


Abstract
Inducing tRNA +1 frameshifting to read a quadruplet codon has the potential to incorporate a non-natural amino acid into the polypeptide chain. While this strategy is being considered for genome expansion in biotechnology and bioengineering endeavors, a major limitation is a lack of understanding of where the shift occurs in an elongation cycle of protein synthesis. Here, we use the high-efficiency +1-frameshifting SufB2 tRNA, containing an extra nucleotide in the anticodon loop, to address this question. Physical and kinetic measurements of the ribosome reading frame of SufB2 identify twice exploration of +1 frameshifting in one elongation cycle, with the major fraction making the shift during translocation from the aminoacyl-tRNA binding (A) site to the peptidyl-tRNA binding (P) site and the remaining fraction making the shift within the P site upon occupancy of the A site in the +1-frame. We demonstrate that the twice exploration of +1 frameshifting occurs during active protein synthesis and that each exploration is consistent with ribosomal conformational dynamics that permits changes of the reading frame. This work indicates that the ribosome itself is a determinant of changes of the reading frame and reveals a mechanistic parallel of +1 frameshifting with –1 frameshifting.
Graphical Abstract
Graphical Abstract.
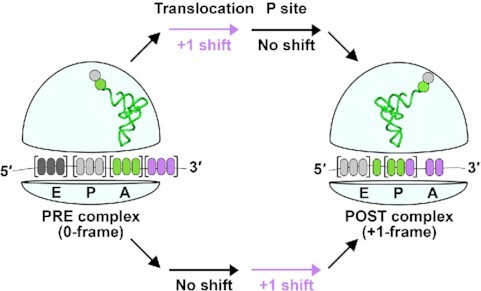
tRNA+1 frameshifting during translocation and in the P site.
INTRODUCTION
A designer tRNA that induces +1 frameshifting at a quadruplet codon is a potential tool for expansion of the genome (1). This designer tRNA could carry a non-natural amino acid and deliver it to protein synthesis by shifting into the +1-frame within the quadruplet codon, allowing site-specific incorporation of the non-natural amino acid into the nascent polypeptide chain. While genome expansion can also be achieved with suppressor tRNAs that deliver a non-natural amino acid to an internal stop codon (2), the capacity is limited, due to the need to reserve one of the three stop codons for termination of protein synthesis. In contrast, genome expansion with +1-frameshifting tRNAs has a much higher capacity, possibly allowing insertion of non-natural amino acids to multiple quadruplet codons at the same time. However, despite the high potential of genome expansion with +1-frameshifting tRNAs, the yield of the shift at a given quadruplet codon has been poor, due to a lack of understanding of where it can be explored in an elongation cycle of protein synthesis. Lacking a clear answer to this question, current efforts have focused on designing +1-frameshifting tRNAs with an extra nucleotide inserted to the anticodon loop in an expanded anticodon–stem–loop (ASL) structure for pairing with a quadruplet codon (1). This design is based on known structures of high-efficiency +1-frameshifting tRNAs that were isolated from genetic studies (3). However, in vitro designed +1-frameshifting tRNAs vary broadly in achieving successful genome expansion (4).
Recent work has shed light on the mechanism of +1 frameshifting. In the terminology of bacterial protein synthesis, each elongation cycle is defined by three key steps: (i) delivery of an aminoacyl-tRNA (aa-tRNA) to the mRNA codon at the ribosomal A site via a ternary complex (TC) with the GTPase EF-Tu and GTP; (ii) synthesis of the next peptide bond in the peptidyl transferase center on the ribosome by transferring the nascent chain from the P-site tRNA to the aa-tRNA at the A site to form a pre-translocation complex (PRE) and (iii) translocation of the mRNA and the P- and A-site tRNAs to the exit (E) site and P site, respectively, to form a post-translocation complex (POST) in a reaction catalyzed by the GTPase EF-G and GTP. Interestingly, crystal structures of several +1-frameshifting tRNAs show that, despite having an expanded ASL, these tRNAs maintain the 0-frame anticodon-codon pairing in the A site (5–8), but that they occupy the +1-frame in the P site (9). These structures emphasize that +1 frameshifting does not occur in the A site. Indeed, a +1-frameshifting tRNA, having an expanded ASL, in our recent study retains all of the kinetic features of a canonical tRNA up to and including the formation of a PRE complex in the A site (10). Instead, we show that it is substantially slowed as it moves from the A site to the P site (10), indicating that it undergoes +1 frameshifting during translocation. This result is consistent with our recent cryo-EM structures of Escherichia coli ProM tRNAPro(UGG) (UGG: the anticodon), which despite having a canonical ASL, is highly prone to +1 frameshifting (11). In these structures, E. coli tRNAPro(UGG) forms the canonical 0-frame anticodon-codon pairing in the A site, but it shifts to the +1-frame during translocation to the P site (12). Separately, we had shown that the N1-methylation of G37 (m1G37) in a different canonical E. coli ProL tRNAPro(GGG) is a major determinant of maintaining the protein synthesis reading frame, and that loss of m1G37 leads to +1 frameshifting when the tRNA is stalled in the P site next to an empty A site (13). However, to explore +1-frameshifting tRNAs for genome expansion, we need to focus on dedicated tRNAs for translation of quadruplet codons, rather than canonical tRNAs, to avoid off-target effects.
Here, we focus on the SufB2 tRNA as a model, which was isolated from Salmonella (14) as a high-efficiency +1-frameshifting tRNA derived from the canonical ProL tRNAPro(GGG) by carrying an extra G37a nucleotide inserted next to m1G37 on the 3'-side of the anticodon GGG (15) (Supplementary Figure S1). In Salmonella that had an insertion of a single C to the Pro codon CCC, resulting in a CCC-C quadruplet codon, SufB2 exhibited a +1-frameshfiting frequency 1–5% above background (14,16), even in the presence of ProM and ProL that would compete for reading of the triplet CCC within the quadruplet codon. SufB2 is trackable model to study the mechanism of +1 frameshifting, because it has been expressed and confirmed in E. coli with a well-characterized +1-frameshifting activity not only in ensemble and single-molecule kinetic assays, but also in cell-based assays (10). However, SufB2 lacks a ribosome-bound structure and, other than kinetic evidence, it has no direct evidence for +1 frameshifting during translocation. Additionally, whether SufB2 can undergo +1 frameshifting within the P site during active protein synthesis is unknown. Further, given that +1 frameshifting is achieved only partially at each step (10,13), whether the shift can reach the full capacity in one elongation cycle remains an open question, for example by occurring first during translocation and second during occupancy within the P site.
In this study, we address the open questions on SufB2 using two different methods. One method uses the force-induced remnant magnetization spectroscopy (FIRMS) as a physical measurement to map the reading frame of a SufB2-bound ribosome on an mRNA, while the other uses kinetic assays to monitor the reading frame of SufB2 after it has translocated into the P site. The results show that SufB2 can twice explore +1 frameshifting in one elongation cycle, with the major fraction making the shift during translocation and the remaining fraction making the shift within the P site, so that it completely shifts to the +1-frame at the end of the cycle. Additionally, we show by both in vitro and in vivo experiments that the exploration of +1 frameshifting of SufB2 in the P site is modulated by the reading frame of the A-site tRNA during ongoing protein synthesis. The insight that has emerged from this work is important for understanding the limitation of genome expansion and for drawing a mechanistic parallel between +1 and –1 frameshifting. It demonstrates that changes of the reading frame occur in collaboration with the dynamics of the ribosome, indicating that the ribosome itself is a determinant of frameshifting in both the +1 and –1 direction.
MATERIALS AND METHODS
Translation reagents for in vitro experiments
Tightly coupled 70S ribosomes were purified by zonal centrifugation as described (17). Briefly, E. coli cells CAN/20-E12, derived from E. coli K12, deficient of RNases BN, II, D and I, and grown in LB (with 20% glucose (w/v)) to A560 = 0.5, were harvested by centrifugation at 30 000 × g at 4°C. Cells were resuspended in the ribosome buffer (20 mM HEPES–KOH, pH 7.5, 6 mM Mg(OAc)2, 30 mM NH4Cl, and 4 mM β-mercaptoethanol (β-Me), removed of debris, and grounded in a pre-chilled mortar and pestle with Alcoa A-305 (Serva, Heidelberg, Germany). The cell paste was removed of Alcoa A-305 by centrifugation at 15 000 × g for 20 min, and then at 30 000 × g for 1 h. Crude ribosomes were collected from the cleared supernatant by centrifugation at 110 000 × g for 17 h, rinsed in the ribosome buffer, incubated in the ribosome buffer by shaking for 1 h, and collected by centrifugation at 10 000 rpm for 5 min in an Eppendorf. The concentration of crude ribosomes is determined, typically at 300–400 A260 units per gram of cells and 500–1000 A260 units/ml. The 70S of the crude ribosomes were collected from a sucrose gradient (10–30%, 48 000 × g, 16 h, 4°C) in the ribosome buffer, pelleted by centrifugation (47 000 × g, 4°C, 24 h), resuspended in the ribosome buffer by gentle shaking for 1 h, 4°C, clarified by a low-speed centrifugation, and the concentration determined by A260. Over-expression clones of E. coli initiation factors IF1, IF2 and IF3, elongation factors EF-Tu and EF-G, each fused with a His-tag, were gifts of Dr Barry Cooperman and purified via binding to and elution from an Ni-NTA column as described (18).
