


Abstract
AaRSs (aminoacyl-tRNA synthetases) group into two ten-member classes throughout evolution, with unique active site architectures defining each class. Most are monomers or homodimers but, for no apparent reason, many bacterial GlyRSs are heterotetramers consisting of two catalytic α-subunits and two tRNA-binding β-subunits. The heterotetrameric GlyRS from Escherichia coli (EcGlyRS) was historically tested whether its α- and β-polypeptides, which are encoded by a single mRNA with a gap of three in-frame codons, are replaceable by a single chain. Here, an unprecedented X-shaped structure of EcGlyRS shows wide separation of the abutting chain termini seen in the coding sequences, suggesting strong pressure to avoid a single polypeptide format. The structure of the five-domain β-subunit is unique across all aaRSs in current databases, and structural analyses suggest these domains play different functions on α-subunit binding, ATP coordination and tRNA recognition. Moreover, the X-shaped architecture of EcGlyRS largely fits with a model for how two classes of tRNA synthetases arose, according to whether enzymes from opposite classes can simultaneously co-dock onto separate faces of the same tRNA acceptor stem. While heterotetrameric GlyRS remains the last structurally uncharacterized member of aaRSs, our study contributes to a better understanding of this ancient and essential enzyme family.
INTRODUCTION
AaRSs establish the rules of the genetic code by covalently linking an amino acid to its cognate tRNA, and therefore play irreplaceable roles in protein synthesis (1,2). Corresponding to the twenty amino acids used in protein synthesis, cellular lives usually have a full set of twenty aaRSs. These aaRSs evolved from two different ancestral active site domains and are thus classified as class I or class II (3,4). Class I enzymes are either monomers or dimers, that are formed from a Rossmann fold that binds the tRNA acceptor stem from the minor groove side, whereas class II aaRSs are either dimers or tetramers with a core active site of antiparallel β strands that binds to the major groove side of the acceptor stem (5). It is believed that a full set of twenty aaRSs emerged before the tree of life diverged from the last common ancestor (LUCA) into three domains (6). After that, aaRSs underwent extensive horizontal gene transfer during evolution which led to the insertion of new sequences or domains (especially in eukaryotic aaRSs) (7,8), endowing the aaRSs with better efficiency, fidelity and regulation and, even more interestingly, abundant non-canonical functions beyond protein translation (9,10). Despite the significant diversity of protein sequences and domain organizations of aaRSs from different organisms, it is clear that, for a specific amino acid, the aaRSs from three kingdoms share a common ancestor and catalytic mechanism in most cases (8).
For no apparent reason, two types of glycyl-tRNA synthetase (GlyRS) exist in different organisms. Eukaryotes, archaea and some bacteria have a homodimeric GlyRS belonging to typical class II aaRSs (11), while other bacteria exclusively utilize a heterotetrameric GlyRS with two α-subunits and two β-subunits (termed (αβ)2 as explained later) to produce glycyl-tRNAGly (12,13). The α-subunits from (αβ)2Campylobacter jejuni GlyRS (CjGlyRS) and Aquifex aeolicus GlyRS (AaGlyRS) have been crystallized as α2 homodimers (14,15). Although the α-subunit of (αβ)2 GlyRS encodes the core catalytic domain with all three signature motifs of class II aaRSs (16), its larger β-subunit is not similar in sequence to α2 GlyRS or to other class II aaRSs (17). The α-subunit alone possesses no or extremely low activity even for amino acid activation, the first step of catalysis, indicating that both α- and β-subunits are indispensable for the aminoacylation of tRNAGly (18). A later study revealed that the N-terminal part of the β-subunit is necessary for adenylate synthesis, while the C-terminal part harbors a tRNA binding domain (19). The central part of the β-subunit was predicted to be a hydrolase domain (HD) that is frequently observed in metal-dependent phosphohydrolases, but it does not have the requisite histidine-aspartate catalytic doublet, which leaves its function unclear (17).
The α2 and (αβ)2 GlyRSs are so unrelated in sequence, domain organization and strategies for amino acid recognition that they ostensibly originated from different ancestors (20), which thus challenges the models for the evolution of the aaRS family (21). Supporting this point, the distribution of GlyRS types in different bacteria also does not correlate with the evolutionary emergence of these bacteria (22). From a practical point of view, its unique features provide a larger chemical space to seek bacteria-specific inhibitors that do not cross-react with human GlyRS and, consequently, lessen the problem of toxicities in the drive to find new antibiotics to combat emerging antibiotic resistances.
To date, the singular (αβ)2 GlyRS overall structure remains undetermined. Herein, we report a crystal structure of Escherichia coli GlyRS (EcGlyRS) with a resolution of 2.68 Å that adopts an unprecedented X-shaped architecture. The structure reveals five domains in the β-subunit. While some of the domains facilitate the formation of the intact active pocket by the α-subunit, some domains may contribute to tRNA binding by showing significant structural similarity to tRNA CCA-adding enzymes and a tRNA recognition domain in alanyl-tRNA synthetase (AlaRS). Moreover, the evolutionary strategy of this peculiar family of GlyRSs appears to select for a large separation of distinct parts of what normally would be a single polypeptide chain, which enables an X-shaped architecture as shown here. Despite this unprecedented structural format, the resulting enzyme appears to conform to a tRNA docking scheme that pairs two specific synthetases on one tRNA, thereby reinforcing an evolutionary model that provides a rationale for the two classes of synthetases and their subclasses.
MATERIALS AND METHODS
Protein expression and purification of EcGlyRS
The genes glyQ and glyS encoding the α- and β-subunits of EcGlyRS were amplified from the genomic DNA of E. coli K-12 by PCR. The full-length α-subunit (res. 1–303) was inserted into the pET20b vector (Novagen) using the NdeI and XhoI sites. For crystallization, the β-subunit fragment (res. 1–575) was subcloned into the pET29b vector (Novagen) with a C-terminal 6 × His tag using the NdeI and XhoI sites. BL21(DE3) E. coli cells (Novagen) carrying both plasmids were grown to OD600 = 0.6 in LB medium supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin. The overexpression of the β-subunit-truncated EcGlyRS (named EcGlyRS575) protein was induced by adding 0.1 mM IPTG and was shaken at 220 rpm at 25°C for 16 h. The cells were harvested by centrifugation at 4000 rpm at 4°C for 30 min. Cells were resuspended and sonicated with ice-cold lysis buffer (200 mM NaCl, 50 mM Tris pH 8.0, 20 mM imidazole). Cell lysates were centrifuged at 18 000 rpm for 30 min, and the supernatant was loaded onto a Ni-NTA column (Qiagen) pre-equilibrated with lysis buffer. The Ni-NTA column was washed with 20 column volumes of lysis buffer to remove impurities, and the EcGlyRS575 protein was eluted with 50 ml of elution buffer (200 mM NaCl, 50 mM Tris–HCl, pH 8.0, 200 mM imidazole). The protein in the elution fraction was concentrated to 15–20 mg/ml using a 50 kDa Ultra-15 centrifugal filter device (Millipore) and was then further purified by size-exclusion chromatography (Cytiva, HiLoad Superdex 200 pg) with SEC buffer (100 mM NaCl, 10 mM Tris–HCl, pH 8.0). The purified EcGlyRS575 protein was desalted and concentrated in a storage buffer (50 mM NaCl, 5 mM Tris–HCl, pH 8.0) and frozen at –80°C before use.
The selenomethionine-substituted EcGlyRS575 (SeMet-EcGlyRS575) protein was expressed with E. coli strain B834 (DE3) (Novagen) using M9 medium supplemented with selenomethione as previously described (23), and then purified the same as the native EcGlyRS575 protein.
The full-length β-subunit (res. 1–689) was also inserted into the pET29b vector (Novagen) the same as the β-subunit fragment (res. 1–575). All of the site-directed mutations of EcGlyRS-FL were constructed using the QuickChange Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer's instructions, while the plasmid for expressing the ΔB2 deletion protein (res. 66–124 was replaced by a -GSGS- linker) was generated through a homologous recombination method. The expression and purification of these EcGlyRS variants were the same as those of EcGlyRS575.
