

The worldwide adaptation of a Western lifestyle is associated with the increased consumption of high glycemia diets and an increased prevalence of obesity, metabolic syndrome, and diabetes. These diets increase the risk for a plethora of age-related diseases including cerebrovascular, cardiovascular, and eye-related disorders, which all share a common pathogenic factor: the accumulation of advanced glycation end-products (AGEs) (Semba et al., 2010; Aragno and Mastrocola, 2017). AGEs are a diverse group of pathogenic compounds formed via a non-enzymatic process called glycation in which dietary sugars, or reactive dicarbonyls formed during carbohydrate metabolism, are covalently attached to different biomolecules, inactivating them (Rabbani and Thornalley, 2015). A growing literature indicates that AGEs impact brain function and contribute to initiate and accelerate neurodegeneration and also interfere with the process of neuroregeneration (Li et al., 2012; Vicente Miranda et al., 2017; Fleitas et al., 2018; Bao et al., 2020). In order to avoid AGEs-derived toxicity, our cells have different anti-AGEs defense mechanisms including the clearance of these detrimental compounds through different proteolytic pathways. To date, the lysosomal system (autophagy) and the ubiquitin-proteasome system (UPS) have been identified as proteolytic routes able to degrade AGEs. Unfortunately, the proteolytic capacity declines with age, making tissues more vulnerable to AGEs-derived damage (Uchiki et al., 2012; Rowan et al., 2017; Aragonès et al., 2020). AGEs-modification also compromises the proteolytic machinery, leading to double jeopardy due to the higher glycemia diets or diabetes. Boosting proteolytic pathways might represent a therapeutic strategy to counteract the deposition of toxic, glycated, proteinaceous aggregates in the brain and other tissues with limited regeneration capacity, those most vulnerable to glycation-derived damage.
Glycation-derived damage in molecular aging and brain disease: Aging is a multifactorial biological phenomenon controlled by both genetic and environmental factors. The aging process is accompanied by gradual changes in multiple molecular and cellular processes whose malfunction leads to age-related diseases. Recent research on the biology of aging stresses diet as one the most profound environmental determinants for health, and a better understanding of the interactions between diet composition, physiology, biochemistry, and genetics is imperative for developing anti-aging interventions to prevent age-related disorders.
Glycation is a non-enzymatic process that plays a pathogenic role in the progression of molecular and cellular aging. AGEs are toxic compounds formed during glycation, a chemical process in which reducing sugars react with different biomolecules, impacting their function, location, and/or solubility (Uchiki et al., 2012; Aragonès et al., 2020). Due to the age-related decline of different anti-AGEs defense mechanisms, glycated biomolecules accumulate during normal aging in our bodies in a tissue-dependent manner. Highly differentiated tissues with a low regenerative capacity, such as brain or ocular tissues, cannot dilute the AGEs-derived cellular damage through cellular division and are more susceptible to glycative-stress induced toxicity (Uchiki et al., 2012; Rowan et al., 2017; Bejarano and Taylor, 2019; Aragonès et al., 2020). Diabetes or consumption of high glycemia diets leads to increased blood glucose levels and triggers the aberrant production of AGEs and associated stress. Thus, hyperglycemia accelerates the age-associated accumulation of glycated proteins and exacerbates the detrimental consequences of AGE deposition on organ function.
The deposition of extracellular and intracellular AGEs contributes to molecular aging and is a molecular mechanism underpinning age-related cellular and tissue deterioration. Extracellular AGEs trigger signaling pathways involved in inflammation, production of reactive oxygen species, changes in vasopermeability, and angiogenesis. Intracellular AGEs modify protein structure and induce conformational changes, leading to a loss of function. These changes include cross-linking of proteins, thereby decreasing their solubility, and accumulation of insoluble aggregates. Different age-related disorders are causally associated with AGEs production including ocular disorders such as cataract, diabetic retinopathy, and age-related macular degeneration (AMD), and neurodegenerative diseases (Semba et al., 2010; Rabbani and Thornalley, 2015; Bejarano and Taylor, 2019).
