

Abstract
Background
Relapsed acute myeloid leukemia (AML) is associated with the acquisition of additional somatic mutations which are thought to drive phenotypic adaptability, clonal selection and evolution of leukemic clones during treatment. We performed high throughput exome sequencing of matched presentation and relapsed samples from 6 cytogenetically normal AML (CN-AML) patients treated with standard remission induction chemotherapy in order to contribute with the investigation of the mutational landscape of CN-AML and clonal evolution during AML treatment.
Result
A total of 24 and 32 somatic variants were identified in presentation and relapse samples respectively with an average of 4.0 variants per patient at presentation and 5.3 variants per patient at relapse, with SNVs being more frequent than indels at both disease stages. All patients have somatic variants in at least one gene that is frequently mutated in AML at both disease presentation and relapse, with most of these variants are classic AML and recurrent hotspot mutations including NPM1 p.W288fs, FLT3-ITD, NRAS p.G12D and IDH2 p.R140Q. In addition, we found two distinct clonal evolution patterns of relapse: (1) a leukemic clone at disease presentation acquires additional mutations and evolves into the relapse clone after the chemotherapy; (2) a leukemic clone at disease presentation persists at relapse without the addition of novel somatic mutations.
Conclusions
The findings of this study suggest that the relapse-initiating clones may pre-exist prior to therapy, which harbor or acquire mutations that confer selective advantage during chemotherapy, resulting in clonal expansion and eventually leading to relapse.
Keywords: Acute myeloid leukemia, Clonal evolution, Mutation, Relapse, Whole exome sequencing
Background
Cytogenetic analysis has been used for more than three decades to define the molecular pathogenesis of acute myeloid leukemia (AML), and remains as the first-tier screening for AML classification. Recurrent chromosomal structural variations such as t(8;21), inv(16), t(15;17), del(5) and del(7) are established diagnostic and prognostic markers suggesting that acquired genomic abnormalities play an essential role in leukemogenesis [1]. However, nearly half of adult AML cases (40–47%) and about 15–30% of pediatric AML cases have a normal karyotype lacking recurrent structural abnormalities [2, 3]. Somatic point mutations affecting numerous genes have been described in AML at presentation, many of which are pathogenic and prognostic, including RUNX1 [4], NPM1 [5], NRAS/KRAS [6] and CEBPA [7]. High throughput sequencing technology has intensified the search for somatic mutations in AML, leading to the discovery of novel mutations in established pathways, such as RAS [8], and identified new pathways through whole genome-based approaches [9]. This approach has also been applied to the study of relapsed leukemia, successfully identifying relapse-specific coding sequence mutations affecting ETV6 and MYO18B [10].
Cancer is an evolutionary process, as evidenced by the accumulation of somatic mutations in cancer cells as a result of continuous mutation acquisition during disease progression. Most mutations at disease presentation in AML are thought to be acquired after initiating genetic lesions such as t(8;21) and t(15;17), although a small number of pre-existing mutations are already present before cells acquire these advantageous initiating mutations. There are some evidences that initiating events promote mutagenesis, predisposing cells to the acquisition of additional somatic mutations [11–14] which may contribute to genomic instability in a cancer genome. Genomic instability can increase the mutation rate in the cancer genome through many different mechanisms, which play an important role in cancer evolution.
While chemotherapy with cytotoxic agents may eliminate cancer cells with dominant clones during remission induction treatment, the majority of AML patients experience disease relapse, which is difficult to treat. The mechanisms driving relapse evolution remain unclear although there is evidence that relapse is driven by novel mutations acquired after the chemotherapy. High throughput sequencing of matched presentation and relapse AML samples have revealed two major patterns of AML relapse evolution [10, 15–17]. Firstly, a leukemic clone at disease presentation acquires additional mutations and evolves into the relapse clone after the chemotherapy and secondly, the pre-leukemic clone from the founding clone acquires additional mutations and evolves into a relapse clone. Importantly, both patterns of relapse are defined by the acquisition of additional somatic mutations.
Therefore, it is thought that there are two major underlying mechanisms driving mutagenesis in relapsing AML. Firstly, the genomic instability of the leukemic clone is a major cause of mutagenesis and contributes to genetic heterogeneity in AML. Secondly, chemotherapy agents used in induction chemotherapy contribute to relapsed AML since most chemotherapy agents are genotoxic, which may lead to mutation. Collectively, both mechanisms are predicted to be responsible for the etiology of relapse-driver mutations. In this study, whole exome sequencing of matched presentation and relapse AML samples was performed to map changes in the mutational landscape between presentation and relapse AML in order to determine the clonal origins of relapsed AML and clonal evolution during treatment of AML in order to better understand disease evolution and heterogeneity in AML.
Results
Identification of somatic variants
Somatic variants including nonsynonymous single nucleotide variant (SNV, both missense and nonsense) and small insertion/deletion (indel, both in-frame and frameshift), as well as splice site variants were identified following several layers of filters using Ingenuity Variant Analysis software and after exclusion of variants with benign and uncertain significance (Table 1). Two mutations at presentation, NPM1 p.W288fs (Patient 1), FLT3-ITD (Patient 1) and one mutation at relapse, NPM1 p.W288fs (Patient 5) detected from initial RT-PCR screening were not detected by exome sequencing. In total, 24 and 32 variants were identified in presentation and relapse samples respectively (Table 2), with an average of 4.0 variants per patient at presentation (range 3–6) and 5.3 variants per patient at relapse (range 3–8). In terms of type of variant, SNVs were more common than indels at both presentation and relapse with 9 indels and 15 SNVs identified at presentation and 12 indels and 20 SNVs identified at relapse.