Native-state E. coli tRNAfMet(CAU), tRNAArg(ICG) (I = inosine), and tRNAVal(*UAC) (*U = cmo5U capable of pairing with all four nucleotides (19)), were over-expressed in E. coli and affinity-purified from total tRNA using biotinylated oligonucleotides attached to streptavidin-sepharose (20). Unmodified transcripts of SufB2 and ProL were synthesized by in vitro transcription and purified by denaturing 12% PAGE/7M urea gels. Each tRNA was charged using the respective cognate aminoacyl-tRNA synthetase (aaRS) and stored in 25 mM NaOAc (pH 5.0) at –70°C. Initiator fMet-tRNAfMet was formylated by including formyl methionyl transferase and 10-formyl-tetrahydrofolate in the charging reaction (21).
Formation of ribosome complexes for FIRMS analysis
PRE and POST complexes were prepared as described (22): four for SufB2 (fMP-SufB2-PRE, fMP-SufB2-POST, fMPV-SufB2-POST and fMPR-SufB2-POST) and two for ProL (fMP-ProL-PRE and fMP-ProL-POST). Reaction components were prepared in 1X TAM buffer (20 mM Tris–HCl, pH 7.5, 10 mM Mg(OAc)2, 30 mM NH4Cl, 70 mM KCl, 0.5 mM EDTA, 7 mM β-Me and 0.05% Tween 20). The molar ratio of Mg2+ to EDTA was maintained at 20:1 throughout the experiments to stabilize ribosome–mRNA–DNA probe complexes. The ribosome 70S initiation complex (70SIC) was prepared by mixing 1 μM 70S ribosome, 1.5 μM each IF1, IF2, and IF3, 2 μM mRNA, 4 μM charged fMet-tRNAfMet, and 4 mM GTP. The factor mixture contained 6 μM EF-Tu, 4 mM GTP, 4 mM phosphoenol pyruvate (PEP), 0.02 mg/ml pyruvate kinase, and 3 μM EF-G for POST complexes or no EF-G for PRE complexes. The mixture of aa-tRNAs contained 100 mM Tris–HCl, pH 7.8, 20 mM Mg(OAc)2, 1 mM EDTA, 4 mM ATP, 0.1 mg/ml of a mixture of all aaRSs, one A260 units/ml of requisite tRNAs and 0.25 mM of the corresponding amino acids to form the respective TCs. The fMPV- and fMPR-POST were made in the presence of tRNAs charged with Pro, Val and Arg. The ribosome, factors and the mixture of aa-tRNAs were incubated separately at 37°C for 25 min, after which they were mixed in a 1:2:2 ratio and incubated 2 min at 37°C to form PRE complexes, or 30 min at 37°C to form POST complexes. The resulting ribosome complexes were purified by a 1.1 M sucrose cushion to remove translation factors and free mRNA.
Probing schemes and sample preparation for FIRMS analysis
The coding mRNA was 5′-biotin-GGC AAC UGU UAA UUA AAU UAA AUU AAA AAG GAA AUA AAA AUG CCC CGU AAG UAC GUA AAU CUA CUG CUG AAC UC-3′, where the initiation codon AUG is in bold face. The oligonucleotide used to probe fMP-PRE and fMP-POST complexes, termed P16c, was 5′-biotin-teg-CAA GTC CAG TAG ATT TAC GTA C-3′, where teg denotes an 18-atom spacer. The oligonucleotide used to probe fMPV and fMPR complexes, termed P16, was 5′-biotin-teg-CT CAA CAG CAG TAG ATT TAC G-3′. Both probes were designed to form duplexes of 12–16 bp with the mRNA downstream of the mRNA entry into the ribosome. These duplexes were in the resolvable range of FIRMS. The oligonucleotides were purchased from IDT and used without further purification.
The sample well with dimensions of 4 × 3 × 2 mm3 (L × W × D) was coated with biotin on the bottom glass surface. A streptavidin solution (0.30 mg/ml) of 20 μl was loaded onto the sample well and incubated for 1 h. The sample well was then rinsed with 1× TAM buffer three times to remove excess streptavidin. A 20 μl ribosome complex (0.1 μM) was immobilized on the surface via the 5′-biotin on the mRNA by incubation for 1 h. After rinsing the surface, 20 μl of a biotinylated oligonucleotide probe (1 μM) was added to hybridize overnight with the accessible region of the mRNA. Streptavidin-coated magnetic beads (0.7 mg/ml) (M280, Invitrogen) were then introduced to the sample well and incubated for 2 h. After extensive washes, the sample was magnetized for 2 min using a permanent magnet (∼0.5 T), where T is tesla, a measurement unit for the magnetic flux density. All workups and analyses were carried out in 1× TAM buffer at room temperature.
FIRMS measurements
FIRMS has been used to identify accessible mRNA nucleotides downstream from the entry of the mRNA into a ribosome complex, with single nucleotide resolution of multiple reading frames (22,23). The FIRMS technique measures the magnetic signal of samples after applying centrifugal force of increasing magnitude. When the force is below that required to dissociate an mRNA-probe duplex, a strong magnetic signal is obtained because the immobilized magnetic beads are attached to the duplex. When the force exceeds that required to dissociate the duplex, a decrease in magnetic signal is observed because the dissociated beads are subsequently removed from the sample well. From the dissociation forces, the number of base pairs in the duplex is obtained which in turn defines the location of the ribosome and its reading frame. Magnetic detection was conducted using an atomic magnetometer with ∼2–3 pT sensitivity. The force was provided by a centrifuge (Eppendorf 5417R) operated at varying speeds. The force values were calculated according to F = mω2r, in which m is the buoyant mass of M280 magnetic beads (4.6 × 10–15 kg), ω is the centrifugal speed, and r is the distance of the magnetic beads from the rotor axis (8 cm for 5417R). FIRMS profiles were obtained by normalizing the overall magnetic signal (B0) as 100% and by plotting the decrease of the relative magnetic signal (B/B0) versus the external force. The typical force resolution was 3–4 pN. Each profile reported in this study was repeated at least three times to assure reproducibility. Standard deviation was obtained by sending a calibration signal of 100 pT to the atomic magnetometer prior to sample measurements to allow calibration of the error. The percentage error was obtained by this error divided by the amplitude of the sample magnetic signal before it was dissociated under force. The high sensitivity of the atomic magnetometer throughout the entire trace of each experiment results in a constant standard deviation, which varies from experiment to experiment. Due to the robustness of our isolation of the ribosome, FIRMS measurements have been highly reproducible, reporting the same products and yields of frameshifting that were performed four years apart (22,24). Thus, all data in this work are reported as technical replicates.
Formation and analysis of di- and tri-peptides
Peptide synthesis was carried out as described (10) in 50 mM Tris–HCl, pH 7.5, 70 mM NH4Cl, 30 mM KCl, 3.5 mM MgCl2, 1 mM dithiothreitol (DTT) and 0.5 mM spermidine at 20°C (21,25). TCs were formed by incubating EF-Tu with GTP for 15 min at 37°C, followed by addition of one or more aa-tRNAs and incubation for 15 min in an ice bath. The 70SIC was prepared by incubating a 70 ribosome with [35S]-fMet-tRNAfMet, IF1, IF2, and IF3, EF-G and GTP, in the presence of mRNA for 25 min at 37°C. The mRNA was prepared by in vitro transcription with the sequence 5′-GGG AAG GAG GUA AAA AUG CCC CGU UCU AAG (CAC)7–3′, where the SD sequence is underlined, the initiation codon in bold face, followed by a CCC-C codon motif. The 70SIC and one or more TCs were mixed in a 1:1 molar ratio to initiate peptide bond formation at the final concentrations (1×) of 70S ribosome (0.4 μM), each initiation factor (0.5 μM), [35S]-fMet-tRNAfMet (0.25 μM), mRNA (0.5 μM), each aa-tRNA (0.5 μM), EF-Tu (0.8 μM per tRNA), EF-G (2 μM) and GTP (1 mM). Kinetics of tripeptide formation were monitored on the bench or in a Kintek RQF-3 chemical quench apparatus in triplicate. Reaction aliquots were quenched with KOH and peptide distribution determined by electrophoretic thin layer chromatography.
Cell-based assays of +1 frameshifting
The proL strain was the E. coli wt JM109 strain, while the sufB2 strain was generated by replacing the proL locus with sufB2 via P1 transduction from a previously constructed sufB2 strain in MG1655 (10). The kanamycin marker was removed by FLP recombination using pCP20. The isogenic proL and sufB2 strains were each transformed with two plasmids, one for expression of a lacZ reporter and the other for an E. coli tRNA gene. The lacZ reporter was expressed from the pZS2R vector (a gift of Roy Kishony) and carried insertion of the 0-frame CCC or the +1-frame CCC-C at the second codon position. Each reporter was previously created (13) in the pKK223-3 plasmid and was introduced into pZS2R at the BspHI site together with the tac promoter and the rrnB terminator. The plasmid for over-expression of an E. coli tRNA gene was derived from pKK223-3, where each gene was cloned between the EcoRI and PstI sites and over-expressed upon induction for 4–6 h at 37°C to allow synthesis of natural post-transcriptional modifications. Biochemical assays have confirmed the presence of selected post-transcriptional modifications in tRNAs that have been over-expressed in this cell model, including m1G37 (10), cmo5U34 (26), dihydrouridine D17 (27), and lysidine L34 (unpublished). The confirmation of the expected post-transcriptional modifications at positions 34 and 37 is important, both of which are determinants of the reading-frame. An overnight culture of the proL and sufB2 strains, harboring both a lacZ reporter plasmid and a tRNA plasmid, was inoculated into fresh LB at a 1:100 dilution and grown at 37°C for 4 h in the presence of 0.4 mM IPTG to induce co-expression of both the reporter and the tRNA gene. The control culture had cells harboring a lacZ reporter plasmid and the empty pKK223-3 plasmid and were induced with 0.4 mM IPTG at 37°C for 4 h. Cells were harvested and an aliquot of the cell lysate was assayed for the frequency of +1 frameshifting by measuring the lacZ activity in cells expressing the CCC-C reporter plasmid relative to cells expressing the CCC control plasmid (13). A separate aliquot of the cell lysate was quantified for tRNA expression in a label-free aminoacylation assay (28), in which total tRNA from cell lysates over-expressing hisT or thrV was aminoacylated by the respective E. coli HisRS and ThrRS, biotinylated with sulfo-NHS-biotin, ethanol precipitated twice, and bound to streptavidin. Charged aa-tRNA was separated from uncharged tRNA by a denaturing 12% PAGE/7 M urea gel and quantified by SYBR Gold. A control lane of the cell lysate without aminoacylation was run in parallel to provide background for subtraction.