Crystallography
SeMet-EcGlyRS575 protein (30 mg/ml) was mixed with 2 mM adenylyl-imidodiphosphate (AMP-PNP, Sigma-Aldrich) and 5 mM glycine (Sigma-Aldrich) and incubated on ice for 30 min. Crystals were grown using the sitting-drop vapor-diffusion method, in which 1 μl of protein solution was mixed with an equal volume of reservoir solution and then equilibrated against 70 μl of reservoir solution at 18°C. Large crystals appeared after 3 days in the wells using the reservoir solution consisting of 0.4 M magnesium acetate, 0.1 M HEPES pH 6.8, 2% (v/v) PEG 3350, 2% (v/v) PEG 5000 MME, 2% (v/v) PEG 4000, 2% (v/v) PEG 2000 and 10% ethylene glycol. Single-wavelength anomalous diffraction (SAD) data were collected at 0.9792 Å using a single crystal at 100 K on beamline BL19U1 of the National Center for Protein Sciences Shanghai (NCPSS) and the Shanghai Synchrotron Radiation Facility (SSRF). The oscillation angle is 1° for each frame, and the whole data set contains 360 frames.
The diffraction data were processed and scaled with HKL3000 (24). The initial phases were calculated and the model was automatically built using the Crank2 pipeline in CCP4 (25). The structure model was further refined in Coot (26) and Refmac5 (27). The final structure model was refined to 2.68 Å with R/Rfree = 23.89%/25.29% and achieved good stereochemistry quality as assessed by the program MolProbity (28). The statistics of data collection and structure refinement are listed in Supplementary Table S1. The structure was analyzed in PyMOL (www.pymol.org), which was also used to create figures.
Measurement of glycine activation activity of EcGlyRS
Glycine activation by EcGlyRS and its variants was detected by employing a coupled-assay as previously described (29,30), with a few modifications. Briefly, in the active site of EcGlyRS, the reactions between an AMP-PNP and a glycine generate a molecule of glycyl-AMP that would be attacked by a pyrophosphate to form glycine and one ATP. Hexokinase consumes one ATP to phosphorylate glucose to glucose 6-phosphate, from which glucose 6-phosphate dehydrogenase then consumes one NADP+ to produce one glucose-6-phosphate and one NADPH. Thus, the formation of one glycyl-AMP will finally cause the production of one NADPH in this coupled continuous assay. The experiment was conducted at room temperature in a clear 96-well microplate (Corning). 90 μl of reaction buffer consisting of 50 mM HEPES pH 7.5, 10 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol, 5 mM AMP-PNP, 5 mM glycine, 10 mM d-glucose, 2.5 mM sodium pyrophosphate, 0.5 mM NADP+, 0.025 U yeast hexokinase and 0.025 U glucose 6-phosphate dehydrogenase was placed in the wells of the microplate. The reactions were started by adding 10 μl of EcGlyRS or its variants (the final concentrations of the enzymes were 200 nM) to each well. The production of NADPH in the coupled-assay was continuously recorded for the first 10 min by monitoring the absorbance at 340 nm using a FlexStation 3 multimode microplate reader (Molecular Devices), and the reactions without adding any EcGlyRS enzyme were used as blank controls. The slope of NADPH formation in the first 10 min (equal to the glycine activation rate) of wild-type EcGlyRS-FL was defined as 100%. The results are from three independent assays, and the error bars are standard deviations (SD).
RNA transcription
Escherichia coli tRNAGly (anticodon: GCC, tRNAdb ID: tdbD00000838) was produced by in vitro transcription using T7 polymerase. The DNA template was generated by PCR using Primer1 (5′-TAATACGACTCACTATAGCGGGAATAGCTCAGTTGGTAGAGCACGACCTTGCCAAG-3′) and Primer2 (5′-TGGAGCGGGAAACGAGACTCGAACTCGCGACCCCGACCTTGGCAAGGTCGTGCTC-3′). The underlined nucleotides overlap between two primers, and the nucleotides in bold are T7 promoters. The PCR product of Primer1 and Primer2 was further amplified by PCR using Primer3 (5′-TAATACGACTCACTATAGCGGGAATAGC-3′) and Primer4 (5′-TGGAGCGGGAAACGAGACTCG-3′), and the PCR product was directly used as the DNA template for in vitro T7 transcription without any further purification. The T7 transcription reaction contained 40 mM Tris–HCl pH 8.0, 1 mM spermidine, 5 mM DTT, 1% (v/v) Triton X-100, 20 mM MgCl2, 50 μg of DNA template, 0.4 mM ATP, 0.4 mM UTP, 0.4 mM GTP and 0.4 mM CTP. 1 ml of the reaction was incubated at 37°C for 3–4 h, and then the transcripts were purified by using denaturing 12% polyacrylamide gel electrophoresis. The concentrations of the purified tRNA products were determined through the UV absorbance at a wavelength of 260 nm.
ATP consumption assay
The ATP consumption assay was employed to evaluate the aminoacylation activity (including both steps of glycine activation and tRNA charging) of EcGlyRS and its variants. The 100 μl reactions consisted of 50 nM EcGlyRS or its variants, 200 μM ATP, 500 μM glycine, 5 mg/ml in vitro transcribed E. coli tRNAGly (anticodon: GCC), 30 mM HEPES pH 7.5, 150 mM NaCl, 30 mM KCl, 40 mM MgCl2, 0.1% BSA and 1 mM DTT. Reactions were incubated at room temperature, a 10 μl aliquots of the reactions were moved at different time points (2, 5, 10, 20, 40 min) to a 384-well microplate with 10 μl of Kinase-Glo™ Reagent (Promega) to stop the reaction, and after 10 min incubation, the luminescence (L) was read on a FlexStation 3 multimode microplate reader (Molecular Devices). The concentrations of remaining ATP at each time point were calculated according to the standard curve generated by different concentrations (10, 25, 50, 100, 200 and 400 μM) of ATP (Sigma-Aldrich). Finally, ATP consumption was obtained from the difference between the molar quantities of remaining ATP in each well and initial ATP (2000 pmol). Each reaction group was repeated three times, and the results were expressed as the mean ± SD (n = 3). Statistical analyses were performed with GraphPad Prism 7.0 software, and a one phase association equation was used for the curve fitting of the ATP consumption assay.
Small-angle X-ray scattering (SAXS) analysis
The SAXS data were collected on beamline BL19U2 of the National Center for Protein Sciences Shanghai (NCPSS) and the Shanghai Synchrotron Radiation Facility (SSRF). X-ray scattering was performed at a wavelength of λ = 1.033 nm (12.0 keV). The distance between the sample and detector was 2.626 m, leading the momentum transfer q to be recorded in the range of 0.076–3.74 nm–1. The scattering profiles were measured using 50 μl of protein solution at 5 mg/ml for EcGlyRS-FL and 8 mg/ml for EcGlyRS575. Both proteins were pre-equilibrated in a SAXS buffer (100 mM NaCl, 50 mM Tris–HCl pH 8.0) and loaded into a fully automated sample system on the BL19U2 beamline. A set of 15 two-dimensional images was recorded for each buffer or sample solution measurement with an exposure time of 1 s per image and was then reduced on-site to one-dimensional scattering profiles using the BioXTAS RAW software package (31).
The SAXS data were analyzed using the ATSAS suite (32). The radius of gyration (Rg) and the scattering intensity I(0) were calculated using PRIMUS (33) according to the Guinier approximation at low q values in the range of qRg < 1.3. The P(r) values and the maximum dimension of the protein (Dmax) were also estimated from the scattering profile using the indirect Fourier transform method implemented in the GNOM program (34). The twenty independent ab initio shape envelopes for each protein were calculated using DAMMIF (35) and represented by an ensemble of densely packed beads. The independent models were averaged and superimposed by DAMAVER (36) to generate the final model. The theoretical scattering intensities of the crystal structure or the assembly model were calculated and fitted to the experimental scattering intensity using CRYSOL (37).
Isothermal titration calorimetry (ITC) assay
To detect the binding of ATP to EcGlyRS-FL and the α-subunit, EcGlyRS-FL (20 μM) or the α-subunit (100 μM) was placed in the sample cell with buffer (20 mM HEPES 7.0, 100 mM NaCl, 2 mM MgCl2 and 20% glycerol), 200 mM ATP was loased in the syringe, and then the titration assays were performed at 28°C, with 5 μl for the first injection and 10 μl for the next 19 injections. The interval between two injections was 150 s. The disassociation constants (Kd) were determined by fitting the calorimetric data to a one-site binding model using Origin for ITC software (version 7).
