

Abstract
While solid-organ transplantation has evolved steadily with many breakthroughs in the past 110 years, many problems remain to be addressed and advanced therapeutic strategies need to be considered. T-cell immunometabolism is a rapidly advancing field that has gathered much attention recently, providing ample mechanistic insight from which many novel therapeutic approaches have been developed. Applications from the field include antitumor and antimicrobial therapies, as well as for reversing graft-versus-host disease and autoimmune diseases. However, the immunometabolism of T-cells remains underexplored in solid-organ transplantation. In this review, we will highlight key findings from hallmark studies centered around various metabolic modes preferred by different T-cell subtypes (categorized into naïve, effector, regulatory, and memory T-cells), including glycolysis, glutaminolysis, oxidative phosphorylation, fatty acid synthesis and oxidation. This review will discuss the underlying cellular signaling components that affect these processes, including the transcription factors Myc, HIF1α, ERRα, SREBPs, along with mechanistic target of rapamycin and AMPK signaling. We will also explore potential therapeutic strategies targeting these pathways, as applied to the potential for tolerance induction in solid-organ transplantation.
INTRODUCTION
Immunometabolism is quickly emerging as an important concept in solid-organ transplantation (SOT). Energy homeostasis and metabolic reprogramming of immune cells have been the center of many recent publications particularly in the context of normothermic versus hypothermic preservation.1–5 Accordingly, different subsets of T-cells, B-cells, dendritic cells, macrophages, and granulocytes adopt specific metabolic modes, depending on their developmental and functional states, as well as the surrounding physiological conditions.6 While the immunometabolism of T-cells has been robustly studied and heavily exploited for therapeutic applications in many subfields of immunology, this concept is scarcely explored in SOT. Therefore, the purpose of this review is to focus on the immunometabolism of T-cells in the context of SOT. In this review, we will describe the current paradigms in T-cell immunometabolism (TABLE 1). Depending on their functional state, T-cells undergo different metabolic reprogramming strategies, and thus it is crucial to understand a T-cell’s different metabolic modes to better modulate their function. Additionally, we will discuss relevant signaling pathways that drive metabolic reprogramming in T-cells and comment on potential therapeutic strategies to target these pathways to improve outcomes in SOT.
Table 1:
Summary of Hallmark Studies In T-Cell Immunometabolism.
| SUMMARY OF HALLMARK STUDIES | ||
|---|---|---|
| RELEVANT TO T-CELL ACTIVATION | ||
| Authors | Year | Major Findings |
| Bailis W et al | 2019 | Mitochondrial complexes I and II control T-cell proliferation, differentiation, and function in parallel and independently of each other |
| Blagih J et al | 2015 | AMPK regulates effector T-cells’ metabolic plasticity in low-glucose environments. |
| Carr EL et al | 2010 | CD28 and TCR signaling increases glutamine uptake and glutaminolysis. |
| Cham et al | 2005 and 2008 | Activated CD8+ T-cells increase glucose uptake and glycolysis. |
| Chang CH et al | 2013 | Aerobic glycolysis disinhibits translation of interferon gamma to initiate T-cell’s effector function. |
| Duvel K et al | 2010 | mTORC1 controls Myc, HIF1α, SREBPs to regulate T-cell metabolism. |
| Frauwirth KA et al | 2002 | CD28 signaling increases glucose metabolism. |
| Kidani Y et al | 2013 | De novo fatty acid synthesis mediated via SREBPs is important for CD8+ T-cell activation. |
| Macintyre AN and Gerriets VA et al | 2014 | Glucose transporter Glut1 is important for CD4+ T-cell’s activation and function. |
| Michalek RD et al | 2011 | Naïve T-cells are metabolically quiescent. Effector CD4+ T-cells favor glycolysis and glucose metabolism while regulatory T-cells favor lipid oxidation. |
| Sena LA et al | 2013 | Mitochondrial reactive oxygen species are important for T-cell activation. |
| Sinclair LV et al | 2013 | T-cell receptor and IL-2 signaling leads to increased uptake of large neutral amino acids, including leucine, using System L1 transporters that can be blocked by cyclosporin A. |
| Wang R et al | 2011 | Myc is the main driver for glycolysis and glutaminolysis during early T-cell activation. HIF1α sustains glycolysis in effector T-cells in the later stage. |
| Yang K et al | 2013 | mTORC1 is necessary for activated T-cells to exit quiescence and to increase glucose metabolism, lipid biosynthesis, and oxidative phosphorylation. |
| Zheng Y et al | 2009 | The impairment of IL-2 production and proliferation is linked to anergic T-cells’ failure to upregulate cellular metabolic machinery. |
| RELEVANT TO REGULATORY T-CELL DEVELOPMENT & FUNCTION | ||
| Authors | Year | Major Findings |
| Beier UH et al | 2015 | Mitochondrial oxidative phosphorylation is required for regulatory T-cells to mediate cardiac allograft acceptance. |
| Berod L et al | 2014 | De novo fatty acid synthesis (FAS) from glycolytic acetyl CoA is important for CD4+ effector T-cells while regulatory T-cells utilize exogenous fatty acids. Blocking de novo FAS with soraphen A favors regulatory T-cell formation. |
| Fu Z et al | 2019 | Loss of mitochondrial transcription factor A decreases FoxP3 expression |
| Hester J et al | 2012 | Inhibiting mTORC1 promotes regulatory T-cell formation and alleviates transplant arteriosclerosis. |
| Michalek RD et al | 2011 | Naïve T-cells are metabolically quiescent. Effector CD4+ T-cells favor glycolysis and glucose metabolism while regulatory T-cells favor lipid oxidation. |
| Michalek RD et al | 2011 | ERRα is necessary for glucose metabolism, and ERRα inhibition induces a metabolic switch to fatty acid oxidation. Addition of lipids to ERRα-deficient T-cells rescues regulatory T-cell differentiation. |
| Pearce EL et al | 2009 | AMPK signaling is crucial for fatty acid oxidation in regulatory and memory T-cells. |
| Rosa VD et al | 2015 | Enolase-1 facilitates the expression of FoxP3-E2 variant and immunosuppressive activity |
