

Abstract
6-Gingerol, a pungent ingredient of ginger, has been reported to possess anti-inflammatory and antioxidant activities, but the effect of 6-gingerol on pressure overload-induced cardiac remodeling remains inconclusive. In this study, we investigated the effect of 6-gingerol on cardiac remodeling in in vivo and in vitro models, and to clarify the underlying mechanisms. C57BL/6 mice were subjected to transverse aortic constriction (TAC), and treated with 6-gingerol (20 mg/kg, ig) three times a week (1 week in advance and continued until the end of the experiment). Four weeks after TAC surgery, the mice were subjected to echocardiography, and then sacrificed to harvest the hearts for analysis. For in vitro study, neonatal rat cardiomyocytes and cardiac fibroblasts were used to validate the protective effects of 6-gingerol in response to phenylephrine (PE) and transforming growth factor-β (TGF-β) challenge. We showed that 6-gingerol administration protected against pressure overload-induced cardiac hypertrophy, fibrosis, inflammation, and dysfunction in TAC mice. In the in vitro study, we showed that treatment with 6-gingerol (20 μM) blocked PE-induced-cardiomyocyte hypertrophy and TGF-β-induced cardiac fibroblast activation. Furthermore, 6-gingerol treatment significantly decreased mitogen-activated protein kinase p38 (p38) phosphorylation in response to pressure overload in vivo and extracellular stimuli in vitro, which was upregulated in the absence of 6-gingerol treatment. Moreover, transfection with mitogen-activated protein kinase kinase 6 expressing adenoviruses (Ad-MKK6), which specifically activated p38, abolished the protective effects of 6-gingerol in both in vitro and in vivo models. In conclusion, 6-gingerol improves cardiac function and alleviates cardiac remodeling induced by pressure overload in a p38-dependent manner. The present study demonstrates that 6-gingerol is a promising agent for the intervention of pathological cardiac remodeling.
Keywords: 6-gingerol, pressure overload, cardiac remodeling, mitogen-activated protein kinase p38, cardiomyocytes, cardiac fibroblasts, phenylephrine, TGF-β
Introduction
Pathological cardiac remodeling, which is characterized by maladaptive alterations in cardiac structure and function, occurs after myocardial infarction, pressure overload, inflammatory heart muscle disease, idiopathic dilated cardiomyopathy, and volume overload [1]. Long-term left ventricular pressure overload is one of the principal clinical pathogenic factors of pathological cardiac remodeling. Although initially functioning as a compensatory response to elevated ventricular wall pressure, cardiac hypertrophy, features of which include cardiomyocyte enlargement, increased protein synthesis, the formation of rigid cytoskeletal actomyosin fibrillar networks and the expression of embryonic genes, results in progressive compromise of cardiomyocyte contractility [2]. In response to proinflammatory cytokines, vasoactive peptides and hormones, cardiac fibroblasts (CFs) transdifferentiate into a myofibroblast phenotype that expresses contractile proteins and exhibits increased migratory, proliferative, and secretory properties. Myofibroblasts play a key role in extracellular matrix (ECM) overproduction and deposition, resulting in altered heart architecture with deleterious effects on cardiac function [3]. Left ventricular pressure overload can result in chronic cardiac inflammation characterized by inflammatory cell infiltration and myocardial release of proinflammatory cytokines, which are associated with the hypertrophic response of cardiomyocytes and increase fibrosis, cardiac contractile dysfunction, and adverse cardiac remodeling [4–7]. Therefore, pharmacological treatments targeting the underlying molecular mechanisms of cardiac hypertrophy, fibrosis, and inflammation may be of great therapeutic interest for improving the prognosis of pathological cardiac remodeling.
The mitogen-activated protein kinase (MAPK) signaling cascade is an attractive intermediate signal transduction cascade for pharmacological intervention in pathological cardiac remodeling because it is typically activated in response to most hypertrophy-associated stimuli [8]. Among the MAPK subfamilies, p38 is a protein kinase that is activated by proinflammatory cytokines, growth factors, mechanical stretch, hemodynamic stresses, and other stress stimuli and modulates and orchestrates a wide array of genes at the transcriptional level in the heart, the majority of which are associated with cell division, inflammation, fibrosis, cell signaling, cell adhesion, and transcription [9]. Although the role of p38 in the short-term and long-term development of cardiac hypertrophy is controversial and multifaceted, it is clear from the results of both in vitro and in vivo studies that p38 activation has a detrimental effect on cardiac function and exacerbates pathological cardiac remodeling after injury [10, 11]. Therefore, pharmacological inhibition of p38 signaling is a novel therapeutic strategy for pathological cardiac remodeling.
6-Gingerol (6G) is a pungent, nontoxic, and biologically active component of ginger rhizomes that has been shown to possess multiple pharmacological activities, including antibacterial [12], anti-inflammatory [13–15], antioxidant [13, 16], cardiotonic [17], and antitumoral [18, 19] properties, demonstrating the substantial medicinal applications of ginger. Several studies have shown that 6G exerts protective effects by attenuating the production of inflammatory cytokines and the expression of inflammatory mediators by inhibiting p38/nuclear factor kappa B (NF-κB) signaling in a variety of animal models, such as rat intestinal ischemia/reperfusion [15], phorbol ester-stimulated mouse skin [14], and rat diabetes-induced kidney damage [13]. In addition, gingerol can prevent vascular smooth muscle cell proliferation, neointimal hyperplasia, endothelial dysfunction, and phenylephrine (PE)-induced vascular contraction by modulating p38 MAPK, suggesting its pharmacological role in protecting against the progression of vascular proliferative diseases, such as atherosclerosis [20]. 6G can also suppress oxidative stress, apoptosis, mitochondrial energy metabolism disorders, and respiratory function impairments during doxorubicin-induced cardiomyocyte toxicity by targeting phosphoinositide 3-kinase/protein kinase B (Akt) and peroxisome proliferator-activated receptor α (PPARα)/PPARγ coactivator 1α (PGC-1α)/sirtuin 3 (SIRT3) signaling [16, 21]. Based on these data, we hypothesized that 6G may have promising protective applications in cardiac remodeling. However, little work has been performed to determine whether 6G has protective effects against chronic pressure overload-induced cardiac remodeling through a direct effect on cardiomyocytes and CFs. Thus, in the present study, we performed in vivo and in vitro experiments to assess the effect of 6G on cardiac remodeling and elucidate the underlying mechanism.
