


Abstract
Numerous studies have shown that repetitive regions in genomes play indispensable roles in the evolution, inheritance and variation of living organisms. However, most existing methods cannot achieve satisfactory performance on identifying repeats in terms of both accuracy and size, since NGS reads are too short to identify long repeats whereas SMS (Single Molecule Sequencing) long reads are with high error rates. In this study, we present a novel identification framework, LongRepMarker, based on the global de novo assembly and k-mer based multiple sequence alignment for precisely marking long repeats in genomes. The major characteristics of LongRepMarker are as follows: (i) by introducing barcode linked reads and SMS long reads to assist the assembly of all short paired-end reads, it can identify the repeats to a greater extent; (ii) by finding the overlap sequences between assemblies or chomosomes, it locates the repeats faster and more accurately; (iii) by using the multi-alignment unique k-mers rather than the high frequency k-mers to identify repeats in overlap sequences, it can obtain the repeats more comprehensively and stably; (iv) by applying the parallel alignment model based on the multi-alignment unique k-mers, the efficiency of data processing can be greatly optimized and (v) by taking the corresponding identification strategies, structural variations that occur between repeats can be identified. Comprehensive experimental results show that LongRepMarker can achieve more satisfactory results than the existing de novo detection methods (https://github.com/BioinformaticsCSU/LongRepMarker).
INTRODUCTION
The genomes of all eukaryotes contain a certain proportion of repetitive elements, particularly mammalians in which repeats account for 25–50% of their entire genomes (1,2). Repetitive regions can be caused by various mechanisms, such as chromosome translocations, transposons, errors in replication and recombination, etc (3). Numerous studies have shown that the repetitive elements in the genome play indispensable roles in the evolution, inheritance, variation, gene expression, transcriptional regulation, chromosome construction, and physiological metabolism of living organisms (4–7), and they are one of the principal causes of genomic instability (8). How to quickly, accurately and completely identify repetitive regions in genomes has become an important research topic in bioinformatics.
According to the arrangement, the repeats in eukaryotic and certain prokaryotic genomes can be divided into two types: tandem repeats and interspersed repeats (9) (Supplementary Table S1). Tandem repeats are arrays in which repeating elements consisting of 1–500 bp sequences are connected end to end to form multiple repeats. They are arranged in clusters in the telomere, the centromere peripheral region or the heterochromatin region on the chromosome arm (10). On the contrary, repeating elements of interspersed repeats are not connected, but are doped with other unrelated repeats or single copy sequences. They are dispersed throughout the genome and are usually referred to as transposons, including retrotransposons and DNA transposons (11). There are two main types of retrotransposons: (i) long-terminal repeat retrotransposons (LTRs), the length of which generally ranges from 100 bp to 25 kb (12,13) and (ii) non-long terminal repeat retrotransposons (Non-LTRs), which are divided into long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) (14). The detailed classification of repeats is shown in Supplementary Section S1.1.
Many computational methods have been proposed to identify repeats in genomes, which can be classified into three categories, including homology-based, structure-based and de novo methods (15–17) (Supplementary Figure S1 and Table S2). The homology-based identification methods are based on a certain database for the homology search, so as to find and mask the repeats. RepeatMasker (18) is a representative method of this category, which performs a similarity search based on local alignment with AB-BLAST (19) or Crossmatch (20). RepeatMasker has its own library (RepBase and Dfam) of repetitive sequences and has become a gold standard of this field in terms of accuracy. Most other similar methods use RepeatMasker as the main reference library. The homology-based methods have high search efficiency and can be used to discover families with small numbers of copies. However, such methods can only be used to search for known repeats, and cannot be used to discover novel ones. Typical methods based on the homologous search also include Censor (21), TESeeker (22), Greedier (23) and T-lex (24). Among them, CENSOR is a program designed to identify and eliminate fragments of DNA sequences homologous to any chosen reference sequence, which uses RepBase as the homologous database; TESeeker implements an automated homology-based approach for identifying transposable elements, which uses Tefam and RepBase as the homologous databases; Greedier effectively solves the problem of embedded duplications by using greedy algorithms and local alignment methods; and T-lex is a tool for fast and accurate assessment of transposable element presence in high-throughput sequencing data, which can use data from a large number of strains and return estimates of population frequencies of individual TE (transposable element) insertions in a reasonable time.
The structure-based identification methods are based on the prior information of the sequence and structure features, using a heuristic algorithm to find and identify the repeat sequences. Typical structure-based identification methods include: LTRharvest (25), MASiVE (26), MGEScan-LTR (27), FINDMITE (28), MUST (29), detectMITE (30), MITE-Hunter (31), MITE-Digger (32) and MITE Tracker (33). Among them, LTRharvest implements several steps of filtering based on structural features of sequences, determines the boundary position of the LTR, and annotates the LTR with LTRdigest. MASiVE is a tool specifically designed to analyze specific LTR transposons in plant genomes. MGEScan-LTR uses approximate string matching and protein domain analysis methods to determine intact LTR retrotransposons. TEs are a type of repeat sequences abundant in eukaryotic genomes. TEs play important roles in genome organization and evolution. Commonly, TEs in genomes can be classified into two major categories, retrotransposons (Class I) and DNA transposons (Class II). Miniature inverted repeat transposable elements (MITEs) are a special type of DNA transposons. MITE-Hunter, detectMITE, FINDMITE, MUST, MITE-Digger and MITE-Hunter are six typical structure-based methods for MITE identification, among which FINDMITE requires users to predefine the TSD sequences, TIR length and the minimum and maximum distances between the TIRs. MITE-Hunter is a program pipeline that can be used to identify MITEs as well as other small Class II non-autonomous TEs from genomic DNA datasets. Compared to FINDMITE and MUST, MUST-Hunter has a much lower false-positive rate and the output is easier to be checked and classified. Both MITE-Hunter and MITE Digger utilize a mixture of both de novo and structural-based methods in MITE detection. Although they have successfully reduced false positive rates in MITE detection, neither of them can detect all MITEs hidden in the genomes.
The de novo methods require no prior information of the repeat structure or similarity to the known repeat sequences, and tend to be more flexible than the other two methods (34). The de novo methods can also be divided into three categories (Supplementary Figure S1). The first category relies on the multiple sequence alignment to identify repeats, which mainly include RPT(Repeat Pattern Toolkit) (35), RECON (36), PILER (37) and LTRdigest (38). Such methods are usually designed based on a search tree structure with a complete genome as input, and the algorithm finds repeat sequences by copying the genome and comparing the similarity between the genome and its copy. The methods in the second category rely on k-mer and space seed extension strategies to identify repetitive sequences. These methods convert the sequences in the genome into k-mers of a certain length, select the k-mers whose frequency exceeds a certain threshold as a seed, search for the locations of these seeds in the genome, and perform the sequence extension to both ends of the genome and get the expended sequences. During the extension process, it always judges whether the extended sequences are consistent at multiple locations in the genome. If yes, it continues the extension, otherwise stops the extension. EDTA (39), RepeatFinder (40), RepeatScout (41), ReAS (42), Generic Repeat Finder (GRF) (43) and Repeatmodelerl2 (44) are representatives of this category. They start with a library of high-frequency k-mers that are used in initial identification, alignment and extension of sequence substrings. The methods in the third category rely on sequence assembly and similarity network to identify repeats, which mainly include RepARK (45), REPdenovo (46) and RepLong (47). Among these three methods, the first two are based on the NGS short reads, and both of them obtain repetitive sequences by the assembly of the high frequency k-mers. The last method is currently the only detection method suitable for the third generation sequencing reads, which constructs the similarity network by getting the overlaps between the long reads, and then uses the community discovery algorithm to get the detection results. The community discovery algorithm in RepLong is developed based on modularity optimization (48–50). Introduction of various tools and the community discovery algorithms are shown in Supplementary Sections S1.2 and S1.4.4, respectively.
In the process of NGS sequence assembly, the paired-end reads with large insert sizes are mainly used to resolve the ambiguity paths generated by the repeated regions in the assembly graph and determine the successive positions of contigs in the process of scaffolding. The assembly-based detection methods are based on the high-frequency k-mer assembly to obtain repetitive sequences. Due to the lack of support for long sequence fragments that can span the repetitive regions, the assembler will inevitably make misassemblies when processing these short and highly repetitive sequences. On the other hand, they depend too much on the threshold of the high frequency k-mers, which is difficult to obtain accurately due to the sequencing bias. The SMS long reads are more likely to cover repetitive regions completely, which are more favorable for recognizing long repeats. However, the high error rate of SMS long reads has a great impact on the accuracy of this method. In addition, such methods construct the similarity network by comparing the long reads, and then use the community discovery algorithm to get the detection results, which has a higher computational complexity when processing large datasets. In summary, it is often difficult for existing de novo detection methods to achieve satisfactory results in terms of both accuracy and size.
In order to overcome these bottlenecks, we propose a novel identification framework called LongRepMarker based on assembly of Illumina short paired-end reads and barcode linked reads or SMS long reads, and multiple sequence alignment for accurately detecting the long repetitive regions in genomes. In addition, as the development of the third generation sequencing, the SMS long reads have been widely applied in various fields of bioinformatics. In order to better comply with the market demand and further expand the application scope of this system, we further develop a detection mode based on only SMS long reads under the LongRepMarker framework (Supplementary Figure S2). The overall workflow of LongRepMarker is shown in Figure 1.
Figure 1.