RESULTS
SufB2 occupies the 0-frame in the A site in FIRMS analysis
While we have used kinetic assays to investigate +1 frameshifting of SufB2 (10), here we applied FIRMS (22,23,29) as a physical method to map the reading frame of SufB2 on a translating mRNA. In FIRMS analysis, the 5′-biotinylated mRNA is immobilized to a streptavidin-coated surface (Figure 1A). The sequence downstream from the mRNA entry site into the translating ribosome is mapped for the accessibility to base-pairing with a complementary DNA probe. The base-pairing accessibility of the sequence determines the ribosome reading frame (Figure 1B). Measurements of the number of base pairs are based on the magnetic dipoles of all duplexes, which are initially aligned with the external magnetic field to produce an overall magnetization (measured as B0). With an external force that dissociates non-stable duplexes, the remaining magnetization (measured as B) represents stable duplexes while the dissociated probes have random magnetic dipoles and make no contribution to the magnetization (Figure 1C). The force profile of magnetization is shown from the maximum magnetic signal (B/B0 = 100%), where all beads are aligned, to the minimum magnetic signal (B/B0 = 0%), where all beads are dissociated (Figure 1D). A single transition from 100% to 0% indicates a homogeneous state of the ribosome complex. The force that causes the transition corresponds to the probe hybridization to the mRNA entry point into the ribosome, from which 11 nucleotides upstream on the mRNA defines the first nucleotide of the P site (30,31). In our design of FIRMS experiments, the 0-frame placement of the AUG start codon in the P site indicates formation of the 0-frame complex, whereas a +1-frame placement indicates formation of the +1-frame complex due to +1 frameshifting at the adjacent CCC-C codon motif. The fraction of each complex is calculated as the fractional decrease of B/B0 from the maximum signal. We have applied centrifugation as an external force and measured the force and the number of base pairs in an mRNA-probe duplex up to the mRNA entry into the ribosome, showing a linear correlation with a reproducible slope of ∼13 pN between the force and the duplex length in different experimental settings (22,23,29). These FIRMS measurements have identified novel features of the ribosomes, including –1 and –2 frameshifting (22), occurrence of an mRNA looping intermediate during translocation (29), and even movement of the ribosome by half of a nucleotide in a PRE complex (32).
Figure 1.

A general scheme of FIRMS experiments. (A) The mRNA carrying a translating ribosome is immobilized to the surface of a streptavidin-coated sample well via its 5′-biotin (blue). The nucleotides downstream from the mRNA entry site into the ribosome are accessible to hybridization to a DNA probe, whereas the nucleotides upstream inside the ribosome are not accessible. (B) Addition of a DNA probe to the immobilized mRNA allows formation of an mRNA-DNA duplex. The DNA probe is conjugated with a magnetic bead, allowing for magnetization of the ribosome complex by a permanent magnet, and showing magnetic ordering of the bead by the black arrow pointing up. The magnetic signal of the ribosome complex is measured by an atomic magnetometer. Because the magnetic bead is immobilized to the surface via the mRNA-DNA duplex, its binding strength depends on the number of base pairs of the duplex. Prior to the force-induced dissociation of the duplex, the ribosome complex gives a high strong magnetic signal. (C) When the applied centrifugal force exceeds the dissociation force of the mRNA-DNA duplex, the DNA probe is dissociated from the duplex, leaving its magnetic bead randomly orientated due to Brownian motion as indicated by the white arrow in the bead. A complete dissociation of the duplex gives a zero magnetic signal. Because the dissociation force of the mRNA-DNA duplex is proportional to the number of base pairs, the position of mRNA is revealed by the dissociation force as indicated by a decrease of the magnetic signal. Single base-pair resolution is routinely achieved by FIRMS to distinguish different reading frames (22). (D) A representative force spectrum that illustrates the high magnetic signal prior to duplex dissociation and the low magnetic signal after dissociation. The value of the dissociation force reveals the ribosome position on the mRNA, hence the reading frame.
We began FIRMS analysis by determining whether SufB2 maintains the 0-frame anticodon-codon pairing in the A site as shown in our kinetic study (10). To allow the possibility of +1 frameshifting, we prepared SufB2 in the transcript form, lacking post-transcriptional modifications and eliminating m1G37 from maintaining the reading frame. An E. coli 70SIC was programmed with the mRNA template AUG-CCC-CGU-AAG to place the AUG start codon and an initiator fMet-tRNAfMet at the P site and the CCC-C quadruplet codon at the A site. We delivered a SufB2-TC or a ProL-TC to the A site to enable transfer of the formyl-methionyl (fM) group from the initiator tRNA to the prolyl group of each TC, generating an fMP-SufB2-PRE or an fMP-ProL-PRE. Each PRE complex was produced with a saturating concentration of the TC relative to the 70SIC and in long enough time (2 min) relative to the rate of fMP formation (1.2 and 0.9 s–1 for fMP-ProL and fMP-SufB2, respectively) (10). Each PRE complex was then immobilized to a streptavidin-coated well, incubated with a DNA probe, and the dissociation force of the mRNA-probe duplex was measured by an atomic magnetometer as a function of the centrifugal force.
The force profile of fMP-SufB2-PRE and of fMP-ProL-PRE is virtually identical, showing a single magnetic transition from 100% to 0% occurring at 80 ± 3 pN (Figure 2A, B). Based on our calibration of force versus the number of base pairs in the duplex (22,23,29), this single transition corresponds to the force required to dissociate a 16-base-pair duplex (Figure 2C). Because FIRMS analysis measured the ribosome in the end-point complex, the single transition indicates that each PRE was positioned at a specific frame on the mRNA after it reaches an equilibrium of exploration of different frames. This single transition required only one minimal force to dissociate the mRNA-probe duplex of 16 base pairs. From the 3′-end of the probe, we found that the position corresponding to 11 nucleotides upstream of the mRNA was localized to the first nucleotide of the AUG codon at the P site, indicating that each PRE was in the 0-frame. Had a fraction of either complex shifted to the +1-frame, FIRMS would have detected a force of 65 pN corresponding to dissociation of a 15-base-pair duplex (Figure 2D). Thus, consistent with our kinetic data (10), SufB2 reads the A-site codon in the 0-frame, without evidence of +1 frameshifting.
Figure 2.
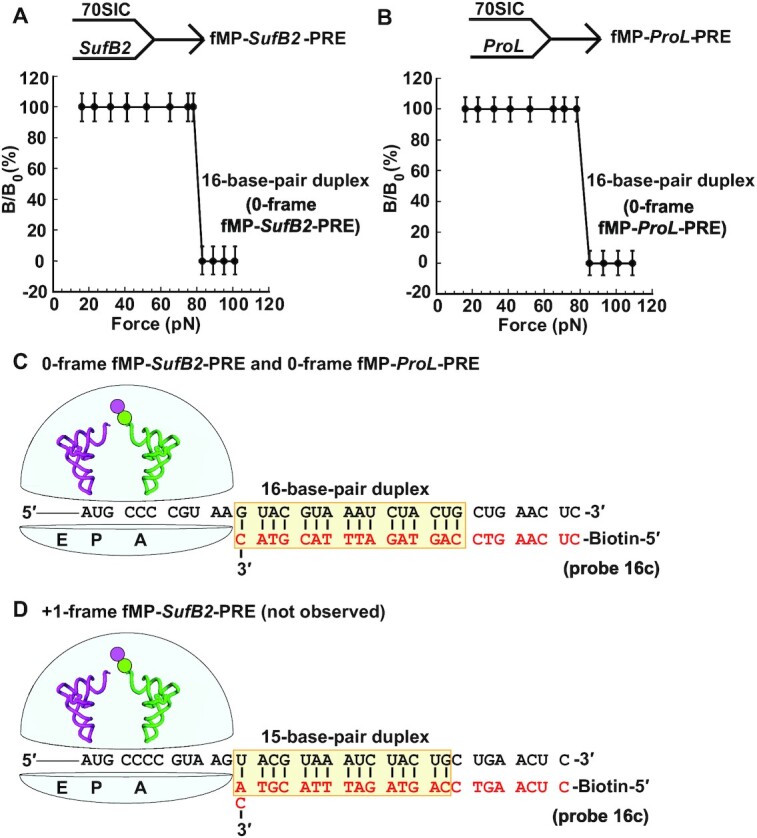
The force profile of fMP-PRE complexes in FIRMS analysis. (A) Formation of an fMP-SufB2-PRE complex by rapid delivery of SufB2-TC to a 70SIC, and (B) formation of an fMP-ProL-PRE complex by rapid delivery of ProL-TC to a 70SIC. A single transition is observed for both (A) and (B), corresponding to dissociation of a 16-base-pair duplex and indicating occupancy of 0-frame of the PRE complex. (C) A scheme representing the immobilized PRE complex in the 0-frame, prior to dissociation of the biotinylated DNA probe, illustrates the sequence of the 16-base-pair mRNA-probe duplex. (D) A scheme representing the immobilized PRE complex in the +1-frame, prior to dissociation of the biotinylated DNA probe, illustrates the sequence of the 15-base-pair mRNA-probe duplex. The duplex up to the entry of the mRNA channel into the ribosome is shaded, while the codons at the A and P site of each complex are marked. Errors were derived from three technical repeats.