Materials and methods
Animals and treatments
Male C57 black 6 (C57BL/6) mice (8–10 weeks old; 23.5–27.5 g) were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). The animals were housed and raised in a pathogen-free environment with an appropriate temperature and humidity. All of the animal care and experimental procedures complied with the guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH publication, 2011 Revision). The animal experiments were approved by the Laboratory Animal Welfare & Ethics Committee of Renmin Hospital of Wuhan University (IACUC Issue No. WDRM20180401). Transverse aortic constriction (TAC) operations were performed to establish the cardiac hypertrophy model [22]. All mice were randomly divided into four groups: the sham + vehicle, sham + 6G, TAC + vehicle, and TAC + 6G groups. During the operations, the mice were first anesthetized by an intraperitoneal injection of 3% pentobarbital sodium at a dose of 50 mg/kg. The left chest of each mouse was then opened at the second intercostal space to expose the thoracic aorta by blunt dissection. The exposed aorta was tied against a 27-gauge (for body weights (BWs) of 24–25 g) or a 26-gauge (for BWs of 26–27 g) needle with a 7–0 silk suture that was later removed. The efficiency and adequacy of aortic constriction were confirmed by Doppler analysis [23]. 6G suspended in 50 μL of 0.002% ethanol was administered to each mouse by gavage three times each week. Three different doses of 6G were used in our study (low dose: 0.1 mg; middle dose: 0.25 mg; high dose: 0.5 mg). The doses of 6G were determined according to a previous article [18]. Consistently, we observed dose-dependent protective effects of 6G, and 0.5 mg 6G was the most efficient at alleviating cardiac hypertrophy, fibrosis, and dysfunction post TAC. Thus, this dose of 6G was used for subsequent experiments. To determine whether the protective effects of 6G on cardiac remodeling depended on p38, the mice were administered an intramyocardial injection of 1 × 109 viral genome particles in three separate locations in the left ventricle (LV) free wall of the heart. Mitogen-activated protein kinase kinase 6 expressing adenoviruses (Ad-MKK6) or β-galactosidase-expressing adenoviruses (Ad-LacZ, as a control) were each diluted in 15 μL of phosphate-buffered saline. One week after adenoviral injection, the mice were subjected to TAC surgery. 6G treatment began 1 week before surgery and continued until the end of the experiment. Four weeks after TAC surgery, the mice were subjected to echocardiography measurements. Finally, the mice were sacrificed with an overdose of sodium pentobarbital (200 mg/kg; intraperitoneal injection), and the hearts were harvested.
Echocardiography
After the mice were anesthetized with 1.5% isoflurane, echocardiography was performed to obtain two-dimensional M-mode echocardiographic images of the papillary muscle for five consecutive cardiac cycles. LV morphology was primarily assessed by measuring the left ventricular internal diastolic diameter (LVIDd), shortening fraction (FS), and left ventricular ejection fraction (LVEF).
Histological analysis
To evaluate myocardial hypertrophy and fibrosis, we performed morphological analyses of heart sections. Mouse hearts were removed after euthanasia, arrested in diastole with a 10% potassium chloride solution and then fixed in 10% formalin, followed by embedding in paraffin according to standard histological protocols. The hearts were then transversely sliced into 5-μm-thick sections in the region close to the apex to visualize the LVs [24]. Sections of at least six heart samples from each group were stained with hematoxylin and eosin (H&E) to evaluate cardiomyocyte cross-sectional areas or with picrosirius red (PSR) to evaluate the extent of interstitial and perivascular fibrosis. Image-Pro Plus version 6.0 was used to observe and calculate the cross-sectional areas of individual cardiomyocytes (at least 200 cardiomyocytes in each group) and measure the collagen volume. For immunohistochemistry, the heart sections underwent a 5-min high-pressure and high-temperature treatment in citrate buffer for antigen retrieval [23]. After being washed, the heart tissues were incubated with 0.3% hydrogen peroxide to block endogenous peroxidase activity and then incubated with 10% goat serum [25]. The sections were then incubated overnight at 4 °C with primary antibodies, including anti-α-smooth muscle actin (α-SMA), anti-cluster of differentiation (CD)45, and antitumor necrosis factor-α (TNF-α) (1:100). On the following day, the sections were incubated with goat anti-rabbit/-mouse immunoglobulin G (IgG) labeled with horseradish peroxidase for 30 min. Finally, a diaminobenzidine detection kit (Gene Technology, Shanghai, China; GK600705) was used to visualize the target proteins.
Western blot analysis
Western blot analysis was performed to examine the expression levels of the target proteins in tissues and cells. Total protein was extracted from heart tissues and cells in lysis buffer containing 720 μL of radioimmunoprecipitation assay buffer, 20 μL of phenylmethyl sulfonyl fluoride, 100 μL of complete protease inhibitor cocktail, 100 μL of PhosStop, 50 μL of NaF, and 10 μL of Na3VO4. A bicinchoninic acid protein analysis kit was used to determine and standardize the protein concentrations of all extracted samples. Markers (4 μL of marker plus 6 μL of loading buffer) and quantitative protein samples (50 μg per well) were loaded into gels and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis before being transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA; IPFL00010). The electrophoretic voltage was set at 75 V at the beginning of electrophoresis and was increased to 100 V when the samples reached the boundary between the stacking gel and separating gel. The PVDF membranes were blocked with 5% skim milk dissolved in Tris-buffered saline for 1 h. Then, after being thoroughly washed in Tris-buffered saline Tween-20 (TBST), the membranes were incubated with primary antibodies (1:1000) diluted in TBST containing 5% skim milk overnight at 4 °C. The following day, the PVDF membranes were washed in TBST 3 times and then incubated with either goat anti‐rabbit IgG or goat anti‐mouse IgG secondary antibodies (1:10,000) diluted in TBST containing 5% skim milk for 1 h at room temperature. A two-color infrared imaging system (Odyssey, LI-COR Biosciences, Lincoln, NE, USA) was used to scan the blots. The specific protein expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control.
Quantitative real-time polymerase chain reaction (PCR)
To examine the messenger ribonucleic acid (mRNA) expression of markers associated with cardiac hypertrophy, fibrosis and inflammation, total RNA was isolated from the LV, neonatal rat cardiomyocytes (NRCMs), and CFs using TRIzol reagent (Invitrogen, Carlsbad, CA, USA; 15596-026). Briefly, 100 mg of tissue samples or 107 cells were homogenized in 1 mL of TRIzol reagent and then mixed with 200 μL of chloroform for RNA extraction. After centrifugation, the supernatants containing RNA were added to isopropanol for RNA precipitation. The precipitated RNA was then washed with 75% alcohol, which was prepared with diethyl pyrocarbonate (DEPC)-treated water, and then dissolved in DEPC-treated water after desiccation. The 260/280 absorbance ratio was measured with a spectrophotometer to estimate the purity of the RNA and calculate the RNA concentration. Then, the total RNA extracted from tissues and cells was reverse transcribed into complementary deoxyribonucleic acid (cDNA) at a concentration of 100 ng/μL using oligo (dT) primers and a Transcriptor First Strand cDNA Synthesis kit (Roche, Basel, Switzerland; 04896866001). The reverse transcription product was diluted to a concentration of 25 ng/μL and used as template cDNA. PCR was performed in a 20-μL reaction system with each well containing 1 μL of template cDNA at a final concentration of 25 ng/μL, 0.5 μL of 10 μM forward primer at a final concentration of 0.25 μM, 0.5 μL of 10 μM reverse primer at a final concentration of 0.25 μM, 8 μL of DEPC-treated water and 10 μL of SYBR Green I Master Mix. The levels of target gene transcripts were normalized to GAPDH. All primer sequences used in this study are listed in Supplementary Table S1.