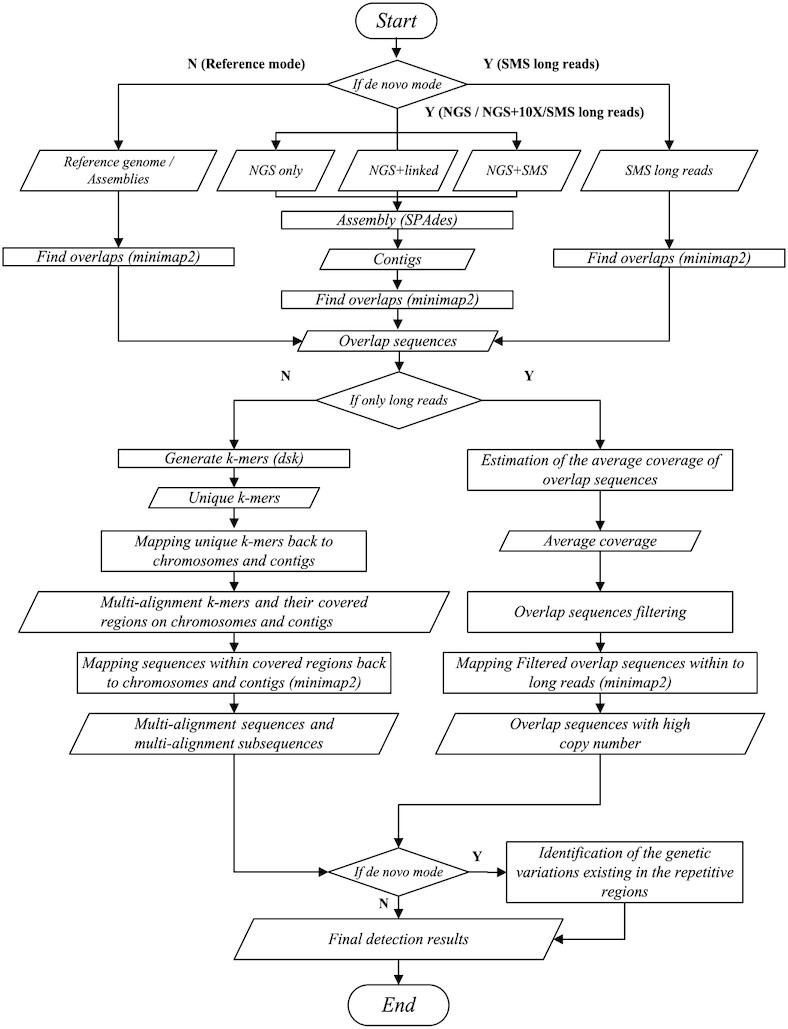
The workflow of LongRepMarker. The directed path on the left shows the pipeline of the reference-assisted mode. The directed path on the right shows the pipeline of the de novo modes which can also be divided into the detection mode based on the mixed sequencing data and the detection mode based on only short reads or long reads.
OVERVIEW
Working modes of LongRepMarker
LongRepMarker provides two different working modes: (i) reference-assisted mode and (ii) de novo mode (Figure 1). The detailed description of these two different working modes are shown in Supplementary Section S1.3.
The reference-assisted mode. Since the sequencing data of large genomes is massive, it is difficult for the de novo methods to handle them. LongRepMarker provides a reference-assisted mode. If there is a reference sequence, a rough assembly of a species or a reference sequence of similar species, it can quickly and accurately derive a repeat library for that species. The detailed description of this mode is shown in Supplementary Section S1.3.1.
-
The de novo mode. Repeats are present in the genomes of all organisms. The DNA sequence organization of eukaryotic genomes consists of numerous repeats, some of which are clustered in structural regions of chromosomes particularly in the cetromeric and telomeric regions. This organization has been elucidated through renaturation rate studies of denatured DNA. Prokaryotic genomes contain a variety of low-copy-number repeated sequences, such as insertion elements, rRNA operons, tRNA genes, and other genes such as those belonging to the rhs gene family. These sequences may contribute to the evolution of chromosome structure through DNA rearrangements such as chromosomal deletions, duplications, and inversions. However, most existing de novo identification methods (such as RepARK, Repdenovo and RepLong) cannot achieve satisfactory results for detecting these repetitive sequences as the NGS reads are too short and the SMS long reads are with high error rates. According to the different input data, de novo mode can be divided into the following sub-modes.
Sub-mode based on only NGS short reads. In this mode, the proposed framework produces detection results based on only NGS short paired-end reads (Supplementary Sections S1.3.2). By calling SPAdes (51) which adopts some better repeat processing strategies and has superior assembly performance than other similar tools (such as SOAPdenovo2 (52), Abyss (53), Velvet (54) and IDBA-UD (55)), the framework can recover the repetitive sequences contained in the sequencing data to the greatest extent. The reasons for choosing SPAdes as the assembler and the performance comparison analysis of it and other similar tools are shown in Section S1.4.3 of the supplementary.
Sub-mode based on NGS + barcode linked reads. In this mode, the proposed framework introduces barcode linked reads into the assembly process of Illumina short paired-end reads, assists the assembler in resolving the ambiguity path caused by repeats in the assembly graph (Supplementary Figure S3), and uses multi-alignment unique k-mers based identification strategy to fully and accurately recover the repeats in the genome. The detailed description of this mode is shown in Supplementary Sections S1.3.3.
Sub-mode based on NGS + SMS long reads. An important advantage of the third generation sequencing is the read length. PacBio RS II system with C4 chemistry boasts average read lengths over 10 kb, with an N50 of more than 20kb and maximum read lengths over 60kb. In this mode, the proposed framework introduces the SMS long reads into the assembly process of Illumina short paired-end reads, and makes full use of the advantages of long reads in span to effectively resolve the ambiguity formed by repetitive sequences in the assembly process of short reads (Supplementary Figure S4). The detailed description of this mode is shown in Supplementary Sections S1.3.3. The advantages of using SPAdes to assemble Illumina short paired-end reads and barcode linked reads or corrected SMS long reads in repetitive sequences identification are described in Section S1.4.2 of the Supplementary.
Sub-mode based on only SMS long reads. In order to further expand the application scope of this system, we have developed a detection mode based on only SMS long reads. The input of this mode is SMS long reads and the output is the repeat library of the genome. The workflow of this mode can be divided into the following steps: (i) getting the overlap sequences between SMS long reads; (ii) estimating the average coverage of the overlap sequences; (iii) filtering the overlap sequences with low coverage; (iv) getting the filtered overlap sequences with the high copy number in SMS long reads (e.g. the copy number is more than 1.5 × AverageCoverage); (v) identifying the genetic variations existing in the detected repetitive regions and (vi) generating the final repeat library. The detailed workflow of this mode is shown in Supplementary Section S1.3.4.
The main differences between the reference-assisted mode and de novo mode, and the advantages of barcode linked reads and SMS long reads in assisting the assembly of Illumina short paired-end reads are shown in Supplementary Sections S1.4.1 and Section S1.4.2, respectively.
Main improvements of LongRepMarker
Compared with the existing de novo detection methods, the major improvements of LongRepMarker are as follows:
-
The repeats obtained by LongRepMarker are more comprehensive and accurate.
▸ By assembling all paired-end reads and barcode linked reads or SMS long reads instead of assembling the high frequency k-mers, the algorithm can identify the repeats in the genomes to a greater extent.
▸ The repetitive sequences are a special kind of overlap sequences, and the overlap sequences occupy only a small partion of the overall sequences (Figure 2). By finding the overlap sequences between assemblies or chomosomes, the algorithm locates the repetitive regions faster and more accurately.
▸ Due to the sequencing bias, the high frequency threshold is often difficult to obtain accurately, which has a great impact on the range of the high frequency k-mers. By using the multi-alignment unique k-mers to identify repeats in overlap sequences, the algorithm can obtain the repeats in the genomes more comprehensively and stably.
The parallel alignment model based on the multi-alignment unique k-mers can greatly optimize the efficiency of data processing in LongRepMarker (Supplementary Figure S19). LongRepMarker has superior computing efficiency when dealing with large genomes such as human and mouse. For example, it takes only 264.05 min to obtain the whole repeat library of the human genome (hg38) in the reference-assisted mode and 2.86 hours to obtain the whole repeat library of the mouse genome in the de novo mode.
The structural variations that occur between repetitive regions can be identified by LongRepMarker. The study and analysis of genomic structural variations that occur within the repetitive regions can provide a new perspective for understanding life processes and analyzing life mechanisms. In order to identify structural variations in the repetitive regions, the proposed algorithm also designs corresponding identification strategies.
The new detection mode based on only SMS long reads has been integrated into LongRepMarker. As the development of the third generation sequencing, the SMS long reads have been widely applied in various fields of bioinformatics. A new detection mode based on only SMS long reads has been developed in the LongRepMarker framework. Compared with the existing detection methods based on SMS long reads, this mode has the advantages of low memory consumption, high speed and high detection accuracy.
Figure 2.
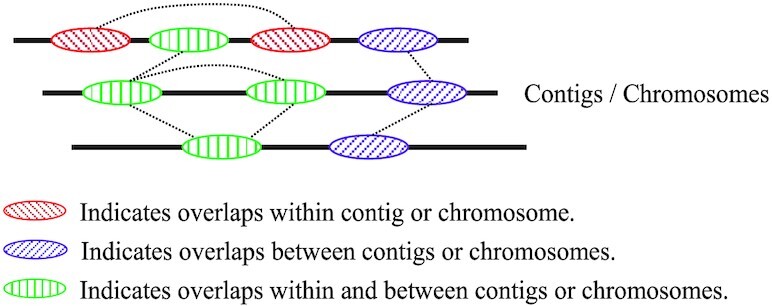
The illustration of overlap relationships within and between chromosomes and contigs. Repetition relation is a special kind of overlap relation. All possible repetition relationships can be found by identifying overlap relationships.
MATERIALS AND METHODS
The pipeline of LongRepMarker is illustrated in Figure 3. The entire workflow of LongRepMarker can be divided into the following modules. The detailed description of each module is shown in Supplementary Section S2.