Size-exclusion chromatographic (SEC) assay
The binding between tRNAGly and two EcGlyRS truncations (EcGlyRSΔB2 and EcGlyRS575) was analyzed by size-exclusion chromatography. in vitro transcribed E. coli tRNAGly (anticodon: GCC, 80 μM) was co-incubated with 20 μM protein at room temperature for 30 min in the solution containing 20 mM Tris–HCl pH 7.0, 100 mM NaCl, 2 mM MgCl2, 5 mM glycine, 5 mM AMP and 5% glycerol. The sample was loaded onto a Superdex 200 Increase 10/300 GL column (Cytiva) and the elution profile was monitored at 260 and 280 nm. As a control, 20 μM EcGlyRS-FL was also co-incubated with 80 μM tRNAGly and the above steps were performed under the same conditions.
RESULTS
Overview of the heterotetrameric structure of EcGlyRS
Organized as a dimer of two protomers
EcGlyRS (EC 6.1.1.14) is operationally a large protein machine of >2000 residues (with molecular weights of 34.7 kDa for the α-subunit and 76.8 kDa for the β-subunit; Figure 1A). Consistent with earlier work (19), we found that a construct that removed 114 amino acids from the C-terminus of the β-subunit was well-expressed and formed an active (αβ)2 heterotetramer. The 114 aa C-terminal deletion construct was named EcGlyRS575 (Figure 1A). In solution, EcGlyRS575 was shown to form a tight heterotetramer (Supplementary Figure S1) and had a SAXS envelope similar to that of full-length EcGlyRS (EcGlyRS-FL) (Supplementary Figure S2). Through reiterative trial and error we obtained high-quality crystals of EcGlyRS575, and a structural model was refined to 2.68 Å (with an R/Rfree factor of 23.9%/25.3%, Supplementary Table S1).
Figure 1.

The overall structure of heterotetrameric EcGlyRS. (A) The domain organization of EcGlyRS. To facilitate crystallization, the C-terminal part of the anticodon binding domain was truncated, resulting in the construct named EcGlyRS575. (B) Cartoon representation of the overall structure of (αβ)2 heterotetrameric EcGlyRS575. The protomer consisting of chains B and D are colored as shown in Figure. 1A, while the other protomer (chains A and C) are colored gray. The glycine, Mg2+, and ATP analog (AMP-PNP) are shown as sphere models in the two aminoacylation pockets. Two protomers are organized through a noncrystallographic 2-fold axis to form the functional heterotetramer. (C) The topology diagram of the α- and β-subunits is colored as depicted in Figure 1A. Arrows represent β-strands, while α- and 310-helices are shown as cylinders. Class II signature motifs 1, 2 and 3 in the α-subunit are highlighted in salmon, yellow and blue, respectively, and the residues Arg326 and Asp329 in the β-subunit B3 domain contributing to ATP binding are labeled.
The crystallographic asymmetric unit contains one heterotetramer, and bound glycine and AMP-PNP, a non-hydrolyzable analog of ATP, are observed in both aminoacylation pockets (Figure 1B). EcGlyRS575 presents an unprecedented X-shape overall structure, with the two α-subunits forming a globular dimer at the center and two β-subunits flanking to each side of the α-subunit dimer (Figure 1B). A protomer is formed by one α-subunit plus one β-subunit, and EcGlyRS is a dimer of two protomers that are organized approximately symmetrically around a 2-fold axis (Figure 1B). The subunit organization formula of heterotetrameric GlyRSs is more accurately expressed as (αβ)2 than the previously used α2β2. As analyzed by PISA (38), the interface between two α-subunits buries an area of 2447.6 Å2, and the interfaces between α- and β-subunits of α-β protomers are 2087.3 and 2107.8 Å2, respectively. Remarkably, no interaction between the two β-subunits, or between the α- and β-subunits from different protomers occurs.
The structures of the two protomers are similar with root mean square deviations (RMSD) of 0.18 Å for the α-subunits (281 superposable Cα atoms) and 0.56 Å for the β-subunits (500 superposable Cα atoms). Thus, unless otherwise indicated, only the structure of the protomer consisting of chain B and D was analyzed due to its better electron density (Supplementary Figure S3A).
Core catalytic domain of the α-subunit
Figure 1C shows the topology of EcGlyRS. Each α-subunit is composed of an N-terminal core catalytic domain (β-strands S1-S7 and helices H1-H6), a C-terminal three-helix domain (H9-H11), and a structured linker between them (β-hairpin S8-S9 and helices H7-H8). The core catalytic domain of the α-subunit contains signature motifs 1–3 of class II aaRSs (Figure 1C, Supplementary Figures S3B and S4), which create a large cavity for ATP and glycine binding. The dimer interface between two α-subunits is mainly formed by their class II-defining motif 1 (H1 and S1) and helices H8-H10. Consistent with a previous analysis (14), the interactions between two α-subunits are mostly provided by H-bonds and salt bridges, which is significantly different from the dimerization interface of the catalytic domain of other class II aaRSs where hydrophobic interactions play a major role (39,40).
Five domains make up the β-subunit
The β-subunit folds into an arch-like structure with the top of the arch attached to the α-subunit at the side opposite to the α2 dimerization interface. The β-subunit can be divided schematically into five domains according to its tertiary structure (Figure 1): the B1 domain (res. 1–65, 125–215), B2 domain (res. 66–124), B3 domain (res. 216–340), HD hydrolase domain (res. 341–540), and anticodon binding domain (ABD, res. 541 to C-terminus). The B1 domain adopts an α-β-α sandwich structure, which is composed of a five-stranded antiparallel β-sheet (S1–S3 and S7–S8) with two α-helices (H1 and H3) packed on one side and two α-helices (H4 and H5) and two short β hairpins (S9–S10 and S11–S12) packed on the other side (Figure 1C). The B2 domain (res. 65–125) is composed of a three-stranded long β-sheet (S4–S6) attached by an α-helix (H2). The B3 domain forms another α–β–α sandwich structure with an antiparallel β-sheet (S13–S16) surrounded by five α-helices (H6-H10). While the B1 and B3 domains form tight contacts with the α-subunit to form the tetrameric GlyRS, the B2 domains are stretched out from the B1 domain. When the two protomers are aligned, the B2 domains are seen to rotate ∼40° (Supplementary Figure S5).
The HD domain consists of twelve α-helices (H11–H22). The C-terminal ABD of the β-subunit was partly truncated, while the remaining residues form two α-helices (H23 and H24) in an extended conformation. Interestingly, crystal packing analysis revealed that these two remaining α-helices of ABD form domain-swapping interactions with the same motif of EcGlyRS form the adjacent asymmetric unit (Supplementary Figure S6). This is believed to be due to artificial interactions in crystal packing, but it well explains why EcGlyRS575 could grow better crystals than EcGlyRS-FL and other variants.
The β-subunit supports substrate recognition by the α-subunit
Capture of ATP and glycine by the α-subunit alone is consistent with its class II active site signature motifs
Previous studies revealed that the active site is located on the α-subunit, which harbors signature motifs 1–3 of class II aaRSs (14). In the EcGlyRS575 structure, glycine and AMP-PNP were observed in both aminoacylation pockets of each protomer (Figure 1B). Glycine was deeply buried in a pocket on the α-subunit that was formed mainly by five residues (Thr39, Gln82, Gln84, Trp121 and Glu162) that are conserved among all (αβ)2 GlyRSs (Figure 2A and Supplementary Figure S4). A water molecule contributes to bridging a hydrogen-bond (H-bond) network between these conserved residues and the substrate glycine (Figure 2A). In addition to recognizing glycine, the α-subunit alone can also capture the ATP substrate in an open cleft on the surface (14). The adenine and ribose moieties of ATP insert into the cleft and are mainly formed by signature motifs 2 and 3 (PDB: 3rgl and 3ufg) (14). In contrast, the phosphate groups of ATP were more exposed to solvent and formed very different conformations when binding to the α-subunit alone (Supplementary Figure S7).
Figure 2.

