

Abstract
Amyotrophic lateral sclerosis is the most common adult-onset neurodegenerative disease affecting motor neurons. Its defining feature is progressive loss of motor neuron function in the cortex, brainstem, and spinal cord, leading to paralysis and death. Despite major advances in identifying genes that can cause disease when mutated and model the disease in animals and cellular models, it still remains unclear why motor symptoms suddenly appear after a long pre-symptomatic phase of apparently normal function. One hypothesis is that age-related deregulation of specific proteins within key cell types, especially motor neurons themselves, initiates disease symptom appearance and may also drive progressive degeneration. Genome-wide in vivo cell-type-specific screening tools are enabling identification of candidates for such proteins. In this minireview, we first briefly discuss the methodology used in a recent study that applied a motor neuron-specific RNA-Seq screening approach to a standard model of TAR DNA-binding protein-43 (TDP-43)-driven amyotrophic lateral sclerosis. A key finding of this study is that synaptogyrin-4 and pleckstrin homology domain-containing family B member 1 are also deregulated at the protein level within motor neurons of two unrelated mouse models of mutant TDP-43 driven amyotrophic lateral sclerosis. Guided by what is known about molecular and cellular functions of these proteins and their orthologs, we outline here specific hypotheses for how changes in their levels might potentially alter cellular physiology of motor neurons and detrimentally affect motor neuron function. Where possible, we also discuss how this information could potentially be used in a translational context to develop new therapeutic strategies for this currently incurable, devastating disease.
Key Words: amyotrophic lateral sclerosis, glucagon-like peptide-1 receptor, motor neuron disease, mouse model, neurodegeneration, phosphatidylserine, pleckstrin homology domain, synaptogyrin, TAR DNA-binding protein-43, vesicle transport
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease featuring progressive loss of motor neuron (MN) function (Brown and Al-Chalabi, 2017). Patients gradually lose control over their muscles, become paralyzed, and typically die within 3–5 years. The two approved ALS therapies are only marginally effective, highlighting an urgent need to better understand what causes disease and to find useful molecular targets for therapeutic intervention. Identifying mutated genes in ALS has enabled development of cellular and animal disease models and intensive investigation of these models, particularly those based on SOD1 mutations, has revealed key molecular pathways deregulated in disease (Cook and Petrucelli, 2019). ALS is known as a non-cell autonomous disease in which glia, in particular astrocytes, have been demonstrated to drive MN degeneration (Taylor et al., 2016). Nevertheless, several lines of evidence highlight cell-autonomous roles in MNs. Overexpression of mutant TAR DNA-binding protein-43 (TDP-43) in MNs was sufficient for disease phenotypes in a rat ALS model (Huang et al., 2012). Studies in mice imply that expression of mutant SOD1 or TDP-43 within MNs themselves is important for timing of onset, with subsequent interaction with surrounding cells driving disease progression (reviewed in Ilieva et al., 2009; Ditsworth et al., 2017). Yet a fundamental issue remains unresolved: what specific molecular changes within the MNs themselves actually cause symptoms to appear? Insight into this question has been provided via cell-type specific profiling in a SOD1 mouse model and this study further supports the idea that major molecular changes begin in MNs (Sun et al., 2015). However, because different underlying genetic causes may lead to the same ALS disease symptoms through distinct molecular pathways, investigation of newer ALS models based on patient mutations in more recently identified genes will be crucial to address this question comprehensively. Focusing exclusively on the original SOD1 mutation-based models might lead to insights relevant only for this patient group (Turner et al., 2013).
Search Strategy and Selection Criteria
Studies cited in this review were identified mainly through two approaches. First, open-ended keyword searches in the PubMed database were performed with no limitation on publication date. The last such search was performed in December 2020. Keywords used (typically in combination) included, but were not limited to: neurodegeneration; neurodegenerative disease; amyotrophic lateral sclerosis; Parkinson's; Alzheimer's; motor neuron; synaptogyrin; cellugyrin; vesicle transport; vesicle recycling; endosome; trans-Golgi network; exocytosis; endocytosis; glucose transporter 4 vesicles; glucagon-like peptide-1 receptor; brain glucose metabolism; CNS insulin signaling; diabetes therapy; drug re-purposing; pleckstrin homology domain; evectin; G-protein coupled receptor; actin; myosin; phosphatidylserine; autophagy; neuronal cell death; synaptic pruning. Second, publications cited by the references identified in the open-ended searches were specifically examined and screened for their relevance to the topic.
