

Abstract
Background & Aims
Patients with simple steatosis (SS) and nonalcoholic steatohepatitis can develop progressive liver fibrosis, which is associated with liver-related mortality. The mechanisms contributing to liver fibrosis development in SS, however, are poorly understood. SS is characterized by hepatocellular free fatty acid (FFA) accumulation without lobular inflammation seen in nonalcoholic steatohepatitis. Because the Hippo signaling transcriptional coactivator YAP1 (YAP) has previously been linked with nonalcoholic fatty liver disease (NAFLD)–related fibrosis, we sought to explore how hepatocyte FFAs activate a YAP-mediated profibrogenic program.
Methods
We analyzed RNA sequencing data from a GEO DataSet (accession: GSE162694) consisting of 143 patients with NAFLD. We also performed immunohistochemical, immunofluorescence, immunoblot, and quantitative reverse-transcription polymerase chain reaction analyses (qRT-PCR) in liver specimens from NAFLD subjects, from a murine dietary NAFLD model, and in FFA-treated hepatic spheroids and hepatocytes.
Results
YAP-target gene expression correlated with increasing fibrosis stage in NAFLD patients and was associated with fibrosis in mice fed a NAFLD-inducing diet. Hepatocyte-specific YAP deletion in the murine NAFLD model attenuated diet-induced fibrosis, suggesting a causative role of YAP in NAFLD-related fibrosis. Likewise, in hepatic spheroids composed of Huh7 hepatoma cells and primary human hepatic stellate cells, Huh7 YAP silencing reduced FFA-induced fibrogenic gene expression. Notably, inhibition of p38 mitogen-activated protein kinase could block YAP activation in FFA-treated Huh7 cells.
Conclusions
These studies provide further evidence for the pathological role of YAP in NAFLD-associated fibrosis and that YAP activation in NAFLD may be driven by FFA-induced p38 MAPK activation.
Keywords: YAP, p38 MAPK, Nonalcoholic Fatty Liver Disease, Fibrosis
Abbreviations used in this paper: α-SMA, α-smooth muscle actin; CDAHFD, choline-deficient; L-amino acid defined, high fat diet; ECM, extracellular matrix; ERK, extracellular signal-regulated kinase; FFA, free fatty acid; GFP, green fluorescent protein; HCC, hepatocellular carcinoma; IL, interleukin; KO, knockout; JNK, c-Jun N-terminal kinase; LD, lipid droplet; MAPK, mitogen-activated protein kinase; MGH, Massachusetts General Hospital; mRNA, messenger RNA; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NT, nontargeting; PHH, primary human hepatocyte; pHSC, primary human hepatic stellate cell; RNAseq, RNA sequencing; shRNA, short hairpin RNA; siRNA, small interfering RNA; SS, simple steatosis; WT, wild-type
Graphical abstract

Nonalcoholic fatty liver disease (NAFLD) is the most common liver disorder in the United States, affecting up to 46% of Americans.1 NAFLD encompasses a spectrum of conditions ranging from simple steatosis (SS) to nonalcoholic steatohepatitis (NASH), characterized by steatosis, concurrent necroinflammation, and varying degrees of fibrosis, which can ultimately lead to cirrhosis and its complications.2 NAFLD is now the leading indication for liver transplantation3 and a rapidly increasing cause of hepatocellular carcinoma in the United States.4 Despite the tremendous burden of NAFLD, there are currently no Food and Drug Administration–approved therapeutic agents for any stage of disease. A comprehensive understanding of the mechanisms underlying NAFLD-related pathology is essential to unearthing novel targets that can be modulated through drug-based intervention.
Hippo signaling, an evolutionarily conserved pathway that controls tissue growth and organ size, has gained particular interest as a potential therapeutic target for NAFLD as its major downstream effectors, transcriptional coactivator YAP1 (YAP) and WW domain-containing transcription regulator protein 1 (WWTR1/TAZ), have been associated with liver fibrosis and NAFLD in mice and humans.5,6 When Hippo signaling is activated or “on,” YAP and TAZ are phosphorylated by serine/threonine-protein kinases LATS1/2, retained in the cytoplasm, and targeted for degradation.7 In the Hippo “off” state, YAP/TAZ are free to translocate to the nucleus and associate with transcription factors, including members of the TEAD family, to activate expression of many different target genes including those involved in hepatic inflammation and fibrogenesis.6,8 However, there is a paucity of information describing how YAP/TAZ activation occurs in NAFLD.
The pathogenesis of NAFLD is still not fully understood but involves a complex interplay of environmental, genetic, and host factors, including diet, central obesity and adipocytes, the microbiome, genetic predisposition and polymorphisms, host cellular responses and epigenetics that culminate in disturbed lipid homeostasis, lipotoxicity, inflammation, and fibrogenesis.9 SS without inflammation has been traditionally considered a benign condition rarely associated with liver disease progression, in contrast to NASH, which confers an increased risk of liver-related mortality and hepatocellular carcinoma (HCC) thought to be mediated via fibrosis progression. However, there are increasing data demonstrating that fibrosis progression rates are similar in patients with SS and NASH.10 After prospective extended follow-up of SS and NASH patients with multiple paired liver biopsies, baseline histological features of steatosis and inflammation did not predict subsequent development of clinically significant disease progression to advanced fibrosis,11 challenging prior assumptions regarding SS in the absence of inflammation.
The hepatic accumulation of free fatty acids (FFAs) and other toxic lipids characteristic of NAFLD can arise from increased de novo lipogenesis, owiung to carbohydrate surplus, and accelerated lipolysis of adipose stores by hormone sensitive lipase—an activity negatively regulated by insulin. In vitro studies have previously revealed that FFAs (oleic acid) promote gluconeogenic gene expression and decrease insulin sensitivity through p38 mitogen-activated protein kinase (MAPK) activation in hepatocytes.12,13 Because insulin resistance likely plays a key role in the development of NAFLD, we sought to (1) determine whether fibrosis progression in NAFLD is dependent on hepatocyte YAP and (2) whether YAP activity is regulated by p38 MAPK in the context of excess FFAs.