Cell culture and treatments
NRCMs and CFs were isolated from 1 to 2-day-old Sprague-Dawley rats. Neonatal rats were disinfected with 75% alcohol, after which the hearts were rapidly excised, gently washed and cut into pieces. Then, the ventricles were digested in 0.125% trypsin prepared in D-Hanks solution four times for 15 min each. Digestion was stopped by the addition of 16 mL of Dulbecco’s modified Eagle’s medium/nutrient mixture F12 (DMEM/F12, Gibco, Grand Island, NY, USA; C11330500BT) containing 4 mL of fetal bovine serum (FBS, Gibco, Grand Island, NY, USA; 10099-141) [26, 27]. NRCMs and CFs were isolated using a differential attachment technique [24]. Specifically, the resuspended cells were incubated in a 100-mm culture dish for 90 min at 37 °C until the CFs adhered to the wall of the dish. The relatively pure CFs were then collected and cultured in DMEM/F12 supplemented with 15% FBS at 37 °C in a humidified incubator and an atmosphere with 5% CO2 [28]. The unattached cardiomyocytes were then seeded onto six-well culture plates coated with gelatin in plating medium at a density of 5 × 105 cells per well. NRCMs were cultured in DMEM/F12 supplemented with 15% FBS, 5-bromodeoxyuridine (0.1 mM, to inhibit fibroblast proliferation) and penicillin/streptomycin for 48 h [24]. The purity of NRCMs and CFs was verified by morphological observations and immunofluorescence staining for α-actinin and vimentin, and only cell samples with purities greater than 95% were used for further experiments. CFs at the second passage were used for subsequent experiments. To assess the effects of 6G on cardiomyocyte hypertrophy and cardiac fibroblast differentiation, NRCMs were treated with different concentrations of 6G (5, 10, and 20 μM) [21] or an equal volume of vehicle in the presence or absence of PE (50 μM) for 24 h, whereas CFs were incubated with the indicated concentration of 6G or vehicle with or without transforming growth factor-β (TGF-β) (10 ng/mL) stimulation for 24 h. 6G was dissolved in dimethyl sulfoxide and then diluted to the indicated concentrations. To evaluate the role of p38 in the protective effect of 6G, the culture medium was changed to serum-free DMEM/F12 for 12 h, after which NRCMs and CFs were infected with Ad-MKK6 at a multiplicity of infection of 100 particles per cell to constitutively activate p38 or with Ad-LacZ as a negative control.
Immunofluorescence staining
Before being incubated with antibodies, the heart tissue sections underwent a 5-min high-pressure and high-temperature treatment in citrate buffer for antigen retrieval, while cardiomyocytes and fibroblasts were fixed with 4% paraformaldehyde and then permeabilized with 0.2% Triton X-100. Then, the sections and cells were blocked in 10% goat serum and incubated with anti-α-actinin, anti-α-SMA, anti-phosphorylated (p) p38, anti-p-NF-κB p65 subunit (p65) and anti-p-mothers against DPP homolog (Smad)3 antibodies (1:100) at 4 °C overnight. The following day, the sections and cells were incubated at 37 °C for 60 min with Alexa Fluor 488 or 568 goat anti-rabbit secondary antibodies (Invitrogen, Carlsbad, CA, USA; A11008, A11011) (1:200). 4′,6-Diamidino-2-phenylindole dihydrochloride was used to visualize the nuclei. The samples were imaged with a fluorescence microscope (Olympus dx51, Tokyo, Japan) and then analyzed with Image-Pro Plus software.
Plasmids and adenovirus production
The Ad-MKK6 used in our study was generated by Hanbio Biotechnology Co. (Shanghai, China). To produce adenovirus expressing MKK6Glu, MKK6Glu in pcDNA3.1 was subcloned into pENTR3C at the Not I and Kpn I sites. The primer sequences were 5′-TTAAGGGTACCGGCGCCATGTCTCAGTCGAAAGGCAA-3′ (forward) and 5′-TTAAGGCGGCCGCTTATCATTAGTCTCCAAGAATCAG-3′ (reverse). The resulting MKK6Glu in pENTR3C was further subcloned into pAD by using LR clonase to generate the adenovirus. MKK6Glu was produced with the ViraPower Adenoviral Expression System according to the manufacturer’s instructions.
Antibodies and reagents
Antibodies against p-c-Jun N-terminal kinase (JNK) (4668P), p-p38 (4511P), p-extracellular signal-regulated kinase (ERK) (4370P), p-Akt (4060), total (t)-JNK (9258), t-p38 (9212P), t-ERK (4695), t-Akt (4691), p-p65 (3033), t-p65 (8242), and GAPDH (2118) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against α-actinin (ab68167), α-SMA (ab5694), p-Smad3 (ab52903), t-Smad3 (ab40854), CD45 (ab10558), collagen I (ab34710), troponin I (ab47003), vimentin (ab92547), and TNF-α (ab6671) were obtained from Abcam (Cambridge, UK). 6G (≥98% purity verified by high-performance liquid chromatography), PE (P1240000) and TGF-β (T7039) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Proteins were measured with assay kits obtained from Pierce (Rockford, IL, USA; 23225). All other chemicals were of analytical grade.
Statistical analysis
All data are presented as the means ± standard error of the mean and were analyzed by statistical products and service solutions 22.0 software. Comparisons between two groups were analyzed by unpaired, two-tailed Student’s t-tests. When the analysis of variance showed that the F value was significant and there was no difference in homogeneity, one-way analysis of variance followed by the least significant difference post hoc test was used for comparisons among multiple groups. A value of P < 0.05 was considered statistically significant.
Results
6-Gingerol alleviated cardiac remodeling and dysfunction post TAC in vivo
To assess the effect of 6G on cardiac remodeling induced by pressure overload, C57BL/6 mice were subjected to either TAC or sham surgery. The TAC operation significantly increased the heart weight-to-BW (HW/BW) ratios and the HW-to-tibia length (HW/TL) ratios of mice in the vehicle-treated group, while these hypertrophic indicators were decreased after the administration of high and middle doses of 6G (Fig. 1a). In addition, mice that were treated with a high dose of 6G exhibited improvements in FS and LVEF and a reduction in LVIDd after the TAC operation (Fig. 1b). Serum levels of alanine aminotransferase and aspartate aminotransferase were not significantly different between vehicle-treated and 6G-treated mice (Supplementary Fig. S1), suggesting that 6G does not induce hepatotoxicity, which was consistent with the results of a previous study [29]. As shown by H&E and PSR staining, the cross-sectional and fibrosis areas of the mice treated with middle/high doses of 6G in response to pressure overload were less than those of the mice treated with vehicle or a low dose of 6G (Fig. 1c–e). Moreover, the mRNA expression levels of hypertrophic markers, including atrial natriuretic peptide, brain natriuretic peptide (BNP), and β-myosin heavy chain, were markedly decreased after treatment with middle/high doses of 6G (Fig. 1f). These data indicated that 6G treatment attenuated TAC-induced cardiac remodeling in a dose-dependent manner.
Fig. 1. 6-Gingerol attenuated pressure overload-induced cardiac remodeling and dysfunction in vivo.

a Statistical analysis of the HW/BW ratio and HW/TL ratio at 4 weeks post TAC operation (n = 12). b Echocardiographic parameters in 6G- and vehicle-treated mice (n = 10). c Representative H&E and PSR staining. d Statistical analysis of the cardiomyocyte cross-sectional area (n = 6). e Statistical analysis of the average collagen volume (n = 6). f The mRNA levels of hypertrophic markers (n = 6). 6G 6-Gingerol. Low dose: 0.1 mg; middle dose: 0.25 mg; high dose: 0.5 mg. *P < 0.05 vs. the corresponding sham group. NS no statistical significance.