Our intention was obviously not to exhaustively review each of the topics searched. Rather, we aimed to provide a compact, yet scholarly, summary of potentially interesting hypotheses for ALS pathobiology arising specifically from our recent work in this area.
Deregulation of SYNGR4 and PLEKHB1 within Motor Neurons at Symptom Onset in Mouse Models of TAR DNA-Binding Protein-43-Driven Amyotrophic Lateral Sclerosis
Recently, we used an MN-selective ribosome purification/RNA-Seq screening approach (Translating Ribosome Affinity Purification, Heiman et al., 2008) to identify changes in MN gene expression at symptom onset in a mouse model of TDP-43-driven ALS (Marques et al., 2020). We used an ALS mouse model based on a well-characterized patient mutation in the RNA-binding protein TDP-43, hTDP-43A315T (Wegorzewska et al., 2009). In addition to using RNA-seq as a sensitive readout, another important novel aspect of our study was inclusion of an asymptomatic transgenic TDP-43 line that expresses the protein at levels insufficient to cause symptoms (Xu et al., 2010). This proved crucial to filter out numerous gene expression changes that are induced by TDP-43 overexpression, but do not correlate with disease. Together with quantitative immunostaining of spinal cord, our approach revealed that synaptogyrin-4 (SYNGR4) and pleckstrin homology domain-containing family B member 1 (PLEKHB1) are proteins whose levels are altered within MNs coincident with the transition to overt motor symptoms (Figure 1).
Figure 1.
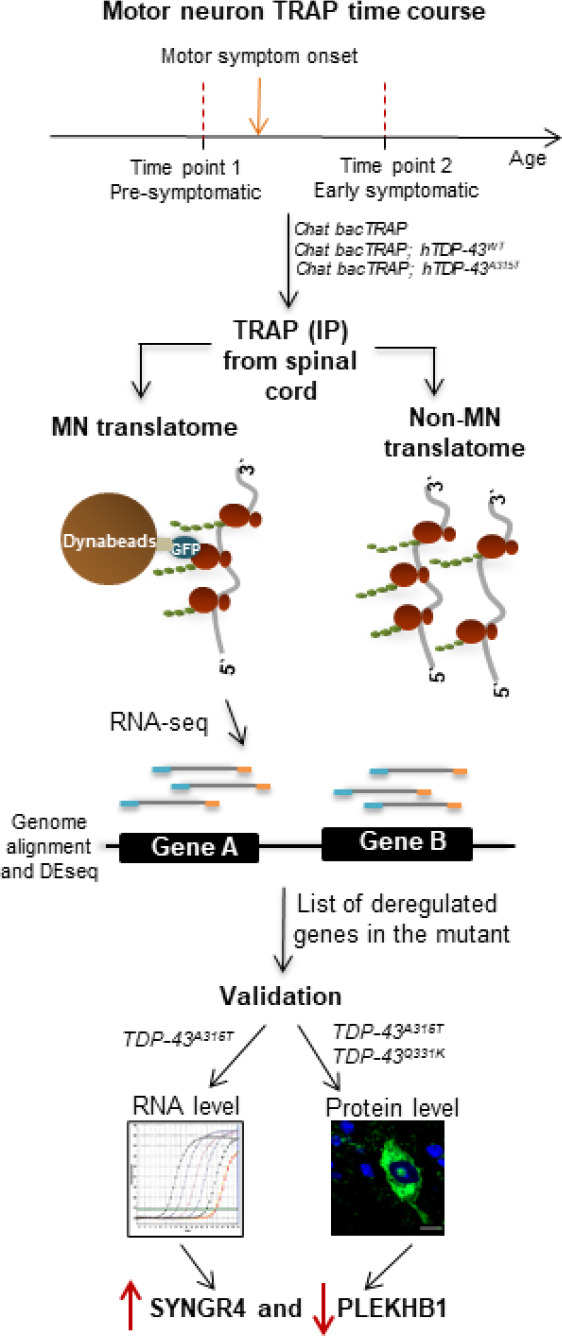
Translatome screening reveals specific proteins deregulated within motor neurons (MNs) at symptom onset in mouse models of mutant TDP-43-driven amyotrophic lateral sclerosis (ALS).
