


Abstract
Tpt1, an essential component of the fungal and plant tRNA splicing machinery, catalyzes transfer of an internal RNA 2′-PO4 to NAD+ yielding RNA 2′-OH and ADP-ribose-1′,2′-cyclic phosphate products. Here, we report NMR structures of the Tpt1 ortholog from the bacterium Runella slithyformis (RslTpt1), as apoenzyme and bound to NAD+. RslTpt1 consists of N- and C-terminal lobes with substantial inter-lobe dynamics in the free and NAD+-bound states. ITC measurements of RslTpt1 binding to NAD+ (KD ∼31 μM), ADP-ribose (∼96 μM) and ADP (∼123 μM) indicate that substrate affinity is determined primarily by the ADP moiety; no binding of NMN or nicotinamide is observed by ITC. NAD+-induced chemical shift perturbations (CSPs) localize exclusively to the RslTpt1 C-lobe. NADP+, which contains an adenylate 2′-PO4 (mimicking the substrate RNA 2′-PO4), binds with lower affinity (KD ∼1 mM) and elicits only N-lobe CSPs. The RslTpt1·NAD+ binary complex reveals C-lobe contacts to adenosine ribose hydroxyls (His99, Thr101), the adenine nucleobase (Asn105, Asp112, Gly113, Met117) and the nicotinamide riboside (Ser125, Gln126, Asn163, Val165), several of which are essential for RslTpt1 activity in vivo. Proximity of the NAD+ β-phosphate to ribose-C1″ suggests that it may stabilize an oxocarbenium transition-state during the first step of the Tpt1-catalyzed reaction.
INTRODUCTION
In fungal and plant tRNA splicing, ligation of exons by tRNA ligase results in a 2′-PO4, 3′,5′-phosphodiester splice junction. This internal 2′-PO4 is converted to a 2′-OH by the action of an NAD+-dependent 2′-phosphotransferase enzyme, Tpt1 (1,2), via a two-step reaction (Supplementary Figure S1). In the first step, a nucleophilic attack on NAD+ by the RNA 2′-PO4 generates an RNA 2′-phospho-(ADP-ribose) intermediate with the expulsion of nicotinamide. A subsequent transesterification reaction yields ADP-ribose-1″,2″-cyclic phosphate and a 2′-OH at the splice site (3). Tpt1 is an essential component of the tRNA splicing machinery in Saccharomyces cerevisiae and its deletion or inactivation by active site mutation is lethal (4,5). Tpt1 homologs are found in bacterial, archaeal and metazoan proteomes, possess NAD+-dependent 2′-phosphotransferase activity in vitro, and can function in lieu of S. cerevisiae Tpt1 in vivo (6–8). Yet, the role of these enzymes in their native taxa is unclear, insofar as these organisms are not known to have physiologic RNA transactions that yield internal 2′-PO4 substrates for Tpt1. Moreover, a mouse Tpt1 knockout elicits no physiological defects (9). Given their essential role in fungal tRNA splicing and the lack of an obvious role in mammalian physiology, Tpt1 enzymes are attractive targets for the development of new anti-fungals.
Understanding how Tpt1 recognizes NAD+ and an internal 2′-PO4-containing RNA and performs its unique 2′-phosphotransferase chemistry hinges on capturing structures of the enzyme at each stage along its reaction pathway. Initial low-resolution structural probing of yeast and Escherichia coli Tpt1 enzymes by limited proteolysis indicated that they consist of folded N- and C-terminal domains separated by a protease-sensitive interdomain linker (5). A 2.8 Å crystal structure of a Tpt1 homolog from the hyperthermophilic archaeon Aeropyrum pernix (ApeTpt1) confirmed its two-lobed fold (10), but did not provide insights into substrate recognition or catalysis. Subsequent biochemical studies verified that ApeTpt1 is a bona fide NAD+-dependent 2′-phosphotransferase (11). Tpt1 from the mesothermophilic bacterium Clostridium thermocellum (CthTpt1) was recently crystallized and its structure was solved at 1.4 Å resolution. The CthTpt1 structure fortuitously contained co-enzyme A (CoA) and ADP-ribose-1″-phosphate engaged to its N- and C-terminal lobes, respectively (12). This structure likely mimics a product state with the pAp moiety of CoA representing the 2′-OH RNA product and the ADP-ribose-1″-phosphate corresponding to the hydrolyzed remnant of the ADP-ribose-1″,2″-cyclic phosphate product (12). Whereas this structure represents a significant advance in understanding the mechanism of the second transesterification step of the Tpt1 pathway, insights on the structural and dynamic interactions in the reactant states bound to an intact NAD+ and/or an intact 2′-PO4-modified RNA prior to step 1 chemistry are still lacking.
Here we utilize solution NMR methodology to determine the structures of the substrate-free state of Tpt1 from the bacterium Runella slithyformis (RslTpt1) (6) and that of its complex with NAD+. RslTpt1 displays significant inter-lobe mobility in both the apoenzyme and NAD+-bound states. Biophysical assays and supporting NMR measurements indicate that a majority of the binding energy for the interaction of RslTpt1 with NAD+, which localizes exclusively to the C-terminal lobe of the enzyme, is provided by the ADP moiety. NMN and nicotinamide do not bind detectably to RslTpt1 in isolation, and these moieties contribute modestly to the affinity for NAD+. NADP+, in contrast to NAD+, binds exclusively to the N-terminal lobe of RslTpt1, in effect behaving as a low-affinity mimetic of the nucleoside 2′-PO4 at the splice junction. The NMR structure of the binary RslTpt1·NAD+ complex provides the first detailed insight into the recognition of NAD+ prior to step 1 chemistry and identifies enzymic contacts to NAD+ by C-lobe amino acid residues conserved among Tpt1 family members. Structure-guided alanine mutagenesis identifies stacking interactions with the adenine nucleobase and nicotinamide moieties of NAD+ as essential for RslTpt1 function in vivo, as gauged by yeast tpt1Δ complementation.
MATERIALS AND METHODS
Protein expression and purification
The plasmid pET28a-His6-Tpt1, encoding residues Val5 to Val178 of RslTpt1, was transformed into E. coli One Shot BL21 Star (DE3) cells (Thermofisher). For the expression of unlabeled RslTpt1, colonies were inoculated into starter Luria-Bertani (LB) medium supplemented with kanamycin (35 mg/l) and grown overnight at 37°C. The starter culture was then used to inoculate 1 L of LB supplemented with kanamycin (35 mg/l) and grown at 37°C until the A600 reached 0.8. Isopropyl-β-d-thiogalactoside (IPTG) was then added to a final concentration of 500 μM together with 2% (v/v) ethanol, and the culture was incubated for 18 h at 17°C with continuous shaking at 250 rpm. Cells were harvested by centrifugation at 4,000 rpm for 30 min using a Fiberlite F12–6 × 500 LEX (Thermo Fisher Scientific). Cell pellets were stored at -80°C.
RslTpt1 samples that were either uniformly 13C,15N-labeled (CN-labeled) or specifically 13CH3-labeled at Val-γ1/2, Leu-δ1/2, Met-ϵ, Ile-δ1 positions in an otherwise 2H (except for Met sidechains that were fully protonated),12C,15N background (ILVM-labeled) were prepared for the NMR studies. Overnight starter cultures in M9 medium supplemented with kanamycin (35 mg/l) were used to inoculate either H2O-based M9 media supplemented with 3 g/l of 13C-glucose (for CN-labeled samples or 12C-glucose for samples that were only uniformly 15N-labeled) and 15NH4Cl or D2O-based M9 media supplemented with 1 g/l 15NH4Cl and 2 g/l of 2H,13C-glucose (for ILVM-labeled samples). For CN (or N) labeling, the cultures were grown at 37°C under gentle agitation at 250 rpm until the A600 reached 0.7. Protein expression was then induced by the addition of 500 μM IPTG and 2% ethanol and the cultures were subsequently incubated for 18 h at 17°C. For ILVM-labeling, the cells were grown at 37°C under gentle agitation at 250 rpm until the A600 reached 0.6, at which point 100 mg/l of methyl-13C, 3,3-D2 α-keto-butyric acid and 250 mg/l of 3–3′-dimethyl-13C2, 3-D α-keto-isovaleric acid were added to the medium and growth was continued at 37°C for an additional hour. The culture was then slowly cooled on ice for 30 min, 120 mg/l of uniformly 13C-labeled methionine was added to the growth medium, and protein expression was induced by the addition of 500 μM IPTG. Overexpression continued for 22 h at 17°C. The cells were harvested as before and stored at -80°C.
Cell pellets were thawed and resuspended in 45 ml of lysis buffer (50 mM Tris pH 7.5, 1 M NaCl, 10% glycerol, 0.5% Triton X100, 0.05% NaN3) and supplemented with one tablet of protease inhibitor (EDTA-free, Pierce) and 1 mg/ml of lysozyme. The samples were kept at 4°C under gentle rotation for 1 h, then lysed by sonication (7 min, pulse on for 0.3 s, pulse off for 0.7 s, power amplitude at 35%) using a Sonic Dismembrator Model 500 (Thermo Fisher Scientific). The cell debris was separated by centrifugation at 15 000 rpm for 30 min (using a Fiberlite F21–8 × 50y rotor). The supernatant was then incubated with nickel NTA agarose beads (GoldBio), which were pre-washed with lysis buffer, for 1 h at 4°C under gentle rotation and the slurry was then poured into an Econo-Pac chromatography column (Bio-Rad). The column flow-through was discarded and beads were washed, first with 9 column volumes of lysis buffer, then with 9 column volumes of washing buffer 1 (50 mM Tris-HCl, pH 7.5, 3 M KCl, 0.05% NaN3) and finally with 9 column volumes of washing buffer 2 (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 20 mM imidazole, 0.05% NaN3). The bound protein was eluted with 50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 300 mM imidazole, 0.05% NaN3 and subsequently dialyzed against a buffer containing 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1.5 mM CaCl2, 5% glycerol, 5 mM β-mercaptoethanol under gentle stirring at 4°C. One unit of human α-thrombin (Enzyme Research Laboratory) per mg of protein was added after 1 h and dialysis was then continued for 16 h at 4°C. Complete cleavage of the N-terminal His6-tag was confirmed by SDS-PAGE. The resulting RslTpt1 protein contained a 4-amino acid tag (Gly-Ser-His-Met) at its N-terminus.
The cleaved RslTpt1 samples were supplemented with 10 mM each of DTT and EDTA, concentrated using Millipore 3 kDa MWCO concentrators, and injected into a HiLoad 16/60 Superdex 75 PG (GE Healthcare) pre-equilibrated with NMR buffer (20 mM HEPES pH 7.0, 200 mM NaCl, 5% glycerol, 2 mM EDTA, 2 mM DTT, 0.04% NaN3). All NMR samples were prepared in NMR buffer containing 5% D2O for field locking and 1 mM of 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, Sigma Aldrich) to reduce protein degradation over time. Samples used to record 13C-edited NOESY and methyl-based experiments were exchanged (using spin columns) against NMR buffer in which glycerol and EDTA were replaced by 2H8-glycerol and 2H16-EDTA (Cambridge Isotope Laboratories), respectively. NMR samples of NAD+-bound complexes were prepared by direct addition of the ligand (the NAD+ stock solution was prepared in NMR buffer appropriate for the experiment type followed by the adjustment of pH as needed) in 4 molar excess into the NMR sample.