6-Gingerol inhibited collagen deposition and myofibroblast differentiation
The extent of cardiac fibrosis was estimated by PSR staining. Although little ECM deposition was detected with or without 6G treatment at baseline, 6G slowed the progression of perivascular and interstitial fibrosis after TAC compared to that observed in the control mice, as shown by the visibly reduced percentage of fibrosis (Fig. 1e). Immunohistochemical analysis of α-SMA as a marker of transformation from interstitial fibroblasts to myofibroblasts was conducted to further investigate the effect of 6G on cardiac fibrosis after TAC. As shown in Fig. 2a, 6G-treated mice exhibited reduced α-SMA staining intensity after TAC. In addition, the mRNA levels of collagen I and collagen III were dramatically suppressed after 6G treatment (Fig. 2b). Consistent with these results, protein quantification by Western blot analysis demonstrated that 6G reduced the expression and production of α-SMA and collagen I (Fig. 2c, d).
Fig. 2. 6-Gingerol protected against cardiac fibrosis post TAC in vivo.

a Representative immunohistochemical images of α-SMA expression and statistical analysis of the α-SMA intensity (n = 6). b The mRNA levels of fibrotic markers (n = 6). c, d Representative Western blots of α-SMA and collagen expression and the statistical analysis of the protein levels (n = 6). 6G 6-Gingerol. *P < 0.05 vs. the corresponding sham group. #P < 0.05 vs. the TAC+vehicle group.
6-Gingerol inhibited cardiac inflammation following chronic pressure overload in vivo
Cardiac inflammation was primarily assessed by the infiltration of inflammatory cells and the release of inflammatory cytokines. The TAC operation stimulated severely excessive inflammatory responses, as reflected by the large increase in the transcription levels of cytokines, including interleukin-6 (IL-6), TNF-α, and monocyte chemoattractant protein-1 (MCP-1), which were sharply reduced by 6G treatment (Fig. 3a). In accordance with the results showing decreased expression of inflammatory factor genes, the immunohistochemical staining densities of CD45-labeled leukocytes and TNF-α also diminished after 6G treatment (Fig. 3b), indicating that the infiltration of inflammatory cells and the production of inflammatory factors were abated by 6G treatment. The NF-κB signaling pathway plays a crucial role in chronic cardiac inflammation, and the p65 subunit is the primary active site involved in promoting inflammation, exacerbating pathological remodeling and increasing mortality [30]. Western blot analysis showed that the phosphorylation of p65 was attenuated by 6G treatment after TAC surgery (Fig. 3c). Furthermore, we performed immunofluorescence colocalization staining analysis of p-p65 in cardiac tissues to assess whether 6G modulated the activation of NF-κB. As shown in Fig. 3d, the percentage of p-p65-positive nuclei was reduced by 6G treatment after TAC, suggesting that 6G alleviated inflammation by downregulating p65 activation.
Fig. 3. 6-Gingerol alleviated pressure overload-induced cardiac inflammation in vivo.

a The mRNA levels of inflammatory cytokines (n = 6). b Representative immunohistochemical images of CD45 and TNF-α and statistical analysis of CD45-positive cells and the TNF-α intensity (n = 6). c Representative Western blots of total (t-) and phosphorylated (p-) p65 and the quantitative analysis of the ratios of phosphorylated-to-total p65 in the indicated groups (n = 6). d Representative images of immunofluorescence staining of p-p65 and fluorescein isothiocyanate-labeled wheat germ agglutinin (WGA) staining and the percentage of p-p65-positive nuclei (n = 6). 6G 6-Gingerol. *P < 0.05 vs. the corresponding sham group. #P < 0.05 vs. the TAC+vehicle group.
6-Gingerol inhibited p38 and Smad3 phosphorylation in mouse hearts stimulated by chronic pressure overload
To elucidate the specific molecular mechanism associated with the cardioprotective effects of 6G, we examined the levels of marker proteins involved in the Akt and MAPK signaling pathways. Although the phosphorylation levels of JNK, ERK1/2 and Akt did not exhibit significant changes between 6G-treated mice and vehicle-treated mice at baseline or after TAC surgery, we observed that p38 phosphorylation post TAC was inhibited by 6G treatment (Fig. 4a, b). Furthermore, we performed immunofluorescence staining of p-p38 in cardiac tissues. The results showed that the cytoplasmic and nuclear densities of p-p38 markedly decreased after 6G treatment, indicating that 6G inhibited TAC-induced p38 activation (Fig. 4c). In addition to the observed downregulation of p-p38, p-Smad3 levels were also decreased by 6G treatment (Fig. 4b), suggesting that the p38 and Smad3 pathways are responsible for the protective effects of 6G.
Fig. 4. Alterations in signaling pathways after 6-gingerol treatment.

a Representative Western blots showing total and phosphorylated JNK, p38, ERK, Smad3, and Akt. b The statistical analysis of the Western blot results. (n = 6). c Representative images of immunofluorescence staining for p-p38 and WGA and the percentage of p-p38-positive nuclei (n = 6). 6G 6-Gingerol. *P < 0.05 vs. the corresponding sham group. #P < 0.05 vs. the TAC + vehicle group. NS no statistical significance.
6-Gingerol protected against cardiomyocyte hypertrophy in a p38-dependent manner in vitro
To further confirm the optimal dose of 6G to protect against cardiac remodeling in vitro, NRCMs, and CFs were incubated with serum-free DMEM/F12 for 12 h to achieve synchronization. Subsequently, different concentrations of 6G (5, 10, and 20 μM) or an equal volume of vehicle was added to the medium. As shown in Supplementary Fig. S2, 6G treatment blocked cardiac hypertrophy and fibrosis marker mRNA levels in a dose-dependent manner in vitro. Therefore, we used 20 μM 6G in our in vitro study. The hypertrophic response of NRCMs induced by PE can be intuitively observed by α-actinin immunofluorescence staining. In line with the in vivo observations, 6G dramatically reduced the extended cell surface area in response to PE challenge (Fig. 5a). Mechanistically, the phosphorylation and nuclear translocation of p38 after PE stimulation were diminished in the 6G-treated NRCMs, as indicated by the reduced expression of p-p38 (Fig. 5b, c), which was consistent with the in vivo data. Thus, some NRCMs were infected with Ad-MKK6 to constitutively activate p38, while the rest were infected with Ad-LacZ as a control. 6G treatment did not modulate p38 phosphorylation in NRCMs infected with Ad-MKK6 (Fig. 5d). The α-actinin immunofluorescence staining results also verified that the antihypertrophic effect of 6G in vitro was neutralized by excessive activation of p38 (Fig. 5e). Moreover, no significant difference was observed in the mRNA transcript levels of hypertrophic markers in PE-stimulated NRCMs treated with 6G or vehicle in the presence of Ad-MKK6 transfection (Fig. 5f). Taken together, these results indicate that the protective effect of 6G on cardiomyocyte hypertrophy in vitro is mediated by p38 pathway suppression.
Fig. 5. 6-Gingerol mitigated PE-induced cardiomyocyte hypertrophy in vitro but did not protect NRCMs expressing constitutively active p38.

