

Abstract
Fatal familial insomnia (FFI) is a dominantly inherited prion disease linked to the D178N mutation in the gene encoding the prion protein (PrP). Symptoms, including insomnia, memory loss and motor abnormalities, appear around 50 years of age, leading to death within two years. No treatment is available. A ten-year clinical trial of doxycycline (doxy) is under way in healthy individuals at risk of FFI to test whether presymptomatic doxy prevents or delays the onset of disease.
To assess the drug's effect in a tractable disease model, we used Tg(FFI-26) mice, which accumulate aggregated and protease-resistant PrP in their brains and develop a fatal neurological illness highly reminiscent of FFI. Mice were treated daily with 10 mg/kg doxy starting from a presymptomatic stage for twenty weeks. Doxy rescued memory deficits and restored circadian motor rhythmicity in Tg(FFI-26) mice. However, it did not prevent the onset and progression of motor dysfunction, clinical signs and progression to terminal disease. Doxy did not change the amount of aggregated and protease-resistant PrP, but reduced microglial activation in the hippocampus. Presymptomatic doxy treatment rescues cognitive impairment and the motor correlates of sleep dysfunction in Tg(FFI-26) mice but does not prevent fatal disease.
Keywords: Genetic prion disease, Prion protein, PrP, Protein misfolding, Transgenic mice, Pharmacological therapy
Abbreviations: AD, Alzheimer's disease; ARRIVE, Animal Research: Reporting of In Vivo Experiments; doxy, doxycycline; DVC®, Digital Ventilated Cages; FFI, fatal familial insomnia; GFAP, glial fibrillary acidic protein; Iba1, ionized calcium binding adapter molecule 1; i.p., intraperitoneal; IVC, individual ventilated cages; non-Tg, nontransgenic; NOR, novel object recognition; PBS, phosphate buffered saline; PK, proteinase-K; PrP, prion protein; PrPSc, scrapie prion protein; r.p.m., rotations per minute; sCJD, sporadic Creutzfeldt-Jakob disease; SDS-PAGE gel, sodium dodecyl sulphate-polyacrylamide gel electrophoresis; SEM, standard error of the mean; WT, wild-type
Highlights
-
•
Early doxycycline treatment improves memory in fatal familial insomnia mice.
-
•
Doxycycline restores normal circadian motor rhythmicity.
-
•
Doxycycline reduces microglial activation in the hippocampus.
-
•
Doxycycline does not reduce mutant prion protein aggregation or protease resistance.
-
•
Doxycycline does not delay disease onset or prolong survival of the mice.
1. Introduction
Human prion diseases are rare degenerative disorders of the central nervous system which can arise sporadically, and may be genetically inherited due to mutations in the PRNP gene encoding the prion protein (PrP), or acquired through infection (Prusiner, 1998). They are caused by the conformational conversion of PrP into scrapie prion protein (PrPSc or prion), an abnormal isoform rich in β-sheet structure, which is detergent-insoluble, partially protease-resistant, and can induce conversion of native PrP. In the sporadic forms, PrP is thought to convert spontaneously to PrPSc; in the genetic forms PrP misfolding is favored by PRNP mutation; in the acquired forms it is triggered by contact with exogenous PrPSc (Prusiner, 1998).
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common human prion disease, accounting for 85–90% of cases. It causes cognitive impairment and motor abnormalities and has a very rapid course with mean survival of six months and over 90% of patients dying within one year from symptom onset (Mackenzie and Will, 2017). Fatal familial insomnia (FFI) is a genetic prion disease linked to aspartic acid (D) to asparagine (N) substitution at codon 178 of the PRNP gene, in association with methionine (M) at polymorphic site 129 (D178N/M129) (Goldfarb et al., 1992). The FFI mutation has a almost complete penetrance, leading to disease onset at approximately 50 years of age; mean survival is 18 months with more than 50% of individuals dying within one year from the first symptom (Minikel et al., 2019; Minikel et al., 2016; Montagna et al., 1998). The most striking clinical feature is progressive insomnia, accompanied by hallucinations and enacted dreams. Other symptoms, which in some cases are the earliest and most marked neurological signs, include ataxia and other motor symptoms such as myoclonus, tremor, dysarthria and pyramidal impairment, loss of attention and memory, and autonomic disturbances such as tachycardia and hypertension (Montagna et al., 2003; Schenkein and Montagna, 2006). Neuropathology includes spongiosis, marked neuronal loss and astrogliosis, especially in the medio-dorsal and anterior-ventral nuclei of the thalamus (Montagna et al., 2003). Microgliosis is also present and can be an early neuropathological change (Iaccarino et al., 2018).
Several compounds have been tested in prion disease models, reducing PrPSc replication in vitro and delaying the appearance of clinical symptoms in vivo, but so far none have been clinically validated. One class of potential anti-prion drugs are the tetracycline antibiotics, particularly the second-generation derivative doxycycline (doxy), which has favorable pharmacokinetics, good capacity to cross the blood-brain barrier, low toxicity, and good tolerability even for long treatment (Stoilova et al., 2013).
Initial in vitro studies indicated that tetracycline interferes with the self-assembly of PrPSc-like synthetic peptides corresponding to residues 106–126 and 82–146 of human PrP, antagonizing their neurotoxic and gliotrophic effects (Tagliavini et al., 2000). Doxy promoted enzymatic proteolysis of PrPSc extracted from CJD brains and reduced prion infectivity when co-incubated with a brain homogenate of hamsters infected with the 263K prion strain (Forloni et al., 2002; Tagliavini et al., 2000). In vivo doxy given soon after infection by the intramuscular or intraperitoneal (i.p.) route significantly prolonged the survival of peripherally 263K prion-infected hamsters (De Luigi et al., 2008). A single high dose of liposome-entrapped doxy infused into the ventricle of intracerebrally infected hamsters, when 50% of them showed initial signs of disease, prolonged survival by 8% (De Luigi et al., 2008). Finally, observational studies suggested prolonged survival of CJD patients given doxy as compassionate treatment (Ponto and Zerr, 2013; Tagliavini, 2008).
In the light of these findings doxy was tested in two randomized, double-blind, placebo-controlled phase II clinical trials in sCJD (Haïk et al., 2014; Varges et al., 2017). The first trial enrolled 121 patients in an advanced stage of disease, and doxy was given orally, 100 mg/day to 62 of them (Haïk et al., 2014). This trial was stopped when interim analysis showed no differences between doxy- and placebo-treated patients in any of the endpoints (survival time, autonomous feeding, akinetic mutism). At autopsy, no differences were seen in the patterns of protease-resistant PrP deposition or in the severity of spongiform degeneration or gliosis (Haïk et al., 2014). The second study enrolled 13 sCJD patients at an earlier stage of disease, and assigned seven to the 100 mg/day doxy group (Varges et al., 2017). Again, doxy gave no beneficial effect over placebo. This study also analyzed a subgroup of patients who received compassionate doxy treatment starting from an early symptomatic stage, and presented a significant increase in survival time compared to a control dataset (Varges et al., 2017). This suggested that treatment should be started as early as possible to attain beneficial effects – ideally in the presymptomatic phase.
Carriers of PRNP mutations may be identified by molecular genetic analysis decades in advance of symptom onset, creating an opportunity for presymptomatic intervention. Thus a prevention trial (DOXIFF; https://www.clinicaltrialsregister.eu/ctr-search/trial/2010-022233-28/IT) was designed to test doxy in healthy individuals at risk of FFI (Forloni et al., 2015). The DOXIFF study involves subjects belonging to a large group of carriers of the PRNP D178N/M129 mutation from north-east Italy, who agreed to be treated with doxy for a long period. Ten asymptomatic individuals were enrolled on the basis of their risk age, considering that historical data indicated that the median age of disease onset was 49 years. The study, which started in 2012, set an observational period of 10 years. The participants are being treated with 100 mg/day doxy (200 mg since June 2019), and are closely monitored, with neurological and neuropsychological assessments and blood tests every six months. According to statistical analysis, doxy will be considered effective if no more than three affected cases arise (Forloni et al., 2015).
In the present study, we tested doxy in the transgenic Tg(FFI-26) mouse model of FFI (Bouybayoune et al., 2015). Tg(FFI-26) mice express the mouse PrP homolog of the D178N/M129 mutation (moPrP D177N/M128) and develop a progressive and invariably fatal neurological disease, which recapitulates key characteristics of FFI, including profound alterations in sleep and circadian motor activity, ataxia and memory deficits (Bouybayoune et al., 2015; Chiesa et al., 2016). Mice were treated daily for twenty weeks with 10 mg/kg doxy, starting from a presymptomatic stage. During and after treatment they were examined in behavioral tests to check the development and progression of motor dysfunction, cognitive function, circadian motor activity, and appearance and progression of neurological signs. Doxy had some beneficial effects but did not modify the natural history of the Tg(FFI-26) disease overall.