Table 2.
Number of somatic variants at presentation and relapse
| Patient | Presentation | Relapse | ||||
|---|---|---|---|---|---|---|
| Indel | SNV | Total no. of variants | Indel | SNV | Total no. of variants | |
| 1 | 2 | 1 | 3 | 3 | 1 | 4 |
| 2 | 1 | 2 | 3 | 1 | 2 | 3 |
| 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| 4 | 2 | 2 | 4 | 2 | 4 | 6 |
| 5 | 1 | 5 | 6 | 3 | 5 | 8 |
| 6 | 2 | 3 | 5 | 2 | 6 | 8 |
| Total no. of variants | 9 | 15 | 24 | 12 | 20 | 32 |
Table 1.
Somatic variants identified from whole exome sequencing
| Patient | Gene | Reference allele | Variant allele | Variation type | Gene region | AA change | Presentation | Relapse | Frequency in COSMIC (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Read depth | VAF (%) | Read depth | VAF (%) | ||||||||
| 1 | PTPRQ | GGGT | Deletion | Exonic |
c.6452_6453 + 2delGGGT; |
– | – | 103 | 17.5 | 0.0 | |
| RB1 | AC | Frameshift deletion | Exonic | p.K123fs*39 | 104 | 15.4 | – | – | 0.0 | ||
| RB1 | ATAC | Frameshift insertion | Exonic | p.P121fs*4 | 103 | 16.5 | – | – | 0.0 | ||
| RB1 | T | Frameshift deletion | Exonic | p.Y120fs*8 | 99 | 17.2 | – | – | 0.0 | ||
| RB1 | TTG | In-frame deletion | Exonic | p.A117_I118delinsV | 99 | 17.2 | – | – | 0.0 | ||
| RB1 | A | Frameshift deletion | Exonic | p.L116fs*12 | 95 | 17.9 | – | – | 0.0 | ||
| RB1 | AATC | Frameshift deletion | Exonic | p.R114fs*13 | 98 | 17.4 | – | – | 0.0 | ||
| SELE | C | T | SNV | Splice site |
c.902-1G>A |
113 | 55.8 | 111 | 55.9 | 0.0 | |
| 2 | ADAM9 | TTTT | Frameshift insertion | Exonic |
c.1883_1884insTTTT; p.C629fs*3 |
43 | 18.6 | – | – | 0.0 | |
| APOB | G | A | Nonsynonymous SNV | Exonic |
c.10,579 C>T; p.R3527W |
278 | 45.0 | 247 | 40.9 | 0.0 | |
| DNAH5 | T | Deletion | Splice site |
c.2578-2delA |
– | – | 17 | 17.7 | 0.0 | ||
| NRAS | C | T | Nonsynonymous SNV | Exonic |
c.35G>A; p.G12D |
390 | 14.4 | 434 | 33.4 | 29.1 | |
| 3 | CEBPA | CTG | In-frame insertion | Exonic |
c.934_936dupCAG; p.Q312dup |
283 | 87.6 | 268 | 47.0 | 2.1 | |
| CFTR | G | A | Nonsynonymous SNV | Exonic |
c.1865G>A; p.G622D |
103 | 50.5 | 94 | 56.4 | 0.0 | |
| MUTYH | C | A | Stop gain SNV | Exonic |
c.1435G>T; p.E479* |
152 | 47.4 | 126 | 48.4 | 0.0 | |
| 4 | ESRRB | C | T | Nonsynonymous SNV | Exonic |
c.1144 C>T; p.R382C |
202 | 49.5 | 236 | 48.7 | 0.0 |
| FLT3 | G | A | Nonsynonymous SNV | Exonic |
c.1577 C>T; p.T526M |
419 | 48.5 | 385 | 48.8 | 0.0 | |
| HBB | AAAG | Frameshift deletion | Exonic |
c.126_129delCTTT; p.F42fs*19 |
241 | 50.2 | 263 | 39.5 | 0.0 | ||
| LFNG | GATG | Frameshift insertion | Exonic |
c.163_166dupGATG; p.E56fs*2 |
104 | 50.0 | 116 | 35.3 | 0.0 | ||
| MAP3K20 | A | T | SNV | Splice site | c.416-2 A>T | – | – | 66 | 15.2 | 0.0 | |
| MAP3K20 | G | T | SNV | Splice site | c.416-1G>T | – | – | 66 | 15.2 | 0.0 | |
| 5 | ESRRB | C | T | Nonsynonymous SNV | Exonic |
c.1144 C>T; p.R382C |
218 | 48.6 | 216 | 50.5 | 0.0 |
| EYS | C | A | Stop gain SNV | Exonic |
c.8170G>T; p.E2724* |
155 | 57.4 | 153 | 54.3 | 0.0 | |
| GUCY2D | C | T | Nonsynonymous SNV | Exonic |
c.164 C>T; p.T55M |
75 | 66.7 | 64 | 53.1 | 0.0 | |
| IDH2 | C | T | Nonsynonymous SNV | Exonic |
c.419G>A; p.R140Q |
124 | 38.7 | 115 | 26.1 | 42.9 | |
| NPM1 | TCTG | Frameshift insertion | Exonic |
c.860_863dupTCTG; p.W288fs |
– | – | 17 | 41.2 | 36.3 | ||
| SLC12A1 | C | A | Stop gain SNV | Exonic |
c.1584 C>A; p.Y528* |
204 | 55.4 | 146 | 38.4 | 0.0 | |
| WRN | AGGG | Deletion | Splice site |
c.840-2_841delAGGG; |
– | – | 31 | 19.4 | 0.0 | ||
| WRN | TTTT | Frameshift insertion | Exonic |
NM_000553.5; c.844_845insTTTT; p.S282fs*24 |
– | – | 31 | 19.4 | 0.0 | ||
| 6 | ATM | G | T | Nonsynonymous SNV | Splice site |
c.497-1G>T |
– | – | 18 | 33.3 | 0.0 |
| ATRX | G | A | Stop gain SNV | Exonic |
c.7219 C>T p.R2407* |
– | – | 120 | 44.2 | 14.29 | |
| CEBPA | TTGGCCTT | Frameshift insertion | Exonic |