In the present study, we found that hepatic expression of YAP target genes was increased in NAFLD patients with fibrosis and in wild-type mice fed the NAFLD-inducing choline-deficient, L-amino acid–defined, high fat diet (CDAHFD). When hepatocyte-specific YAP conditional knockout (KO) mice were fed the CDAHFD, immunohistochemical, immunoblot, and reverse-transcription polymerase chain reaction analyses revealed that fibrosis-related parameters were significantly reduced, suggesting that YAP is necessary for NAFLD-related fibrosis progression. In vitro experiments in hepatic spheroids composed of Huh7 hepatoma cells and primary human hepatic stellate cells (pHSCs) similarly demonstrated that hepatocyte YAP depletion could preclude FFA-induced pHSC activation and extracellular matrix (ECM) deposition. We also provide evidence that p38δ MAPK (MAPK13) regulates hepatocyte YAP activation in response to exposure to exogenous FFAs. Collectively, these data suggest distinct antifibrotic approaches to targeting YAP activation to reverse NAFLD-related pathology.
Results
YAP Is Activated in the Livers of Humans With NAFLD-Related Fibrosis
We used a previously described RNA sequencing (RNAseq) dataset from liver samples of 143 individuals to evaluate YAP-regulated gene expression in relation to NAFLD-associated fibrosis progression. In our analyses, patients were categorized into 4 groups based on fibrosis stage: no evidence of steatosis or fibrosis (G1), steatosis with no evidence of fibrosis (G2), steatosis and minimal or moderate fibrosis (METAVIR F1 or F2; G3), and steatosis and advanced fibrosis or cirrhosis (METAVIR F3 or F4; G4). Twenty-two of 89 YAP-related genes were found to be differentially expressed in pairwise comparisons between each group and across fibrosis stages. To further probe YAP activation, expression patterns of YAP-dependent genes were analyzed. Following normalization of data count to variance-stabilized transformation values, distinctive clustering patterns were observed as shown in the metricized heatmap (Figure 1A). There is a dominant cluster of upregulated genes and only 3 genes that were downregulated in G2 or G3 but upregulated in G4 (Figure 1B). As shown in Figure 1B, 19 of the 22 genes from the bulk analysis were consistently upregulated in steatosis and increasing fibrosis stage. We then performed immunohistochemistry (Figure 1C) and immunofluorescence (Figure 2) analyses on liver specimens from the same cohort of patients and observed markedly enhanced YAP staining in patients with steatosis and fibrosis (G2, G3, and G4) relative to patients who had neither steatosis nor fibrosis (G1). More importantly, nuclear (active) YAP (brown) was more prevalent in the samples from patients with fibrosis (Figure 1C). In addition to nuclear localization, YAP activity can be assessed by the degree of phosphorylation on serine residue 127 (S127). Phosphorylation at this site generates a 14-3-3 binding site, which promotes YAP cytoplasmic sequestration and targeting for degradation.7,14,15 Immunoblot analyses revealed that phosphorylated S127 YAP (P-YAP) levels were reduced in G2, G3, and G4 patients compared with G1 patients (Figure 1D). This reduction in P-YAP was accompanied by an increase in total p38 MAPK levels in patients with steatosis and moderate fibrosis (Figure 1D). Therefore, our human data confirm that hepatic YAP activity is upregulated in NAFLD-related fibrosis and is associated with p38 MAPK expression.
Table 1.
Demographic and Patient Characteristics
| All Patients (N = 182) | Normal Histology (n = 41) | Steatosis (n = 45) | F1–2 (n = 81) | F3–4 (n = 33) | |
|---|---|---|---|---|---|
| Male | 35 | 22.5 | 33.3 | 36.2 | 53.7 |
| BMI, kg/m2 | 41.4 ± 10.3 | 41.4 ± 4.4 | 46.6 ± 10.2 | 38.8 ± 11 | 36.1 ± 11.7 |
| Diabetes | 33.5 | 15 | 24.4 | 37.7 | 85.7 |
| ALT, U/L | 36.6 ±19.2 | 22.9 ± 12.1 | 29.9 ± 9.6 | 41.8 ± 21 | 53.3 ± 18 |
| AST, U/L | 30 ± 15.8 | 19.1 ± 5 | 22.7 ± 4.8 | 33.1 ± 16 | 48.8 ± 19.5 |
Values are % or mean ± SD.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index.
Figure 1.

YAP is activated in the livers of humans with NAFLD. (A) Variance-stabilized transformation metric heatmap showing expression patterns of differentially expressed YAP-related genes (using genes with abs(log2FC) >0.3 and false discovery rate <0.05) from the indicated groups. The adjacent legend indicates the scale of expression based on variance-stabilized transformation count, with red signifying upregulation and black signifying downregulation. (B) Venn diagram of gene expression patterns of YAP DE genes. (C) YAP immunohistochemistry (brown) in liver specimens from the patient groups in Table 1 (original magnification ×20, inset ×40). Percentage of YAP-positive cells are graphed as mean ± SD (n = 3–5 subjects per group; ∗P < .05). (D) Immunoblot and densitometric analysis of phosphorylated YAP, YAP, and p38 MAPK from patient liver samples, as in panel C.
Figure 2.

YAP is activated in the livers of humans with NAFLD. YAP (purple) immunofluorescence with DAPI (blue) counterstain and Masson’s trichrome staining of liver sections from indicated groups.
YAP Is Activated in the Livers of Mice With NAFLD
We next evaluated YAP expression in a widely accepted dietary mouse model of NAFLD, the CDAHFD model.9,16 Analyses of liver sections from mice fed the CDAHFD for 12 weeks showed a substantial increase in YAP immunostaining when compared with chow-fed mice (Figure 3A). Assessment of fibrosis by Picrosirius red staining, lipid content by hematoxylin and eosin, and expression of ECM-related genes (Col1a1 and Timp1) showed that CDAHFD-fed mice developed fibrosis and increased lipid content after 4 weeks of feeding and bridging fibrosis by 12 weeks (Figures 3B–D and 4). Furthermore, these fibrosis-related parameters were associated with YAP activation, as indicated by decreased P-YAP levels relative to total YAP, and increased YAP target gene expression (Ctgf and Cyr61) (Figure 3C and D). Collectively, our in vivo mouse data confirm that hepatic YAP activity is induced in the CDAHFD mouse model and is associated with fibrosis.