Isothermal titration calorimetry measurements
The protocol used to prepare unlabeled RslTpt1 samples for isothermal titration calorimetry (ITC) measurements was the same as that for NMR, described above, except that no DTT was added after the overnight dialysis step. For the final purification step, a HiLoad 16/60 Superdex 75 PG column was pre-equilibrated with ITC buffer (20 mM HEPES pH 7.0, 200 mM NaCl, 5% glycerol, 2 mM EDTA, 0.04% NaN3). The same buffer was used to dissolve each of the six ligands used in the titrations: NAD+, ADPR (ADP-ribose), ADP, NADP+, nicotinamide and NMN (β-nicotinamide mononucleotide). Stock solution were 40 mM for NAD+, ADP and NADP+, and 20 mM for the other ligands. The pH in each case was adjusted to within ±0.1 units after dissolution of the ligand in ITC buffer. All ITC measurements utilized 100 μM of RslTpt1 in the cell and 3 mM of each of the ligands in the syringe with the exception of ADPR and NADP, for which concentrations of 4.5 and 6 mM respectively, were used. All experiments were performed on an ITC200 (Malvern) at 17°C using a 400 rpm stirring speed, a reference power of 10 μCal/s, and the following injection parameters: 20 injections of 2 μl with 4.0 s injection times, 210 s injection delays and filter periods of 5 s. All titrations were recorded at least in duplicate to confirm reproducibility of the parameters obtained. Reference isotherms using the same experimental parameters, in which the same concentration of ligand was injected into buffer alone, were obtained for each case. The thermograms were analyzed in Origin 5.0 (OriginLab) and thermodynamic parameters were obtained by fits to a one-site binding model.
NMR resonance assignments
All NMR experiments were performed at 17°C on Bruker Avance spectrometers operating at 600, 700, 800 or 900 MHz, all equipped with cryogenic probes capable of applying pulsed-field gradients along the z-axis. Sample concentrations ranged from 220–320 μM for apo-RslTpt1 (unliganded RslTpt1 was unstable in solution at higher concentrations) and 650–720 μM for the RslTpt1·NAD+ complex in 4 mm Shigemi tubes. All NMR data were processed using NMRpipe (13) and analyzed using NMRviewJ (14). 1H, 13C, 15N resonance assignments for both apo-RslTpt1 and the RslTpt1·NAD+ complex were achieved using standard gradient-enhanced, backbone-directed triple-resonance strategies and included HNCO, HN(CA)CO, HNCA, HN(CO)CA, CBCA(CO)NH, HNCACB experiments (15). Aliphatic sidechain resonances were assigned using H(CCO)NH, (H)C(CO)NH, HC(C)H-TOCSY and (H)CCH-TOCSY experiments (16). Resonance assignments for the aromatic sidechains were obtained using a 13C-edited NOESY-HSQC optimized for the aromatic region, while Met-ϵ resonances were assigned using 13C-NOESY-HMQC optimized for methyl resonances using ILVM-labeled apo-RslTpt1 or RslTpt1·NAD+ samples. Non-uniform sampling was used for some experiments and the corresponding sampling schemes were generated using the Poisson gap sampling method (17). Parameters for all experiments used in the resonance assignment procedure are provided in Supplementary Table S1 (apo-RslTpt1) and Supplementary Table S2 (RslTpt1·NAD+ complex). The resonance assignments for apo-RslTpt1 and the RslTpt1·NAD+ complex have been deposited into the BMRB with accession codes 30819 and 30820, respectively.
Measurement of ligand-induced chemical shift perturbations
Backbone chemical shift perturbations (900 MHz) induced on RslTpt1 by NAD+, NADP+, ADP, ADPR, NMN or nicotinamide were derived from 15N, 1H HSQC experiments (using 512 and 128 complex points for the direct and indirect dimensions, respectively, with corresponding spectral windows of 12.5 ppm and 34 ppm). The chemical shift perturbations induced on a 75 μM sample of RslTpt1 by 4 molar equivalents each of NAD+ and NADP+, or 12 molar equivalents of each of ADP and ADPR, or 10 molar equivalents of each of NMN and nicotinamide, were calculated using Equation (1) below. Perturbations (800 MHz) on the methyl resonances on Ile (δ1), Leu, Val and Met residues were obtained from 13C, 1H SOFAST-HMQC experiments (18) (using 512 complex points and 64 complex points for the direct and indirect dimensions, respectively, with corresponding spectral windows of 12.5 ppm and 18 ppm, utilizing a 0.2 s recycling delay). Measurements utilized a 70 μM sample of ILVM-labeled RslTpt1 and 4 molar equivalents of NAD+, or 8 molar equivalents of each of ADP or ADPR, or 10 molar equivalents of each of NMN or nicotinamide. 4-mm Shigemi tubes were used for all experiments. In all cases, the chemical shift perturbations values (Δδ, in ppm) were calculated using Equation (1):
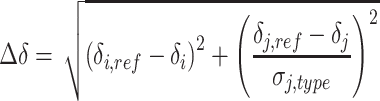 |
(1) |
where i and j index the 1H (amide or methyl) and the corresponding heteronucleus (15N or 13C), respectively. The reference shifts for apo-RslTpt1 in each case are indexed by ‘ref’ and the heteronuclear shift is normalized by the standard deviation (σj,type) obtained from the BMRB for the specific position (amide or methyl). In addition, a full titration set using both amide and methyl-based experiments was collected for NAD+, using 0, 0.1, 0.25, 0.5, 1, 2, 3 and 4 molar equivalents of the ligand.
Measurement of hydrogen/deuterium exchange
Amide-based hydrogen/deuterium exchange NMR experiments were performed at 700 MHz, using 4-mm Shigemi tubes at 17°C. Two 160 μl samples for each of apo-RslTpt1 or the RslTpt1·NAD+ complex at initial concentrations of 280 and 680 μM, respectively, were lyophilized and dissolved in either 160 μl of ddH2O (as reference) or 160 μl of 100% D2O. Then, a series of 10 1H,15N SOFAST HMQC spectra (using 512 complex points and 128 complex points for the direct and indirect dimensions, respectively, with corresponding spectral windows of 12.5 ppm and 34 ppm, utilizing a 0.2 second recycling delay) were collected over 3 h to monitor the loss of signal due to the exchange of the amide 1H with the solvent D2O.
{1H}-15N steady-state NOE measurements
Steady-state {1H}-15N NOE experiments (19) were performed at 800 MHz using uniformly 15N-labeled samples of apo-RslTpt1 (350 μM) or the RslTpt1·NAD+ complex (600 μM). 512 and 128 complex points with sweep-widths of 13 and 34 ppm for the 1H 15N dimensions, respectively. A set of two experiments were acquired with or without (reference) 5 s of 1H saturation in a total pre-delay period of 7 s.
Methyl relaxation dispersion measurements
13C-1H multiple-quantum dispersion CPMG experiments (20) were performed at 17°C (800 MHz) for both apo-RslTpt1 and the RslTpt1·NAD+ complex using 300 μM of uniformly 15N, 2H, ILVM-labeled sample of RslTpt1 in NMR buffer (see above) prepared in 100% D2O without or with 1.2 mM of NAD+. Experiments utilized sweep-widths of 12.5 and 18 ppm with 512 and 64 complex points in the 1H and 13C dimensions, respectively, using a recovery delay of 3 s and the relaxation delay, TCPMG set to 32 ms. The following RF fields 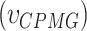 : 0, 40, 80, 120, 200, 320, 400, 480, 600, 720, 800, 920, 1000 Hz were used. The effective relaxation rate for a given RF field (R2,eff) was obtained using:
: 0, 40, 80, 120, 200, 320, 400, 480, 600, 720, 800, 920, 1000 Hz were used. The effective relaxation rate for a given RF field (R2,eff) was obtained using:
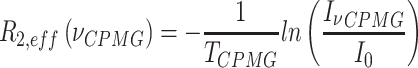 |
(2) |
Where 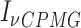 and I0 are the peak intensities at a field of νCPMG or in the absence of a CPMG block, respectively. The curves were visually inspected and Rex values for those resonances that displayed clear dispersion behavior were obtained using the following expression:
and I0 are the peak intensities at a field of νCPMG or in the absence of a CPMG block, respectively. The curves were visually inspected and Rex values for those resonances that displayed clear dispersion behavior were obtained using the following expression:
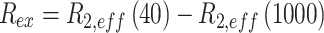 |
(3) |
Measurement of structural restraints
Distance restraints used to calculate the solution structures of apo-RslTpt1 and of the RslTpt1·NAD+ complex were obtained from a set of 3D 15N-edited NOESY-HSQC and 3D 13C-edited NOESY-HSQC experiments, the latter experiments were separately optimized either for aliphatic or aromatic resonances (see Supplementary Tables S1 and S2 for experimental details). Additional distance restraints involving methyl groups were derived from 3D 13C-NOESY-HMQC and 3D 13C-HMQC-NOESY-HMQC experiments. Mixing times of 150 ms were used in all NOESY experiments except for those optimized for methyl groups for which mixing times of 300 ms were used. Additional intra-molecular distance restraints for NAD+ were obtained from a set of 2D-NOESY spectra recorded using mixing times of either 150 or a 300 ms, performed on a 15N, 2H, ILVM-labeled sample of the RslTpt1·NAD+ complex in NMR buffer (see above) prepared in 100% D2O. Experimental parameters utilized for these experiments are provided in Supplementary Tables S1 and S2. The distance restraints were supplemented with backbone dihedral angle restraints obtained from chemical shifts utilizing the TALOS-N suite (21).
Structure calculations
Structure calculations for apo-RslTpt1 were performed using the ARIA2.3 suite (22,23) utilizing experimentally obtained distance and dihedral angle restraints. The PARALLHDG force field with PROLSQ (24) for non-bonded parameters was used. A simulated annealing protocol comprising of 20 000 steps (27 fs integration time) was carried out at high temperature (10 000 K) followed by two Cartesian cooling phases of 1000 and 50 K of 40 000 steps each (3 fs integration times). A network anchoring protocol was introduced for the first three iterations of the protocol using default parameters, and floating chirality was implemented for prochiral moieties. Starting with the fourth iteration, hydrogen bonding restraints were added for residues predicted by TALOS-N to be part of defined secondary structure elements and for which amide resonances persisted in 15N, 1H HSQC spectra 3 h after dissolution in pure D2O.
For calculation of the structure of the RslTpt1·NAD+ complex, the topology and parameter files for NAD+ were extracted from the PHENIX ligand database (25) and incorporated into the ARIA2.3 force-field, which was also manually modified to enable automatic assignment of NAD+ resonances. Structure calculations of the complex were performed using same strategy as described above with activation of the non-crystallographic symmetry and with a slight change of the simulated annealing protocol where the integration time was reduced from 3 to 2 fs during the two Cartesian cooling phases of the calculation. To ensure correct positioning of NAD+ a set of intermolecular distance restraints based on unambiguous assignments were used at the beginning of the structure calculations using broad upper and lower bounds (1.8 to 5.5 Å) to minimize packing artifacts. These intermolecular restraints were obtained from manually assigned cross-peaks of the methyl-based 3D 13C-NOESY-HMQC recorded on a uniformly 15N, ILVM-labeled sample of the RslTpt1·NAD+ complex, therefore the only detectable 1H-1H cross-peaks involved protons of methyl groups from RslTpt1 or protons from NAD+. After few runs, the ARIA protocol was able to assign a sufficient number of intermolecular NOEs, and therefore the loose manual distance restraints were no longer necessary and were removed in the final production run.