The aminoacylation pocket of EcGlyRS. Glycine (A) and AMP-PNP (B) binding to the aminoacylation pocket. The bound glycine (magenta) and AMP-PNP (cyan) are shown as ball-and-stick representations, and Mg2+ (light green) ions are shown as spheres. The yellow dashed lines indicate the polar contacts. (C) Structural comparison of the aminoacylation pockets between EcGlyRS and CjGlyRS (PDB: 3grl, colored yellow), and the major conformation differences in the two structures are indicated by red arrows. (D) The activities of glycine adenylation of wild-type EcGlyRS-FL and its variants. The activity of the wild-type enzyme was defined as 100%. (E) The aminoacylation activities of EcGlyRS and its variants were detected by measuring ATP consumption. The enzymatic assays here and later were repeated three times, and the error bars indicate the standard deviation (SD).
Support from the β-subunit further stabilizes substrate binding in α-subunit
In our structure of heterotetrameric EcGlyRS, the B3 domain of the β-subunit folds against the α-subunit and partially covers the surface cavity, resulting in the formation of an intact pocket for holding AMP-PNP (Figure 2B and Supplementary Figure S8). The glycine and the adenine and ribose moieties of AMP-PNP are coordinated in this intact pocket with the same manner that they are coordinated by the α-subunit alone (PDB: 3rgl and 3ufg) (14). In contrast, the phosphate groups of AMP-PNP rotate toward the β-subunit (Figure 2C and Supplementary Figure S7). As a result, α-subunit Arg76 and Arg169 form additional charge interactions with the γ-phosphate of AMP-PNP. Asp67, Glu134, and Glu141 also contribute to stabilizing β- and γ- phosphates by coordinating two Mg2+ which are necessary for catalysis by class II aaRSs (41,42) (Figure 2B). These interactions and Mg2+ were not observed in the structure of the ATP-bound α-subunit of CjGlyRS (Figure 2C). Importantly, Arg326 from the β-subunit B3 domain was observed to form direct charge interactions with the γ-phosphate of AMP-PNP (Figure 2B and C), and Asp329 from the β-subunit B3 domain is involved in stabilizing the conformations of residues Arg76 and Arg69 from the α-subunit which, in turn, contributes to AMP-PNP binding (Figure 2B and C).
The activity of the α-subunit in isolation is extremely low, so the Km value for ATP could not be determined. Instead, we tried to measure the disassociation constant (Kd) of ATP binding to the α-subunit alone and to the full enzyme by employing isothermal titration calorimetry (ITC). The results showed that ATP binds to EcGlyRS-FL with a Kd of about 6 μM, but no significant binding between ATP and the α-subunit alone could be detected (Supplementary Figure S9).
Mutational analysis further demonstrates the role of α- and β-subunit cooperation
To further study the recognition of ATP, a set of residues considered to contact the Mg2+ or phosphates of AMP-PNP were substituted with alanine, and the resulting variants were measured for their activities for both glycine activation and tRNAGly aminoacylation (Figure 2D and E). Mutation of Arg64 on the α-subunit, a key residue of motif 2 that contacts an oxygen atom of the β-phosphate, severely decreased the activity of glycine activation (∼985-fold) (Figure 2D). Consistently, the aminoacylation of tRNAGly by R64A was fully abolished compared to that of wild-type EcGlyRS (Figure 2E). Significant activity decreases were also observed for two other groups of mutations. The first are mutants of three arginine residues proximal to the γ-phosphate of AMP-PNP (Arg76 and Arg169 on the α-subunit and Arg326 on the β-subunit). These mutations decreased glycine activation by ∼47-, ∼123- and ∼15-fold, respectively. The second are mutants of three α-subunit residues—sp67, Glu134 and Glu141—associated with Mg2+ binding. These mutations decrease glycine activation by ∼109-, ∼11- and ∼19-fold, respectively (Figure 2D). All of the mutations also significantly decreased the aminoacylation activity of EcGlyRS (Figure 2E).
Thus, although the α-subunit alone is sufficient to bind glycine and ATP in co-crystallization, our structural and biochemical data show that, by directly interacting with the γ-phosphate of ATP and reshaping the conformation of some residues on the α-subunit, the β-subunit facilitates ATP binding and rearranges the conformation of ATP in the aminoacylation pocket.
The B1-B2 domains of the β-subunit contribute to tRNA binding
Searching for structural homologs
To gain more information to understand the evolution and functions of each domain of EcGlyRS, we employed the DALI server to search their structural homologs. For the α-subunit, the best structural homologs in the PDB are α2 GlyRSs, followed by the class II AlaRSs AsnRS, LysRS, PheRS, AspRS, etc. (Supplementary Table S2). This result confirms that the α-chain is most similar to other synthetases in class II. The top results also included the ATP phosphoribosyltransferase regulatory subunit (an inactive class II HisRS paralog; PDB: 6r02) and a mitochondrial DNA polymerase accessory subunit (PDB: 1g5h).
For the β-subunit B1-B2 domains, the DALI server identified that the best structural homologs are a class I CCA-adding enzyme (AfCCA, an enzyme responsible for the maturation or repair of the functional 3′ end of tRNAs by adding the 3′-essential nucleotides CCA, PDB: 3ovs), a putative transposase (PDB: 2fyx), and the Klenow fragment of DNA polymerase I (PDB: 4yfu) (Supplementary Table S3), but the latter two proteins showed similarity only to the B1 domain (a common fold of the RNA Recognition Motif, RRM) (Supplementary Figure S10).
Structural homologs suggested that the B1–B2 domains of the β-subunit facilitate tRNA binding
The body and tail domains of AfCCA have significant similarity to the B1–B2 domains of the EcGlyRS β-subunit, respectively (43). When the B1 domain was superimposed to the body domain of AfCCA in complex with tRNAPhe (PDB: 1sz1), the B1 domain formed contacts with the acceptor stem of tRNA, and the B2 domain, after a domain shift, could bind to the tRNA elbow region similar to that of the AfCCA tail domain (43) (Figure 3A). We analyzed the conserved surface patches on the β-subunit. The β-subunits of twelve GlyRSs with overall sequence identity lower than 35% were aligned (Supplementary Figure S11), and the output was rendered as a surface coloring according to the level of sequence conservation (Figure 3B). In total, three large conserved patches on the β-subunit were identified: a region on B1 and B3 domains that interact with the α-subunit, a region in and around the cavity of the HD domain, and a region on the B2 domain. Importantly, the conserved region on the B2 domain is located on the distal end of B2, which corresponds to the region of AfCCA that interacts with the tRNA elbow region. Perhaps of relevance, the elbow was thought to be important for tRNA recognition of some aaRSs (44).
Figure 3.

Structural and functional analysis of the B1–B2 domains of the β-subunit. (A) Structural superposition of the B1–B2 domains of the β-subunit (colored the same as Figure 1A) and the class I CCA-adding enzyme (AfCCA; PDB: 1sz1; yellow) in complex with tRNAPhe (gray). (B) The surface conservation profile of the β-subunit of EcGlyRS. The β-subunit is represented as a surface model and colored according to its conservation score, which varies from white (highly variable) to purple (highly conserved). (C) Glycine activation of wild-type EcGlyRS and its truncates. (D) The aminoacylation activities of EcGlyRS-FL and its truncates.
As shown in Figure 3D, removal of the B2 domain (EcGlyRSΔB2) decreased charging activity even more severely than truncation of the ABD (EcGlyRS575). When the B2 and ABD domains were removed simultaneously (EcGlyRS575ΔB2), aminoacylation activity was completely lost (Figure 3D). However, formation of the adenylate intermediate was not strongly impaired in these truncates (Figure 3C), suggesting that the domain truncation mainly affects the tRNAGly-dependent step of aminoacylation rather than glycine activation. Consistently, size-exclusion chromatographic (SEC) assays showed that the EcGlyRS-FL protein can form a small but clear peak of an enzyme-tRNA complex, but the complex peak was not observed for EcGlyRS575 or EcGlyRSΔB2 under the same conditions (Supplementary Figure S12), indicating that truncation of the B2 domain disrupts the EcGlyRS-tRNAGly interaction.
