

Exploring the Alzheimer's disease neuroepigenome: recent advances and future trends: Alzheimer's disease (AD) is a chronic neurodegenerative disease and the most common cause of dementia. After decades of ongoing efforts by scientists, many hallmarks of AD, such as amyloid-β (Aβ) and tau pathologies, have finally been understood. But these milestone discoveries still failed to help us find a cure. In recent years, based on advances in genomics, researchers have discovered more than 20 AD-associated alleles. Three of these alleles can cause autosomal dominant AD: amyloid precursor protein and presenilin 1/2 genes. The rest of these alleles can increase the risk to AD, such as the apolipoprotein E gene. These risk loci implicate Aβ, tau, immunity, and lipid processing, which have helped us accurately understand the complex changes in AD patients’ molecular networks.
However, genetic alterations are irreversible, limiting their potential to become drug targets. Unlike genetics, epigenetic alterations are reversible, giving AD epigenetic studies a vast prospect of clinical application. Epigenetics mainly studies the mechanism of heritable phenotypic variation without changing the gene sequence. It covers DNA modifications (transcriptional level), histone modifications (transcriptional level), and non-coding RNAs (post-transcriptional level). Thus far, the best-characterized DNA modifications are DNA methylation and demethylation at the 5-position of the cytosine, and the link between global DNA methylation and AD has proven to be a negative correlation (Mastroeni et al., 2010). Multiple forms of histone modifications exist, and histone acetylation dysregulation in AD, in particular, has been most vigorously researched. It is now well-accepted that histone acetylation is associated with transcriptional activation and histone deacetylation correlates with transcriptional repression. Compelling evidence from these studies (Sanchez-Mut and Graff, 2015; Patel et al., 2019) reveals a dramatic decrease in global histone acetylation (Figure 1A) and consistent downregulation of overall gene expression in AD (Figure 1B). Research on non-coding RNAs, particularly microRNAs, has also flourished in the context of AD based on detection methods and genetic manipulation feasibility. Researchers have been screening alterations of microRNAs in various AD patients’ specimens, including brain tissue, cerebrospinal fluid, and blood. They have provided us a picture of “microRNA signatures” that may serve as promising biomarkers for AD (Nagaraj et al., 2019). With the aforementioned concrete advances in AD epigenetics, here in this perspective, we focus our discussion on the AD epigenome and summarize the future trends in AD epigenomic studies and corresponding transcriptomic investigations as “4S”: single genes, single disease stages, single brain regions, and single cells (Figure 1C).
Figure 1.
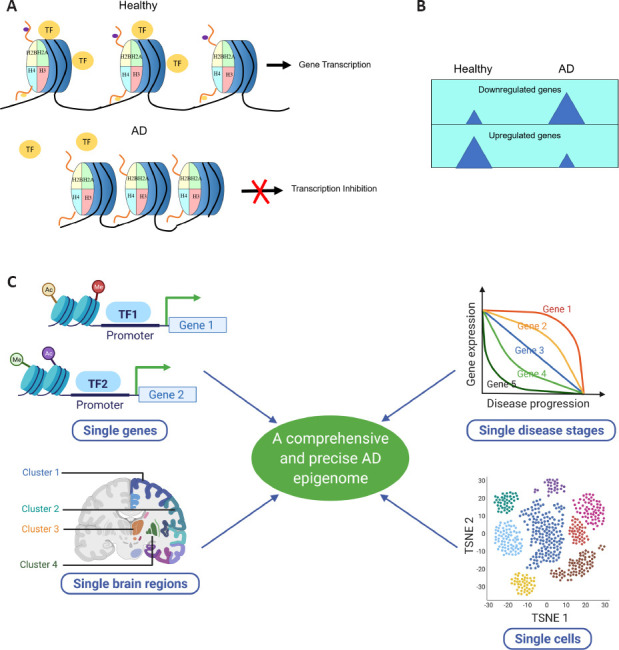
The elucidation of the AD neuroepigenome is evolving from global to specific levels.