Figure 3.

The pipeline of LongRepMarker. (A) shows five working modes of LongRepMarker, which are reference-assisted mode, de novo mode based on only NGS short paired-end reads, de novo mode based on NGS short paired-end reads + barcode linked reads, de novo mode based on NGS short paired-end reads + SMS long reads and de novo mode based on only SMS long reads. (B) shows the principle of finding overlaps between chromosomes and contigs by using minimap2. (C) Transforming overlaps into unique k-mers by DSK. (D) Using minimap2 to obtain multi-alignment unique k-mers and the regions on chromosomes and contigs that can be covered by these unique k-mers. (E) Using minimap2 to obtain multi-alignment regions and single-alignment regions on chromosomes, contigs and long reads, and the sequences marked in multi-alignment regions are saved in the final repeat library. (F) Single-alignment regions are cut into several smaller segments, and some multi-alignment segments are saved in the final repeat library. (G) Analyzing the relationship and spacing between these saved sequences, combining some saved sequences and their gaps that meet certain conditions to a complete fragment and replacing the corresponding saved sequences in the final detection results by this fragment. (H) Components of the final repeat library. (A), (B), (B1), (C1), (D1), (F), (G) and (H) illustrate the workflow of the detection mode based on only the SMS long reads.
Identification of overlap sequences
As illustrated in Figure 2, the repetition relation is a special case of the overlap relation. Thus all possible repetition relationship can be found by searching overlap sequences. By searching for overlaps between and within chromosomes or assemblies (contigs), the search space of the algorithm can be greatly narrowed, and the computational complexity of the algorithm can also be greatly reduced. In this step, minimap2 (56) is used for generating the overlap sequences between and within chromosomes or contigs. The specific commands and parameters for obtaining the overlap sequences are shown in Supplementary Section S2.1.
Conversion of overlap sequences into unique k-mers
DSK (disk streaming of k-mers) (57) is a new streaming algorithm for k-mer counting, which only requires a fixed, user-defined amount of memory and disk space. In this step, DSK is used for generating the unique k-mers. Assuming that there are n overlap sequences, which respectively correspond to n fragments in an overlap file. Let ci be the ith fragment (i =1, 2, ..., n) and lci be the length of ci. Given a fix length k of k-mers (k << lci), ci can be represented as a list of (lci-k + 1) k-mers. Therefore, the total number of k-mers (Numk) that are transferred from all overlap sequences can be expressed as 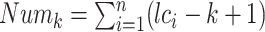 .
.
When the value of lc is large and the value of k is small, the total number of k-mer generated from these overlap sequences is very large (58). In order to further reduce the total number of k-mers, DSK converts all k-mers to their canonical representation with respect to reverse-complementation which is called the unique k-mers, so that the actual number of converted unique k-mers is much smaller than the actual number of k-mers directly converted from the original overlap sequences. Therefore, using unique k-mers instead of k-mers for mapping can further greatly reduce the complexity of the alignment. The detailed analysis of the quantitative relationship among reads, k-mers and the unique k-mers, and the complexity of the alignment is shown in Supplementary Section S2.2.
Generation of multi-alignment unique k-mers and their coverage regions on overlap sequences
LongRepMarker uses the multiple sequence alignment to find the unique k-mers which can be aligned to different locations on overlap sequences and the regions on overlap sequences that can be covered by these multi-alignment unique k-mers. The process of generating the multi-alignment unique k-mers is described in Supplementary Algorithm S1, and the process of generating the regions on overlap sequences that can be covered with these multi-alignment unique k-mers is described in Supplementary Algorithm S2. The time complexity of these two algorithms is O(n). The results of the multiple sequence alignment are stored in a sam file. Once LongRepMarker receives the sam file, it first filters the file, and keeps the multiple alignment records and the ID of multi-alignment unique k-mers. It then converts the filtered sam file into a depth file via the samtools (59). Finally, based on the information provided by the depth file and the ID records of multi-alignment unique k-mers, it extracts the regions on overlap sequences that can be covered with these multi-alignment unique k-mers, and forms several sequence fragments which are called sequence fragments with high probability of being repetitive regions. This procedure is illustrated in (B), (C), (D) and (E) of Figure 3. The detailed description of generation of multi-alignment unique k-mers and their coverage regions on overlap sequences is shown in Supplementary Section S2.3, and the detailed description of the combination of multiple threads parallel computing model and k-mer based multiple sequence alignment is shown in Supplementary Section S2.4.
Classification of regions on overlap sequences that can be covered by multi-alignment k-mers
The regions on original sequences (chromosomes or contigs) covered by the multi-alignment k-mers can be divided into two categories (Supplementary Figure S19). The regions in the first category can be aligned to different locations (≥2 locations) of the overlap sequences, which are highly likely to be repeats, so they are stored into the final repeat library directly. The regions in the second category cannot be aligned to the overlap sequences mutiple times, but some sub-segments of them can, which are probably caused by coupling matches due to sequencing errors (e.g. the two sequences are originally not repetitive sequences, due to sequencing errors that form some coupled alignments under error-tolerant conditions, resulting in multiple subsequences within them that can be aligned with each other. Thus the two sequences should be removed) or the genetic variations (e.g. the two sequences are originally repetitive sequences, due to structural variations, multiple subsequences within them cannot be aligned with each other. Thus the two sequences should be retained). The characteristic of coupling alignment due to the sequencing errors is that the alignment region is short and scattered, and it accounts for a relatively small proportion of the entire sequence fragment. On the contrary, the distribution of structural variation regions on the sequence fragment is relatively concentrated, and all have a certain length (e.g. greater than 50bp). Based on these obvious features, we can further filter these non-multiple aligned sequences. The detailed description of classification of regions on overlap sequences that can be covered by multi-alignment k-mers is shown in Supplementary Section S2.5.
Identification of the genetic variations existing in the repetitive regions
The genomic variations between repeating segments are also an important component of repeating regions, and also an important manifestation of repetitive regions polymorphism (60). In addition, the study and analysis of genomic structural variations that occur within the repetitive regions can provide a new perspective for understanding life processes and analyzing life mechanisms. Therefore, we designed a module in LongRepMarker to detect genomic variations that occur in the repetitive regions. The detailed description of identification of genetic variations existing in detected repetitive sequences are shown in Supplementary Sections S2.5 and S3.7.
Merging fragments with duplication or inclusion relationships
There may exist some duplication and inclusion relationships between the detected fragments obtained by multiple sequence alignment. The repetitive sequences generated from the detection tool should be as pure as possible without any impurities and redundancy. In order to achieve this goal, LongRepMarker merges the detected repetitive fragments with duplication and inclusion relationships, and remains only one copy of them in the final detection results.
RESULTS
We use the reference genomes of six species to evaluate the performance of LongRepMarker in the reference-assisted mode: Homo sapiens (hg38), Gallus, Mouse, Drosophila melanogaster, Glycine max and Leafcutter ant. The reference genome sequences of these six species are downloaded from the NCBI website (https://www.ncbi.nlm.nih.gov/). Details of the genomes of these six species are shown in Supplementary Table S13. In order to illustrate the effectiveness of the de novo mode of LongRepMarker, five groups of NGS short reads (Leafcutter Ant, D.melanogaster, Mouse, Human-chr14 and HG003_24149_father), three groups of barcode linked reads (HG003_24149_father, HG004_NA24143_mother and HG002_NA24385_son), three groups of CCS long reads (HG003_24149_father, HG004_NA24143_mother and HG002_NA24385_son) and four groups of PacBio long reads (dro_100k, human_100k, dmel_filtered and human_polished) are used to test the performance of the four different de novo modes of LongRepMarker (Supplementary Table S13). The main evaluation results are shown in Figures 4–10 and Tables 1, 2, 3 and 4. The detailed evaluation results are shown in Supplementary Section S3.5.
Figure 4.

The demonstration of an practical example of alignment between the reference genome of Human(hg38) and 20 repetitive fragments randomly selected from the detection results of the Human dataset which were generated from the reference-assisted mode of LongRepMarker.
Figure 5.

Comparison between the detection results generated from the reference-assisted mode of LongRepMarker based on the six species (Drosophila, Ant, Gallus, Soybean, Human and Mouse) and the corresponding detection results of benchmarking methods (RepeatScout and RepeatModeler2) in terms of the proportion of covering the RepBase library and the repetitive regions on the reference genome. The label All represents the total coverage ratio, which is the sum of the proportion of detection results covering all kinds of repetitive sequences in the corresponding library. The label SINEs indicates the proportion of the detection results covering the SINEs-type repetitive sequences in the corresponding library, label LINEs indicates the proportion of the detection results covering the LINEs-type repetitive sequences in the corresponding library, label LTR indicates the proportion of the detection results covering the LTR-type repetitive sequences in the corresponding library, and label DNA indicates the proportion of the detection results covering the DNA transposon elements-type repetitive sequences in the corresponding library. Sub-figures (A) to (F) show the comparison of the ratio of the detection results of the three tools on the 6 species covering the corresponding RepBase libraries. Sub-figures (G) to (L) show the comparison of the ratio of the detection results of the three tools on the 6 species covering the repetitive sequences on the corresponding reference genomes.
Figure 6.