a Cell surface area was determined by α-actinin immunofluorescence staining and the quantitative analysis of NRCMs (n = 6). b Representative immunofluorescence staining of p-p38 and the percentage of p-p38-positive nuclei in NRCMs (n = 6). c Representative Western blots showing total and phosphorylated p38 and the statistical analysis of the ratios of phosphorylated-to-total p38 in NRCMs (n = 6). d The effect of 6G on p38 phosphorylation in NRCMs infected with Ad-MKK6 (n = 6). e, f The effect of 6G on PE-induced cardiomyocyte hypertrophy in NRCMs with sustained p38 activation (n = 6). 6G 6-Gingerol. *P < 0.05 vs. the corresponding control group. #P < 0.05 vs. the PE + vehicle group. NS no statistical significance.
6-Gingerol suppressed the activation of cardiac fibroblasts by inhibiting p38/Smad3 activation in vitro
CFs have long been regarded as dominant factors in the process of cardiac fibrosis. Induced by pressure overload or growth factors/cytokines, CFs transdifferentiate into myofibroblasts and generate excessive amounts of ECM proteins, such as collagen I, collagen III, and other glycoproteins, inducing the disordered arrangement and maladjusted mechanoelectrical coupling of cardiomyocytes and subsequent impairment of myocardial contraction [31]. To assess whether 6G blocks CF differentiation in vitro, neonatal rat CFs were isolated and passaged to the second generation for further experiments. Consistent with the in vivo data, 6G treatment inhibited the significant increase in α-SMA expression in CFs induced by TGF-β (Fig. 6a, d). We also demonstrated that 6G blocked TGF-β-mediated phosphorylation and nuclear translocation of Smad3 in CFs (Fig. 6b, d). Therefore, we further explored whether p38 was involved in the antifibrotic effect of 6G in vitro. As shown in Fig. 6c, the phosphorylation and nuclear translocation of p38 were inhibited by 6G treatment. In addition, to confirm whether the antifibrotic effect of 6G could be reversed by p38 activation, we randomly transfected CFs with Ad-MKK6 or Ad-LacZ as a control. As shown in Supplementary Fig. S4, α-SMA expression and collagen biosynthesis were markedly exacerbated in CFs transfected with Ad-MKK6 regardless of 6G treatment. Interestingly, 6G inhibited the phosphorylation and nuclear translocation of Smad3, which was counteracted by p38 overexpression (Fig. 6e, f). In summary, these results demonstrate that 6G can block the activation of CFs in vitro by suppressing the p38-Smad3 pathway.
Fig. 6. 6-Gingerol blocked TGF-β-induced cardiac fibroblast activation and differentiation in a p38-dependent manner in vitro.

a Representative immunofluorescence staining of α-SMA and statistical analysis of the α-SMA intensity in cardiac fibroblasts (CFs). b, c Representative immunofluorescence staining of phosphorylated Smad3 and p38 and the statistical analysis of the positive nuclei percentage (n = 6). d Representative Western blots showing p-Smad3, t-Smad3, and α-SMA in CFs and the statistical analysis (n = 6). e, f The effects of 6G on α-SMA intensity and Smad3 activation induced by TGF-β in CFs infected with Ad-MKK6 (n = 6). 6G 6-Gingerol. *P < 0.05 vs. the corresponding control group. #P < 0.05 vs. the TGF-β + vehicle group. NS no statistical significance.
p38 overexpression reversed 6-gingerol-mediated cardioprotection in vivo
To ascertain whether the 6G-mediated cardioprotective effects against pressure overload were p38 dependent, mice were infected with adenoviral vectors carrying MKK6 to induce constitutively active p38. The activation of p38 by adenoviral transfection is shown in Supplementary Fig. S3. Consistent with the above findings, Ad-MKK6-infected mice were insensitive to the antihypertrophic and antifibrotic effects of 6G, as indicated by indistinct reductions in HW/BW, HW/TL, echocardiographic alterations, the myocyte cross-sectional area, collagen volume, and mRNA levels of hypertrophic and fibrotic markers after 6G treatment (Fig. 7a–g). In addition, the anti-inflammatory effect of 6G in remodeled hearts was abrogated by p38 activation, since transcript levels of IL-6, TNF-α, and MCP-1 and inflammatory cell invasion and infiltration were dramatically reversed after infection with Ad-MKK6 despite 6G treatment (Fig. 8a, d). Furthermore, Ad-MKK6 transfection substantially promoted p65 phosphorylation and nuclear translocation, which were inhibited by 6G treatment (Fig. 8b, c). Taken together, these results provide the first clear evidence that the protective effects of 6G against cardiac remodeling in vivo are mediated by the inhibition of the p38 MAPK pathway (Fig. 9).
Fig. 7. 6-Gingerol lost its antihypertrophic and antifibrotic effects in mice with constitutively active p38.

a Statistical analysis of the HW/BW ratio and HW/TL ratio (n = 10). b Echocardiographic parameters in the indicated groups (n = 10). c–e Representative images of H&E and PSR staining and statistical analysis of the cardiomyocyte cross-sectional area and average collagen volume (n = 10). f, g Relative mRNA levels of hypertrophic and fibrotic markers (n = 10). 6G 6-Gingerol. NS no statistical significance.
Fig. 8. The effects of 6-gingerol against cardiac inflammation were blocked by Ad-MKK6 transfection in vivo.

a Representative immunohistochemical images of CD45 and TNF-α and the statistical analysis of CD45-positive cells and the TNF-α intensity (n = 6). b, c Western blots and immunofluorescence staining showing the phosphorylation levels of p65 in mouse hearts (n = 6). d Relative mRNA levels of inflammatory cytokines in mouse hearts (n = 6). 6G 6-Gingerol. NS no statistical significance.
Fig. 9. Illustrative diagram of the protective effect of 6-gingerol on pressure overload-induced cardiac remodeling.
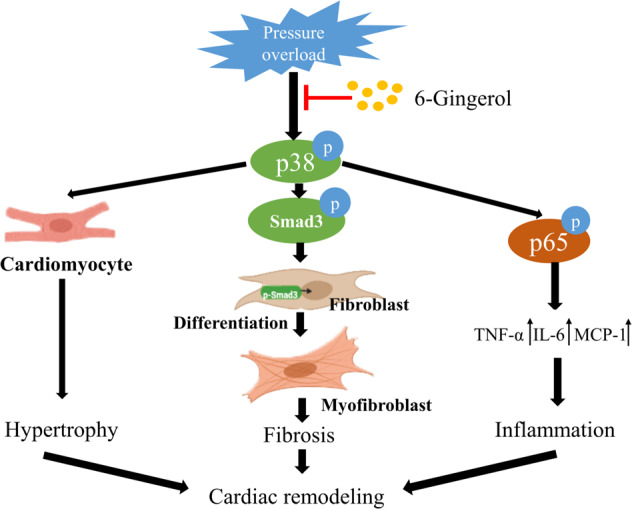
6-Gingerol treatment protected against pressure overload-induced cardiac remodeling by blocking p38 activation in vitro and in vivo without cell specificity. In addition, 6-gingerol also blocked the phosphorylation of p65 and subsequently decreased inflammatory cytokines by inhibiting the p38 pathway. Thus, 6-gingerol plays a critical protective role in cardiac remodeling by alleviating cardiac hypertrophy, fibrosis, and inflammation in a p38-dependent manner.