| Schenk U et al | 2011 | P2RX7 activation by ATPs destabilizes regulatory T-cells |
| Shi LZ et al | 2011 | Impaired glycolysis due to HIF1α deficiency prevents Th17 differentiation and favors regulatory T-cells formation. |
| Shrestha S et al | 2015 | PTEN is necessary to stabilize regulatory T-cells by maintaining their preferred metabolism. |
| Tanimine N et al | 2019 | Natural regulatory T-cells are more dependent on glycolysis early after activation than induced counterparts |
| Weinberg SE et al | 2019 | Lacking mitochondrial complex III causes regulatory T-cell to lose function while still stably expressing FoxP3 |
| RELEVANT TO MEMORY T-CELL FORMATION | ||
| Authors | Year | Major Findings |
| Bantug GR et al | 2018 | mTORC2 increases localization of hexokinase I to mitochondrial voltage-dependent anion channel, increasing pyruvate influx into mitochondria and facilitating rapid reactivation of CD8+ memory T-cells |
| Borges de Silva H et al | 2018 | ATP plays a regulatory role in formation and maintenance of long-lived CD8+ memory T-cells through an autocrine loop |
| Buck MD et al | 2016 | Forcing mitochondrial fusion and inhibiting mitochondrial fission skews T-cell’s phenotype into memory. |
| Fraser KA et al | 2013 | High mitochondrial spare respiratory capacity in CD8+ memory T-cells positively correlates to the number of antigen restimulation. |
| O’Sullivan D et al | 2014 | Memory CD8+ T-cells utilize fatty acids synthesized de novo via glycolytic-lipogenic pathway. |
| Pearce EL et al | 2009 | AMPK signaling is crucial for fatty acid oxidation in regulatory and memory T-cells. |
| Sukumar M et al | 2013 | CD8+ memory T-cell formation and function are impaired when glycolysis is forced and are enhanced when glycolysis is inhibited. |
| Van der Windt GJ et al | 2012 and 2013 | CD8+ memory T-cells have increased mitochondrial spare respiratory capacity in favor of fatty acid oxidation. |
| EXEMPLAR THERAPEUTIC APPLICATIONS | ||
| Authors | Year | Major Findings |
| Beier UH et al | 2015 | Mitochondrial oxidative phosphorylation is required for regulatory T-cells to mediate cardiac allograft acceptance. |
| Ledderose C et al | 2018 | Antagonizing P2RX4 receptors prevents CD4+ T-cell recruitment and infiltration into lung allografts. |
| Lee CF et al | 2015 | Concurrently blocking glycolysis, glutaminolysis, and activating AMPK pharmacologically delays skin graft rejection and prolongs cardiac allograft survival |
| Liu Z et al | 2012 | Targeting System L1 amino acid transporters in T-cells to block amino acid influx prolongs cardiac allograft survival. |
| Yin Y et al | 2015 | Concurrently blocking glycolysis and activating AMPK pharmacologically in T-cells reverses autoimmune cytotoxicity. |
Glucose metabolism is often considered to be the center of cellular metabolism.7 As the first step, glucose is converted by glycolysis into pyruvate, which is transported into the mitochondria where the compound is further converted to acetyl-CoA, a key metabolite for mitochondrial metabolism. Acetyl-CoA fuels the tricyclic acid (TCA) cycle which feeds into oxidative phosphorylation (OXPHOS) at the mitochondrial electron transport chain (ETC) to generate ATP for cellular physiology. Fatty acid oxidation (FAO) can break down high-carbon molecules into acetyl-CoA and acts as another source besides glucose to drive mitochondrial metabolism. On the other hand, acetyl-CoA can be used to build fatty acids in fatty acid synthesis (FAS) for anabolic processes during cellular proliferation. Additionally, many amino acids and their metabolites (ie, glutamine and alpha-ketoglutarate) also feed into the TCA cycle and drive mitochondrial metabolism.7 Together, these metabolic processes are regulated by multiple cellular signaling factors, such as mechanistic target of rapamycin (mTOR) and AMPK along with their downstream transcription factors.8,9
CURRENT PARADIGMS IN T-CELL IMMUNOMETABOLISM
1. Naïve T-cells:
Cellular metabolism is an old field, but the relationship between metabolism and T-cell function has been the center of substantial focus in the past 2 decades.10 This relationship is well illustrated by anergic T-cells, of which the impairment of IL-2 production and proliferation are linked to their failure to upregulate machinery to support cellular metabolism.11 During the course of an immune response, T-cells exist in 3 main functional states: naïve, effector, and memory,12 and the levels of individual metabolic pathways decrease or increase according to these states (FIGURE 1). Before activation, naïve T-cells are functionally and bioenergetically quiescent. These resting T-cells maintain a low-level metabolic profile, with basal nutrient uptake and basal glycolytic rate, producing ATP mainly from OXPHOS to accommodate the basal energy demand necessary for housekeeping cellular activities.13–16 The intracellular metabolome of naïve T-cells include more medium-chained and long-chained fatty acids for catabolism and significantly less glucose and glycolytic metabolites compared to activated T-cells.17
Figure 1: Summary of Metabolic Features of T-cells.
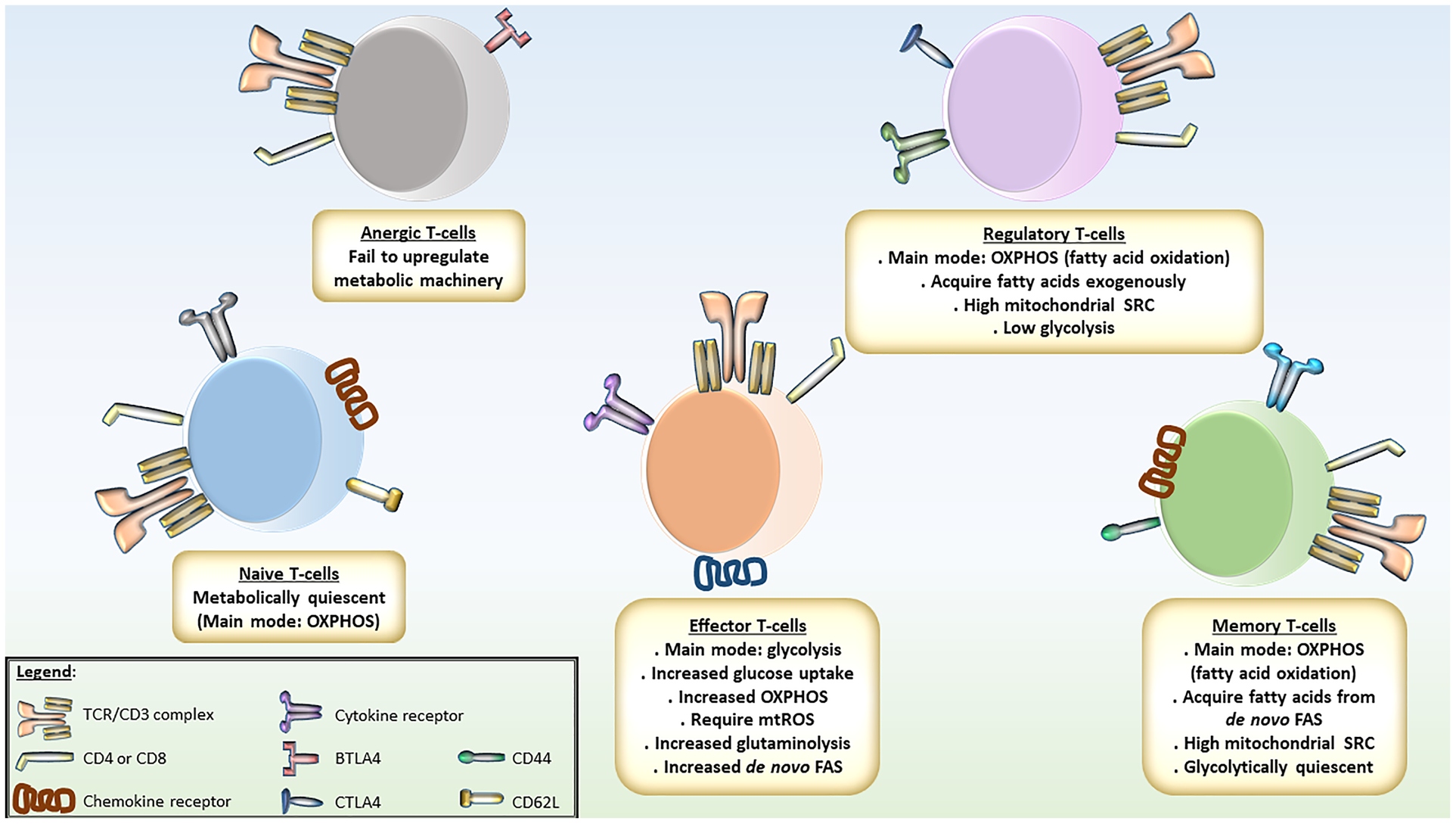
Naïve T-cells are metabolically quiescent, using oxidative phosphorylation (OXPHOS) to generate ATPs for basal cellular activities. Upon activation, T-cells increase glucose uptake and commit to glycolysis to rapidly generate ATPs and metabolites for anabolism, including precursors for de novo fatty acid synthesis (FAS) for membrane expansion. OXPHOS is also increased to generate mitochondrial reactive oxygen species (mtROS) necessary for activation. Amino acids, such as glutamine, are fed into the tricarboxylic acid cycle for anaplerosis. After effector phase, memory T-cells adopt fatty acid oxidation (FAO) as the main metabolic mode, metabolizing fatty acids generated from de novo FAS. These cells have high mitochondrial spare respiratory capacity, metabolically ready for reactivation. When becoming anergic, T-cells exhaust their metabolic machinery. Regulatory T-cells are metabolically similar to memory T-cells. They have high mitochondrial capacity and utilize FAO as the main metabolic mode, albeit metabolizing exogenous fatty acids.