SufB2 occupies the +1-frame in the P site in FIRMS analysis
We next used FIRMS to determine whether SufB2 occupies the +1-frame in the P site and the level of this occupancy, taking advantage of the ability of FIRMS to identify multiple frames at the same time (22). An fMP-SufB2-PRE or an fMP-ProL-PRE was formed as above and was converted to the POST by adding EF-G and GTP. The conversion was achieved in 30 min, based on the rate constant of each under an ensemble experimental condition (kfMP-PRE→fMP-POST of 0.09 and 0.04 s–1 for the SufB2 and ProL complex, respectively) (10). Each POST was then purified through a sucrose cushion, immobilized to a streptavidin bead, and hybridized to a DNA probe. The force profile of fMP-SufB-POST exhibited two transitions – the first occurring at the force of 21 ± 2 pN and corresponding to a 12-base-pair duplex, while the second occurring at the force of 32 ± 4 pN and corresponding to a 13-base-pair duplex (Figure 3A). In contrast, the force profile of fMP-ProL-POST exhibited only one transition, occurring at the force of 32 ± 4 pN and corresponding to a 13-base-pair duplex (Figure 3B). The 13-base-pair duplex positioned the ribosome in the 0-frame, placing the triplet CCC of the CCC-C codon in the P site, whereas the 12-base-pair duplex positioned the ribosome in the +1-frame, placing the quadruplet CCC-C codon in the P site (Figure 3C, D). These results demonstrate that fMP-SufB2-POST occupies both the 0-frame and the +1-frame, whereas fMP-ProL-POST occupies only the 0-frame, consistent with our kinetic data (10).
Figure 3.
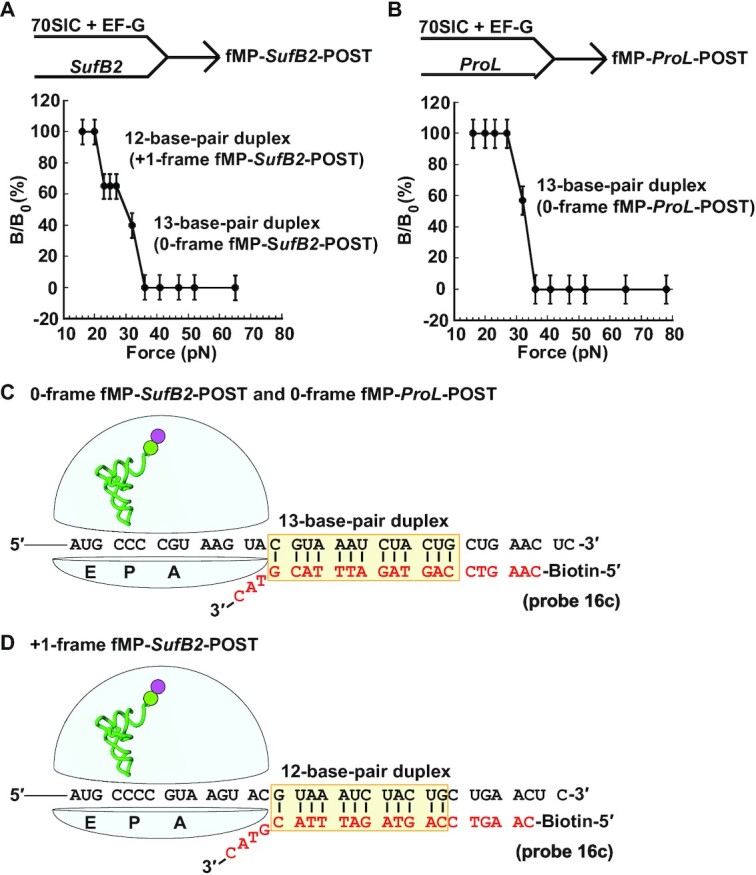
The force profile of dipeptidyl-POST complexes in FIRMS analysis. (A) Formation of the fMP-SufB2-POST complex by rapid delivery of SufB2-TC to a 70SIC in the presence of EF-G and GTP, showing two transitions corresponding to dissociation of a 12-base-pair and a 13-base-pair duplex and indicating occupancy of the POST in the +1-frame and 0-frame, respectively. (B) Formation of the fMP-ProL-POST complex by rapid delivery of ProL-TC to a 70SIC in the presence of EF-G and GTP, showing a single transition corresponding to dissociation of a 13-base-pair duplex and indicating occupancy of the POST in the 0-frame. (C) A scheme representing the immobilized POST complex in the 0-frame, prior to dissociation of the biotinylated DNA probe, illustrates the sequence of the 13-base-pair mRNA-probe duplex. (D) A scheme representing the immobilized POST complex in the +1-frame, prior to dissociation of the biotinylated DNA probe, illustrates the sequence of the 12-base-pair mRNA-probe duplex. The codons at the A, P and E site of each complex are marked, and errors were derived from three technical repeats.
We found that the fractional distribution of fMP-SufB2-POST in the +1 frame was 35%, which was lower than the distribution at 90% in our kinetic data (10). We tested whether the low occupancy in the +1-frame was due to the high Mg2+ concentration in FIRMS analysis (10 mM) relative to that in our kinetic assay (3.5 mM). The 10 mM Mg2+ in FIRMS was required to stabilize the mRNA-probe interaction during centrifugation in an magnetic field, whereas the 3.5 mM in kinetic assay was in the physiological range to provide high-fidelity discrimination against near-cognate tRNAs (33). Recent work has shown that higher Mg2+ concentrations inhibit translation by preventing the ribosome from conformational rearrangements necessary to complete an elongation cycle (34). Indeed, in our kinetic measurements of the fractional distribution of SufB2 in the +1-frame as a function of Mg2+ concentration, we found a linear decrease (Supplementary Figure S2). In this decrease, we found a distribution of 70% in the +1 frame at 10 mM Mg2+, which is still higher than the observed 35% in FIRMS analysis. Thus, although Mg2+ is a key determinant that contributes to the discrepancy of the +1-frameshifting efficiency between FIRMS and kinetic assays, it does not completely resolve the discrepancy.
Additional factors contribute to the discrepancy. A major one besides Mg2+ is the physical method of measurements, which is surface-based in FIRMS but solution-based in kinetic assays. The surface-based FIRMS measures the end point of each reaction, after a long incubation time that allows the ribosome to explore both the 0-frame and +1-frame to establish an equilibrium between the two. The long incubation time in FIRMS incurs other factors that contribute to the discrepancy. First, FIRMS analysis is performed with an overnight incubation of the mRNA-probe complex and involves several steps of hour-long incubations (e.g. ribosome binding to the coated well (1 h) and magnetic beads binding to the mRNA-probe complex (2 h)), followed by extensive washes, which could challenge the stability of the SufB2-ribosome complex and reduce the +1-frame fractional distribution. In contrast, kinetic assays capture the SufB2 reading frame instantaneously. Second, the fMP-SufB2 complex in the P site is likely prone to drop-off, due to the instability of the expanded ASL. Previous structural work of a related SufA6 tRNA with an expanded ASL demonstrates inefficient binding to the ribosome relative to a canonical tRNA (5). Additionally, our recent cryo-EM structures of the +1-frameshift-prone E. coli tRNAPro(UGG) demonstrates instability near the P site, involving the bulge-out of the first nucleotide of the quadruplet codon after the tRNA has shifted to the +1-frame (12). Third, the instability and propensity of drop-off of fMP-SufB2 from the P site is consistent with additional FIRMS data. For example, we showed that the tripeptidyl fMPV-tRNAVal has a higher occupancy of the +1-frame (50%) relative to the dipeptidyl fMP-SufB2 (35%) (Figure 4A below), indicating that fMPV-tRNAVal with a normal ASL is more stably accommodated in the P site by the 30S subunit relative to fMP-SufB2 with an expanded ASL. Combined, the high Mg2+ concentration required for FIRMS, and the associated long processing time, exacerbates the inherent instability of SufB2 in the P site, providing a reasonable explanation for the low occupancy in the +1-frame compared to kinetic measurements.
Figure 4.
