

Abstract
Renal fibrosis is characterized by glomerulosclerosis and tubulointerstitial fibrosis in diabetic nephropathy (DN). We aimed to evaluate the effects of PP2 on renal fibrosis of DN. GSE33744 and GSE86300 were downloaded from the GEO database. Firstly, 839 DEGs were identified between nondiabetic and diabetic mice renal glomerular samples. COX-2 was selected to assess the effects of PP2 on renal glomerulosclerosis. In db/db mice, PP2 decreased the expression of COX-2, phosphorylated p65, and fibrotic proteins, accompanied with attenuated renal glomerulosclerosis. In cultured glomerular mesangial cells, high glucose- (HG-) induced p65 phosphorylation and COX-2 expression were attenuated by PP2 or NF-κB inhibitor PDTC. PP2, PDTC, or COX-2 inhibitor NS-398 ameliorated abnormal proliferation and expression of fibrotic proteins induced by HG. Secondly, 238 DEGs were identified between nondiabetic and diabetic mice renal cortex samples. UCP2 was selected to assess the effects of PP2 on renal tubulointerstitial fibrosis. In db/db mice, PP2 decreased the expression of PPARγ and UCP2, accompanied with attenuated renal tubulointerstitial fibrosis and EMT. In cultured proximal tubular cells, HG-induced PPARγ and UCP2 expression was inhibited by PP2 or PPARγ antagonist GW9662. PP2, GW9662, or UCP2 shRNA ameliorated HG-induced EMT. These results indicated that PP2 ameliorated renal fibrosis in diabetic mice.
1. Introduction
Diabetic nephropathy (DN), a major microvascular complication of diabetes mellitus, is the leading cause of end-stage renal disease. The pathogenesis and development of DN are extremely complicated. Clinically, it is initially characterized by an increase in urinary albumin excretion, followed by a gradual decline in glomerular filtration rate, eventually progressing to end-stage renal disease [1]. Morphologically, it is featured by thickening glomerular basement membrane and renal tubular basement membrane, widening mesangial matrix, glomerular sclerosis, podocyte loss, tubular atrophy and increased apoptosis, renal inflammatory infiltration, and renal interstitial fibrosis. In the meantime, periosteal capillaries become sparse, and the wall of efferent arteriole and afferent arteriole is hyaline degeneration, especially the efferent arteriole [2]. DN affects approximately one third of patients with either type I or type II diabetes, contributing to increased mortality and economic burden [3]. Therefore, it is extremely important to understand the molecular mechanisms of DN and identify novel therapeutic targets.
Renal fibrosis is the final common pathway for a variety of chronic kidney diseases, including DN. Chronic progressive renal fibrosis leads to loss of renal function and, eventually, end-stage renal disease. Generally, renal fibrosis is a complex process and refers to glomerulosclerosis and tubulointerstitial fibrosis, especially the increase of matrix protein synthesis and the inhibition of matrix degradation contributing to abnormal accumulation of extracellular matrix (ECM, e.g., collagen fibers and fibronectin) [4]. Many cells in the kidney have been reported to participate in this pathogenesis, especially renal mesangial cells and tubular epithelial cells. On the one hand, abnormal and excessive growth of mesangial cells plays a prominent role in the pathophysiologic processes of glomerulosclerosis in DN [5]. Under the diabetic conditions, mesangial cells are activated, leading to cell proliferation, glomerular hypertrophy, and excess renal ECM deposition in the glomerular mesangial region. Moreover, mesangial cells also secrete various inflammatory cytokines, adhesion molecules, chemokines, and enzymes, all of which contribute significantly to the process of renal glomerulosclerosis and reduced kidney function eventually [6]. On the other hand, tubular injury is another crucial determinant of progressive renal injury in DN, while epithelial-to-mesenchymal transition (EMT) of tubular cells contributes to tubulointerstitial fibrosis and glomerular tubular dissociation [7]. EMT, characterized by loss of epithelial phenotype and gain of profibrotic features that are characteristic of mesenchymal cells, enables cell cycle arrest of renal tubular epithelial cells, elevated migration capacity, resistance to apoptosis, the ability to produce inflammatory cytokines, and even direct production of ECM [8]. Nevertheless, the exact role of mesangial cells and tubular epithelial cells on the renal fibrosis and pathogenesis of DN has not been completely understood yet.
PP2 [4-amino-5-(4-chlorophenyl)-(t-butyl)pyrazolo[3,4-d]pyrimidine] is a selective Src family kinase inhibitor. c-Src is a member of the Src tyrosine kinase family that is involved in many cellular events such as mitosis, cell growth, and tumorigenesis [9, 10]. In response to a variety of external stimuli, c-Src can be activated (phosphorylated at Tyrosine 416) and subsequently mediates intracellular signal transduction by phosphorylating tyrosine residues of numerous cellular cytosolic, nuclear, and membrane proteins [2]. We and other investigators have verified the critical role of c-Src in pathological conditions including DN. In glomerular mesangial cells, astaxanthin treatment or overexpression of Cx43 attenuated the high glucose- (HG-) induced interaction between Nrf2 and c-Src, thereby enhancing the Nrf2 accumulation in the nuclei [11, 12]. In podocytes, c-Src mediated the translocation of diacylglycerol kinase α and contributed to the amelioration of DN upon stimulation by epigallocatechin gallate [13]. In addition, pretreatment of the differentiated podocytes with the c-Src kinase inhibitor, PP2, not only inhibited phosphorylation of c-Src but also restricted phosphorylation of EGFR and the activation of TGFβ-Smad2/3 signaling pathway in response to HG [14]. In tubular epithelial cells, we have previously demonstrated that PP2 reduces c-Src activity and therefore inhibits cell apoptosis, which at least in part, is due to suppressed p38 MAPK activation [15]. However, the exact role of c-Src on renal fibrosis is yet to be elucidated.
In the present study, we aimed to investigate the role of c-Src on proliferation and fibrosis of mesangial cells as well as EMT of tubular epithelial cells in DN and identify its novel downstream genes. Therefore, we performed two separate studies to address these items. In the first study, by performing bioinformatics analysis based on Gene Expression Omnibus (GEO) data, we identified the differentially expressed genes (DEGs) between nondiabetic and diabetic renal glomerular samples. We identified COX-2 as the common hub gene of DM. Then, we performed a serial experiment to investigate the correlation between c-Src activation and COX-2 expression and evaluated the effects of c-Src/COX-2 pathway on proliferation and fibrosis of mesangial cells under in vivo and in vitro diabetic conditions. In the second study, by performing bioinformatic analysis based on GEO data, we identified the DEGs between nondiabetic and diabetic mice renal kidney cortex samples. We identified UCP2 as the common hub gene of DN. Then, we performed a serial experiment to investigate the correlation between c-Src activation and UCP2 expression and evaluated the effects of c-Src/UCP2 pathway on EMT of tubular epithelial cells under in vitro and in vivo diabetic conditions. The present study may provide an effective therapeutic target for DN.
2. Materials and Methods
2.1. Bioinformatic Analysis
Microarray gene expression profiles GSE33744 [16] and GSE86300 [17] were used to perform bioinformatic analysis, respectively. Probe IDs of two datasets were matched to gene symbol on the platform. Probe IDs matching to none or multiple genes symbol were deleted. If multiple IDs match a single gene, the highest expression value of these probes would be set for this gene. The limma package of R software program (version 4.0.3, https://www.r-project.org/) was used to normalize and screen for DEGs in each dataset, respectively. Adjusted p value <0.05 and ∣log FC | ≥1 were selected as the cut-off criteria of DEGs. The upregulated and downregulated DEGs were saved for subsequent analysis.
In order to explore the biological function and pathway of the above DEGs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed with the Database for Annotation, Visualization, and Integrated Discovery (DAVID v6.8, https://david.ncifcrf.gov/). The results of enrichment analyses were downloaded for subsequent analysis. p value <0.05 was considered statistically significant.
The STRING database (https://www.string-db.org/) was used to construct protein-protein interaction (PPI) network for the identified DEGs with the restriction of a minimum required interaction score of 0.400. The Cytoscape software (http://www.cytoscape.org/) was used to visualize the PPI network. The CytoHubba plugin in Cytoscape software was employed to identify the top 50 hub genes among DEGs. The Molecular Complex Detection (MCODE) plugin in Cytoscape software was employed to select the significant modules.
2.2. Materials
Antibodies against c-Src, phospho-Src (Tyr416), NF-κB p65, phospho-NF-κB p65 (Ser536), COX-2, PPARγ, and UCP2 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against fibronectin and collagen IV were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against E-cadherin, α-SMA, DRP1, and FIS1 were purchased from the Proteintech Group (Chicago, USA). The β-actin antibody was purchased from Abcam (Cambridge, UK). The goat anti-rabbit IgG-HRP secondary antibody was purchased from Zhongshan Golden bridge Biotechnology (Beijing, China). c-Src inhibitor PP2, NF-κB inhibitor PDTC, COX-2 inhibitor NS-398, and PPARγ antagonist GW9662 were purchased from Selleck Chemicals (Houston, TX, USA). The pIRES2-ZsGreen1-UCP2 overexpression plasmid and PLV-UCP2-shRNA plasmid were purchased from Yingrun Biotechnology Inc. (Changsha, China). PhosSTOP phosphatase inhibitor cocktail tablets were purchased from Roche Ltd. (Mannheim, Germany). The protease inhibitor cocktail was purchased from Calbiochem (San Diego, CA, USA). All culture media were purchased from Gibco-BRL (Grand Island, NY, USA). D-glucose was purchased from Sigma (St. Louis, MO, USA). The polyvinylidene difluoride (PVDF) membrane was purchased from Millipore (Billerica, MA, USA).
2.3. Animals and Groups
The animal experiment was performed as previously described [15]. Briefly, male diabetic db/db mice and nondiabetic db/m mice were purchased from the Model Animal Research Center of Nanjing University. According to the guidelines of National Institute of Health, all experimental animals were housed in the specific pathogen-free conditions with free access to food and water. At eight weeks of age, the animals were randomly divided into three groups: control group (db/m mice, n = 10), diabetes group (db/db mice, n = 10), and PP2 group (db/db mice treated with PP2, n = 10). The PP2 group received 2 mg/kg PP2 dissolved in DMSO every other day by i.p. injection. Mice in the control group and diabetes group received the same amount of normal saline. At the age of 16 weeks, the animals that were sacrificed by exsanguination and the kidneys were harvested for further analysis.
2.4. Cell Culture
Immortalized mouse mesangial cells (MMCs) were obtained from the Chinese Academy of Sciences, Shanghai Institute for Biological Sciences Cell Resource Center and stored in our laboratory. MMCs were cultured in DMEM/F12 (3 : 1) medium supplemented with 10% fetal bovine serum, 1% streptomycin-penicillin mixture in a 95% air, and 5% CO2 atmosphere at 37°C. The culture medium was replaced every two days. After reaching 70-80% confluence, the cells were washed once with serum-free DMEM medium and then growth arrested in serum-free DMEM medium for 24 h to synchronize the cell growth. The media was then changed to fresh serum-free media containing normal glucose (NG, 5.6 mmol/l) or HG (30 mmol/l) for an additional 48 h. PP2 (10 μM), PDTC (10 μM), or NS-398 (20 μM) was added to the culture medium 2 h prior to HG.
Human renal tubule epithelial (HK-2) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM medium supplemented with 10% fetal bovine serum, 1% streptomycin-penicillin mixture in a 95% air and 5% CO2 atmosphere at 37°C. The culture medium was replaced every two days. After reaching 70-80% confluence, the cells were washed once with serum-free DMEM medium, and then growth-arrested in serum-free DMEM medium for 24 h to synchronize the cell growth. The media was then changed to fresh serum-free media containing NG (5.6 mmol/l) or HG (30 mmol/l) for an additional 48 h. PP2 (10 μM) and GW9662 (20 μM) were added to the culture medium 2 h prior to HG. Stable transfections of HK-2 cells with pIRES2-ZsGreen1-UCP2 plasmid or PLV-UCP2-shRNA plasmid were performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
2.5. Protein Extraction and Western Blotting
Whole protein extraction was performed according to the method used in the previous research [15]. In detail, after the treatment, renal cortex tissues and cells were washed twice with ice cold phosphate-buffered saline (PBS), lysed in protein lysis buffer with protease and phosphatase inhibitors for 30 min on ice. The cell lysates were centrifuged at 12,000 rpm for 20 min at 4°C. Protein concentration was determined with the BCA protein assay kit with BSA as the standard. Protein samples (40 mg/lane) were separated with SDS-PAGE and transferred to PVDF membranes which were then blocked with 5% (v/v) nonfat milk at 37°C for 2 h. The membranes were subsequently incubated overnight at 4°C with primary antibodies against c-Src, phospho-Src (Tyr416), p65, phospho-p65 (Ser536), COX-2, PPARγ, UCP2, fibronectin, collagen IV, E-cadherin, α-SMA, DRP1, FIS1, and β-actin, respectively. The next morning, the membranes were rinsed with TBST and incubated with secondary antibody for 1 h at room temperature. Imaging was performed with the Odyssey Fc System (LICOR, USA). The intensity of the bands was performed with NIH ImageJ software.