2. Effector T-cells:
Upon activation, T-cells undergo substantial metabolic changes. The most significant metabolic hallmark of activation is their commitment to aerobic glycolysis, a term that describes increased glycolytic activity in the presence of oxygen, known in oncology as the Warburg effect.18 Two studies conducted by Cham et al have shown that activated CD8+ T-cells increase glucose uptake, expression of glycolytic enzymes, glycolytic rate, and lactate production. Production of IFNγ and cytolytic proteins, as well as cell-cycle progression and cytolytic production in these cells are impaired, if glucose is deprived or if glycolysis is inhibited.19,20 CD4+ T-cells, on the other hand, are divided into several effector subsets and 1 regulatory subset,12 with the effector T-cells (Teffs) sharing the same metabolic mode as activated CD8+ T-cells. T-helper 1 (Th1), T-helper 2 (Th2), T-helper 17 (Th17) cells elevate glycolytic rate much more significantly than naïve CD4+ T-cells and regulatory T-cells (Treg). Their glycolytic capacity and glycolytic reserve are also much higher than Treg.15,21 In addition, blocking glycolysis in Th1 and Th17 cells also leads to a decrease in their proliferative capacity and viability.21 Consistent with this view, Glut1, the most abundant glucose transporter in the Glut family in naïve and activated T-cells, is crucial for the activation of murine CD4+ and human T-cells, but not for the survival of quiescent naïve T-cells. When activated in vitro with CD3/CD28 agonists, CD4+ T-cells isolated from Glut1-deficient mice have impaired glucose uptake, glycolysis, lactate production, and glycolytic capacity. Similarly, knocking down Glut1 in human T-cells decreases both glucose uptake and glycolysis.22 To further link aerobic glycolysis and T-cell activation, another study conducted by the same group showed that the increase in expression of Glut1 was mediated via the PI3K-Akt signaling pathway as a consequence of CD28 costimulation, suggesting that a commitment to aerobic glycolysis is integrated into T-cell activation by sharing the same signaling pathway.23
These studies lead to the question: why do T-cells adopt such specific metabolic modes after they become activated? The answer lies in what we already know about proliferating cancer cells. Although aerobic glycolysis is inefficient to produce ATP for cells that are in high demand for energy compared to OXPHOS, it allows cells to produce energy more quickly. In addition, aerobic glycolysis allows proliferating activated T-cells to generate important metabolites and reducing agents necessary for anabolic reactions, including nucleotide, amino acid, and fatty acid biosynthesis.18 Chang et al demonstrated the necessity of aerobic glycolysis for effector function of CD4+ T-cells by showing that the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) can bind to the 3’ UTR region of IFNγ transcripts when inactive. However, if T-cells are activated in media containing galactose instead of glucose, there is a significant increase in GAPDH binding to IFNγ transcripts, preventing the translation of this protein. Thus, aerobic glycolysis allows activated T-cells to disinhibit the translation of IFNγ and initiate important effector functions.16
While aerobic glycolysis is the metabolic hallmark of T-cell activation, OXPHOS is also crucial in many regards. Firstly, OXPHOS is increased along with aerobic glycolysis in activated T-cells compared to naïve T-cells, but the degree of increase in OXPHOS is not as great as the degree of increase in aerobic glycolysis, as demonstrated by a decrease in oxygen consumption rate/extracellular acidification rate (OCR/ECAR) ratio in activated T-cells compared to naïve T-cells.16,22 Interestingly, the ATP generated by OXPHOS is required from the time of activation until somewhere between 1 and 2 days postactivation.16 These findings suggest that OXPHOS is important for the early period of T-cell activation, perhaps serving as a “jump start,” and is not required to maintain the activated state during the course of Teffs life in normal conditions. Secondly, reactive oxygen species (ROS) generated by the ETC via OXPHOS are necessary for T-cell activation. Sena et al showed that mitochondrial ROS increases 10 minutes after activation via TCR-mediated calcium flux. IL-2 production is impaired when T-cells are activated in the presence of the antioxidant vitamin E targeted to mitochondria or when T-cells are deficient of ETC’s complex III. T-cell activation shown via CD69 and CD25 markers also decreases in complex III–deficient T-cells when they are activated with CD3/CD28 agonists. These defects are rescued when ROS are biochemically induced or when they are generated in the cells via calcium-induced complex I & complex II.24 Thirdly, OXPHOS has been suggested to be capable of sustaining T-cell viability and activation when glucose is replaced by another source of nutrient such as galactose or pyruvate, although complete withdrawal of sugar is detrimental to T-cells.16,19,24 Lastly, recent loss-of-function studies have shown that mitochondrial complexes I and II control T-cell proliferation, differentiation, and function in parallel and independently of each other during early and late activation.25 Together, these findings suggest that OXPHOS is an important ancillary process that supports aerobic glycolysis, the main metabolic mode, during T-cell activation.