The B1 domain forms extensive interactions with the α-subunit as well as with the B3 domain of the β-subunit; therefore, the removal of the B1 domain or both B1-B2 domains disrupted the formation of the (αβ)2 tetramer (data not shown). Instead, we made single-site mutations on the B1 domain for its positively charged residues that face the tRNA molecule. These residues are mainly located on two loops: the loop between β-strand S1 and α-helix H1 (Lys18 and Arg21) and the loop between H3 and S7 (Lys146, Arg149, Trp150 and Arg159) (Supplementary Figure S13A). While K18A and R21A showed little effect on the tRNAGly aminoacylation activity of EcGlyRS, mutations of W150A and R159A significantly abolished tRNAGly aminoacylation (Supplementary Figure S13C). As a control, all mutations were fully active in forming the adenylate intermediate (Supplementary Figure S13B). Thus, the H3–S7 loop of the B1 domain is likely to contribute to tRNA binding.
Evidence for structural and functional convergence of evolutionarily distinct GlyRSs
Structural comparisons and mutagenesis analyses suggest the importance of the B1–B2 domains in the productive binding of tRNAGly to EcGlyRS. More interestingly, the human α2 GlyRS (HsGlyRS), although independent in evolution, has an extended and flexible insertion domain (Ins3) that interacts with the elbow region of tRNAGly. Deletion of Ins3 markedly reduced its activity to less than 1% of wild-type activity (45). Remarkably, the EcGlyRS B2 domain and HsGlyRS Ins3 share significant structural similarities (Supplementary Figure S14), and although coming from opposite directions, both approach the elbow region of tRNAGly (Supplementary Figure S14). This structural and functional convergence in evolution highlights the importance of the elbow region in tRNAGly binding and aminoacylation.
Structure and function features of the HD domain
Similarities to the HD superfamily of hydrolases
Although the catalytic histidine-aspartate doublet) is changed to Ser393 and Lys394 in EcGlyRS (17), the HD domain (res. 341–540) of the β-subunit showed significant similarity to the HD superfamily of hydrolases, such as the PgpH HD domain (PDB: 4s1b-A, DALI-score: 11.1), the HD-GYP domain cyclic-di-GMP phosphodiesterase (PDB: 4mcw), the drosophila MESH1 (PDB: 3nqw) and the Thermus thermophilus (P)PPGPP synthetase Spot/RelA (PDB: 6s2t) (Supplementary Table S4). These homologs are related to the accumulation/degradation of endogenous nucleotides. Accordingly, we tested some nucleotides, including c-Di-GMP, cAMP, cGMP, and ppGpp, for binding to the HD domain. They neither increased the Tm values of the purified HD domain (res. 340–540) in TSA nor affected ATP consumption of EcGlyRS-FL (data not shown).
Interestingly, the region in and around the large cavity of the HD domain is one of the most conserved surface patches on the β-subunit of (αβ)2 GlyRS (Figure 3B). This cavity is mainly formed by residues Lys364, Arg367, Lys394, Tyr434, Asp456, Lys457 and Asp459, and faces the aminoacylation pocket (Figure 4A). Remarkably, although the cavity is more than 30 Å away from the aminoacylation site, mutations of some of the residues did affect the aminoacylation of tRNAGly (Figure 4B) and did not affect the activation of glycine (Supplementary Figure S15), suggesting a role of the HD domain cavity in the second step of catalysis. It is possible that this cavity functions as an allosteric regulating site. From the practical side, this cavity could be used to target (αβ)2 GlyRS-specific inhibitors for antimicrobial drug discovery. The uniqueness of this cavity would diminish possibilities for cross-reaction with the host human cytoplasmic and mitochondrial GlyRSs, which are both of the α2 type. It is worth noting that many human pathogens such as Helicobacter pylori, Pseudomonas aeruginosa, Streptococcus pneumoniae, etc. rely on the (αβ)2 GlyRS.
Figure 4.

Structural and functional analysis of the HD domain. (A) The cavity on the HD domain. The conserved residues in the cavity are colored purple, while other regions are colored white. (B) The aminoacylation activities of wild-type EcGlyRS and its variants with single mutations in the HD domain. (C) The structural superposition between the HD domain and the Mid1 subdomain of Archaeoglobus fulgidus AlaRS (AfAlaRS) in complex with tRNAAla (PDB: 3wqy). Mid 1 (cyan) in the tRNA-recognition domain of AfAlaRS and tRNAAla(black) are shown as cartoons. The discriminator base A73 and the G3·U70 pair in tRNAAla are presented as sticks. The polar contact between Asp459 and U70 is indicated by the yellow dashed line.
C-terminal half of the HD domain may play a role in acceptor arm binding
The Mid1 subdomain of Archaeoglobus fulgidus AlaRS (AfAlaRS, PDB: 3wqy-A) appeared in the top list when we searched for structural homologs of the HD domain, and the C-terminal half (res. 446–540) of the HD domain was able to superpose with the AfAlaRS Mid1 subdomain with an RMSD of 3.6 Å for 126 Cα pairs (Supplementary Figure S16). The Mid1 (res. 258–419) and the Mid2 subdomain (res. 420–484) are thought to clamp the major and minor grooves of the acceptor stem and to play an essential role in the recognition of the G3·U70 identity element of tRNAAla (46). When the Mid1 subdomain of the AfAlaRS-tRNAAla complex was superimposed to the EcGlyRS β-subunit HD domain, the C-terminal half of the HD domain was located close to the acceptor arm of the tRNA molecule (Figure 4C), implying a function of the HD domain in the binding and recognition of tRNAGly. Interestingly, the discriminate U73 of prokaryotic tRNAGly located beside the N-terminus of helix H19, where amino acid sequences are highly conserved among all heterotetrameric GlyRSs aligned (Supplementary Figure S11).
Notably, C-terminal half of the HD domain was observed to approach the tRNA acceptor arm in a similar manner when the tail and body domains of the AfCCA-tRNAPhe complex were superimposed to the EcGlyRS β-subunit B1-B2 domains (Figure 3A), indicating that the EcGlyRS-tRNA binding models suggested by the AfCCA-tRNAPhe complex and AfAlaRS-tRNAAla are consistent with each other.
Structure analysis reveals a large distance between the α-subunit C- and β-subunit N-termini
Persistence across bacteria for breaking apart a single chain
For EcGlyRS, both subunits are encoded in one reading frame in the genome, with an in-frame spacer of 9 nucleotides after a TAA stop interruption at the end of the α-subunit coding region and an ATG start at the beginning of the β-subunit coding region (Figure 5A). Thus, we imaged that in the structure of EcGlyRS, the N-terminus of the β-subunit would be juxtaposed with the C-terminus of the α-subunit. With this in mind, an artificial α–β fusion protein was attempted in an earlier study, but the results were inconclusive (47). Indeed, in our structure, the distance between the C-terminus of the α-subunit and the N-terminus of the β-subunit is ∼55 Å within the same protomer and is even further (∼85 Å) across two protomers (Figure 5B). This observation can explain what appeared to be an unstable fusion protein in an earlier study.
Figure 5.
