10.1002/term.2464. [DOI] [PubMed] [Google Scholar]
- 81.Bowers DT, Olingy CE, Chhabra P, Langman L, Merrill PH, Linhart RS, et al. An engineered macroencapsulation membrane releasing FTY720 to precondition pancreatic islet transplantation. J Biomed Mater Res B Appl Biomater. 2018;106:555–68. doi: 10.1002/jbm.b.33862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harding K, Williams O, Willis M, Hrastelj J, Rimmer A, Joseph F, et al. Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol. 2019;76:536–41. doi: 10.1001/jamaneurol.2018.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 84.Hu Y, Jin P, Peng J, Zhang X, Wong FS, Wen L. Different immunological responses to early-life antibiotic exposure affecting autoimmune diabetes development in NOD mice. J Autoimmun. 2016;72:47–56. doi: 10.1016/j.jaut.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.