Comparison between the detection results generated from the de novo mode of LongRepMarker based on only NGS short reads over five species (Drosophila, Ant, Mouse, Human-chr14 and HG003_NA14149_father) and the corresponding detection results of benchmarking methods (RepARK and REPdenovo) in terms of the proportion of covering the RepBase library and the repetitive regions on the reference genome. The label All represents the total coverage ratio, which is the sum of the proportion of detection results covering all kinds of repetitive sequences in the corresponding library. The label DNA indicates the proportion of the detection results covering the DNA transposon elements-type repetitive sequences in the corresponding library, label LINEs indicates the proportion of the detection results covering the LINEs-type repetitive sequences in the corresponding library, label LTR indicates the proportion of the detection results covering the LTR-type repetitive sequences in the corresponding library, label RC? indicates the proportion of the detection results covering the RC?-type repetitive sequences in the corresponding library, and label Satellite indicates the proportion of the detection results covering the Satellite-type repetitive sequences in the corresponding library. Sub-figures (A) to (E) show the comparison of the ratio of the detection results of the three tools on the 5 groups of NGS short reads covering the corresponding RepBase libraries. Sub-figures (F) to (J) show the comparison of the ratio of the detection results of the three tools on the 5 groups of NGS short reads covering the repetitive sequences on the corresponding reference genomes. Sub-figures (K) and (L) show the comparison of the number of repetitive fragments detected by LongRepMarker and RepeatMasker on Drosophila and Ant datasets, in which ‘LongRepMarker-Ref’ represents the detection results are generated based on the reference-assisted mode of LongRepMarker, and ‘LongRepMarker-DeNovo’ represents the detection results are generated based on the de novo mode of LongRepMarker.
Figure 7.

Comparison between the detection results generated from the de novo mode of LongRepMarker based on three groups of NGS short reads + barcode linked reads (HG004_NA24143_father, HG004_NA24143_mother and HG002_NA24385_son) and the detection results generated from the de novo mode of LongRepMarker based on three groups of NGS short reads + SMS long reads (CCS) in terms of the proportion of covering the human RepBase library and the repetitive regions on the reference genome of human. The label All represents the total coverage ratio, which is the sum of the proportion of detection results covering all kinds of repetitive sequences in the corresponding library. The label DNA indicates the proportion of the detection results covering the DNA transposon elements-type repetitive sequences in the corresponding library, label LINEs indicates the proportion of the detection results covering the LINEs-type repetitive sequences in the corresponding library, label LTR indicates the proportion of the detection results covering the LTR-type repetitive sequences in the corresponding library, label lc indicates the proportion of the detection results covering the low complexity-type repetitive sequences in the corresponding library, and label Satellite indicates the proportion of the detection results covering the Satellite-type repetitive sequences in the corresponding library. Sub-figures (A) to (C) show the comparison of the ratio of the detection results of these two models on the three groups of hybrid sequencing data covering the human RepBase library. Sub-figures (D) to (F) show the comparison of the ratio of the detection results of these two models on the three groups of hybrid sequencing data covering the repetitive sequences on the reference genome of human.
Figure 8.

Comparison between the detection results generated from the de novo mode of LongRepMarker based on four groups of SMS long reads (dro_100k, dmel_filtered, human_100k and human_polished) and the corresponding detection results of benchmarking method RepLong in terms of the proportion of covering the RepBase library and the repetitive regions on the reference genome. The label All represents the total coverage ratio, which is the sum of the proportion of detection results covering all kinds of repetitive sequences in the corresponding library. The label DNA indicates the proportion of the detection results covering the DNA transposon elements-type repetitive sequences in the corresponding library, label LINEs indicates the proportion of the detection results covering the LINEs-type repetitive sequences in the corresponding library, label LTR indicates the proportion of the detection results covering the LTR-type repetitive sequences in the corresponding library, label low complexity indicates the proportion of the detection results covering the low complexity-type repetitive sequences in the corresponding library, and label Satellite indicates the proportion of the detection results covering the Satellite-type repetitive sequences in the corresponding library. Sub-figures (A) to (D) show the comparison of the ratio of the detection results of two tools on four groups of SMS long reads covering the corresponding RepBase libraries. Sub-figures (E) to (H) show the comparison of the ratio of the detection results of two tools on four groups of SMS long reads covering the repetitive sequences on the corresponding reference genomes. Sub-figure (I) shows the comparison of the total number of bases in the corresponding RepBase library masked by the detection results of the two tools on four detasets.
Figure 9.

Comparison between the size distribution range of the detected fragments generated from the five detection modes of LongRepMarker on 21 groups of real datasets and the size distribution range of the detected fragments of benchmarking methods. For the hybrid mode (i.e. NGS short reads + barcode linked/SMS reads), since there is no existing methods take the same type of inputs, we only compared the two modes of LongRepMarker.
Figure 10.

Visualization of the longest detection fragment obtained by LongRepMarker on the dataset of Human_100k. Sub-graph(a) shows the alignment of the longest fragment (NODE_6520_Length_25778) detected by LongRepMarker with the reference genome of human (hg38) using MUMmer and LINKVIEW. Sub-graph(b) shows the self-alignment of chromosome NC_000011.10 of the reference genome of human (hg38) using MUMmer and LINKVIEW. It can be seen from the sub-graph(a) that the longest detection fragment can be aligned to multiple different locations on the chromosome NC_000011.10 many times, and the intricate cross lines in the sub-graph(b) also indicate that there are a large number of complex repetitive sequences within chromosome NC_000011.10. This experiment proved that the longest detection fragment obtained by LongRepMarker is a real repeating unit which appears repeatedly inside the chromosome NC_000011.10.
Table 1.
Compared with RepeatMasker, LongRepMarker found new repeat families and their detailed numbers on the Human-chr14 dataset
| Human-chr14 | ||
|---|---|---|
| Super-Family | Family | amount |
| LTR | ERVL-MaLR | 2 |
| LTR | ERV1 | 4 |
| LTR | Gypsy | 6 |
| LTR | Pao | 1 |
| LTR | Copia | 4 |
| LTR | ERVK | 1 |
| LTR | ERVL | 1 |
| LINE | L1 | 7 |
| LINE | R2-NeSL | 1 |
| LINE | L2 | 3 |
| DNA | MULE-MuDR | 1 |
| DNA | hAT-Charlie | 3 |
| DNA | PiggyBac | 1 |
| DNA | CMC-EnSpm | 2 |
| DNA | MuLE-MuDR | 1 |
| DNA | Ginger | 1 |
| DNA | Other | 1 |
| SINE | MIR | 2 |
| scRNA | - | 1 |
| Simple_repeat | - | 2 |
| Satellite | telomeric | 1 |
| Unknown | - | 2551 |
Table 2.
Compared with RepeatMasker, LongRepMarker found repeat families and their detailed numbers on the Ant dataset
| LongRepMarker (reference-assisted mode) | LongRepMarker (de novomode) | RepeatMasker | ||||||
|---|---|---|---|---|---|---|---|---|
| Super-Family | Family | Amount | Super-Family | Family | Amount | Super-Family | Family | Amount |
| LTR | Copia | 150 | LTR | Copia | 18 | LTR | Copia | 4 |
| LTR | Gypsy | 1608 | LTR | Gypsy | 145 | LTR | Gypsy | 17 |
| LTR | Pao | 981 | LTR | Pao | 69 | LTR | Pao | 17 |
| LTR | DIRs | 4 | LTR | DIRs | 1 | LTR | DIRs | 0 |
| LTR | ERV1 | 2 | LTR | ERV1 | 1 | LTR | ERV1 | 3 |
| LTR | Gypsy-Cigr | 1 | LTR | Gypsy-Cigr | 1 | LTR | Gypsy-Cigr | 1 |
| LTR | Caulimovirus | 1 | LTR | Caulimovirus | 1 | LTR | Caulimovirus | 0 |
| LTR | Ngaro | 1 | LTR | Ngaro | 1 | LTR | Ngaro | 1 |
| LTR | ERVL-MaLR | 1 | LTR | ERVL-MaLR | 1 | LTR | ERVL-MaLR | 1 |
| LTR | Other | 4 | LTR | Other | 1 | LTR | Other | 0 |
| LINE | Penelope | 1007 | LINE | Penelope | 172 | LINE | Penelope | 5 |
| LINE | CR1 | 6 | LINE | CR1 | 5 | LINE | CR1 | 9 |
| LINE | I | 40 | LINE | I | 6 | LINE | I | 0 |
| LINE | R1 | 269 | LINE | R1 | 34 | LINE | R1 | 5 |
| LINE | RTE-X | 20 | LINE | RTE-X | 12 | LINE | RTE-X | 6 |
| LINE | R2-NeSL | 7 | LINE | R2-NeSL | 11 | LINE | R2-NeSL | 0 |
| LINE | Other | 5 | LINE | Other | 1 | LINE | Other | 3 |
| DNA | Maverick | 2381 | DNA | Maverick | 136 | DNA | Maverick | 4 |
| DNA | Kolobok-Hydra | 81 | DNA | Kolobok-Hydra | 5 | DNA | Kolobok-Hydra | 0 |
| DNA | Kolobok-T2 | 1122 | DNA | Kolobok-T2 | 97 | DNA | Kolobok-T2 | 26 |
| DNA | TcMar-Mariner | 2275 | DNA | TcMar-Mariner | 257 | DNA | TcMar-Mariner | 2228 |
| DNA | TcMar-Tc4 | 185 | DNA | TcMar-Tc4 | 78 | DNA | TcMar-Tc4 | 3 |
| DNA | TcMar | 49 | DNA | TcMar | 21 | DNA | TcMar | 1 |
| DNA | TcMar-Tc1 | 1317 | DNA | TcMar-Tc1 | 227 | DNA | TcMar-Tc1 | 136 |
| DNA | Merlin | 16 | DNA | Merlin | 18 | DNA | Merlin | 1 |
| DNA | hAT | 14 | DNA | hAT | 16 | DNA | hAT | 8 |
| DNA | hAT-Blackjack | 37 | DNA | hAT-Blackjack | 41 | DNA | hAT-Blackjack | 2 |
| DNA | Other | 119 | DNA | Other | 96 | DNA | Other | 10 |
| RC | Helitron | 153 | RC | Helitron | 37 | RC | Helitron | 16 |
| Unknown | Other | 13308 | Unknown | Other | 7400 | Unknown | Other | 8332 |
Table 3.