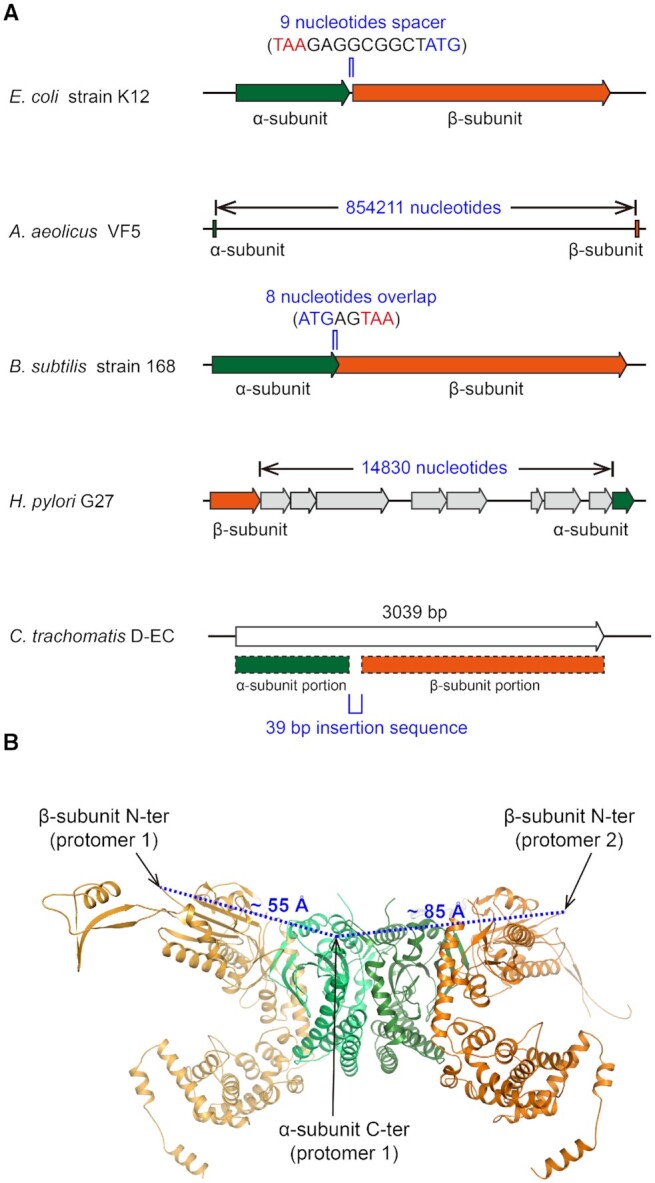
Strong evolutionary pressure separates bacterial-type GlyRSs into two chains. (A) Diagrammatic representation of the arrangements of the GlyRS encoding genes found in the genomes of Escherichia coli strain K-12 (GenBank: CP009685.1), Aquifex aeolicus VF5 (GenBank: AE000657.1), Bacillus subtilis strain 168 (GenBank: NC_000964.3), Helicobacter pylori G27 (GenBank: CP001173.1) and Chlamydia trachomatis D-EC (GenBank: CP002052.1). Green, genes encoding the α-subunit or the α-subunit portion in CtGlyRS; orange, genes encoding the β-subunit or the β-subunit portion in CtGlyRS; gray, genes between the α- and β-subunit in the genome of Helicobacter pylori G27. (B) The dashed lines (blue) indicate the distance between the C-terminus of the α-subunit and the N-terminus of the β-subunit. The α-subunit (green) and β-subunit (orange) of EcGlyRS in one protomer are shown as cartoons, and the C-terminus of the α-subunit and the N-terminus of the β-subunit are indicated by arrows.
The organization of the α- and β-subunit genes was quite different among different bacteria (48). The distance between the two subunits is 854211 nucleotides in Aquifex aeolicus VF5 (Figure 5A). In contrast, in Bacillus subtillis strain 168, the C-terminus of the α-subunit coding sequence has an 8-nucleotide overlap with the N-terminus of the β-subunit coding region. In H. pylori G27, the β-subunit coding sequence is located upstream of the α-subunit, with several other genes between them. Thus, in all of these examples, the α- and β-subunit coding sequences are separated in each respective genome. Strong evolutionary pressure is likely responsible for this separation.
An exception that further supports apparent evolutionary pressure to break the chain
Chlamydia trachomatis GlyRS (CtGlyRS) is encoded by a single open reading frame (ORF), and the deduced polypeptide is 1013 residues (21,49) (Figure 5A). The N- and C-terminal regions of the polypeptide show significant similarity to the α- and β-subunits of EcGlyRS, respectively, but not eukaryotic α2 GlyRSs, and between the α- and β-subunit coding regions of CtGlyRS is an in-frame linker of 39 nucleotides coding for 13 amino acids. This linker and the tail of the α-subunit are proline-rich and predicted to be flexible, thus they accommodate the separation of subunit termini seen in our structure of EcGlyRS. The fused α- and β-subunits are also observed in other prokaryote-type GlyRSs from chloroplasts of A. thaliana and Phaseolus vulgaris (21,50). These results further suggest strong evolutionary pressure to separate GlyRSs into two subunits, even when joined together through a linker that facilitates the spacing required for the chain termini to match up with other prokaryote GlyRSs.
Heterotetrameric PheRSs from different bacteria are also organized in either an open or a distal location in the genome (Supplementary Figure S17A). Interestingly, mitochondrial PheRS (mtPheRS) is a monomer evolved from bacterial PheRS and is fully active (51). This polypeptide of mtPheRS is a fusion of the catalytic module (CAM) of the bacterial α-subunit and the C-terminal domain of the bacterial β-subunit, and many domains involved in the binding of tRNA and in protein quality control have lost during evolution (Supplementary Figure S17B) (51). In contrast, the fused GlyRSs from Chlamydia trachomatis and chloroplasts of A. thaliana and Phaseolus vulgaris retain all the domains of heterotetrameric GlyRS, supporting the proposal that (αβ)2 GlyRS is a uniquely ‘obligate’ heterotetramer.
EcGlyRS supports the subclass-specific pairwise docking of aaRSs on tRNA
Specificity of pairing of two aaRSs on one acceptor stem
The two classes of tRNA synthetases are conserved with rare exceptions through evolution. These two classes, which are based on two distinct architectures for their respective catalytic domains, can be broken down into subclasses (Ia, b, c) and (IIa, b, c) with amino acids of similar size being grouped into the same subclass (Figure 6A) (52). From the early work of Ohno and Rodin and further work by Carter et al. on urzymes (53,54), these two classes could in principle arise from opposite strands of early RNA genomes. Co-crystal structures showed that the two classes approach the tRNA acceptor stems from opposite sides (55,56). These observations led to the speculation that primitive tRNA synthetases, among their catalytic roles, served as chaperones to protect the acceptor stems from degradation by nucleases and the high temperatures of early thermophiles (56). To do so, the enzymes need to be paired together in a specific way, to avoid clashing by steric interference. Thus, class Ia enzymes would pair with class IIa enzymes, Ib with IIb, and Ic with IIc. Using structures available at the time, strong support was obtained for this hypothesis.
Figure 6.

The modeled complexes of aaRS catalytic domains bound simultaneously to a tRNA acceptor stem. (A) Classification of class I and II aaRSs. Each enzyme is referred to by the name of its cognate amino acid. The exceptional cases of GlyRS and LysRS where two distinct origins exist for the same enzyme are labeled accordingly. (B) The docking result showing that the EcGlyRS and IleRS active sites bound simultaneously to a tRNA acceptor stem. The figure shows the molecules along the axis of the anticodon stem, from the acceptor stem side. (C) The docking between the core regions of EcGlyRS, AspRS and TyrRS will result in large steric clashes. (D) Class I and II LysRSs, which are the only synthetases known to date that can be of either class I or class II architecture. These two LysRSs can bind to opposite sides of the tRNA acceptor stem without a clash.
GlyRS is in class IIa, as part of the six amino acids in that class are matched by rough size with others in class Ia. Of the twelve tRNA synthetases that correspond to those amino acids, as an unusual (αβ)2 tRNA synthetase, GlyRS is the only one that is neither a monomer nor a homodimer, and has a completely idiosyncratic structure. Thus, our unusual structure of EcGlyRS presented a singular opportunity to further test the hypothesis concerning the coverage of tRNA acceptor stems.
Unusual X-shaped (αβ)2 GlyRSs fit the paradigm for specific synthetases pairing on tRNA acceptor stems
As shown in Figure 6B, EcGlyRS fits onto the acceptor stem without a clash with class Ia IleRS or with any of the other class Ia enzymes (Supplementary Figure S18). In contrast, EcGlyRS cannot fit without a steric clash with the class Ib or Ic enzymes (Figure 6C), that is, class I enzymes of a different subclass, suggesting that the (αβ)2 GlyRSs belong to class IIa synthetases like the conventional α2 GlyRS. Interestingly, however, we found that the tRNA molecules bound from different directions to EcGlyRS and HsGlyRS when their catalytic core domains were aligned (Supplementary Figure S19A). When the tRNA molecules were aligned, the catalytic domains of these two types of GlyRSs were found to bind the acceptor stem of tRNA from different directions (Supplementary Figure S19B). Although the orientations of HsGlyRS and EcGlyRS on the acceptor stem are different, both GlyRSs could dock to tRNA without a clash with its partner class Ia aaRSs(Supplementary Figure S19B).
LysRS is the only aaRS that has examples of class Ib and class IIb architectures. The prediction at the time of the original report was that, when both structures were available, the two enzymes would pair across each other on an acceptor stem, and without a steric clash (55). This expectation was fulfilled (57,58), as shown in Figure 6D.
These results further illustrate how synthetases from opposite subclasses can protect the acceptor stem of the target tRNA. Separately, they are consistent with the idea that the synthetases were ‘born’ in pairs—for example, class Ia enzymes being coded by one strand of a primitive gene and class IIa by the opposite strand (5,53,59). Here, the possibility is raised that the early genetic code was enabled by primitive synthetases that recognized amino acids by size before specificity was developed for side chain fine structure.