2. Materials and methods
2.1. Study design
The aim of the study was to test whether presymptomatic doxy treatment prevents the disease in Tg(FFI-26) mice. Mice were assigned to the experimental groups using simple randomization (allocation ratio 1:1). The randomization list was generated using the Excel RAND function. Treatment was planned to overlap the onset of motor deficits and progressive decline in motor performance, assessed with the rotarod and beam walking tests, for 20 weeks starting from a presymptomatic stage (from ~70 to ~220 days of age) (Bouybayoune et al., 2015). The NOR task, which relies on the animal's natural preference for novelty (Grayson et al., 2015), was used to assess memory function during treatment. At the end of treatment, four animals of each experimental group were culled for biochemical and histological analysis, and the remaining mice were monitored for the appearance of clinical signs of the disease according to pre-specified criteria (see below) until the terminal stage. These mice were also caged for three months in Tecniplast DVC® cages, which allowed 24/7 monitoring of their movements, for assessment of circadian motor activity.
All animal experiments were designed in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (Kilkenny et al., 2010), with a commitment to refinement, reduction and replacement, minimizing the number of mice, and using biostatistics to optimize mouse numbers. Thus, for statistical validity, the sample size was calculated from previous assessment of rotarod performance (primary endpoint) (Bouybayoune et al., 2015). Assuming a normal distribution of latency to fall, a linear progressive reduction of mean time on the rotarod for 10 equidistant and consecutive time points (mean latency to falls: 247 s, time 1; 239 s, time 2; 231 s, time 3; 223 s, time 4; 215 s, time 5; 207 s, time 6; 199 s, time 7; 191 s, time 8; 183 s, time 9; 175 s, time 10), an AR(1) correlation of 0.80 and residual variance of 2500 s2, 12 animals per experimental group were necessary to obtain a 33% improvement of the mean latency to falls for mice treated with doxy (type 1 error: 0.05, power: 0.85). No sex-related differences were observed in motor performance, so both male and female mice were included. Doxy 10 mg/kg was given daily by i.p. injection, based on previous pharmacokinetic analysis and therapeutic doxy treatment in mouse models of Alzheimer's disease (AD) (Balducci et al., 2018; Lucchetti et al., 2019).
2.2. Mice
Transgenic Tg(FFI-26+/−)/Prnp0/0 and Tg(WT-E1+/−)/Prnp0/0 mice, expressing respectively moPrP D177N/M128 and moPrP WT at approximately 2× on a PrP knockout background have been described previously (Bouybayoune et al., 2015; Chiesa et al., 1998). These mice were maintained by backcrossing to an inbred strain of C57BL/6J/Prnp0/0 mice (European Mouse Mutant Archive, Monterotondo, Italy; EM:01723). Non-Tg mice were nontransgenic littermates of Tg(FFI-26+/−)/Prnp0/0 mice. The animals were housed at controlled temperature (22 ± 2 °C) with a 12/12-h light/dark cycle and free access to pelleted food and water. The health and home-cage behavior were monitored daily, according to guidelines for health evaluation of experimental laboratory animals (Burkholder et al., 2012). Animal facilities meet international standards and are regularly checked by a certified veterinarian who is responsible for health monitoring, animal welfare supervision, experimental protocols and review of procedures.
2.3. Treatment
Doxycycline (doxycycline hyclate, D9891-5G, Sigma) was dissolved in saline solution (Fresenius Kabi Italia; 5 mg/mL) and injected i.p. daily at a dose of 10 mg/kg. Vehicle-treated animals were injected with saline solution. Treatments were done by an operator blind to the mouse genotype.
2.4. Accelerated rotarod test
The accelerating Rotarod 7650 model (Ugo Basile) was used: mice were trained three times the week before official testing. They were positioned on the rotating bar and allowed to become acquainted with the environment for 30 s. The rod motor was started at an initial setting of 7 r.p.m. and accelerated to 40 r.p.m. at a constant rate of 0.11 r.p.m./s for a maximum of 300 s. Performance was scored as the latency to fall, in seconds. Animals were given three trials, and the average was used for statistical analysis. The investigator was blind to the mouse genotype and treatment allocation.
2.5. Beam walking test
For three days before testing mice were trained to walk along a metal beam 0.8 cm wide, 100 cm long, suspended 30 cm above bedding. On the day of testing they were given three trials. Mice were video-recorded and the number of hindfoot missteps, falls and time to traverse the beam during the three trials were counted by an investigator blinded to the experimental group. The average number of missteps and time to traverse the beam during the three trials were used for statistical analysis.
2.6. Novel object recognition test
Mice were tested in an open square gray arena (40 × 40 cm), 30 cm high, with the floor divided into 25 squares by black lines. The following objects were used: a black plastic cylinder (4 × 5 cm), a glass vial with a white cap (3 × 6 cm) and a metal cube (3 × 5 cm). The task started with a habituation trial during which the animals were placed in the empty arena for 5 min and their movements were recorded as the number of line-crossings. The next day, mice were placed in the same arena containing two identical objects (familiarization phase). Exploration was recorded in a 10-min trial by an investigator blinded to the experimental group. Sniffing, touching and stretching the head toward the object at a distance of not more than 2 cm were scored as object investigation. Twenty-four hours later (test phase) mice were placed in the arena containing two objects: one of the objects presented during the familiarization phase (familiar object), and a new, different one (novel object), and the time spent exploring the two objects was recorded for 10 min. Memory was expressed as a discrimination index:
where Tn is the time (in seconds) the mice spent on the new object, and To the time spent on the old object. This ranges from +1 to −1, a positive score indicating more time spent on the new object, and a negative one more time on the old object. Mice that did not reach the minimum 6.5 s threshold of total exploration time required to evaluate memory, were excluded from the analysis (d'Isa et al., 2014).
2.7. Circadian motor activity
The spontaneous motor activity of the mice was monitored using Digital Ventilated Cages (DVC®) manufactured by Tecniplast SpA (Buguggiate, Italy). This system is designed to collect information from individual ventilated cages (IVC) directly from the home cage rack. The system builds up on the top of a standard IVC rack by installing an electronic sensing board underneath each cage position, mechanically connected to the rack without influencing conventional IVC operations. The sensing board is composed of 12 electrodes connected to an integrated circuit that continuously measures their electrical capacitance. Since capacitance is influenced by the matter present in each electrode's surrounding, its measurements are affected by the mouse's movements (Iannello, 2019). Mice were individually housed in DVC® cages and data were collected 24/7 for three months. Animal activity was captured similarly to the activation density metric defined in (Iannello, 2019), which gave an average percentage of the amount of activations of the animal over the 12 electrodes of the cage floor in a given interval (Golini et al., 2020; Iannello, 2019; Voikar and Gaburro, 2020).
2.8. Clinical evaluation
Clinical signs of disease were scored by an investigator blind to the treatment and mouse genotype. The signs and their scores were: kyphosis (0 no kyphosis; 0.5 kyphosis visible when the mouse was still; 1 kyphosis visible also when the mouse was moving); ruffled coat (0 fur is healthy looking and shiny; 0.5 fur does not look shiny; 1 fur is ruffled and untidy); foot-clasp reflex when the mouse is suspended by the tail (0 the mouse struggles to free itself and spreads the hind limbs apart; 1 the mouse is passive and keeps the hind limbs close together, but does not make a proper foot clasp; 2 the mouse is passive and clasps the hind limbs); ability to walk on a horizontal grid (0 the mouse can walk without any problem; 1 the mouse can walk on the grid despite the limbs slipping through the rods; 2 the mouse is unable to walk with the limbs dangling through the grid); ability to remain on a vertical grid for at least 30 s (the mouse is placed on the grid facing down: 0 the animal turns up and climbs the grid; 1 the mouse stays still on the grid facing downward; 2 the mouse falls within 30 s); and twitches (0 no twitches; 0.5 the mouse displays a light but continuous tremor; 1 mouse displays strong twitches). A score was assigned for each sign, for a maximum daily score of 9.
2.9. Brain homogenates
Frozen brains were homogenized using a glass/Teflon tissue homogenizer in 1:10 volume of ice-cold PBS, pH 7.4 (phosphate buffered saline w/o CaCl2 and MgCl2; Gibco) and the protein concentration was measured with a BCA Protein Assay (Pierce™ BCA Assay Kit by ThermoFisher).
2.10. Proteinase-K (PK) resistance
Brain homogenate corresponding to 200 μg of total proteins was diluted to 1 μg/mL in TND 1× buffer (20 mM Tris-HCl, 0.5% NP-40, 0.5% Na deoxycholate), and incubated at 4 °C for 20 min on a rotating wheel. After 3-min centrifugation at 16,000 xg at 4 °C, supernatants were incubated at 4 °C for 1 h with 0, 0.5, 1 or 2 μg/mL of PK in a final volume of 250 μL. Digestion was stopped with Pefabloc (1.6 mM final concentration, 10 min in ice), and proteins were precipitated by adding four volumes of ice-cold CH3OH, then incubated overnight at −20 °C. After centrifugation at 16,000 xg for 30 min at 4 °C, the supernatant was removed and the pellet was resuspended in Laemmli Sample Buffer, and PK-resistant PrP was analyzed by western blot with antibody 6D11.