An overview of the experimental strategy used to screen for proteins that are deregulated within MNs at symptom onset in mutant TDP-43-driven amyotrophic lateral sclerosis. Spinal cords from mice of three different genotypes: Chat bacTRAP, Chat bacTRAP; hTDP-43A315T, and Chat bacTRAP; hTDP-43WT were extracted and lysed. MN ribosomes and associated mRNAs were purified via translating ribosome affinity purification (TRAP) and mRNAs were processed for RNA sequencing. This generated gene level datasets of specifically translated mRNAs in MNs (the “translatome”) which were analyzed for differential expression. Two candidate genes found to be exclusively deregulated in the mutant cohort were further validated at both the RNA and protein levels using qRT-PCR and tissue immunostaining, respectively. Collectively, this approach revealed upregulation of SYNGR4 and downregulation of PLEKHB1 within spinal MNs coincident with the onset of motor symptoms in mouse ALS models based on patient mutations in TDP-43 (Marques et al., 2020).
Importantly, protein-level changes in MNs were also observed in a completely independent mouse model based on the hTDP-43Q331K patient mutation (Arnold et al., 2013), demonstrating that the effects are not model- or mutation-specific. Thus, SYNGR4 and PLEKHB1 are promising candidates for molecular regulators of this key transition in disease, although this needs to be confirmed by additional rescue experiments. It will also be important to determine whether their deregulation is specific to ALS with TDP-43 mutations or is a more general pathobiological feature of disease. Interestingly, altered PLEKHB1 levels were found in a recent proteomic study of olfactory tissue of human ALS patients lacking TDP-43 mutations (Lachen-Montes et al., 2020). This observation raises the possibility that PLEKHB1 deregulation may be a general feature of ALS that apparently extends beyond the motor system.
Assuming changes in SYNGR4 and PLEKHB1 protein levels within MNs do lead to degeneration and ALS symptoms, how might they do this? A definitive answer is currently impossible, as neither protein has a well-understood function. Nevertheless, both proteins are highly similar to other proteins whose cellular functions are much better characterized and there is strong reason to believe that these functions are likely to be conserved. Here, we synthesize the relevant literature and outline testable hypotheses for cellular processes that may be altered by changes in SYNGR4 and PLEKHB1 levels and the impact this might have on MN health. Our working model features altered trafficking of specific vesicles, likely roles for phosphatidylserine (PS) and unconventional myosins, and possible effects on cell-surface levels of specific receptors implicated in insulin signaling and neurodegeneration (Figure 2).
Figure 2.
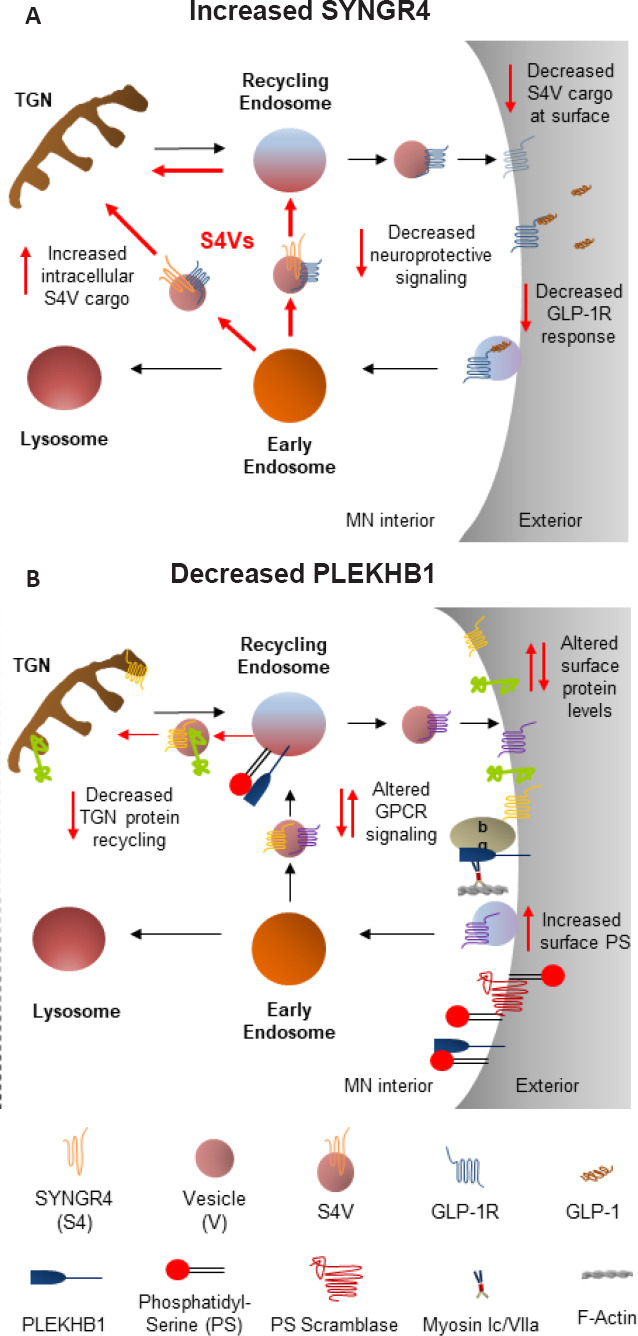
Possible cellular and pathobiological effects of altered SYNGR4 and PLEKHB1 protein levels in motor neurons (MNs).