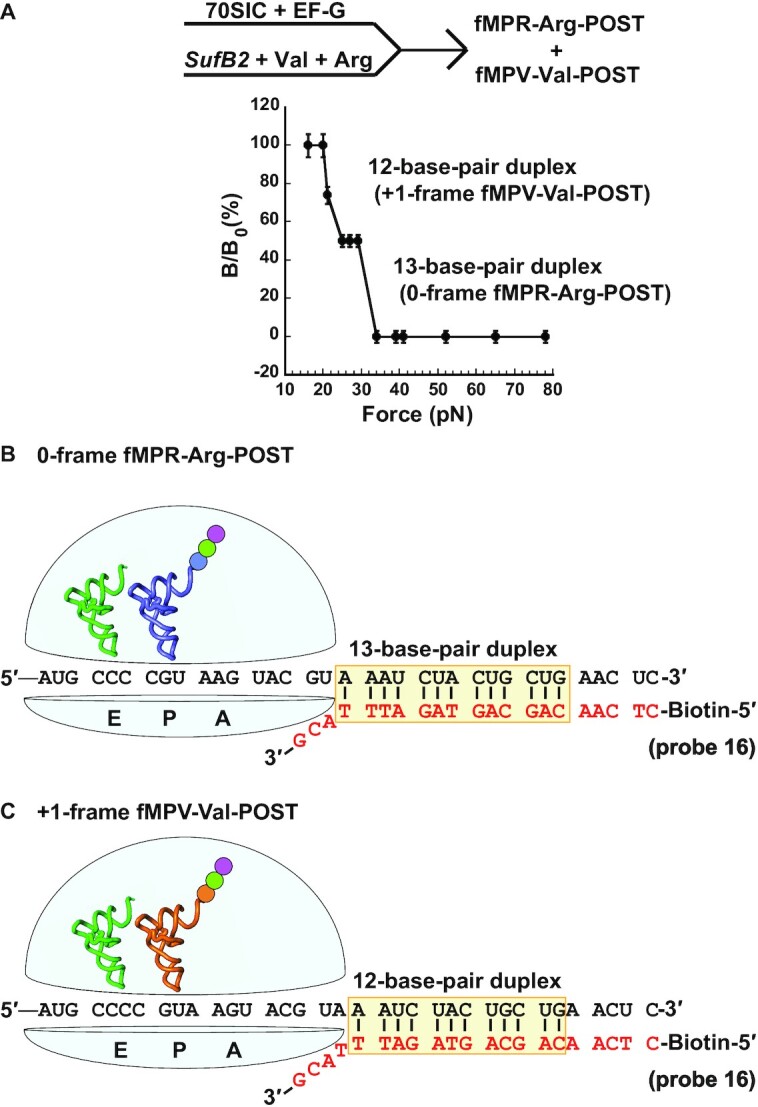
The force profile of a tripeptidyl-POST complex in FIRMS analysis. (A) Formation of fMPV- and fMPR-SufB2-POST complexes by rapid delivery of an equal molar mixture of SufB2-, Val- and Arg-TC to a 70SIC in the presence of EF-G and GTP, showing two transitions corresponding to dissociation of a 12-base-pair and a 13-base-pair duplex, and indicating occupancy of the +1-frame and 0-frame of the POST, respectively. (B) A scheme representing the immobilized POST complex in the 0-frame, prior to dissociation of the biotinylated DNA probe, illustrates the sequence of the 13-base-pair mRNA-probe duplex. (C) A scheme representing the immobilized POST complex in the +1-frame, prior to dissociation of the biotinylated DNA probe, illustrates the sequence of the 12-base-pair mRNA-probe duplex. The codons at the A, P and E site of each complex are marked, and errors were derived from three technical repeats.
SufB2 shifts to the +1-frame during translocation in FIRMS analysis
We tested whether the shift of fMP-SufB2-POST to the +1-frame in the P site occurred during translocation from the A site to the P site. To test this possibility, we performed FIRMS analysis by delivering a mixture of equal concentrations of SufB2-TC, Val-TC (tRNAVal(*UAC), where *U = cmo5U) and Arg-TC (tRNAArg(ICG), where I = inosine), to a 70SIC in the presence of EF-G and GTP to catalyze translocation. In this design based on the mRNA sequence AUG-CCC-CGU-A, the fractional distribution of SufB2 between the +1-frame and 0-frame that occurred during translocation would be immediately captured in the P site, because the simultaneous inclusion of all three TCs would leave no time for SufB2 to shift in the P site. As we showed recently (10), this capture would be represented by the fractional occupancy of Val-TC in the +1-frame and Arg-TC in the 0-frame of the A site, generating tripeptides fMPV and fMPR, respectively. We chose SufB2 in the transcript-state, but tRNAVal and tRNAArg in the native-state with the full complement of natural post-transcriptional modifications to prevent the latter two from unwanted frameshifting.
FIRMS analysis of SufB2 showed two transitions, one occurring at 21 ± 2 pN and corresponding to dissociation of a 12-base-pair duplex, while the other occurring at 32 ± 2 pN and corresponding to dissociation of a 13-base-pair duplex (Figure 4A). The 13-base-pair duplex placed the Arg CGU codon in the P site and the triplet CCC codon in the E site, indicating a 0-frame POST, whereas the 12-base-pair duplex placed the Val GUA codon in the P site and the CCC-C codon in the E site, indicating a +1-frame POST (Figure 4B, C). This result supports the notion that +1 frameshifting had occurred during translocation. Fractional calculation showed 50% occupancy in the +1-frame during translocation, lower than the observed 90% in our kinetic assay (10). We again attribute it to the high Mg2+ concentration required to stabilize the mRNA-DNA complex in FIRMS and to the technical differences from the kinetic assay.
SufB2 can shift to the +1-frame after translocation to the P site
We next asked if the fraction of SufB2 that remained in the 0-frame after translocation could attempt to shift in the P site. To quantify not only the yield but also the rate of +1 frameshifting, the latter of which cannot be achieved by FIRMS analysis, we used kinetic assays to monitor the shift in the P site. In our recent kinetic assay with simultaneous delivery of all three TCs to a 70SIC, while the assay isolated the fraction of SufB2 undergoing +1 frameshifting during translocation (10) (Figure 5A), it was not designed to determine whether the 0-frame fraction could shift in the P site. Separately, in our kinetic assay with simultaneous delivery of only Val-TC and Arg-TC to a POST complex of SufB2 (10) (Figure 5B), it was also not designed to address the question. In the latter, the POST complex of SufB2 is stalled over time to allow the distribution between the +1-frame and the 0-frame to reach an equilibrium. This equilibrium distribution is then monitored by co-addition of Val-TC and Arg-TC to the A site. The observation of no change of the fractional distribution of the two pairing frames in the P site, as reported in our previous assay (10), indicates that the two sub-populations of SufB2 in the P site after translocation have reached an equilibrium in the stalled POST complex. To capture +1 frameshifting of the 0-frame SufB2 within the P site, before the equilibrium, we performed the kinetic assay in a modified form, in which only Val- or Arg-TC was co-delivered with SufB2-TC to a 70SIC in the presence of EF-G and GTP. If the post-translocation fraction of SufB2 that remained in the 0-frame attempted +1 frameshifting in the P site, we would expect an increase of the overall yield of the fMPV tripeptide relative to that observed during translocation.
Figure 5.
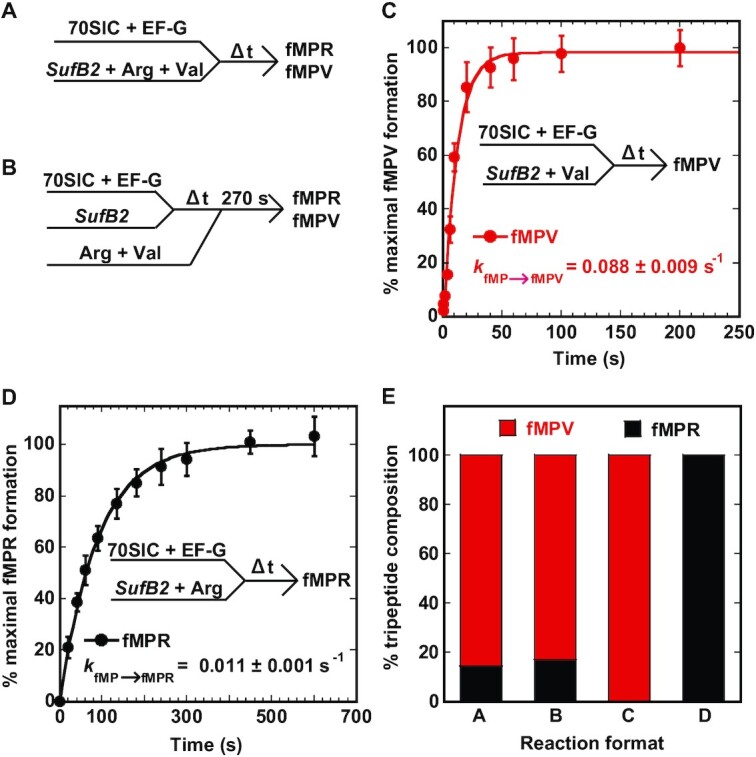
Kinetic analysis of +1 frameshifting of SufB2 by analysis of fMP to fMPV and fMPR. (A) A kinetic scheme of rapid delivery of an equal mixture of SufB2-, Val- and Arg-TC to a 70SIC as described previously (10). (B) A kinetic scheme of rapid delivery of an equal mixture of Val- and Arg-TC over time to a stalled fMP-SufB2-POST complex as described previously (10). (C) A kinetic scheme of rapid delivery of an equal mixture of SufB2- and Val-TC to a 70SIC. The fractional conversion of fMP to fMPV was monitored over time, showing the yield and rate of conversion. (D) A kinetic scheme of rapid delivery of an equal mixture of SufB2- and Arg-TC to a 70SIC. The fractional conversion of fMP to fMPR was monitored over time, showing the yield and rate of conversion. (E) Summary of the fractional conversion of fMP to fMPV and fMPR from reactions (A), (B), (C) and (D) in bar graphs. The bar graphs in (A) and (B) were taken from previously published results (10) that were recalculated as the conversion from fMP to fMPV and fMPR. Each value in panels (C) and (D) represents mean ± SD, n = 3, of technical replicates, whereas values in panel (E) are means.