Compared with RepeatMasker, LongRepMarker found repeat families and their detailed numbers on the Drosophila dataset
| LongRepMarker (reference-assisted mode) | LongRepMarker (de novomode) | RepeatMasker | ||||||
|---|---|---|---|---|---|---|---|---|
| Super-Family | Family | Amount | Super-Family | Family | Amount | Super-Family | Family | Amount |
| LTR | Gypsy | 7941 | LTR | Gypsy | 560 | LTR | Gypsy | 171 |
| LTR | Pao | 2076 | LTR | Pao | 130 | LTR | Pao | 5 |
| LTR | Copia | 577 | LTR | Copia | 44 | LTR | Copia | 7 |
| LTR | Viper | 39 | LTR | Viper | 0 | LTR | Viper | 0 |
| LTR | ERVK | 1 | LTR | ERVK | 1 | LTR | ERVK | 1 |
| LTR | Gypsy-Cigr | 1 | LTR | Gypsy-Cigr | 0 | LTR | Gypsy-Cigr | 0 |
| LTR | ERVL-MaLR | 1 | LTR | ERVL-MaLR | 1 | LTR | ERVL-MaLR | 8 |
| LTR | ERV1 | 1 | LTR | ERV1 | 1 | LTR | ERV1 | 6 |
| LTR | ERVL | 1 | LTR | ERVL | 1 | LTR | ERVL | 3 |
| LTR | Other | 12 | LTR | Other | 2 | LTR | Other | 2 |
| LINE | I-Jockey | 1483 | LINE | I-Jockey | 152 | LINE | I-Jockey | 62 |
| LINE | CR1 | 398 | LINE | CR1 | 24 | LINE | CR1 | 11 |
| LINE | Jockey | 1396 | LINE | Jockey | 94 | LINE | Jockey | 52 |
| LINE | R1 | 2178 | LINE | R1 | 174 | LINE | R1 | 304 |
| LINE | R1-LOA | 55 | LINE | R1-LOA | 11 | LINE | R1-LOA | 0 |
| LINE | I | 170 | LINE | I | 19 | LINE | I | 5 |
| LINE | LOA | 100 | LINE | LOA | 20 | LINE | LOA | 1 |
| LINE | R2 | 223 | LINE | R2 | 2 | LINE | R2 | 0 |
| DNA | hAT-hobo | 100 | DNA | hAT-hobo | 4 | DNA | hAT-hobo | 0 |
| DNA | TcMar-Tc1 | 231 | DNA | TcMar-Tc1 | 17 | DNA | TcMar-Tc1 | 5 |
| DNA | hAT-Tip100 | 5 | DNA | hAT-Tip100 | 1 | DNA | hAT-Tip100 | 2 |
| DNA | P | 812 | DNA | P | 101 | DNA | P | 1 |
| DNA | CMC-Transib | 144 | DNA | CMC-Transib | 7 | DNA | CMC-Transib | 6 |
| DNA | TcMar-Pogo | 46 | DNA | TcMar-Pogo | 4 | DNA | TcMar-Pogo | 8 |
| DNA | MULF-NOF | 67 | DNA | MULF-NOF | 4 | DNA | MULF-NOF | 0 |
| RC | Helitron | 173 | RC | Helitron | 12 | RC | Helitron | 2 |
| Unknown | Other | 2967 | Unknown | Other | 933 | Unknown | Other | 2863 |
Table 4.
Partial INDEL variation statistics of detection results generated by the de novo mode of LongRepMarker on the Mouse dataset
| Repeat Fragment id | Location on fragment | Fragment length (bp) | Repeat family | Reference id | Location on Ref. | Variation/length |
|---|---|---|---|---|---|---|
| NODE_1612_10359 | 7177 | 10359 | LTR / ERV1 | CM001014.2 | 2839743 | Deletion/513bp |
| NODE_1612_10359 | 3281 | 10359 | LTR / ERV1 | CM001014.2 | 3296769 | Deletion/509bp |
| NODE_1612_10359 | 7179 | 10359 | LTR / ERV1 | CM001014.2 | 3780992 | Deletion/469bp |
| NODE_6694_4510 | 4027 | 4510 | LINE / L1 | CM000995.2 | 176220701 | Deletion/483bp |
| NODE_6694_4510 | 493 | 4510 | LINE / L1 | CM000995.2 | 176933206 | Deletion/483bp |
| NODE_6694_4510 | 490 | 4510 | LINE / L1 | CM000995.2 | 177403768 | Deletion/483bp |
| NODE_6694_4510 | 4026 | 4510 | LINE / L1 | CM000995.2 | 177753322 | Deletion/483bp |
| NODE_820_17868 | 5685 | 17868 | Unknown | KQ030486.1 | 22319 | Deletion/454bp |
| NODE_820_17868 | 12222 | 17868 | Unknown | GL456077.1 | 69596 | Deletion/454bp |
| NODE_820_17868 | 5679 | 17868 | Unknown | CM000997.2 | 60686584 | Deletion/453bp |
| NODE_820_17868 | 5685 | 17868 | Unknown | CM000997.2 | 61171930 | Deletion/454bp |
| NODE_3948_6574 | 3828 | 6574 | Unknown | JH584324.1 | 2589865 | Deletion/471bp |
| NODE_3948_6574 | 3827 | 6574 | Unknown | CM000994.2 | 8193887 | Deletion/471bp |
| NODE_3948_6574 | 3827 | 6574 | Unknown | CM001001.2 | 90385973 | Deletion/471bp |
| NODE_3948_6574 | 3828 | 6574 | Unknown | CM000997.2 | 131225987 | Deletion/471bp |
| NODE_4884_6041 | 1320 | 6041 | Unknown | JH584293.1 | 28699 | Deletion/467bp |
| NODE_4884_6041 | 4901 | 6041 | Unknown | CM000997.2 | 42146528 | Deletion/467bp |
| NODE_4884_6041 | 4774 | 6041 | Unknown | CM000997.2 | 42643739 | Deletion/467bp |
| NODE_9286_2926 | 508 | 2926 | Unknown | CM001013.2 | 124364290 | Deletion/521bp |
| NODE_9286_2926 | 508 | 2926 | Unknown | CM001013.2 | 125562337 | Deletion/521bp |
| NODE_9286_2926 | 510 | 2926 | Unknown | CM001013.2 | 125299304 | Deletion/521bp |
| NODE_1162_13541 | 4229 | 13541 | LINE / L1 | KZ289068.1 | 113232 | Deletion/588bp |
| NODE_1162_13541 | 4229 | 13541 | LINE / L1 | CM000997.2 | 146196060 | Deletion/588bp |
| NODE_1162_13541 | 4210 | 13541 | LINE / L1 | CM000997.2 | 146718666 | Deletion/587bp |
| NODE_1162_13541 | 4262 | 13541 | LINE / L1 | GL456053.2 | 123786 | Deletion/587bp |
| NODE_1790_9471 | 1929 | 9471 | Unknown | GL456350.1 | 180728 | Insertion/467bp |
| NODE_1790_9471 | 7117 | 9471 | Unknown | CM000997.2 | 41935405 | Insertion/467bp |
| NODE_1790_9471 | 7141 | 9471 | Unknown | CM000997.2 | 42287518 | Insertion/467bp |
| NODE_5808_5209 | 3213 | 5209 | LINE / L1 | KQ030486.1 | 22317 | Deletion/454bp |
| NODE_5808_5209 | 2013 | 5209 | LINE / L1 | GL456077.1 | 69568 | Deletion/454bp |
| NODE_5808_5209 | 3208 | 5209 | LINE / L1 | CM000997.2 | 60845802 | Deletion/454bp |
| NODE_5808_5209 | 3216 | 5209 | LINE / L1 | CM000997.2 | 60686589 | Deletion/453bp |
Benchmarking methods
In order to illustrate the effectiveness of LongRepMarker, we compared the reference-assisted mode with RepeatScout (41), RepeatModeler2 (44) and RepeatMasker (20), and compared the de novo mode with RepARK (45), REPdenovo (46) and RepLong (47). RepARK and REPdenovo are used as the benchmarking methods in effectiveness evaluation of the de novo mode based NGS short reads, and RepLong is used as the benchmarking method in effectiveness evaluation of the de novo mode based on only SMS long reads (Supplementary Table S11). The detailed configurations of hardware (Supplementary Figure S21), benchmarking methods and evaluation metrics are shown in Supplementary Sections S3.1 , S3.2 and S3.3.
Performance of LongRepMarker in the reference-assisted mode
It is well known that repeat sequences are present in the genomes of all living organisms. Identifying repetitive sequences in the eukaryotic and prokaryotic genomes provides important basic information for the research of evolution. Repeated genes also provide mechanisms to enhance bacterial virulence, such as antigenic variation (61). However, due to the lack of a known library of eukaryote repetitive sequences, homology-based and structure-based identification methods do not work well. In addition, most existing de novo detection methods are not well suited for large and complex genomes such as mammalian and plant genomes.
In order to overcome these bottlenecks, LongRepMarker provides a reference-assisted mode. In this mode, users only need to input the reference sequence of some organisms, and LongRepMarker can identify the repetitive sequences comprehensively, accurately and rapidly. We evaluated the performance of LongRepMarker in the reference-assisted mode on the six eukaryote genomes (Supplementary Table S13). The reference sequence sizes of these six species are 3.196Gb (H.sapiens(hg38)), 2.752Gb (Mouse), 289Mb (Leafcutter Ant), 168Mb (D.melanogaster), 956Mb (soybean) and 1.040Gb (Gallus). We compared the performance of reference-assisted mode of LongRepMarker with RepeatScout, RepeatMasker and RepeatModeler2, and the representative detection results are shown in Figures 4, 5, and Tables 1, 2 and 3. The complete experimental results are shown in Supplementary Section S3.5.1.