(A) Neuropigenomic studies reveal dramatic loss of histone acetylation in AD, suggesting a global transcriptional inhibition state. (B) Transcriptional repression in AD. Transcriptomic studies indicate an overall reduced gene expression state in AD, suggesting a general repressed chromatin state. (C) “4S” of future AD epigenomic studies. AD: Alzheimer's disease; TF: transcription factor. TSNE: t-distributed stochastic neighbor embedding. C was created with BioRender.com.
Single genes: While a compendium of studies implicate global histone deacetylation with concomitant gene repression as contributors to AD pathology (Sanchez-Mut and Graff, 2015; Patel et al., 2019), it is important to keep in mind that while informative, these findings represent a net outcome. In contrast, by examining single genes and their adjacent chromatin states, epigenetic profiles can be obtained at higher resolution. For example, an epigenome-wide association study by Klein et al. using H3K9ac ChIP-Seq in the dorsolateral prefrontal cortex of brains from AD and non-AD participants, enabled these researchers to identify 26,384 H3K9ac peaks, characterize AD-associated peak alterations, and map these alterations genome-wide and at the level of chromatin architecture and transriptome gene changes. (Klein et al., 2019). Future studies may entail researchers combining epigenomic (Chromatin immunoprecipitation followed by sequencing; ChIP-Seq) and transcriptomic (RNA sequencing; RNA-Seq) methods to uncover how arrays of epigenetic marks such as histone acetylation, methylation, phosphorylation and ubiquitination at a single gene loci affects its transcriptional control under AD conditions (Gjoneska et al., 2015; Klein et al., 2019). Thus far, our knowledge of potential histone methylation, phosphorylation, and ubiquitination alterations in AD and the related mechanisms remains limited. Future investigations into these areas may ultimately provide researchers with a big picture of a potential “histone code” that is associated with AD-affected specific gene expression profiles.
Single disease stages: One critical question with significant clinical relevance is whether the epigenetic profiles and the corresponding gene expression are stable or dynamic during AD progression. If the neuroepigenome and corresponding transcriptome are highly dynamic during AD progression, elucidating these changes would help identify specific therapeutic targets for a particular disease stage. Nevertheless, whether gene alterations are dynamic during early and late disease stages remains to be determined as neurodegenerative gene expression studies predominantly focus on aged brain samples. In light of the necessity for this information, a recent study for the first time characterized the dynamic epigenome and corresponding transcriptomic changes during early and late stages of AD-associated progression using the CK-p25 neurodegenerative mouse model. The results of this work revealed and classified transient, consistent, and late changes in histone acetylation domains with concomitant gene expression changes (Gjoneska et al., 2015). Recent work from our lab using an AD-associated Drosophila model expressing human Aβ42 also uncovered distinct modes of neuroepigenetic gene changes in early versus late AD stages that can be potentially useful early biomarkers for AD (Zhang et al., 2020). From these studies, a scenario begins to emerge by which a transcriptional activity switch that is controlled by the neuroepigenome exists in which “AD early-response epigenome” and “AD late-response epigenome” exist during different stages of disease progression. We speculate that post-translational modifications of histones are likely differentially affected during early and late AD stages that contribute to potentially distinct early and late AD-associated epigenetic landscapes that contribute to stage-specific transcriptional changes.