2.6. Histology and Immunohistochemistry
Kidneys were fixed in 4% paraformaldehyde overnight and embedded in paraffin. Paraffin sections were cut at 2 mm and processed for periodic acid Schiff (PAS) and Masson's trichrome staining according to the standard protocol. For immunohistochemical analysis, paraffin sections were cut at 4 mm and deparaffinized with xylene and rehydrated in graded ethanol. Internal peroxidase was inactivated with 3% hydrogen peroxide in 100% methanol for 30 min. Antigen retrieval was performed with microwave oven in 10 mM citrate buffer for 15 min. 10% normal goat serum in PBS was then added to the sections for 30 min at room temperature to block nonspecific antibody binding. The sections were then incubated with primary antibodies for fibronectin, collagen IV, E-cadherin, α-SMA, and COX-2 (diluted 1 : 300) overnight at 4°C. After rinsed in PBS, the sections were then incubated with biotinylated secondary antibody and horseradish peroxidase-conjugated streptavidin. Labeling was visualized with diaminobenzidine to produce a brown color, and sections were counterstained with hematoxylin. For negative controls, the primary antibody was omitted. The area percent of staining was calculated using ImageJ software.
2.7. Transmission Electron Microscopy
The ultramicrostructure of glomerulus was observed with electron microscopy. Kidney tissue was immerged in 2.5% (v/v) glutaraldehyde and 1% (v/v) osmium tetroxide. Then, the kidney tissue was dehydrated in graded alcohol solutions and embedded in Epon 812 resin. Ultrathin sections (70-80 nm) were cut and double-stained with uranyl acetate and lead citrate. Sections in ultrastructure were observed and photographed using Hitachi H7500 transmission electron microscope (Hitachi, Tokyo, Japan).
2.8. Cell Proliferation Assay
Cell proliferation of MMCs was estimated with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) assay according to the manufacturer's instructions (Beyotime Biotechnology, Shanghai, China). In brief, MMCs (2000 cells/well) were inoculated into 96-well plates. After adherence, the medium was replaced with serum-free medium for 24 h. The media was then changed to fresh serum-free media containing NG (5.6 mmol/l) or HG (30 mmol/l) for an additional 24 h. PP2 (10 μM), PDTC (10 μM), or NS-398 (20 μM) was added to the culture medium 2 h prior to HG. Next, 10 μl MTT solution (5 mg/ml) was added to each well and incubated for 4 h at 37°C. Then, 100 μl formazan solution was added to each well and incubated for 4 h at 37°C to fully dissolve the crystals. Finally, the absorbance was measured at 570 nm using BioTek's Microplate spectrophotometer (Gene Company Limited, USA). All conditions were carried out in triplicate (n = 6), and the experiments were replicated three times.
2.9. Mitochondrial Membrane Potential (MMP) Detection
MMP was detected with the MMP Assay Kit with JC-1 (Solarbio, Beijing, China). After cultured in 6-well plates under the different experimental conditions for 48 h, HK-2 cells were washed in PBS and incubated with JC-1 dye for 20 min at 37°C. After the dye was removed by washing, the cells were immediately imaged using a TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany). JC-1 monomers emit a green fluorescence, while aggregates of JC-1 show intense red fluorescence. In apoptotic cells, JC-1 does not form aggregates in mitochondria with low MMP and exhibit green fluorescence.
2.10. Mitochondrial Morphology Analysis
After cultured in 6-well plates under the different experimental conditions for 48 h, HK-2 cells were washed in PBS and incubated with MitoTracker Red CMXRos (Beyotime Biotechnology, Shanghai, China) using a working concentration of 200 nM at 37°C for 30 min. After the dye was removed by washing, the cells were immediately imaged using a TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany).
2.11. Statistical Analysis
Data are expressed as means ± SD. Statistical analysis was performed by one-way ANOVA. Statistical significance was defined as p < 0.05.
3. Results
3.1. Identification of COX-2 as a DEG between Nondiabetic and Diabetic Mice Renal Glomerular
Microarray gene expression profile GSE33744 was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The platform for GSE33744 is GPL1261 [Mouse430_2] Affymetrix Mouse Genome 430 2.0 Array. This dataset contains 39 samples, and we selected 5 glomeruli samples from nondiabetic BKS db/m mouse and 5 glomeruli samples from diabetic BKS db/db mouse to perform bioinformatic analysis.
Based on the criteria of adjusted p value <0.05 and ∣log FC | ≥1, a total of 839 DEGs were identified from GSE33744. Among them, 579 were upregulated, and 260 were downregulated in diabetic mice renal glomerular. The DEGs are visualized by the cluster heat map (Figure 1(a)) and the volcano plot (Figure 1(b)). GO functional enrichment analysis of DEGs was categorized based on biological processes, cellular components, and molecular functions. The top 10 enriched GO terms of biological processes, cellular components, and molecular functions for the upregulated and downregulated DEGs were displayed in Figures 1(c)–1(f). The detailed information on the GO analysis of the upregulated and downregulated DEGs are list in Table 1. In the upregulated DEGs, the most enriched GO terms of biological processes, cellular components, and molecular functions were associated with signal transduction, membrane, and hydrolase activity, respectively. In the downregulated DEGs, the most enriched GO terms of biological processes, cellular components, and molecular functions were associated with oxidation-reduction process, membrane, and metal ion binding, respectively. KEGG pathway enrichment analysis demonstrated that the DEGs were enriched in 23 pathways (Figure 1(g)). The detailed information on the KEGG enrichment analysis of DEGs are listed in Table 2.
Figure 1.
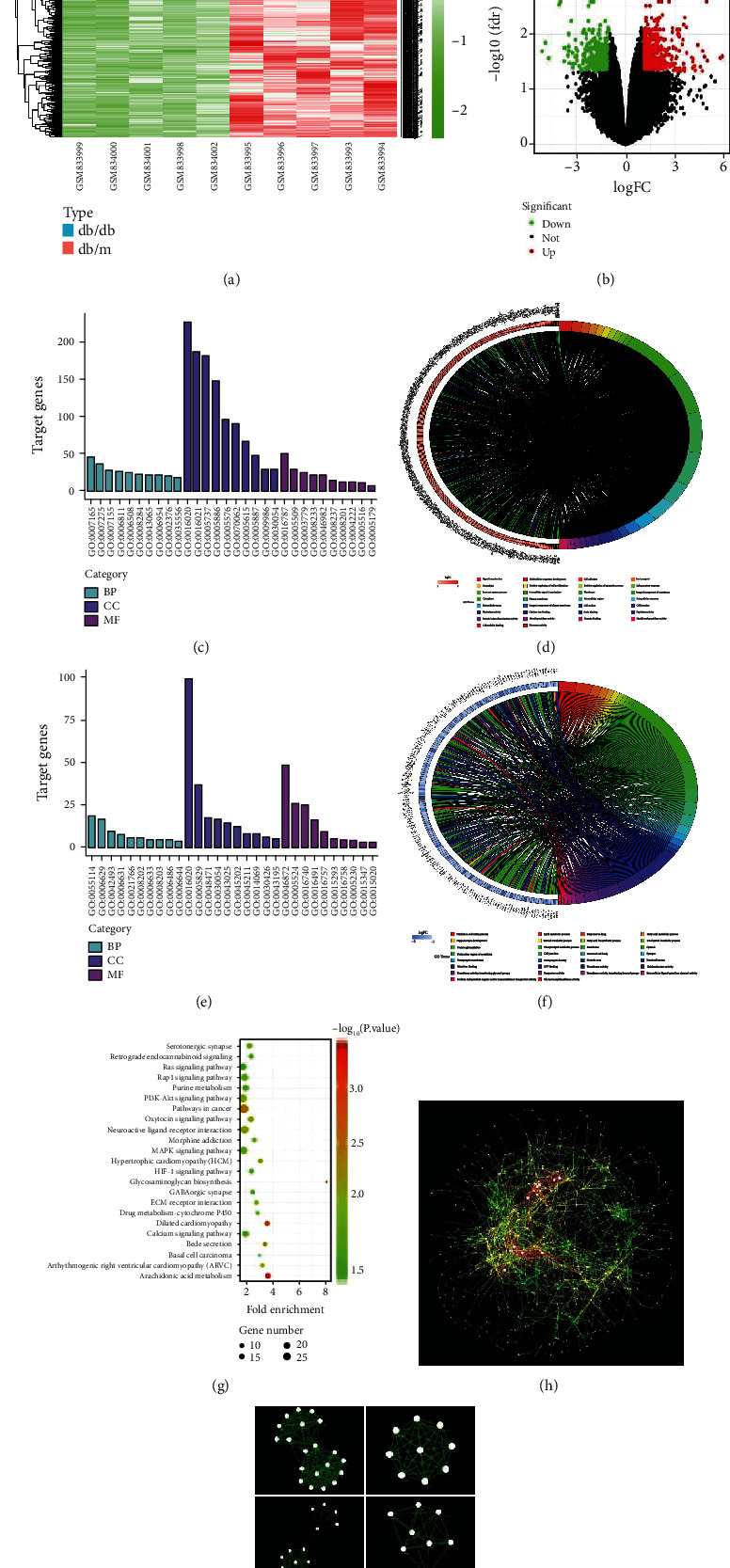
Identification of DEGs and biological pathways between nondiabetic and diabetic mice renal glomerular in GSE33744. (a) Heatmap of DEGs. Red represents an upregulated gene, and green represents a downregulated gene. Legend on the top right indicates the log fold change of the genes. (b) Volcano plot of DEGs. Red represents an upregulated gene, and green represents a downregulated gene. Black represents no significant change in gene expression. (c) Top 30 enriched GO terms of upregulated DEGs. (d) Distribution of upregulated DEGs in different GO-enriched functions. (e) Top 30 enriched GO terms of downregulated DEGs. (f) Distribution of downregulated DEGs for different GO-enriched functions. (g) KEGG pathway enrichment analysis of DEGs. (h) PPI network of DEGs. Every node represents a gene-encoded protein, and each edge represents the interaction between them. The edge color represents the core degree of protein. (i) Top 6 functional modules identified from the PPI network.
Table 1.