Figure 3.

YAP is activated in the livers of mice with NAFLD. (A) YAP immunohistochemistry (brown) in liver specimens from mice fed either standard chow or the CDAHFD for 10 weeks (original magnification ×20, inset ×40). Percentage of YAP-positive cells are graphed as mean ± SD (n = 3 mice per group; ∗∗∗P < .001). (B) Hematoxylin and eosin (H&E) and picrosirius red staining in liver specimens from mice fed standard chow or CDAHFD (original magnification ×10). Percent of picrosirius red–stained liver tissue is graphed as mean ± SD (n = 3 mice per group; ∗∗P < .01). (C, D) Immunoblot and reverse-transcription polymerase chain reaction analyses of Yap, Ctgf, Cyr61, Col1a1, and Timp1. Mean ± SD are graphed (n = 3 mice per group; ∗P < .05, ∗∗P < .01). W, week.
Figure 4.
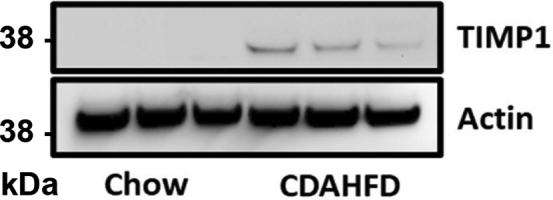
TIMP1 (metalloproteinase inhibitor 1) is induced in the livers of mice with NAFLD. Immunoblot analysis of TIMP1 and Actin in liver specimens from mice fed standard chow or CDAHFD.
FFAs Promote YAP Activation in Human Hepatocytes
We next developed an in vitro model to evaluate whether FFAs specifically induce YAP activation in hepatocytes. We exposed Huh7 cells to linoleic acid-oleic acid or palmitic acid. Fluorescence microscopy after 24 hours revealed an increase in neutral lipid staining in a dose-responsive manner (Figure 5A and B). As Huh7 cells are a well-differentiated hepatoma cell line, we performed analogous studies in primary human hepatocytes (PHHs) isolated from healthy human liver donors to determine if the results would be congruous. After exposure to linoleic acid-oleic acid for 24 hours, we observed a similar appreciable increase in intracellular lipid staining with BODIPY (Figure 5C). Cell viability assays in Huh7 cells indicated that the 150 μg/mL linoleic acid-oleic acid concentration induced stable lipid accumulation while having a negligible effect on cell viability (Figure 5D); this maximum tolerated dose was used for all subsequent experiments.
Figure 5.

Validation of in vitro NAFLD model. (A) Lipid staining (red) in Huh7 cells treated with FFAs for 24 hours (scale bar = 50 μm). (B) Lipid staining (green) in Huh7 cells treated with palmitic acid for 24 hours (scale bar = 50 μm). (C) Lipid staining (green) in PHHs treated with FFAs for 24 hours (scale bar = 50 μm). (D) Cell viability assessed using the CellTiter-Glo Luminescent Cell Viability assay (Promega) in Huh7 and pHSCs treated with FFAs. (E) Representative images of Huh7-ARE-GFP reporter cells untreated and treated with FFAs (37.6 μg/mL) or the positive control tert-Butylhydroquinone (TBHQ) for 48 hours.
Increased levels of reactive oxygen species due to mitochondrial β-oxidation of fatty acids17 can activate fibrotic pathways.18 We therefore utilized a Huh7 green fluorescent protein (GFP) reporter cell line driven by functional antioxidant response element/nuclear factor erythroid 2–related factor 2 (ARE/Nrf2) transcription factor response elements, to study dynamic transcriptional regulation of oxidative stress-related genes in living cells. Using this reporter, we found that ARE-driven transcriptional activity was upregulated following FFA administration (Figure 5E). Together, these studies suggest that our in vitro model can be utilized to elucidate the mechanisms underlying fundamental cellular processes in NAFLD.
After exposing Huh7 cells to linoleic acid-oleic acid (hereafter referred to as FFAs) or palmitic acid for 72 hours, we observed no changes in YAP messenger RNA (mRNA) levels but appreciable increases in mRNA expression of the YAP-dependent genes ANKRD1, CYR61, CTGF, JUB, TGFB1, and WTIP relative to untreated cells (Figure 6A). FFAs induced a similar phenotype in HepG2 cells, another transformed hepatocyte cell line (Figure 6A). At the protein level, FFA treatment in Huh7 cells induced a dose-dependent decrease in P-YAP protein levels while total YAP levels remained relatively constant (Figure 6B). This was also associated with a FFA dose-dependent increase in CYR61 and p38 MAPK protein expression (Figure 6B). Furthermore, YAP nuclear immunostaining was enhanced in FFA-treated Huh7 cells compared with untreated cells (Figure 6C). In PHHs, we similarly observed significant upregulation of the direct YAP target genes ANKRD1, CYR61, CTGF, and JUB (Figure 6D). At the protein level, we found that CYR61 expression increased following FFA treatment in PHHs, while the ratio of P-YAP to total YAP decreased, reflecting increased YAP nuclear localization and transcriptional activity (Figure 6E). Because we were able to successfully recapitulate hepatic YAP activation observed in NAFLD patients and in our dietary murine NAFLD model, we were encouraged that our monoculture model would permit deeper dissection of the pathways that regulate YAP.
Figure 6.

FFAs activate YAP in human hepatocytes. (A) mRNA expression of ANKRD1, CYR61, CTGF, JUB, TGFB1, and WTIP in Huh7 and HepG2 cells treated with linoleic acid-oleic acid (FFAs) or palmitic acid (PA). Mean ± SD are graphed (n = 3 biological replicates; ∗P < .05, ∗∗P < .01). (B) Immunoblot of phosphorylated YAP, YAP, CYR61, and p38 MAPK in FFA-treated Huh7 cells. (C) YAP (yellow) and DAPI (blue) staining in Huh7 cells treated with FFAs. (D) mRNA expression of ANKRD1, CYR61, CTGF, and JUB in PHHs treated with FFAs. Mean ± SD are graphed (n = 3 biological replicates; ∗P < .05, ∗∗P < .01). (E) Immunoblot and densitometric analysis of phosphorylated YAP, YAP, and CYR61 in Huh7 cells treated with FFAs. Mean ± SD are graphed (n = 3 biological replicates; ∗∗P < .01).