c.929_930insAAGGCCAA p.Q346fs*10 |
303 | 42.9 | 349 | 49.6 | 0.0 | ||
| CEBPA | CTCCA | Frameshift deletion | Exonic |
c.923_927delTGGAG; p.V343fs*11 |
329 | 43.5 | 350 | 47.4 | 0.0 | ||
| MPST | C | A | Stop gain SNV | Exonic |
c.52G>T; p.E18* |
184 | 51.1 | 193 | 49.7 | 0.0 | |
| RYR2 | C | T | Nonsynonymous SNV | Exonic |
c.13,739 C>T; p.T4580M |
– | – | 29 | 51.7 | 0.0 | |
| TBXAS1 | C | T | Stop gain SNV | Exonic |
c.997 C>T; p.R333* |
148 | 41.2 | 150 | 52.0 | 0.0 | |
| VWF | G | A | Nonsynonymous SNV | Exonic |
c.4195 C>T; p.R1399C |
161 | 40.4 | 154 | 37.7 | 0.0 | |
A total of 23 shared variants occurred at both presentation and relapse (Fig. 1). Moreover, 1 variant were presentation-specific, and 9 variants were relapse-specific. Additional somatic variants were acquired in five relapse samples (Patient 1, 2, 4, 5 and 6) which gained between 1 and 3 new variants when compared to their corresponding presentation samples. Two patients had the same number of variants at both presentation and relapse samples (Patient 2 and 3), of which 1 patient (Patient 2) acquired and lost equivalent numbers of variants.
Fig. 1.
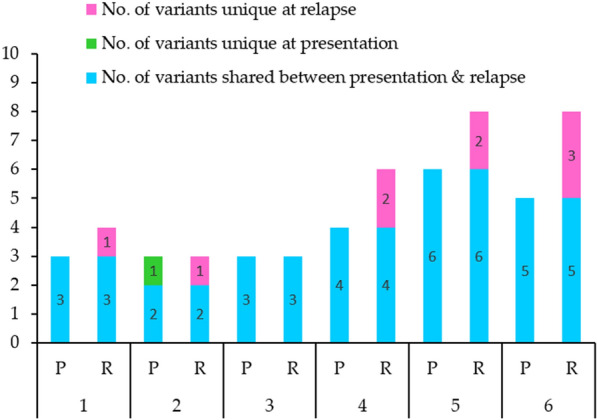
Total number of somatic variants specific to presentation or relapse and somatic variants shared at both disease stages
Evaluation of somatic variants at presentation and relapse
The filtered somatic variants were further evaluated to define the oncogenic effect of the variants in determining their potential importance as AML driver mutations via interrogation of the COSMIC database. The mutated genes were ranked according to frequency of genes implicated in AML as reported in the COSMIC database regardless of type and position of the variants in genes, as either classical types of variants, or previously reported and/or recurrent hotspots variants, or novel variants (Fig. 2). Somatic variants in recurrently mutated genes in AML (NPM1, FLT3, NRAS, ATM, CEBPA, IDH2) were identified in all patients. Six of these variants are classic AML driver and recurrent hotspot mutations whereas variants such as FLT3 p.T526M (Patient 4), ATM c.497-1G>T (Patient 6), CEBPA p.Q346fs*10 (Patient 6) and CEBPA p.V343fs*11 (Patient 6) were not previously reported in AML (Table 1). In addition to somatic variants in recurrently mutated genes, there were 9 mutated genes that have been previously reported in a small number of AML cases in COSMIC (less than 2%) and an additional 11 mutations in genes not previously reported in AML.
Fig. 2.

Mutational spectrum of somatic variants at presentation and relapse characterized according to frequency in AML as reported in COSMIC. The mutated genes identified in each patient were ranked according to frequency of genes implicated in AML as reported in the COSMIC database
To further analyze identified mutations, the mutated genes were clustered into three groups based on their putative effect that may relevant for leukemic transformation according to existing model of leukemogenesis [18, 19]: (1) mutations affecting genes that contribute to cell proliferation (class I mutations); (2) mutations affecting genes involved in myeloid differentiation (class II mutations); and (3) mutations affecting genes involved in epigenetic modification (class III mutations) (Fig. 3). All patients have mutations in at least one gene that either affect cell proliferation, are involved in myeloid differentiation or involved in epigenetic modification at presentation, with 1 patient (Patient 1) having both class I and II mutations at presentation. At relapse, acquisition of both classes of mutations were observed in 2 patients (Patient 1 and 6), where Patient 6 that lack complementary class of mutation at presentation acquired a class I mutation at relapse. In contrast, the remaining 20 mutated genes identified in this cohort have yet unknown functions in leukemogenesis.
Fig. 3.