The detailed information on the GO analysis of the upregulated and downregulated DEGs in GSE33744.
| Category | Term | Count | p value |
|---|---|---|---|
| Upregulated | |||
| BP | Signal transduction | 45 | 0.007753334 |
| BP | Multicellular organism development | 37 | 0.016000738 |
| BP | Cell adhesion | 28 | 4.97064E-05 |
| BP | Ion transport | 27 | 0.002019604 |
| BP | Proteolysis | 25 | 0.007536866 |
| BP | Positive regulation of cell proliferation | 23 | 0.012198374 |
| BP | Positive regulation of apoptotic process | 22 | 6.66135E-05 |
| BP | Inflammatory response | 22 | 9.69908E-05 |
| BP | Immune system process | 21 | 0.001040971 |
| BP | Intracellular signal transduction | 18 | 0.017213392 |
| CC | Membrane | 226 | 1.18309E-07 |
| CC | Integral component of membrane | 187 | 0.038634939 |
| CC | GO: 0005737 ~ cytoplasm | 180 | 0.045971674 |
| CC | Plasma membrane | 148 | 0.001626062 |
| CC | Extracellular region | 95 | 1.44303E-13 |
| CC | Extracellular exosome | 90 | 0.001132912 |
| CC | Extracellular space | 65 | 7.97336E-06 |
| CC | Integral component of plasma membrane | 46 | 0.000743085 |
| CC | Cell surface | 29 | 0.001618609 |
| CC | Cell junction | 29 | 0.009876677 |
| MF | Hydrolase activity | 49 | 0.042624904 |
| MF | Calcium ion binding | 29 | 0.00687708 |
| MF | Actin binding | 24 | 9.88949E-06 |
| MF | Peptidase activity | 22 | 0.015404845 |
| MF | Protein heterodimerization activity | 21 | 0.027455776 |
| MF | Metallopeptidase activity | 13 | 0.000561141 |
| MF | Heparin binding | 12 | 0.001196627 |
| MF | Metalloendopeptidase activity | 11 | 0.000858787 |
| MF | Calmodulin binding | 10 | 0.034548521 |
| MF | Hormone activity | 8 | 0.0197492 |
| Downregulated | |||
| BP | Oxidation-reduction process | 19 | 0.000443777 |
| BP | Lipid metabolic process | 17 | 4.48872E-05 |
| BP | Response to drug | 10 | 0.012221837 |
| BP | Fatty acid metabolic process | 8 | 0.001618978 |
| BP | Hippocampus development | 6 | 0.001202789 |
| BP | Steroid metabolic process | 6 | 0.002245487 |
| BP | Fatty acid biosynthetic process | 5 | 0.008687078 |
| BP | Cholesterol metabolic process | 5 | 0.016263715 |
| BP | Protein glycosylation | 5 | 0.038138181 |
| BP | Phospholipid metabolic process | 4 | 0.010702838 |
| CC | Membrane | 99 | 0.000388674 |
| CC | Cytosol | 37 | 0.000119979 |
| CC | Perinuclear region of cytoplasm | 18 | 0.001140012 |
| CC | Cell junction | 17 | 0.004153908 |
| CC | Neuronal cell body | 15 | 0.001706257 |
| CC | Synapse | 12 | 0.018930533 |
| CC | Postsynaptic membrane | 8 | 0.009523669 |
| CC | Postsynaptic density | 8 | 0.013861599 |
| CC | Growth cone | 6 | 0.027381838 |
| CC | Terminal Bouton | 5 | 0.032102973 |
| MF | Metal ion binding | 48 | 0.045332685 |
| MF | ATP binding | 26 | 0.026765145 |
| MF | Transferase activity | 25 | 0.035427208 |
| MF | Oxidoreductase activity | 16 | 0.00299816 |
| MF | Transferase activity, transferring glycosyl groups | 9 | 0.002247304 |
| MF | Symporter activity | 5 | 0.035838809 |
| MF | Transferase activity, transferring hexosyl groups | 4 | 0.004770367 |
| MF | Extracellular ligand-gated ion channel activity | 4 | 0.010436623 |
| MF | Sodium-independent organic anion transmembrane transporter activity | 3 | 0.03565372 |
| MF | Glucuronosyltransferase activity | 3 | 0.040654245 |
BP: biological process; CC: cellular component; MF: molecular function.
Table 2.
The detailed information on the KEGG enrichment analysis of DEGs in GSE33744.
| Term | Pathway | Count | p value | Genes |
|---|---|---|---|---|
| mmu05200 | Pathways in cancer | 27 | 0.003017125 | ITGB1, CDKN1A, LAMA1, LPAR1, ADCY1, LPAR4, RASGRP2, PTGS2, EGFR, GLI3, ADCY5, FGF6, GNG2, BID, RUNX1T1, FZD3, FZD2, PTCH1, MMP2, PTCH2, VEGFB, BMP2, TRAF4, CCNE1, COL4A6, GNB4, CYCS |
| mmu04151 | PI3K-Akt signaling pathway | 23 | 0.010735996 | ITGB1, CDKN1A, ANGPT2, ANGPT1, LAMA1, NOS3, FLT4, TNC, VEGFB, LPAR1, IL4RA, LPAR4, EPOR, EGFR, FGF6, NR4A1, GNG2, CCNE1, GNB4, COL4A6, ITGB6, TLR4, ITGA9 |
| mmu04080 | Neuroactive ligand-receptor interaction | 20 | 0.009368143 | GABRB3, GABRB1, CHRNA4, GRID1, CHRNA7, P2RY14, GABRA4, C5AR1, LPAR1, LPAR4, NR3C1, ADRA1A, P2RX7, GLRA2, CALCR, CNR1, HRH2, P2RX1, ADORA3, TSHB |
| mmu04010 | MAPK signaling pathway | 17 | 0.024073268 | NTRK2, MEF2C, GADD45B, PLA2G4B, CACNA2D4, RASGRP2, EGFR, CACNA1G, FGF6, NR4A1, CACNG7, CACNB3, MAP3K8, MAP3K6, HSPA1B, MAP4K4, HSPA1A |
| mmu04015 | Rap1 signaling pathway | 15 | 0.027634917 | ITGB1, ANGPT2, ANGPT1, FLT4, VEGFB, LPAR1, ADCY1, LPAR4, RASGRP2, EGFR, ADCY5, FGF6, CNR1, PRKD3, RAPGEF5 |
| mmu04014 | Ras signaling pathway | 15 | 0.044994767 | PLA2G12B, ANGPT2, PLA2G2E, ANGPT1, PLA2G4B, FLT4, VEGFB, GAB2, RASGRP2, EGFR, FGF6, GNG2, GNB4, RIN1, RAPGEF5 |
| mmu04921 | Oxytocin signaling pathway | 13 | 0.009629191 | RYR1, MEF2C, CDKN1A, NOS3, PLA2G4B, NFATC2, CACNA2D4, ADCY1, COX-2, EGFR, ADCY5, CACNG7, CACNB3 |
| mmu00230 | Purine metabolism | 13 | 0.033776013 | ENTPD1, ENTPD2, PDE6H, GDA, PDE1A, AK1, NME4, NME5, ADCY1, ADCY5, ADSSL1, ENPP3, PDE8B |
| mmu04020 | Calcium signaling pathway | 13 | 0.03503903 | RYR1, NOS3, PDE1A, CHRNA7, ADCY1, ADRA1A, EGFR, CACNA1G, P2RX7, SLC8A3, GNA14, HRH2, P2RX1 |
| mmu00590 | Arachidonic acid metabolism | 12 | 0.000415 | PTGIS, PLA2G2E, PLA2G12B, CYP2J13, PLA2G4B, GPX7, ALOX12B, CYP4A14, LTC4S, COX-2, PTGDS, PTGS1 |
| mmu05414 | Dilated cardiomyopathy | 11 | 0.000926 | ITGB1, CACNG7, CACNB3, TNNT2, TPM1, DAG1, CACNA2D4, ITGB6, ADCY1, ADCY5, ITGA9 |
| mmu04726 | Serotonergic synapse | 11 | 0.024426847 | GABRB3, GABRB1, GNG2, CYP2J13, KCND2, PLA2G4B, GNB4, ALOX12B, COX-2, ADCY5, PTGS1 |
| mmu04976 | Bile secretion | 9 | 0.004518526 | ABCC3, SLCO1A1, SLCO1A5, ADCY1, SLC51A, ABCB1A, SLC22A7, ABCB1B, ADCY5 |
| mmu05410 | Hypertrophic cardiomyopathy | 9 | 0.008596921 | ITGB1, CACNG7, CACNB3, TNNT2, TPM1, DAG1, CACNA2D4, ITGB6, ITGA9 |
| mmu04512 | ECM-receptor interaction | 9 | 0.015941382 | ITGB1, SV2B, LAMA1, DAG1, TNC, COL4A6, GP1BA, ITGB6, ITGA9 |
| mmu05032 | Morphine addiction | 9 | 0.021582369 | GABRB3, GABRB1, GNG2, GABRA4, PDE1A, GNB4, ADCY1, PDE8B, ADCY5 |
| mmu04066 | HIF-1 signaling pathway | 9 | 0.035057542 | CDKN1A, ANGPT2, PFKFB3, ANGPT1, NOS3, TRF, TLR4, EGFR, PDK1 |
| mmu04723 | Retrograde endocannabinoid signaling | 9 | 0.036841276 | GABRB3, GABRB1, GNG2, CNR1, GABRA4, GNB4, ADCY1, COX-2, ADCY5 |
| mmu05412 | Arrhythmogenic right ventricular cardiomyopathy | 8 | 0.011585663 | ITGB1, CACNG7, CACNB3, DAG1, DSG2, CACNA2D4, ITGB6, ITGA9 |
| mmu04727 | GABAergic synapse | 8 | 0.04190812 | GABRB3, GABRB1, GNG2, GABRA4, GNB4, ADCY1, HAP1, ADCY5 |
| mmu00982 | Drug metabolism-cytochrome P450 | 7 | 0.034801983 | ALDH1A3, ADH1, AOX4, GSTO1, UGT2B37, AOX1, UGT2A3 |
| mmu00532 | Glycosaminoglycan biosynthesis | 6 | 0.000661 | CHST7, CHST11, CHST12, CHPF2, CHST15, CHSY3 |
| mmu05217 | Basal cell carcinoma | 6 | 0.048717123 | BMP2, FZD3, FZD2, PTCH1, PTCH2, GLI3 |
PPI network of DEGs was constructed with Cytoscape software, and the results are shown in Figure 1(h). Furthermore, 23 functional modules were identified from the PPI network with MCODE plugin of Cytoscape, and the 6 most important modules are shown in Figure 1(i). Afterward, the top 50 hub genes were identified from the PPI network with the CytoHubba plugin of Cytoscape based on their degree value, and the results are shown in Table 3. The top 2 hub genes were epidermal growth factor receptor (EGFR) and COX-2 (also known as prostaglandin-endoperoxide synthase 2, PTGS2). The role of the c-Src/EGFR signaling pathway in DN has been previously reported [18]. COX-2 is an inducible enzyme responsible for prostaglandin synthesis and primarily associated with inflammation. There is also increasing evidence that COX-2 is upregulated in pathological conditions [19]. The GO pathway enrichment showed that COX-2 was related to positive regulation of apoptotic process, inflammatory response, positive regulation of cell proliferation, response to lipopolysaccharide, oxidation-reduction process, response to oxidative stress, angiogenesis, negative regulation of cell proliferation, regulation of blood pressure, cellular response to mechanical stimulus, memory, regulation of cell proliferation, maintenance of blood-brain barrier, prostaglandin biosynthetic process, positive regulation of NF-kappaB import into nucleus, positive regulation of synaptic transmission, glutamatergic, positive regulation of vascular endothelial growth factor production, positive regulation of smooth muscle contraction, positive regulation of nitric oxide biosynthetic process, positive regulation of vasoconstriction, positive regulation of cell death, positive regulation of phosphatidylinositol 3-kinase signaling, sensory perception of pain, positive regulation of smooth muscle cell proliferation, cyclooxygenase pathway, positive regulation of fever generation, response to lithium ion, negative regulation of calcium ion transport, prostaglandin metabolic process, membrane, endoplasmic reticulum, intracellular membrane-bounded organelle, neuron projection, organelle membrane, metal ion binding, oxidoreductase activity, heme binding, and prostaglandin-endoperoxide synthase activity. Therefore, COX-2 pathway was selected for further investigation.
Table 3.