2.11. Insolubility
A volume of brain homogenate corresponding to 90 μg of total protein was diluted in 300 μL of TND 1× buffer containing protease inhibitors (1× SigmaFast™ Protease Inhibitor Tablets), incubated 20 min at 4 °C on a rotating wheel. After centrifugation at 16,000 xg for 5 min at 4 °C, the supernatant was collected, and the pellet (P0) was resuspended in Laemmli sample buffer. The supernatant was ultracentrifuged at 186,000 x g for 45 min at 4 °C, the pellet (P) resuspended in Laemmli sample buffer, and proteins in the supernatant were precipitated by adding four volumes of ice-cold CH3OH. After overnight incubation at −20 °C, samples were centrifuged at 16,000 xg for 30 min at 4 °C. The supernatant was discarded and the precipitated proteins (S) were resuspended in Laemmli Sample Buffer. PrP in the supernatant and pellets was analyzed by western blot.
2.12. Western blot
Protein samples in Laemmli Sample Buffer were boiled 10 min at 100 °C and run on a 12% SDS-PAGE gel (sodium dodecyl sulphate-polyacrylamide gel electrophoresis). Proteins were transferred onto a nitrocellulose membrane using the Bio-Rad Trans-Blot® Turbo™ Transfer System. Membranes were blocked in 5% non-fat dry milk in TBST (Tris-buffered saline and 0.1% Tween-20) before incubation for 1 h at room temperature or overnight at 4 °C with anti-PrP antibody 6D11 (1:5000), followed by incubation with the secondary antibody (Bio-Rad Goat Anti-Mouse IgG (H + L)-HRP Conjugate, 1:5000) for 1 h at room temperature. Signals were revealed using enhanced chemiluminescence (Clarity Western ECL Subtrate, Bio-Rad), and visualized with a ChemiDoc Imaging System (Bio-Rad). Quantitative densitometry of protein bands was done using Image Lab 6.0 software (Bio-Rad).
2.13. Doxycycline bioanalysis
Doxycycline in the plasma and brain of the treated mice (culled 4 h after the last dose) was quantified by HPLC-MS/MS, as described (Lucchetti et al., 2019), in parallel with quality control samples (two replicates at three concentrations, 0.3, 3 and 7.5 μg/mL or 0.18, 3.0 and 4.5 μg/g) and freshly prepared calibration curves linear in the range 0.1–10 μg/mL for plasma and 0.06–6.0 μg/g for brain. The GraphPad Prism® program (GraphPad Software, Inc. La Jolla, CA, USA) was used for plotting the calibration curves and quantifying the unknown concentrations of doxycycline in plasma and brain.
2.14. Immunohistochemistry
Mice were euthanized by CO2 inhalation, brains were removed and fixed in Carnoy's fixative (ethanol, chloroform, acetic acid, 6:3:1), dehydrated in graded ethanol solutions, cleared in xylene, and embedded in paraffin. Midsagittal sections (8 μm thick) were cut with a Leica microtome onto glass slides. Slides were deparaffinized and boiled in citrate buffer (10 mM pH 6.0, Dako) for antigen retrieval, followed by incubation (1 h in humid environment) with 10% formalin. After 15 min in 3% H2O2, the slides were incubated for 1 h in blocking solution (10% FBS, 1% Triton X-100 in PBS), then overnight at 4 °C with rabbit polyclonal anti-glial fibrillary acidic protein (GFAP; DAKO, Z0334; 1:2500) or rabbit polyclonal anti-ionized calcium binding adapter molecule 1 (Iba1; Wako Reagent; 1:2500). After washing, slices were incubated with biotinylated goat anti-rabbit IgG antibody (Vector Laboratories, 1:200) for 1 h at room temperature, then visualized with the Vectastain ABC kit (Vector Laboratories) for the GFAP antibody and with the TSA Biotin System (Perkin Elmer) for Iba1, using DAB as chromogen. Slices were examined with an Olympus BX-61 Virtual Stage microscope, interfaced with VS-ASW-FL software (Olympus). For quantification of the GFAP- and Iba1-positive staining, the hippocampal area was selected from the sagittal sections. Slices were matched at comparable sagittal levels for comparison of the different experimental groups. Images were acquired using a 20× objective and analyzed using Fiji software, applying a home-made macro and normalizing on the quantified area (Balducci et al., 2018).
2.15. Statistics
All graphs present the mean ± standard error of the mean (SEM). SAS version 9.4 and R version 3.5.0 software were used for analyses. Data were analyzed by parametric and non-parametric ANOVA or mixed-effects models for repeated measures, followed by Bonferroni's post-hoc tests, as detailed in the figure legends. Assumption of normally distributed residuals and homoscedasticity were checked using graphical and formal methods (Q-Q plots and Kolmogorov-Smirnov test for normality of residuals and histograms and Levene's test for homoscedasticity). For mixed-effects models correlation structure was chosen based on the plot of covariance as a function of lag in time between pairs of observations within each mouse and the AIC, AICC and BIC indexes. The Kenward-Roger standard error and degree of freedom correction was used with repeated measures analysis. In case of groups with n < 5 non-parametric analyses were applied. Survival times were plotted as Kaplan-Meier curves. The exact Wilcoxon rank-sum test was used to compare survival distributions. A p-value ≤0.05 was considered statistically significant.
2.16. Study approval
Procedures involving animals were conducted in conformity with the institutional guidelines at the Istituto di Ricerche Farmacologiche Mario Negri IRCCS, in compliance with national (D.lgs 26/2014; Authorization n. 19/2008-A issued March 6, 2008 by Ministry of Health) and international laws and policies (EEC Council Directive 2010/63/UE; the NIH Guide for the Care and Use of Laboratory Animals, 2011 edition). They were reviewed and approved by the Mario Negri Institute Animal Care and Use Committee, which includes ad hoc members for ethical issues, and by the Italian Ministry of Health (Decreto no. 370/2016-PR and 321/2015-PR).
3. Results
3.1. Doxycycline does not modify the development and progression of motor dysfunction in Tg(FFI) mice
The Tg(FFI-26) mice used in this study express moPrP D177N/M128 on a PrP knockout background, therefore they express mutant but not endogenous wild-type PrP. These mice develop a progressive and invariably fatal neurological disease, with profound alterations of sleep, deficits in recognition and spatial working memory, and ataxia (Bouybayoune et al., 2015; Chiesa et al., 2016). As controls for all the behavioral experiments, we used nontransgenic (non-Tg) littermates of Tg(FFI-26) mice because they have the same genetic background as Tg(FFI-26) mice and were born and raised together, avoiding potential confounding effects of different genetic makeups and/or rearing conditions. We found that non-Tg mice did not differ from C57BL/6 in motor behavior, cognitive function, sleep and motor circadian rhythmicity (Bouybayoune et al., 2015). Tg(WT-E1) mice, expressing transgenically encoded WT PrP at the same level as D177N/M128 PrP in Tg(FFI-26) mice (Bouybayoune et al., 2015), were used as controls for the biochemical analysis of PrP.
Groups of Tg(FFI-26) and non-Tg mice were trained in the accelerating rotarod and beam walking tests, and assigned to daily i.p. saline (vehicle) or 10 mg/kg doxy. Basal rotarod and beam walking performances were evaluated before starting treatment (week 0), at 66 ± 10 days of age; then the tests were repeated every two weeks. There was a statistically significant difference in rotarod performance between Tg(FFI-26) and non-Tg mice at each week of treatment (p-value for the main effect of strain: <0.0001; Bonferroni adjusted p-values from week 2 of treatment: <0.05), with Tg(FFI-26) performance worsening progressively during treatment as the mice aged (p-value for interaction strain x time: 0.048; Fig. 1A). There was no difference between vehicle- and doxy-treated mice (p-value for the main effect of treatment: 0.37; p-value for interaction treatment x time: 0.095; p-value for interaction treatment x strain: 0.94; Fig. 1A).
Fig. 1.

Doxycycline has no effect on the progression of Tg(FFI-26) motor dysfunction assessed on the accelerating rotarod. (A) Non-Tg and Tg(FFI-26) mice treated with doxy or the vehicle were tested on the accelerating rotarod at the weeks of treatment indicated. Data are the mean ± SEM latency to fall (s) of 13 vehicle- and doxy-treated non-Tg, 12 vehicle-treated Tg(FFI-26) and 13 doxy-treated Tg(FFI-26) mice. A linear mixed-effects model with a first-order autoregressive correlation structure was used. In case of main effects, Bonferroni's post-hoc tests were done. (B) Mice were tested on the accelerating rotarod 6 and 24 weeks after the treatment had been stopped. Data are the mean ± SEM latency to fall (s) of 9 vehicle- and doxy-treated non-Tg, 8 vehicle-treated Tg(FFI-26) and 9 doxy-treated Tg(FFI-26) mice. A three-way analysis of variance for repeated measures (RM ANOVA) was used. In case of interaction effects, additional Bonferroni's post-hoc tests were done.