Hepatocyte YAP Depletion Hinders Fibrosis Progression in NAFLD
Recent evidence has demonstrated that hepatocyte YAP silencing precludes fibrosis development in chemically induced murine liver injury.8 However, this function has not yet been evaluated in the CDAHFD mouse model. As a result, Yapflox/flox mice were crossed with mice harboring Albumin-Cre (Alb-Cre) recombinase to obtain Alb-Cre;Yapflox/flox mice, as previously described.19 These mice are subsequently referred to as YAP-KO. For all experiments and analyses, Alb-Cre;YapWT/WT mice were used as control animals and are referred to as YAP-wild-type (WT). Genotyping results for Alb-Cre;Yapflox/flox mice are displayed in (Figure 7A and B). We additionally confirmed hepatocyte-specific YAP deletion by costaining for albumin/YAP in livers of Alb-Cre;Yapflox/flox mice (Figure 7C). YAP was not expressed in hepatocytes (albumin+) cells. All mice were fed the CDAHFD for 12 weeks. YAP silencing was confirmed by immunoblot analyses of P-YAP/YAP and Cyr61 expression from whole liver lysates (Figure 8B). At the mRNA and protein levels, we found that ECM gene expression was markedly lower in YAP-KO mice compared with YAP-WT mice (Figures 8A and B and 9). Furthermore, proinflammatory (Il6) and profibrogenic (Pdgfb and Vegfb) genes were downregulated in YAP-KO mice (Figure 8B). We also observed a nonsignificant reduction in lipid droplet (LD) staining by hematoxylin and eosin and Oil Red O (Figure 8A).
Figure 7.

Genotyping for Alb-Cre;Yapflox/floxmice. Genomic DNA samples from tail biopsies were amplified via polymerase chain reaction (PCR). (A) The Cre transgene PCR product can be observed in Alb-Cre;Yapflox/flox mice (clones 1, 2, and 4), while there is no PCR product visible in clone 3 (Yapflox/flox). (B) The ~270-bp PCR product representing exon 4 of Yap flanked by 2 loxP sites can be seen in all 4 clones. Expected bands from Cre recombinase and loxP sequence (floxed allele) are shown in ethidium bromide-stained agarose gel. The genotype of #1, #2, and #4 Alb-Cre; Yapflox/flox (Yap KO) mice; #3 Yapflox/flox (WT) mice. (C) Representative albumin (green), YAP (purple), and DAPI (blue) staining in liver tissue from Albumin-Cre;Yapflox/flox (YAP-KO) mice (original magnification ×20).
Figure 8.

Hepatocyte YAP depletion hinders fibrosis progression in NAFLD. (A) Representative microscopic photographs of liver sections stained with picrosirius red, Oil red O, and hematoxylin and eosin in liver tissue from Albumin-Cre;YapWT/WT (YAP-WT) or Albumin-Cre;Yapflox/flox (YAP-KO) mice fed standard chow or CDAHFD for 14 weeks (original magnification ×10). Percent of picrosirius red–stained liver tissue is graphed as mean ± SD (n = 3 mice per group; ∗∗P < .01, ∗∗∗P < .001). (B) Immunoblot and reverse-transcription polymerase chain reaction analyses of extracellular matrix-related (Acta2, Col1a1), fibrogenic (Ctgf, Pdgf, Vegfb), proinflammatory (Cyr61, Il6), and YAP-dependent (Ctgf, Cyr61, Il6) gene expression in liver samples from mice in (A). Mean ± SD are graphed (n = 3 mice per group, ∗P < .05, ∗∗P < .01). (C) Immunoblot and reverse-transcription polymerase chain reaction analyses of YAP, ANKRD1, CYR61, CTGF, JUB, and TGFB1 expression in Huh7 cells stably transduced with NT or YAP shRNA (shYAP-2 or 5) lentiviral particles. Mean ± SD are graphed (n = 3 biological replicates; ∗P < .05, ∗∗P < .01). (D) mRNA expression of COL1A1, CO3A1, ACTA2, and TIMP1 in spheroids consisting of Huh7 cells stably transduced with shYAP-5 (shY5) and pHSCs. Mean ± SD are graphed (n = 3 biological replicates; ∗P < .05, ∗∗P < .01).
Figure 9.

Hepatocyte YAP depletion lowers TIMP1 expression in the murine NAFLD model. Representative immunoblot for TIMP1 and Actin in total liver lysates from Alb-Cre;YapWT/WT (YAP-WT) or Alb-Cre;Yapflox/flox (YAP-KO) mice fed standard chow or CDAHFD for 14 weeks.
We next developed a reproducible spheroid co-culture model comprised of pHSCs and either Huh7 cells or PHHs to determine whether hepatocyte YAP knockdown could directly prevent HSC activation (Figure 10A). As in our monoculture model, FFAs activated YAP in both Huh7/pHSC and PHH/pHSC spheroids, evidenced by increased expression of YAP target genes (ANKRD1, CYR61, CTGF, and JUB) relative to untreated spheroids (Figure 10C). We then performed BODIPY staining and α-smooth muscle actin (SMA) immunofluorescence on Huh7/pHSC spheroids treated with FFAs for 72 hours. When untreated, these cells were α-SMA negative, but when stimulated with exogenous FFAs, we observed enhanced lipid (BODIPY) staining in the hepatocyte core and increased α-SMA immunostaining in the spheroid exterior, corresponding with activated pHSCs (Figure 10B). mRNA expression of the following fibrosis-related genes was also upregulated: ACTA2 (5-fold change; P < .05), TIMP1 (3.5-fold change; P < .005), COL3A1 (8.5-fold change; P < .05), and COL1A1 (12-fold change; P < .05) (Figure 10D).
Figure 10.