Classification of somatic variants at presentation and relapse samples based on their known or predicted role in leukemogenesis
Correlation of presentation variant allele fraction (VAF) and relapse
Variant allele fraction (VAF) was used to track each somatic variant from presentation to relapse to map clones that persisted, those that resolved from presentation to relapse as well as novel mutations that emerged at relapse in order to delineate the clonal architecture of leukemic cells and to identify driver mutations that promote clonal expansion in relapse AML (Fig. 4). NRAS p.G12D (Patient 2), CEBPA p.Q312dup (Patient 3), FLT3 p.T526M (Patient 4), IDH2 p.R140Q (Patient 5), CEBPA p.Q346fs*10 (Patient 6) and CEBPA p.V343fs*11 (Patient 6) were among mutations in known AML driver genes that were retained after chemotherapy with VAF of 14.4%, 87.6%, 48.5%, 38.7%, 42.9 and 43.5%, respectively, in the presentation samples and 33.4%, 47.0%, 48.8%, 26.1%, 49.6 and 47.4% in the relapse samples. On the other hand, ATM c.497-1G>T (Patient 6) was the only mutation in known AML driver genes acquired at relapse with VAF of 33.3%. VAF score for two mutations at both presentation and relapse in Patient 1 (NPM1 p.W288fs and FLT3-ITD) and one mutation at presentation in Patient 5 (NPM1 p.W288fs) detected by molecular diagnostic screening, were not available as they were not detectable by exome sequencing. In addition, mutations in other genes acquired at relapse include PTPRQ c.6452–6453 + 2del (Patient 1), DNAH5 c.2578-2delA (Patient 2), MAP3K20 c.416-2 A>T (Patient 4), MAP3K20 c.416-1G>T (Patient 4), WRN c.840-2_841delAGGG (Patient 5), WRN p.S282fs*24 (Patient 5), ATRX p.R2407* (Patient 6) and RYR2 p.T4580M (Patient 6) with a range of 15.2–80.0% VAF score (Table 1).
Fig. 4.

Clonal evolution in a single patient based on comparison between variant allele fraction (VAF) score at presentation and relapse. Each line corresponds to an individual mutation and illustrates the presence of the mutation at both time points. Each circle corresponds to an individual cell clone, defined by harboring the identical set of mutations
Discussion
Cytogenetic findings have contributed to the understanding of genetic heterogeneity in AML, which plays an important role in establishing clonality and determining prognosis. However, this genetic heterogeneity is a major concern in cancer especially AML as it drives phenotypic adaptability which may contribute to clonal selection during treatment. One of the important causes of genetic heterogeneity is genomic instability. This instability leads to an increased mutation rate which can shape the evolution of the cancer genome through many mechanisms. Moreover, the diversity of genetic events in a cancer genome poses a significant challenge to identifying the oncogenic effect of specific genetic abnormalities. The advent of next generation sequencing (NGS) technology has allowed for more comprehensive analysis of somatic variants which has facilitated understanding of AML initiation and progression [10, 15–17, 20–22].
The present study compared the mutational landscape of 6 matched presentation and relapse CN-AML samples using high-throughput paired-end exome sequencing. The advantage of paired-end sequencing is that it allows for sequencing of both ends of a DNA fragment, resulting in high-quality and alignable sequence data that enables confident detection of genomic variants. However, one caveat with this approach is that genome coverage was very low (the human exome represents ~ 1.2% of the total genome) and mutations in non-coding sequence were not identified. Despite this, because of the high read depth achieved in this study as a result of exonic sequence enrichment, it was possible to identify somatic variants present in minor leukemic cell clones at presentation and relapse.
Somatic variants at presentation and relapse
SNVs and indels are the most abundant type of genetic variation in the human genome with ratios of indels to SNVs were 0.19–0.22 in WGS and 0.14 in WES [23, 24]. In this study, the ratio of indels to SNVs was 0.60 at both presentation and relapse, with SNVs being approximately twice as common as indels. In addition, the average number of somatic variants (SNVs and indels) identified in this study were fewer compared to average number of mutations found in previous somatic mutation studies [16, 17, 21, 22, 25]. However, some previous studies had greater coverage of the genome (whole genome sequencing) and larger sample sizes whereas SNVs and indels identified in this study were strictly limited to somatic variants with potential biological implications in AML pathogenesis. Despite this, the number of somatic mutations in AML is relatively low with an average of 13 mutations per genome as opposed to other cancers occurring in adults which often have hundreds of somatic mutations, especially solid tumors such as breast, lung or pancreas [21]. Nevertheless, accurate identification of pathogenic SNVs and indels is one of the most important challenges in cancer genome analysis and requires detailed information of their impact on disease development. The use of analysis software equipped with linked databases that provides comprehensive information of each variant is very useful to discern functionality and a role in leukemogenesis.
Coexisting variant within individual patients
The two-hit model of leukemogenesis has served as a basis to understand how mutations in certain genes drive AML [18]. Based on this model, mutations were classified according to two fundamental characteristics of cancer cells: mutations that confer cellular proliferation (class I mutations) and mutations that impair myeloid differentiation (class II mutations). The model predicts that both classes of mutation are required for AML transformation. However, genomic studies have revealed the presence of mutations in genes associated with epigenetic modification (ASXL1, DNMT3A, IDH1/2, TET2, EZH2 and MLL) in a significant proportion of AML patients [26]. Disruption of epigenetic processes including DNA methylation and histone modification can lead to altered function in genes involve in key cellular pathways such as DNA repair, RAS signaling, cell cycle and apoptosis which may cause malignant cellular transformation in cancer [27]. In addition, a genome-wide DNA methylation profiling study has demonstrated that specific methylation profiles are associated with specific AML subtypes [28], which identified oncogenic cooperativity between somatic alterations in epigenetic regulators and known AML driver mutations. However, determining oncogenic cooperativity between mutations is difficult as it requires prior knowledge of the functions of the genes and the impact of the mutated genes on the translated protein. Therefore, given the abundant number of mutated genes with uncertain significance identified in this cohort, analysis was limited to the genes most recurrently mutated in AML as reported in COSMIC and to genes not previously implicated in AML pathogenesis but with significant functions potentially relevant for leukemogenesis.