The degree values of the top 50 hub genes in GSE33744.
| Gene | Gene title | Score | LogFC |
|---|---|---|---|
| EGFR | Epidermal growth factor receptor | 52 | -1.4 |
| COX-2 | Cyclooxygenase-2 | 34 | 2.019 |
| TLR4 | Toll-like receptor 4 | 31 | 1.361 |
| GNG2 | Guanine nucleotide binding protein (G protein) gamma 2 | 30 | 1.84 |
| GNB4 | Guanine nucleotide binding protein (G protein) beta 4 | 28 | 1.235 |
| ADCY5 | Adenylate cyclase 5 | 22 | 1.038 |
| ADCY1 | Adenylate cyclase 1 | 21 | 1.206 |
| CCL6 | Chemokine (C-C motif) ligand 6 | 21 | 2.879 |
| ITGB1 | Integrin beta 1 | 21 | 1.11 |
| NTRK2 | Neurotrophic tyrosine kinase, receptor, type 2 | 19 | 1.575 |
| IL18 | Interleukin 18 | 19 | 1.324 |
| QSOX1 | Quiescin Q6 sulfhydryl oxidase 1 | 19 | 1.103 |
| HSPA1B | Heat shock protein 1B | 18 | -3.42 |
| ENTPD1 | Ectonucleoside triphosphate diphosphohydrolase 1 | 18 | 1.853 |
| MMP2 | Matrix metallopeptidase 2 | 18 | 1.698 |
| PF4 | Platelet factor 4 | 18 | 2.06 |
| SLCO1A1 | Solute carrier organic anion transporter family, member 1a1 | 16 | -4.82 |
| UGT2B37 | UDP glucuronosyltransferase 2 family, polypeptide B37 | 16 | -1.48 |
| ANGPT1 | Angiopoietin 1 | 16 | -1.12 |
| LPAR1 | Lysophosphatidic acid receptor 1 | 16 | 1.231 |
| KALRN | Kalirin, RhoGEF kinase | 16 | 1.014 |
| CYP4A14 | Cytochrome P450, family 4, subfamily a, polypeptide 14 | 16 | 3.522 |
| ISL1 | ISL LIM Homeobox 1 | 16 | 1.93 |
| NOS3 | Nitric oxide synthase 3 | 16 | 1.159 |
| C5AR1 | Complement component 5a receptor 1 | 16 | 3.044 |
| CP | Veruloplasmin | 16 | 1.134 |
| TRF | Transferrin | 15 | 1.563 |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 | 15 | -1.68 |
| NES | Nestin | 15 | 1.083 |
| EXO1 | Exonuclease 1 | 15 | 1.913 |
| FBN1 | Fibrillin 1 | 15 | 1.168 |
| UGT2A3 | UDP glucuronosyltransferase 2 family, polypeptide A3 | 14 | -1.95 |
| GPR143 | G protein-coupled receptor 143 | 14 | 1.294 |
| PTGS1 | Prostaglandin-endoperoxide synthase 1 | 14 | 1.212 |
| P2RY14 | Purinergic receptor P2Y, G-protein coupled, 14 | 14 | 1.788 |
| ENPP3 | Ectonucleotide pyrophosphatase/phosphodiesterase 3 | 13 | -1.31 |
| CNR1 | Cannabinoid receptor 1 (brain) | 13 | -1.58 |
| GNA14 | Guanine nucleotide binding protein, alpha 14 | 13 | -1.38 |
| P2RX1 | Purinergic receptor P2X 1 | 13 | 1.223 |
| EDN2 | Endothelin 2 | 13 | 1.312 |
| GRP | Gastrin releasing peptide | 13 | 2.796 |
| CYP2J13 | Cytochrome P450, family 2, subfamily j, polypeptide 13 | 13 | -2.43 |
| TNNT3 | Troponin T3, fast skeletal type | 13 | 3.247 |
| HP | Haptoglobin | 13 | 3.071 |
| BMP2 | Bone morphogenetic protein 2 | 13 | 1.283 |
| SNAI2 | Snail family zinc finger 2 | 13 | 1.057 |
| NEGR1 | Neuronal growth regulator 1 | 13 | -1.84 |
| FSTL1 | Follistatin-like 1 | 13 | 1.575 |
| ADM | Adrenomedullin | 12 | 1.308 |
| ABCB1A | ATP-binding cassette, subfamily B (MDR/TAP), member 1A | 12 | 2.014 |
3.2. Inhibition of c-Src Ameliorates ECM Accumulation and Fibrosis via NF-κB/COX-2 Pathway in Renal Glomerulus of Diabetic Mice
Our previous study has shown that PP2 treatment can attenuate diabetes induced c-Src activation and therefore ameliorates renal tubular epithelial cells apoptosis in db/db mice [15]. To understand whether c-Src inhibition has a protective effect in renal fibrosis, we first explored the role of PP2 treatment on the histopathological changes of kidney. As shown in Figure 2(a), HE staining showed that compared with the nondiabetic mice, glomerular hypertrophy, partial glomerulosclerosis, mesangial cells hyperplasia and hypertrophy, and inflammatory cell infiltration were significantly increased in diabetic mice. These pathological changes were significantly attenuated by PP2 treatment. Meanwhile, the results of PAS staining showed that there was prominent mesangial expansion in diabetic mice compared with the nondiabetic mice, which was ameliorated with PP2 treatment. Masson's trichrome staining and quantitative assessment confirmed that PP2 treatment resulted in a significant reduction in collagen deposition of renal glomerular when compared with the diabetic mice. Likewise, immunohistochemistry staining showed that ECM components, including fibronectin and collagen IV, were markedly increased in db/db mice than that in the db/m mice, whereas PP2 treatment significantly reversed these changes (Figure 2(b)). Western blotting results were consistent with the immunofluorescence observation (Figure 2(c)). These results indicate that c-Src plays an important role in the renal fibrosis and injury of diabetic mice.
Figure 2.
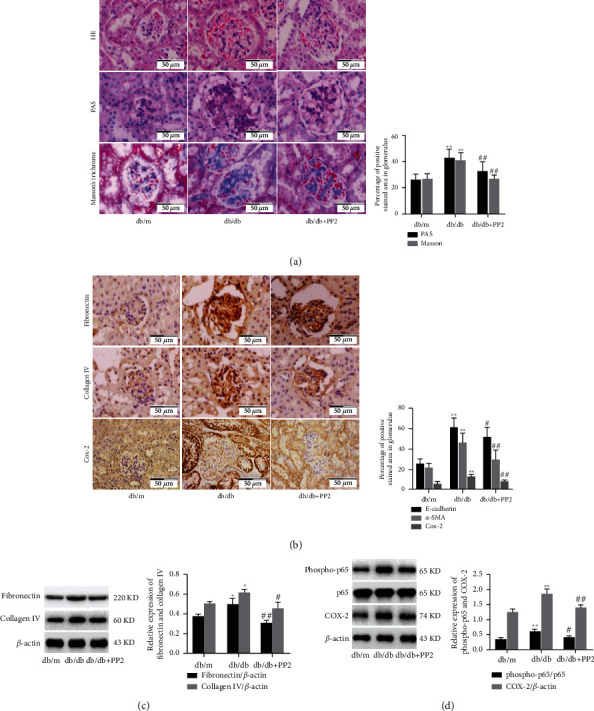
Effect of c-Src inhibiton on ECM accumulation and fibrosis in the renal glomerulus of diabetic mice. (a) HE, PAS, and Masson's trichrome staining of kidney sections (original magnification ×400). (b) Immunohistochemical staining of kidney sections with fibronectin and collagen IV (original magnification ×400). (c) The expression levels of fibronectin and collagen IV analyzed by western blotting. (d) The expression levels of phospho-p65, p65, and COX-2 analyzed by western blotting. The number of mice in every group was 10. 10 visual fields were randomly selected from each group, and the area percent of staining was calculated for statistical analysis. All western blotting experiments were performed independently at least in triplicate. Values are expressed as means ± SD. ∗p < 0.05 versus db/m group. ∗∗p < 0.01 versus db/m group. #p < 0.05 versus db/db group. ##p < 0.01 versus db/db group.
As mentioned above, the results of the bioinformatic analysis indicated that COX-2 showed significantly different expression between nondiabetic and diabetic mice renal glomerular,and was implicated in the biological processes of positive regulation of NF-κB import into nucleus. Then, we evaluated the relationship between c-Src inhibition and NF-κB/COX-2 pathway in db/db mice. Western blotting showed that the phosphorylation levels of NF-κB p65 (activation) and expression of COX-2 were markedly increased in db/db mice than that in the db/m mice, whereas PP2 treatment significantly reversed these changes (Figure 2(d)). Likewise, immunohistochemistry staining showed that the COX-2 expression was statistically increased in renal glomeruli of db/db mice than in the db/m mice, whereas PP2 treatment statistically reversed these changes (Figure 2(e)). Collectively, these data indicate that c-Src inhibition can attenuate renal ECM accumulation and fibrosis through regulation of NF-κB activation and COX-2 expression in diabetic mice.
3.3. Inhibition of c-Src Ameliorates HG-Induced ECM Accumulation via NF-κB/COX-2 Pathway in MMCs
To determine whether the activation of c-Src is indispensable for HG-induced NF-κB activity and COX-2 expression, we treated MMCs with the c-Src inhibitor PP2 or NF-κB inhibitor PDTC. As shown in Figure 3(a), compared with NG groups, exposure of MMCs to HG for 48 h significantly increased phosphorylation of c-Src at Tyr416 and NF-κB p65 at Ser536, which were significantly inhibited by PP2 or PDTC, respectively. PP2 and PDTC also markedly reduced the HG-induced COX-2 expression. We further assessed whether the c-Src/NF-κB/COX-2 pathway was involved in HG-induced ECM accumulation in MMCs. As shown in Figure 3(b), compared with NG groups, exposure of MMCs to HG for 48 h significantly increased the expression of fibronectin and collagen IV, which could be significantly attenuated by PP2, PDTC, and NS-398, respectively. We further assessed whether the c-Src/NF-κB/COX-2 pathway was involved in HG-induced MMCs proliferation with the MTT assay. As shown in Figure 3(c), exposure of MMCs to HG increased the cells' proliferation. The effects of HG on MMC proliferation were abolished by PP2, PDTC, and NS-398 treatment, respectively. Collectively, inhibition of c-Src/NF-κB/COX-2 pathway significantly inhibited HG-induced ECM accumulation, inflammation, and proliferation in mesangial cells. In addition, as a solvent control for PP2, DMSO had no effect on activation of c-Src (Figure 3(d)).
Figure 3.
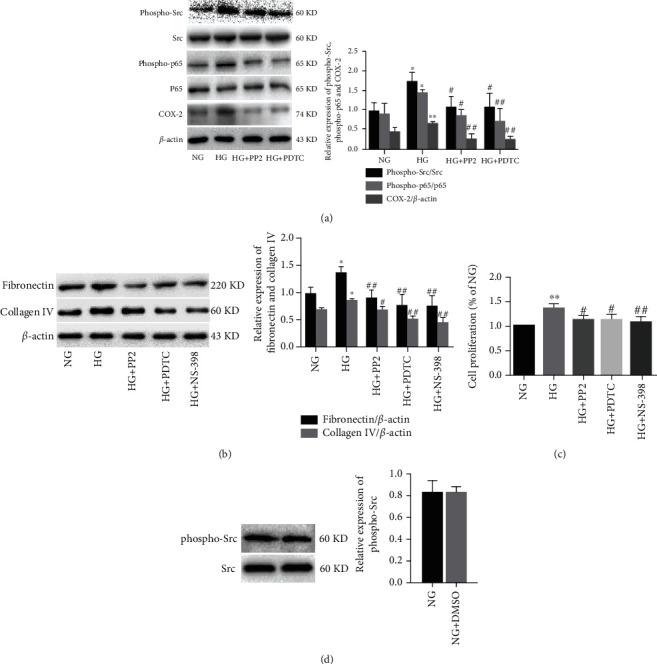
Effect of c-Src inhibiton on HG-induced ECM accumulation in MMCs. (a) The expression levels of phospho-Src, Src, phospho-p65, p65, and COX-2 analyzed by western blotting. (b) The expression levels of fibronectin and collagen IV analyzed by western blotting. (c) Cell proliferation of MMCs detected by the MTT assay. (d) The expression levels of phospho-Src and Src analyzed by western blotting. All experiments were performed independently at least in triplicate. Values are expressed as means ± SD. ∗p < 0.05 versus NG group. ∗∗p < 0.01 versus NG group. #p < 0.05 versus HG group.##p < 0.01 versus HG group.
3.4. Identification of UCP2 as a DEG between Nondiabetic and Diabetic Mice Renal Cortex
Microarray gene expression profile GSE86300 was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The platform for GSE86300 is GPL7546 Affymetrix GeneChip Mouse Genome 430 2.0 Array [CDF: Mm_ENTREZG_10]. This dataset contains 10 samples, and 5 renal cortex samples from nondiabetic BKS db/m mouse and 5 renal cortex samples from diabetic BKS db/db mouse were used to perform bioinformatic analysis.
Based on the criteria of adjusted p value <0.05 and ∣log FC | ≥1, a total of 238 DEGs were identified from GSE33744. Among them, 112 were upregulated, and 126 were downregulated in diabetic mice renal cortex. The DEGs are visualized by the cluster heat map (Figure 4(a)) and the volcano plot (Figure 4(b)). GO functional enrichment analysis of DEGs was categorized based on biological processes, cellular components, and molecular functions. The top 10 enriched GO terms of biological processes, cellular components, and molecular functions for the upregulated and downregulated DEGs were displayed in Figures 4(c)–4(f). The detailed information on the GO analysis of the upregulated and downregulated DEGs are list in Table 4. In the upregulated DEGs, the most enriched GO terms of biological processes, cellular components, and molecular functions were associated with signal transduction, extracellular exosome, and oxidoreductase activity, respectively. In the downregulated DEGs, the most enriched GO terms of biological processes, cellular components, and molecular functions were associated with lipid metabolic process, membrane, and ATP binding, respectively. KEGG pathway enrichment analysis demonstrated that the DEGs were enriched in 12 pathways (Figure 4(g)). The detailed information on the KEGG enrichment analysis of DEGs are listed in Table 5.
Figure 4.