Results were similar for motor function in the beam walking test. There was a statistically significant difference in missteps between Tg(FFI-26) and non-Tg mice, with the number of missteps increasing progressively in Tg(FFI-26) mice compared to non-Tg controls (p-value for the main effect of strain: <0.0001; p-value for interaction strain x time: <0.0001; Bonferroni adjusted p-values from week 12 of treatment: <0.0001; Fig. 2A). There was no difference in missteps between vehicle- and doxy-treated Tg(FFI-26) mice (p-value for the main effect of treatment: 0.11; p-value for interaction treatment x time: 0.064; p-value for interaction treatment x strain: 0.16; Fig. 2A).
Fig. 2.
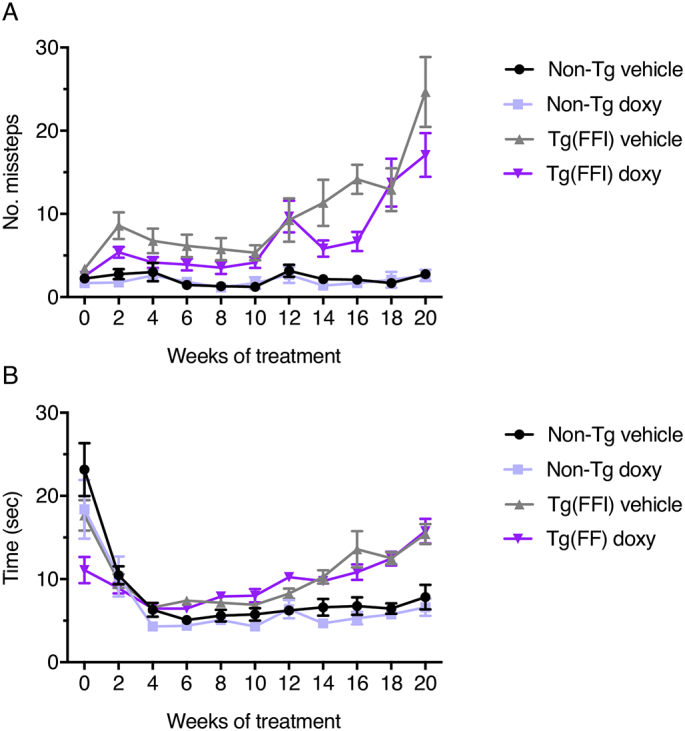
Doxycycline does not modify Tg(FFI-26) motor dysfunction in the beam walking test. Mice were tested for their ability to walk on a suspended beam at the weeks of treatment indicated. Each mouse was tested three times and the mean number of hindfoot missteps (A) and mean time to cross the beam (B) were scored. Data are the mean ± SEM of 13 vehicle- and doxy-treated non-Tg, 12 vehicle-treated Tg(FFI-26) and 13 doxy-treated Tg(FFI-26) mice. A three-way RM ANOVA was used. In case of interaction effects, additional Bonferroni's post-hoc tests were done.
During the first four weeks of treatment all groups of mice took less and less time to cross the beam as they became acquainted with the test (Fig. 2B). At later times, Tg(FFI-26) mice took significantly longer than non-Tg controls (p-value for the main effect of strain: <0.0001; p-value for interaction strain x time: <0.0001; Bonferroni adjusted p-values from week 12 of treatment: <0.05; Fig. 2B). There was no difference in the time to cross the beam for vehicle- and doxy-treated Tg(FFI-26) mice (p-value for the main effect of treatment: 0.21; p-value for interaction treatment x time: 0.30; p-value for interaction treatment x strain: 0.28; Fig. 2B).
3.2. Doxycycline rescues Tg(FFI-26) memory impairment in the novel object recognition test
The effect on memory deficit was assessed in the novel object recognition (NOR) test after 5 and 13 weeks of treatment. There was a main effect of strain (p-value: 0.014; p-value for interaction strain x time: 0.72) and an interaction effect between treatment and time (p-value: 0.005), consistent with progressive deterioration of memory in Tg(FFI-26) mice (Fig. 3A). Post-hoc tests detected an effect of treatment at 13 (Bonferroni adjusted p-value: 0.004) but not at 5 weeks (Bonferroni adjusted p-value: 0.48). At 13 weeks there was no significant interaction between genotype and treatment, indicating a beneficial effect of doxy on memory function in mice of both genotypes. However, the estimated effect was larger in Tg(FFI-26) than non-Tg mice (mean difference: 0.27 vs. 0.09; standardized effect size: 1.42 vs. 0.40) (Fig. 3A). Overall Tg(FFI-26) mice moved less during the test than non-Tg mice (main effect of strain, p-value: 0.014), but there was no effect of the treatment on the distance moved (main effect of treatment, p-value: 0.45), indicating that the beneficial effect of doxy in the NOR test was not due to differences in locomotor activity (Fig. 3B).
Fig. 3.
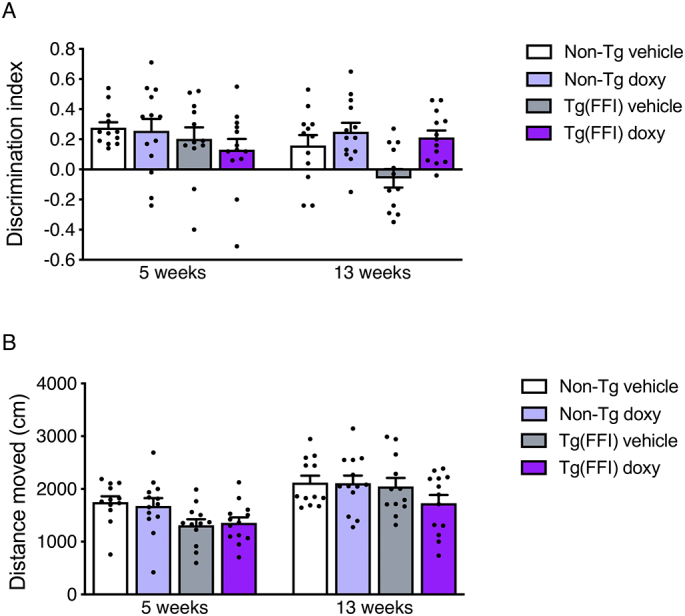
Doxycycline rescues the memory impairment of Tg(FFI-26) mice. (A) Recognition memory was investigated in the novel object recognition (NOR) test at 5 and 13 weeks of doxy treatment. Performance in the NOR task was expressed as a discrimination index (the higher the index, the better the performance). Bars indicate the mean ± SEM. (B) Distance moved during the test between the different groups of mice. A three-way RM ANOVA was used. In case of interaction effects, additional Bonferroni's post-hoc tests were done.
Treatment was stopped after 20 weeks and four mice from each group were culled for biochemical and histological analyses (see below). Doxy concentrations in the plasma and brain were respectively 1.195 ± 0.380 μg/mL and 0.201 ± 0.076 μg/g (mean ± SD; n = 8); brain/plasma ratio was 0.17 ± 0.05. There were no differences in doxy levels between non-Tg and Tg(FFI-26) mice. This confirmed that the compound had penetrated the brain of the mice, reaching concentrations similar to those reported previously (Lucchetti et al., 2019).
3.3. Doxycycline restores circadian motor activity but has no effect on the development of neurological illness or survival of Tg(FFI-26) mice
The mice remaining after the treatment were monitored throughout their life to assess whether the drug had any effect on the natural history of their disease.
Circadian motor activity is disrupted in Tg(FFI-26) mice (Bouybayoune et al., 2015; Chiesa et al., 2016). To measure home cage motor activity, the mice were individually housed in Tecniplast DVC®, which permit continuous automated, non-intrusive monitoring of the animal's activity. Mice were monitored 24/7 for 3 months, from weeks 8 through 20 post-treatment, during which they were undisturbed most of the time, with only two cage changes, and assessment of clinical signs of disease only once, at week 16 (see below).
Mice, as nocturnal animals, are more active during the dark than the light phase of the light-dark cycle. For instance, in the present study, the average activity of the non-Tg mice was 0.908 ± 0.303% during the 12 h of dark and 0.244 ± 0.076% in the light. As already described (Bouybayoune et al., 2015; Chiesa et al., 2016), in Tg(FFI-26) mice circadian distribution of motor activity is flattened, as they move less than controls during the dark phase. In the present study, there was a statistically significant difference between Tg(FFI-26) and non-Tg mice (p-value for the main effect of strain: 0.005; p-value for interaction strain x time: <0.0001; Fig. 4), and an interaction between genotype and treatment (p-value for interaction strain x treatment: 0.050). The behavior of vehicle-treated Tg(FFI-26) mice was significantly different from that of vehicle-treated non-Tg mice (Bonferroni adjusted p-value: 0.002). This difference was not seen in doxy-treated mice (Bonferroni adjusted p-value: 0.92). For example during the first quarter of the dark phase (corresponding to the first response to lights off), the average activity of vehicle-treated Tg(FFI-26) mice was 0.615 ± 0.147%, significantly less than vehicle-treated non-Tg mice (Bonferroni adjusted p-value: 0.020), whereas doxy-treated Tg(FFI-26) mice displayed average activity of 0.723 ± 0.276%, not significantly different from doxy-treated non-Tg mice (Bonferroni adjusted p-value: 1.00; Fig. 4 and Supplementary Table 1). The same patterns were observed in the other time blocks of the dark phase. Doxycycline treatment did not alter motor circadian distribution in non-Tg mice (Fig. 4).