Each patient in this study had a completely unique mutational spectrum, demonstrating the heterogeneous nature and complexity of the AML genomic landscape. Despite this variability, all patients had mutations in at least one known AML driver gene that is recurrently mutated in AML. NPM1, FLT3, NRAS, CEBPA and IDH2 were the mutated genes identified in these patients at presentation which are well reported for their importance and contribution to AML pathogenesis. However, only one patient (Patient 1) presented with co-occurrence of class I and II mutations. The FLT3-ITD and NPM1 p.W288fs (TCTG insertion) mutations are inarguably defined as AML driver mutations, as numerous studies have demonstrated their co-occurrence in AML patients [17, 20, 29, 30]. In Patient 3, CEBPA mutation appeared to be a driver mutation as it is recurrently mutated in AML patients [31, 32] and has been shown to play a key role in AML initiation in murine models [33, 34]. Although no additional mutations in known AML driver genes were detected in this patient, mutation in MUTYH gene appeared to cooperate with the CEBPA mutation at presentation due to its involvement in oxidative DNA damage repair [35, 36], compared to another accompanying mutation in the CFTR gene that was commonly reported in patients with cystic fibrosis [37, 38]. The WRN gene was recognized as a tumor-suppressor gene and evidence suggests that it plays a role in promoting oncogenic proliferation [39, 40] which may likely contribute to AML transformation in Patient 5, in addition to the NPM1 and IDH2 mutations.
Collectively, these findings emphasize the necessity of further investigation of mutation cooperativity, especially for mutations in genes that does not belong to either class I or class II mutations. Indeed, it is particularly important to identify mutation cooperativity in patients that lack known recurrent combinations of mutations as this might identify novel mechanisms of AML leukemogenesis.
Clonal evolution from presentation to relapse
Indeed, flow cytometry is now routine practice in clinical laboratories for the determination of AML clonality essential to the diagnosis, prognosis and monitoring of disease as well as to direct treatment programs. However, one major advantage of NGS approaches over flow cytometry is the ability to quantify the proportion of variant reads for any given mutation, also known as the variant allele fraction (VAF), which indicates the percentage of tumor cells that harbor a specific mutation assuming a relatively pure leukemic sample. Using VAF score, each mutation at presentation can be tracked to map clones that persisted, those that resolved from presentation to relapse as well as novel mutations that emerged at relapse in order to delineate clonal evolution from presentation to relapse. A study demonstrated that multicolor flow cytometry (MFC) and NGS are complementary in detecting abnormal blast populations (via MFC) and somatic mutations (via NGS); however, a multigene NGS approach provides additional information on the mutational profiles of leukemic cells and aids in the better understanding of AML clonal evolution [41].
In most cases, mutations detected at presentation were also present at relapse with additional mutations, except for one case (Patient 3) which had no additional mutation acquired at relapse (Fig. 1). However, almost all of these additional mutations acquired at relapse are mutations in genes that have yet unknown function in leukemogenesis. By excluding these mutations, variants were analyzed to define the effect of chemotherapy on the prevalence of somatic variants at relapse specifically in known AML driver genes based on comparison between VAF score at presentation and relapse (Fig. 4). Except for NPM1 p.W288fs and FLT3-ITD mutations in which no VAF scores were available (not detected by exome sequencing), FLT3 p.T526M, CEBPA p.Q346fs*10 and CEBPA p.V343fs*11 mutations were found to be persist after treatment by having similar values at presentation and relapse (less than 10% difference). NRAS p.G12D mutation is predicted to confer relative resistance to standard combination chemotherapy treatment based on their increased VAF (greater than 10% difference) at relapse, whereas CEBPA p.Q312dup mutation was predicted to confer sensitivity to chemotherapy based on a reduced VAF score (greater than 10% difference) at relapse. On the other hand, ATM c.497-1G>T is predicted to be induced by or selected by chemotherapy since this mutation was not detected at presentation.
Further analysis was performed to determine the clonal evolution pattern from presentation to relapse in this cohort. Based on the models of clonal evolution of AML [42], a linear evolution pattern was clearly observed in one patient (epitomized by Patient 6; Fig. 4) which is characterized by the persistent of major clone at relapse with additional mutations. However, in some cases, relapse might be driven by the same set of mutations acquired at presentation, suggesting these mutations confer selective advantage during AML treatment, resulting in clonal expansion and eventually leading to relapse (epitomized by patient 1, 4 and 5; Fig. 4). In addition, despite the absence of additional mutations at relapse, the VAF score for CEBPA p.Q312dup was significantly decreased, suggesting a major clone with a CEBPA p.Q312dup mutation failed to survived chemotherapy (epitomized by patient 3; Fig. 4).
In summary, the findings from this study suggests two possible mechanisms of relapse: (1) a major clone at disease presentation survived chemotherapy and re-emerged at relapse after acquired additional mutations; (2) a major clone survived chemotherapy and re-emerged at relapse with the same set of mutations without additional mutations. The former showed that relapse is driven by the acquisition of additional somatic mutations, consistent with other previous studies investigating clonal evolution of AML [10, 15–17, 43], while the latter is associated with clonal evolution with stable mutations only [42], suggesting the presence of non-genetic alterations, such as epigenetic alterations, that might confer chemotherapy resistance. Sequence analysis of a remission sample is therefore essential to determine the VAF of these mutations at remission in order to confidently identify the pattern of clonal evolution. Furthermore, this study revealed significant variability in the mutational profile which demonstrates the heterogeneity of genomic landscape in AML; however, consists of classic AML driver and recurrent hotspot mutations as well as mutations in leukemia relevant genes. The mutational profile has important implications in treatment assignment, with a more precise, genomic guided therapy can be administered, thus limiting the use of genotoxic pro-mutagenic chemotherapy.