Since RepeatMasker can only be used to mask the repeats in the genome, it cannot classify the masked repeats in detail, so we can only compare the performance of LongRepMarker with RepeatMasker by detecting the size and alignment rate of detected fragments as shown in Supplementary Table S14. LongRepMarker is superior to RepeatMasker in terms of running time, memory consumption, fragment size and alignment rate. For example, the N50 of fragments detected by LongRepMarker on the human dataset is 1034kb, while the corresponding value of RepeatMasker is only 7.228 kb. In addition, the multiple alignment rate of the fragments detected by LongRepMarker on this dataset is 88.25%, while the corresponding value of RepeatMasker is only 7.37% . In order to further analyze the difference between the detection results of these two tools, we carried out two comparative experiments, the representative results are shown in Tables 1, 2 and 3, and the complete results are shown in Supplementary Tables S61 and S62 of Section S3.8. Among them, Table 1 shows the repeat families found by LongRepMarker on Human-chr14 dataset that cannot be found by RepeatMasker, and Tables 2 and 3 show the comparison of some repeats found by LongRepMarker and RepeatMasker on Drosophila and Ant datasets and their classification. Comparative experiments show that LongRepMaker can find some new repeat families which cannot be found by RepeatMasker. For example, LongRepMarker found 4 repeated families labeled LTR/DIRs, 40 repeated families labeled LINE/I, 7 repeated families labeled LINE/R2-NeSL and 81 repeated families labeled DNA/Kolobok-Hydra on the Ant dataset. These repeated families did not appear in the detection results of RepeatMasker. The analysis of the difference between the detection results of LongRepMarker and RepeatMasker is carried out in the discussion section.
Comparison of the detection results of LongRepMaker with that of RepeatScout and RepeatModeler2 is shown in Figures 4, 5, 9, Supplementary Figures S22 to S23 and Tables S15 to S26. From Figure 4, we can find that most of the detected fragments can be aligned to several different locations on the reference genome of human (hg38). For example, the fragment labeled ‘0’ can be aligned to chr1 and chr19, respectively. Figure 5 shows the proportion and representative classification of the detection results generated from the three tools on the six species covering the corresponding RepBase library and the repetitive regions on the reference genome. From the perspective of the coverage of the total base ratio, LongRepMarker has certain advantages compared with the latter two tools. For example, LongRepMarker’s detection results on the Human dataset cover 82.45% of the bases in the Human’s RepBase library as compared to 73.70% for RepeatScout, and 63.33% for RepeatModeler2. Figure 9 and Supplementary Figure S22 show that the repetition frequency and length distribution of the fragments detected by LongRepMarker have significant advantages over the latter two methods. For example, the length of the longest fragment in LongRepMarker’s detection results on the Drosophila dataset is 32.600 kb, as compared to 20.200 kb for RepeatScout, and 10.000 kb for RepeatModeler2.
Detection results of the de novo mode based on only NGS short reads
The representative detection performance of LongRepMarker based on only NGS short reads is shown in Figures 6 and 9, and the detailed detection results of this mode are shown in Supplementary Figure S24 and Supplementary Tables S30 to S39. Five NGS datasets (Drosophila, Ant, Mouse, Human-chr14 and HG003_NA24149_father (WGS)) are used in this test, and the performance of LongRepMarker is compared with two state-of-the-art tools (RepARK and REPdenovo). From Figure 6, we can see that LongRepMarker has certain advantages compared with the latter two tools in the coverage of the total base ratio. For example, the detection results of LongRepMarker on the Mouse dataset cover 69.48% of the bases in the corresponding RepBase library, while the corresponding ratios of RepARK and REPdenovo are 51.62% and 22.70%, respectively. In addition, from the perspective of repetitive sequence classification, LongRepMarker can find more repetitive fragments and families in most datasets than the latter two methods. For example, on the mouse dataset, the detection result of LongRepMarker can cover 27.05% of the total length of the LINEs-type repetitive sequence in the corresponding RepBase library, while the proportions of the latter two tools are only 19.88% and 17.85%, respectively. Detailed classification of detection results of LongRepMarker based on only NGS short reads is shown in Supplementary Section S3.5.3.
From Figure 9 and Supplementary Figure S24, we can find that the distribution range of length and repetition frequency of the repetitive sequences found by LongRepMarker is larger than that of those two compared tools, which also implies that the detection results of LongRepMarker are more comprehensive and complete than that of the two compared tools. For example, the detected repetitive fragment length of LongRepMarker on the Mouse dataset ranges from 1bp to 23.6 kb, while that of RepARK and REPdenovo ranges from 1 bp to 16.4kp and from 1 bp to 6.1 kp, respectively. Tables S30 to S39 show the proportion and detailed classification of the detection results generated from the three tools on these five NGS datasets covering the corresponding RepBase library and reference genome. Some practical examples show the completeness and coverage of the repetitive sequences detected by LongRepMarker, RepARK and REPdenovo in the same region of the mouse genome (Supplementary Figure S25 to S28).
Performance of the de novo mode based on NGS short reads + barcode linked reads/SMS long reads
In order to verify that long sequencing fragments can effectively resolve the problem of repetitive regions that cannot be solved during the assembly of short sequencing fragments, we used four well-known assemblers to perform the assembly task on three real datasets of HG003_24149_father, HG004_NA24143_mother and HG002_NA24385_son. The test results are shown in Supplementary Tables S40 to S45. Assembly effect comparison of several tools before and after using barcode linked reads is shown in Supplementary Section S3.5.2.
Up to date, de novo detection methods are all proposed based on a single type of sequencing data (e.g. RepARK and REPdenovo are proposed based on NGS short reads, and RepLong is proposed based on the third-generation sequencing long reads). The de novo mode of LongRepMarker is currently the only detection method proposed based on the multi-source sequencing data fusion strategies (e.g. NGS short reads + barcode linked reads or NGS short reads+SMS long reads). To verify the performance of the de novo mode based on NGS short reads + barcode linked reads/SMS long reads, we tested these two types of de novo detection modes using three sets of real hybrid sequencing datasets respectively (Supplementary Table S13). The NGS short paired-end reads, barcode linked reads and SMS long reads used in this experiment are downloaded from the NCBI website (https://ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data). The detection results are shown in Figures 7, 9 and Supplementary Figure S29, and Supplementary Tables S40 to S45.
To our knowledge, LongRepMarker is the first method that can be taken both short and long fragment reads (barcode linked and SMS reads) as inputs. Therefore, we did not compare LongRepMarker with other methods in this experiment. However, the experimental results here demonstrate that (i) LongRepMarker provides more flexible options to users and cope better with the increasing popularity of long reads. (ii) This detection mode can make full use of the advantages of mixed sequencing data and make the detection results more superior than those obtained by using the single source sequencing data. For example, we can take HG003_NA24149_father dataset as an example to compare and analyze the test results of single source sequencing data and that of mixed sequencing data. The test results on single-source data cover the number and base length of DNA transposon elements in the human RepBase library as 448 and 121.106 kp, respectively, whereas the corresponding test results on mixed data are 529 and 157.331 kp, respectively (Supplementary Tables S34 and S40).
Performance of the de novo mode based on only SMS long reads
In order to better comply with the market demand and further expand the application scope of this system, we have developed a new detection mode based on only the SMS long reads under the LongRepMarker framework. Compared with the existing detection methods based on the SMS long reads, this mode has the advantages of longer fragments, lower memory consumption, higher speed and higher detection accuracy. The input of this mode is only SMS long reads, and the output is the detection results which contain the final repeat library and some reports.
RepLong is a novel de novo repeat element identification method based on PacBio long reads. RepLong can handle lower coverage data and serve as a complementary solution to the existing methods to promote the repeat identification performance on long read sequencing data. In order to verify the detection performance of the de novo detection mode based on only the SMS long reads, we carried out a performance comparison between LongRepMarker and RepLong on four sets of real Pacbio datasets, and the representative detection results are shown in Figures 8 and 9. The complete experimental results are shown in Supplementary Section S3.5.5.
From the results shown in Figure 9, the longest fragment of detected results generated from LongRepMarker based on the human_100k dataset is 25.800 kb, while the corresponding value of RepLong is 14.600 kb, and the proportion of detected fragments of LongRepMarker covering the RepBase library is 73.24%, as compared to 20.90% for RepLong. From the results shown in Figure 9 and Supplementary Figure S24, we can find that the longest fragment of detected results generated from LongRepMarker based on the drosophila_100k dataset is 31.400kb, while the corresponding value of RepLong is 14.800 kb, and the proportion of detected fragments of LongRepMarker covering the RepBase library is 37.29%, as compared to 19.56% for RepLong. The data selected in the experiment comes from the RepLong website (Supplementary Table S13), where the coverage of the first two datasets is low, and the coverage of the latter two datasets is relatively high. In order to compare with RepLong under the low and high coverage conditions, we also chose the same datasets for testing. The evaluation results displayed in Figures 8, 9, Supplementary Figure S30, and Supplementary Tables S46 to S53 all show that LongRepMarker can produce superior detection performance than RepLong on both low-coverage sequencing data and high-coverage sequencing data. Furthermore, to verify the authenticity of the longest repetitive sequence detected by LongRepMarker, we selected the longest fragment of detected repeat from the dataset of Human_100k. The visualization is shown in Figure 10, and the alignment tool MUMmer (62) and the visualization tool LINKVIEW (https://github.com/YangJianshun/LINKVIEW) are used in this test. The visualization proved that the longest detected repeat fragment by LongRepMarker is a real repeating unit which appears repeatedly inside the chromosome NC_000011.10.