(A) By analogy to SYNGR2/cellugyrin, we propose the existence of intracellular SYNGR4 vesicles (S4Vs) engaged in storage and/or retrograde transport between endosomal compartments and the Trans-Golgi network (TGN) of specific cell-surface cargo proteins. Increased SYNGR4 leads to increased intracellular retention of specific S4V cargos, reducing their cell-surface levels, as shown for the candidate S4V cargo, GLP-1R, a key regulator of neuroprotective central nervous system insulin signaling. (B) Assuming PLEKHB1 plays a similar role to evectin-2 (evt-2)/PLEKHB2 in sorting of specific endosomal cargos for recycling to the TGN, reducing PLEKHB1 would shift some TGN proteins to the MN cell surface, potentially affecting relative levels of other proteins there as well. Additionally, two potential direct effects of reducing PLEKHB1 levels at the MN plasma membrane are shown. First, reduced PLEKHB1 binding to beta gamma subunits of yet-to-be-identified MN G-protein coupled receptors (GPCRs) could directly affect MN GPCR signaling. Second, reduced PLEKHB1 binding to phosphatidylserine (PS) could enable better access for scramblases, leading to increased PS on the outer leaflet of the MN membrane to serve as an “eat me” signal. In principle, any of these effects might also involve the direct physical interaction of PLEKHB1 with myosin 1c/VIIa motors. Note that while MN soma are shown for simplicity, synaptic and axonal effects are also possible. Since effects shown in A and B presumably occur simultaneously, future studies should consider potential interaction points (e.g. overlap in endosomal cargo proteins; potential PLEKHB1 interaction with GLP-1R, a GPCR). Red arrows reflect specific cellular pathways hypothesized to be affected by upregulation of SYNGR4 (A) and downregulation PLEKHB1 (B).
How Increased SYNGR4 Might Conceivably Affect Motor Neuronal Health
Synaptogyrins are conserved integral membrane proteins associated with cellular vesicles
SYNGR4 is the least characterized of the four-member synaptogyrin family, which all have four membrane-spanning domains (where conservation is highest), connecting intra- and extracellular loops, and short cytoplasmic N-and C-terminal tails (Sugita et al., 1999; data not shown for SYNGR4). Despite their conserved structural core, individual synaptogyrins differ in the specific cellular membrane populations that they associate with and their functions. SYNGR1 and SYNGR3, expressed mainly in the nervous system, are abundant components of classical small synaptic vesicles (SVs) and apparently function as negative regulators of neurotransmission (Raja et al., 2019; and references therein). Conversely, ubiquitously expressed SYNGR2/cellugyrin is not associated with classical SVs, but is a component of synaptic-like microvesicles found in many cell types and can induce their formation (Janz and Sudhof, 1998; Kupriyanova and Kandror, 2000; Belfort and Kandror, 2003).