Single brain regions: Studies using brain specimens from patients and animals with advanced AD stages have revealed that the AD brain exhibits global epigenomic alterations at the whole-brain level as well as more specific changes when assessing the hippocampus and cortex individually. These results support the importance of characterizing a more comprehensive and precise AD neuroepigenome that encompasses brain region variations. AD affects brain regions differently. Is this phenomenon related to spatial epigenomic properties? Aβ plaques only appear in abundance in the cortex and hippocampus. Is this potentially because in these areas, genes that facilitate Aβ aggregation are active, and genes responsible for Aβ clearance are silent? To answer these questions, we need a new multi-dimensional sequencing technique that can collect epigenomic data and at the same time preserve positional information. For this goal, spatial transcriptomics (ST) was born. In July 2016, Ståhl et al. first developed the method of ST and applied it to mouse coronal brain sections (Ståhl et al., 2016). This method successfully captured at least twice as many transcripts as laser capture microdissection and yielded decent sensitivity compared with single-molecule fluorescent in situ hybridization. Most importantly, homologous brain regions displayed very similar gene expression patterns, and genes with previously known restricted expression were specifically detected in particular brain regions. In June 2020, Ortiz et al. described a spatial transcriptome feature during the mouse brain map construction. Spots in the same brain region are more likely to be clustered because of the similarity in transcriptomic characteristics. In each coronal section of the brain, the spot subgroups’ positions and anatomical annotations’ positions are highly similar. These solid pioneering works paved the way for further AD epigenomic investigations with a spatially resolved technique. Although these foundation experiments focused on ST, these findings elicit promise that spatial epigenomics technology will soon emerge.
Single cells: Previous AD epigenomic studies typically use mixed populations of cells from a given brain sample. Thus, the results only reflect the averaged profiles of changes in all different cell types. Because neuronal cell composition displays a significant shift during AD progression, such cellular bulk approaches have limited the comparability of epigenomic profiles between different disease stages. In early AD stages, neuronal apoptosis is minimal, and the neuroimmune response is not adequately initiated. With disease progression, in late AD stages, neurons decrease dramatically due to massive neuronal apoptosis, and glial cells increase significantly due to hyperactivity of the neuroimmune response. Thus, the components of cells used for bulk ChIP-Seq and RNA-Seq are different between early and late AD stages. The early-stage epigenome is more neuronal, and the late-stage epigenome is more glial. To solve this problem, it is necessary to develop a technology that can effectively separate subtypes of cells for RNA isolation and sequencing. After more than ten years of efforts, scientists have made tremendous progress in this field. The results of the latest single-cell sequencing technologies have even refreshed our understanding of known cell types because they have identified even more subtypes than was previously envisioned. Cellular heterogeneity proves to be more diverse than what we have imagined and primary cell subtypes already characterized can be further divided still into secondary or even tertiary subtypes according to epigenomic profiles. Before single-cell RNA-Sequencing (scRNA-Seq) became prevalent, Harzer et al. (2013) developed a method that allows RNA-Seq of FACS-purified neural stem cells and their differentiated neurons from Drosophila larval brains. By combining the utilization of cell-type-specific drivers and identification of differences in cell size and green fluorescent protein expression levels, this method reaches 98% purity in cell populations, suitable for subsequent cell-type-specific gene expression analyses. Later in 2015, Macosko et al. (2015) developed the first massive parallel scRNA-Seq technique using nanoliter droplets. They analyzed more than four thousand mouse retinal cells and identified 39 transcriptionally distinct cell populations, including known retinal cell classes, and additional novel cell subtypes. Single-cell chromatin immunoprecipitation followed by sequencing (scChIP-Seq) was reported in the same year. Rotem et al. (2015) utilized microfluidics and DNA barcoding and performed ChIP-Seq in single cells. This technique successfully distinguished mouse embryonic stem cells from embryonic fibroblasts and hematopoietic progenitors by their distinct chromatin H3K4me3 profiles. Furthermore, embryonic stem cell subpopulations carrying differential pluripotency were identified by different chromatin signatures. At present, AD epigenomic studies with single-cell resolution are still in their infancy, but predictably, the application of scChIP-Seq and scRNA-Seq in the near future is expected to bring about major breakthroughs in AD research.