Regarding brain function, glycation seems to play a role in the pathogenesis of central nervous system debilities such as peripheral diabetic polyneuropathies, Alzheimer's disease, Parkinson's disease, Huntington's disease, Creutzfeldt-Jakob disease, and amyotrophic lateral sclerosis (Münch et al., 2012). AGEs are found in intracytoplasmic inclusions in Lewy bodies in subcortical neurons of Parkinson's patients, and it has been suggested that AGEs trigger Lewy body formation. Also, the AGEs content in plaques extracted from Alzheimer's disease patients is higher compared to age-matched control individuals (Münch et al., 2012).
Accumulation of AGEs is thought to contribute to diabetes-associated cognitive decline by inducing neuronal differentiation defects in neural stem cells. Glycation is proposed to participate in the aggregation of different neuropathological proteins and also impacts on neurite regeneration. For example, recent literature highlights the ability of AGEs to cross-link neuropathological proteins such as α-synuclein, amyloid β, and tau, promoting aggregation and limiting the clearance of toxic oligomers. Multiple experiments in different cellular and animal models, including dopaminergic neurons, SHSY5Y cells, C. elegans and D. melanogaster, support the hypothesis that lowering AGEs accumulation is neuroprotective.
The pathways by which AGEs-derived stress contribute to neurodegeneration are complex and it remains unclear how glycation-derived damage promotes pathology. Extracellular AGEs bind AGEs-receptors in the membrane of microglial cells, triggering inflammation and apoptosis of different brain cells including dopaminergic neurons. One of the major concerns regarding glycative stress is that glycation leads to decreased proteostasis, which can drive neuronal cell death. Also, glycation can take place directly on neurotransmitters such as dopamine, leading to toxic metabolites that are enriched both in diabetic animal models and in Parkinson's disease patients.
Feeding animals high glycemic index diets, a dietary manipulation that leads to glycative stress and development of AMD, was shown to cause enhanced AGEs accumulation in the brain, and ocular tissue toxicity (Uchiki et al., 2012; Rowan et al., 2017; Aragonès et al., 2020). Finding AGEs in both tissues is not unexpected since the retina is considered an extension of the brain. Demonstrating the impact of dietary interventions, it has been reported that changing from high glycemic index to lower glycemic index diets slows the deposition of AGEs in ocular tissues and protects against AMD (Rowan et al., 2017). The impact of this high glycemic index diet on glycation-derived damage in brain tissues is still unknown. Importantly, every human clinical epidemiologic study published to date documents a relationship between consuming higher glycemia diets and enhanced risk for AMD and progress of AMD to clinically significant stages. These human and mouse study results are consistent with the formation of AGEs being enhanced when sugar levels are high. But, AGEs also accumulate when the proteolytic pathways that target and remove them are insufficient. Clearly, limiting the accumulation of AGEs might be a therapeutic avenue to preserve brain function during aging.
Different pharmaceutical agents including aminoguanidine and pyridoxamine limit AGEs formation and were tested in animal models to counteract glycative burden. Despite the largely successful results in preclinical studies with those drugs, most of the clinical trials were discontinued due to unanticipated complications and drug toxicity. Recently, different publications highlighted the possibility of enhancing proteolytic capacities as a means of lowering AGEs levels, but limited information about AGEs clearance is available. A better understanding of the degradation of AGEs is imperative to design strategies that could be exploited for clinical application in order to extend healthspan.
Proteolytic pathways as the last line of defense against glycation-derived damage: Exogenous AGEs, those directly ingested in the food and released into the blood circulation upon digestion, only represent 30% of the total AGEs content in our bodies due to inefficient absorption in the digestive system and efficient renal secretion. The major source of AGEs is the endogenous production of reactive dicarbonyls during glycolysis. Glycation is proportional to blood sugar concentration, particularly in tissues in which glucose transporters are constitutively open, such as most parts of the eye.
Among the mechanisms protecting against the formation of AGEs are the glyoxalase system and the DJ-1/Park7 deglycases. These detoxify glycation intermediates such as aldehydes formed from sugars or fats. Unfortunately, the efficiency of these anti-glycation activities declines with age, leading to accelerated accumulation of AGEs in older tissues (Uchiki et al., 2012; Rowan et al., 2017). To date, there is no effective way to enhance these systems to more efficiently clear glycation intermediates.