Discussion
Pathological hypertrophy, fibrosis and inflammation are cardiac maladjustments induced by pressure overload, such as hypertension and aortic stenosis, leading to alterations in gene transcription and protein expression that trigger cardiac remodeling. Our results are the first to demonstrate the antihypertrophic effect of 6G in response to pressure overload in vivo. Furthermore, we demonstrated that 6G could mitigate collagen deposition and thereby prevent cardiac fibrosis. Another novel finding of the present study is that 6G was shown to have an anti-inflammatory effect on post-TAC heart tissues. The results of our in vitro experiments suggested that 6G could directly block cardiomyocyte hypertrophy and cardiac fibroblast activation via p38 inhibition. Based on the in vivo and in vitro data, we conclude that 6G protects against cardiac remodeling during pressure overload stimulation in a p38-dependent manner (Fig. 9).
Previous studies have shown the protective effects of 6G in a number of cardiovascular diseases. One study demonstrated that the combined application of 6G and higenamine could promote myocardial energy metabolism through the liver kinase B1/adenosine monophosphate-activated protein kinase α/SIRT1 pathway during doxorubicin-induced cardiotoxicity [17]. Other studies have reported the antifibrotic effect of 6G on isoproterenol-induced myocardial fibrosis [32] or carbon tetrachloride-induced liver fibrosis [29] by reducing oxidative stress, inflammation, and apoptosis through the Toll-like receptor 4/MAPKs/NF-κB pathway. Numerous studies have shown that 6G has anti-inflammatory, antioxidant, and antiapoptotic effects by inhibiting the p38 MAPK pathway [13–15, 33]. Although previous studies have shown that 6G can regulate the p38 signaling pathway, our study demonstrated for the first time that 6G treatment protected against cardiac remodeling induced by pressure overload via a p38-dependent pathway. Moreover, 6G treatment simultaneously inhibited the activation of p38 in cardiomyocytes and fibroblasts after cardiac remodeling stimulation. In addition, 6G also blocked the phosphorylation of p65 and Smad3 and subsequently decreased inflammatory cytokines by inhibiting the p38 pathway.
Hypertrophic growth in cardiomyocytes is typified by dramatic increases in cell size, a high degree of sarcomere organization, and the enhanced expression of a number of cardiac-specific genes [34]. The results of our present study demonstrated that 6G could attenuate the hypertrophic index and fetal gene expression in mouse hearts induced by TAC and in isolated cardiomyocytes stimulated by PE. We also observed a marked increase in p38 phosphorylation and nuclear translocation in hypertrophic hearts and cardiomyocytes, which was dramatically decreased after 6G treatment, indicating the involvement of p38 in 6G activity. Our in vitro results further verified that MKK6 transfection promoted hypertrophic responses in cardiomyocytes in a manner similar to that of PE and neutralized the protective effect of 6G. Gain-of-function and loss-of-function studies through genetic manipulation or molecular inhibitors provided evidence that p38 induced cardiomyocyte hypertrophy [34, 35], corroborating our findings. However, the role of p38 in cardiac hypertrophy in intact hearts remains inconclusive. The results of one study using mitogen-activated protein kinase kinase 6b(E) (MKK6bE) transgenic mice demonstrated that ventricular-specific expression of MKK6bE induced p38 MAPK activation and resulted in several detrimental changes in the heart, including marked interstitial fibrosis, profound depression of systolic contractility and compromised diastolic function without significant hypertrophy [36], while another study affirmed that TGF-β-activated kinase 1 activation under chronic pressure overload was responsible for p38 phosphorylation, myocardial hypertrophy, and fulminant heart failure [11]. The partial differences between p38 overexpression-induced hypertrophic phenotypes may be ascribed to the application of molecules affecting components upstream of p38 and different experimental models and settings. Inconsistent with the findings of Liao et al. [36], the data in our study showed that MKK6 overexpression did not affect cardiac function or fibrosis at baseline. Liao et al. found that targeted activation of p38 in ventricular myocytes using sustained activation of MKK induced marked interstitial fibrosis and the expression of fetal marker genes characteristic of cardiac failure but no significant hypertrophy at the organ level. Transgene expression resulted in significant induction of p38 kinase activity and premature death at 7–9 weeks. However, adenovirus was administered by intramyocardial injections in three separate locations in the LV free wall of the heart 1 week before TAC surgery in our study. Therefore, we thought that the duration and expression of p38 kinase activity induced by the adenovirus delivery system are not as powerful as those in transgenic mice. However, our results showed that MKK6 transfection in vivo worsened cardiac hypertrophy, fibrosis and dysfunction when mice underwent TAC stimulation and blocked the protective effect of 6G treatment, suggesting that 6G prevents hypertensive cardiac injury and premature loss of function in a p38-dependent manner.
Interstitial fibrosis is an indispensable feature of pathological pressure overload-induced cardiac remodeling that leads to the deterioration of cardiac function and life-threatening heart failure. Our results demonstrated that 6G could reduce the excessive deposition of ECM and cardiac fibrosis induced by TAC in vivo. CFs are widely recognized as playing a crucial role in cardiac fibrosis [37]. Once stimulated by extracellular neurohumoral or mechanical signals, quiescent fibroblasts transdifferentiate into myofibroblasts with an α-SMA-positive phenotype, and this transdifferentiation triggers the biosynthesis and secretion of ECM proteins, as well as excessive interstitial and perivascular fibrosis. Therefore, we conducted in vitro experiments to verify whether 6G directly affected CFs. Phenotypically, 6G inhibited myofibroblast formation and collagen synthesis in the presence of profibrotic TGF-β signaling. Mechanistically, 6G treatment weakened the activation of p38 and Smad3. The canonical TGF-β–Smads signaling pathway is characterized by activation of TGF-β type I receptor, which initiates recruitment, resulting in the phosphorylation and release of Smad2/3, which binds with Smad4 and translocates to the nucleus to activate the transcription of profibrotic genes [38]. Although the results of numerous studies have shown that Smad3-deficient mice exhibit marked decreases in stress-induced myocardial fibrosis [38, 39], TGF-β-mediated α-SMA stress fiber positivity and myofibroblast differentiation can be entirely inhibited by blocking p38 signaling but not by overexpressing Smad2/3 inhibitors [40], indicating that additional effectors are sufficient to promote CF differentiation and function beyond the canonical TGF-β–Smad signaling pathway. TGF-β is well known to promote myofibroblast differentiation through another noncanonical pathway involving the activation of MAPKs such as MKK3/6 and their direct downstream molecule p38 [41]. Numerous studies have verified that pharmacological inhibition or transgenic deficiency of p38 or its upstream regulators MKK3/6 can inhibit myofibroblast differentiation in CFs, human tenon fibroblasts and dermal wound healing [42, 43]. In contrast, the results of our study confirmed that Ad-MKK6 transfection primed and intensified cardiac myofibroblast differentiation and fibrotic gene expression in vitro and subsequently induced superfluous fibrosis in mouse hearts in vivo, which was consistent with the findings of a previous study [44]. Notably, after Ad-MKK6 transfection, 6G failed to promote resistance to fulminant α-SMA-positive myofibroblasts and cardiac fibrosis both in vitro and in vivo, suggesting that p38 inhibition plays a crucial and indispensable role in the antifibrotic activity of 6G.