Fig. 4.

Doxycycline restores circadian motor activity in Tg(FFI-26) mice. Bars indicate the mean ± SEM of activation density metric (average activations over the 12 electrodes of the board and within the selected time interval, expressed as percentage) of 9 vehicle-treated non-Tg, 8 doxy-treated non-Tg, 8 vehicle-treated Tg(FFI-26) and 9 doxy-treated Tg(FFI-26) mice, during the whole period of monitoring throughout the DVC® system and aggregated in 3-h bins. A generalized linear mixed-effects model with a heterogeneous first-order autoregressive correlation structure and lognormally distributed residuals was used. In case of interaction effects, additional Bonferroni's post-hoc tests were done. The key post-hoc comparisons within each time block are indicated in gray (Tg(FFI) vehicle vs. non-Tg vehicle) or magenta (Tg(FFI) doxy vs. non-Tg doxy); *P < 0.05, ns, non-significant. All multiple comparisons are reported in Supplementary Table 1. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The appearance and progression of clinical illness were scored according to a series of objective criteria, including kyphosis, foot-clasp reflex, piloerection and twitches, the ability to walk on a horizontal grid and to remain on a vertical grid for at least 30 s (see Section 2 for details). This scoring method showed a significant difference between Tg(FFI-26) and non-Tg mice, first evident at 16 weeks post-treatment (p-value for the main effect of strain: <0.0001; Bonferroni adjusted p-values: <0.0001); clinical signs progressively worsened in Tg(FFI-26) mice (p-value for interaction strain x time: <0.0001), with no significant difference between vehicle- and doxy-treated mice (p-value for the main effect of treatment: 0.53; p-value for interaction treatment x time: 0.72; p-value for interaction treatment x strain: 0.22; Fig. 5A).
Fig. 5.

Quantitative scores of clinical signs and survival. (A) Clinical signs were rated using objective criteria, each assigned a score (see Section 2). Bars indicate the mean ± SEM of 9 vehicle- and 6 doxy-treated non-Tg, 6 vehicle-treated Tg(FFI-26) and 9 doxy-treated Tg(FFI-26) mice. A linear mixed-effects model with a SP(POW) correlation structure was used. In case of interaction effects, additional Bonferroni's post-hoc tests were done. Three non-Tg and two Tg(FFI-26) mice died of intercurrent, non-neurological illness at 312, 359, 461, 399 and 513 days of age, and were excluded from the analyses. (B) Percentage survival of doxy- and vehicle-treated Tg(FFI-26) mice. The exact Wilcoxon rank-sum test was used. With the exception of three non-Tg mice that died of intercurrent illness, all other non-Tg mice remained healthy and were culled between 587 and 612 days of age.
At six and 24 weeks post-treatment mice were also tested on the rotarod. A difference in rotarod performance was detected between Tg(FFI-26) and non-Tg mice (p-value for the main effect of strain: <0.0001; Bonferroni adjusted p-values at each time: <0.0001; p-value for interaction strain x time: <0.001; Fig. 1B), but not between the vehicle- and doxy-treated groups (p-value for the main effect of treatment: 0.34; p-value for interaction treatment x time: 0.21; p-value for interaction treatment x strain: 0.84; Fig. 1B). As the disease progressed Tg(FFI-26) mice lost weight, and were euthanized when unable to reach food or water. There was no difference in survival between vehicle- and doxy-treated Tg(FFI-26) mice (exact p-value: 0.28; Fig. 5B).
3.4. Doxycycline does not alter the biochemical properties of mutant PrP
Tg(FFI-26) mice synthesize a misfolded form of mutant PrP in their brains that is detergent-insoluble and mildly protease-resistant, yielding a 19 kDa C-terminal fragment after enzymatic deglycosylation like in FFI patients (Bouybayoune et al., 2015). To test whether doxy altered the biochemical properties of mutant PrP, we assayed the solubility and protease resistance of PrP extracted from the brains of vehicle- and doxy-treated Tg(FFI-26) mice at the end of treatment. As a control, we analyzed brain extracts of hemizygous Tg(WT-E1) mice, which overexpress wild-type (WT) PrP at the same level as D177N PrP in Tg(FFI-26) mice (Bouybayoune et al., 2015; Chiesa et al., 1998).
To assay detergent-insoluble PrP, brain extracts were centrifuged at 16,000 xg for 5 min, the supernatant was collected and ultracentrifuged at 186,000 x g for 45 min. The proteins in the first and second pellets (P0 and P), and in the final supernatant (S) were analyzed by western blot, and PrP in the different fractions was quantified by densitometry and expressed as percentages of the amount of PrP in all the fractions. PrP was mostly in the S fraction of brain extracts from Tg(WT-E1) mice; approximately 40% was in the insoluble P and P0 fractions of Tg(FFI-26) brain extracts, with no significant differences between vehicle- and doxy-treated mice (Fig. 6).
Fig. 6.
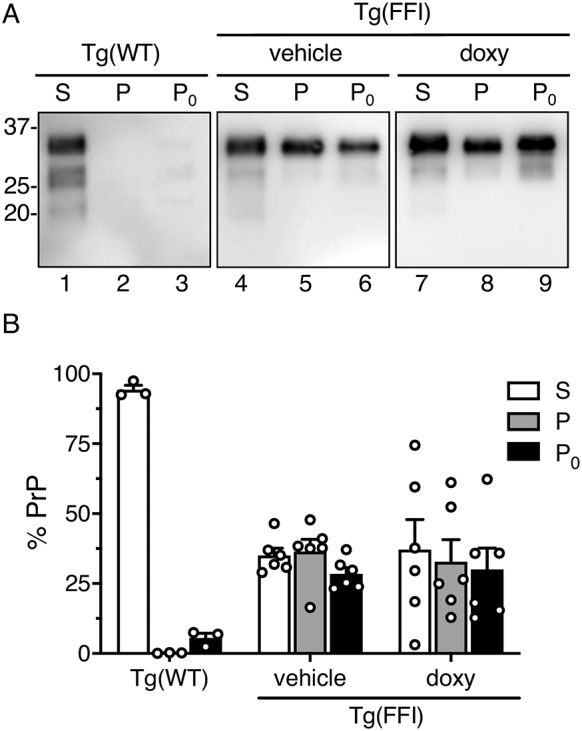
Doxycycline does not change the amount of detergent-insoluble PrP. (A) Brain extracts from mice sacrificed at the end of the treatment (249 ± 9 days of age) were centrifuged at 18,000 xg for 2 min. The pellet (P0) was recovered and the supernatant was ultracentrifuged at 186,000 x g for 45 min. PrP in each fraction [final supernatant (S), first (P0) and second (P) pellets] was analyzed by western blot using the monoclonal antibody 6D11. (B) PrP in the different fractions was quantified by densitometry and expressed as a percentage of the total. No significant differences in the distribution of PrP in the S, P and P0 fractions were found between vehicle- and doxy-treated Tg(FFI-26) mice. Data are the mean ± SEM of 3–5 replicate experiments using brains from 2 to 4 mice for each genotype/treatment.
To assay protease resistance the brain extracts were digested with proteinase-K (PK), and PK-resistant PrP was analyzed by western blot. Wild-type PrP from Tg(WT-E1) mice was completely degraded at PK concentrations as low as 0.5 μg/mL (Fig. 7A, top panel, and Fig. 7B), whereas mutant PrP in the brains of Tg(FFI-26) mice yielded PK-resistant fragments whose amounts or levels of resistance to PK were not affected by doxy (Fig. 7A, compare middle and lower panels, and Fig. 7B).
Fig. 7.

Doxycycline does not affect the protease resistance of mutant PrP in the brains of Tg(FFI-26) mice. (A) Brain lysates from mice sacrificed at the end of the treatment (249 ± 9 days of age) (200 μg of total proteins) were incubated with 0–2 μg/mL PK for 1 h at 4 °C, and PrP was visualized by western blotting using monoclonal antibody 6D11. Each sample represents 100 μg of protein. (B) The percentage of PrP remaining after PK digestion was quantified by densitometric analysis of western blots. Each point represents the mean ± SEM of 3–4 replicates.
3.5. Doxycycline reduces microglial activation in Tg(FFI-26) mice
Doxycycline improved memory function in the transgenic APP/PS1 mouse model of Alzheimer's disease (AD) and in a mouse model of AD-associated memory deficits in which recognition memory is acutely impaired by intracerebroventricularly injected Aβ oligomers (Balducci et al., 2018; Balducci et al., 2010). In both models the protective effect of doxy was reflected in reduced neuroinflammation in the hippocampus, particularly reduced microglial activation (Balducci et al., 2018). We wondered whether doxy reduced neuroinflammation in Tg(FFI-26) mice too.