For the final production run, a similar simulated annealing protocol was used as described above except that 1000 structures were generated for each iteration. At the final step, the 100 lowest energies structures were submitted to a short restrained molecular dynamics simulation in explicit solvent using XPLOR-NIH (26). For the final NMR ensembles corresponding to apo-RslTpt1 or the RslTpt1·NAD+ complex, 20 lowest energies structures displaying the lowest RDC Q values (see below) that showed no distance restraint violations larger than 0.5 Å and no dihedral angle violations larger than 5° were analyzed using PROCHECK-NMR (27), WHATIF (28) and PSVS (29). The final structural ensembles, comprising 20 structures each for apo-RslTpt1 and the RslTpt1·NAD+ complex, were deposited in the PDB with accession codes 7KW8 and 7KW9, respectively. The regions of specific secondary structure (α-helix, β-strand and 310 helix) were determined using STRIDE (30) for consistency and the consensus regions were defined as those that were maintained in at least 80% of the structures in the final NMR ensemble (see Supplementary Table S3).
Measurement of 15N-1H residual dipolar couplings
Residual dipolar couplings (RDCs) were measured utilizing aligned media generated by the direct addition of Pf1 phage (Asla Biotech) into the NMR samples of apo-RslTpt1 (250 μM) or the RslTpt1·NAD+ complex (400 μM RslTpt1 containing a 4-fold excess of NAD+) to final phage concentrations of 16 and 15 mg/ml, respectively. In order to optimize the degree of alignment for the apo-RslTpt1 or the RslTpt1·NAD+ complex samples, the corresponding NaCl concentrations were adjusted by the addition of 5.4 and 5.8 μl of 5 M NaCl, respectively. Amide 15N,1H RDC values were extracted from a set of 2D HSQC-IPAP (31) experiments (512 and 300 complex points using sweep-widths of 12.5 ppm and 34 ppm in the 1H and 15N dimensions respectively in both cases). The alignment tensor values were obtained using PALES (32). Only regions with well-defined secondary structure values and those with {1H}-15N steady-state NOE values > 0.75 (800 MHz) were used in the analysis. Analyses were performed with the intact individual structures and separately for the N-lobe and C-lobe. The Q values were defined as (33):
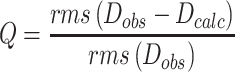 |
(4a) |
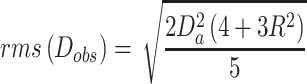 |
(4b) |
Where Dobs and Dcalc are the observed and calculated values of the 15N,1H RDC values; Da and R are the anisotropy and rhombicity of the alignment tensor. In order to determine the relative flexibility between the N- and C-terminal lobes, a generalized degree of order (GDO, 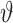 N/C) (34) was determined for each lobe using:
N/C) (34) was determined for each lobe using:
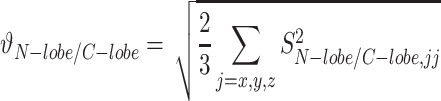 |
(5) |
Where Sjj are the eigenvalues of the alignment tensor for each of the N- or C-lobe. These GDO values were then used to define an order parameter given by (35):
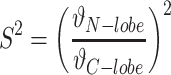 |
(6) |
Complementation of Saccharomyces cerevisiae tpt1Δ by RslTpt1 mutants
The S. cerevisiaetpt1Δ haploid strain YBS501 (MATa ura3–1 ade2–1 trp1–1 his3–11,15 leu2–3,11–2 can1–100 tpt1::LEU2 p360-TPT1), in which the TPT1 ORF was deleted and replaced by LEU2, is dependent for viability on the p360-TPT1 plasmid (CEN URA3 SceTPT1) (5). YBS501 was transformed with: (i) a p413-SceTPT1 plasmid (CEN HIS3 SceTPT1) plasmid as a positive control; (ii) the empty CEN HIS3 vector as negative control; and (iii) p413 (CEN HIS3) plasmids expressing wild-type and mutated RslTPT1 open reading frames under the control of a constitutive yeast TPI1 promoter. Transformants were selected at 30°C on His– agar medium. Three individual His+ colonies from each transformation were patched to His– agar medium and cells from each isolate were then streaked on agar medium containing 0.75 mg/ml 5-FOA (5-fluoroorotic acid). The plates were incubated at 30°C. The SceTPT1, RslTPT1, RslTPT1-(S125A-Q126A) and RslTPT1-(N105A-D112A) plasmids supported the formation of FOA-resistant colonies within 3 d. By contrast, the vector and the RslTPT1-(I109A-M117A) plasmids did not allow formation of FOA-resistant colonies after 8 d; accordingly, the I109A-M117A mutation was judged to be lethal. The RslTPT1-(N163A-V165A) plasmid sustained the formation of sporadic tiny FOA-resistant colonies after 6 d. Viable FOA-resistant tpt1Δ p413-RslTPT1 wild-type, S125A-Q126A and N105A-D112A strains were grown in YPD-Ad (yeast extract, peptone, 2% dextrose, 0.1 mg/ml adenine) liquid medium at 30°C to mid-log phase (A600 0.4–0.6), then diluted to attain A600 of 0.1, and aliquots (3 μl) of serial 10-fold dilutions were spotted on YPD agar plates and incubated at 20°C for 4 d, 25°C for 3 d, and 30, 34, and 37°C for 2 d. FOA-resistant tpt1Δ p413-RslTPT1-(N163A-V165A) colonies were patched to YPD agar and, after 2 d incubation at 30°C, transferred to YPD-Ad liquid medium; serial 10-fold dilutions were spot-tested for growth on YPD agar at 20, 25, 30, 34 and 37°C as above. No growth was detected at any temperature, from which we surmise that the N163A-V165A mutation was effectively lethal in vivo.
RESULTS
Structure of apo-RslTpt1
Resonance assignments for 94% of the backbone and 86% of the sidechains of RslTpt1 were obtained using a variety of labeling schemes (Supplementary Figure S2), as described in the ‘Materials and Methods’ section. These assignments were subsequently utilized to generate a sufficient number of NOE-based distance restraints that were supplemented with chemical shift-derived backbone dihedral angle constraints (21) to obtain the solution structure of apo-RslTpt1. Experimental constraints and structure statistics are shown in Table 1. RslTpt1 consists of two globular αβ-domains, the N-lobe (Val5-Gln78) and the C-lobe (Pro93-Val178) that are connected by a flexible linker (Figure 1A). The disorder in the linker, defined by the Gly79-Val92 segment, is reflected by the relatively low steady-state {1H}-15N NOE values (average ± standard deviation over the segment: 0.63 ± 0.12 at 800 MHz; 0.56 ± 0.05 over the conserved Gly79-Val82 segment; Supplementary Figure S3) compared to the values for the N-lobe (0.80 ± 0.10) and the C-lobe (0.81 ± 0.11). To independently validate the structure of apo-RslTpt1, we measured a set of one-bond 15N-1H residual dipolar couplings (RDCs) utilizing a Pf1 phage alignment medium (36). While good quality factors (Q) (33) are obtained for the individual lobes (N-lobe: 0.17 ± 0.03, C-lobe: 0.28 ± 0.04; Table 1), the corresponding values for the intact structures are significantly worse (0.38 ± 0.04). The lack of inter-lobe NOEs and low RDC-based S2 values (35) (0.36 ± 0.03) for the structures comprising the NMR ensemble suggests the absence of a well-defined orientation between the two lobes. As illustrated in Figure 2, superposition of the constituent structures of the ensemble on either their N-lobes or their C-lobes reveals a high degree of positional variation of the respective non-superimposed lobes.
Table 1.
Constraints, refinement and structure statistics for apo-RslTpt1 and the RslTpt1·NAD+ complex
| Pairwise Cartesian RMS deviation (Å) | ||||
|---|---|---|---|---|
| Apo-RslTpt1 | RslTpt1·NAD+ | |||
| PDB ID: | 7KW8 | 7KW9 | ||
| N-lobe | C-lobe | N-lobe | C-lobe | |
| Global heavy atoms | 1.61 ± 0.16 | 2.18 ± 0.58 | 1.73 ± 0.17 | 2.14 ± 0.55 |
| Global backbone atomsa | 0.75 ± 0.12 | 1.15 ± 0.35 | 0.67 ± 0.13 | 1.06 ± 0.30 |
| Ordered heavy atoms | 1.37 ± 0.12 | 1.70 ± 0.44 | 1.37 ± 0.35 | 1.76 ± 0.17 |
| Ordered backbone atomsb | 0.56 ± 0.10 | 0.79 ± 0.22 | 0.41 ± 0.13 | 0.74 ± 0.14 |
| Restraint Information | ||||
| NOE-derived distance restraints | ||||
| Intra-residue | 895 | 886 | ||
| Inter-residue | 1180 | 1547 | ||
| Sequential | 441 | 552 | ||
| Medium | 292 | 362 | ||
| Long | 447 | 633 | ||
| Inter-molecular | - | 29 | ||
| Ambiguous | 512 | 906 | ||
| Dihedral angle restraints | 278 | 269 | ||
| Hydrogen bond restraints | 132 | 136 | ||
| Energies (kcal/mol) | ||||
| Total | −5811.7 ± 78.1 | −5702.2 ± 110.9 | ||
| NOE | 3.8 ± 1.0 | 4.5 ± 1.2 | ||
| Dihedral angles | 97.6 ± 8.8 | 235.7 ± 11.6 | ||
| Ramachandran statistics (%) c | ||||
| Most favored regions | 85.1 ± 1.3 | 81.4 ± 1.5 | ||
| Additional allowed regions | 13.4 ± 1.3 | 16.6 ± 1.5 | ||
| Generously allowed regions | 1.1 ± 0.6 | 2.0 ± 1.2 | ||
| Disallowed regions | 0.4 ± 0.5 | 0.0 ± 0.0 | ||
| Restraint violations | ||||
| Distance restraints (>0.5 Å) | 0 | 0 | ||
| Dihedral angle restraints (>5°) | 0 | 0 | ||
| Average RMS deviation from experimental restraints | ||||
| Distance restraints (Å) | 0.027 ± 0.001 | 0.043 ± 0.001 | ||
| Dihedral angle restraints (°) | 0.471 ± 0.060 | 0.517 ± 0.072 | ||
| Average RMS deviation from idealized geometries | ||||
| Distance restraints (Å) | 0.017 ± 0.001 | 0.019 ± 0.001 | ||
| Dihedral angle restraints (°) | 1.528 ± 0.038 | 1.656 ± 0.032 | ||
| Average RMS Z-scores for deviation from current reliable structures d | ||||
| Bond lengths | 0.813 ± 0.014 | 0.910 ± 0.020 | ||
| Bond angles | 0.875 ± 0.024 | 0.914 ± 0.020 | ||
| Omega angle | 0.822 ± 0.035 | 0.903 ± 0.043 | ||
| Side-chain planarity | 0.782 ± 0.126 | 0.880 ± 0.106 | ||
| Improper dihedral distribution | 1.022 ± 0.032 | 1.165 ± 0.027 | ||
| Inside/Outside distribution | 0.980 ± 0.015 | 0.961 ± 0.013 | ||
| Average Z-scores for deviation from current reliable structures d | ||||
| First-generation packing quality | −1.2 ± 0.2 | −1.1 ± 0.2 | ||
| Second-generation packing quality | −2.2 ± 0.3 | −2.4 ± 0.3 | ||
| χ1/χ2 rotamer normality | −3.9 ± 0.4 | −4.3 ± 0.5 | ||
| Backbone conformation | −1.2 ± 0.3 | −1.7 ± 0.4 | ||
| Residual Dipolar Couplings e | ||||
| N-lobe | C-lobe | N-lobe | C-lobe | |
| Q for individual lobes | 0.17 ± 0.03 | 0.28 ± 0.04 | 0.25 ± 0.03 | 0.28 ± 0.04 |
| Pearson correlation co-efficient | 0.97 ± 0.01 | 0.93 ± 0.18 | 0.95 ± 0.01 | 0.92 ± 0.02 |
| S 2 | 0.36 ± 0.03 | 0.40 ± 0.06 | ||
aThe global RMSD calculations include residues 3–77 for the N-lobe and residues 93–178 for the C-lobe for both apo-RslTpt1 and the RslTpt1·NAD+ complex.
bThe ordered RMSD calculations include residues 4–14, 20–21, 32–33, 34–44, 50–59, 66–67, 73–75 for the N-lobe; residues 100–102, 103–105, 106–112, 122–124, 128–135, 136–137, 141–146, 148–153, 158–161, 165–168, 173–175, 176–177 for the C-lobe.
cBased on PROCHECK analysis.
dValues based on WHATIF.
eThe 15N-1H RDCs were only used to validate the structural ensembles and not utilized for structure refinement.