Fascinatingly, we also observed that p38 overexpression notably enhanced the phosphorylation and nuclear translocation of Smad3 and counteracted the 6G-mediated suppression of Smad3 activation. The results of a previous study showed that the p38 MAPK pathway promoted Smad3 phosphorylation at the linker region to induce a myofibroblast phenotype in cultured hepatic stellate cells when TGF-β receptor I-mediated activation was inhibited, irrespective of Smad2 activation [45], which was consistent with our observations. These results reveal that p38 promotes cardiac fibrosis through a novel pathway that modulates Smad3 activation independent of TGF-β. In summary, our study is the first to demonstrate that 6G attenuates myofibroblast differentiation and cardiac fibrosis via the p38 MAPK pathway, and the underlying mechanism is partially attributable to the crosstalk between p38 and Smad3 signaling.
p38 signaling is widely known to play a crucial role in immune reactions by promoting the expression of proinflammatory cytokines, including TNF-α, IL-6, and MCP-1, and contributes to cardiomyocyte hypertrophy, ECM remodeling and the recruitment and proliferation of inflammatory cells. A previous study provided direct in vivo and in vitro evidence that p38 activation was sufficient to induce inflammatory cytokine expression in cardiomyocytes and that there was a positive feedback loop between p38 and TNF-α, whereas the p38 inhibitor SB23906 was shown to block the secretion of TNF-α and IL-6 and attenuate ECM remodeling [46]. Our results showed that 6G attenuated the mRNA levels of TNF-α, IL-6, and MCP-1, as well as inflammatory cell infiltration, post TAC, while Ad-MKK6 transfection reversed these inflammatory responses back to their peak levels, indicating that p38 MAPK was a crucial regulator of the anti-inflammatory activity of 6G in cardiac remodeling. NF-κB exists as an inactive dimeric complex composed of the protein subunits p65 and p50, which are bound to the inhibitor protein inhibitor of NF-κB (IκB). The loss of IκBα exposes the nuclear localization motif on the p65 NF-κB subunit, permitting its phosphorylation and nuclear targeting [47]. The NF-κB pathway is believed to be involved in promoting cardiac hypertrophy and inflammatory responses in cardiomyocytes [48]. Intriguingly, several studies have shown that the p38 inhibitor SB203580 directly represses the transactivation potential of the p65 subunit [49]. The results of another study indicated that p38 signals activate the transcription of human BNP through NF-κB, indicating hypertrophic marker activation [50]. Correspondingly, our data suggested that the inhibitory effect of 6G on NF-κB relied on p38 MAPK downregulation, providing strong evidence for the central role of p38 in 6G-mediated cardioprotection after chronic pressure overload.
Conclusion
In summary, our findings provide adequate evidence for the protective role of 6G in attenuating cardiac hypertrophy, fibrosis, inflammation, and dysfunction following chronic pressure overload, as well as in blocking PE-induced cardiomyocyte hypertrophy and TGF-β-induced cardiac fibroblast activation in vitro. We further indicated that p38 mediated the antihypertrophic, antifibrotic, and anti-inflammatory effects of 6G and that the cardioprotective effect of 6G on pathological remodeling occurred in a p38-dependent manner. Our study provides novel insight into the pharmacological function of 6G and a theoretical basis for the application of 6G in the future treatment of pathological cardiac remodeling.
Supplementary information
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (81900219, 81530012, and 81800216), the Fundamental Research Funds for the Central Universities (2042019kf0062 and 2042018kf1032), the Development Center for Medical Science and Technology National Health and Family Planning Commission of China (2016ZX-008-01), and the National Key R&D Program of China (2018YFC1311300).
Author contributions
SQM, ZG, DF, and QZT contributed to the conception and design of the study. SQM, ZG, SGH, NT, PA, DY, and MYW performed the experiments and collected the data. FYL, DF, SQM, ZG, and SGH contributed to the data analysis and interpretation. SQM, DF, ZG, and FYL drafted and wrote the manuscript. QZT, ZY and, HMW provided experimental sources and financial support. SQM, ZG, FYL, and SGH contributed equally to this manuscript. All authors read and approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Shu-qing Ma, Zhen Guo, Fang-yuan Liu, Shahzad-Gul Hasan
Contributor Information
Di Fan, Email: drfanti@yeah.net.
Qi-zhu Tang, Email: qztang@whu.edu.cn.
Supplementary information
The online version of this article (10.1038/s41401-020-00587-z) contains supplementary material, which is available to authorized users.
References
- 1.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling-concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/S0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 2.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 3.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–78. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. 2018;138:2530–44. doi: 10.1161/CIRCULATIONAHA.118.034621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail. 2017;19:1379–89. doi: 10.1002/ejhf.942. [DOI] [PubMed] [Google Scholar]
- 6.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–24. doi: 10.1161/CIRCULATIONAHA.113.007101. [DOI] [PubMed] [Google Scholar]
- 7.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velázquez F, Aronovitz M, et al. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail. 2015;8:776–87. doi: 10.1161/CIRCHEARTFAILURE.115.002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molkentin JD, Dorn GW. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 9.Tenhunen O, Rysä J, Ilves M, Soini Y, Ruskoaho H, Leskinen H. Identification of cell cycle regulatory and inflammatory genes as predominant targets of p38 mitogen-activated protein kinase in the heart. Circ Res. 2006;99:485–93. doi: 10.1161/01.RES.0000238387.85144.92. [DOI] [PubMed] [Google Scholar]
- 10.Westermann D, Rutschow S, Van Linthout S, Linderer A, Bücker-Gärtner C, Sobirey M, et al. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia. 2006;49:2507–13. doi: 10.1007/s00125-006-0385-2. [DOI] [PubMed] [Google Scholar]
- 11.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, et al. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–63. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 12.Lee JH, Kim YG, Choi P, Ham J, Park JG, Lee J. Antibiofilm and antivirulence activities of 6-gingerol and 6-shogaol against candida albicans due to hyphal inhibition. Front Cell Infect Microbiol. 2018;8:299. doi: 10.3389/fcimb.2018.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song S, Dang M, Kumar M. Anti-inflammatory and renal protective effect of gingerol in high-fat diet/streptozotocin-induced diabetic rats via inflammatory mechanism. Inflammopharmacology. 2019;27:1243–54. doi: 10.1007/s10787-019-00569-6. [DOI] [PubMed] [Google Scholar]