Conclusion and future perspectives: Researchers have made many exciting discoveries about the AD neuroepigenome in the past decade. The depiction of the AD neuroepigenome has evolved from global levels to single genes and single cells. H3K9ac ChIP-Seq in human AD patients suggests H3K9ac profile changes as an early epigenetic biomarker of tau pathology but not Aβ pathology, primarily in neuronal populations (Klein et al., 2019). Developmental time course studies tracking AD progression have revealed clinically significant early disease biomarkers. For example, Gjoneska et al. reported an early and persistent increase in immune and inflammatory response gene expression that included genes INPP5D and SPI1 (Gjoneska et al., 2015) with concomitant increased-levels of H3K4me3 and H3K27ac peaks flanking these genes (Gjoneska et al., 2015). Further, recent findings from our laborary identified distinct modes of epigenetic gene changes and Tip60 histone acetyltransferase mediated neuroprotection that occur in early pre-Aβ plaque stages versus late stages of AD that can serve as early biomarkers for AD, and support the therapeutic potential of Tip60 over the course of AD progression (Zhang et al., 2020). But despite these advances, certain challenges remain. For example, the ST achieves the transcript's anatomical position but cannot definitively associate those transcripts with individual cells because each ST spot contains at least several cells. Therefore, through the spatial transcriptome, we only know where the transcript is, but not to which cell type it belongs. Vice versa, scRNA-Seq carries cell type information but lacks spatial information. This necessitates the combination of ST and scRNA-Seq, and potentially the combination of spatial epigenomics and scChIP-Seq in the future, for a more high resolution and detailed characterization of AD -omic features. Despite these challenges, major breakthrough accomplishments in the development of AD neuroepigenomic tools to explore these areas serve as a strong foundation for future promising research elucidating AD neuroepigenomic profiles at more specific levels. Such studies should ultimately aid in the design and development of therapeutics that precisely target a particular subset of genes, disease stage, brain region and cell population for high resolution elucidation of their specific AD-associated neuroepigenomes.
This work was supported by a National Institutes of Health (NIH) R01 grant (No. 5R01NS095799) (to FE).
Footnotes
P-Reviewers: Yang C, Yue C; C-Editors: Zhao M, Zhao LJ, Qiu Y; T-Editor: Jia Y
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
References
- 1.Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harzer H, Berger C, Conder R, Schmauss G, Knoblich JA. FACS purification of Drosophila larval neuroblasts for next-generation sequencing. Nat Protoc. 2013;8:1088–1099. doi: 10.1038/nprot.2013.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein HU, McCabe C, Gjoneska E, Sullivan SE, Kaskow BJ, Tang A, Smith RV, Xu J, Pfenning AR, Bernstein BE, Meissner A, Schneider JA, Mostafavi S, Tsai LH, Young-Pearse TL, Bennett DA, De Jager PL. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer's human brains. Nat Neurosci. 2019;22:37–46. doi: 10.1038/s41593-018-0291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol Aging. 2010;31:2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagaraj S, Zoltowska KM, Laskowska-Kaszub K, Wojda U. microRNA diagnostic panel for Alzheimer's disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res Rev. 2019;49:125–143. doi: 10.1016/j.arr.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Ortiz C, Navarro JF, Jurek A, Martin A, Lundeberg J, Meletis K. Molecular atlas of the adult mouse brain. Sci Adv. 2020;6:eabb3446. doi: 10.1126/sciadv.abb3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel H, Dobson RJB, Newhouse SJ. A meta-analysis of Alzheimer's disease brain transcriptomic data. J Alzheimers Dis. 2019;68:1635–1656. doi: 10.3233/JAD-181085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, Bernstein BE. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez-Mut JV, Graff J. Epigenetic alterations in Alzheimer's disease. Front Behav Neurosci. 2015;9:347. doi: 10.3389/fnbeh.2015.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg Å, Pontén F, Costea PI, Sahlén P, Mulder J, Bergmann O, Lundeberg J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Karisetty BC, Bhatnagar A, Armour EM, Beaver M, Roach TV, Mortazavi S, Mandloi S, Elefant F. Tip60 protects against amyloid-beta-induced transcriptomic alterations via different modes of action in early versus late stages of neurodegeneration. Mol Cell Neurosci. 2020;109:103570. doi: 10.1016/j.mcn.2020.103570. [DOI] [PMC free article] [PubMed] [Google Scholar]