The metabolic demand of proliferating Teffs is considerable, and thus, other metabolic modes are upregulated during T-cell activation to satisfy the cellular energy demands. In proliferating Teffs, the TCA cycle functions not only as a source of NADH, FADH2 for OXPHOS, but also as a source of metabolites that feed into the biosynthesis of many macromolecules, including cholesterol and fatty acids for membrane generation. Thus, anaplerosis is important to replenish this cycle.26 While many amino acids have been found to be important for T-cell physiology, glutamine, a nonessential amino acid that can be converted into alpha-ketoglutarate and enter the TCA cycle, is the most critical and is specifically required for the metabolic demand of T-cell activation.27 During this process, the expression of glutamine transporters, along with glutaminolysis enzymes, and glutamine uptake are upregulated. Also, IFNγ and IL-2 production, as well as proliferation of activated T-cells, are impaired if they are stimulated in a glutamine-free media. These results are thought to be mediated by both CD28 and T-cell receptor (TCR) signaling.27
In addition, de novo lipid biosynthesis is also critical for proliferating cells to build cellular membrane and expand their machinery.26 Indeed, gene expression for cholesterol and FAS enzymes are much higher when compared to their nonactivated counterparts. Deficiency in the signaling pathways controlling lipid biosynthesis in activated CD8+ T-cells prevents them from proliferating, and adding exogenous cholesterol, restores their proliferation.28 When naïve CD4+ T-cells are activated under Th17-polarizing conditions, they also upregulate gene expression for fatty acid biosynthesis enzymes and for the citrate-pyruvate shuttle system that links glycolytic-lipogenic pathways. Blocking either pathway compromises Th17 differentiation and proliferation, with the same effects also observed in Th1- or Th2-polarizing activation. Additionally, in the same study, using radioisotope-labeled glucose, the authors have demonstrated that fatty acids are synthesized de novo in Th17 cells using acetyl-CoA derived from glycolysis rather than being taken up exogenously from the culture media. Interestingly, blocking this de novo fatty acid biosynthesis pathway not only negatively affects glycolysis but also increases uptake of exogenous fatty acids in Th17 cells, suggesting a close relationship between FAS and glycolysis, as well as the importance of lipid for T-cell activation.29
3. Regulatory T-cells:
Functionally distinct from Teffs, Treg unsurprisingly have a different metabolic profile. Upon polarization, Treg increase both glycolytic rate and palmitate oxidation compared to naïve T-cells. Their glycolytic rate is significantly lower, while their palmitate oxidation is significantly higher than those of Teffs. Interestingly, exogenous fatty acids suppress the function of Teffs but not Treg, which presents a therapeutic opportunity to enrich Treg while eliminating Teffs in SOT.15 These findings are echoed in another study performed by the same group, in which both glycolytic capacity and glycolytic reserve of Treg are lower than those of Th1 and Th17 cells, while their mitochondrial spare respiratory capacity (SRC) is significantly higher. Importantly, an increase in OCR/ECAR ratio indicates Treg preferentially use OXPHOS, in this case FAO, over glycolysis. In addition, while blocking OXPHOS via ETC inhibition is shown to be detrimental to Treg proliferation, blocking glycolysis in Treg does not affect their proliferation, but rather increases it.21 Consistent with the fact that glycolysis is not a favored metabolic mode in Treg, Glut1 expression in these cells is significantly lower than that of Teffs, similar to the level of naïve T-cells, and deficiency in Glut1 does not affect Treg differentiation and function.22 Treg that are induced by TGFβ (iTreg),30 similarly to natural Treg described above, also display a strong preference for FAO as their main metabolic mode.31 Glucose deprivation can skew the differentiation of conventional T-cells into iTreg, suggesting the less important need of glycolysis this subtype of Treg.22,32 Furthermore, the need for glycolysis in natural Treg and iTreg are temporally different, with natural Treg more dependent on glycolysis very early after activation.33 From a therapeutic standpoint, these findings suggest that glycolysis is not necessary for Treg enrichment, and thus glycolysis inhibition could be used to selectively target Teffs while still sparing Treg. In fact, treatment of mixed lymphocyte reactions with dichloroacetate, a glycolysis inhibitor, has been shown to have increased IL-10 secretion and FoxP3 expression.34 It is thought that during glycolysis inhibition, the glycolytic enzyme Enolase-1 is free to enter the nucleus, affecting FoxP3 expression and increasing immunosuppressive activity in Treg.35
The fact that Treg depend on FAO begs the question: what are their sources of fatty acids? Pharmacologically blocking de novo FAS increases Treg generation and has no impact on intracellular fatty-acid levels. Moreover, radioisotope-labeled carbon tracing experiments indicate that Treg do not utilize glucose to build fatty acids as much as Th17 cells but rather use it to build other metabolites such as amino acids. More importantly, exogenous palmitate is incorporated into Treg intracellular long-chained fatty acids at a higher level than Th17, indicating Treg preference for exogenous fatty acid uptake over de novo biosynthesis.29
Studies in a heterotopic mouse cardiac transplant model further illustrate the importance of mitochondrial FAO in Treg by employing genetically modified Treg. In these experiments, conventional T-cells and either wild-type Treg or Treg deficient of PCG1α or Sirt3 (transcription factors that regulate mitochondrial respiration/biogenesis and FAO, respectively) were adoptively transferred into an immunodeficient recipient. The survival of cardiac allografts was reduced when Treg possess either deficiency, compared to the wild-type counterparts.36 These results suggest the importance of OXPHOS and FAO in Treg immunometabolism and present potentials for therapeutic strategies that enhance mitochondrial metabolism, via PCG1α or Sirt3 pharmacologic agonists, or other mitochondrial biogenesis induction methods.
With a strong dependence on FAO and OXPHOS, Treg stability can be affected by various extracellular environment conditions. Pathological inflammatory niches, as in posttransplant allograft inflammation, are hypoxic due to decreased blood flow by vascular damage, increased oxygen consumption by the influx of inflammatory cells and activation of oxygenases expressed by multiple cell types.37 Under hypoxia, OXPHOS is undoubtedly compromised, and therefore Treg suppressive function is likely affected. Mitochondrial complex III, an important component of OXPHOS, is known to be crucial for Treg suppressive function. Complex III–deficient Treg have been shown to decrease expression of genes encoding important functional proteins, including NRP1, PD-1, CD73, and TIGIT, while still stably expressing FoxP3.38 The mechanism is linked to increased cellular levels of 2-hydroxyglutarate and succinate, which antagonize DNA demethylases, resulting in hypermethylation of the downregulated genes.38 Furthermore, modifications to mitochondrial genes have been shown to alter FoxP3 expression, thus affecting Treg stability. A loss of mitochondrial transcription factor A, which normally controls mitochondrial respiration and mitochondria-specific protein expression, leads to decreased Treg proliferation and FoxP3 expression.39 Additionally, similarly to Tmems (discussed below) but in the opposite way, the purinergic P2RX7 receptor signaling activated by ATPs can destabilize Treg, prevent their immunosuppressive function, and facilitate Treg conversion into Th17.40 In an inflamed transplanted organ, release of intracellular ATP from dying cells is unavoidable, and therefore provides an explanation for why Treg become unstable in proinflammatory conditions through metabolic reprogramming.
Recent studies have also indicated that mitochondrial fusion/fission dynamics play important roles in Treg development. In CD4+ T-cells, treatment with Mdivi1, a pharmacologic agent that inhibits mitochondrial fission, leads to a decrease in phosphorylation of dynamin-related protein 1, a fission machinery protein, resulting in a significant reduction of Th1 and Th17, and a significant augmentation of IL-10+ CD4+ Foxp3+ T-cells in the central nervous system.41 Furthermore, treatment with Mdivi1 significantly inhibits the production of IFNγ, IL-17, and GM-CSF in activated T-cells while it promotes Treg formation in the spleen, as well as attenuates ROS production.41 These interesting observations are consistent with the fact that mitochondrial OXPHOS is the crucial metabolic mode in Treg, as inhibiting mitochondrial fission is known to enhance mitochondrial OXPHOS efficiency in T-cells.42 These findings also suggest that Mdivi1 can be a potential therapeutic agent to be further exploited in SOT.