We monitored the conversion from the dipeptidyl fMP to the tripeptidyl fMPV or fMPR and calculated the fractional distribution between the two tripeptides. With addition of only Val-TC and SufB2-TC, we showed that the entire population of fMP was converted to fMPV at the rate (kfMP→fMPV) of 0.088 ± 0.009 s–1. In contrast, with addition of only Arg-TC and SufB2-TC, the entire population of fMP was converted to fMPR at the rate (kfMP→fMPR) of 0.011 ± 0.001 s–1 (Figure 5C, D). These results, when compared with the distribution of SufB2 after translocation between fMPV at 90% and fMPR at 10% (Figure 5A), indicate redistribution of SufB2 in the P site (Figure 5E). The increase of fMPV from 90% to 100% upon delivery of only Val-TC to the A site indicates that the fraction that arrived in the 0-frame of the P site had shifted to the +1-frame. Conversely, the increase of fMPR from 10% to 100% upon delivery of only Arg-TC indicates that the fraction that arrived in the +1-frame of the P site had shifted to the 0-frame. Combined, these results demonstrate an unexpected reading-frame flexibility of SufB2 in the P site, showing that the sub-population that occupies the 0-frame after translocation can shift to the +1-frame when the +1-frame Val-TC is in the A site, whereas the sub-population that occupies the +1-frame after translocation can shift back to the 0-frame when the 0-frame Arg-TC is in the A site. To account for some fMP that was not converted to tripeptides in these experiments, we showed that 78% of fMP was converted to fMPV with addition of only Val-TC and that 71% was converted to fMPR with addition of only Arg-TC (Supplementary Figure S3), indicating that approximately the same amount of fMP was active in synthesis of either tripeptide.
The reading frame of the A-site tRNA modulates +1 frameshifting of SufB2 in vivo
The results above demonstrate that the reading frame of the A-site tRNA can modulate levels of +1 frameshifting, suggesting a potential strategy to improve the yield of genome recoding at the P site. Notably, +1 frameshifting that occurs during translocation is a uni-molecular reaction that is unlikely affected by the reading frame of the A-site tRNA. We therefore tested if +1 frameshifting in the P site can be modulated by the reading frame of the A-site tRNA in vivo during active protein synthesis. We used a previously developed cell-based reporter assay (13), in which a CCC-C codon motif was inserted into the second codon position of the reporter lacZ gene. In this assay, a +1-frameshifting event at the motif is necessary to maintain the natural Thr ACC codon at the third codon position and enable full-length synthesis of β-galactosidase (β-Gal), whereas lack of +1 frameshifting would position the His CAC codon at the third position and lead to premature termination of protein synthesis. The efficiency of +1 frameshifting was calculated as the ratio of β-Gal expressed in cells containing the CCC-C insertion relative to cells containing a 0-frame CCC insertion (Figure 6A).
Figure 6.

Modulation of +1 frameshifting in vivo by altering the abundance of the A-site tRNA. (A) The reporter lacZ gene harboring an inserted CCC-C codon at the second codon position. A +1 frameshifting at the CCC-C codon would generate a full-length β-Gal, whereas lack of +1 frameshifting would lead to premature termination of protein synthesis. (B) The lacZ reporter with the CCC-C insertion at the second codon position places the ACC codon at the +1-frame, requiring thrV tRNAThr for translation to synthesize the full-length β-Gal. In contrast, the insertion places the CAC codon at the 0-frame, requiring hisT tRNAHis for translation, which leads to premature termination of protein synthesis. (C) Quantification of thrV tRNAThr and hisT tRNAHis in cells by a label-free aminoacylation assay, showing separation of biotin-streptavidin (SA) conjugated aa-tRNA from uncharged tRNA by a 12% PAGE/7M urea gel. Subtraction of biotinylated tRNA (%) in samples without aminoacylation from that in samples with aminoacylation yielded the level of charged tRNA (%). The fold-change of each tRNA upon IPTG induction is shown at the bottom. (D) The frequency of +1 frameshifting in E. coli strains proL and sufB2 harboring an empty vector, or the vector over-expressing thrV tRNAThr or hisT tRNAHis, respectively. Each value represents mean ± SD, n = 3, of biological replicates.
We tested if increasing the cellular level of E. coli thrV tRNAThr cognate to the +1-frame ACC codon, relative to E. coli hisT tRNAHis cognate to the 0-frame CAC codon, would modulate the frequency of +1 frameshifting (Figure 6B). We used plasmid expression of each tRNA to increase its cellular level in an isogenic pair of E. coli strains that we created recently (10). In this pair, the proL strain expresses the ProL tRNA from the natural chromosomal locus, while the sufB2 strain expresses the SufB2 tRNA from the sufB2 gene that replaced the proL locus. We showed that, upon induction, thrV tRNAThr was over-expressed above background by 3.1-fold, and that hisT tRNAHis was over-expressed by 2.2-fold (Figure 6C).
In control experiment without over-expression of any tRNA, SufB2 showed an elevated +1 frameshifting frequency at 2.64% relative to ProL at 0.77% by more than 3-fold (Figure 6D, bars for cells with an empty vector), demonstrating its higher propensity of +1 frameshifting that is consistent with the previous report in Salmonella (16). While this elevated +1 frameshifting frequency is not as high as that measured in kinetic assays (Figure 5D), it is due to the presence of m1G37 in cell-based assay, which inhibits +1 frameshifting (10,13), and due to the cellular co-existence of SufB2 with competing ProM tRNAPro(UGG), the latter of which can read the CCC codon in the CCC-C codon motif. Upon over-expression of thrV tRNAThr, SufB2 displayed an even higher +1-frameshifting frequency relative to ProL (5.45 versus 0.87%) by more than 6-fold, supporting the notion that increasing the A-site tRNA in the +1-frame has elevated +1 frameshifting. Conversely, upon over-expression of hisT tRNAHis, SufB2 and ProL appeared to show a decrease in +1 frameshifting relative to the control (2.07 versus 2.64% for SufB2, and 0.47 versus 0.77% for ProL) (Figure 6D). Thus, consistent with the results of kinetic assays (Figure 5D), the results of the cell-based assays support the notion that the frequency of +1 frameshifting of SufB2 in the P site can be modulated by intracellular levels of the aa-tRNA that enters the A site in the +1-frame or in the 0-frame. This notion agrees with earlier genetic work, which demonstrates that starvation of intracellular aa-tRNAs alters the frequency of ribosome frameshifting (35,36).
DISCUSSION
Recent advance in genome expansion has seen success in engineering of a new bacterial genome with a minimal set of codons for all amino acids (37). However, the exploration of a +1-frameshifting tRNA for genome expansion has remained an option, making the question relevant of where the shift can occur in an elongation cycle of protein synthesis. Answering this question would provide a better understanding of +1 frameshifting and its limitations, and improve the yield of +1 frameshifting by targeting the specific steps where the shift occurs. Here we use SufB2 to show that it can shift to the +1-frame twice in one elongation cycle, with the major fraction of the shift occurring during translocation (90%) and the remaining fraction of the shift occurring during occupancy in the P site (10%) (Figure 7). The conceptual advance of this work to the field is on multiple fronts. First, while the observation of +1 frameshifting during translocation is consistent with that of our recent kinetic study (10), we provide here physical evidence of the shift by mapping the position of a translating ribosome on its mRNA template. This is important to clarify the reading frame of SufB2, given that it has no ribosome-bound structure to date. Second, while the observation of +1 frameshifting in the P site is consistent with that of our previous kinetic study of a stalled ribosome (13), we show here that the shift can occur during active protein synthesis, which is demonstrated in both kinetic (Figure 5D) and cell-based assays (Figure 6D). Third, while +1 frameshifting during translocation and during P-site occupancy was shown previously, but in separate kinetic models (10,13), we show here that it can occur consecutively and sequentially, first during translocation and second within the P site, leading to a complete shift of the tRNA into the +1-frame at the end of the elongation cycle (Figure 7).
Figure 7.

A model of twice exploration of SufB2 for +1 frameshifting in an elongation cycle. A PRE complex carrying fMP-SufB2 in the A site occupies the 0-frame. Upon initiation of translocation catalyzed by EF-G-GTP, SufB2 makes the first exploration of +1 frameshifting and arrives in the P site in a fractional distribution between the +1-frame (90%) and 0-frame (10%). While in the P site, in response to the entry of the +1-frame Val-TC in the A site, the fraction SufB2 that remains in the 0-frame makes a second exploration of +1 frameshifting to move to the +1-frame, synthesizing fMPV-Val-PRE in the A site. In contrast, the fraction that exists in the +1-frame in the P site readily reacts with Val-TC to synthesize fMPV-Val-PRE in the A site. This model demonstrates that SufB2 twice explores +1 frameshifting in one elongation cycle of protein synthesis, with the potential to reach 100% occupancy in the +1-frame.
While we use SufB2 as a model in this study, which has an expanded ASL, key features of the model are observed in our recent cryo-EM structures of the canonical E. coli tRNAPro(UGG) that is prone to +1 frameshifting (12). In these cryo-EM structures, the tRNA anticodon UGG is in a normal ASL and it pairs with the 0-frame codon during decoding at the A site, but it undergoes +1 frameshifting during translocation and pairs with the +1-frame codon as it moves into the P site (12). Notably, as the tRNA occupies the +1-frame near the P site, the first nucleotide of the quadruplet codon, within which the shift takes place, is bulged out from the mRNA into the narrow space between the P site and E site (12), indicating a disruption that would destabilize the codon-anticodon pairing interaction in the P site. This instability is expected to alter the fractional distribution of the ribosome between the +1-frame and the 0-frame in the thermodynamic equilibrium of the two, which is readily captured in FIRMS analysis of SufB2 (Figure 4A). Thus, the principles underlying the mechanism of +1 frameshifting are consistent between SufB2 and tRNAPro(UGG), validating that SufB2 is an informative and meaningful model to study, particularly due to its well-characterized activities both in kinetic assays and in cell-based assays that provide experimental tractability (10).