SYNGR2/cellugyrin affects insulin-regulated secretion of specific vesicle populations and their cargo
Several papers from the Kandror lab defined a function for SYNGR2/cellugyrin in trafficking of glucose transporter GLUT4-containing vesicles, a key aspect of glucose homeostasis in many tissues. Their central finding was that SYNGR2/cellugyrin marks the significant subpopulation of GLUT4 vesicles that is not recruited to the plasma membrane in response to insulin signaling (Kupriyanova and Kandror, 2000). These vesicles are believed to mediate protein transport between sorting endosomes and the endocytic recycling compartment and/or trans-golgi network (TGN) (reviewed in Kandror and Pilch, 2011). Collectively, these studies indicate that SYNGR2 is a component of intracellular transport vesicles found in diverse cell types and imply an inhibitory role in insulin-stimulated exocytosis of GLUT4 and associated insulin-regulated plasma membrane proteins, reminiscent of the role described for SYNGR1/3 with SV release.
Hypotheses for how SYNGR4 deregulation might affect motor neuronal health in ALS
What can we infer from the studies discussed above about SYNGR4 and how its upregulation in the transition from the pre-symptomatic to symptomatic phase might affect MN pathobiology in ALS? First, it seems extremely likely that SYNGR4 will be present in the membrane of a specific population of small vesicles. However, since SYNGR4 did not colocalize with a classical SV marker in MNs (Marques et al., 2020), we propose that SYNGR4's MN function might be similar to that described for SYNGR2 in other cell types: promoting internalization and retrograde trafficking of “SYNGR-4 vesicles” (S4Vs) from early endosomes to recycling endosomes and the TGN, effectively inhibiting exocytic release to the plasma membrane of associated protein cargo (Figure 2). In principle this cargo might also be GLUT4, but there is currently no evidence that SYNGR4 is a component of GLUT4 vesicles, which have been extensively examined by proteomics. Can potential alternative membrane-protein cargoes for S4Vs be proposed? We are aware of only one published potential protein-protein interaction partner of SYNGR4: the glucagon-like peptide-1 receptor (GLP-1R), which was identified via a membrane-bound yeast two hybrid approach (Dai et al., 2015). While this interaction requires verification, SYNGR3 was independently identified as a possible GLP-1R interacting protein by this approach with a human brain library (Huang et al., 2013). Shared interaction partners for these two proteins seem especially interesting, since SYNGR3 mediates pre-synaptic deficits resulting from its interaction with toxic variants of Tau protein that drive neurodegeneration in flies, mice, and human patients (McInnes et al., 2018).
By analogy to SYNGR2's well described function in GLUT4 trafficking, upregulation of SYNGR4 at symptom onset would presumably shift GLP-1R away from the cell surface to an internal vesicular “storage” pool marked by the presence of SYNGR4 (S4Vs, Figure 2). In addition to being an important therapeutic target in Type II diabetes, GLP-1R mediates central nervous system insulin signaling, which is heavily in preventing at least two neurodegenerative diseases. GLP-1R agonists have already shown promise for restoring function in both preclinical models of Parkinson's and Alzheimer's disease models, as well as in human patients (Holscher, 2020). Thus, it is straightforward to imagine how reduced neuroprotective GLP-1R signaling due to upregulation of SYNGR4 could have a deleterious impact on MN health, as well as how GLP-1R could potentially be targeted therapeutically for benefit in ALS patients. Time will tell whether this admittedly speculative “GLP-1R” hypothesis for ALS etiology driven by SYNGR4 upregulation gains support. Even if GLP-1R ultimately proves not to be relevant for MN degeneration in ALS, identifying membrane interaction partners of SYNGR4 seems a promising strategy to delineate both its cellular function and potential role in MN pathobiology in ALS.