A major concern with glycative stress is the irreversibility of this non-enzymatic process. The only alternative to avoid the accumulation of AGEs, and associated tissue malfunction, is the efficient removal of AGEs. In summary, the accumulation of AGEs depends on both the rate of biogenesis and the rate of AGEs removal by the proteolytic systems. Thus, the proteolytic pathways represent the last line of defense against glycation-derived damage. Recent literature indicates that both the UPS and the autophagy-lysosome pathway (ALP), the two major intracellular proteolytic pathways, contribute to the efficient clearance of these toxic glycated proteins. Each pathway can degrade some AGEs, and their activities are complementary (Figure 1). The UPS operates primarily on soluble substrates, while autophagy can clear insoluble substrates, aggregates, and glycated organelles that cannot be degraded in the proteasome. Both pharmacological and genetic inhibition of these pathways boosts the accumulation of AGEs in vitro and in vivo, and enhanced autophagic activity reduces both AGEs deposition and the development of AGEs-derived damage such as retinal degeneration (Uchiki et al., 2012; Aragonès et al., 2020). In spite of the coordinated action of the UPS and ALP in maintaining homeostatic levels of AGEs, AGEs accumulate over time due to the age-related decline of these proteolytic capacities.
Figure 1.
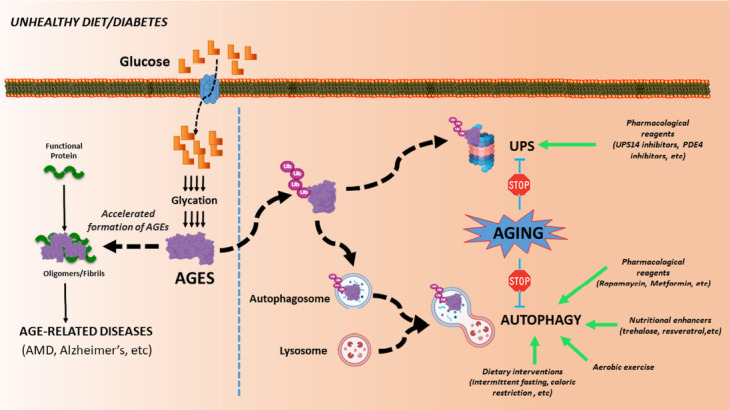
Schematic representation of how glycation promotes cellular toxicity.
(Left) High glycemia diets or diabetes lead to accelerated formation of AGEs, which induces protein aggregation and contributes to the onset of multiple age-related disorders. (Right) In order to avoid the glycation-derived proteotoxicity, AGEs are targeted to the proteasome or nascent autophagosome for degradation via the UPS or autophagy, respectively. The age-related decline of proteolytic pathways contributes to increased AGEs accumulation and AGEs-derived malfunction over time. The enhancements of these proteolytic routes (green arrows) could provide a therapeutic strategy against glycation-derived damage. AGES: Advanced glycation end products; AMD: age-related macular degeneration; UPS: ubiquitin-proteasome system.
Enhancing the UPS and ALP could be beneficial to preserving tissue fitness. The UPS recognizes and degrades damaged or obsolete soluble proteins. UPS substrates are tagged with ubiquitin and then degraded in the proteasome, a barrel-like structure containing multiple proteolytic enzymes. While it was shown that AGEs are ubiquitinated (Uchiki et al., 2012), the mechanistic details including AGEs recognition, which ubiquitin ligases are involved, and the process of AGEs targeting to the proteasome remain an enigma. Strategies that facilitate the proteasomal degradation of misfolded proteins such as PDE1 inhibition or activation of cGMP-dependent protein kinase have been reported to protect against proteinopathies. Of note, these strategies were shown to enhance the degradation of proteins related to neurodegenerative diseases. It is unknown if glycated neuropathological proteins could be efficiently destroyed via the UPS or if enhanced proteasomal activity could accelerate the degradation of glycated proteins.