4. Memory T-cells:
After the antigens are cleared in a cell-mediated immune response, the Teffs population contracts into a small number of long-lived Tmems.43 CD8+ Tmems have low glycolytic activity, similar to that of naïve T-cells, while they have a significantly higher mitochondrial SRC as well as OXPHOS activity than effector and naïve T-cells via increased mitochondrial biogenesis.44 Interestingly, the hallmark high SRC of CD8+ Tmems has been noticed to positively correlate to the number of restimulations.45 This finding suggests that when antigen loads increase, CD8+ Tmems adapt themselves to be metabolically fitter for higher functional efficiency to clear the antigens.
Furthermore, CD8+ Tmems rely on FAO for OXPHOS, demonstrated by an increase in the expression of carnitine palmitoyl transferase 1a (CPT1a), a rate-limiting enzyme that transports fatty acids into mitochondria for FAO. In contrast, Teffs have low CPT1a expression.44 Forced expression of CPT1a (and thus enhancing FAO) leads to increased persistence and memory formation in a bacterial infection model and vice versa when CPT1a is knocked down.44 More importantly, the active lipolytic phenotype of CD8+ Tmems is regulated by lysosome acid lipase, an enzyme that releases fatty acids from lysosomal triacylglycerol (TAG), as T-cells store TAG in lysosomes and do not accumulate cytosol lipid droplets like adipocytes.46 These findings lead to an important question: what is the source of lysosomal fatty acids for FAO in Tmems? O’Sullivan et al demonstrated that CD8+ Tmems do not take up exogenous fatty acids as much as Teffs and have a lower surface expression of receptors for long-chain fatty acids. In addition, inhibition of fatty acid synthase impairs survival of Tmems. More importantly, de novo FAS in Tmems utilizes glucose via glycolytic-lipogenic pathway as the main source of carbon, indicated by a high uptake of glucose during regular memory development and an impaired FAO when Tmems are cultured in low-glucose media.46 Taken together, while CD8+ Tmems and Treg both utilize FAO as a preferred metabolic mode, the sources of fatty acids are different, with Tmems synthesizing them de novo, while Treg utilize exogenous fatty acids.29 When considering SOT, a postoperative therapeutic strategy that targets de novo FAS while enriching exogenous fatty acids could be devised to promote Treg formation while suppressing Tmems in transplant recipients.
With Tmems mitochondrial profile and metabolic phenotype described above, the question becomes: How do a high mitochondrial SRC and a FAO-dominant metabolic profile confer advantages to Tmems during reactivation? With a metabolic hallmark of high mitochondrial SRC that increases the limit of OXPHOS activity fueled by FAO, Tmems have metabolic advantages to proliferate faster and produce more IFNγ than naïve T-cells when challenged with antigens.47 When Tmems are reactivated, they require mitochondrial ATPs (which is similar to activated T-cell’s requirement for mitochondrial ATP during the first 2 days after stimulation16) and an induction of glycolysis.47,48 By having a higher SRC, Tmems are able to produce more mitochondrial ATP and thus become reactivated faster. Also, producing more mitochondrial ATP increases the function of hexokinase and its association with mitochondria, as this enzyme catalyzes the first ATP-dependent reaction in glycolysis and needs to be within a close vicinity to mitochondria for their ATPs, thus leading to a more efficient induction of glycolysis. Consistent with this model, Tmems have an increase in both OCR and ECAR compared to naïve T-cells when being reactivated while their partition of OXPHOS versus glycolysis is the same as naïve T-cells.47,48 Furthermore, ATP has been reported to play a regulatory role in formation and maintenance of long-lived CD8+ Tmems through an autocrine loop. P2RX7, a purinergic receptor on T-cell surface, is normally activated by extracellular ATPs released from dying cells of the surrounding tissues. Healthy Tmems also release ATPs, and these ATP molecules initiate P2RX7 signaling that leads to a mitochondrial phenotype favoring FAO. As illustrated in an LCMV-infection model, genetic deletion of P2RX7 leads to CD8+ Tmems mitochondrial dysregulation, including a reduction in mitochondrial mass, membrane potential, and respiratory capacity, resulting in a decrease in CD8+ Tmems number compared to the wild-type control, and an increased susceptibility to infection.49 On the other hand, in CD4+ T-cells, the purinergic autocrine signaling through P2RX4 receptors affects their migration. As a result of CXCL12-mediated recruitment, the P2RX4 receptors and mitochondria localize near the front of the T-cells, creating a feedforward loop of calcium influx that sustains mitochondrial ATP production to fuel pseudopod protrusion and migration.50
Finally, recent efforts have been focused on the modulation of a T-cell’s phenotype via metabolic reprogramming. Enforcing glycolysis by overexpressing its enzyme phosphoglycerate mutase-1 has been shown to reduce long-term survival of CD8+ T-cells in vivo. In contrast, inhibiting glycolysis with 2-DG during activation not only leads to a transcriptional decrease of glycolytic genes but also enhances CD8+ memory formation, shown by increased memory markers and in vivo persistence of these cells.17 In another novel study, Teffs are shown to have fission-associated round distinct mitochondria while Tmems have fusion-associated packed tubular mitochondria. Importantly, mitochondrial fusion and cristae morphology remodeling is necessary for the survival of Tmems and are the underlying mechanisms for their metabolic fitness with increased OXPHOS activity and higher SRC. When mitochondrial fusion is pharmacologically forced in Teffs, they adopt the characteristics of memory phenotype, such as being long-lived, having increased mitochondrial mass, high OXPHOS activity, as well as high SRC. As a result, these enhanced Teffs have been shown to be more effective at eradicating tumors.42 Although these studies aim to improve T-cell immunity for antitumor purposes, they point transplant immunologists to strategies that could be employed to achieve the opposite outcomes: reducing memory phenotype and preventing organ rejection.
SIGNALING PATHWAYS AND T-CELL IMMUNOMETABOLISM
Advances in immunometabolism have linked many intracellular signaling pathways to T-cell metabolism, which has been much more appreciated recently (FIGURE 2).
Figure 2: Signaling Pathways Driving T-cell Metabolism.