DISCUSSION
The structure of (αβ)2 GlyRS is distinct from that of PheRS
Herein, the first crystal structure of (αβ)2 GlyRS from E. coli was solved in complex with glycine and the cofactor analog AMP-PNP. Indeed, EcGlyRS shows dramatic structural differences from the classical α2 GlyRS in the overall architecture and design of the aminoacylation pockets. Eubacterial and eukaryotic cytoplasmic PheRS is the only other heterotetrameric member in the aaRS family, and also shows an (αβ)2 subunit organization pattern (60,61). The overall structure of EcGlyRS is not similar to that of PheRS (60). EcGlyRS forms an X-shape, with only two α-subunits contributing to the oligomerization of the two protomers. No interaction occurs between the two β-subunits or between the α- and β-subunits across two protomers. And, tRNA apparently is engaged by the α- and β-subunits within the same protomer. In contrast, PheRS is shaped as a ‘leatherback turtle’ with large flippers, with each subunit of PheRS contacting more or less with the other three subunits to contribute to tetramerization (60). Thus, although the α- and β-subunits are mainly responsible for catalysis and tRNA recognition, respectively, in both (αβ)2 GlyRS and PheRS, the domain organization and the folding of these domains are clearly different in the subunits of the respective synthetases.
The possible connections of (αβ)2 GlyRS with AlaRS and CCA-adding enzymes in evolution
Previous studies revealed that the sequence of the α-subunit of bacterial GlyRS presents some features that resemble AlaRS (e.g. the same sequence insertion in motif 2) (13). Their active sites also share similar overall architectures for recognizing glycine and alanine, the two smallest amino acids in protein synthesis (1,15). This observation suggested that the (αβ)2 GlyRS evolved in part from either AlaRS or from an ancestor of AlaRS that was able to aminoacylate both alanine and glycine, while the α2 GlyRS evolved from another independent ancestor (15). In our structure, the C-terminal part of the HD domain of EcGlyRS is structurally superposable to Mid1 of AlaRS, a subdomain involved in interacting with the acceptor arm of substrate tRNAAla (46). This structural feature of EcGlyRS provides additional suggestive evidence, from the side of the β-subunit, that the (αβ)2 GlyRS partly evolved in a way related to AlaRS (Supplementary Figure S16 and Figure 4C).
We also showed that mutations in the B1 and B2 domains of the β-subunit affected the tRNA binding of EcGlyRS (Supplementary Figure S13 and Figure 3D). The closest structural homologs of B1-B2 domains in the PDB are the body and tail domains of the class I CCA-adding enzymes, which add CCA to the 3′ ends of all tRNAs for their maturation (43). While the body domain of class I CCA-adding enzymes interacts with the acceptor stem of substrate tRNA, the tail domain is a key binder of the tRNA elbow region (62). Moreover, while the C-terminal part of the EcGlyRS HD domain is structurally superimposable on the Mid1 subdomain of AlaRS, the N-terminal part of the HD domain exhibits significant similarity to the body domain of the class II CCA-adding enzyme (Supplementary Figure S20) (63). With their essential roles in the maturation of tRNAs for protein translation, CCA-adding enzymes are also among the most ancient proteins (62). The potential evolutionary connection between (αβ)2 GlyRS and both types of CCA-adding enzymes deserve further investigation.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data bank under accession number 7EIV.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff of beamlines BL19U1 and BL19U2 at National Center for Protein Sciences Shanghai (NCPSS) and Shanghai Synchrotron Radiation Facility (SSRF) for assistance during data collection.
Notes
Present address: Yingchen Ju, Shenzhen Proxybio Therapeutics Co., Ltd, Shenzhen 584000, China.
Contributor Information
Yingchen Ju, Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China; Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Lu Han, Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China; Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Bingyi Chen, Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China; Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Zhiteng Luo, Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China; Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Qiong Gu, Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Jun Xu, Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Xiang-Lei Yang, Department of Molecular Medicine, The Scripps Research Institute, La Jolla, CA 92037, USA.
Paul Schimmel, Department of Molecular Medicine, The Scripps Research Institute, La Jolla, CA 92037, USA; Department of Molecular Medicine, The Scripps Research Institute, Jupiter, FL 33458, USA.
Huihao Zhou, Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China; Research Center for Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81773636, 81803435]; Guangdong Natural Science Foundation [2017A030313123]; Guangdong Basic and Applied Basic Research Foundation [2019A1515011571]; Program for Guangdong Introducing Innovative and Entrepreneurial Teams [2016ZT06Y337]; Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery [2019B030301005]; US National Institutes of Health [R35 GM139627]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1.Rubio Gomez M.A., Ibba M.. Aminoacyl-tRNA synthetases. RNA. 2020; 26:910–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibba M., Soll D.. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000; 69:617–650. [DOI] [PubMed] [Google Scholar]
- 3.Eriani G., Delarue M., Poch O., Gangloff J., Moras D.. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990; 347:203–206. [DOI] [PubMed] [Google Scholar]
- 4.Cusack S., Berthet-Colominas C., Hartlein M., Nassar N., Leberman R.. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature. 1990; 347:249–255. [DOI] [PubMed] [Google Scholar]
- 5.Carter C.W. JrCoding of Class I and II Aminoacyl-tRNA Synthetases. Adv. Exp. Med. Biol. 2017; 966:103–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribas de Pouplana L.Biology of Aminoacyl-tRNA Synthetases. 2020; 11–37. [DOI] [PubMed] [Google Scholar]
- 7.Berthonneau E., Mirande M.. A gene fusion event in the evolution of aminoacyl-tRNA synthetases. FEBS Lett. 2000; 470:300–304. [DOI] [PubMed] [Google Scholar]
- 8.Woese C.R., Olsen G.J., Ibba M., Soll D.. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000; 64:202–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon N.H., Fox P.L., Kim S.. Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discov. 2019; 18:629–650. [DOI] [PubMed] [Google Scholar]
- 10.Wei N., Zhang Q., Yang X.L.. Neurodegenerative Charcot-Marie-Tooth disease as a case study to decipher novel functions of aminoacyl-tRNA synthetases. J. Biol. Chem. 2019; 294:5321–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiba K., Schimmel P., Motegi H., Noda T.. Human glycyl-tRNA synthetase. Wide divergence of primary structure from bacterial counterpart and species-specific aminoacylation. J. Biol. Chem. 1994; 269:30049–30055. [PubMed] [Google Scholar]
- 12.Ostrem D.L., Berg P.. Glycyl-tRNA synthetase: an oligomeric protein containing dissimilar subunits. Proc. Natl. Acad. Sci. U.S.A. 1970; 67:1967–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webster T.A., Gibson B.W., Keng T., Biemann K., Schimmel P.. Primary structures of both subunits of Escherichia coli glycyl-tRNA synthetase. J. Biol. Chem. 1983; 258:10637–10641. [PubMed] [Google Scholar]
- 14.Tan K., Zhou M., Zhang R., Anderson W.F., Joachimiak A.. The crystal structures of the alpha-subunit of the alpha(2)beta (2) tetrameric Glycyl-tRNA synthetase. J. Struct. Funct. Genomics. 2012; 13:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valencia-Sanchez M.I., Rodriguez-Hernandez A., Ferreira R., Santamaria-Suarez H.A., Arciniega M., Dock-Bregeon A.C., Moras D., Beinsteiner B., Mertens H., Svergun D.et al.. Structural insights into the polyphyletic origins of glycyl tRNA synthetases. J. Biol. Chem. 2016; 291:14430–14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toth M.J., Schimmel P.. A mutation in the small (alpha) subunit of glycyl-tRNA synthetase affects amino acid activation and subunit association parameters. J. Biol. Chem. 1990; 265:1005–1009. [PubMed] [Google Scholar]