We previously reported marked astrocytosis and microgliosis in the hippocampus of terminally ill Tg(FFI-26) mice (Bouybayoune et al., 2015). We checked whether gliosis was already detectable in the pre-terminal Tg(FFI-26) mice culled at the end of treatment, and whether treatment with doxy had modified the glial response. Brain sections from vehicle- and doxy-treated Tg(FFI-26) and non-Tg mice were immunostained with anti-glial fibrillar acidic protein (GFAP) or anti-ionized calcium binding adapter molecule 1 (Iba1) antibodies to mark respectively astrocytes and microglia, and the GFAP- and Iba1-positive areas in the hippocampus were measured. The GFAP-positive area was similar in all groups, indicating no astrocytic response in Tg(FFI-26) mice at this stage of disease (p-value for the main effect of strain: 0.33; p-value for the main effect of treatment: 0.71; p-value for interaction strain x treatment: 0.27; Fig. 8A and B). The Iba1-positive area was significantly larger in vehicle-treated Tg(FFI-26) mice than in vehicle- and doxy-treated non-Tg controls, indicating that microglia were already activated in the mutant mice (p-value for the main effect of strain: 0.009; p-value for the main effect of treatment: 0.022; p-value for interaction strain x treatment: 0.055). In Tg(FFI-26) mice treated with doxy the area occupied by microglia was significantly smaller than in vehicle-treated Tg(FFI-26) mice (Bonferroni adjusted p-value: 0.047) and comparable to that of non-Tg controls (Fig. 8C and D).
Fig. 8.

Doxycycline reduces microglial activation in Tg(FFI-26) mice. (A) Representative images of GFAP-immunostained hippocampus of doxy- and vehicle-treated Tg(FFI-26) and non-Tg mice culled at the end of treatment (249 ± 9 days of age). (B) Quantitative analysis of GFAP immunostaining. Data are the mean ± SEM of 2–4 mice per group. The R function raov (Kloke and McKean, 2012) was used for the rank-based analysis. (C) Representative images of Iba1-immunostained hippocampus of the same mice shown in A. (D) Quantitative analysis of Iba1 staining. Data are the mean ± SEM of 3–4 mice per group. The R function raov and oneway.rfit were used for the rank-based analysis (Kloke and McKean, 2012). *P < 0.05 by Bonferroni's post-hoc tests.
4. Discussion
There is no disease-modifying treatment for prion disease. The potential efficacy of doxy was suggested by experimental investigations and observational studies in sCJD patients, but has not been confirmed by clinical trials in symptomatic sCJD patients. This illustrates the challenges of treating such a rapidly progressive disease once the neurodegenerative process is under way, and calls for tests of efficacy in prevention trials in carriers of PRNP mutations. A ten-year preventive study is now ongoing in healthy individuals at risk of FFI to determine whether doxy given before irreversible neurodegeneration prevents or delays disease.
In the present study, we gave doxy to presymptomatic Tg(FFI-26) mice, which carry the mouse homolog of the FFI mutation and mimic key aspects of the human disease. Doxy had no effect on onset or progression of motor dysfunction, or any other clinical manifestations of the disease, and did not prolong survival. There was a positive effect on memory function, associated with diminished microglia activation in the hippocampus, which is reminiscent of the effects of doxy in mouse models of AD. In addition, doxy restored normal motor circadian activity.
The bacteriostatic tetracycline antibiotics were proposed for the treatment of prion disease on the basis of their ability to inhibit amyloidogenesis of synthetic PrP peptides, destabilize their aggregates, and promote enzymatic degradation of PrPSc (Forloni et al., 2002; Tagliavini et al., 2000). Doxy, a semi-synthetic second-generation derivative with a safe toxicological profile and good penetration of the blood-brain barrier, has been tested in preclinical and clinical settings. Preclinical trials in prion-infected hamsters showed prolonged survival in the doxy-treated group when treatment was started well before disease onset, but only modest effects when the drug was given at a symptomatic stage (De Luigi et al., 2008). In line with this, treatment of symptomatic sCJD patients did not prolong survival, with only a slight increase in a subset of patients treated from an early stage (Haïk et al., 2014; Varges et al., 2017). This suggested that doxy should be given presymptomatically for robust beneficial effects.
Carriers of highly penetrant PRNP mutations, who are at risk of developing genetic prion disease, can be given treatment before neurological damage has occurred. These individuals manifest no symptoms until adulthood (Minikel et al., 2019), even though PrP is expressed from early embryogenesis and misfolded/aggregated mutant PrP starts accumulating early postnatally in the brain, as inferred from studies in mice (Bouybayoune et al., 2015; Chiesa et al., 2000; Chiesa et al., 1998; Manson et al., 1992; McKinley et al., 1987). This suggests that disease ensues when a critical toxic PrP threshold is reached, perhaps because misfolded protein clearance declines with age (Chiesa and Harris, 2001; Hipp et al., 2019; Martinez-Vicente et al., 2005). Tetracyclines, by hindering PrP aggregation and promoting PrP aggregate disassembly and proteolysis (Tagliavini et al., 2000), may prevent this toxic threshold being reached.
Based on this rationale, the DOXIFF study was designed to test the effect of preventive doxy treatment in healthy carriers of the highly penetrant FFI mutation. The rarity of the disease and ethical reasons excluded the possibility to design a randomized, double-blind, placebo-controlled trial. The lack of prodromal biomarkers and surrogate outcome measures of treatment efficacy required starting the treatment well in advance of the presumed age of disease onset, and comparing the incidence of the disease in the treated group and in a historical dataset during ten years of observation (Forloni et al., 2015).
In Tg(FFI-26) mice, we mimicked a randomized placebo-controlled clinical trial by assigning presymptomatic mice to either the doxy- or vehicle-treatment group, and used performance on the accelerated rotarod, a sensitive and robust indicator of motor dysfunction, as primary outcome measure. Longitudinal assessment of rotarod performance found no difference in progressive decline between doxy- and vehicle-treated Tg(FFI-26) mice. Results were similar with the beam walking test which detects subtle deficits in motor skills and balance (Grande et al., 2018). Compared to non-Tg mice the number of missteps on the beam increased significantly in Tg(FFI-26) mice starting from 12 weeks of treatment, with no overall difference in the progressive increase between doxy- and vehicle-treated mice.
Consistent with a lack of any disease-modifying effect of doxy, the deficit in rotarod performance progressed similarly in vehicle- and doxy-treated mice during the post-treatment phase. Doxy also had no effect on neurological signs which appear at later stages in Tg(FFI-26) mice, such as hind-foot clasping, kyphosis or the ability to walk on a horizontal metal grid or climb a vertical grid, and did not prolong survival.
This is clearly different from the effect of doxy in other transgenic mouse models of neurodegenerative disease (Santa-Cecília et al., 2019, p.). For example, presymptomatic doxy treatment improved rotarod performance in the R6/2 mouse model of Huntington's disease (Paldino et al., 2020). In these mice, doxy also improved the distance travelled and the speed of locomotion in the open field, ameliorated the hind-foot clasping phenotype, reduced body weight loss, and prolonged survival (Paldino et al., 2020). In R6/2 mice doxy was injected 10 mg/kg i.p. twice a day, raising the possibility that a higher daily dose or more frequent doses may be necessary for beneficial effects. However, the pharmacokinetic profiles of doxy in mice given 10 mg/kg i.p. once or twice a day did not differ much, giving a similar brain Cmax of ~0.5 μM, which is comparable to the brain concentration of doxy in CJD patients chronically treated with 100 mg/day (Haïk et al., 2014; Lucchetti et al., 2019). This, and the fact that other aspects of the Tg(FFI-26) phenotype improved with a single 10 mg/kg daily dose (see discussion below), indicate that doxy reached therapeutically useful concentrations (Balducci et al., 2018; Lucchetti et al., 2019).
Tg(FFI-26) mice have alterations in long-term recognition and spatial working memory, detectable with the NOR task and the eight-arm radial maze (Bouybayoune et al., 2015). To assess the drug's effect on memory, we used the NOR test because it relies on a spontaneous behavior and, unlike the radial maze, it is not necessary to water deprive the mice, avoiding a potential source of stress which might have invalidated other behavioral tests. Moreover, the NOR test has been used before to assess the effect of doxy on memory deficits in mouse models of AD (Balducci et al., 2018). Compared to vehicle-treated Tg(FFI-26) mice, doxy-treated Tg(FFI-26) mice distinguished the new from the familiar object, indicating rescue of recognition memory. A beneficial effect of doxy was also observed in non-Tg (PrP knockout) mice, whose poor performance in the NOR test was unexpected on the basis of our previous results and requires further investigation (Balducci et al., 2010; Bouybayoune et al., 2015; Dossena et al., 2008; La Vitola et al., 2019). The development of motor impairment in Tg(FFI) mice meant we could not repeat the NOR test at later times to establish whether the beneficial effect of doxy on memory persisted.