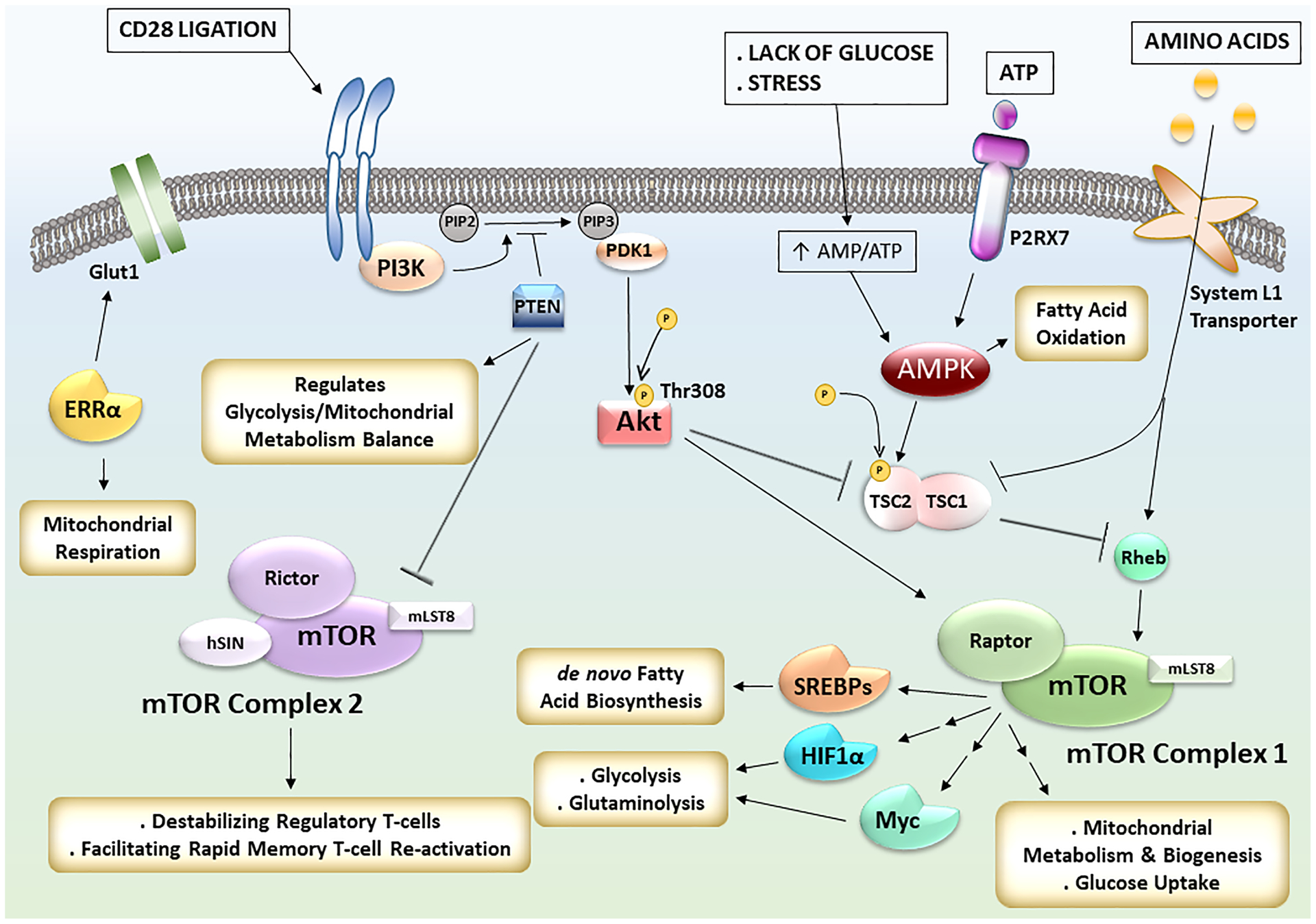
Upon CD28 ligation during T-cell activation, the mTOR pathway is activated through PI3K-Akt axis. mTORC1 regulates mitochondrial metabolism, mitochondrial biogenesis, glucose uptake, and activates the transcriptional factors SREBPs for de novo fatty acid synthesis, HIF1α and Myc for glycolysis and glutaminolysis. Amino acids transported into T-cells by System L1 transporters also contribute to mTORC1 activation. The transcription factor ERRα acts in parallel to increase glucose uptake via Glut1 and regulates mitochondrial respiration. During stress and glucose-limiting conditions, as encountered in the core of the inflammatory microenvironment, AMPK is activated, suppresses mTORC1 pathway, and activates fatty acid oxidation to generate ATPs. PTEN acts as an upstream negative regulator of both mTORC1 and mTORC2. It regulates the balance between glycolysis and mitochondrial metabolism, and its presence facilitates regulatory T-cells stability via mTORC2 inhibition.
1. Important Transcription Factors: Myc, HIF1α, ERRα, and SREBPs
As described above, key metabolic changes during naïve-to-effector transition in T-cells activated in vitro include a significant increase in glycolysis and glutaminolysis as well as a decrease in pyruvate and FAO. In a hallmark study, Wang et al have shown that both Myc and HIF1α are induced within the first 24 hours postactivation when T-cells increase in size but have not yet proliferated. Interestingly, Myc appears to be the main driver for activation-induced glycolysis and glutaminolysis upregulation while HIF1α is not required during the early stage but becomes essential to sustain glycolytic activities after T-cells start to proliferate.51 Indeed, HIF1α deficiency leads to impaired glycolytic activities, prevents Th17 differentiation, and promotes Treg differentiation. Blocking glycolysis achieves the same effect tipping the Th17/ Treg balance.32 At the molecular level, HIF1α directly activates the transcription of RORγt, an indispensable transcription factor of Th17 cells, while targeting FoxP3, a signature transcription factor of Treg, for proteosomal degradation.52 In certain conditions, HIF1α drives differentiation of Treg into a Teff functional phenotype with preserved FoxP3 expression, a concept termed Treg plasticity.53 In response to IL-12 stimulation, Treg become Th1-like, produce IFNγ, and lose their suppressive function.54 Increased glycolysis, driven by HIF1α, has been shown to mediate Treg differentiation into Th1-like phenotype.55
Besides glycolysis and glutaminolysis, mitochondrial respiration is also important for T-cell activation, as mentioned above. Pharmacological inhibition and genetic deficiency studies of the orphan receptor ERRα indicate this protein is necessary for CD4+ T-cell’s glucose uptake via Glut1, pyruvate flux into TCA cycle, and mitochondrial respiration. Interestingly, CD4+ T-cells without ERRα activity appear to switch to FAO for energy production. Adding exogenous lipids into cultures of ERRα-deficient T-cells rescues their differentiation into Treg but not Teffs.56 This presents a therapeutic opportunity to enrich Treg by pharmacologically inhibiting ERRα and adding exogenous lipids at the same time.
Additionally, glycolysis, glutaminolysis, and OXPHOS are shown to be impaired when the signaling pathway mediated by SREBPs is genetically ablated. Normally, the main role of SREBPs is to mediate de novo fatty acid and cholesterol biosynthesis, which is required for proliferating T-cells to generate membrane and expand cellular machinery. Broad deficiencies in metabolism of activated T-cells is observed independently of Myc and HIF1α alteration when activity of SREBPs is compromised, suggesting SREBPs to be important transcription factors for T-cell activation.28
2. mTOR Pathway
As described above, Myc, HIF1α, SREBPs are important transcription factors that regulate T-cell metabolism. Upstream to these transcription factors is the mTORC1, a central component of the mTOR network that plays cardinal roles in cellular physiology.8,32 Evolutionarily conserved, the mTOR pathway is comprised of 2 complexes, mTORC1 and mTORC2, with the former being the most elaborated to date. Both complexes contain the serine/threonine kinase mTOR and a specific adaptor protein, Raptor for mTORC1 and Rictor for mTORC2. The canonical upstream signaling of mTORC1 is through the PI3K-PDK1-Akt axis. Upon phosphorylation by PDK1, Akt directly or indirectly activates mTORC1, which then phosphorylates its downstream effectors and initiates many cellular programs, including glycolysis, pentose phosphate pathway, glutaminolysis, lipid biosynthesis, ribosome biogenesis and protein translation. mTORC1 activity can also be activated independently from PI3K-Akt axis via amino acids or can be suppressed in hypoxic and glucose-limited conditions via AMPK.8,9 The signaling of mTORC2 has been less vigorously characterized than mTORC1.