How Decreased Levels of PLEKHB1 Might Affect Motor Neuronal Health
Although it is better characterized than SYNGR4, cellular functions of PLEKHB1 (aka evectin-1, PHR1 or PKL1) also remain mysterious. PLEKHB1 is a single-pass transmembrane protein with an N-terminal Pleckstrin homology domain, preferentially expressed in mammalian brain and sensory systems (Krappa et al., 1999; Xu et al., 1999). In mouse retina, antibody staining detected strong signal in the outer segment of both cones and rods, mainly localized to the plasma membrane (Xu et al., 1999). Conversely, pulse-chase immunoblotting of frog retina supported mainly intracellular localization in a “post Golgi” membrane compartment (Krappa et al., 1999). Thus, the relative proportion of PLEKHB1 in specific membrane pools may be regulated differentially in a cell/species-specific manner.
PLEKHB1 interacts with GPCRs and myosins 1c and VIIa
In vitro studies probing PLEKHB1 interactions revealed none with phosphoinositides, but binding to the beta-gamma subunits of the G-protein-coupled receptor, transducin, a key mediator of signal transduction in the visual system (Xu et al., 1999). This suggests that PLEKHB1 might also interact with G-protein-coupled receptors in MNs and motivates systematically searching for them and establishing assays for modulation of their function. An interesting possibility is that PLEKHB1 might interact with GLP-1R to modulate its function, reflecting a potential point of convergence with SYNGR4 deregulation (Figure 2). In addition, based on multiple lines of evidence for direct interaction of PLEKHB1 with the C-terminal tails of the unconventional myosins 1c and VIIa, PLEKHB1 was proposed to anchor the actin cytoskeleton to the plasma membrane in sensory cells (Etournay et al., 2005). However, mice lacking PLEKHB1 do not display any obvious sensory deficits or other phenotypes (Xu et al., 2004). Thus, despite its interesting localization and molecular interactions, PLEKHB1's cellular and organismal functions remain mysterious.
PLEKHB1 is likely to bind directly to phosphatidylserine, an “eat me” signal
Arguably the strongest clue regarding a possible PLEKHB1 function that could be relevant to ALS pathobiology comes from studies of its more broadly expressed ortholog, PLEKHB2/evectin-2. In cultured cell lines, evectin-2 showed a peri-nuclear localization coincident with PS and markers of recycling endosomes (Uchida et al., 2011). This localization depended on specific amino acid residues in the N-terminus of the Pleckstrin homology domain that make direct contact with the PS head group in a co-crystal structure. Importantly, all of these residues are conserved in PLEKHB1, strongly suggesting that it too will specifically interact with PS. Assuming it does, what might be the impact for MN health of having reduced levels of PLEKHB1, as we observed in the ALS models at symptom onset? An obvious possibility would be the same effect observed when PLEKHB2/evectin-2 is depleted in cultured cells: a selective reduction in retrograde trafficking from recycling endosomes to the TGN (Uchida et al., 2011). Why exactly this would lead to neurodegeneration is not clear, but shifting recycled TGN proteins towards the cell surface would result in changes in cell-surface levels of specific proteins and might thereby affect the function of those mediating MN health (Figure 2). Alternatively, the pathobiological role for reduced PLEKHB1-PS interaction might involve another PS function. In healthy cells PS is found almost exclusively on the inner leaflet of the plasma membrane. Conversely, PS on the cell surface serves as an “eat me” signal in apoptosis and synaptic pruning during development, in response to injury, and in various neurodegenerative disease models (Sapar et al., 2018; Scott-Hewitt et al., 2020). How might this relate to ALS? In healthy MNs, intracellular PLEKHB1 binding to PS in the plasma membrane might compete with other PS-interacting proteins that can catalyze its exposure on the cell surface (e.g. scramblases). If so, reduced PLEKHB1 in comparison to a healthy MN at symptom onset would effectively enhance access of those enzymes to PS. This would, in turn, lead to increased cell surface exposure and targeting of MN regions by scavenging cells recognizing the “eat me” signal, with resulting detrimental effects on MN health (Figure 2).