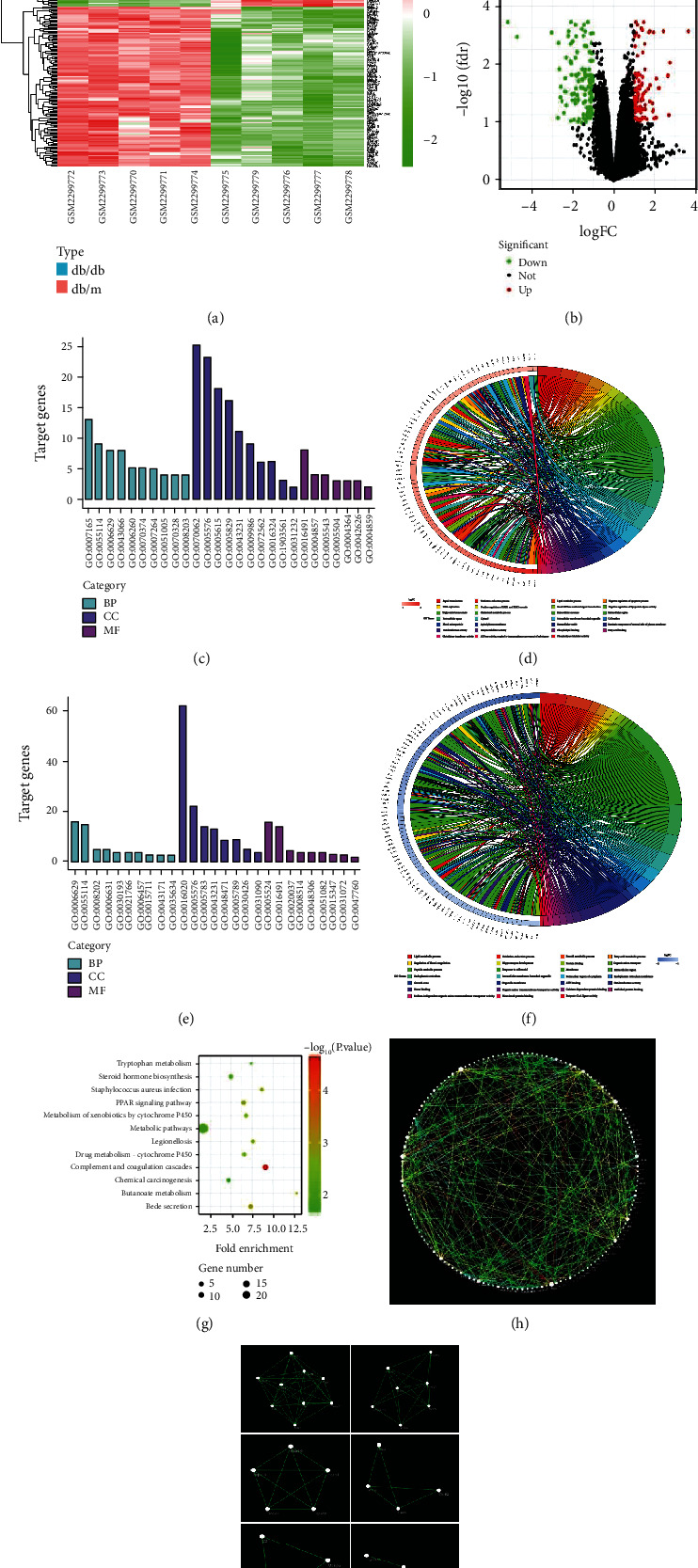
Identification of DEGs and biological pathways between nondiabetic and diabetic mice renal glomerular in GSE86300. (a) Heatmap of DEGs. Red represents an upregulated gene, and green represents a downregulated gene. Legend on the top right indicates the log fold change of the genes. (b) Volcano plot of DEGs. Red represents an upregulated gene, and green represents a downregulated gene. Black represents no significant change in the gene expression. (c) Top 30 enriched GO terms of upregulated DEGs. (d) Distribution of upregulated DEGs in different GO-enriched functions. (e) Top 30 enriched GO terms of downregulated DEGs. (f) Distribution of downregulated DEGs for different GO-enriched functions. (g) KEGG pathway enrichment analysis of DEGs. (h) PPI network of DEGs. Every node represents a gene-encoded protein, and each edge represents the interaction between them. The edge color represents the core degree of protein. (i) Top 6 functional modules identified from the PPI network.
Table 4.
The detailed information on the GO analysis of the upregulated and downregulated DEGs in GSE86300.
| Category | Term | Count | p value |
|---|---|---|---|
| Upregulated | |||
| BP | Signal transduction | 13 | 0.029093438 |
| BP | Oxidation-reduction process | 9 | 0.024404362 |
| BP | Lipid metabolic process | 8 | 0.00998742 |
| BP | Negative regulation of apoptotic process | 8 | 0.028039906 |
| BP | Intracellular signal transduction | 6 | 0.057525068 |
| BP | DNA replication | 5 | 0.003880893 |
| BP | Positive regulation of ERK1 and ERK2 cascade | 5 | 0.01671187 |
| BP | Small GTPase mediated signal transduction | 5 | 0.034683472 |
| BP | Negative regulation of lipoprotein lipase activity | 4 | 2.69E-06 |
| BP | Triglyceride homeostasis | 4 | 3.64E-04 |
| CC | Extracellular exosome | 25 | 0.00346128 |
| CC | Extracellular region | 23 | 5.66E-05 |
| CC | Extracellular space | 18 | 0.00145449 |
| CC | Cytosol | 16 | 0.035864717 |
| CC | Intracellular membrane-bounded organelle | 11 | 0.004880998 |
| CC | Cell surface | 9 | 0.014877768 |
| CC | Blood microparticle | 6 | 5.91E-04 |
| CC | Apical plasma membrane | 6 | 0.026095659 |
| CC | Basolateral plasma membrane | 4 | 0.084936357 |
| CC | Extracellular vesicle | 3 | 0.025790832 |
| MF | Oxidoreductase activity | 8 | 0.048891279 |
| MF | Calcium ion binding | 8 | 0.090469254 |
| MF | Enzyme inhibitor activity | 4 | 0.001278238 |
| MF | Phospholipid binding | 4 | 0.012646826 |
| MF | Heparin binding | 4 | 0.05071775 |
| MF | Heme binding | 4 | 0.072179828 |
| MF | ATPase activity | 4 | 0.098135899 |
| MF | Fatty acid binding | 3 | 0.008936699 |
| MF | Glutathione transferase activity | 3 | 0.014161492 |
| MF | ATPase activity, coupled to transmembrane movement of substances | 3 | 0.029804054 |
| Downregulated | |||
| BP | Lipid metabolic process | 16 | 9.41E-08 |
| BP | Oxidation-reduction process | 15 | 0.0000495 |
| BP | Cell differentiation | 9 | 0.094964371 |
| BP | Spermatogenesis | 6 | 0.096883326 |
| BP | Steroid metabolic process | 5 | 0.001609187 |
| BP | Fatty acid metabolic process | 5 | 0.014290964 |
| BP | Regulation of blood coagulation | 4 | 0.000187 |
| BP | Hippocampus development | 4 | 0.009520707 |
| BP | Protein folding | 4 | 0.041356989 |
| BP | Organic anion transport | 3 | 0.003069172 |
| CC | Membrane | 62 | 3.31E-05 |
| CC | Extracellular region | 22 | 8.29E-04 |
| CC | Endoplasmic reticulum | 14 | 0.037877677 |
| CC | Extracellular space | 14 | 0.085953795 |
| CC | Intracellular membrane-bounded organelle | 13 | 0.001184947 |
| CC | Perinuclear region of cytoplasm | 9 | 0.044162931 |
| CC | Endoplasmic reticulum membrane | 9 | 0.04998487 |
| CC | Cell surface | 8 | 0.068104588 |
| CC | Growth cone | 5 | 0.013026472 |
| CC | Organelle membrane | 4 | 0.016577263 |
| MF | ATP binding | 16 | 0.038783118 |
| MF | Oxidoreductase activity | 14 | 7.50E-05 |
| MF | Calcium ion binding | 9 | 0.06266387 |
| MF | Heme binding | 5 | 0.022041571 |
| MF | Organic anion transmembrane transporter activity | 4 | 3.09E-04 |
| MF | Calcium-dependent protein binding | 4 | 0.00854366 |
| MF | Unfolded protein binding | 4 | 0.010726793 |
| MF | Metallopeptidase activity | 4 | 0.073836747 |
| MF | Sodium-independent organic anion transmembrane transporter activity | 3 | 0.011626112 |
| MF | Heat shock protein binding | 3 | 0.041228079 |
BP: biological process; CC: cellular component; MF: molecular function.
Table 5.
The detailed information on the KEGG enrichment analysis of DEGs in GSE86300.
| Term | Pathway | Count | P value | Genes |
|---|---|---|---|---|
| mmu01100 | Metabolic pathways | 24 | 0.013965513 | RRM2, MAOB, ACSM3, UGT8A, ST8SIA1, ALOX15, CYP4A14, LTC4S, CYP51, HSD11B1, CYP24A1, BDH1, ALDH1B1, CYP2J13, MTHFD1L, KYNU, HSD17B2, AGPS, UGT2A3, PLCH1, HMGCS2, DEGS2, IDO2, ACOT3 |
| mmu04610 | Complement and coagulation cascades | 8 | 2.33E-05 | C3, CPB2, FGG, MASP2, MASP1, C8A, CD55, MBL2 |
| mmu04976 | Bile secretion | 6 | 0.001258086 | ABCC4, SLCO1A4, SLCO1A1, EPHX1, ABCB1A, SLC22A7 |
| mmu03320 | PPAR signaling pathway | 6 | 0.002143511 | FABP1, LPL, CYP4A14, ANGPTL4, CD36, RXRG |
| mmu05150 | S. aureus infection | 5 | 0.002467676 | C3, FGG, MASP2, MASP1, MBL2 |
| mmu05134 | Legionellosis | 5 | 0.003983622 | PYCARD, C3, NAIP1, HSPA1B, HSPA1A |
| mmu00980 | Metabolism of xenobiotics by cytochrome P450 | 5 | 0.006031624 | HSD11B1, GSTO1, EPHX1, UGT2A3, GSTM6 |
| mmu00982 | Drug metabolism-cytochrome P450 | 5 | 0.006724748 | MAOB, GSTO1, UGT2A3, FMO5, GSTM6 |
| mmu00140 | Steroid hormone biosynthesis | 5 | 0.017339009 | HSD11B1, SRD5A2, HSD17B2, UGT2A3, CYP7B1 |
| mmu05204 | Chemical carcinogenesis | 5 | 0.02085394 | HSD11B1, GSTO1, EPHX1, UGT2A3, GSTM6 |
| mmu00650 | Butanoate metabolism | 4 | 0.003471535 | BDH1, ACSM3, HMGCS2, AACS |
| mmu00380 | Tryptophan metabolism | 4 | 0.016335965 | MAOB, ALDH1B1, KYNU, IDO2 |
PPI network of DEGs was constructed using Cytoscape software, and the results are shown in Figure 4(h). Furthermore, 7 functional modules were identified from the PPI network using MCODE plugin of Cytoscape, and the 6 most important modules are shown in Figure 4(i). Afterward, the top 50 hub genes were identified from the PPI network with the CytoHubba plugin of Cytoscape based on their degree value, and the results are shown in Table 6. In these hub genes, UCP2 was annotated as an inner mitochondrial membrane protein that belongs to the uncoupling protein family and has recently been demonstrated in the control of mitochondria dysfunction [20]. Considering the key role of mitochondrial dysfunction in DN, we focused our attention to UCP2. The GO pathway enrichment showed that UCP2 was related to the biological processes of negative regulation of apoptotic process, liver regeneration. Therefore, the UCP2 pathway was selected for further investigation.
Table 6.