The twice exploration of +1 frameshifting is consistent with the dynamics of the ribosome structure in an elongation cycle. The lack of +1 frameshifting in the A site is attributed to the tight structure of the 30S subunit that uses movement of 16S rRNA nucleotides (G530, C1054, A1492 and A1493) to select for the 0-frame triplet anticodon-codon pairing scheme (38). In contrast, translocation consists of a series of large conformational rearrangements that facilitates the PRE complex to move into the POST complex (39). This series begins with thermally driven rotation of the 30S subunit relative to the 50S subunit of the A-site-bound PRE complex and closure of the L1 stalk to move the two tRNA acceptor stems in the A and P sites into the P and E sites of the 50S subunit (40). Upon EF-G binding to the A site, the PRE complex is transiently stabilized, while EF-G promotes swiveling of the 30S head domain and opening of the L1 stalk (41,42) to translocate the ASLs of the two tRNAs and the associated mRNA codons in the A and P sites into the P and E sites of the 30S subunit, respectively. While the ribosome dynamics in these late steps of translocation maintains anticodon-codon pairings in a canonical reaction, we showed by single-molecule analysis that the SufB2-bound ribosome is severely impeded from moving through these late steps (10), indicating a distinct ribosome conformation that induces +1 frameshifting. Subsequently, in the POST complex with a canonical tRNA in the P site, the 30S subunit uses only one nucleotide (C1400) to inspect the quality of the anticodon-codon pairing, but three nucleotides (A790, A1338, A1339) to stabilize the anticodon stem (43). In contrast, once a frameshift-prone tRNA moves into the P site, the 30S head domain undergoes a large swiveling-like rotation relative to the body domain to position the anticodon–codon pair in the +1-frame (44). This large movement of the 30S head domain is similar to that occurring during late steps of translocation and is observed for both a natural ASL lacking post-transcriptional modifications (44), and for an expanded ASL (9), indicating that it can provide a structural basis for the remaining fraction of SufB2 in the 0-frame to shift to the +1-frame. Thus, the two steps of the elongation cycle that allow SufB2 to explore +1 frameshifting are supported by the ribosome dynamics at each, suggesting that the ribosome itself is a determinant of +1 frameshifting.
While +1 frameshifting during translocation and during P-site occupancy both involve swiveling of the 30S head domain, the mechanism is different, based on kinetic comparison of SufB2 and a frameshift-prone ProL lacking m1G37 (13). While the ribosome dynamics during translocation accompanying SufB2 to move into the P site is significantly slower relative to ProL by 2–3 orders of magnitude (10), the trend is opposite within the P site. Compared to the rate of SufB2 shifting into the +1-frame in the P site (kobs = 0.088 s–1, Figure 5C), the rate of ProL in our previous study with a similar kinetic method is significantly slower (kobs = 0.001 s–1) by 1–2 orders of magnitude (13). This difference upholds even considering the rate of SufB2 shifting back to the 0-frame in the P site (kobs = 0.011 s–1, Figure 5D). Thus, while +1 frameshifting of SufB2 during translocation is slow relative to ProL, it is accelerated in the P site, indicating the distinct response of ribosome dynamics to the expansion of ASL in SufB2.
The two steps of ribosome exploration of +1 frameshifting have mechanistic parallel with –1 frameshifting (45). While +1 frameshifting tRNAs often contain mutations of the ASL (e.g. insertion of an extra nucleotide, and/or loss of post-transcriptional modifications), previously isolated –1 frameshifting tRNAs from genetic studies also contain mutations of the ASL. For example, one of the most efficient –1 frameshifting tRNAs isolated from genetic studies was derived from Salmonella tRNAGly(UCC), by substituting the wobble nucleotide mnm5U34 with C34 (46). Relative to the insertion mutation in ProL that confers +1 frameshifting of SufB2, this wobble mutation in tRNAGly(UCC) is different in nature, but it still involves alteration of the ASL that results in re-pairing in the –1-frame. The –1 frameshifting of the mutant tRNAGly(UCC) is thought to occur within the P site (3), suggesting a parallel with the +1 frameshifting of SufB2 in the P site. Importantly, previous studies (47–50) have shown that the level of +1 or –1 frameshifting at the P site, some of which are associated with ASL mutations, can be successfully improved by modulating the codon-anticodon interaction in the +1-frame or –1-frame of the A site. This notion is supported by our cell-based assay in vivo (Figure 6D). Separately, a different type of –1 frameshifting, which is genetically programmed, is induced by specific stimulatory signals outside of the ASL (51,52), such as a slippery mRNA sequence (53) and a downstream mRNA secondary structure that hinders the ribosome from moving forward and shifts the reading frame backward by one nucleotide. Programmed –1 frameshifting occurs in all domains of life but is most prevalent in viruses and mobile genetic elements. Notably, the physical hindrance that induces programmed –1 frameshifting occurs during the 30S head domain swiveling in the translocation reaction (54–58), similar to the involvement of the head domain swiveling during +1 frameshifting (10). Thus, exploration of frameshifting either in the +1 or –1 direction occurs in the same two steps of the elongation cycle, supporting the notion that the ribosome itself is a determinant of changes of the reading frame.
This study of SufB2 improves our understanding of +1 frameshifting at a quadruplet codon. While it highlights the potential of twice exploration of +1 frameshifting to reach the full capacity of the shift, it also identifies limitations of the approach for genome expansion. Specifically, the low efficiency of +1 frameshifting in cell-based assays with the native-state of SufB2 (Figure 6D) relative to the high efficiency in kinetic assays with the transcript form (Figure 5E) identifies the limitations. These limitations are due to the presence of the post-transcriptional m1G37 modification in SufB2, and due to the cellular competition of SufB2 with native isoacceptors that read a triplet of the quadruplet codon. Because almost all native tRNAs contain a purine at position 37 that is post-transcriptionally modified, it is unlikely that evolution of native tRNAs into a +1 frameshifting tRNA can readily escape the post-transcriptional purine modification at position 37. Instead, a de novo approach may be needed to generate designer +1 frameshifting tRNAs with an unmodified pyrimidine. Additionally, genetic engineering is needed to construct host bacterial strains that lack some or all of the non-essential isoacceptors to reduce their competition with a designer tRNA. Finally, between the two steps that permit +1 frameshifting, this work shows that the exploration of the shift in the P site is promising, which can be achieved by increasing cellular levels of tRNAs in the +1-frame for the A site. These considerations, when taken together, should improve the yield of +1 frameshifting at a quadruplet codon and also guide the development of strategies that improve –1 frameshifting for genome expansion.
DATA AVAILABILITY
All data supporting the findings of this study are presented within this article.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Jinwei Zhang for help with presentation of figures.
Contributor Information
Howard Gamper, Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA 19107, USA.
Yujia Mao, Department of Chemistry, University of Houston, Houston, TX 77204, USA.
Isao Masuda, Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA 19107, USA.
Henri McGuigan, Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA 19107, USA.
Gregor Blaha, Department of Biochemistry, University of California, Riverside, CA 92521, USA.
Yuhong Wang, Department of Biology and Biochemistry, University of Houston, Houston, TX 77204, USA.
Shoujun Xu, Department of Chemistry, University of Houston, Houston, TX 77204, USA.
Ya-Ming Hou, Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA 19107, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [R01 GM126210, GM134931, AI139202 to Y.M.H., R01 GM111452 to Y.W. and S.X.]; Welch Foundation [E-1721 to Y.W.]. Funding for open access charge: NIH [GM126210].
Conflict of interest statement. None declared.