Figure 1.

Solution structure of unliganded RslTpt1. (A) Stereo view of the structure of apo-RslTpt1 as illustrated by a representative member selected from the NMR ensemble. The secondary structure elements are labeled; β-strands are colored magenta; α-helices are colored cyan; 310 helices are colored blue. The N-lobe (Val5-Gln78) and the C-lobe (Pro93-Val178) are connected by a flexible linker. The structures of the individual lobes are well defined in solution, but their relative orientation is not (see Supplementary Figure S3). (B) Alignment of the amino acid sequences of Tpt1 proteins from Runella slithyformis (Rsl, Genbank: WP_013927919.1), Candida auris (Cau, Genbank: PIS54536.1), Candida albicans (Cal, Genbank: AOW28085.1), Saccharomyces cerevisiae (Sce, Genbank: NP_014539.1) and Clostridium thermocellum (Cth, Genbank: ABN54255.1). Positions of amino acid identity/similarity in all five proteins are indicated by dots above the alignment. Gaps in the alignment are indicated by dashes. The peptide linker between the N-terminal RNA lobe and the C-terminal NAD+ lobe of RslTpt1 is indicated by the bidirectional arrow above the alignment. The conserved Arg-His-Arg-Arg catalytic tetrad is highlighted in cyan shading. Conserved amino acids that contact NAD+ in the RslTpt1·NAD+ structure reported here are highlighted in gold shading.
Figure 2.
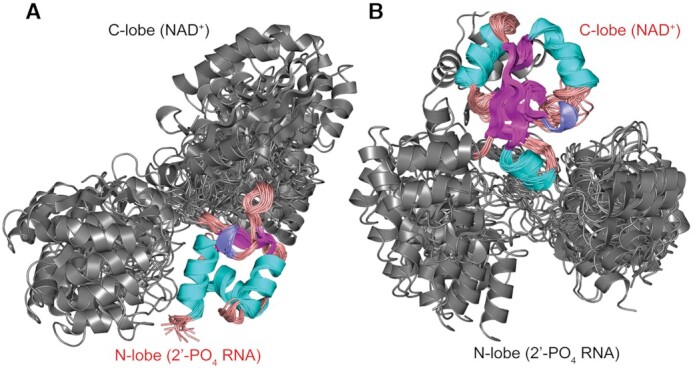
Structures of the NMR ensemble of apo-RslTpt1 overlaid on the N-lobe (that engages 2′-PO4 RNA, panel A) and the C-lobe (that engages NAD+, panel B). The lobe on which the structures are aligned is labeled in red font. The inability to align the structures on both lobes simultaneously illustrates the fact that while the structures of the individual lobes are well-defined in solution, their relative orientation is not, due to significant inter-lobe flexibility. Given that there are no experimental inter-lobe constraints, the apparent parsing into two sub-families for the unaligned lobe is likely an artifact of the force-field and the limited number of structures (20) selected to represent the NMR ensemble. Elements of secondary structure are indicated with α-helices, β-strands and 310 helices colored cyan, magenta and blue, respectively. Loops are colored salmon.
The RslTpt1 N-terminal lobe (Figure 1A), which has been termed the RNA lobe (12), has a winged helix fold comprising three α-helices (see Supplementary Table S3 for consensus secondary structures), α1 (5-14), α2 (34-44), and α3 (50–60) and a three-stranded anti-parallel β-sheet (β1: 32–33, β2: 65–67 and β3: 73–76). The 20–22 segment forms a 310 helix. The C-terminal lobe, also termed the NAD+lobe, forms a mixed αβ-fold (Figure 1A) resembling that of NAD+-utilizing toxins that catalyze ADP ribosylation of proteins and DNA (12). The C-lobe is composed of three α-helices (see Supplementary Table S3), α4 (106–112), α5 (128–136), and α6 (148–154), and two 310 helices (103–105 and 173–175). A 310 helix also forms in the 135–137 segment in about 40% of the structures in the NMR ensemble. The C-lobe also contains five β-strands - β4 (96–102), β5 (122–124), β6 (141–147), β7 (158–161) and β8 (165–168), that are organized as an antiparallel β-sheet. The C-terminus is well ordered, as indicated by high {1H}-15N NOE values (0.88 ± 0.07 for Ile176 and 0.74 ± 0.09 for Lys177) and forms a sixth β-strand (β9) in about 40% of the structures in the NMR ensemble. This last β-strand is also present in the structure of CthTpt1 (12).
Figure 1B shows an alignment of the RslTpt1 primary structure to those of biochemically validated Tpt1 orthologs from the bacterium Clostridium thermocellum and three species of fungi: the model yeast Saccharomyces cerevisiae (Sce) and the human fungal pathogens Candida albicans (Cal) and Candida auris (Cau). The alignment highlights 55 positions of sidechain identity/similarity in all five Tpt1 proteins. RslTpt1 residues Arg16, His17, Arg64, and Arg119 comprise an active site catalytic tetrad (highlighted in cyan in Figure 1B) (5) that is essential for the activity of RslTpt1, SceTpt1, and EcoTpt1 and is conserved in all Tpt1 orthologs (5,6). The catalytic tetrad directly engages the RNA 2′-PO4 to promote the transesterification step of the Tpt1 pathway (12). Arg16, His17 and Arg64 present a positively charged solvent-exposed surface at the interface between the two lobes of the apo-RslTpt1 structure. These sidechains show significant disorder in the NMR ensemble (Supplementary Figure S4A) in the absence of the RNA substrate. Arg119 is housed in the C-lobe and its sidechain is solvent-exposed and disordered in the NMR ensemble in the absence of NAD+ (Supplementary Figure S4B).
As mentioned above, crystal structures of two Tpt1 enzymes have been reported: a 1.4 Å structure of CthTpt1 in complex with CoA/ADP-ribose-1″-phosphate (12) and a 2.8 Å structure of ApeTpt1 in the unliganded state (10). There is a high degree of similarity between the N-lobe of RslTpt1 and the N-lobes of CthTpt1 (PDB: 6E3A, 6EDE) and ApeTpt1 (PDB: 1WFX) with RMSDs of 1.5 ± 0.1 Å and 1.4 ± 0.1 Å, respectively (Supplementary Figure S5), determined over regions of well-defined secondary structure. The corresponding C-lobes align with RMSDs of 1.9 ± 0.1 Å (Rsl versus Cth) and 2.2 ± 0.2 Å (Rsl versus Ape), respectively (Supplementary Figure S6), with the regions comprising β5 and α6 being the largest sources of these deviations. β5 in RslTpt1 is shorter than in both CthTpt1 and ApeTpt1; α6 displays a closed conformation in ApeTpt1 and a more open conformation in RslTpt1, with the α6 helix in CthTpt1 lying between these two extremes (Supplementary Figure S6). The degree of similarity between the C-lobes of RslTpt1, CthTpt1 and ApeTpt1 improves with the exclusion of these two structural elements, lowering the RMSDs to 1.4 ± 0.1 Å in both cases; these values are comparable to those obtained for the corresponding N-lobes.
Interaction of RslTpt1 with nucleotide ligands
We used isothermal titration calorimetry (ITC) measurements to gauge the affinity of RslTpt1 for NAD+ (Table 2, see Figure 3 for representative traces). NAD+ binds RslTpt1 with an apparent KD of 31 ± 1 μM (Table 2; Figure 3A shows a representative plot). It is notable that the intracellular concentrations of NAD+ of ∼1.0–1.6 mM in S. cerevisiae (37) and 2.6 mM in E. coli (38) are well in excess of this apparent KD. RslTpt1 is also able to bind ADP-ribose (ADPR, Figure 3B shows a representative plot) and ADP (Figure 3C shows a representative plot) with apparent KD values of 96 ± 6 μM and 123 ± 10 μM (Table 2), respectively. These observations suggest that the ADP moiety provides most of the binding energy for the interaction between RslTpt1 and NAD+; the nicotinamide riboside moiety makes only a small contribution. Indeed, no binding of nicotinamide (Figure 3D) or β-nicotinamide mononucleotide (NMN, Figure 3E) to RslTpt1 can be detected by ITC.
Table 2.
ITC-determined affinities of NAD+ and variants for RslTpt1
| Ligand | KD (μM)1 |
|---|---|
| NAD+ | 31 ± 1 |
| ADPR | 96 ± 6 |
| ADP | 123 ± 10 |
| Nicotinamide | NB2 |
| NMN | NB2 |
| NADP+ | 1021 ± 53 |
1Average and standard deviations obtained from duplicate measurements made at various times.
2NB implies no observable binding.
Figure 3.

Interaction of RslTpt1 with a variety of nucleotide ligands. Representative thermograms from isothermal titration calorimetry (ITC) measurements for the interaction of RslTpt1 with (A) NAD+, (B) ADP-ribose (ADPR), (C) ADP, (D) nicotinamide and (E) β-nicotinamide mononucleotide (NMN), are shown. In each case, the top panel shows the heat corresponding to each injection and the bottom panel shows the corresponding fit of the integrated intensity to a one-site binding model. The KD values and errors indicated on the thermograms correspond to those obtained from the fits for the specific traces that are shown. Averages (and corresponding standard deviations) over multiple measurements are listed in Table 2.
Next, we utilized solution NMR to determine the nature of the interaction between NAD+ and RslTpt1 in residue-specific detail. As shown in Figure 4A, significant chemical shift perturbations (CSPs) are observed for backbone 15N, 1H resonances in the presence of NAD+ and are localized exclusively on the C-lobe (Δδ = 0.14 ± 0.12 ppm, averaged over the 72 C-lobe amide resonances for which numerical values could be obtained from 2D correlation spectra) while no significant CSPs are noted in the N-lobe, indicating that NAD+ partitions exclusively to the C-lobe. Further, the lack of CSPs on the N-lobe in the presence of NAD+ is indicative of the fact that this ligand is unable to induce stable closure between the two lobes. A closer inspection of the C-lobe CSPs shows that the most significant Δδ values (>0.25 ppm) are centered on β4 (Thr101: 0.44 ppm, Ala102: 0.47 ppm), α4 (Leu108: 0.36 ppm), the α4-β5 loop (Ser118: 0.39 ppm), β5 (Val122: 0.35 ppm), α5 (Thr130: 0.39 ppm, Ala131: 0.42 ppm, Lys133: 0.27 ppm, Val134: 0.31 ppm, Gly135: 0.35 ppm) and near the C-terminus (Val171: 0.27 ppm). In addition, specific resonances on β4 (Gly100), on the α4-β5 loop (Lys115, Arg119), on the β5-α5 loop (Asp127), on α5 (Ile128), and on β6 (Val143, Val146) are significantly shifted (or fully broadened out), their NAD+-bound states are not available in the 2D correlation spectra for analysis. Most residues that show large CSPs appear to be in slow exchange on the chemical shift timescale (see Supplementary Figure S7 for representative examples). This is, however, not surprising given the extremely large chemical shift differences between the free and NAD+-bound states of RslTpt1 that enable the slow exchange condition (39) to be maintained despite the modest affinity. CSPs in the presence of ADPR or ADP are also limited to the C-lobe with no significant perturbations seen on the N-lobe (Supplementary Figure S8). As expected in light of the ITC data, no significant CSPs are observed in the presence of nicotinamide or NMN (not shown).