Generation of spheroid co-culture NAFLD model. (A) Schematic diagram of spheroid generation. (B) α-SMA (yellow) and lipid (green) staining in Huh7/pHSC spheroids after FFA treatment (image capture at ×10 magnification). (C) mRNA expression of YAP targets (ANKRD1, CYR61, CTGF, JUB) and fibrosis-related genes (COL1A1, COL3A1, ACTA2, TIMP1) in Huh7/pHSC spheroids treated with FFAs. ∗P < .05; ∗∗P < .005; mean ± SD; n = 3.
To knock down YAP, we created lentiviral vectors expressing short hairpin RNAs (shRNAs) targeting YAP mRNA (shY2/shY5). When compared with Huh7 cells transduced with a control vector expressing a nontargeting control shRNA (NT shControl), Huh7-shY2/Y5 cells exhibited profound YAP silencing and the expression of YAP-dependent genes was significantly reduced (Figure 8C). YAP silencing did not affect FFA entry into cells, LD accumulation or LD shape (data not shown). We then adapted our 3D NAFLD co-culture model to incorporate the Huh7-shY5 cell line and found that FFA-induced ACTA2, TIMP1, COL3A1, and COL1A1 upregulation could be suppressed by YAP silencing (Figure 8D). These in vitro and in vivo mechanistic studies provide clear evidence that hepatocyte YAP is necessary for HSC activation and fibrosis progression in NAFLD.
p38 MAPK Regulates FFA-Induced YAP Activation in Huh7 cells
Prior studies have demonstrated that p38δ MAPK expression is increased in the livers of NAFLD/NASH patients.20 Indeed, in our studies p38 MAPK was elevated in NAFLD patients with fibrosis and MAPK13 (p38δ MAPK) was significantly upregulated (3.8-fold change; P < .005) in FFA-treated Huh7 cells (Figure 11A). To investigate whether FFA-induced p38 MAPK expression regulates YAP activity, we independently depleted the p38 MAPK isoforms (MAPK11/12/13) expressed in the liver by small interfering RNA (siRNA) and then assessed expression of YAP-dependent genes in Huh7 cells. Gene knockdown was confirmed by immunoblot (Figure 12). Silencing of MAPK13 had the greatest effect in suppressing ANKRD1 mRNA expression (2-fold reduction; P < .005) following FFA treatment (Figure 11B). Furthermore, MAPK13 siRNA transfection increased S127 YAP phosphorylation and reduced CYR61 protein expression in FFA-treated Huh7 cells (Figure 11C). RNA interference-mediated depletion of p38δ MAPK (MAPK11) also resulted in moderate suppression of YAP activity, as evidenced by increased P-YAP levels and reduced ANKRD1 mRNA levels relative to NT siRNA-transfected Huh7 cells (Figure 11B and C). Chemical inhibition of MAPK11/14 using SB203580 also reduced FFA-induced expression of ANKRD1, CYR61, CTGF, TGFB1, and JUB compared with mock-treated cells (Figure 11D). To confirm the specificity of FFAs in activating p38 MAPK and not other classes of MAPKs, we also treated Huh7 cells with siRNAs targeting extracellular signal-regulated kinases 1/2 (ERK1/2) and c-Jun N-terminal kinases (JNK1/2).21 Inhibition of ERK1/2 had no effect on the upregulation of YAP target genes in response to treatment with exogenous FFAs (Figure 11E). However, silencing of JNK1 (MAPK8) and JNK2 (MAPK9) modestly suppressed CYR61 expression in FFA-treated Huh7 cells (Figure 11C). These data suggest that FFAs predominantly activate p38 MAPK, which in turn regulates hepatocyte YAP activation and fibrosis progression.
Figure 11.

p38 MAPK regulates FFA-induced YAP activation in Huh7 cells. (A) MAPK13 mRNA expression in Huh7 cells treated with FFAs. Mean ± SD are graphed (n = 3 biological replicates; ∗∗P < .01). (B) ANKRD1 mRNA expression in Huh7 cells transfected with MAPK8, MAPK9, MAPK11, MAPK12, MAPK13, or NT siRNAs. Mean ± SD are graphed (n = 3 biological replicates; ∗P < .05, ∗∗P < .01). (C) Immunoblot of phosphorylated YAP, total YAP, and CYR61 in Huh7 cells transfected with siRNAs as in panel B. (D) mRNA expression of ANKRD1, CYR61, CTGF, JUB, and TGFB1 in Huh7 cells preincubated with the p38γ/δ MAPK inhibitor SB203580 and then treated with FFAs. Mean ± SD are graphed (n = 3 biological replicates; ∗P < .05, ∗∗P < .01). (E) ANKRD1, CYR61, and CTGF mRNA expression in Huh7 cells transfected with siRNA targeting ERK1/2 and subsequently treated with FFAs. Mean ± SD are graphed.
Figure 12.

Confirmation of JNK/p38 MAPK siRNA knockdown in Huh7 cells. Huh7 cells were transfected with an NT control siRNA (NT siControl) or an siRNA-targeting MAPK8/9/11/12/13 (siMAPK8, 9, 11, 12, and 13). The panel shows the Western blot results from whole cell lysates 48 hours posttransfection.
Discussion
NAFLD encompasses a spectrum of disorders that range from SS to NASH and eventually end-stage liver disease and HCC. NASH has traditionally been considered the more severe progressive form of NAFLD that can eventually culminate in cirrhosis or cirrhosis-related complications such as HCC, while SS has been considered a more benign condition. However, these conclusions were based on small studies with limited duration of follow-up. Emerging data now suggest that a similar proportion of patients with SS compared with NASH exhibit disease progression, defined as an increase in fibrosis stage or development of cirrhosis or end-stage liver disease. Furthermore, baseline histological features do not necessarily predict which patients will subsequently progress. Together, these data suggest that SS is not necessarily a bland condition and leads to activation of pathways that lead to progressive fibrosis. Given the significant and rising global burden of NAFLD, and the fact that there are currently no Food and Drug Administration–approved treatments for NAFLD, a key focus of research in the field is the dissection of signal transduction pathways underlying the pathogenesis of NAFLD to identify drug targets that reverse or impede disease progression.