Conclusions
The data from this study provide detailed information of mutations in CN-AML patients which is fundamental in identifying somatic variants with putative functional impact on AML pathogenesis, both at disease presentation and relapse, and facilitates better understanding of clonal evolution from presentation to relapse. Ultimately, this approach will allow for the identification of relapse-driver mutations that could be exploited for the development of synthetic lethal interactions and novel therapies to improve outcomes and reduce death from disease.
Materials and methods
Patient samples
A cohort of 6 matched presentation and relapsed CN-AML cases were retrospectively selected from the diagnostic cases within the National Referral Centre for Bone Marrow Cytogenetics and Molecular Genetics, Cancer Research Centre, Institute for Medical Research (IMR, Kuala Lumpur, Malaysia). Patients were selected based on the normal cytogenetic finding and the availability of DNA from the bone marrow samples with leukemic blasts > 20% at both disease presentation and relapse. Patients achieved clinical remission after treatment with combination chemotherapy that included ara-C and an anthracycline (daunorubicin or idarubicin), followed by consolidation chemotherapy that included high-dose of ara-C. The median age was 36.5 years (range: 17–51 years).
G-banding karyotype analysis of 20 metaphase chromosomal spreads from the bone marrow samples was performed to rule out numerical and structural chromosomal abnormalities. Multiplex RT-PCR using HemaVision®-28 N kit (DNA Diagnostic, Risskov, Denmark) was also performed for all samples to screen for 28 leukemia causing translocations including common translocations in AML such as t(8;21)(q22;q22), t(15;17)(q24;q21) and inv(16)(p13;q22) in order to exclude cryptic chromosomal abnormalities. This qualitative test uses reverse transcription of RNA to cDNA followed by multiplex nested polymerase chain reaction and agarose gel electrophoresis to identify chromosomes, genes and exons at the breakpoint in fusion genes. In addition, all samples were further screened for AML prognostic markers including gene mutations in NPM1 and FLT3 via targeted PCR using specific primers targeted to regions of interest. The characteristics and detailed information of these patients with cytogenetics and molecular profiles are further described in Table 3.
Table 3.
Clinical and molecular characteristics of study cohort
| Patient | Status | Age at Dx (years) | Blast% | G-banding Karyotype | Molecular diagnostic (28 translocations) | AML prognostic marker |
|---|---|---|---|---|---|---|
| 1 | P | 26 | 55 | 46,XX [20] | Negative |
NPM1 (MutA) positive, FLT3-ITD positive |
| R | 30 | 46,XX [20] | Negative |
NPM1 (MutA) positive, FLT3-ITD negative |
||
| 2 | P | 17 | > 80 | 46,XX [20] | Negative | NPM1 negative, FLT3-ITD negative |
| R | 60 | 46,XX [20] | Negative | NPM1 negative, FLT3-ITD negative | ||
| 3 | P | 43 | 91 | 46,XY [20] | Negative | NPM1 negative, FLT3-ITD negative |
| R | 94 | 46,XY [20] | Negative | NPM1 negative, FLT3-ITD negative | ||
| 4 | P | 40 | 11 | 46,XY [20] | Negative | NPM1 negative, FLT3-ITD negative |
| R | 70 | 46,XY [20] | Negative | NPM1 negative, FLT3-ITD negative | ||
| 5 | P | 51 | 54 | 46,XY [20] | Negative |
NPM1 (MutA) positive, FLT3-ITD negative |
| R | 20 | 46,XY [20] | Negative |
NPM1 (MutA) positive, FLT3-ITD negative |
||
| 6 | P | 33 | 61 | 46,XX [20] | Negative | NPM1 negative, FLT3-ITD negative |
| R | 91 | 46,XX [20] | Negative | NPM1 negative, FLT3-ITD negative |
Whole exome sequencing
DNA from all primary patient samples was extracted using QIAamp DNA Blood Mini Kit (Qiagen) according to manufacturer’s protocol. Quantitation of the extracted DNA was carried out using a Nanodrop ND-1000 spectrophotometer and the required concentration and volume of DNA for exome sequencing was prepared according to sequencing service provider instruction. Sample preparation, library construction, sequencing and bioinformatics analysis were performed offsite by the Beijing Genome Institute (BGI) (Hong Kong, China). SureSelect Human All Exon V5 + UTR kit (Agilent Technologies, Santa Clara, USA) was used for enrichment of exon sequence including untranslated regions (UTRs). Samples were sequenced to give an average read depth (reads per base) of 100X, supporting the identification of minor leukemic cell clones. Paired-end sequencing was performed using Illumina HiSeq 4000 platform and analysis of raw data was processed according to BGI analysis pipeline to map reads against the hg19 build of the human genome and identify single nucleotide variants (SNVs) and insertion/deletions (indels). Paired-end whole exome sequencing was performed for each sample to identify somatic variants. By enriching for exonic sequence, high quality whole exome sequencing reads with average coverage of 93.8% of exome (range: 77.3–96.5%) and average sequencing depth/read depth of 151X (range: 15–434X) was achieved.