In APP/PS1 mice chronic doxy treatment did not modify the Aβ plaque burden, but reduced the amount of oligomeric Aβ, consistent with the drug's ability to hinder synthetic Aβ aggregation (Balducci et al., 2018; Forloni et al., 2001). In wild-type mice, acute intracerebroventricular injection of synthetic Aβ oligomers activated microglia in the hippocampus and impaired recognition memory, and systemic doxy injection prevented microgliosis and the memory deficit (Balducci et al., 2018). Thus, the drug's beneficial effect on memory in AD models may result from reduction of Aβ oligomers and neuroinflammation (Balducci and Forloni, 2019). In Tg(FFI-26) mice doxy did not modify the amount of detergent-insoluble or protease-resistant PrP but reduced hippocampal microglial activation, measured by Iba-1 immunostaining, suggesting that the effect on memory might be related to its anti-inflammatory activity. Poly(ADP-ribose)polymerase-1 (PARP-1) influences microglia response to Aβ (Kauppinen et al., 2011), and submicromolar concentrations of doxy similar to those measured in the brain of Tg(FFI-26) mice potently inhibit PARP-1 activity (Alano et al., 2006). This raises the possibility that doxy may have damped microglial activation in Tg(FFI-26) mice through an effect on PARP-1.
During the dark phase of the light-dark cycle, vehicle-treated Tg(FFI-26) animals moved significantly less than non-Tg controls, while doxy-treated Tg(FFI-26) animals moved as much as controls. When animals were housed in DVC cages for analysis of motor activity, both vehicle- and doxy-treated Tg(FFI-26) mice were clearly impaired in the accelerated rotarod test. This suggests that doxy preserved the animal's general ability to move but not the motor skill necessary for more complex tasks. Moreover, doxy made Tg(FFI-26) mice move more during the appropriate circadian phase, i.e. during the dark phase of the light-dark cycle, indicating rescue of circadian motor rhythmicity. Future studies may clarify whether doxy rescues other circadian rhythms, such as circadian distribution of sleep and/or body temperature. Interestingly the tetracycline antibiotic minocycline, which acts directly on microglia, influences sleep and the response to sleep loss in rodents (Wisor et al., 2011).
5. Conclusions
Our data indicate that presymptomatic doxy treatment improves key aspects of the Tg(FFI-26) phenotype but does not rescue fatal neurological disease. Should these findings be applicable to humans, they would not uphold the efficacy of preventive doxy treatment in FFI, although some aspects of the symptomatology may be improved. However, current models of genetic prion disease do not fully recapitulate the whole spectrum of clinical, pathological, and biochemical hallmarks of the human disease, and Tg(FFI-26) mice are no exception (Vorberg and Chiesa, 2019). For example, although these mice present FFI-specific clinical and pathological features, the mutant PrP that accumulates in their brains cannot self-propagate like a bona fide prion (Bouybayoune et al., 2015; Chiesa et al., 2016). On the other hand, transgenic mice expressing the D178N/M129 mutation in the context of bank vole PrP show biochemical PrP features and neuropathological changes less representative of the human phenotype, but develop a rapidly progressive fatal disease that is horizontally transmissible to other mice by intracerebral inoculation, indicating spontaneous generation of prion infectivity (Watts et al., 2016). It will be interesting to see whether doxy hampers genetically determined prion formation and prevents disease in this model.
Declaration of Competing Interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by grants from Fondazione Telethon Italy (GGP10208), the EC (ERARE14-fp-097 - CHAPRION) and the Italian Ministry of Health (RF-2016-02362950).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2021.105455.
Appendix A. Supplementary data
S1 Data. Excel spreadsheet containing, in separate sheets, the underlying numerical data and key descriptive statistics for all Figure panels.
Supplementary Table S1
References
- Alano C.C., Kauppinen T.M., Valls A.V., Swanson R.A. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9685–9690. doi: 10.1073/pnas.0600554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C., Forloni G. Doxycycline for Alzheimer’s disease: fighting β-amyloid oligomers and neuroinflammation. Front. Pharmacol. 2019;10:738. doi: 10.3389/fphar.2019.00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C., Beeg M., Stravalaci M., Bastone A., Sclip A., Biasini E., Tapella L., Colombo L., Manzoni C., Borsello T., Chiesa R., Gobbi M., Salmona M., Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U. S. A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C., Santamaria G., La Vitola P., Brandi E., Grandi F., Viscomi A.R., Beeg M., Gobbi M., Salmona M., Ottonello S., Forloni G. Doxycycline counteracts neuroinflammation restoring memory in Alzheimer’s disease mouse models. Neurobiol. Aging. 2018;70:128–139. doi: 10.1016/j.neurobiolaging.2018.06.002. [DOI] [PubMed] [Google Scholar]
- Bouybayoune I., Mantovani S., Del Gallo F., Bertani I., Restelli E., Comerio L., Tapella L., Baracchi F., Fernández-Borges N., Mangieri M., Bisighini C., Beznoussenko G.V., Paladini A., Balducci C., Micotti E., Forloni G., Castilla J., Fiordaliso F., Tagliavini F., Imeri L., Chiesa R. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder T., Foltz C., Karlsson E., Linton C.G., Smith J.M. Health evaluation of experimental laboratory mice. Curr. Protoc. Mouse Biol. 2012;2:145–165. doi: 10.1002/9780470942390.mo110217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R., Harris D.A. Prion diseases: what is the neurotoxic molecule? Neurobiol. Dis. 2001;8:743–763. doi: 10.1006/nbdi.2001.0433. [DOI] [PubMed] [Google Scholar]
- Chiesa R., Piccardo P., Ghetti B., Harris D.A. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. S0896-6273(00)80653-4 (pii) [DOI] [PubMed] [Google Scholar]
- Chiesa R., Drisaldi B., Quaglio E., Migheli A., Piccardo P., Ghetti B., Harris D.A. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5574–5579. doi: 10.1073/pnas.97.10.5574. 97/10/5574 (pii) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R., Restelli E., Comerio L., Del Gallo F., Imeri L. Transgenic mice recapitulate the phenotypic heterogeneity of genetic prion diseases without developing prion infectivity: role of intracellular PrP retention in neurotoxicity. Prion. 2016;10:93–102. doi: 10.1080/19336896.2016.1139276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luigi A., Colombo L., Diomede L., Capobianco R., Mangieri M., Miccolo C., Limido L., Forloni G., Tagliavini F., Salmona M. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Isa R., Brambilla R., Fasano S. Behavioral methods for the study of the Ras-ERK pathway in memory formation and consolidation: passive avoidance and novel object recognition tests. Methods Mol. Biol. 2014;1120:131–156. doi: 10.1007/978-1-62703-791-4_9. [DOI] [PubMed] [Google Scholar]
- Dossena S., Imeri L., Mangieri M., Garofoli A., Ferrari L., Senatore A., Restelli E., Balducci C., Fiordaliso F., Salio M., Bianchi S., Fioriti L., Morbin M., Pincherle A., Marcon G., Villani F., Carli M., Tagliavini F., Forloni G., Chiesa R. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron. 2008;60:598–609. doi: 10.1016/j.neuron.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Forloni G., Colombo L., Girola L., Tagliavini F., Salmona M. Anti-amyloidogenic activity of tetracyclines: studies in vitro. FEBS Lett. 2001;487:404–407. doi: 10.1016/s0014-5793(00)02380-2. [DOI] [PubMed] [Google Scholar]
- Forloni G., Iussich S., Awan T., Colombo L., Angeretti N., Girola L., Bertani I., Poli G., Caramelli M., Grazia Bruzzone M., Farina L., Limido L., Rossi G., Giaccone G., Ironside J.W., Bugiani O., Salmona M., Tagliavini F. Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10849–10854. doi: 10.1073/pnas.162195499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forloni G., Tettamanti M., Lucca U., Albanese Y., Quaglio E., Chiesa R., Erbetta A., Villani F., Redaelli V., Tagliavini F., Artuso V., Roiter I. Preventive study in subjects at risk of fatal familial insomnia: innovative approach to rare diseases. Prion. 2015;9:75–79. doi: 10.1080/19336896.2015.1027857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb L.G., Petersen R.B., Tabaton M., Brown P., LeBlanc A.C., Montagna P., Cortelli P., Julien J., Vital C., Pendelbury W.W. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- Golini E., Rigamonti M., Iannello F., De Rosa C., Scavizzi F., Raspa M., Mandillo S. A non-invasive digital biomarker for the detection of rest disturbances in the SOD1G93A mouse model of ALS. Front. Neurosci. 2020;14:896. doi: 10.3389/fnins.2020.00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande V., Ornaghi F., Comerio L., Restelli E., Masone A., Corbelli A., Tolomeo D., Capone V., Axten J.M., Laping N.J., Fiordaliso F., Sallese M., Chiesa R. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjögren syndrome. Hum. Mol. Genet. 2018;27:2477–2489. doi: 10.1093/hmg/ddy152. [DOI] [PubMed] [Google Scholar]