REFERENCES
- 1.Wang K., Schmied W.H., Chin J.W.. Reprogramming the genetic code: from triplet to quadruplet codes. Angew. Chem. Int. Ed. Engl. 2012; 51:2288–2297. [DOI] [PubMed] [Google Scholar]
- 2.Kato Y.Translational control using an expanded genetic code. Int. J. Mol. Sci. 2019; 20:887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkins J.F., Bjork G.R.. A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignment. Microbiol Mol. Biol. Rev. 2009; 73:178–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lajoie M.J., Rovner A.J., Goodman D.B., Aerni H.R., Haimovich A.D., Kuznetsov G., Mercer J.A., Wang H.H., Carr P.A., Mosberg J.A.et al.. Genomically recoded organisms expand biological functions. Science. 2013; 342:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maehigashi T., Dunkle J.A., Miles S.J., Dunham C.M.. Structural insights into +1 frameshifting promoted by expanded or modification-deficient anticodon stem loops. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:12740–12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fagan C.E., Maehigashi T., Dunkle J.A., Miles S.J., Dunham C.M.. Structural insights into translational recoding by frameshift suppressor tRNASufJ. RNA. 2014; 20:1944–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunham C.M., Selmer M., Phelps S.S., Kelley A.C., Suzuki T., Joseph S., Ramakrishnan V.. Structures of tRNAs with an expanded anticodon loop in the decoding center of the 30S ribosomal subunit. RNA. 2007; 13:817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen H.A., Hoffer E.D., Dunham C.M.. Importance of a tRNA anticodon loop modification and a conserved, noncanonical anticodon stem pairing in tRNACGGProfor decoding. J. Biol. Chem. 2019; 294:5281–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong S., Sunita S., Maehigashi T., Hoffer E.D., Dunkle J.A., Dunham C.M.. Mechanism of tRNA-mediated +1 ribosomal frameshifting. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:11226–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamper H., Li H., Masuda I., Miklos Robkis D., Christian T., Conn A.B., Blaha G., Petersson E.J., Gonzalez R.L. Jr, Hou Y.M.. Insights into genome recoding from the mechanism of a classic +1-frameshifting tRNA. Nat. Commun. 2021; 12:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamper H.B., Masuda I., Frenkel-Morgenstern M., Hou Y.M.. The UGG isoacceptor of tRNAPro is naturally prone to frameshifts. Int. J. Mol. Sci. 2015; 16:14866–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demo G., Gamper H.B., Loveland A.B., Masuda I., Carbone C.E., Svidritskiy E., Hou Y.M., Korostelev A.A.. Structural basis for +1 ribosomal frameshifting during EF-G-catalyzed translocation. Nat. Commun. 2021; 12:4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamper H.B., Masuda I., Frenkel-Morgenstern M., Hou Y.M.. Maintenance of protein synthesis reading frame by EF-P and m(1)G37-tRNA. Nat. Commun. 2015; 6:7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riddle D.L., Roth J.R.. Suppressors of frameshift mutations in Salmonella typhimurium. J. Mol. Biol. 1970; 54:131–144. [DOI] [PubMed] [Google Scholar]
- 15.Sroga G.E., Nemoto F., Kuchino Y., Bjork G.R.. Insertion (sufB) in the anticodon loop or base substitution (sufC) in the anticodon stem of tRNA(Pro)2 from Salmonella typhimurium induces suppression of frameshift mutations. Nucleic Acids Res. 1992; 20:3463–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riddle D.L., Roth J.R.. Frameshift suppressors. 3. Effects of suppressor mutations on transfer RNA. J. Mol. Biol. 1972; 66:495–506. [DOI] [PubMed] [Google Scholar]
- 17.Blaha G., Stelzl U., Spahn C.M., Agrawal R.K., Frank J., Nierhaus K.H.. Preparation of functional ribosomal complexes and effect of buffer conditions on tRNA positions observed by cryoelectron microscopy. Methods Enzymol. 2000; 317:292–309. [DOI] [PubMed] [Google Scholar]
- 18.Pan D., Zhang C.M., Kirillov S., Hou Y.M., Cooperman B.S.. Perturbation of the tRNA tertiary core differentially affects specific steps of the elongation cycle. J. Biol. Chem. 2008; 283:18431–18440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allner O., Nilsson L.. Nucleotide modifications and tRNA anticodon-mRNA codon interactions on the ribosome. RNA. 2011; 17:2177–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yokogawa T., Kitamura Y., Nakamura D., Ohno S., Nishikawa K.. Optimization of the hybridization-based method for purification of thermostable tRNAs in the presence of tetraalkylammonium salts. Nucleic Acids Res. 2010; 38:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C., Gamper H., Liu H., Cooperman B.S., Hou Y.M.. Potential for interdependent development of tRNA determinants for aminoacylation and ribosome decoding. Nat. Commun. 2011; 2:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai T.W., Yang H., Yin H., Xu S., Wang Y.. High-efficiency ‘-1’ and ‘-2’ ribosomal frameshiftings revealed by force spectroscopy. ACS Chem. Biol. 2017; 12:1629–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao L., Li Y., Tsai T.W., Xu S., Wang Y.. Noninvasive measurement of the mechanical force generated by motor protein EF-G during ribosome translocation. Angew. Chem. Int. Ed. Engl. 2013; 52:14041–14044. [DOI] [PubMed] [Google Scholar]
- 24.Mao Y., Lin R., Xu S., Wang Y.. High-resolution DNA dual-rulers reveal a new intermediate state in ribosomal frameshifting. ChemBioChem. 2021; 22:1775–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C., Gamper H., Shtivelband S., Hauenstein S., Perona J.J., Hou Y.M.. Kinetic quality control of anticodon recognition by a eukaryotic aminoacyl-tRNA synthetase. J. Mol. Biol. 2007; 367:1063–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masuda I., Takase R., Matsubara R., Paulines M.J., Gamper H., Limbach P.A., Hou Y.M.. Selective terminal methylation of a tRNA wobble base. Nucleic Acids Res. 2018; 46:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betteridge T., Liu H., Gamper H., Kirillov S., Cooperman B.S., Hou Y.M.. Fluorescent labeling of tRNAs for dynamics experiments. RNA. 2007; 13:1594–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gamper H., Hou Y.M.. A label-free assay for aminoacylation of tRNA. Genes (Basel.). 2020; 11:1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin H., Xu S., Wang Y.. Dual DNA rulers reveal an ‘mRNA looping’ intermediate state during ribosome translocation. RNA Biol. 2018; 15:1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takyar S., Hickerson R.P., Noller H.F.. mRNA helicase activity of the ribosome. Cell. 2005; 120:49–58. [DOI] [PubMed] [Google Scholar]
- 31.Qu X., Wen J.D., Lancaster L., Noller H.F., Bustamante C., Tinoco I. Jr. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature. 2011; 475:118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia H., Wang Y., Xu S.. Super-resolution force spectroscopy reveals ribosomal motion at sub-nucleotide steps. Chem. Commun. (Camb). 2018; 54:5883–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gromadski K.B., Rodnina M.V.. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell. 2004; 13:191–200. [DOI] [PubMed] [Google Scholar]
- 34.Mohammad F., Green R., Buskirk A.R.. A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. Elife. 2019; 8:e42591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsley D., Gallant J.. On the directional specificity of ribosome frameshifting at a ‘hungry’ codon. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:5469–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Connor M.Imbalance of tRNA(Pro) isoacceptors induces +1 frameshifting at near-cognate codons. Nucleic Acids Res. 2002; 30:759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang K., de la Torre D., Robertson W.E., Chin J.W.. Programmed chromosome fission and fusion enable precise large-scale genome rearrangement and assembly. Science. 2019; 365:922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogle J.M., Brodersen D.E., Clemons W.M. Jr, Tarry M.J., Carter A.P., Ramakrishnan V.. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001; 292:897–902. [DOI] [PubMed] [Google Scholar]
- 39.Fei J., Bronson J.E., Hofman J.M., Srinivas R.L., Wiggins C.H., Gonzalez R.L. Jr. Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:15702–15707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noller H.F., Lancaster L., Zhou J., Mohan S.. The ribosome moves: RNA mechanics and translocation. Nat. Struct. Mol. Biol. 2017; 24:1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pulk A., Cate J.H.. Control of ribosomal subunit rotation by elongation factor G. Science. 2013; 340:1235970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ratje A.H., Loerke J., Mikolajka A., Brunner M., Hildebrand P.W., Starosta A.L., Donhofer A., Connell S.R., Fucini P., Mielke T.et al.. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature. 2010; 468:713–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selmer M., Dunham C.M., Murphy F.V.t., Weixlbaumer A., Petry S., Kelley A.C., Weir J.R., Ramakrishnan V.. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006; 313:1935–1942. [DOI] [PubMed] [Google Scholar]
- 44.Hoffer E.D., Hong S., Sunita S., Maehigashi T., Gonzalez R.L.J., Whitford P.C., Dunham C.M.. Structural insights into mRNA reading frame regulation by tRNA modification and slippery codon-anticodon pairing. Elife. 2020; 9:e51898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korniy N., Samatova E., Anokhina M.M., Peske F., Rodnina M.V.. Mechanisms and biomedical implications of -1 programmed ribosome frameshifting on viral and bacterial mRNAs. FEBS Lett. 2019; 593:1468–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Mahony D.J., Mims B.H., Thompson S., Murgola E.J., Atkins J.F.. Glycine tRNA mutants with normal anticodon loop size cause -1 frameshifting. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:7979–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jager G., Nilsson K., Bjork G.R.. The phenotype of many independently isolated +1 frameshift suppressor mutants supports a pivotal role of the P-site in reading frame maintenance. PLoS One. 2013; 8:e60246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pande S., Vimaladithan A., Zhao H., Farabaugh P.J.. Pulling the ribosome out of frame by +1 at a programmed frameshift site by cognate binding of aminoacyl-tRNA. Mol. Cell Biol. 1995; 15:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Caulfield T., Coban M., Tek A., Flores S.C.. Molecular dynamics simulations suggest a non-doublet decoding model of -1 frameshifting by tRNA(Ser3). Biomolecules. 2019; 9:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atkins J.F., Gesteland R.F., Reid B.R., Anderson C.W.. Normal tRNAs promote ribosomal frameshifting. Cell. 1979; 18:1119–1131. [DOI] [PubMed] [Google Scholar]
- 51.Atkins J.F., Loughran G., Bhatt P.R., Firth A.E., Baranov P.V.. Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016; 44:7007–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Advani V.M., Dinman J.D.. Reprogramming the genetic code: the emerging role of ribosomal frameshifting in regulating cellular gene expression. Bioessays. 2016; 38:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blinkowa A.L., Walker J.R.. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res. 1990; 18:1725–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caliskan N., Katunin V.I., Belardinelli R., Peske F., Rodnina M.V.. Programmed -1 frameshifting by kinetic partitioning during impeded translocation. Cell. 2014; 157:1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen J., Petrov A., Johansson M., Tsai A., O’Leary S.E., Puglisi J.D.. Dynamic pathways of -1 translational frameshifting. Nature. 2014; 512:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H.K., Liu F., Fei J., Bustamante C., Gonzalez R.L. Jr, Tinoco I. Jr. A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:5538–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Napthine S., Ling R., Finch L.K., Jones J.D., Bell S., Brierley I., Firth A.E.. Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun. 2017; 8:15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bock L.V., Caliskan N., Korniy N., Peske F., Rodnina M.V., Grubmuller H.. Thermodynamic control of -1 programmed ribosomal frameshifting. Nat. Commun. 2019; 10:4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are presented within this article.