Conclusions and Outlook
In conclusion, our recently published work identified two transmembrane proteins whose levels change in MNs of mouse models of TDP-43 driven ALS at a crucial transition in disease: the age-dependent appearance of motor symptoms. As such, they are promising candidates to mediate this crucial transition as disease “drivers”. However, they could equally well be passive “markers” or even play a positive “compensator” role to restore homeostasis. Distinguishing among these alternatives will be crucial to understanding how SYNGR4 and PLEKHB1 contribute to disease etiology and their potential as therapeutic targets. This can be done initially by using a genetic approach to assess the impact of offsetting the increase or decrease in disease models and monitoring the impact on disease onset and progression using quantitative behavioral assays and cellular pathological readouts. Assuming the proteins do play an active role in driving or compensating for disease, it will be important to understand their precise functions in MNs and how changing their levels affects MN health. This may prove challenging, as in vivo functional studies in adult MNs are necessarily limited and it is not clear how much of the relevant MN cell biology can be properly recapitulated in culture models. In any case, the working hypotheses that we have outlined here provide several interesting experimental directions to pursue. We find possible connections to GLP-1R trafficking (via SYNGR4) and PS membrane regulation (via PLEKHB1), especially worthy of future investigation.
Footnotes
C-Editors: Zhao M, Qiu Y; T-Editor: Jia Y
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:Work in the Duncan lab on this topic was supported in part by funding from the Else Kröner Fresenius Stiftung (Co-PI) and the Werner Otto Stiftung (PI) (to KED).
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Funding:Work in the Duncan lab on this topic was supported in part by funding from the Else Kröner Fresenius Stiftung (Co-PI) and the Werner Otto Stiftung (PI) (to KED).
References
- 1.Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, Da Cruz S, Clutario KM, Swing D, Tessarollo L, Marsala M, Shaw CE, Yeo GW, Cleveland DW. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110:E736–745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belfort GM, Kandror KV. Cellugyrin and synaptogyrin facilitate targeting of synaptophysin to a ubiquitous synaptic vesicle-sized compartment in PC12 cells. J Biol Chem. 2003;278:47971–47978. doi: 10.1074/jbc.M304174200. [DOI] [PubMed] [Google Scholar]
- 3.Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 4.Cook C, Petrucelli L. Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron. 2019;101:1057–1069. doi: 10.1016/j.neuron.2019.02.032. [DOI] [PubMed] [Google Scholar]
- 5.Dai FF, Bhattacharjee A, Liu Y, Batchuluun B, Zhang M, Wang XS, Huang X, Luu L, Zhu D, Gaisano H, Wheeler MB. A novel GLP1 receptor interacting protein ATP6ap2 regulates insulin secretion in pancreatic beta cells. J Biol Chem. 2015;290:25045–25061. doi: 10.1074/jbc.M115.648592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ditsworth D, Maldonado M, McAlonis-Downes M, Sun S, Seelman A, Drenner K, Arnold E, Ling SC, Pizzo D, Ravits J, Cleveland DW, Da Cruz S. Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133:907–922. doi: 10.1007/s00401-017-1698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Etournay R, El-Amraoui A, Bahloul A, Blanchard S, Roux I, Pezeron G, Michalski N, Daviet L, Hardelin JP, Legrain P, Petit C. PHR1, an integral membrane protein of the inner ear sensory cells, directly interacts with myosin 1c and myosin VIIa. J Cell Sci. 2005;118:2891–2899. doi: 10.1242/jcs.02424. [DOI] [PubMed] [Google Scholar]
- 8.Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holscher C. Brain insulin resistance: role in neurodegenerative disease and potential for targeting. Expert Opin Investig Drugs. 2020;29:333–348. doi: 10.1080/13543784.2020.1738383. [DOI] [PubMed] [Google Scholar]