The degree values of the top 50 hub genes in GSE86300.
| Gene | Gene title | Score | LogFC |
|---|---|---|---|
| Fgg | Fibrinogen gamma chain | 3781 | 1.945439 |
| C8a | Complement component 8, alpha polypeptide | 3614 | -2.32194 |
| Gc | Group specific component | 3517 | 2.661948 |
| Hrg | Histidine-rich glycoprotein | 3437 | -2.73542 |
| Tm4sf4 | Transmembrane 4 superfamily member 4 | 2904 | -1.7815 |
| Mbl2 | Mannose-binding lectin (protein C) 2 | 1824 | 1.632683 |
| Cpb2 | Carboxypeptidase B2 (plasma) | 1815 | 1.143985 |
| Fabp1 | Fatty acid binding protein 1, liver | 1498 | 2.670807 |
| Angptl3 | Angiopoietin-like 3 | 1489 | 1.208791 |
| C3 | Complement component 3 | 562 | 1.982437 |
| Pzp | Pregnancy zone protein | 290 | -2.58665 |
| Masp1 | Mannan-binding lectin serine peptidase 1 | 246 | 2.101058 |
| Masp2 | Mannan-binding lectin serine peptidase 2 | 246 | -1.38516 |
| Dscc1 | DNA replication and sister chromatid cohesion 1 | 162 | 1.272575 |
| Mcm5 | Minichromosome maintenance complex component 5 | 162 | 1.07099 |
| Apoe | Apolipoprotein E | 158 | 1.551564 |
| Ncapg2 | Non-SMC condensin II complex, subunit G2 | 152 | 1.352226 |
| Rrm2 | Ribonucleotide reductase M2 | 150 | 1.020213 |
| Trf | Transferrin | 144 | 1.059667 |
| Kif20b | Kinesin family member 20B | 132 | -1.50486 |
| Arhgap11a | Rho GTPase activating protein 11A | 129 | 1.365888 |
| Apoc1 | Apolipoprotein C-I | 54 | 1.28042 |
| Ugt2a3 | UDP glucuronosyltransferase 2 family, polypeptide A3 | 39 | -1.37089 |
| Hsph1 | Heat shock 105 kDa/110 kDa protein 1 | 38 | -1.06227 |
| Pcsk9 | Proprotein convertase subtilisin/kexin type 9 | 38 | -1.11691 |
| Hspa1a | Heat shock protein 1A | 36 | -2.39622 |
| Hspa1b | Heat shock protein 1B | 34 | -1.87672 |
| Chaf1b | Chromatin assembly factor 1 subunit B (p60) | 34 | 1.093252 |
| Dnaja4 | DnaJ heat shock protein family (Hsp40) member A4 | 33 | -1.03221 |
| Lpl | Lipoprotein lipase | 32 | -1.02081 |
| Dnajb6 | DnaJ heat shock protein family (Hsp40) member B6 | 24 | -1.08687 |
| Cyp4a14 | Cytochrome P450, family 4, subfamily a polypeptide 14 | 16 | 2.444874 |
| Cd36 | CD36 antigen | 15 | -2.11604 |
| Kynu | Kynureninase (L-kynurenine hydrolase) | 13 | 1.90505 |
| Hmgcs2 | 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 | 12 | 3.639891 |
| Blm | Bloom syndrome, RecQ helicase-like | 12 | 1.117173 |
| Slco1a1 | Solute carrier organic anion transporter family member 1a1 | 10 | -4.71252 |
| Angptl4 | Angiopoietin-like 4 | 10 | 1.562673 |
| Slc22a7 | Solute carrier family 22 (organic anion transporter) member 7 | 9 | -5.16673 |
| Ucp2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 9 | 1.274927 |
| Fmo5 | Flavin containing monooxygenase 5 | 9 | -2.31674 |
| Slco1a4 | Solute carrier organic anion transporter family member 1a4 | 7 | -1.87469 |
| Cyp2j13 | Cytochrome P450 family 2 subfamily j polypeptide 13 | 7 | -1.10038 |
| Angptl8 | Angiopoietin-like 8 | 7 | 1.356146 |
| Ido2 | Indoleamine 2,3-dioxygenase 2 | 7 | -2.04659 |
| Hist2h3c2 | Histone cluster 2 H3c2 | 7 | -3.04114 |
| Abcb1a | ATP-binding cassette, subfamily B (MDR/TAP) member 1A | 6 | 1.424367 |
| Ahsa2 | AHA1, activator of heat shock protein ATPase 2 | 6 | -1.45842 |
| Maob | Monoamine oxidase B | 6 | 1.392949 |
| Gas2l3 | Growth arrest-specific 2 like 3 | 6 | 1.321103 |
3.5. Inhibition of c-Src Ameliorates EMT and Tubulointerstitial Fibrosis via PPARγ/UCP2 Pathway in Renal Tubule of Diabetic Mice
Our previous studies have indicated that EMT in renal tubular epithelial cells, characterized by loss of epithelial phenotype and gain of profibrotic features that are characteristic of mesenchymal cells, is directly involved in the progression of tubulointerstitial fibrosis that eventually contributed to renal fibrosis in DN [7, 21]. To understand whether c-Src inhibition has a protective effect in renal EMT and tubulointerstitial fibrosis, we first explore the role of PP2 treatment on the histopathological changes of kidney. As shown in Figure 5(a), HE staining showed that compared with the nondiabetic mice, interstitial fibrous tissue proliferation and inflammatory cell infiltration, tubular ectasia, and cell swelling were significantly increased in diabetic mice. These pathological changes were significantly attenuated by PP2 treatment. Masson's trichrome staining and quantitative assessment confirmed that PP2 treatment resulted in a significant reduction in interstitial fibrosis when compared with the diabetic mice. Additionally, compared with the nondiabetic mice, transmission electron microscopy showed nuclear volume reduction (pyknosis), chromatin condensation, and nuclear membrane shrinkage in renal tubular epithelial cell of db/db mice. PP2 treatment could partially reverse these morphological changes of apoptosis in renal tubular epithelial cells. Likewise, immunohistochemistry staining showed that the expression of epithelial marker E-cadherin was markedly decreased, and mesenchymal marker α-SMA was markedly increased in db/db mice than that in the db/m mice, whereas PP2 treatment significantly reversed these changes (Figure 5(b)). Western blotting results were consistent with the immunofluorescence observation (Figure 5(c)). These results indicate that c-Src plays an important role in the renal EMT and tubulointerstitial fibrosis of diabetic mice.
Figure 5.
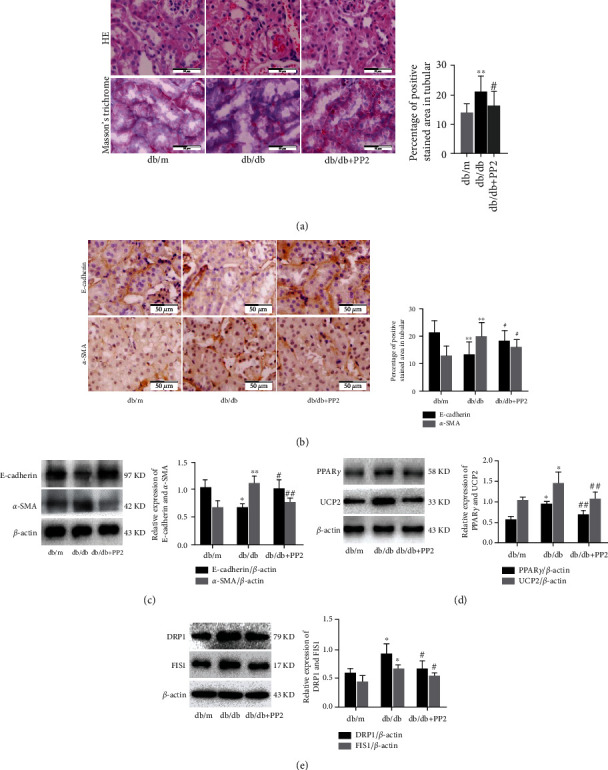
Effect of c-Src inhibiton on EMT and tubulointerstitial fibrosis in renal tubule of diabetic mice. (a) HE and Masson's trichrome staining of kidney sections (original magnification ×400). (b) Immunohistochemical staining of kidney sections with E-cadherin and α-SMA (original magnification ×400). (c) The expression levels of E-cadherin and α-SMA analyzed by western blotting. (d) The expression levels of PPARγ and UCP2 analyzed by western blotting. (e) The expression levels of DRP1 and FIS1 analyzed by western blotting. The number of mice in every group was 10. 10 visual fields were randomly selected from each group, and the area percent of staining was calculated for statistical analysis. All western blotting experiments were performed independently at least in triplicate. Values are expressed as means ± SD. ∗p < 0.05 versus db/m group. ∗∗p < 0.01 versus db/m group. #p < 0.05 versus db/db group. ##p < 0.01 versus db/db group.
As mentioned above, the results of the bioinformatic analysis indicated that UCP2 showed significantly different expression between nondiabetic and diabetic mice renal cortex. Interestingly, PPARγ was previously highlighted as the major regulator of UCP2 under physiological conditions [22, 23]. Therefore, we evaluated the relationship between c-Src inhibition and PPARγ/UCP2 pathway in db/db mice. Western blotting showed that the expression of PPARγ and UCP2 was markedly increased in db/db mice than that in the db/m mice, whereas PP2 treatment significantly reversed these changes (Figure 5(d)). Moreover, western blotting also showed that the expression of DRP1 and FIS1, two mitochondrial fission-related proteins, was markedly increased in db/db mice than that in the db/m mice, whereas PP2 treatment significantly reversed these changes (Figure 5(e)). Collectively, these data indicate that c-Src inhibition can attenuate renal EMT and tubulointerstitial fibrosis through regulation of PPARγ and UCP2 expression in diabetic mice.
3.6. Inhibition of c-Src Ameliorates HG-Induced EMT via PPARγ/UCP2 Pathway in HK-2 Cells
To determine whether the activation of c-Src is indispensable for HG-induced PPARγ and UCP2 expression, we treated HK-2 cells with the c-Src inhibitor PP2 or PPARγ antagonist GW9662. Compared with NG groups, exposure of HK-2 cells to HG for 48 h significantly increased PPARγ and UCP2 expression, which were significantly inhibited by PP2 or GW9662, respectively (Figure 6(a)). We further assessed whether the c-Src/PPARγ/UCP2 pathway was involved in HG-induced EMT in HK-2 cells. As shown in Figure 6(b), compared with NG groups, exposure of HK-2 cells to HG or UCP2 overexpression plasmid for 48 h significantly decreased the expression of E-cadherin and increased the expression of α-SMA, respectively. Furthermore, HG-induced EMT could be significantly attenuated by transfection with UCP2 shRNA plasmid. Next, to elucidate the mechanisms by which blockage of UCP2 ameliorated HG-induced EMT, we examined the role of UCP2 on the MMP in HK-2 cells by the confocal microscope. As shown in Figure 6(c), HG or UCP2 overexpression plasmid significantly induced decreased MMP. HG-induced MMP loss was rescued by cotreatment with UCP2 shRNA plasmid. We further explored the role of UCP2 on the mitochondrial morphology using Mitotracker. As shown in Figure 6(d), HG or UCP2 overexpression plasmid induced mitochondrial fragmentation, indicating the increased mitochondrial fission and dysfunction. In contrast, UCP2 shRNA plasmid treatment attenuated this trend induced by HG. Collectively, inhibition of c-Src/PPARγ/UCP2 pathway significantly inhibited HG-induced EMT and mitochondrial dysfunction in HK-2 cells.
Figure 6.
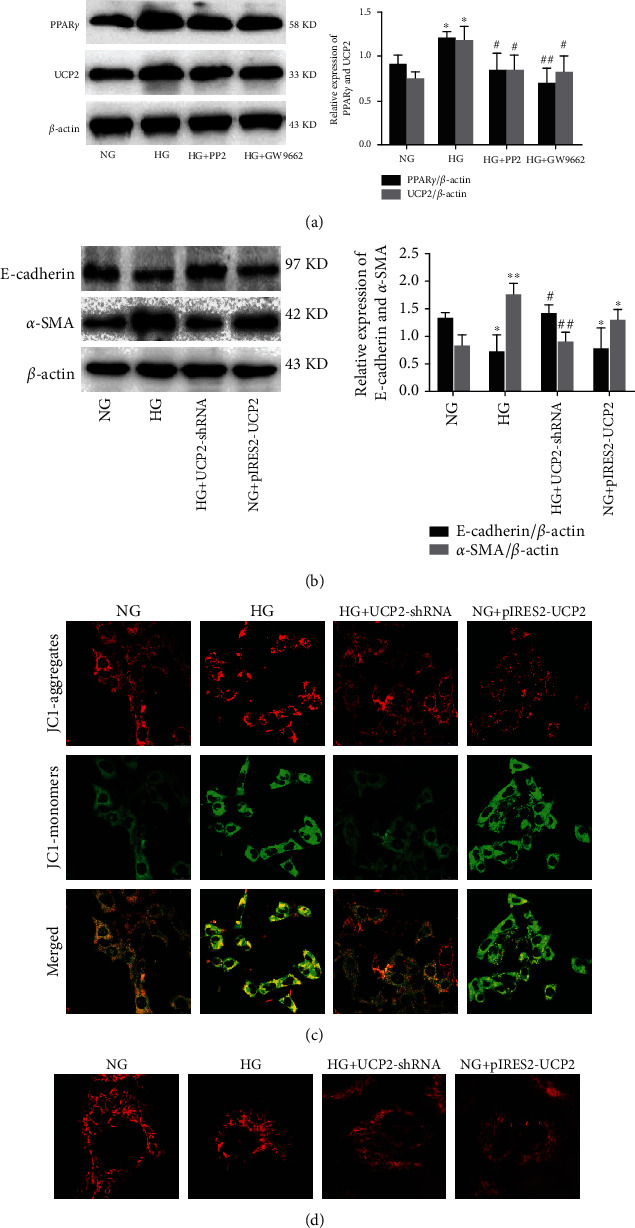
Effect of c-Src inhibiton HG-induced EMT in HK-2 cells. (a) The expression levels of PPARγ and UCP2 analyzed by western blotting. (b) The expression levels of E-cadherin and α-SMA analyzed by western blotting. (c) MMP detected by the JC-1 membrane potential assay (magnification ×100). The shift of fluorescence from red to green after HG exposure indicates a decrease in mitochondria membrane potential. (d) The mitochondrial morphology detected by Mitotracker (magnification×400). All western blot experiments were performed independently at least in triplicate. Values are expressed as means ± SD. ∗p < 0.05 versus NG group. ∗∗p < 0.01 versus NG group. #p < 0.05 versus HG group.##p < 0.01 versus HG group.