- 14.Kim SO, Kundu JK, Shin YK, Park JH, Cho MH, Kim TY, et al. [6]-Gingerol inhibits COX-2 expression by blocking the activation of p38 MAP kinase and NF-κB in phorbol ester-stimulated mouse skin. Oncogene. 2005;24:2558–67. doi: 10.1038/sj.onc.1208446. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Xu B, Xu M, Chen D, Xiong Y, Lian M, et al. 6-Gingerol protects intestinal barrier from ischemia/reperfusion-induced damage via inhibition of p38 MAPK to NF-κB signalling. Pharmacol Res. 2017;119:137–48. doi: 10.1016/j.phrs.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 16.Chen YL, Zhuang XD, Xu ZW, Lu LH, Guo HL, Wu WK, et al. Higenamine combined with [6]-gingerol suppresses doxorubicin-triggered oxidative stress and apoptosis in cardiomyocytes via upregulation of PI3K/Akt pathway. Evid Based Complement Altern Med. 2013;2013:970490. doi: 10.1155/2013/970490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen J, Zhang L, Wang J, Wang J, Wang L, Wang R, et al. Therapeutic effects of higenamine combined with [6]-gingerol on chronic heart failure induced by doxorubicin via ameliorating mitochondrial function. J Cell Mol Med. 2020;24:4036–50. doi: 10.1111/jcmm.15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong CH, Bode AM, Pugliese A, Cho YY, Kim HG, Shim JH, et al. [6]-Gingerol suppresses colon cancer growth by targeting leukotriene A4 hydrolase. Cancer Res. 2009;69:5584–91. doi: 10.1158/0008-5472.CAN-09-0491. [DOI] [PubMed] [Google Scholar]
- 19.Ju SA, Park SM, Lee YS, Bae JH, Yu R, An WG, et al. Administration of 6-gingerol greatly enhances the number of tumor-infiltrating lymphocytes in murine tumors. Int J Cancer. 2012;130:2618–28. doi: 10.1002/ijc.26316. [DOI] [PubMed] [Google Scholar]
- 20.Jain M, Singh A, Singh V, Maurya P, Barthwal MK. Gingerol inhibits serum-induced vascular smooth muscle cell proliferation and injury-induced neointimal hyperplasia by suppressing p38 MAPK activation. J Cardiovasc Pharmacol Ther. 2016;21:187–200. doi: 10.1177/1074248415598003. [DOI] [PubMed] [Google Scholar]
- 21.Wen J, Wang J, Li P, Wang R, Wang J, Zhou X, et al. Protective effects of higenamine combined with [6]-gingerol against doxorubicin-induced mitochondrial dysfunction and toxicity in H9c2 cells and potential mechanisms. Biomed Pharmacother. 2019;115:108881. doi: 10.1016/j.biopha.2019.108881. [DOI] [PubMed] [Google Scholar]
- 22.Weng LQ, Zhang WB, Ye Y, Yin PP, Yuan J, Wang XX, et al. Aliskiren ameliorates pressure overload-induced heart hypertrophy and fibrosis in mice. Acta Pharmacol Sin. 2014;35:1005–14. doi: 10.1038/aps.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, et al. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1-dependent signalling. Nat Commun. 2016;7:11267. doi: 10.1038/ncomms11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, et al. Suppressor of IKKɛ is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. doi: 10.1038/ncomms11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, et al. Temporal response and localization of integrins β1 and β3 in the heart after myocardial infarction: regulation by cytokines. Circulation. 2003;107:1046–52. doi: 10.1161/01.CIR.0000051363.86009.3C. [DOI] [PubMed] [Google Scholar]
- 26.Roberts AB, Roche NS, Winokur TS, Burmester JK, Sporn MB. Role of transforming growth factor-β in maintenance of function of cultured neonatal cardiac myocytes. Autocrine action and reversal of damaging effects of interleukin-1. J Clin Invest. 1992;90:2056–62. doi: 10.1172/JCI116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu QQ, Xiao Y, Duan MX, Yuan Y, Jiang XH, Yang Z, et al. Aucubin protects against pressure overload-induced cardiac remodelling via the β3 -adrenoceptor-neuronal NOS cascades. Br J Pharm. 2018;175:1548–66. doi: 10.1111/bph.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li N, Zhou H, Ma ZG, Zhu JX, Liu C, Song P, et al. Geniposide alleviates isoproterenol-induced cardiac fibrosis partially via SIRT1 activation in vivo and in vitro. Front Pharmacol. 2018;9:854. doi: 10.3389/fphar.2018.00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Algandaby MM, El-Halawany AM, Abdallah HM, Alahdal AM, Nagy AA, Ashour OM, et al. Gingerol protects against experimental liver fibrosis in rats via suppression of pro-inflammatory and profibrogenic mediators. Naunyn Schmiedebergs Arch Pharm. 2016;389:419–28. doi: 10.1007/s00210-016-1210-1. [DOI] [PubMed] [Google Scholar]
- 30.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–38. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, et al. Mechanisms contributing to cardiac remodelling. Clin Sci. 2017;131:2319–45. doi: 10.1042/CS20171167. [DOI] [PubMed] [Google Scholar]
- 32.Han X, Liu P, Liu M, Wei Z, Fan S, Wang X, et al. [6]-Gingerol ameliorates ISO-induced myocardial fibrosis by reducing oxidative stress, inflammation, and apoptosis through inhibition of TLR4/MAPKs/NF-κB pathway. Mol Nutr Food Res. 2020;64:e2000003. doi: 10.1002/mnfr.202000003. [DOI] [PubMed] [Google Scholar]
- 33.Kang C, Kang M, Han Y, Zhang T, Quan W, Gao J. 6-Gingerols (6G) reduces hypoxia-induced PC-12 cells apoptosis and autophagy through regulation of miR-103/BNIP3. Artif Cells Nanomed Biotechnol. 2019;47:1653–61. doi: 10.1080/21691401.2019.1606010. [DOI] [PubMed] [Google Scholar]
- 34.Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J Cell Biol. 1997;139:115–27. doi: 10.1083/jcb.139.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clerk A, Michael A, Sugden PH. Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G protein-coupled receptor agonists, endothelin-1 and phenylephrine: a role in cardiac myocyte hypertrophy? J Cell Biol. 1998;142:523–35. doi: 10.1083/jcb.142.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA. 2001;98:12283–8. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–83. doi: 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Divakaran V, Adrogue J, Ishiyama M, Entman ML, Haudek S, Sivasubramanian N, et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ Heart Fail. 2009;2:633–42. doi: 10.1161/CIRCHEARTFAILURE.108.823070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–15. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Li Z, Zhang C, Li P, Wu Y, Wang C, et al. Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3-p38 signaling. Circulation. 2017;135:2041–57. doi: 10.1161/CIRCULATIONAHA.116.024599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer-Ter-Vehn T, Gebhardt S, Sebald W, Buttmann M, Grehn F, Schlunck G, et al. p38 inhibitors prevent TGF-β-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47:1500–9. doi: 10.1167/iovs.05-0361. [DOI] [PubMed] [Google Scholar]
- 44.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. 2017;136:549–61. doi: 10.1161/CIRCULATIONAHA.116.026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–89. doi: 10.1002/hep.1840380414. [DOI] [PubMed] [Google Scholar]
- 46.Li M, Georgakopoulos D, Lu G, Hester L, Kass DA, Hasday J, et al. p38 MAP kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation. 2005;111:2494–502. doi: 10.1161/01.CIR.0000165117.71483.0C. [DOI] [PubMed] [Google Scholar]
- 47.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, et al. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation. 2015;131:643–55. doi: 10.1161/CIRCULATIONAHA.114.011079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–86. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-κB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–90. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 50.Liang F, Gardner DG. Mechanical strain activates BNP gene transcription through a p38/NF-κB-dependent mechanism. J Clin Invest. 1999;104:1603–12. doi: 10.1172/JCI7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.