Recent mechanistic studies demonstrated that hepatocyte YAP deletion potently inhibits fibrosis development in carbon tetrachloride-induced liver injury in mice.8 However, whether this relationship exists in dietary-induced NAFLD had not been determined. In the present study, we observed a reduction in fibrosis-related parameters in hepatocyte-specific YAP conditional KO mice fed a NAFLD-inducing diet. It has previously been proposed that YAP exerts its profibrogenic effects through transcriptional activation of CYR61, which functions to increase macrophage trafficking to the injured liver.8,22 Indeed, we show that diet-induced NAFLD increased hepatic CYR61 expression in mice in a YAP-dependent manner and was associated with fibrosis development.
The hepatic accumulation of FFAs is central to the pathogenesis of NAFLD; however, the effects of FFAs on the molecular events underlying fibrosis progression have not been well studied. Our work suggests p38 MAPK is activated by FFAs, and in turn promotes a YAP-mediated profibrogenic program. As stress-responsive kinases, p38 MAPKs have diverse functions in hepatic metabolism and pathophysiology. Notably, p38γ/δ MAPKs have been implicated in the development of hepatic steatosis, inflammation, and fibrosis in the methionine- and choline-deficient diet murine NAFLD model.20 Huang et al23 also previously established cross-talk between Hippo and p38 MAPK signaling in mammalian cell lines, demonstrating that p38 MAPK silencing reduced CTGF and CYR61 expression.
There are a number of limitations to our work. First, we did not investigate how hepatocyte YAP silencing in CDAHFD-fed mice or hepatocyte or HSC spheroids affected its paralogue TAZ, which regulates fibrosis development through activation of the Hedgehog pathway.6 Furthermore, it has been reported that TAZ is only upregulated in NASH and not SS,6 but in the present study, we observed increased YAP activity in patients with steatosis alone. Thus, describing the precise mechanism through which YAP and TAZ become activated in NAFLD, in addition to their possible functional interactions, will remain important for future studies. Furthermore, immune cells such as macrophages (both liver-resident Kupffer cells and monocyte-derived macrophages) secrete cytokines and growth factors that contribute to HSC activation. Therefore, our co-culture model could benefit in future studies from the incorporation of macrophages or Kupffer cells to render it more physiologically accurate. Alternatively, the use of more sophisticated and anatomically authentic models that permit the investigation of the cell-to-cell interactions involved in the pathogenesis of NAFLD, like organoids, may be required.
While we show that hepatocyte-specific YAP expression is necessary for fibrosis progression in NAFLD, previous studies of NAFLD in humans reported enhanced YAP immunostaining only in reactive ductular cells and not in hepatocytes.5 Similarly, in mice fed a high fructose-palmitate-cholesterol diet, researchers observed elevated hepatic YAP levels but not in hepatocytes, casting doubt over the significance of YAP activation in hepatocytes.6 However, lineage tracing following mouse hepatocyte-specific merlin (upstream negative regulator of YAP) deletion showed that hepatocytes dedifferentiated into YAP-positive ductular-like cells,24 explaining why YAP immunostaining may not colocalize with hepatocytes. Therefore, perhaps p38 MAPK–mediated YAP activation is an early event in NAFLD that promotes the de-differentiation of hepatocytes into reactive ductular cells. However, given the short duration of our in vitro experiments and primary use of immortalized carcinoma cell lines, we could not confirm this hypothesis, but further studies should evaluate longer treatment durations to assess for hepatocyte dedifferentiation and interactions between YAP and other developmental pathways. Moreover, single-cell RNAseq of human NAFLD liver may permit more rigorous characterization of YAP-positive cell populations, as recent evidence suggests YAP expression in HSCs and Kupffer cells are also critical for fibrosis and NASH development.25,26
In summary, we confirm that the Hippo signaling transcription coactivator YAP is upregulated in human and murine NAFLD liver and is associated with fibrosis progression. Furthermore, hepatocyte YAP silencing attenuated fibrosis severity in a dietary-induced murine NAFLD model. We also show that FFAs activate hepatocyte YAP and promote HSC activation and ECM deposition through a p38 MAPK–dependent pathway. While further mechanistic work is warranted, our data reveal potentially novel therapeutic targets for intervention in the natural progression of NAFLD. In addition, we show here that our 3-dimensional co-culture model can be used to study the pathogenesis of NAFLD and may have applications beyond NAFLD in other models of liver diseases.
Materials And Methods
Human Samples and Histopathological Analysis
We utilized GEO data (accession: GSE162694) (G. Agyapongm, MD et al, 2020, unpublished data), which includes RNAseq data for 143 NAFLD patients with various stages of fibrosis. Clustering of gene expression patterns across the 4 examined groups were obtained using the R package DEGreport.
Tissue samples were obtained from wedge liver biopsies of 39 patients with fatty liver disease seen at the Massachusetts General Hospital (MGH) Fatty Liver Clinic between 2012 and 2018. Serum samples were collected from these patients within a few days of the date of biopsy. Fibrosis was scored as previously described.27 The study was approved by the local ethics committee at MGH as part of an approved protocol (MGH IRB#2010P000220).
Animal Studies
Alb-Cre and Yapflox/flox mice were purchased from the Jackson Laboratory (Bar Harbor, ME). WT C57BL/6 male mice (5 weeks old) were purchased from Charles River Laboratories (Wilmington, MA) and adapted to a specific environment for 1 week. To achieve liver-specific gene deletion, Yapflox/flox mice were bred to Alb-Cre transgenic mice as previously described.19 Mice were then fed the following diets for 4, 10, and 12 weeks: standard chow (Prolab Isopro 3000; Scotts Distributing #8670) and CDAHFD (Research Diets A06071302; L-amino acid diet with 60 kcal% fat, 0.1% methionine, and without added choline). At the conclusion of feeding, mice were weighed and euthanized. Livers were excised and weighed, and portions were snap-frozen in liquid nitrogen for protein and biochemical assays and preserved in 10% neutral-buffered formalin for histopathological analysis and RNA extraction. All mice were housed in a pathogen-free, temperature-controlled animal facility with a 12-hour light-dark cycle. All animal protocols were approved by the Institutional Animal Care and Use Committee of MGH.