Figure 4.
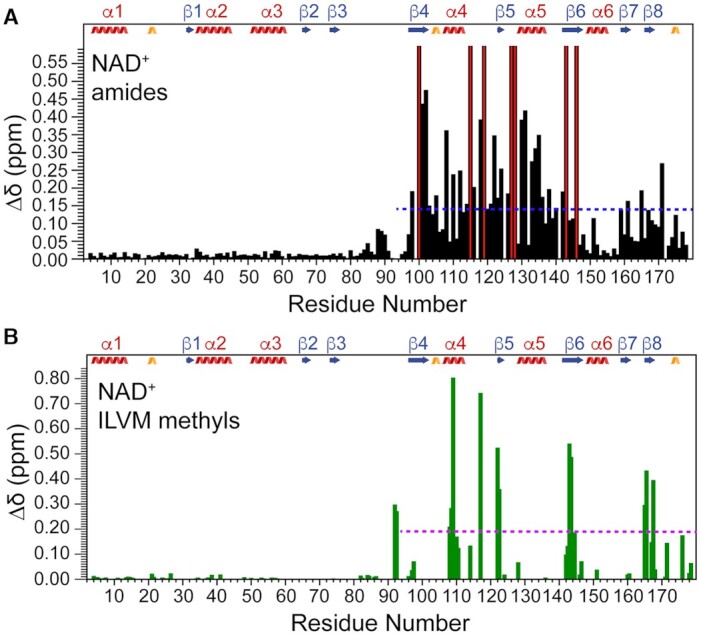
Spectral perturbations induced by NAD+ on RslTpt1. Chemical shift perturbations induced on (A) amide 15N, 1H resonances or (B) methyl 13C, 1H resonances of Ile (δ1 only), Leu, Met and Val residues in the presence of 4 molar equivalents of NAD+, are shown. The red bars indicate residues for which the corresponding resonances were perturbed but the chemical shifts for their final NAD+-bound states could not be analyzed due to vanishing resonances, missing assignments, or spectral crowding. The blue and magenta dashed lines represent the average Δδ values for the C-lobe amide (0.14 ppm) and ILVM methyl (0.19 ppm) resonances, respectively. Secondary structural elements (definitions are as per apo-RslTpt1, see Supplementary Table S3) are indicated; α-helices, β-strands and 310-helices are colored red, blue and gold, respectively.
Next, we analyzed the CSPs induced on the 13C,1H resonances of Ile (δ1 only), Leu, Val and Met methyl residues, given that such residues are candidates to take part in hydrophobic interactions with NAD+. As in the case of the backbone resonances, the largest methyl CSPs in the presence of NAD+ (Figure 4B; methyl CSPs in the presence of ADPR and ADP are shown in Supplementary Figure S9) are seen for the C-lobe (Δδ=0.19 ± 0.21 ppm over 37 Ile-δ1, Leu, Val and Met methyl groups) with no significant perturbations seen in the N-lobe (<0.01 ppm). The largest CSPs (>0.39 ppm) are noted for Ile109, δ1 (0.80 ppm), Met117, ϵ (0.74 ppm), Val122, γ1/2 (0.52/0.36 ppm), Val143, γ1/2 (0.54/0.49 ppm), Val165, γ1/2 (0.43/0.29 ppm) and Leu167, δ1/2 (0.39/0.15 ppm).
The interaction of NAD+ with RslTpt1 can also be gauged by the observed perturbations induced on the 1H spectrum of NAD+ by the presence of RslTpt1. While analyses of the perturbations of the 1H nuclei of ribose moieties (except H1D, Δδ = 7 Hz; see Supplementary Figure S10) is complicated by extensive line-broadening in an already crowded region, measurable CSPs (>5 Hz) are noted for 1H nuclei of both the adenosine (H2A, 12 Hz) and nicotinamide moieties (H4N, 10 Hz; H5N, 6 Hz). Some representative examples of these CSPs are shown in Supplementary Figure S10.
In line with the large CSPs described above, increased protection from solvent is seen for the RslTpt1 C-lobe in the presence of NAD+. Indeed, the backbone amides for apo-RslTpt1 show rapid exchange with solvent and few amide resonances from either the N- or the C-lobe are observable after ∼3 h following dissolution in D2O. While the protection of resonances corresponding to the N-lobe remains largely unchanged in the presence of an excess of NAD+, a substantial increase in the protection of C-lobe amide resonances is noted (Supplementary Figure S11).
The recognition of the internal 2′-PO4 of an RNA substrate by Tpt1 is proposed to occur through the N-lobe (12). We measured CSPs induced by NADP+ that contains a 2′-PO4 and which we suspected might bind to the RNA substrate site. ITC assays show that RslTpt1 binds NADP+ with an apparent KD of ∼1 mM (Table 2; Figure 5A), that is, with ∼30-fold less affinity than NAD+. The observed backbone amide CSPs indicate that NADP+ interacts exclusively with the N-lobe (Δδ = 0.02 ± 0.03 ppm) of RslTpt1 (Figure 5B). No significant NADP+-induced CSPs are seen in the C-lobe. The largest CSPs (>0.08 ppm) are seen for N-lobe residues Lys6 (0.09 ppm), Ser12 (0.17 ppm) and Arg64 (0.11 ppm). A large CSP is also noted for Val82 (0.10 ppm) in the inter-lobe linker peptide. It is notable that the N-lobe constituents of the RslTpt1 catalytic tetrad all show significant perturbations, as follows: Arg16 (0.07 ppm), His17 (0.07 ppm) and Arg64 (0.11 ppm). Arg16 and Arg64 have been proposed to engage the 2′-PO4 of the RNA-2′-phospho-ADPR intermediate and catalyze step 2 transesterification (Supplementary Figure S1) to form the RNA 2′-OH product. Indeed, in the structure of CthTpt1, the 1′-PO4 product (corresponding to a 2′-PO4 in the reactant) is encased within a mesh of hydrogen bonds involving the corresponding residues Arg18 and Arg68 (12). In the case of RslTpt1, alanine mutations of Arg16 and Arg64 were found to reduce the step 2 transesterification rate constant by 710-fold and 210-fold, respectively (6).
Figure 5.

Interaction of RslTpt1 with NADP+. (A) Representative ITC thermogram and corresponding fit of the integrated intensity to a one-site binding model are shown in the top and bottom panels, respectively. (B) Chemical shift perturbations induced on the amide 15N, 1H resonances of RslTpt1 in the presence of 4 molar equivalents of NADP+. The magenta dashed line indicates the average Δδ value for the N-lobe amides (0.02 ppm). The red bar represents the Val7 resonance that is fully broadened out in the presence of NADP+. (C) NADP+-induced chemical shift perturbations for amide resonances mapped onto a surface representation of the RslTpt1 N-lobe. Residues that display the most significant perturbations are labeled. Residues for which resonances are missing and those corresponding to the 4-residue N-terminal tag are colored black and yellow, respectively.
As described above, NAD+ and NADP+ partition exclusively to the C- and N-lobes of RslTpt1, respectively, with the ligand binding to a given lobe having no observable effect on the other. To further probe this effect, we measured by ITC the binding affinity of NAD+ for RslTpt1 pre-saturated with NADP+. Only a small change in the NAD+ binding affinity KD = 47 ± 3 μM (compared to 31 ± 1 μM, Table 2) was noted (Supplementary Figure S12A). The affinity of NADP+ toward RslTpt1 was virtually the same with (951 ± 33 μM, see Supplementary Figure S12B) or without (1021 ± 53 μM, Table 2) pre-saturation with NAD+. CSPs induced by each ligand, NAD+ on the C-lobe and NADP+ on the N-lobe, are also independent of the presence of the other (Supplementary Figure S13). These observations confirm that binding of NADP+ and/or NAD+ is insufficient to induce stable interaction between the two lobes of RslTpt1 or influence the affinities for the corresponding ligands. It is, however, possible that a more substantial engagement of the RNA-binding groove on the N-lobe, beyond simple low affinity 2′-PO4 recognition (as in NADP+), is required to elicit cross-talk between the two lobes.
Structure of the RslTpt1·NAD+ complex
Our NMR studies on apo-RslTpt1 gave us confidence that we would be able to solve the structure of the RslTpt1·NAD+ complex using solution NMR methodology. However, significant changes in resonance positions evident from the amide and methyl CSPs in the presence of NAD+ (Figure 4) necessitated extensive reassignment of the backbone and sidechain resonances of RslTpt1. The near complete assignments of backbone (92%) and sidechain (83%) resonances of RslTpt1 bound to NAD+ (Supplementary Figure S2) could then be leveraged to yield a sufficient number of intra-molecular and inter-molecular NOE-based distance restraints (see Supplementary Figure S14 for some representative examples) that were combined with chemical shift based dihedral angle restraints (Table 1) to obtain a structure of the RslTpt1·NAD+ complex (Figure 6A).
Figure 6.
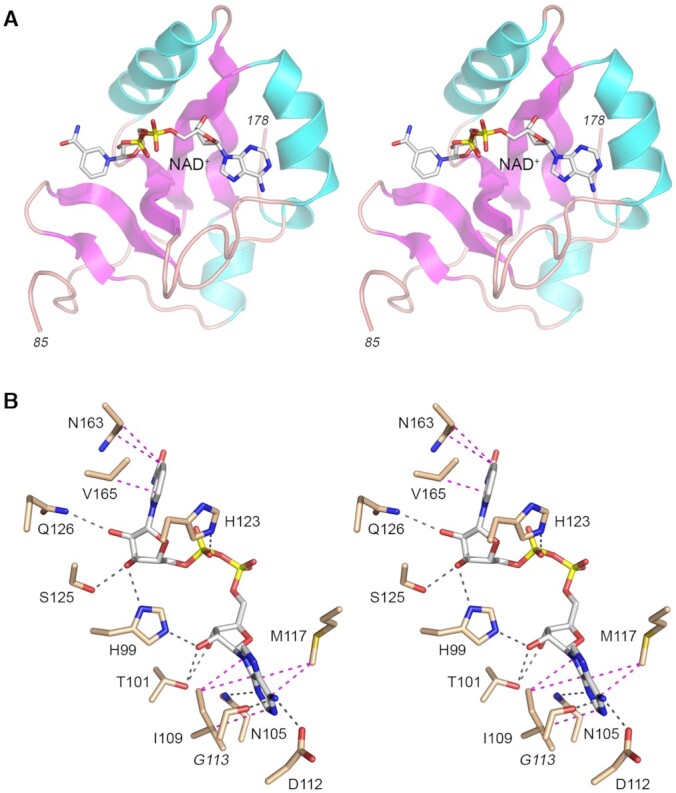
Structural basis for NAD+ recognition. (A) Stereo view of a representative structure of NAD+ (stick model with gray carbons and yellow phosphorous atoms) bound to the C-lobe of RslTpt1 (cartoon model colored as in Figure 1A). (B) Detailed stereo view of the enzymic contacts to NAD+. RslTpt1 amino acids are depicted as stick models with beige colored carbons. Side chains are labeled in plain font; the Gly113 main chain carbonyl is labeled in italics. Hydrogen bonds are denoted by black dashed lines and van der Waals contacts by magenta dashed lines. The hydrogen-bond between His123 and the pyrophosphate backbone (disordered in the NMR ensemble) is presumed based on proximity.