4. Discussion
DN is the leading cause of end-stage renal disease and contributes to increased mortality and economic burden worldwide. Multiple factors, such as metabolic abnormalities, inflammation, immunoregulation, and genetic predisposition, are involved in the process of DN, but the exact mechanism is not quite clarified [24]. It is now widely accepted that renal fibrosis is the most vital process for a variety of chronic kidney diseases that eventually turn into end-stage renal disease, including DN. Renal fibrosis, characterized by the abnormal accumulation of ECM, can manifest as glomerulosclerosis in the glomerulus and tubulointerstitial fibrosis mainly in the tubules [25, 26]. c-Src, a member of the Src kinase family, is activated by a number of stresses and implicated in the regulation of cellular processes in multiple tissues. c-Src can be activated in the kidneys of diabetic animals, glomerular mesangial cells, podocytes, and tubular epithelial cells exposed to HG [14, 15, 27]. Indeed, accumulating evidence suggested a strong correlation between c-Src and DN pathogenesis. Our previous study has demonstrated that c-Src activity was increased in kidneys of db/db mice and involved in renal tubular epithelial cells apoptosis in response to hyperglycemia in DN [15]. However, the role and molecular mechanisms of c-Src on renal fibrosis are yet to be elucidated. In this study, we explored the effect of c-Src inhibition on glomerulosclerosis and tubulointerstitial fibrosis in vivo and in vitro, respectively. In vivo, c-Src inhibition improved diabetes-induced glomerular hypertrophy, partial glomerulosclerosis, mesangial cells hyperplasia and hypertrophy, inflammatory cell infiltration, mesangial expansion, collagen deposition, tubular ectasia and cell swelling, and interstitial fibrous tissue proliferation in the diabetic kidneys. Similarly, c-Src inhibition could significantly alleviate renal fibrosis through decreasing protein levels of fibronectin, collagen IV, and α-SMA, as well as increasing protein level of E-cadherin. In vitro, HG-induced ECM accumulation, inflammation, and proliferation were inhibited in PP2-treated MMCs. Similarly, HG-induced EMT was also inhibited in PP2-treated HK-2 cells. These findings suggest that inhibition of c-Src can ameliorate diabetes-induced renal fibrosis.
Bioinformatic analysis is an efficient and reliable tool to identify potential therapeutic targets and diagnostic biomarkers for precision medicine. With the widespread application of high-throughput sequencing technology and public databases, bioinformatic analysis has been gradually used to study the molecular mechanisms of kidney diseases, such as membrane nephropathy, lupus, IgA nephropathy, and DN [28]. In this study, we performed bioinformatic analysis to screen the key genes and pathways related to DN and elucidated their molecular mechanism based on two GEO datasets. The GSE33744 dataset was used to identify the DEGs between nondiabetic and diabetic mice renal glomerular. A total of 839 DEGs were identified from GSE33744, including 579 upregulated genes and 260 downregulated genes. GO functional enrichment analysis showed that the upregulated genes were mainly enriched in signal transduction, membrane, and hydrolase activity, and the downregulated genes were mainly enriched in oxidation-reduction process, membrane, and metal ion binding. KEGG pathway enrichment analysis showed that the DEGs were mainly enriched in pathways in cancer, PI3K-Akt signaling pathway, neuroactive ligand-receptor interaction, MAPK signaling pathway, and Rap1 signaling pathway. Following PPI network analysis identified 23 functional modules and top 50 hub genes. In these 50 hub genes, the COX-2 pathway was selected for further investigation based on GO and KEGG enrichment analyses. Furthermore, the GSE86300 dataset was used to identify the DEGs between nondiabetic and diabetic mice renal cortex. A total of 238 DEGs were identified from GSE86300, including 112 upregulated genes and 126 downregulated genes. GO functional enrichment analysis showed that the upregulated genes were mainly enriched in signal transduction, extracellular exosome, and oxidoreductase activity, and the downregulated genes were mainly enriched in lipid metabolic process, membrane, and ATP binding. KEGG pathway enrichment analysis showed that the DEGs were mainly enriched in pathways in metabolic pathways, complement and coagulation cascades, Bile secretion, and PPAR signaling pathway. Following PPI network analysis identified 7 functional modules and top 50 hub genes. In these 50 hub genes, the UCP2 pathway was selected for further investigation based on GO and KEGG pathway enrichment analyses.
COX-2 is an inducible enzyme that catalyzes the conversion of arachidonic acid into prostaglandins which in turn cause pain, fever, and inflammation responses. Its expression can be induced by a number of inflammatory cytokines, mitogenic factors, and physical stimuli [29]. Previously, numerous studies revealed that COX-2 was increased in podocyte, glomerular mesangial cells, and renal tubular cells under HG condition, which contributed to the pathogenesis of DN [30–33]. Interestingly, the promoter region of the COX-2 gene contains several putative binding motifs for the binding of NF-κB [34]. NF-κB is a family of inducible transcription factors that mediates signal-induced expression of numerous genes involved in different biological processes, including immune response, inflammation, cellular transformation, cell proliferation, angiogenesis, invasion, and metastasis [35]. The activated and phosphorylated form of NF-κB subunits (p65 and p50) translocates to the nucleus and thereby induces the expression of target genes related to DN [36]. However, almost nothing is known about the role of NF-κB in modulating the COX-2 expression in DN. In this study, we found that the phosphorylation of NF-κB subunit p65 and its downstream target gene COX-2 was significantly elevated in diabetic kidneys. These changes were reversed by treatment of c-Src inhibitor PP2. Meanwhile, c-Src inhibitor PP2 also inhibited HG-induced the phosphorylation of NF-κB p65 and COX-2 expression in MMCs. In addition, NF-κB inhibitor PDTC not only reduced HG-induced COX-2 expression but also dramatically abrogated HG-induced phosphorylation of c-Src. These results reveal c-Src or NF-κB could act as an upstream regulator of COX-2 and a complex crosstalk between NF-κB and c-Src. We then further examined the role of the c-Src/NF-κB/COX-2 pathway on HG-induced ECM accumulation in MMCs. Our data demonstrated that pharmacological inhibition of c-Src with PP2, NF-κB with PDTC, or COX-2 with NS-398 also resulted in the decreased expression of fibronectin and collagen IV. On the mechanism, prostaglandin E2 (PGE2) was reported to be involved in numerous physiological and pathological processes of the kidney via its four receptors [37]. PGE2 receptor (EP1 or EP2) deficiency markedly suppressed the extracellular matrix accumulation and COX-2 expression in mouse mesangial cells induced by TGF-β1 which is a pivotal mediator of renal fibrosis and glomerulosclerosis in diabetic nephropathy [38, 39]. Importantly, inhibition of COX-2 via a specific inhibitor celecoxib markedly blocked PGE2 production [40]. These findings suggested not only the important role of COX-2 in the expressions of extracellular matrix proteins in mesangial cells but also a complex crosstalk among COX-2, PGE2 and PGE2 receptors. Collectively, these results suggest that inhibition of the c-Src pathway ameliorated renal ECM accumulation and fibrosis at least in part through modulating the activity of the NF-κB and expression of COX-2.
UCP2 is a mitochondrial inner membrane protein that uncouples the oxidative phosphorylation from ATP synthesis by dissipating the proton gradient generated across the mitochondrial inner membrane, therefore regulating glucose and lipid metabolism, immune cell activation, and ROS formation by mitochondria [41]. Consistently, previous studies have demonstrated that downregulation of UCP2 exacerbated oxidative stress damage as well as diabetes or HG-induced renal tubular epithelial cells apoptosis [42, 43]. However, a conflicting report suggested that UCP2 may act as a double-edged sword in renal tubular epithelial cell injury. Increased mitochondrial UCP-2 expression in renal proximal tubular cells increased O2 consumption and reduced O2 availability which subsequently contributed to diabetes-induced progressive kidney damage [44]. In this study, we found that the expression of UCP2 and its upstream gene PPARγ was significantly elevated in diabetic kidneys. These changes were reversed by treatment with c-Src inhibitor PP2. Meanwhile, PP2 also inhibited HG-induced UCP2 and PPARγ expression in HK-2 cells. In addition, PPARγ antagonist GW9662 also reduced the HG-induced UCP2 expression. We further examined the role of the c-Src/PPARγ/UCP2 pathway on HG-induced EMT in HK-2 cells. Our data demonstrated that HG induced EMT in HK-2 cells by increasing the mesenchymal marker and decreasing epithelial marker expression. The effect of HG on EMT was abolished by UCP2 shRNA plasmid. Additionally, UCP2 shRNA plasmid also prevented MMP loss and mitochondrial fragmentation induced by HG.
5. Conclusions
In conclusion, we identified COX-2 as an important hub gene in renal glomerular between nondiabetic and diabetic mice through bioinformatic analysis. Administration of PP2 attenuated renal ECM accumulation and fibrosis via the NF-κB/COX-2 pathway in the kidneys of diabetic mice. Moreover, PP2 ameliorates HG-induced ECM accumulation in MMCs by inhibiting NF-κB activation and COX-2 expression in vitro. Furthermore, we identified UCP2 as an important hub gene in renal cortex between nondiabetic and diabetic mice through bioinformatic analysis. Administration of PP2 attenuated renal EMT and tubulointerstitial fibrosis via the PPARγ/UCP2 pathway in the kidneys of diabetic mice. Moreover, PP2 ameliorated HG-induced EMT in HK-2 cells by inhibiting PPARγ and UCP2 expression in vitro (Figure 7). Thus, c-Src inhibition may provide a potential therapeutic target for the treatment of DN.
Figure 7.
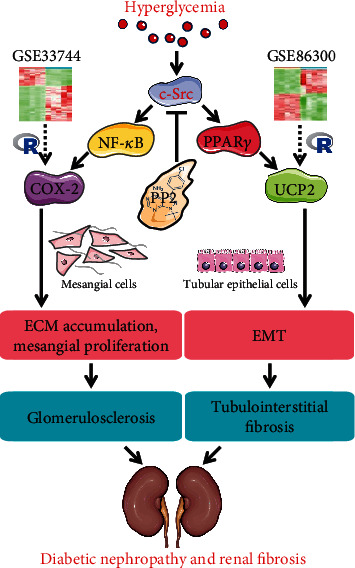
Schematic illustration of the mechanisms responsible for the PP2 effects on renal fibrosis in diabetic mice. PP2 decreased the expression of COX-2, phosphorylated p65, and fibrotic proteins, accompanied with attenuated renal glomerulosclerosis. PP2 also decreased the expression of PPARγ and UCP2, accompanied with attenuated renal tubulointerstitial fibrosis and EMT.
Acknowledgments
This study was supported by the Hebei Science and Technology Department Program (Nos. H2018206110 and H2020206326) and Youth TopNotch Talents Project of Education Department of Hebei Province (No. BJ2020006).
Data Availability
This study benefited from the Cancer Genome Atlas (TCGA) and GEO databases. We appreciate the data platform, and the authors uploaded their data.
Ethical Approval
The experimental animal protocol was approved by the Ethics Committee of the Fourth Hospital of Hebei Medical University (approval number: 4TH-HOS-HEBMU-2014013).