Data analysis
Variant calling files (.VCF) were received from the BGI. Analysis of genetic variants was performed using Ingenuity Variant Analysis™ software (Qiagen Bioinformatics) to compare the somatic mutation profile of matched AML samples at presentation and relapse in order to identify somatic mutations that drive disease progression. A series of filters were applied to select variants most likely to impact on gene function or biological processes known to be implicated in disease progression. Variants with a read depth < 10 were excluded from the analysis as were variants with an allele frequency (VAF) ≥ 5% in the healthy population [databases used include the 1000 Genomes Project, ExAC, gnomAD, NHLBI ESP exomes; minor allele frequency (MAF) < 1%)]. Identification of putative AML driver mutations were determined by identification of variants in established AML genes. Specifically, the Catalogue of Somatic Mutations in Cancer (COSMIC) database was interrogated to identify the most frequently recurrently mutated genes in AML.
Acknowledgements
The authors thank the Director General of Health, Ministry of Health Malaysia for approval to publish this scientific paper. We would also like to thank the Deputy Director General of Health (Research and Technical Support) and the Director of the Institute for Medical Research (IMR) for their support. We are also grateful to Professor James M Allan from Northern Institute for Cancer Research, Newcastle University, United Kingdom for aiding in the whole exome sequencing data analysis.
Authors’ contributions
YMY and FA designed the study and drafted the manuscript. YMY and EE collected and reviewed the clinical data. YMY, ZAS, JA and NRK carried out the translocation and mutation studies and performed data analysis. FA performed the WES data analysis. ZZ helped draft and reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This research was funded by Ministry of Health Malaysia (JPP-IMR 15–018).
Availability of data and materials
Data available for sharing upon request.
Declarations
Ethics approval and consent to participate
The study was approved by Medical Research and Ethics Committee (MREC), Ministry of Health, Malaysia (Ref. no. KKM/NIHSEC/P15-1298; date of approval 21/10/2015).
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuslina Mat Yusoff and Fadly Ahid: Joint lead authors
References
- 1.Betz BL, Hess JL. Acute myeloid leukemia diagnosis in the 21st century. Arch Pathol Lab Med. 2010;134(10):1427–33. doi: 10.5858/2010-0245-RA.1. [DOI] [PubMed] [Google Scholar]
- 2.Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood. 1998;92(7):2322–33. doi: 10.1182/blood.V92.7.2322. [DOI] [PubMed] [Google Scholar]
- 3.Stark B, Jeison M, Gabay LG, Mardoukh J, Luria D, Bar-Am I, Avrahami G, Kapeliushnik Y, Sthoeger D, Herzel G, et al. Classical and molecular cytogenetic abnormalities and outcome of childhood acute myeloid leukaemia: report from a referral centre in Israel. Br J Haematol. 2004;126(3):320–37. doi: 10.1111/j.1365-2141.2004.05038.x. [DOI] [PubMed] [Google Scholar]
- 4.Tang J-L, Hou H-A, Chen C-Y, Liu C-Y, Chou W-C, Tseng M-H, Huang C-F, Lee F-Y, Liu M-C, Yao M, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114(26):5352–61. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
- 5.Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D, Whitman SP, Wu Y-Z, Schwind S, Paschka P, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a cancer and leukemia group B study. J Clin Oncol. 2010;28(4):596–604. doi: 10.1200/JCO.2009.25.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen JW, Steenvoorden AC, Lyons J, Anger B, Böhlke JU, Bos JL, Seliger H, Bartram CR. RAS gene mutations in acute and chronic myelocytic leukemias, chronic myeloproliferative disorders, and myelodysplastic syndromes. Proc Natl Acad Sci USA. 1987;84(24):9228–32. doi: 10.1073/pnas.84.24.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green CL, Koo KK, Hills RK, Burnett AK, Linch DC, Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol. 2010;28(16):2739–47. doi: 10.1200/JCO.2009.26.2501. [DOI] [PubMed] [Google Scholar]
- 8.Tyner JW, Erickson H, Deininger MWN, Willis SG, Eide CA, Levine RL, Heinrich MC, Gattermann N, Gilliland DG, Druker BJ, et al. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113(8):1749–55. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Araten DJ, Krejci O, DiTata K, Wunderlich M, Sanders KJ, Zamechek L, Mulloy JC. The rate of spontaneous mutations in human myeloid cells. Mutat Res/Fundam Mol Mech Mutagen. 2013;749(1–2):49–57. doi: 10.1016/j.mrfmmm.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krejci O, Wunderlich M, Geiger H, Chou F-S, Schleimer D, Jansen M, Andreassen PR, Mulloy JC. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood. 2008;111(4):2190. doi: 10.1182/blood-2007-06-093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E, Casciari C, et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Investig. 2003;112(11):1751–61. doi: 10.1172/JCI17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forster VJ, Nahari MH, Martinez-Soria N, Bradburn AK, Ptasinska A, Assi SA, Fordham SE, McNeil H, Bonifer C, Heidenreich O, et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia. 2015;30:250–253. doi: 10.1038/leu.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welch John S, Ley Timothy J, Link Daniel C, Miller Christopher A, Larson David E, Koboldt Daniel C, Wartman Lukas D, Lamprecht Tamara L, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sood R, Hansen NF, Donovan FX, Carrington B, Bucci D, Maskeri B, Young A, Trivedi NS, Kohlschmidt J, Stone RM, et al. Somatic mutational landscape of AML with inv(16) or t(8;21) identifies patterns of clonal evolution in relapse leukemia. Leukemia. 2015;30:501–504. doi: 10.1038/leu.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garg M, Nagata Y, Kanojia D, Mayakonda A, Yoshida K, Haridas Keloth S, Zang ZJ, Okuno Y, Shiraishi Y, Chiba K, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126(22):2491–501. doi: 10.1182/blood-2015-05-646240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly LM, Gilliland DG. Genetics of myeloid leukaemias. Annu Rev Genomics Hum Genet. 