A comparison of secondary structure elements between the apo and NAD+-bound states of RslTpt1 shows a few changes (Supplementary Table S3). Strand β6 appears to be slightly distorted at Leu144 in some of the structures; the Glu103-Asn105 segment that forms a 310-helix in apo-RslTpt1 integrates into α4 in the complex that now encompasses residues Glu103-Asp112 in the complex. Additionally, the terminal β9-strand (Ile176-Lys177) appears to be better defined in the structural ensemble representing the complex, where it is formed in all of the structures compared to only 40% of the structures for apo-RslTpt1. Other changes are restricted to single-residue variations at the termini of the secondary structural elements (Supplementary Table S3). While their positions remain largely unchanged, the relative orientations of these secondary structure elements appear to be altered upon engaging NAD+. The RMSD over the secondary structural elements for the C-lobe comparing the NMR ensembles of apo-RslTpt1 and the RslTpt1·NAD+ complex is found to be significant (Supplementary Figure S15A) at 1.9 ± 0.2 Å in line with the large CSPs induced by the ligand. A smaller but significant RMSD of 1.4 ± 0.2 Å is also noted for the N-lobe (Supplementary Figure S15B). Given the limited CSPs induced on the N-lobe by NAD+ (<0.01 ppm, Figure 4), this difference is likely related to the overall precision of the two NMR-determined ensembles.
As suggested by the lack of CSPs on the N-lobe, no significant change is seen in the degree of inter-lobe mobility and the two lobes remain disordered with respect to each other (Supplementary Figure S16). The presence of significant inter-lobe flexibility is also validated by analyses of the 15N-1H RDCs. As in the case of apo-RslTpt1, the Q-factors corresponding to the experimental RDC values for the individual lobes (N-lobe: 0.25 ± 0.03, C-lobe: 0.28 ± 0.04) suggest good agreement between the experimental and predicted values. In contrast, the Q-factors for the intact structures are significantly higher (0.69 ± 0.23) suggesting poor correlation between the predicted and experimentally determined RDCs. Additionally, the RDC-based S2 values also confirm substantial inter-lobe mobility (S2 = 0.40 ± 0.06).
Recognition of NAD+
The NAD+ moiety makes several contacts with the C-lobe of RslTpt1 (Figure 6B); no stable contacts with the N-lobe are evident. The adenine ring stacks against the hydrophobic residues Ile109 and Met117 (the methyl groups of both hydrophobic residues show substantial CSPs; see Supplementary Figure S17) and engages in a pair of hydrogen bonds to enzymic sidechains: adenine-N3 with Asn105-Nδ2 and adenine-N6 with Asp112-Oδ2. The backbone carbonyl oxygen of Gly113 is also within hydrogen-bonding distance of adenine-N6 in many of the structures. The contacts of the α4-β5 loop (Gly113, Ile114, Met117) with NAD+ lead to a greater degree of closure of this loop in the NAD+-bound state compared to apo-RslTpt1 (Supplementary Figure S15A). This conformational change is reflected in large amide CSPs (0.20 ± 0.10 ppm; in addition to the disappearance of the resonances of Lys115 and Arg119). The adenosine ribose 2′-OH forms a bifurcated hydrogen-bond with His99-Nϵ2 and Thr101-Oγ1. This latter interaction would be disrupted by the presence of a 2′-PO4, which would also elicit numerous steric clashes, most notably with His99 and Gly100, explaining the inability of the C-lobe to engage NADP+. The pyrophosphate that links the adenosine and the nicotinamide riboside moieties is relatively disordered in the NMR ensemble (Supplementary Figure S18) either due to the absence of specific constraints resulting from a lack of 1H nuclei within this segment, or dynamics within the complex, or both. However, His123 or His138, both located in close proximity to the phosphates, could potentially serve as hydrogen-bond donors to stabilize this segment. The nicotinamide moiety of NMN stacks against Val165 (the methyl groups of which show substantial CSPs, see Supplementary Figure S17) and Asn163. The NMN ribose is stabilized by a pair of hydrogen-bonds of the 2′-OH with Gln126-Nϵ2 and the 3′-OH with Ser125-Oγ.
Conformational dynamics in the RslTpt1·NAD+ complex
As seen in Supplementary Figure S18, the NMR structural ensemble of the RslTpt1·NAD+ complex suggests some disorder in the orientation of NAD+. The adenine moiety appears to be better ordered (RMSD: 0.89 ± 0.44 Å) compared to the nicotinamide moiety (RMSD: 1.33 ± 0.68 Å). The relatively stable interaction of the RslTpt1 C-lobe with the adenosine moiety of ADP is in line with the ITC results that indicate the dominant role of the latter in the interaction compared to nicotinamide. In order to test whether this disorder reflects, at least in part, conformational dynamics within the solution ensemble, we conducted relaxation dispersion measurements on multiple-quantum coherences involving the methyl groups of Ile (δ1), Leu, Val and Met residues. For apo-RslTpt1, only three residues, Ile109, Ile114 and the C-terminal Val178, display significant exchange contributions (Rex > 5 s−1; Figure 7A) suggesting limited conformational flexibility on the millisecond timescale in the unliganded state. In contrast, Rex values exceeding 5 s−1 are seen for 13 sets of methyl resonances, all of them belonging to the C-lobe of RslTpt1, upon formation of the RslTpt1·NAD+ complex (Figure 7A). Val165, which shows large Rex values for both methyl groups (16.6 and 12.1 s−1), stacks against the nicotinamide ring (Figure 7B). This exchange contribution likely results from fluctuations in the relative orientations of nicotinamide and the Val165 sidechain leading to large transient variations in the chemical shifts of the latter and consequently to large Rex values. In contrast, the sidechains of Ile109 (33.6 s−1) and Ile114 (18.2 s−1) that show substantial Rex values in the unliganded state of RslTpt1, appear to be stabilized through their contacts with adenine ring of NAD+ (Figure 7B) in the RslTpt1·NAD+ complex.
Figure 7.
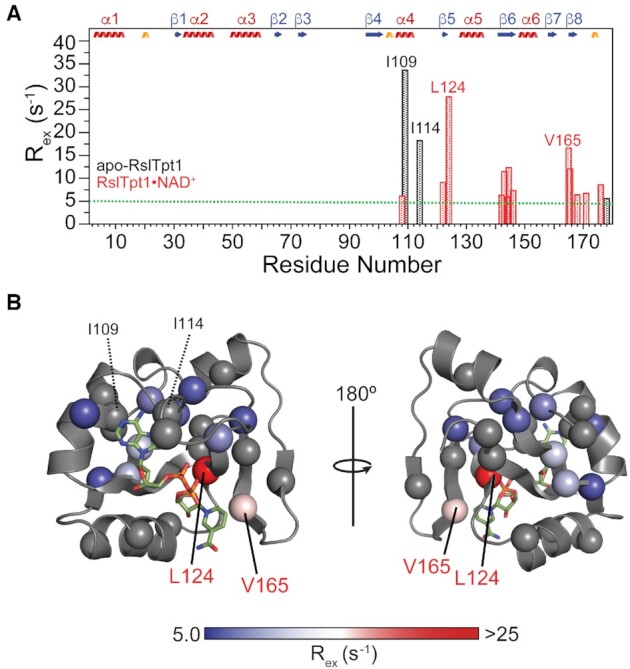
Conformational exchange in the RslTpt1 C-lobe when bound to NAD+. (A) Exchange contributions [Rex = 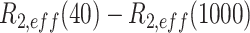 > 5 s−1] to the multiple-quantum relaxation rates of Ile (δ1), Leu, Val and Met methyl resonances for apo-RslTpt1 (black) and the RslTpt1·NAD+ complex (red) are plotted against residue number. Those residues that display the largest Rex values for the apo (Ile109: 33.6 s−1 and Ile114: 18.2 s−1) and the NAD+-bound states (Val124: 27.8 s−1 and Val165: 16.6/12.1 s−1) are labeled. (B) The Rex values of methyl groups (denoted by spheres) are mapped onto the structure of the C-lobe in its NAD+-bound state and shown using a blue to red gradient; methyl groups without any detectable Rex values are colored gray. Leu124 that shows the largest exchange contribution in the bound state lies in close spatial proximity to the phosphate backbone; Val165, whose methyl groups display substantial Rex values contact the nicotinamide moiety. Also indicated (in black font) are Ile109 and Ile114 that show substantial Rex values in the unliganded state. These residues, that engage the adenosine moiety of NAD+, have no detectable exchange contributions in the NAD+-bound state.
> 5 s−1] to the multiple-quantum relaxation rates of Ile (δ1), Leu, Val and Met methyl resonances for apo-RslTpt1 (black) and the RslTpt1·NAD+ complex (red) are plotted against residue number. Those residues that display the largest Rex values for the apo (Ile109: 33.6 s−1 and Ile114: 18.2 s−1) and the NAD+-bound states (Val124: 27.8 s−1 and Val165: 16.6/12.1 s−1) are labeled. (B) The Rex values of methyl groups (denoted by spheres) are mapped onto the structure of the C-lobe in its NAD+-bound state and shown using a blue to red gradient; methyl groups without any detectable Rex values are colored gray. Leu124 that shows the largest exchange contribution in the bound state lies in close spatial proximity to the phosphate backbone; Val165, whose methyl groups display substantial Rex values contact the nicotinamide moiety. Also indicated (in black font) are Ile109 and Ile114 that show substantial Rex values in the unliganded state. These residues, that engage the adenosine moiety of NAD+, have no detectable exchange contributions in the NAD+-bound state.
Structure-guided mutagenesis of the NAD+ binding site
Many of the RslTpt1 amino acids that contact NAD+ are conserved in other Tpt1 orthologs (Figure 1B; highlighted in gold shading), suggesting a common mode of NAD+ recognition by members of the Tpt1 family. In particular, the enzymic contacts to the NAD+ adenosine nucleoside are common to the RslTpt1·NAD+ complex described here and the CthTpt1·ADP-ribose-1″-PO4 complex reported previously (12). The adenosine ribose 2′-OH forms a bifurcated hydrogen-bond with the histidine and threonine side chains of the His-Xxx-Thr motif found in all Tpt1 enzymes. Alanine scanning mutagenesis of budding yeast SceTpt1 established that the ribose 2′-OH contacts are functionally redundant, insofar as single His-Ala and Thr-Ala mutations had no effect on SceTpt1 function in vivo, whereas a His-Ala/Thr-Ala double mutation of the His-Xxx-Thr motif was lethal (5). Guided by the CthTpt1·ADP-ribose-1″-PO4 structure, an equivalent H99A-T101A double-mutation was made in RslTpt1 and found to be lethal when tested for tpt1Δ complementation (12).