Cell Culture
We used Huh7 and HepG2 human hepatoma cells, Human Embryonic Kidney 293T (HEK293T) cells, PHHs, and pHSCs. pHSCs were isolated from the nonparenchymal cell fraction of 3 different HIV/hepatitis B virus/hepatitis C virus–free donor livers (Triangle Research Labs, Durham, NC) and selected as previously described.28 Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 1% penicillin/streptomycin, 0.4% L-glutamine, and 10% heat-inactivated fetal bovine serum and maintained at 37°C in humidified air containing 5% CO2. Cells were harvested for reverse-transcription polymerase chain reaction and immunoblot studies at the specified time points, and data were acquired from at least 3 independent experiments.
Generation of Hepatocyte/HSC Spheroids
The commonly used hanging drop method was employed for spheroid generation.29 In brief, 10-μL media drops (Dulbecco's modified Eagle's medium 10% fetal bovine serum with Pen/Strep and L-glutamine) each consisting of 20,000 Huh7 cells or 50,000 PHHs were deposited evenly over the inside surface of a 100-mm cell culture dish lid. The lid was then inverted onto the bottom chamber that had been filled with phosphate-buffered saline. The cells were incubated at 37°C in humidified air containing 5% CO2. After 2 days, 20,000 pHSCs suspended in 10-μL media were added over each drop to create the outer layer of the spheroid. After 24-hour incubation, multicell-type spheroids (Huh7 or PHHs with pHSCs) had formed and were subsequently placed on a shaker for 2 more days to take shape.
Production of Stable Lentiviral-Transduced Huh7 Cell Lines
The ARE/NRF2 GFP reporter lentiviral plasmid was generated as previously described.30 YAP shRNA plasmids were purified from YAP shRNA bacterial glycerol stocks TRCN0000300282 (shYAP-2) and TRCN0000107265 (shYAP-5) purchased from MilliporeSigma (Burlington, MA). VSV-G pseudotyped lentiviral vectors were produced by cotransfection of HEK293T cells with the aforementioned plasmids, the packaging plasmid psPAX2 (Addgene #12260; Addgene, Watertown, MA), and the envelope plasmid pMD2.G (Addgene #12259) using FuGENE HD Transfection Reagent (Promega, Madison, WI). Lentiviral supernatants were harvested at 48 and 72 hours posttransfection and stored at −80°C. Huh7 cells were transduced with lentiviral particles encoding the 2 different shRNAs targeting YAP or the ARE GFP reporter for 4 hours in the presence of 8 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO) and selected with puromycin to generate stable cell lines.
siRNA Transfection
Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA) was used for siRNA transfection. Dharmacon ON-TARGETplus SMARTpool human siRNAs (Horizon Discovery, Lafayette, CO) were used for MAPK8, MAPK9, MAPK11, MAPK12, and MAPK13 knockdown and an NT negative control (siControl). Gene knockdown was confirmed by immunoblot.
Statistical Analysis
Statistical analyses were performed using Microsoft Excel 2019 and GraphPad Prism 8 (GraphPad Software, San Diego, CA). Densitometric analyses of immunoblot images were performed using Image Studio Lite (LI-COR Biosciences, Lincoln, NE). Data are presented as a mean ± SD from at least 3 independent experiments unless otherwise stated in the figure legends. P values were calculated using Student’s t test for normally distributed data; P < .05 was considered statistically significant.
Human data were compared using Fisher’s exact test.
Acknowledgments
CRediT Authorship Contributions
Shadi Salloum (Conceptualization: Lead; Data curation: Lead; Formal analysis: Lead; Funding acquisition: Lead; Investigation: Lead; Methodology: Lead; Project administration: Lead; Software: Lead; Supervision: Lead; Validation: Lead; Visualization: Lead; Writing – original draft: Lead; Writing – review & editing: Lead)
Andre Jeyarajan (Conceptualization: Supporting; Data curation: Supporting; Formal analysis: Supporting; Funding acquisition: Supporting; Investigation: Supporting; Methodology: Supporting; Project administration: Supporting; Resources: Supporting; Supervision: Supporting; Validation: Supporting; Visualization: Supporting; Writing –original draft: Supporting; Writing – review & editing: Supporting)
Annie J Kruger (Investigation: Supporting; Writing – original draft: Supporting; Writing –review & editing: Supporting)
Jacinta A Holmes (Data curation: Supporting; Formal analysis: Supporting; Writing –original draft: Supporting; Writing – review & editing: Supporting)
Tuo Shao (Investigation: Supporting; Methodology: Supporting)
Mozhdeh Sojoodi (Investigation: Supporting; Methodology: Supporting)
Myung-Ho Kim (Investigation: Supporting; Methodology: Supporting)
Zhu Zhuo (Data curation: Supporting; Formal analysis: Supporting)
Stuti G Shroff, MD (Data curation: Supporting; Resources: Supporting)
Andrew Kassa (Investigation: Supporting)
Kathleen E E Corey (Resources: Supporting)
Sanjoy K Khan (Investigation: Supporting; Methodology: Supporting)
Wenyu Lin (Investigation: Supporting)
Nadia Alatrakchi (Investigation: Supporting)
Esperance A. K. Schaefer (Investigation: Supporting)
Raymond T. Chung, MD (Data curation: Supporting; Investigation: Supporting; Resources: Lead; Supervision: Lead; Writing – original draft: Supporting; Writing –review & editing: Supporting)
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding This work was supported by the National Institutes of Health AI136715 (to Raymond T. Chung). Raymond T. Chung is supported by the MGH Research Scholars Program.