DISCUSSIONS
Linked-reads provide the long range information missing from standard approaches, which builds on the Illumina sequencing technology to provide indexing and barcoding information along with short reads to localize the latter on long DNA fragments (barcode linked-reads), thus benefiting the economies of a high throughput platform. There have been some barcode linked genomics datasets. For example, one can download the real barcode linked reads of human from the NCBI website (ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/). However, the available barcode linked data is still limited. In order to fully validate the performance of LongRepMarker based on the NGS short reads + real barcode linked reads, we can also use the method introduced in Supplementary Section S1.4.5 to simulate the required barcode linked reads.
LongRepMarker can discover some new repetition types (include new families and novel repetitive sequences) that RepeatMasker cannot find. In order to prove this, we conducted two experiments: (i) classifying the detection results of the two tools by RepeatModeler2, and then comparing the classification results, and (ii) removing LongRepMarker’s detection results that are covered by RepeatMasker, and classifying the remainders by RepeatModeler2. Those two specific experiments are carried on the three species including Drosophila, Ant and Human-chr14. In order to fully demonstrate the high specificity of repeat sequences detected by LongRepMarker, the working modes are set to reference-assisted and de novo, respectively. The input of these two modes are reference genome and sequencing reads, respectively. The detailed steps of experiments 1 and 2 are described in Supplementary Section S3.8.
The results in Tables 1 to 3, and Supplementary Tables S61 and S62 show that LongRepMarker can find some new repetitive sequence types that cannot be found by RepeatMasker. For example, the results in Supplementary Table S61 show that LongRepMarker found DNA transposon elements such as hAT-hATm, IS, MULE-NOF and hAT-hobo on the Drosophila dataset, which are not found by RepeatMasker. In addition, in terms of the number of repeats in some categories, LongRepMarker can find more repeats than that of RepeatMasker under the same condition. For example, LongRepMarker found 277 DNA transposon elements with subclass name tcmar-tc1 in the ant dataset, whereas RepeatMasker only found 136 such elements. Furthermore, it can be seen from the results shown in Supplementary Table S62 that LongRepMarker can find many unique repetitive sequences which do not appear in RepeatMasker’s detection results. For example, LINE elements such as R1, R1-LOA, Jockey, I-Jockey, CR1, I, LOA, R2 and L2 on the Drosophila dataset only appear in the detection results of LongRepMarker. It is worth noting that the results of the two experiments here are different from the classification of the detection results on the two sppecies of Ant and Drosophila in the supplementary. The main reason is that the two experiments here are based on the RepeatClassifier module in RepeatModeler2 to classify the detection results of LongRepMarker and RepeatMasker, that is, to label the attribution of each detected fragment based on RepBase and Dfam libraries (each fragment has a unique repetition type corresponding to it). The classification in supplementary refers to the number and ratio of the corresponding types of repetitive sequences in the RepBase and Dfam libraries, and reference genome covered by the detection results (each fragment is RepBase may have multiple detection fragments corresponding to it, which means that as long as the detection fragment can be aligned with the sequence in the library, it will be counted). Further more, from the data shown in these tables, we also found that many repeated fragments are labeled as unknown. It means that these fragments cannot find their category in the corresponding RepBase and Dfam libraries. By definition, these fragments should belong to the novelly discovered repetitive sequences, but it is still unclear what type they should be. According to the number of the unknown fragments, LongRepMarker can find more such kind of fragments than RepeatMasker. It can be seen from Table 4 that these fragments can also be aligned to different locations in the genome.
The size of k-mers has a certain impact on the processing efficiency of LongRepMarker, because the smaller the size of k, the easier it is for k-mers to aggregate into unique k-mers (k-mers to their canonical representation with respect to reverse-complementation which are called the unique k-mers), which makes the final unique k-mer set smaller, thus reducing the time and computational overhead of the subsequent alignment process. Theoretically, the influence of k-mers size on the accuracy of the test results is not significant, because LongRepMarker finds candidate repetitive sequences by looking for multiple alignment unique k-mers and their coverage regions on the reference genome or assembly results. Theoretically, the size of k-mers does not affect the acquisition of multiple alignment unique k-mers and their coverage regions on the reference genome or assemblies. However, in fact, due to the existence of sequencing errors, the size of k-mers will have a certain impact on the accuracy of detection results, which is mainly manifested in the small size of k (such as less than 11 bp). The main reason for this effect is that when the size of k-mer is small, it is easy to cause coupling alignment under the combined effect of sequencing error and alignment fault tolerance strategy, which leads to the ordinary unique k-mer which are not in the range of multiple alignment unique k-mers to be screened into the process of detection, resulting in the final detection results containing a large number of non-repetitive elements. In order to solve this problem, we need to limit the minimum value of k. In practice, the formula in Supplementary Section S1.4.7 is usually used to limit the size of k.
There are some reports generated in the detection results of LongRepMarker (Supplementary Section S1.4.6). First of all, LongRepMarker generates a repetitive sequence library with annotation information, as shown in Supplementary Figure S8. In this file, the first line starting with the angle bracket records the fragment ID and the repeat type of this fragment (e.g. the repeat type of the 4603-th fragment is satellite DNA). The second line is composed of A–T–G–C bases, which records the specific repetitive sequence. Secondly, LongRepMarker generates a report that records the detailed distribution of repetitive sequences in the genome, as shown in Supplementary Figure S9. The report includes the fragment ID, the starting position and ending position of the repetitive region on the fragment, the starting position and ending position of the repetitive region aligned to the reference sequence, the detailed alignment (cigar string), and the identity value of the alignment. The multiple occurrence of the same fragment ID in the report indicates that there are multiple copies of the fragment in the genome, and the number of occurrences is the number of copies. Thirdly, LongRepMarker generates a statistical report which records the details about the number of repeats, the proportion and the covered bases of each type of repeats, as shown in Supplementary Figure S10. This report is obtained by mapping the records in the RepBase and Dfam libraries to the detection results generated by LongRepMarker through RepeatMasker. Finally, LongRepMarker generates a VCF format structural variation statistical report in the detection results, just as shown in Supplementary Figure S11. VCF (Variant Calling Format) is a tab-delimited text file that is used to describe single nucleotide variants (SNVs) as well as insertions, deletions, and other sequence variations.
The genomic variation regions between repeating segments generate a chimeras, which can negatively affect the alignment of the entire segment. Chimeras consist of two or more repetitive regions and some genomic variation regions, which cannot be aligned to overlap sequences many times. However, the genomic variation regions between repeating segments are also the important component of repeating regions, and they are an important manifestation of repetitive regions polymorphism (63). In addition, the study and analysis of genomic structural variations that occur within the repetitive regions can provide a new perspective for understanding life processes and analyzing life mechanisms (64). LongRepMarker is designed based on technologies of the de novo sequence assembly and multiple sequence alignment to identify repetitive regions in a genome. From the perspective of implementation principles, it can identify the genomic structural variations contained in the repetitive sequences, as shown in Table 4. In this table, the number of repetitions of a fragment tag in the first column is equal to the number of copies of the repetitive sequence that the tag corresponds to in the genome, and the last column records the detailed type and size of the variation in each repetitive sequence. Based on the above reasons, we have completely preserved the genomic variations that occur inside the repetitive fragments (Supplementary Section S3.7).
CONCLUSION
Various studies have demonstrated the important of repetitive elements in genomes. However, existing methods are not able to provide robustly satisfactory performance because NGS reads are too short and long reads often have high error rates. In this study, we proposed a novel identification framework, LongRepMarker, based on the global de novo assembly of Illumina short paired-end reads and barcode linked reads or SMS long reads, and the k-mer-based multiple sequence alignment for precisely marking long repetitive sequences in genomes. LongRepMarker provides three different workflows: (i) the reference-assisted mode can quickly and accurately derive a repeat library for large species when the reference genomes are provided; (ii) the de novo modes based on NGS short reads + barcode linked reads/SMS long reads can identify the repeats in the genomes to a greater extent by assembling mixed sequencing reads of different spans; (iii) the de novo mode based on only SMS long reads is one of the few tools that only rely on third generation sequencing reads for repetitive sequence detection, and has the advantages of low memory consumption, high speed and high detection accuracy. Our comprehensive experimental results show that LongRepMarker can not only identify the repetitive sequences comprehensively, accurately and rapidly in the reference-assisted mode, but also achieve more satisfactory results than state-of-the-art de novo detection methods.
Supplementary Material
Contributor Information
Xingyu Liao, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China; Computational Bioscience Research Center (CBRC), Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia.
Min Li, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
Kang Hu, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
Fang-Xiang Wu, Department of Mechanical Engineering and Division of Biomedical Engineering, University of Saskatchewan, Saskatoon, SK S7N5A9, Canada.
Xin Gao, Computational Bioscience Research Center (CBRC), Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia.