- Grayson B., Leger M., Piercy C., Adamson L., Harte M., Neill J.C. Assessment of disease-related cognitive impairments using the novel object recognition (NOR) task in rodents. Behav. Brain Res. 2015;285:176–193. doi: 10.1016/j.bbr.2014.10.025. [DOI] [PubMed] [Google Scholar]
- Haïk S., Marcon G., Mallet A., Tettamanti M., Welaratne A., Giaccone G., Azimi S., Pietrini V., Fabreguettes J.-R., Imperiale D., Cesaro P., Buffa C., Aucan C., Lucca U., Peckeu L., Suardi S., Tranchant C., Zerr I., Houillier C., Redaelli V., Vespignani H., Campanella A., Sellal F., Krasnianski A., Seilhean D., Heinemann U., Sedel F., Canovi M., Gobbi M., Di Fede G., Laplanche J.-L., Pocchiari M., Salmona M., Forloni G., Brandel J.-P., Tagliavini F. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:150–158. doi: 10.1016/S1474-4422(13)70307-7. [DOI] [PubMed] [Google Scholar]
- Hipp M.S., Kasturi P., Hartl F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019;20:421–435. doi: 10.1038/s41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- Iaccarino L., Presotto L., Bettinardi V., Gianolli L., Roiter I., Capellari S., Parchi P., Cortelli P., Perani D. An in vivo 11C-PK PET study of microglia activation in fatal familial insomnia. Ann. Clin. Transl. Neurol. 2018;5:11–18. doi: 10.1002/acn3.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannello F. Non-intrusive high throughput automated data collection from the home cage. Heliyon. 2019;5 doi: 10.1016/j.heliyon.2019.e01454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen T.M., Suh S.W., Higashi Y., Berman A.E., Escartin C., Won S.J., Wang C., Cho S.-H., Gan L., Swanson R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid β. J. Neuroinflammation. 2011;8:152. doi: 10.1186/1742-2094-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C., Browne W.J., Cuthill I.C., Emerson M., Altman D.G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloke J.D., McKean J.W. Rfit: Rank-based estimation for linear models. R Journal. 2012;4:57. doi: 10.32614/RJ-2012-014. [DOI] [Google Scholar]
- La Vitola P., Beeg M., Balducci C., Santamaria G., Restelli E., Colombo L., Caldinelli L., Pollegioni L., Gobbi M., Chiesa R., Forloni G. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain. 2019;142:249–254. doi: 10.1093/brain/awy318. [DOI] [PubMed] [Google Scholar]
- Lucchetti J., Fracasso C., Balducci C., Passoni A., Forloni G., Salmona M., Gobbi M. Plasma and brain concentrations of doxycycline after single and repeated doses in wild-type and APP23 mice. J. Pharmacol. Exp. Ther. 2019;368:32–40. doi: 10.1124/jpet.118.252064. [DOI] [PubMed] [Google Scholar]
- Mackenzie G., Will R. Creutzfeldt-Jakob disease: recent developments. F1000Res. 2017;6 doi: 10.12688/f1000research.12681.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson J., West J.D., Thomson V., McBride P., Kaufman M.H., Hope J. The prion protein gene: a role in mouse embryogenesis? Development. 1992;115:117–122. doi: 10.1242/dev.115.1.117. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M., Sovak G., Cuervo A.M. Protein degradation and aging. Exp. Gerontol. 2005;40:622–633. doi: 10.1016/j.exger.2005.07.005. [DOI] [PubMed] [Google Scholar]
- McKinley M.P., Hay B., Lingappa V.R., Lieberburg I., Prusiner S.B. Developmental expression of prion protein gene in brain. Dev. Biol. 1987;121:105–110. doi: 10.1016/0012-1606(87)90143-6. [DOI] [PubMed] [Google Scholar]
- Minikel E.V., Vallabh S.M., Lek M., Estrada K., Samocha K.E., Sathirapongsasuti J.F., McLean C.Y., Tung J.Y., Yu L.P., Gambetti P., Blevins J., Zhang S., Cohen Y., Chen W., Yamada M., Hamaguchi T., Sanjo N., Mizusawa H., Nakamura Y., Kitamoto T., Collins S.J., Boyd A., Will R.G., Knight R., Ponto C., Zerr I., Kraus T.F., Eigenbrod S., Giese A., Calero M., de Pedro-Cuesta J., Haik S., Laplanche J.L., Bouaziz-Amar E., Brandel J.P., Capellari S., Parchi P., Poleggi A., Ladogana A., O’Donnell-Luria A.H., Karczewski K.J., Marshall J.L., Boehnke M., Laakso M., Mohlke K.L., Kahler A., Chambert K., McCarroll S., Sullivan P.F., Hultman C.M., Purcell S.M., Sklar P., van der Lee S.J., Rozemuller A., Jansen C., Hofman A., Kraaij R., van Rooij J.G., Ikram M.A., Uitterlinden A.G., van Duijn C.M., Daly M.J., MacArthur D.G. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aad5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minikel E.V., Vallabh S.M., Orseth M.C., Brandel J.-P., Haïk S., Laplanche J.-L., Zerr I., Parchi P., Capellari S., Safar J., Kenny J., Fong J.C., Takada L.T., Ponto C., Hermann P., Knipper T., Stehmann C., Kitamoto T., Ae R., Hamaguchi T., Sanjo N., Tsukamoto T., Mizusawa H., Collins S.J., Chiesa R., Roiter I., de Pedro-Cuesta J., Calero M., Geschwind M.D., Yamada M., Nakamura Y., Mead S. Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology. 2019;93:e125–e134. doi: 10.1212/WNL.0000000000007745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna P., Cortelli P., Avoni P., Tinuper P., Plazzi G., Gallassi R., Portaluppi F., Julien J., Vital C., Delisle M.B., Gambetti P., Lugaresi E. Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol. 1998;8:515–520. doi: 10.1111/j.1750-3639.1998.tb00172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna P., Gambetti P., Cortelli P., Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003;2:167–176. doi: 10.1016/s1474-4422(03)00323-5. S1474442203003235 (pii) [DOI] [PubMed] [Google Scholar]
- Paldino E., Balducci C., La Vitola P., Artioli L., D’Angelo V., Giampà C., Artuso V., Forloni G., Fusco F.R. Neuroprotective effects of doxycycline in the R6/2 mouse model of Huntington’s disease. Mol. Neurobiol. 2020;57:1889–1903. doi: 10.1007/s12035-019-01847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponto C., Zerr I. In|Fo|Neurologie & Psychiatrie. 2013. Prionerkrankungen—welche Rolle spielen sie heute? pp. 2–8. [Google Scholar]
- Prusiner S.B. Prions. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa-Cecília F.V., Leite C.A., Del-Bel E., Raisman-Vozari R. The neuroprotective effect of doxycycline on neurodegenerative diseases. Neurotox. Res. 2019;35:981–986. doi: 10.1007/s12640-019-00015-z. [DOI] [PubMed] [Google Scholar]
- Schenkein J., Montagna P. Self management of fatal familial insomnia. Part 1: what is FFI? Medscape Gen. Med. 2006;8:65. [PMC free article] [PubMed] [Google Scholar]
- Stoilova T., Colombo L., Forloni G., Tagliavini F., Salmona M. A new face for old antibiotics: tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013;56:5987–6006. doi: 10.1021/jm400161p. [DOI] [PubMed] [Google Scholar]
- Tagliavini F. Alzheimers Dement. 2008. Prion therapy: tetracyclic compounds in animal models and patients with Creutzfeldt-Jakob disease; pp. T149–T150. [Google Scholar]
- Tagliavini F., Forloni G., Colombo L., Rossi G., Girola L., Canciani B., Angeretti N., Giampaolo L., Peressini E., Awan T., De Gioia L., Ragg E., Bugiani O., Salmona M. Tetracycline affects abnormal properties of synthetic PrP peptides and PrPSc in vitro. J. Mol. Biol. 2000;300:1309–1322. doi: 10.1006/jmbi.2000.3840. [DOI] [PubMed] [Google Scholar]
- Varges D., Manthey H., Heinemann U., Ponto C., Schmitz M., Schulz-Schaeffer W.J., Krasnianski A., Breithaupt M., Fincke F., Kramer K., Friede T., Zerr I. Doxycycline in early CJD: a double-blinded randomised phase II and observational study. J. Neurol. Neurosurg. Psychiatry. 2017;88:119–125. doi: 10.1136/jnnp-2016-313541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voikar V., Gaburro S. Three pillars of automated home-cage phenotyping of mice: novel findings, refinement, and reproducibility based on literature and experience. Front. Behav. Neurosci. 2020;14:575434. doi: 10.3389/fnbeh.2020.575434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorberg I., Chiesa R. Experimental models to study prion disease pathogenesis and identify potential therapeutic compounds. Curr. Opin. Pharmacol. 2019;44:28–38. doi: 10.1016/j.coph.2019.02.002. [DOI] [PubMed] [Google Scholar]
- Watts J.C., Giles K., Bourkas M.E.C., Patel S., Oehler A., Gavidia M., Bhardwaj S., Lee J., Prusiner S.B. Towards authentic transgenic mouse models of heritable PrP prion diseases. Acta Neuropathol. 2016;132:593–610. doi: 10.1007/s00401-016-1585-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor J.P., Schmidt M.A., Clegern W.C. Evidence for neuroinflammatory and microglial changes in the cerebral response to sleep loss. Sleep. 2011;34:261–272. doi: 10.1093/sleep/34.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Data. Excel spreadsheet containing, in separate sheets, the underlying numerical data and key descriptive statistics for all Figure panels.
Supplementary Table S1