Here, we relied on the NMR structure of the RslTpt1·NAD+ complex to guide a new round of RslTpt1 alanine mutagenesis, targeting pairs of amino acid sidechains that make atomic contacts to the same constituent of the NAD+ substrate. Thus, we mutated Ile109 and Met117 that sandwich the adenine nucleobase, Asn105 and Asp112 that make hydrogen bonds to the adenine nucleobase, Ser125 and Glu126 that make hydrogen bonds to the NMN ribose hydroxyl, and Asn163 and Val165 that make van der Waals contacts with nicotinamide (Figure 6B). The wild-type and mutant RslTPT1 alleles were placed on yeast CEN plasmids under the control of the constitutive TPI1 promoter and tested for complementation of tpt1Δ by plasmid shuffle (5). We thereby found that RslTPT1-(S125A-Q126A) and RslTPT1-(N105A-D112A) cells were viable and grew as well as wild-type RslTPT1 cells on YPD agar at all temperatures tested, as gauged by colony size (Figure 8, bottom panel; scored as +++ growth in Figure 8, top panel). Thus, we surmise that the pairs of hydrogen bond contacts to the NMN ribose hydroxyls and the adenine N3 and N6 atoms are not critical for RslTpt1 function in vivo. In this light of this observation, it is noteworthy that RslTpt1 residues Asn105, Asp112, and Gln126 are not conserved in other Tpt1 orthologs (Figure 1B).
Figure 8.

Contacts to nicotinamide and the adenine nucleobase of NAD+ are essential for RslTpt1 function. The activity of the indicated double-alanine RslTpt1 mutants in S. cerevisiae tpt1Δ complementation was tested by plasmid shuffle as described in the ‘Materials and Methods’ section. Results are summarized in the top panel. Serial dilutions of the viable tpt1Δ RslTPT1 strains expressing wild-type (WT) RslTpt1 and the N105-D112A or S125A-Q126A mutants were spot-tested for growth on YPD agar at the temperatures specified (bottom panel).
In contrast, the N163A-V165A and I109A-M117A mutations were lethal when tested for tpt1Δ complementation (Figure 8), signifying that the stacking interactions with the nicotinamide and the adenine moieties of NAD+ observed in the NMR structure are essential for RslTpt1 activity. The essential Ile-Met pair that sandwiches adenine is strictly conserved in the three fungal Tpt1 orthologs shown in Figure 1B and corresponds to an Ile-Gln pair in CthTpt1. The essential Asn-Val pair that contacts nicotinamide is conserved as Asn-Val in the fungal Tpt1 proteins and is replaced by the conservative by Asp-Val in CthTpt1 (Figure 1B).
DISCUSSION
Our application of solution NMR methods to RslTpt1 has provided significant new insights into its structure and mechanism, as follows: (i) by highlighting the highly dynamic spatial orientations of the component N- and C-lobes, irrespective of occupancy of the NAD+ pocket on the C-lobe; (ii) by solving the solution structure of a Tpt1·NAD+ binary complex and thereby identifying the network of enzymic contacts to the phosphoacceptor substrate; and (iii) by suggesting clues to how Tpt1 might catalyze phosphorolytic displacement of nicotinamide during the first step of the Tpt1 reaction pathway.
The NMR structural ensembles underscore how the separately well-folded and N- and C-lobe modules of RslTpt1 are arrayed in solution with a high degree of positional freedom with respect to one another. NAD+ binding to the C-lobe does not suffice to impose order on the system. This domain mobility resonates with, and likely accounts for, our inability to grow crystals of RslTpt1, in unliganded form or in the presence of NAD+. We were similarly thwarted in our efforts to grow crystals of several other Tpt1 orthologs that we produced and purified (11), the exception being CthTpt1 by virtue of the fact that a sub-population of the recombinant enzyme molecules purified from E. coli were stably complexed with ADP-ribose-1″-PO4 and it was this state of CthTpt1 that was selectively captured in crystallo (12). The key distinction between the ‘closed’ domain conformation seen in the product-like complex of CthTpt1·ADP-ribose-1″-PO4 and the conformationally mobile ‘open’ arrangements of the RslTpt1·NAD+ ensemble is that the former structure is locked down by multiple atomic contact between N-lobe active site amino acids and the phosphate of ADP-ribose-1″-PO4 (Figure 9), whereas no such interactions are possible with NAD+. Since the phosphate of ADP-ribose-1″-PO4 would have derived from the 2′-PO4 of an RNA substrate (note its proximity to the pAp 2′-OH in Figure 9), we envision that domain closure in RslTpt1 (and other orthologs) is contingent on binding of a 2′-PO4 RNA substrate, per se or simultaneously with NAD+.
Figure 9.
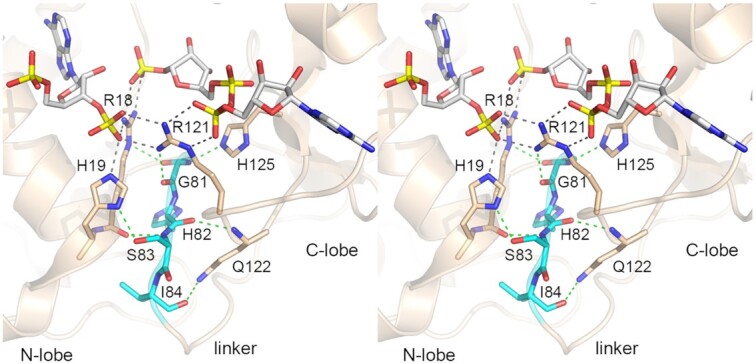
Contributions of the interdomain linker in stabilizing a closed conformation in the presence of ligands. The product state mimic structure of CthTpt1 illustrates the numerous contacts between the N- and C-lobes. The conserved N-terminus of the variable linker 81GHSI84 (the corresponding region in RslTpt1 is 79GHSV82) shown in cyan plays a major role in maintaining inter-lobe contacts some of which are mediated by the ADP-ribose-1″-phosphate and pAp products.
The freedom of movement of the RslTpt1 lobes in solution owes to the conformational flexibility of the inter-domain linker, 79GHSVEVDLGYEKQV92. The linker segment varies in length among Tpt1 homologs and its primary structure is not well conserved, except for the Gly-His-Ser-(Val/Leu/Ile) tetrapeptide at the junction to the N-lobe (Figure 1A). In the product complex of CthTpt1 with pAp in the RNA site and ADPR-1″-PO4 in the NAD+ site, the 81GHSI84 linker tetrapeptide forms a key part of the inter-domain interface of the closed enzyme conformation (Figure 9). The linker makes multiple backbone hydrogen bonds to moieties in the N- and C-lobes. Gly81-N coordinates His125 (part of the NAD+ pocket equivalent to His123 in RslTpt1). Gly81-O hydrogen bonds to Arg121 (Arg119 in RslTpt1), an essential active site residue that engages the ribose 3′-PO4 at the RNA splice junction and the adenylate-phosphate of ADPR-1″-PO4 (Figure 9). The main-chain carbonyl preceding Gly81 hydrogen bonds with Arg18 (Arg16 in RslTpt1), an essential catalytic residue that engages the ribose 3′-PO4 and 2′-PO4 at the RNA splice junction. His82-O and Ile84-O receive hydrogen bonds from the Gln122 mainchain amide and sidechain amide, respectively. Ser83-N makes a hydrogen bond to the Arg81 carbonyl. Ser83-Oγ donates a hydrogen bond to active site constituent His19-Nδ, thereby ensuring a proper tautomer for His19 interaction with the pAp 3′-PO4 (Figure 9). We envision that the conserved linker segment will play a similar stabilizing role in promoting the closed catalytic conformation of other Tpt1 orthologs. This linker segment (79GHSV82) in RslTpt1 is highly disordered in the unliganded state ({1H}-15N NOE = 0.56 ± 0.05) and becomes somewhat more ordered (0.65 ± 0.07) when bound to NAD+, suggesting the possibility of transient contacts in the NAD+-bound state. One may speculate that these contacts are stabilized by the formation of a ternary Michaelis complex prior to Step 1 chemistry.
In addition to the variable inter-lobe orientation observed here for RslTpt1, there is some disorder noted in the position/conformation of NAD+ within the C-lobe of RslTpt1. As ascertained by relaxation dispersion measurements on methyl-bearing sidechains of RslTpt1, this disorder can be attributed, at least in part, to dynamics on the millisecond timescale involving the NAD+ moiety and/or the RslTpt1 sidechains in its close spatial proximity. It is conceivable that there are local changes in NAD+ conformation or NAD+ contacts upon formation of a Step 1 Michaelis complex with 2′-PO4 RNA.
Nine of the twelve RslTpt1 amino acids that contact NAD+ are conserved in other Tpt1 orthologs (Figure 1B). Alanine scanning of the pairs of non-glycine residues that contact the constituents of the NAD+ substrate has now identified hydrogen-bonding to the adenosine 2′-OH (by His99 and Thr101), bilateral stacking on the adenine nucleobase (by Ile109 and Met117), and van der Waals interactions with one face of the nicotinamide ring (by Asn163 and Val165) as essential for RslTpt1 activity in vivo.
Beyond delineating the principles of NAD+ recognition via the structure of a RslTpt1·NAD+ binary complex, we hoped to extract insights into the mechanism of the first step of the Tpt1 reaction pathway, which entails attack by the RNA 2′-PO4 on the C1″ atom of NAD+ leading to the expulsion of nicotinamide and the formation of an RNA-2′-phospho-(ADP-ribose) intermediate. We hypothesized, based on studies of related enzymatic reactions involving nucleoside N-glycosides, that Tpt1 Step 1 chemistry proceeds via a dissociative SN1-like mechanism involving an oxocarbenium ion transition state, as depicted in Figure 10. The oxocarbenium ion mechanism applies to nucleoside hydrolases (40), uracil DNA glycosylase (41), purine and pyrimidine nucleoside phosphorylases (42–44), 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase (45), Sir2 NAD+-dependent lysine deacetylase (46), CD38 NAD+ glycohydrolase (47) and Clostridium perfringens ADP-ribosylating toxin (48). A key factor in driving the SN1-like reaction is electrostatic stabilization of the oxocarbenium ion. Yet, our structures of the RslTpt1·NAD+ binary complex reveal no negatively charged enzymic groups in the vicinity that could directly stabilize the positive charge on an oxocarbenium ion transition state. In the case of purine nucleoside phosphorylase, which also lacks anionic enzymic moieties in proximity to the ribose, it is proposed that the phosphate anion substrate (the nucleophile that attacks ribose C1 in the phosphorylase reaction) plays this vital role, in an example of substrate-assisted catalysis (42,43). Although we envision that the RNA 2′-PO4 plays an analogous role in the Tpt1 step reaction, there is as of yet no structure of a Tpt1 enzyme in a ternary complex with NAD+ and a 2′-PO4 nucleophile. However, the present RslTpt1·NAD+ binary complex structure does suggest an additional source of catalytic power for stabilizing an oxocarbenium ion transition state, that is, via the negative charge on the NMN phosphate of NAD+. In the binary complex depicted in Figure 6B, the non-bridging NMN phosphate oxygens are situated roughly 3.0 Å from the O4″ atom of the NMN ribose. Assuming such proximity is maintained in the Step 1 Michaelis complex, we suggest NAD+ substrate-assisted catalysis as a novel feature of the Tpt1 mechanism.
Figure 10.

Proposed chemical mechanism of Step 1 of the Tpt1 reaction pathway that proceeds via an oxocarbenium ion transition state.