- 17.Wolf Y.I., Aravind L., Grishin N.V., Koonin E.V.. Evolution of aminoacyl-tRNA synthetases–analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999; 9:689–710. [PubMed] [Google Scholar]
- 18.Nagel G.M., Cumberledge S., Johnson M.S., Petrella E., Weber B.H.. The beta subunit of E. coli glycyl-tRNA synthetase plays a major role in tRNA recognition. Nucleic. Acids. Res. 1984; 12:4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toth M.J., Schimmel P.. Deletions in the large (beta) subunit of a hetero-oligomeric aminoacyl-tRNA synthetase. J. Biol. Chem. 1990; 265:1000–1004. [PubMed] [Google Scholar]
- 20.Freist W., Logan D.T., Gauss D.H.. Glycyl-tRNA synthetase. Biol. Chem. Hoppe Seyler. 1996; 377:343–356. [PubMed] [Google Scholar]
- 21.Ibba M., Francklyn C., Cusack S.. The Aminoacyl-tRNA Synthetases. 2005; Georgetown, Texas: Landes Bioscience. [Google Scholar]
- 22.Giege R., Springer M.. Aminoacyl-tRNA synthetases in the bacterial world. EcoSal Plus. 2016; 7: 10.1128/ecosalplus.ESP-0002-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ju Y., Tong S., Gao Y., Zhao W., Liu Q., Gu Q., Xu J., Niu L., Teng M., Zhou H.. Crystal structure of a membrane-bound l-amino acid deaminase from Proteus vulgaris. J. Struct. Biol. 2016; 195:306–315. [DOI] [PubMed] [Google Scholar]
- 24.Minor W., Cymborowski M., Otwinowski Z., Chruszcz M.. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 25.Potterton L., Agirre J., Ballard C., Cowtan K., Dodson E., Evans P.R., Jenkins H.T., Keegan R., Krissinel E., Stevenson K.et al.. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr D Struct Biol. 2018; 74:68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B.et al.. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lloyd A.J., Potter N.J., Fishwick C.W., Roper D.I., Dowson C.G.. Adenosine tetraphosphoadenosine drives a continuous ATP-release assay for aminoacyl-tRNA synthetases and other adenylate-forming enzymes. ACS Chem. Biol. 2013; 8:2157–2163. [DOI] [PubMed] [Google Scholar]
- 30.Roy S.A continuous spectrophotometric assay for Escherichia coli alanyl-transfer RNA synthetase. Anal. Biochem. 1983; 133:292–295. [DOI] [PubMed] [Google Scholar]
- 31.Hopkins J.B., Gillilan R.E., Skou S.. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017; 50:1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M.et al.. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I.. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003; 36:1277–1282. [Google Scholar]
- 34.Svergun D.I.Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992; 25:495–503. [Google Scholar]
- 35.Franke D., Svergun D.I.. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009; 42:342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Volkov V.V., Svergun D.I.. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003; 36:860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svergun D., Barberato C., Koch M.H.J.. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995; 28:768–773. [Google Scholar]
- 38.Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 39.Yaremchuk A., Cusack S., Tukalo M.. Crystal structure of a eukaryote/archaeon-like protyl-tRNA synthetase and its complex with tRNAPro(CGG). EMBO J. 2000; 19:4745–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnez J.G., Harris D.C., Mitschler A., Rees B., Francklyn C.S., Moras D.. Crystal structure of histidyl-tRNA synthetase from Escherichia coli complexed with histidyl-adenylate. EMBO J. 1995; 14:4143–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belrhali H., Yaremchuk A., Tukalo M., Berthet-Colominas C., Rasmussen B., Bosecke P., Diat O., Cusack S.. The structural basis for seryl-adenylate and Ap4A synthesis by seryl-tRNA synthetase. Structure. 1995; 3:341–352. [DOI] [PubMed] [Google Scholar]
- 42.Arnez J.G., Augustine J.G., Moras D., Francklyn C.S.. The first step of aminoacylation at the atomic level in histidyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7144–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong Y., Steitz T.A.. Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature. 2004; 430:640–645. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J., Ferre-D’Amare A.R.. The tRNA elbow in structure, recognition and evolution. Life (Basel). 2016; 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng X., Qin X., Chen L., Jia Q., Zhang Y., Zhang Z., Lei D., Ren G., Zhou Z., Wang Z.et al.. Large conformational changes of insertion 3 in human glycyl-tRNA synthetase (hGlyRS) during catalysis. J. Biol. Chem. 2016; 291:5740–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naganuma M., Sekine S., Chong Y.E., Guo M., Yang X.L., Gamper H., Hou Y.M., Schimmel P., Yokoyama S.. The selective tRNA aminoacylation mechanism based on a single G*U pair. Nature. 2014; 510:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keng T., Webster T.A., Sauer R.T., Schimmel P.. Gene for Escherichia coli glycyl-tRNA synthetase has tandem subunit coding regions in the same reading frame. J. Biol. Chem. 1982; 257:12503–12508. [PubMed] [Google Scholar]
- 48.Tang S.N., Huang J.F.. Evolution of different oligomeric glycyl-tRNA synthetases. FEBS Lett. 2005; 579:1441–1445. [DOI] [PubMed] [Google Scholar]
- 49.Wagar E.A., Giese M.J., Yasin B., Pang M.. The glycyl-tRNA synthetase of Chlamydia trachomatis. J. Bacteriol. 1995; 177:5179–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duchene A.M., Peeters N., Dietrich A., Cosset A., Small I.D., Wintz H.. Overlapping destinations for two dual targeted glycyl-tRNA synthetases in Arabidopsis thaliana and Phaseolus vulgaris. J. Biol. Chem. 2001; 276:15275–15283. [DOI] [PubMed] [Google Scholar]
- 51.Bullard J.M., Cai Y.C., Demeler B., Spremulli L.L.. Expression and characterization of a human mitochondrial phenylalanyl-tRNA synthetase. J. Mol. Biol. 1999; 288:567–577. [DOI] [PubMed] [Google Scholar]
- 52.Schimmel P.Classes of aminoacyl-tRNA synthetases and the establishment of the genetic code. Trends Biochem. Sci. 1991; 16:1–3. [DOI] [PubMed] [Google Scholar]
- 53.Rodin S.N., Ohno S.. Two types of aminoacyl-tRNA synthetases could be originally encoded by complementary strands of the same nucleic acid. Orig. Life Evol. Biosph. 1995; 25:565–589. [DOI] [PubMed] [Google Scholar]
- 54.Carter C.W. JrCognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu. Rev. Biochem. 1993; 62:715–748. [DOI] [PubMed] [Google Scholar]
- 55.Ribas de Pouplana L., Schimmel P.. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001; 104:191–193. [DOI] [PubMed] [Google Scholar]
- 56.Ribas de Pouplana L., Schimmel P.. Aminoacyl-tRNA synthetases: potential markers of genetic code development. Trends Biochem. Sci. 2001; 26:591–596. [DOI] [PubMed] [Google Scholar]
- 57.Terada T., Nureki O., Ishitani R., Ambrogelly A., Ibba M., Soll D., Yokoyama S.. Functional convergence of two lysyl-tRNA synthetases with unrelated topologies. Nat. Struct. Biol. 2002; 9:257–262. [DOI] [PubMed] [Google Scholar]
- 58.Onesti S., Miller A.D., Brick P.. The crystal structure of the lysyl-tRNA synthetase (LysU) from Escherichia coli. Structure. 1995; 3:163–176. [DOI] [PubMed] [Google Scholar]
- 59.Carter C.W. Jr, Wills P.R.. Hierarchical groove discrimination by Class I and II aminoacyl-tRNA synthetases reveals a palimpsest of the operational RNA code in the tRNA acceptor-stem bases. Nucleic. Acids. Res. 2018; 46:9667–9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mosyak L., Reshetnikova L., Goldgur Y., Delarue M., Safro M.G.. Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat. Struct. Biol. 1995; 2:537–547. [DOI] [PubMed] [Google Scholar]
- 61.Michalska K., Jedrzejczak R., Wower J., Chang C., Baragana B., Gilbert I.H., Forte B., Joachimiak A.. Mycobacterium tuberculosis Phe-tRNA synthetase: structural insights into tRNA recognition and aminoacylation. Nucleic Acids Res. 2021; 49:5351–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiong Y., Steitz T.A.. A story with a good ending: tRNA 3′-end maturation by CCA-adding enzymes. Curr. Opin. Struct. Biol. 2006; 16:12–17. [DOI] [PubMed] [Google Scholar]
- 63.Yamashita S., Tomita K.. Mechanism of 3′-matured tRNA discrimination from 3′-Immature tRNA by Class-II CCA-adding enzyme. Structure. 2016; 24:918–925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data bank under accession number 7EIV.