- 10.Huang C, Tong J, Bi F, Zhou H, Xia XG. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122:107–118. doi: 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Dai FF, Gaisano G, Giglou K, Han J, Zhang M, Kittanakom S, Wong V, Wei L, Showalter AD, Sloop KW, Stagljar I, Wheeler MB. The identification of novel proteins that interact with the GLP-1 receptor and restrain its activity. Mol Endocrinol. 2013;27:1550–1563. doi: 10.1210/me.2013-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janz R, Sudhof TC. Cellugyrin, a novel ubiquitous form of synaptogyrin that is phosphorylated by pp60c-src. J Biol Chem. 1998;273:2851–2857. doi: 10.1074/jbc.273.5.2851. [DOI] [PubMed] [Google Scholar]
- 14.Kandror KV, Pilch PF. The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic. 2011;12:665–671. doi: 10.1111/j.1600-0854.2011.01175.x. [DOI] [PubMed] [Google Scholar]
- 15.Krappa R, Nguyen A, Burrola P, Deretic D, Lemke G. Evectins: vesicular proteins that carry a pleckstrin homology domain and localize to post-Golgi membranes. Proc Natl Acad Sci U S A. 1999;96:4633–4638. doi: 10.1073/pnas.96.8.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kupriyanova TA, Kandror KV. Cellugyrin is a marker for a distinct population of intracellular Glut4-containing vesicles. J Biol Chem. 2000;275:36263–36268. doi: 10.1074/jbc.M002797200. [DOI] [PubMed] [Google Scholar]
- 17.Lachen-Montes M, Mendizuri N, Ausin K, Andres-Benito P, Ferrer I, Fernandez-Irigoyen J, Santamaria E. Amyotrophic lateral sclerosis is accompanied by protein derangements in the olfactory bulb-tract axis. Int J Mol Sci. 2020;21:8311. doi: 10.3390/ijms21218311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marques RF, Engler JB, Kuchler K, Jones RA, Lingner T, Salinas G, Gillingwater TH, Friese MA, Duncan KE. Motor neuron translatome reveals deregulation of SYNGR4 and PLEKHB1 in mutant TDP-43 amyotrophic lateral sclerosis models. Hum Mol Genet. 2020;29:2647–2661. doi: 10.1093/hmg/ddaa140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McInnes J, Wierda K, Snellinx A, Bounti L, Wang YC, Stancu IC, Apostolo N, Gevaert K, Dewachter I, Spires-Jones TL, De Strooper B, De Wit J, Zhou L, Verstreken P. Synaptogyrin-3 mediates presynaptic dysfunction induced by Tau. Neuron. 2018;97:823–835. doi: 10.1016/j.neuron.2018.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Raja MK, Preobraschenski J, Del Olmo-Cabrera S, Martinez-Turrillas R, Jahn R, Perez-Otano I, Wesseling JF. Elevated synaptic vesicle release probability in synaptophysin/gyrin family quadruple knockouts. Elife. 2019;8:e40744. doi: 10.7554/eLife.40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sapar ML, Ji H, Wang B, Poe AR, Dubey K, Ren X, Ni JQ, Han C. Phosphatidylserine externalization results from and causes neurite degeneration in drosophila. Cell Rep. 2018;24:2273–2286. doi: 10.1016/j.celrep.2018.07.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, Tamborini M, Bizzotto M, Passoni L, Filipello F, Jahn R, Stevens B, Matteoli M. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 2020;39:e105380. doi: 10.15252/embj.2020105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugita S, Janz R, Sudhof TC. Synaptogyrins regulate Ca2+-dependent exocytosis in PC12 cells. J Biol Chem. 1999;274:18893–18901. doi: 10.1074/jbc.274.27.18893. [DOI] [PubMed] [Google Scholar]
- 24.Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S, Kopelevich A, Kaspar BK, Lagier-Tourenne C, Cleveland DW. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A. 2015;112:E6993–7002. doi: 10.1073/pnas.1520639112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchida Y, Hasegawa J, Chinnapen D, Inoue T, Okazaki S, Kato R, Wakatsuki S, Misaki R, Koike M, Uchiyama Y, Iemura S, Natsume T, Kuwahara R, Nakagawa T, Nishikawa K, Mukai K, Miyoshi E, Taniguchi N, Sheff D, Lencer WI, et al. Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc Natl Acad Sci U S A. 2011;108:15846–15851. doi: 10.1073/pnas.1109101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu S, Wang Y, Zhao H, Zhang L, Xiong W, Yau KW, Hiel H, Glowatzki E, Ryugo DK, Valle D. PHR1, a PH domain-containing protein expressed in primary sensory neurons. Mol Cell Biol. 2004;24:9137–9151. doi: 10.1128/MCB.24.20.9137-9151.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu S, Ladak R, Swanson DA, Soltyk A, Sun H, Ploder L, Vidgen D, Duncan AM, Garami E, Valle D, McInnes RR. PHR1 encodes an abundant, pleckstrin homology domain-containing integral membrane protein in the photoreceptor outer segments. J Biol Chem. 1999;274:35676–35685. doi: 10.1074/jbc.274.50.35676. [DOI] [PubMed] [Google Scholar]
- 31.Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]