In an elegant study, Yang et al showed that both CD4+ and CD8+ T-cells require mTORC1 for their activation, proliferation, and effector functions using T-cells isolated from Raptor and Rictor conditional knockout mouse models. More specifically, mTORC1 is necessary for T-cells to exit quiescence, enter the cell cycle, and to initiate metabolic reprogramming to increase glycolysis, lipid biosynthesis, and OXPHOS.57 During T-cell activation, CD28 costimulation leads to an increase in surface Glut1, glucose uptake, and glycolytic flux. This observation has been linked to the activation of PI3K-Akt pathway and thus with mTORC1 activation as a consequence of CD28 ligation.23 Also, as described, cellular demand for many amino acids are increased during T-cell activation.27,58 Notably, TCR and IL-2 signaling leads to an increase in transportation of large neutral amino acids, including leucine, using System L1 transporters to activate mTORC1 and its downstream transcription factor Myc.58 Leucyl-tRNA synthetase, an enzyme that normally couples leucine to its corresponding tRNA, has a noncanonical role facilitating the activation of mTORC1 in the presence of leucine.59 Together, these studies illustrate a close relationship between molecular events occurring during T-cell activation and mTORC1 signaling for metabolic reprogramming.
Lastly, the relationship between mTOR signaling and Treg has been established in transplantation. Shrestha et al showed that the stability of Treg require PTEN, a negative regulator of PI3K-Akt axis, and more importantly deletion of this protein increases glycolysis while decreases OXPHOS, mitochondrial mass, mitochondrial membrane potentials, and ROS formation, suggesting PTEN to be the metabolic checkpoint to deter Teffs metabolic mode and to maintain preferred metabolism of Treg. Interestingly, loss of PTEN activity increases mTORC2 activity and destabilizes Treg, whereas genetic deletion of Rictor rescues Treg defects.60 In other words, mTORC2 likely acts as a negative regulator of mitochondrial metabolism that is predominantly used in both memory and Treg phenotypes. Furthermore, recent studies have demonstrated that mTORC2 plays a significant role in rapid reactivation of CD8+ Tmems. Through a cascade of events, mTORC2 increases localization of hexokinase-1, an important glycolysis enzyme, to mitochondrial voltage-dependent anion channel, facilitating pyruvate influx into mitochondria to satisfy metabolic demands for Tmem reactivation.61 With mTORC2 both destabilizing Treg and facilitating rapid reactivation of Tmems, a specific inhibitor for this complex could potentially favor immune tolerance in SOT.
3. AMPK Signaling
Part of the mTOR network, AMPK acts as an upstream negative regulator of mTORC1. During stress and glucose-limited conditions, high AMP/ATP ratio activates AMPK to suppress energy-consuming processes mediated by mTORC1, such as lipid biosynthesis, and to facilitate energy-generating processes, such as FAO.9 In T-cells, AMPK plays an important role in facilitating the formation of Treg and Tmems via promoting FAO, which is a preferred metabolic mode of these 2 types of T-cells.15,62 Recently, AMPK has been shown to regulate the metabolic plasticity of Teffs in vivo. In high-glucose media which is commonly used in most in vitro culture systems, AMPK is dispensable for Teffs as nutrients are in excess, and Teffs rely on the Warburg effect for cellular demands. However, when glucose level is at the physiological concentration, or when glucose is limited, such as within the tumor or inflammatory zone, AMPK activity increases T-cell mitochondrial bioenergetics, glucose uptake, and especially increases glutaminolysis to replenish TCA cycle intermediates and to generate pyruvate for ATP production via OXPHOS. Consistent with this view, genetic deletion of AMPK impairs effector T-cell response in vivo due to inability to adapt to metabolic stress.63
Furthermore, AMPK is the main mediator in P2RX7 signaling that is cardinal in formation and maintenance of CD8+ Tmems, as described above. In transplantation, after cold storage and ischemia-reperfusion injury, dying cells of injured donor tissues release ATPs as a form of danger signals. Healthy T-cells also release ATPs into the extracellular environment via P2RX7-mediated PANX1 channels. These ATPs activate P2RX7 and initiate a signaling cascade that activates AMPK, which drives FAO. Pharmacologic inhibition and genetic deletion of P2RX7 leads to accumulation of ATPs inside CD8+ Tmems, resulting in a decrease in AMP/ATP ratio and consequently AMPK inactivation. Consequently, mitochondrial dysregulation due to a loss of AMPK-mediated downstream effects destabilizes Tmems.49
T-CELL METABOLIC REPROGRAMMING IN SOLID-ORGAN TRANSPLANTATION
1. Effects of Current Immunosuppressive Agents on T-cell Metabolism
Given what is known about T-cell metabolism, metabolically reprogramming T-cells could be a fruitful therapeutic strategy in SOT. The current standard of care mandates that during and after the operation, patients are administered a variety of immunosuppressive drugs to suppress T-cell-mediated alloimmune rejection. The main classes of maintenance drugs include steroids, calcineurin inhibitors (such as tacrolimus), mTOR inhibitors (sirolimus and rapalogs), and mycophenolic acid (MPA).64,65 While these drugs have different targets and pharmacologic mechanisms (TABLE 2), they modulate T-cell metabolism via interacting with key components of the important signaling pathways. The most apparent examples are sirolimus and rapalogs that suppress mTOR activity.64 As mTORC1 is upstream to HIF1α, Myc, and SREBPs that control glycolysis, glutaminolysis, lipid biosynthesis,8,28,32,51 inhibiting mTORC1 blocks all metabolic pathways necessary for naïve T-cell activation as well as reactivation of Tmems. Besides, inhibiting mTOR pathway also promotes Treg which plays a key role in promoting graft tolerance and suppressing transplant arteriosclerosis.66,67 In addition, as described above, amino acid transportation via System L1 transporters is mediated by TCR signaling and necessary for mTORC1 activation. Treatment of T-cells with cyclosporin A impairs the activity of these transporters via blocking TCR signaling and thus incidentally mediates the same effects as sirolimus and the rapalogs.58
Table 2:
Summary of Molecules That Potentially Impact T-cell Immunometabolism
| MOLECULES POTENTIALLY IMPACTING T-CELL IMMUNOMETABOLISM | |
|---|---|
| Molecule | Impact |
| sirolimus and rapalogs | inhibit glycolysis, glutaminolysis, lipid biosynthesis |
| cyclosporin A | block amino acid import via System L1 transporters |
| calcineurin inhibitors | inhibit glycolysis, glutaminolysis |
| anti-System L1 transporters antibodies | block amino acid import via System L1 transporters |
| metformin | facilitate fatty acid oxidation, inhibit mitochondrial complex I |
| soraphen A | inhibit de novo fatty acid synthesis |
| P2RX4 and P2RX7 inhibitors | block purinergic autocrine signaling mediated by ATP |
| 2-DG | block glycolysis, pentose phosphate pathway |
| 6-diazo-5-oxo-L-norleucine | block glutaminolysis |
| M1 | promote mitochondrial fusion |
| Mdivi1 | inhibit mitochondrial fission |
Cyclosporin A and other calcineurin inhibitors, such as tacrolimus, have the ability to block NFAT activation, a common pathway triggered by CD3 complex signaling during T-cell activation.64,65 In pancreatic cancer cells, NFAT is known to facilitate the transcription of Myc by binding to its promoter.68 Because Myc is important for glycolysis and glutaminolysis,51 this class of drugs may inhibit these 2 important metabolic processes in T-cells by blocking the signaling pathway underlying them. Similarly, the expression of HIF1α is upregulated via NFAT-dependent mechanisms in mast cells upon stimulation by ionomycin, and this process is sensitive to both cyclosporin A and tacrolimus.69 In T-cells, HIF1α not only sustains glycolysis after clonal expansion begins but also promotes Th17 differentiation while inhibiting Treg formation.32,51,52 Therefore, if in T-cells, HIF1α is similarly upregulated by NFAT pathway, and is also sensitive to cyclosporin A and tacrolimus, the calcineurin inhibitors may modulate T-cell metabolism toward tolerogenic induction through these mechanisms.