Jianxin Wang, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [62002388, 61772557]; The NSFC-Zhejiang Joint Fund for the Integration of Industrialization and Informatization [U1909208]; Hunan Provincial Science and Technology Program [2018wk4001]; 111 Project [B18059]; King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) [BAS/1/1624-01, FCC/1/1976-18-01, FCC/1/1976-23-01, FCC/1/1976-25-01, FCC/1/1976-26-01, REI/1/0018-01-01, REI/1/4216-01-01, REI/1/4437-01-01, REI/1/4473-01-01, URF/1/4352-01-01, REI/1/4742-01-01, URF/1/4098-01-01]. Funding for open access charge: The NSFC-Zhejiang Joint Fund for the Integration of Industrialization and Informatization [U1909208]; Hunan Provincial Science and Technology Program [2018wk4001].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kazazian H.H. JrMobile elements: drivers of genome evolution. Science. 2004; 303:1626–1632. [DOI] [PubMed] [Google Scholar]
- 2.Liao X., Li M., Luo J., Zou Y., Wu F.-X., Pan Y., Luo F., Wang J.. Improving de novo assembly based on read classification. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2020; 17:177–188. [DOI] [PubMed] [Google Scholar]
- 3.Treangen T.J., Salzberg .L.. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2012; 13:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu Q., Wallrath L.L., Granok H., Elgin S.C.. (CT)n(GA)n repeats and heat shock elements have distinct roles in chromatin structure and transcriptional activation of the Drosophila hsp26 gene. Mol. Cell. Biol. 1993; 13:2802–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kundu T.K., Rao M.R.. CpG islands in chromatin organization and gene expression. J. Biochem. 1999; 125:217–222. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro J.A., von Sternberg R.. Why repetitive DNA is essential to genome function. Biol. Rev. 2005; 80:227–250. [DOI] [PubMed] [Google Scholar]
- 7.Kaltenegger E., Leng S., Heyl A.. The effects of repeated whole genome duplication events on the evolution of cytokinin signaling pathway. BMC Evol. Biol. 2018; 18:76–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu S., Wang G., Bacolla A., Zhao J., Spitser S., Vasquez K.M.. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 2015; 10:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pavlicek A., Kapitonov V.V., Jurka J.. Human Repetitive DNA. Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine. 2005; Berlin, Heidelberg: Springer Inc; 822–831. [Google Scholar]
- 10.Benson G.Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999; 27:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruce R.K., Achara S.. Chapter 19 - Introduction to Human Genetics. Clinical and Translational Science. 2009; Elsevier Inc; 265–287. [Google Scholar]
- 12.Wicker T., Francois S., Aurelie H.-V., Bennetzen J.L., Pierre C., Boulos C., Andrew F., Philippe L., Michele M., Olivier P.et al.. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007; 8:973–982. [DOI] [PubMed] [Google Scholar]
- 13.Du D., Du X., Mattia M.R., Wang Y., Yu Q., Huang M., Yu Y., Grosser J.W., Gmitter F.G.. LTR retrotransposons from the Citrus x clementina genome: characterization and application. Tree Genet. Genomes. 2018; 14:43–57. [Google Scholar]
- 14.Schmidt T.LINEs, SINEs and repetitive DNA: non-LTR retrotransposons in plant genomes. Plant Mol. Biol. 1999; 40:903–910. [DOI] [PubMed] [Google Scholar]
- 15.Lerat E.Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity. 2010; 104:520–533. [DOI] [PubMed] [Google Scholar]
- 16.Romero J.R., Carballido J.A., Garbus I., Echenique V.C., Ponzoni I.. A bioinformatics approach for detecting repetitive nested motifs using pattern matching. Evol. Bioinform. Online. 2016; 12:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergman C.M., Quesneville H.. Discovering and detecting transposable elements in genome sequences. Brief. Bioinform. 2007; 8:382–392. [DOI] [PubMed] [Google Scholar]
- 18.Smit A.F.A., Hubley R., Green P.. RepeatMasker Open-4.0. 2015; http://www.repeatmasker.org.
- 19.Tarailo-Graovac M., Chen N.. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2009; 25:4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- 20.Tempel S.Bigot Y.Using and understanding RepeatMasker. Mobile Genetic Elements. Methods in Molecular Biology (Methods and Protocols). 2012; 859:Humana Press; 29–51. [DOI] [PubMed] [Google Scholar]
- 21.Jurka J., Klonowski P., Dagman V., Pelton P.. CENSOR—a program for identification and elimination of repetitive elements from DNA sequences. Computers & chemistry. 1996; 20:119–121. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy R.C.Identification and Annotation of Transposable Elements and Agent-and GIS-based Modeling of Pathogen Transmission. 2011; University of Notre Dame. [Google Scholar]
- 23.Joseph A.B., Ian K., Warren G.. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics. 2000; 16:1040–1041. [DOI] [PubMed] [Google Scholar]
- 24.Fiston-Lavier A.S., Carrigan M., Petrov D.A., González J.. T-lex: a program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 2010; 39:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellinghaus D., Kurtz S., Willhoeft U.. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008; 9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darzentas N., Bousios A., Apostolidou V., Tsaftaris A.S.. MASiVE: mapping and analysis of SireVirus elements in plant genome sequences. Bioinformatics. 2010; 26:2452–2454. [DOI] [PubMed] [Google Scholar]
- 27.Rho M., Choi J.H., Kim S., Lynch M., Tang H.. De novo identification of LTR retrotransposons in eukaryotic genomes. BMC Genomics. 2007; 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhijian T.Eight novel families of miniature inverted repeat transposable elements in the African malaria mosquito Anopheles gambiae. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:1699–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y., Zhou F., Li G., Xu Y.. MUST: a system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene. 2009; 436:1–7. [DOI] [PubMed] [Google Scholar]
- 30.Ye C., Ji G., Liang C.. detectMITE: a novel approach to detect miniature inverted repeat transposable elements in genomes. Sci. Rep. 2016; 6:19688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han Y., Wessler S.R.. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010; 38:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang G.MITE Digger, an efficient and accurate algorithm for genome wide discovery of miniature inverted repeat transposable elements. BMC Bioinformatics. 2013; 14:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crescente J.M., Zavallo D., Helguera M., Vanzetti L.S.. MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinformatics. 2018; 19:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerat E.Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity. 2010; 104:520–533. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal P., States D.J.. The Repeat Pattern Toolkit (RPT): analyzing the structure and evolution of the C. elegans genome. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:1–9. [PubMed] [Google Scholar]
- 36.Chen G.-L., Chang Y.-J., Hsueh C.-H.. PRAP: an ab initio software package for automated genome-wide analysis of DNA repeats for prokaryotes. Bioinformatics. 2013; 29:2683–2689. [DOI] [PubMed] [Google Scholar]
- 37.Edgar R.C., Myers E.W.. PILER: identification and classification of genomic repeats. Bioinformatics. 2005; 21:i152–i158. [DOI] [PubMed] [Google Scholar]
- 38.Nicolas J., Peterlongo P., Tempel S.. Finding and characterizing repeats in plant genomes. Plant Bioinformatics. 2016; 1374:293–337. [DOI] [PubMed] [Google Scholar]
- 39.Ou S., Su W., Liao Y., Chougule K., Agda J.R.A., Hellinga A.J., Lugo C., Santiago B., Elliott T.A., Ware D.et al.. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019; 20:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saha S., Bridges S., Magbanua Z.V., Peterson D.G.. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008; 36:2284–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price A.L., Jones N.C., Pevzner P.A.. De novo identification of repeat families in large genomes. Bioinformatics. 2005; 21:i351–i358. [DOI] [PubMed] [Google Scholar]
- 42.Ruiqiang L., Jia Y., Songgang L., Jing W., Yujun H., Chen Y., Jian W., Huanming Y., Jun Y., Wong G.et al.. ReAS: Recovery of ancestral sequences for transposable elements from the unassembled reads of a whole genome shotgun. PLoS Comput. Biol. 2005; 1:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jieming S., Liang C.. Generic repeat finder: a high-sensitivity tool for genome-wide de novo repeat detection. Plant Physiol. 2019; 180:1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jullien M.F., Robert H., Clément G., Jeb R., Andrew G.C., Cédric F., Arian F.S.. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koch P., Platzer M., Downie B.R.. RepARK-de novo creation of repeat libraries from whole-genome NGS reads. Nucleic Acids Res. 2014; 42:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu C., Nielsen R., Wu Y.. REPdenovo: inferring de novo repeat motifs from short sequence reads. PLoS one. 2016; 11:e0150719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo R., Li Y.R., He S., Ou-Yang L., Sun Y., Zhu Z.. RepLong: de novo repeat identification using long read sequencing data. Bioinformatics. 2017; 34:1099–1107. [DOI] [PubMed] [Google Scholar]
- 48.Newman M.E.J.Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:8577–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E.. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008; 2008:P10008. [Google Scholar]
- 50.Yang Z., Algesheimer R., Tessone C.J.. Comparative analysis of community detection algorithms on artificial networks. Scientific Rep. 2016; 6:30750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D.et al.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y.et al.. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012; 1: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J., Birol I.. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009; 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zerbino D.R., Birney E.. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008; 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng Y., Leung H.C.M., Yiu S.M., Chin F.Y.L.. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012; 28:1420–1428. [DOI] [PubMed] [Google Scholar]
- 56.Li H.Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rizk G., Lavenier D., Chikhi R.. DSK: k-mer counting with very low memory usage. Bioinformatics. 2013; 29:652–653. [DOI] [PubMed] [Google Scholar]
- 58.Liao X., Li M., Zou Y., Wu F.-X., Pan Y., Wang J.. An efficient trimming algorithm based on multi-feature fusion scoring model for NGS data. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2020; 17:728–738. [DOI] [PubMed] [Google Scholar]
- 59.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000 Genome Project Data Processing Subgroup. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee H., Popodi E., Foster P.L., Tang H.. Detection of structural variants involving repetitive regions in the reference genome. J. Comput. Biol. 2014; 21:219–233. [DOI] [PubMed] [Google Scholar]
- 61.Smirnov G.B.Repeats in bacterial genome: evolutionary considerations. Mol. Gen. Mikrobiol. Virusol. 2010; 25:56–65. [PubMed] [Google Scholar]
- 62.Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L.. Versatile and open software for comparing large genomes. Genome Biol. 2015; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Minh D.C., Sureshkumar B., Mikael B.. Sequencing technologies and tools for short tandem repeat variation detection. Brief. Bioinform. 2015; 16:193–204. [DOI] [PubMed] [Google Scholar]
- 64.Lupski J.R., Weinstock G.M.. Short, interspersed repetitive DNA sequences in prokaryotic genomes. J. Bacteriol. 1992; 174:4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.