2002;3(1):179–98. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- 19.Conway O’Brien E, Prideaux S, Chevassut T. The epigenetic landscape of acute myeloid leukemia. Adv Hematol. 2014;2014:103175. doi: 10.1155/2014/103175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel JL, Schumacher JA, Frizzell K, Sorrells S, Shen W, Clayton A, Jattani R, Kelley TW. Coexisting and cooperating mutations in NPM1-mutated acute myeloid leukemia. Leuk Res. 2017;56:7–12. doi: 10.1016/j.leukres.2017.01.027. [DOI] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research N Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farrar JE, Schuback HL, Ries RE, Wai D, Hampton OA, Trevino LR, Alonzo TA, Guidry Auvil JM, Davidsen TM, Gesuwan P, et al. Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Can Res. 2016;76(8):2197. doi: 10.1158/0008-5472.CAN-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills RE, Pittard WS, Mullaney JM, Farooq U, Creasy TH, Mahurkar AA, Kemeza DM, Strassler DS, Ponting CP, Webber C, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;21(6):830–9. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shigemizu D, Fujimoto A, Akiyama S, Abe T, Nakano K, Boroevich KA, Yamamoto Y, Furuta M, Kubo M, Nakagawa H, et al. A practical method to detect SNVs and indels from whole genome and exome sequencing data. Sci Rep. 2013;3:2161. doi: 10.1038/srep02161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartmann L, Dutta S, Opatz S, Vosberg S, Reiter K, Leubolt G, Metzeler KH, Herold T, Bamopoulos SA, Bräundl K, et al. ZBTB7A mutations in acute myeloid leukaemia with t(8;21) translocation. Nat Commun. 2016;7:11733. doi: 10.1038/ncomms11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 27.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81(4):303–11. doi: 10.1111/j.1399-0004.2011.01809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alpermann T, Schnittger S, Eder C, Dicker F, Meggendorfer M, Kern W, Schmid C, Aul C, Staib P, Wendtner C-M, et al. Molecular subtypes of NPM1 mutations have different clinical profiles, specific patterns of accompanying molecular mutations and varying outcomes in intermediate risk acute myeloid leukemia. Haematologica. 2016;101(2):e55. doi: 10.3324/haematol.2015.133819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krönke J, Bullinger L, Teleanu V, Tschürtz F, Gaidzik VI, Kühn MWM, Rücker FG, Holzmann K, Paschka P, Kapp-Schwörer S, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–8. doi: 10.1182/blood-2013-01-479188. [DOI] [PubMed] [Google Scholar]
- 31.Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C, Weissmann S, Dicker F, Kohlmann A, Schindela S, et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia. 2013;28:794. doi: 10.1038/leu.2013.273. [DOI] [PubMed] [Google Scholar]
- 32.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat Genet. 2001;27:263. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 33.Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, Theilgaard-Mönch K, Månsson R, Pedersen T, Pabst T, et al. Modeling of C/EBPα mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13(4):299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 34.Bereshchenko O, Mancini E, Moore S, Bilbao D, Månsson R, Luc S, Grover A, Jacobsen SEW, Bryder D, Nerlov C. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPα mutant AML. Cancer Cell. 2009;16(5):390–400. doi: 10.1016/j.ccr.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 35.Banda DM, Nuñez NN, Burnside MA, Bradshaw KM, David SS. Repair of 8-oxoG:A mismatches by the MUTYH glycosylase: mechanism, metals and medicine. Free Radic Biol Med. 2017;107:202–15. doi: 10.1016/j.freeradbiomed.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dumanski JP, Rasi C, Björklund P, Davies H, Ali AS, Grönberg M, Welin S, Sorbye H, Grønbæk H, Cunningham JL, et al. A MUTYH germline mutation is associated with small intestinal neuroendocrine tumors. Endocr Relat Cancer. 2017;24(8):427–43. doi: 10.1530/ERC-17-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortiz SC, Aguirre SJ, Flores S, Maldonado C, Mejía J, Salinas L. Spectrum of CFTR gene mutations in Ecuadorian cystic fibrosis patients: the second report of the p.H609R mutation. Mol Genet Genom Med. 2017;5(6):751–7. doi: 10.1002/mgg3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furgeri DT, Marson FAL, Correia CAA, Ribeiro JD, Bertuzzo CS. Cystic fibrosis transmembrane regulator haplotypes in households of patients with cystic fibrosis. Gene. 2018;641:137–43. doi: 10.1016/j.gene.2017.10.052. [DOI] [PubMed] [Google Scholar]
- 39.Opresko PL, Calvo JP, von Kobbe C. Role for the Werner syndrome protein in the promotion of tumor cell growth. Mech Ageing Dev. 2007;128(7):423–36. doi: 10.1016/j.mad.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Agrelo R, Cheng W-H, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA. 2006;103(23):8822–7. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Getta BM, Devlin SM, Levine RL, Arcila ME, Mohanty AS, Zehir A, Tallman MS, Giralt SA, Roshal M. Multicolor flow cytometry and multigene next-generation sequencing are complementary and highly predictive for relapse in acute myeloid leukemia after allogeneic transplantation. Biol Blood Marrow Transplantation. 2017;23(7):1064–71. doi: 10.1016/j.bbmt.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosom Cancer. 2019;58(12):839–49. doi: 10.1002/gcc.22806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greif PA, Hartmann L, Vosberg S, Stief SM, Mattes R, Hellmann I, Metzeler KH, Herold T, Bamopoulos SA, Kerbs P, et al. Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: an exome sequencing study of 50 patients. Clin Cancer Res. 2018;24(7):1716. doi: 10.1158/1078-0432.CCR-17-2344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available for sharing upon request.