The ALP is an intracellular catabolic process that mediates the degradation of different biomolecules in the lysosomal lumen. We can subdivide the autophagic process into 3 types depending on how the autophagic cargoes are delivered into the lysosome: macroautophagy (autophagic cargo is sequestered into double-membrane structures called autophagosomes that fuse with the lysosomal compartment where cargo is degraded), microautophagy (autophagic cargo is engulfed directly by invagination of lysosomal membranes) and chaperone-mediated autophagy (soluble cytosolic proteins with lysosomal targeting motifs are translocated across the lysosomal membrane). To date, only macroautophagy has been linked to the targeting of AGEs for degradation. The mechanisms of this recognition and degradation remain to be elucidated (Figure 1). A significant effort to identify novel targets should be revealing since recent literature demonstrates that enhanced autophagy ameliorates the neuronal degeneration associated with aging in rodent models. Natural and pharmacological compounds that accelerate the formation of autophagosomes through changes in signaling cascades such as the AMPK/mTOR/ULK1 axis were reported to protect against neurodegeneration. However, the information about the role of macroautophagy in the clearance of AGEs is scanty.
Macroautophagy is a proteolytic pathway that transports a highly diverse set of cargoes into autophagosomes, degradative vesicles that engulf portions of the cytosol. The engulfment of cytosolic cargo into autophagosomes can take place randomly, but there is also a dedicated set of receptors that enrich the forming autophagosomes with selectively ubiquitinated cargoes. This seems to be the case for the autophagic degradation of AGEs. A recent report from our laboratory revealed a protective role for the autophagic receptor p62 (Aragonès et al., 2020). The presence of p62 diminishes AGEs accumulation, and blocking p62-selective autophagy leads to accumulation of AGEs and increased vulnerability to glycative stress. Thus, our study identified, for the first time, an autophagy-related protein that could be a therapeutic target against AGEs-associated disorders. It remains to be clarified if other autophagic receptors contribute to the clearance of AGEs or if tissue-dependent expression of those receptors determines the ability of the tissue to clear toxic AGEs. We found that different cell types including retinal, lens, and fibroblasts or different tissues such as brain, retina, heart or liver showed different capacities to deal with glycative stress damage (Uchiki et al., 2012; Rowan et al., 2017; Aragonès et al., 2020), suggesting that cell- and tissue-dependent differences should be considered for future interventions. Furthermore, it has been reported that chronic glycative stress or high levels of acute glycative stress can compromise proteolytic pathways by glycating components of these degradative routes and promoting proteostatic impairment. This places the aging organism in double jeopardy.
In sum, deficits in proteolytic pathways during aging or in diabetic conditions contribute to enhanced accumulation of glycated proteins and AGEs deposition, which ultimately leads to AGEs-derived tissue malfunction. We propose that boosting the UPS and autophagy might represent a useful strategy to limit glycation-derived proteotoxicity in older individuals.
Concluding remarks: Insoluble proteinaceous aggregation is accelerated through glycation and is associated with multiple age-related diseases, including neurological disorders. Therefore, the identification of a means to lower AGEs could provide enormous benefits to human health. Due to the lack of suitable pharmacological tools to reduce the formation of AGEs or reverse this process, the potential of boosting proteolytic capacities remains attractive and should be validated in a human context. Pharmacological or nutritional compounds could be helpful to enhance proteolytic capacity along with lifestyle changes (healthy diet and regular exercise) (Figure 1). Dietary management might represent the cheapest and safest alternative to modulate blood sugar levels and prolong the proteolytic capacities with age. Even two isocaloric diets with the same carbohydrate content can impact our bodies differently: a diet with a more readily-digested branched carbohydrate will increase circulating blood sugar faster, triggering the production of AGEs and leading to detrimental consequences. Nutritional interventions might be implemented to boost the proteolytic capacities involved in AGEs clearance. In this regard, research indicates that fasting can promote autophagy within the brain. A better understanding of how AGEs are targeted to degradative compartments will provide essential molecular clues towards developing therapeutic interventions to prevent glycation-derived damage and improve tissue and organ function. Further research should be conducted to establish nutritional recommendations/interventions to enhance proteolytic capacities.