References
- 1.Williams C.D., Stengel J., Asike M.I., Torres D.M., Shaw J., Contreras M., Landt C.L., Harrison S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 2.Diehl A.M., Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. 2017;377:2063–2072. doi: 10.1056/NEJMra1503519. [DOI] [PubMed] [Google Scholar]
- 3.Kim W.R., Lake J.R., Smith J.M., Schladt D.P., Skeans M.A., Noreen S.M., Robinson A.M., Miller E., Snyder J.J., Israni A.K., Kasiske B.L. OPTN/SRTR 2017 Annual Data Report: liver. Am J Transplant. 2019;19:184–283. doi: 10.1111/ajt.15276. [DOI] [PubMed] [Google Scholar]
- 4.Estes C., Razavi H., Loomba R., Younossi Z., Sanyal A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123–133. doi: 10.1002/hep.29466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machado M.V., Michelotti G.A., Pereira T.A., Xie G., Premont R., Cortez-Pinto H., Diehl A.M. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol. 2015;63:962–970. doi: 10.1016/j.jhep.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X., Zheng Z., Caviglia J.M., Corey K.E., Herfel T.M., Cai B., Masia R., Chung R.T., Lefkowitch J.H., Schwabe R.F., Tabas I. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24:848–862. doi: 10.1016/j.cmet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao B., Zhao B., Wei X., Wei X., Li W., Li W., Udan R.S., Udan R.S., Yang Q., Yang Q., Kim J., Kim J., Xie J., Xie J., Ikenoue T., Ikenoue T., Yu J., Yu J., Li L., Li L., Zheng P., Zheng P., Ye K., Ye K., Chinnaiyan A., Chinnaiyan A., Halder G., Halder G., Lai Z., Lai Z., Guan K., Guan K. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mooring M., Fowl B.H., Lum S.Z.C., Liu Y., Yao K., Softic S., Kirchner R., Bernstein A., Singhi A.D., Jay D.G., Kahn C.R., Camargo F.D., Yimlamai D. Hepatocyte stress increases expression of yes-associated protein and transcriptional coactivator with PDZ-binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology. 2019;0:1–18. doi: 10.1002/hep.30928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman S.L., Neuschwander-Tetri B.A., Rinella M., Sanyal A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–922. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McPherson S., Hardy T., Henderson E., Burt A.D., Day C.P., Anstee Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J Hepatol. 2015;62:1148–1155. doi: 10.1016/j.jhep.2014.11.034. [DOI] [PubMed] [Google Scholar]
- 11.Nasr P., Ignatova S., Kechagias S., Ekstedt M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol Commun. 2018;2:199–210. doi: 10.1002/hep4.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins Q.F., Xiong Y., Lupo E.G., Liu H.-Y., Cao W. p38 mitogen-activated protein kinase mediates free fatty acid-induced gluconeogenesis in hepatocytes. J Biol Chem. 2006;281:24336–24344. doi: 10.1074/jbc.M602177200. [DOI] [PubMed] [Google Scholar]
- 13.Liu H.-Y., Collins Q.F., Xiong Y., Moukdar F., Lupo E.G., Liu Z., Cao W. Prolonged treatment of primary hepatocytes with oleate induces insulin resistance through p38 mitogen-activated protein kinase. J Biol Chem. 2007;282:14205–14212. doi: 10.1074/jbc.M609701200. [DOI] [PubMed] [Google Scholar]
- 14.Lei Q.-Y., Zhang H., Zhao B., Zha Z.-Y., Bai F., Pei X.-H., Zhao S., Xiong Y., Guan K.-L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh H., Irvine K.D. In vivo regulation of Yorkie phosphorylation and localization. Development. 2008;135:1081–1088. doi: 10.1242/dev.015255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farrell G., Schattenberg J.M., Leclercq I., Yeh M.M., Goldin R., Teoh N., Schuppan D. Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology. 2019;69:2241–2257. doi: 10.1002/hep.30333. [DOI] [PubMed] [Google Scholar]
- 17.Neuschwander-Tetri B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 18.Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 19.Zhang N., Bai H., David K.K., Dong J., Zheng Y., Cai J., Giovannini M., Liu P., Anders R.A., Pan D. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.González-Terán B., Matesanz N., Nikolic I., Verdugo M.A., Sreeramkumar V., Hernández-Cosido L., Mora A., Crainiciuc G., Sáiz M.L., Bernardo E., Leiva-Vega L., Rodríguez E., Bondía V., Torres J.L., Perez-Sieira S., Ortega L., Cuenda A., Sanchez-Madrid F., Nogueiras R., Hidalgo A., Marcos M., Sabio G. p38γ and p38δ reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 2016;35:536–552. doi: 10.15252/embj.201591857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson G.L., Lapadat R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 22.Bian Z., Peng Y., You Z., Wang Q., Miao Q., Liu Y., Han X., Qiu D., Li Z., Ma X. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res. 2013;54:44–54. doi: 10.1194/jlr.M026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang D., Li X., Sun L., Huang P., Ying H., Wang H., Wu J., Song H. Regulation of Hippo signalling by p38 signalling. J Mol Cell Biol. 2016;8:328–337. doi: 10.1093/jmcb/mjw036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yimlamai D., Christodoulou C., Galli G.G., Yanger K., Pepe-Mooney B., Gurung B., Shrestha K., Cahan P., Stanger B.Z., Camargo F.D. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mannaerts I., Leite S.B., Verhulst S., Claerhout S., Eysackers N., Thoen L.F.R., Hoorens A., Reynaert H., Halder G., Van Grunsven L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679–688. doi: 10.1016/j.jhep.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Song K., Kwon H., Han C., Chen W., Zhang J., Ma W., Dash S., Gandhi C.R., Wu T. Yes-associated protein in Kupffer cells enhances the production of proinflammatory cytokines and promotes the development of nonalcoholic steatohepatitis. Hepatology. 2020;0:1–16. doi: 10.1002/hep.30990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Almpanis Z., Demonakou M., Tiniakos D. Evaluation of liver fibrosis: “something old, something new….”. Ann Gastroenterol. 2016;29:445–453. doi: 10.20524/aog.2016.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitani H., Takenouchi T., Sato M., Yoshioka M., Yamanaka N. A Simple and efficient method to isolate macrophages from mixed primary cultures of adult liver cells. J Vis Exp. 2011:1–5. doi: 10.3791/2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foty R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J Vis Exp. 2011;20:4–7. doi: 10.3791/2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salloum S., Holmes J.A., Jindal R., Bale S.S., Brisac C., Alatrakchi N., Lidofsky A., Kruger A.J., Fusco D.N., Luther J., Schaefer E.A., Lin W., Yarmush M.L., Chung R.T. Exposure to human immunodeficiency virus/hepatitis C virus in hepatic and stellate cell lines reveals cooperative profibrotic transcriptional activation between viruses and cell types. Hepatology. 2016;64:1951–1968. doi: 10.1002/hep.28766. [DOI] [PMC free article] [PubMed] [Google Scholar]