DATA AVAILABILITY
The chemical shifts of apo-RslTpt1 and the RslTpt1·NAD+ complex have been deposited in the BMRB with accession numbers 30819 and 30820, respectively; the corresponding structures have been deposited in the PDB with accession codes 7KW8 and 7KW9, respectively. All other data are available from the authors upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr James Aramini (CUNY-ASRC Biomolecular NMR facility) and Dr Shibani Bhattacharya (New York Structural Biology Center) for assistance with the NMR experiments, Dr Charles Schwieters (NIH) for help in parameterizing NAD+ for structure calculations, and Dr Beate Schwer (Weill Cornell Medical College) for assistance with yeast complementation experiments. The NMR measurements were carried out in the CUNY ASRC Biomolecular NMR facility and at the New York Structural Biology Center (NYSBC).
Contributor Information
Sébastien Alphonse, Department of Chemistry and Biochemistry, The City College of New York, New York, NY 10031, USA.
Ankan Banerjee, Molecular Biology Program, Sloan-Kettering Institute, New York, NY 10021, USA.
Swathi Dantuluri, Molecular Biology Program, Sloan-Kettering Institute, New York, NY 10021, USA.
Stewart Shuman, Molecular Biology Program, Sloan-Kettering Institute, New York, NY 10021, USA.
Ranajeet Ghose, Department of Chemistry and Biochemistry, The City College of New York, New York, NY 10031, USA; Graduate Program in Chemistry, The Graduate Center of CUNY, New York, NY 10016, USA; Graduate Program in Biochemistry, The Graduate Center of CUNY, New York, NY 10016, USA; Graduate Program in Physics, The Graduate Center of CUNY, New York, NY 10016, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB 1412007 to R.G.]; National Institutes of Health [R35-GM126945 to S.S.]; Deutsche Forschungsgemeinschaft [394320208 to A.B.]; NIH [OD016432, OD018509 to NYSBC]; Funding for open access charge: Institutional Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.McCraith S.M., Phizicky E.M.. A highly specific phosphatase from Saccharomyces cerevisiae implicated in tRNA splicing. Mol. Cell. Biol. 1990; 10:1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCraith S.M., Phizicky E.M.. An enzyme from Saccharomyces cerevisiae uses NAD+ to transfer the splice junction 2′-phosphate from ligated tRNA to an acceptor molecule. J. Biol. Chem. 1991; 266:11986–11992. [PubMed] [Google Scholar]
- 3.Steiger M.A., Jackman J.E., Phizicky E.M.. Analysis of 2′-phosphotransferase (Tpt1p) from Saccharomyces cerevisiae: evidence for a conserved two-step reaction mechanism. RNA. 2005; 11:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spinelli S.L., Kierzek R., Turner D.H., Phizicky E.M.. Transient ADP-ribosylation of a 2′-phosphate implicated in its removal from ligated tRNA during splicing in yeast. J. Biol. Chem. 1999; 274:2637–2644. [DOI] [PubMed] [Google Scholar]
- 5.Sawaya R., Schwer B., Shuman S.. Structure-function analysis of the yeast NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA. 2005; 11:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munir A., Abdullahu L., Damha M.J., Shuman S.. Two-step mechanism and step-arrest mutants of Runella slithyformis NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA. 2018; 24:1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spinelli S.L., Malik H.S., Consaul S.A., Phizicky E.M.. A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14136–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dantuluri S., Schwer B., Abdullahu L., Damha M., Shuman S.. Activity and substrate specificity of Candida, Aspergillus, and Coccidioides Tpt1: essential tRNA splicing enzymes and potential anti-fungal targets. RNA. 2021; doi:10.1261/rna.078660.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harding H.P., Lackey J.G., Hsu H.C., Zhang Y., Deng J., Xu R.M., Damha M.J., Ron D.. An intact unfolded protein response in Trpt1 knockout mice reveals phylogenic divergence in pathways for RNA ligation. RNA. 2008; 14:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato-Murayama M., Bessho Y., Shirouzu M., Yokoyama S.. Crystal structure of the RNA 2′-phosphotransferase from Aeropyrum pernix K1. J. Mol. Biol. 2005; 348:295–305. [DOI] [PubMed] [Google Scholar]
- 11.Munir A., Banerjee A., Shuman S.. NAD+-dependent synthesis of a 5′-phospho-ADP-ribosylated RNA/DNA cap by RNA 2′-phosphotransferase Tpt1. Nucleic Acids Res. 2018; 46:9617–9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee A., Munir A., Abdullahu L., Damha M.J., Goldgur Y., Shuman S.. Structure of tRNA splicing enzyme Tpt1 illuminates the mechanism of RNA 2′-PO4 recognition and ADP-ribosylation. Nat. Commun. 2019; 10:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 14.Johnson B.A.From Raw data to protein backbone chemical shifts using nmrfx processing and NMRViewJ analysis. Meth. Mol. Biol. 2018; 1688:257–310. [DOI] [PubMed] [Google Scholar]
- 15.Sattler M., Schleucher J., Griesinger C.. Heteronuclear multidimensi onal NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. NMR Spect. 1999; 34:93–158. [Google Scholar]
- 16.Cavanagh J., Fairbrother W.J. III, Mark R.A.J.P., Skelton N.J.. Protein NMR Spectroscopy. 2007; 2nd ed.San Diego: Academic Press. [Google Scholar]
- 17.Hyberts S.G., Takeuchi K., Wagner G.. Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J. Am. Chem. Soc. 2010; 132:2145–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gal M., Schanda P., Brutscher B., Frydman L.. UltraSOFAST HMQC NMR and the repetitive acquisition of 2D protein spectra at Hz rates. J. Am. Chem. Soc. 2007; 129:1372–1377. [DOI] [PubMed] [Google Scholar]
- 19.Ferrage F., Cowburn D., Ghose R.. Accurate sampling of high-frequency motions in proteins by steady-state 15N-{1H} nuclear Overhauser effect measurements in the presence of cross-correlated relaxation. J. Am. Chem. Soc. 2009; 131:6048–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korzhnev D.M., Kloiber K., Kanelis V., Tugarinov V., Kay L.E.. Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme. J. Am. Chem. Soc. 2004; 126:3964–3973. [DOI] [PubMed] [Google Scholar]
- 21.Shen Y., Bax A.. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR. 2013; 56:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bardiaux B., Malliavin T., Nilges M.. ARIA for solution and solid-state NMR. Meth. Mol. Biol. 2012; 831:453–483. [DOI] [PubMed] [Google Scholar]
- 23.Habeck M., Rieping W., Linge J.P., Nilges M.. NOE assignment with ARIA 2.0: the nuts and bolts. Meth. Mol. Biol. 2004; 278:379–402. [DOI] [PubMed] [Google Scholar]
- 24.Engh R., Huber R.. Accurate bond and angle parameters. Acta Crystallogr. A. 1991; 47:392–400. [Google Scholar]
- 25.Liebschner D., Afonine P.V., Baker M.L., Bunkoczi G., Chen V.B., Croll T.I., Hintze B., Hung L.W., Jain S., McCoy A.J.et al.. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019; 75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bermejo G.A., Schwieters C.D.. Protein structure elucidation from NMR data with the program Xplor-NIH. Meth. Mol. Biol. 2018; 1688:311–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M.. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996; 8:477–486. [DOI] [PubMed] [Google Scholar]
- 28.Vriend G.WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990; 8:52–56. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya A., Tejero R., Montelione G.T.. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007; 66:778–795. [DOI] [PubMed] [Google Scholar]
- 30.Frishman D., Argos P.. Knowledge-based protein secondary structure assignment. Proteins. 1995; 23:566–579. [DOI] [PubMed] [Google Scholar]
- 31.Yao L., Ying J., Bax A.. Improved accuracy of 15N-1H scalar and residual dipolar couplings from gradient-enhanced IPAP-HSQC experiments on protonated proteins. J. Biomol. NMR. 2009; 43:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zweckstetter M.NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc. 2008; 3:679–690. [DOI] [PubMed] [Google Scholar]
- 33.Cornilescu G., Marquardt J.L., Ottiger M., Bax A.. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 1998; 120:6836–6837. [Google Scholar]
- 34.Tolman J.R., Al-Hashimi H.M., Kay L.E., Prestegard J.H.. Structural and dynamic analysis of residual dipolar coupling data for proteins. J. Am. Chem. Soc. 2001; 123:1416–1424. [DOI] [PubMed] [Google Scholar]
- 35.Lee K.H., Alphonse S., Piserchio A., Tavares C.D.J., Wellmann R.M., Dalby K.N., Ghose R.. Structural Basis for the recognition of eukaryotic elongation factor 2 kinase by calmodulin. Structure. 2016; 24:1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen M.R., Mueller L., Pardi A.. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 1998; 5:1065–1074. [DOI] [PubMed] [Google Scholar]
- 37.Gancedo J.M., Gancedo C.. Concentrations of intermediary metabolites in yeast. Biochimie. 1973; 55:205–211. [DOI] [PubMed] [Google Scholar]
- 38.Bennett B.D., Kimball E.H., Gao M., Osterhout R., Van Dien S.J., Rabinowitz J.D.. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009; 5:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuiderweg E.R.P.Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry. 2002; 41:1–7. [DOI] [PubMed] [Google Scholar]
- 40.Versees W., Steyaert J.. Catalysis by nucleoside hydrolases. Curr. Opin. Struct. Biol. 2003; 13:731–738. [DOI] [PubMed] [Google Scholar]
- 41.Werner R.M., Stivers J.T.. Kinetic isotope effect studies of the reaction catalyzed by uracil DNA glycosylase: evidence for an oxocarbenium ion-uracil anion intermediate. Biochemistry. 2000; 39:14054–14064. [DOI] [PubMed] [Google Scholar]
- 42.Erion M.D., Stoeckler J.D., Guida W.C., Walter R.L., Ealick S.E.. Purine nucleoside phosphorylase. 2. Catalytic mechanism. Biochemistry. 1997; 36:11735–11748. [DOI] [PubMed] [Google Scholar]
- 43.Fedorov A., Shi W., Kicska G., Fedorov E., Tyler P.C., Furneaux R.H., Hanson J.C., Gainsford G.J., Larese J.Z., Schramm V.L.et al.. Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry. 2001; 40:853–860. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz P.A., Vetticatt M.J., Schramm V.L.. Transition state analysis of the arsenolytic depyrimidination of thymidine by human thymidine phosphorylase. Biochemistry. 2011; 50:1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Namanja-Magliano H.A., Stratton C.F., Schramm V.L.. Transition state structure and inhibition of Rv0091, a 5′-Deoxyadenosine/5′-methylthioadenosine nucleosidase from Mycobacterium tuberculosis. ACS Chem. Biol. 2016; 11:1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu P., Wang S., Zhang Y.. Highly dissociative and concerted mechanism for the nicotinamide cleavage reaction in Sir2Tm enzyme suggested by ab initio QM/MM molecular dynamics simulations. J. Am. Chem. Soc. 2008; 130:16721–16728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Q., Kriksunov I.A., Graeff R., Munshi C., Lee H.C., Hao Q.. Structural basis for the mechanistic understanding of human CD38-controlled multiple catalysis. J. Biol. Chem. 2006; 281:32861–32869. [DOI] [PubMed] [Google Scholar]
- 48.Tsuge H., Nagahama M., Oda M., Iwamoto S., Utsunomiya H., Marquez V.E., Katunuma N., Nishizawa M., Sakurai J.. Structural basis of actin recognition and arginine ADP-ribosylation by Clostridiumperfringens ι-toxin. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:7399–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The chemical shifts of apo-RslTpt1 and the RslTpt1·NAD+ complex have been deposited in the BMRB with accession numbers 30819 and 30820, respectively; the corresponding structures have been deposited in the PDB with accession codes 7KW8 and 7KW9, respectively. All other data are available from the authors upon request.