We are grateful to Dr. Jasper Weinberg (Human Nutrition Research Center on Aging-Tufts University) and Dr. Gemma Aragonès (Human Nutrition Research Center on Aging-Tufts University) for valuable comments to the manuscript.
This work was supported by NIH RO1EY028559, RO1EY026979 (to AT), USDA NIFA 2016–08885 (to AT), USDA 8050-51000-089-01S (to AT), Kamada (to AT), Thome Memorial Foundation (to AT) and a grant from the Human Nutrition Research Center on Aging (to EB). This material is based upon work supported by the US Department of Agriculture—Agricultural Research Service (ARS), under Agreement No. 58-1950-4-003.
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
References
- 1.Aragno M, Mastrocola R. Dietary sugars and endogenous formation of advanced glycation endproducts: emerging mechanisms of disease. Nutrients. 2017;9:385. doi: 10.3390/nu9040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aragonès G, Dasuri K, Olukorede O, Francisco SG, Renneburg C, Kumsta C, Hansen M, Kageyama S, Komatsu M, Rowan S, Volkin J, Workman M, Yang W, Daza P, Ruano D, Dominguez-Martín H, Rodríguez-Navarro JA, Du XL, Brownlee MA, Bejarano E, et al. Autophagic receptor p62 protects against glycation-derived toxicity and enhances viability. Aging Cell. 2020;19:e13257. doi: 10.1111/acel.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao Y, Chen H, Cai Z, Zheng J, Zou J, Shi Y, Jiang L. Advanced glycation end products inhibit neural stem cell differentiation via upregualtion of HDAC3 expression. Brain Res Bull. 2020;159:1–8. doi: 10.1016/j.brainresbull.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Bejarano E, Taylor A. Too sweet: problems of protein glycation in the eye. Exp Eye Res. 2019;178:255–262. doi: 10.1016/j.exer.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleitas C, Piñol-Ripoll G, Marfull P, Rocandio D, Ferrer I, Rampon C, Egea J, Espinet C. proBDNF is modified by advanced glycation end products in Alzheimer's disease and causes neuronal apoptosis by inducing p75 neurotrophin receptor processing. Mol Brain. 2018;11:68. doi: 10.1186/s13041-018-0411-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li XH, Lv BL, Xie JZ, Liu J, Zhou XW, Wang JZ. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging. 2012;33:1400–1410. doi: 10.1016/j.neurobiolaging.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Münch G, Westcott B, Menini T, Gugliucci A. Advanced glycation endproducts and their pathogenic roles in neurological disorders. Amino Acids. 2012;42:1221–1236. doi: 10.1007/s00726-010-0777-y. [DOI] [PubMed] [Google Scholar]
- 8.Rabbani N, Thornalley PJ. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem Biophys Res Commun. 2015;458:221–226. doi: 10.1016/j.bbrc.2015.01.140. [DOI] [PubMed] [Google Scholar]
- 9.Rowan S, Jiang S, Korem T, Szymanski J, Chang ML, Szelog J, Cassalman C, Dasuri K, McGuire C, Nagai R, Du XL, Brownlee M, Rabbani N, Thornalley PJ, Baleja JD, Deik AA, Pierce KA, Scott JM, Clish CB, Smith DE, et al. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc Natl Acad Sci U S A. 2017;114:E4472–4481. doi: 10.1073/pnas.1702302114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semba RD, Nicklett EJ, Ferrucci L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A Biol Sci Med Sci. 2010;65:963–975. doi: 10.1093/gerona/glq074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, Pawlak D, Handa JT, Brownlee M, Nagaraj R, Taylor A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics) Aging cell. 2012;11:1–13. doi: 10.1111/j.1474-9726.2011.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vicente Miranda H, Szego ÉM, Oliveira LMA, Breda C, Darendelioglu E, de Oliveira RM, Ferreira DG, Gomes MA, Rott R, Oliveira M, Munari F, Enguita FJ, Simões T, Rodrigues EF, Heinrich M, Martins IC, Zamolo I, Riess O, Cordeiro C, Ponces-Freire A, et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain. 2017;140:1399–1419. doi: 10.1093/brain/awx056. [DOI] [PubMed] [Google Scholar]