Conflicts of Interest
All authors declare that no conflicts of interest exist.
Authors' Contributions
Jinying Wei and Xinna Deng contributed equally to this work.
References
- 1.Tian N., Gao Y., Wang X., et al. Emodin mitigates podocytes apoptosis induced by endoplasmic reticulum stress through the inhibition of the PERK pathway in diabetic nephropathy. Drug Design, Development and Therapy. 2018;12:2195–2211. doi: 10.2147/dddt.s167405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang F., Hao Y., Zhang X., Qin J. Effect of echinacoside on kidney fibrosis by inhibition of TGF-β1/Smads signaling pathway in the db/db mice model of diabetic nephropathy. Drug Design, Development and Therapy. 2017;Volume 11:2813–2826. doi: 10.2147/DDDT.S143805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu H., Deng X., Shi Y., Su Y., Wei J., Duan H. PGC-1α, glucose metabolism and type 2 diabetes mellitus. The Journal of Endocrinology. 2016;229(3):R99–R115. doi: 10.1530/JOE-16-0021. [DOI] [PubMed] [Google Scholar]
- 4.Ren L., Guo X. Y., Gao F., Jin M. L., Song X. N. Identification of the perturbed metabolic pathways associating with renal fibrosis and evaluating metabolome changes of pretreatment with Astragalus polysaccharide through liquid chromatography quadrupole time-of-flight mass spectrometry. Frontiers in Pharmacology. 2019;10 doi: 10.3389/fphar.2019.01623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon J. J., Lee H. K., Kim H. Y., et al. Sauchinone protects renal mesangial cell dysfunction against angiotensin II by improving renal Fibrosis and inflammation. International Journal of Molecular Sciences. 2020;21(19):p. 7003. doi: 10.3390/ijms21197003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J. H. Mesangial cells and renal fibrosis. Advances in Experimental Medicine and Biology. 2019;1165:165–194. doi: 10.1007/978-981-13-8871-2_9. [DOI] [PubMed] [Google Scholar]
- 7.Song S., Qiu D., Luo F., et al. Knockdown of NLRP3 alleviates high glucose or TGFB1-induced EMT in human renal tubular cells. Journal of Molecular Endocrinology. 2018;61(3):101–113. doi: 10.1530/JME-18-0069. [DOI] [PubMed] [Google Scholar]
- 8.Qi R., Wang J., Jiang Y., et al. Snai1-induced partial epithelial-mesenchymal transition orchestrates p53-p21-mediated G2/M arrest in the progression of renal fibrosis via NF-κB- mediated inflammation. Cell Death & Disease. 2021;12(1):p. 44. doi: 10.1038/s41419-020-03322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Summy J. M., Gallick G. E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Reviews. 2003;22(4):337–358. doi: 10.1023/A:1023772912750. [DOI] [PubMed] [Google Scholar]
- 10.Shalloway D., Bagrodia S., Chackalaparampil I., Shenoy S., Lin P. H., Taylor S. J. c-Src and mitosis. Ciba Foundation Symposium 170 - Regulation of the Eukaryotic Cell Cycle. 1992;170:248–265. doi: 10.1002/9780470514320.ch15. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q., Tao J., Li G., et al. Astaxanthin ameliorates experimental diabetes-induced renal oxidative stress and fibronectin by upregulating connexin43 in glomerular mesangial cells and diabetic mice. European Journal of Pharmacology. 2018;840:33–43. doi: 10.1016/j.ejphar.2018.09.028. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z., Xie X., Huang J., et al. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-β1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radical Biology & Medicine. 2017;102:77–86. doi: 10.1016/j.freeradbiomed.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi D., Wang L., Ueda S., Yamanoue M., Ashida H., Shirai Y. The mechanisms of ameliorating effect of a green tea polyphenol on diabetic nephropathy based on diacylglycerol kinase α. Scientific Reports. 2020;10(1):p. 11790. doi: 10.1038/s41598-020-68716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J., Chen J. K., Harris R. C. EGF receptor deletion in podocytes attenuates diabetic nephropathy. Journal of the American Society of Nephrology. 2015;26(5):1115–1125. doi: 10.1681/ASN.2014020192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H., Shi Y., Deng X., et al. Inhibition of c-Src/p38 MAPK pathway ameliorates renal tubular epithelial cells apoptosis in db/db mice. Molecular and Cellular Endocrinology. 2015;417:27–35. doi: 10.1016/j.mce.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Hodgin J. B., Nair V., Zhang H., et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes. 2013;62(1):299–308. doi: 10.2337/db11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sas K. M., Kayampilly P., Byun J., et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. 2016;1(15):p. e86976. doi: 10.1172/jci.insight.86976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taniguchi K., Xia L., Goldberg H. J., et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62(11):3874–3886. doi: 10.2337/db12-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rawat C., Kukal S., Dahiya U. R., Kukreti R. Cyclooxygenase-2 (COX-2) inhibitors: future therapeutic strategies for epilepsy management. Journal of Neuroinflammation. 2019;16(1):p. 197. doi: 10.1186/s12974-019-1592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pierelli G., Stanzione R., Forte M., et al. Uncoupling protein 2: a key player and a potential therapeutic target in vascular diseases. Oxidative Medicine and Cellular Longevity. 2017;2017:11. doi: 10.1155/2017/7348372.7348372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei J., Shi Y., Hou Y., et al. Knockdown of thioredoxin-interacting protein ameliorates high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Cellular Signalling. 2013;25(12):2788–2796. doi: 10.1016/j.cellsig.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Bazwinsky-Wutschke I., Zipprich A., Dehghani F. Endocannabinoid system in hepatic glucose metabolism, fatty liver disease, and cirrhosis. International Journal of Molecular Sciences. 2019;20(10):p. 2516. doi: 10.3390/ijms20102516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou M. C., Yu P., Sun Q., Li Y. X. Expression profiling analysis: uncoupling protein 2 deficiency improves hepatic glucose, lipid profiles and insulin sensitivity in high-fat diet-fed mice by modulating expression of genes in peroxisome proliferator-activated receptor signaling pathway. Journal of Diabetes Investigation. 2016;7(2):179–189. doi: 10.1111/jdi.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H., Ni W. J., Meng X. M., Tang L. Q. MicroRNAs as regulators of immune and inflammatory responses: potential therapeutic targets in diabetic nephropathy. Frontiers in Cell and Development Biology. 2020;8 doi: 10.3389/fcell.2020.618536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colon S., Luan H., Liu Y., Meyer C., Gewin L., Bhave G. Peroxidasin and eosinophil peroxidase, but not myeloperoxidase, contribute to renal fibrosis in the murine unilateral ureteral obstruction model. American Journal of Physiology. Renal Physiology. 2019;316(2):F360–F371. doi: 10.1152/ajprenal.00291.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edeling M., Ragi G., Huang S., Pavenstädt H., Susztak K. Developmental signalling pathways in renal fibrosis: the roles of notch, Wnt and hedgehog. Nature Reviews. Nephrology. 2016;12(7):426–439. doi: 10.1038/nrneph.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie X., Lan T., Chang X., et al. Connexin43 mediates NF-κB signalling activation induced by high glucose in GMCs: involvement of c-Src. Cell Communication and Signaling: CCS. 2013;11(1):p. 38. doi: 10.1186/1478-811X-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang W., Wang Z., Cao Y., Zhang D., Mi X. Bioinformatic analysis reveals novel immune-associated hub genes in human membranous nephropathy. Genetic Testing and Molecular Biomarkers. 2019;23(1):23–31. doi: 10.1089/gtmb.2018.0137. [DOI] [PubMed] [Google Scholar]
- 29.Kiritoshi S., Nishikawa T., Sonoda K., et al. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes. 2003;52(10):2570–2577. doi: 10.2337/diabetes.52.10.2570. [DOI] [PubMed] [Google Scholar]
- 30.Abdallah M. S., Kennedy C. R. J., Stephan J. S., et al. Transforming growth factor-β1 and phosphatases modulate COX-2 protein expression and TAU phosphorylation in cultured immortalized podocytes. Inflammation Research. 2018;67(2):191–201. doi: 10.1007/s00011-017-1110-y. [DOI] [PubMed] [Google Scholar]
- 31.Lee W. J., Liu S. H., Chiang C. K., et al. Aryl hydrocarbon receptor deficiency attenuates oxidative stress-related mesangial cell activation and macrophage infiltration and extracellular matrix accumulation in diabetic nephropathy. Antioxidants & Redox Signaling. 2016;24(4):217–231. doi: 10.1089/ars.2015.6310. [DOI] [PubMed] [Google Scholar]
- 32.Cai J., Liu B., Guo T., et al. Effects of thromboxane prostanoid receptor deficiency on diabetic nephropathy induced by high fat diet and streptozotocin in mice. European Journal of Pharmacology. 2020;882:p. 173254. doi: 10.1016/j.ejphar.2020.173254. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y., Liu J., Zhou Z., Liu K., Liu C. Fangchinoline protects against renal injury in diabetic nephropathy by modulating the MAPK signaling pathway. Experimental and Clinical Endocrinology & Diabetes. 2020;128(8):499–505. doi: 10.1055/a-0636-3883. [DOI] [PubMed] [Google Scholar]
- 34.Li Q., Guo Y., Tan W., et al. Cardioprotection afforded by inducible nitric oxide synthase gene therapy is mediated by cyclooxygenase-2 via a nuclear factor-kappaB dependent pathway. Circulation. 2007;116(14):1577–1584. doi: 10.1161/CIRCULATIONAHA.107.689810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abd Wahab N. A., Lajis N. H., Abas F., Othman I., Naidu R. Mechanism of anti-cancer activity of curcumin on androgen-dependent and androgen-independent Prostate cancer. Nutrients. 2020;12(3):p. 679. doi: 10.3390/nu12030679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J., Ling Y., Yin S., Yang S., Kong M., Li Z. Baicalin serves a protective role in diabetic nephropathy through preventing high glucose-induced podocyte apoptosis. Experimental and Therapeutic Medicine. 2020;20(1):367–374. doi: 10.3892/etm.2020.8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., Yin J., Xu Y., Qiu Z., Liu J., Chen X. Effect of selective inhibition or activation of PGE2 EP1 receptor on glomerulosclerosis. Molecular Medicine Reports. 2020;22:2887–2895. doi: 10.3892/mmr.2020.11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X., Jiang D., Wang J., et al. Prostaglandin E2 EP1 receptor enhances TGF-β1-induced mesangial cell injury. International Journal of Molecular Medicine. 2015;35(1):285–293. doi: 10.3892/ijmm.2014.1979. [DOI] [PubMed] [Google Scholar]
- 39.Li N., Xu Y., Chen X., Fan Y., Wu J. Role of the prostaglandin E2/E-prostanoid 2 receptor signalling pathway in TGFβ-induced mice mesangial cell damage. Bioscience Reports. 2014;34(6):p. e00159. doi: 10.1042/BSR20140130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X., Yang Y., Yuan H., et al. Inhibition of COX-2/PGE2 cascade ameliorates cisplatin-induced mesangial cell apoptosis. American Journal of Translational Research. 2017;9(3):1222–1229. [PMC free article] [PubMed] [Google Scholar]
- 41.Crispim D., Rodrigues M., da Silva L. P. A., et al. The a allele of the UCP2 -866G/A polymorphism changes UCP2 promoter activity in HUVECs treated with high glucose. Molecular Biology Reports. 2019;46(5):4735–4741. doi: 10.1007/s11033-019-04918-0. [DOI] [PubMed] [Google Scholar]
- 42.Zhu X., Wu S., Guo H. Active vitamin D and vitamin D receptor help prevent high glucose induced oxidative stress of renal tubular cells via AKT/UCP2 signaling pathway. BioMed Research International. 2019;2019 doi: 10.1155/2019/9013904.9013904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X. L., Tang W. X., Tang X. H., Qin W., Gong M. Downregulation of uncoupling protein-2 by genipin exacerbates diabetes-induced kidney proximal tubular cells apoptosis. Renal Failure. 2014;36(8):1298–1303. doi: 10.3109/0886022X.2014.930650. [DOI] [PubMed] [Google Scholar]
- 44.Friederich M., Fasching A., Hansell P., Nordquist L., Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochimica et Biophysica Acta. 2008;1777(7-8):935–940. doi: 10.1016/j.bbabio.2008.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study benefited from the Cancer Genome Atlas (TCGA) and GEO databases. We appreciate the data platform, and the authors uploaded their data.