Taken together, the current immunosuppressive agents may reduce allograft rejection by modulating cellular pathways affecting T-cell metabolism. Yet, these agents are known to cause multiple side effects in patients and lower both quality of life and compliance rate.64,65 As activated T-cells have been viewed by many immunometabolism researchers to metabolically behave like tumor cells, a metabolism-targeted approach may offer advantages by selectively targeting the preferred metabolic mode of alloreactive T-cells, thus inhibiting their alloimmune function, while sparing or promoting Treg development. This leads to the question, what strategies could we use to target T-cell metabolism specifically in transplantation?
2. Potential Strategies to Modulate T-cell Metabolism
Activity of the System L1 amino-acid transporters is increased during T-cell activation to transport leucine and other amino acids into cells and activate mTORC1.58 By targeting these transporters with specific antibodies and hence blocking the amino acid influx, cardiac allograft survival can be prolonged.70 From this study, it could be envisioned that using antibodies targeting Glut1 or glutamine transporters in solid-organ transplant models would achieve the same effects, as glucose and glutamine are required for T-cell activation, proliferation and functions.15,22,27
In addition, in the experimental autoimmune encephalomyelitis (EAE) model, genetic deletion of HIF1α protects mice and improves their pathological conditions by skewing the Th17/ Treg balance.32,52 Importantly, HIF1α is a pro-oncogenic protein that has been linked to mediating metabolic adaptation of tumor cells in hypoxic microenvironment and thus is a therapeutic target of considerable interest.71 Thus, targeting this protein in the solid-organ transplant context would yield dual effects: blocking glycolysis of alloreactive T-cells and preventing immunosuppression-associated development of malignancy.
Furthermore, currently Tmems are still a major challenge in SOT because they are minimally affected by the current immunosuppressive agents.72 One common feature between Tmems and Treg is that they both utilize FAO as the main mode for energy production.15,44,46 Yet, their source of fatty acids is different, with Tmems relying on de novo biosynthesis while Treg relying on exogenous uptake.29,46 Importantly, Teffs also need to synthesize lipids for membrane expansion and cellular machinery generation.28 Such difference in sources of fatty acids between Treg and Teffs, Tmems presents a unique therapeutic opportunity to modulate T-cell metabolism. A dual drug regimen to facilitate FAO (such as using metformin) while simultaneously inhibiting de novo fatty acid biosynthesis (such as using soraphen A) is a promising strategy to selectively promote Treg while suppressing both Tmems and Teffs. In fact, treatment of EAE with soraphen A alone improves outcomes by preferentially promoting Treg over Th17 cells.29 Thus, such strategy could potentially be applied to SOT. Another strategy to target Tmems includes pharmacologic inhibition of the purinergic receptor P2RX7. As described, genetic deletion or pharmacologic inhibition of P2RX7 leads to destabilization of CD8+ Tmems, and more importantly, favors the Treg stability.40,49 Currently, inhibitors of P2RX7 have been advanced to clinical trials to reduce neuropathic pain, and thus, targeting P2RX7 and other purinergic signaling, such as P2RX4, could be a potentially successful avenue to reduce allograft rejection.50,73 As a proof of concept, in a mouse lung transplant model, treatment with P2RX4 receptor antagonists decreases CD4+ T-cell recruitment and infiltration into the lung allografts, improving posttransplant respiratory function.50
As T-cells in a certain functional state prefer a specific metabolic mode to others, modulating metabolism using combined drugs may deliver synergistic effects. Indeed, administering both glycolysis inhibitor 2-DG and AMPK activator metformin reverses disease markers by suppressing cytotoxic functions of T-cells in systemic lupus erythematosus mice.74 Additionally, inhibiting glycolysis while simultaneously depleting intracellular leucine and activating AMPK can successfully block T-cell activation.11
In the context of SOT, in vivo success has been achieved using synergistic drugs to modulate T-cell metabolism. In 1 of the first and few studies that target T-cell immunometabolism in SOT, using triple-drug regimen: 2-DG to inhibit glycolysis, metformin to activate AMPK, and 6-diazo-5-oxo-L-norleucine to inhibit glutaminolysis, Lee et al successfully delayed mouse skin graft rejection and prolonged cardiac allograft survival. Importantly, this study has shown that a triple-drug therapy is able to synergistically reduce IFNγ production, T-cell proliferation, decrease de novo fatty acid biosynthesis in vitro as well as to skew the Teffs/Treg balance toward Treg.75 This study is an exciting proof-of-concept demonstrating that T-cell immunometabolism can and perhaps should be exploited to achieve transplant tolerance. Further investigation, however, is still needed to dissect detailed mechanisms of the impact of these molecules on T-cell metabolism, as many have indirect and/or off-target effects. For example, 2-DG inhibition of glucose metabolism could increase AMP/ATP ratio, therefore activating AMPK signaling,9 and besides glycolysis, 2-DG also inhibits the pentose phosphate pathway.33 Metformin is also known to inhibit mitochondrial complex I.76 The promising results achieved by these molecules despite their indirect and/or off-target effects reflect an intricate metabolic network that presents potentials to be harnessed for clinical applications.
CONCLUDING REMARK
As the field of immunology has recently witnessed many advances in the mechanistic understanding of T-cell metabolism, such knowledge can be applied to SOT to improve transplant outcomes. While current immunosuppressive agents are helpful in reducing alloimmune rejection and increasing survival, modulating T-cell metabolism may offer advantages in selectively eliminating alloreactive Teffs and Tmems while promoting Treg. T-cell immunometabolism in SOT may still be in its infancy, but the field is growing and success in achieving organ tolerance could well be within reach in the near future.
Financial Disclosure:
NIH/NIAID R01 AI142079 01A1 (S.N.)
NIH/NHLBI R01 HL140470 (C.A.)
NIH/NIBIB K08 EB019495-01A1 (S.N.)
NIH/NHLBI Predoctoral Fellowship T32 HL007260 (D.T.)
NIH/NIGMS MSTP Predoctoral Fellowship T32 GM008716 (D.T.)
Patterson Barclay Memorial Foundation (S.N., C.A.)
Abbreviations:
- ECAR
extracellular acidification rate
- ETC
electron transport chain
- FAO
fatty acid oxidation
- FAS
fatty acid synthesis
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- iTreg
induced regulatory T-cells
- mTOR
mechanistic target of rapamycin
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- SOT
solid-organ transplantation
- SRC
spare respiratory capacity
- TAG
triacylglycerol
- TCA
tricarboxylic acid
- TCR
T-cell receptor
- Teffs
effector T-cells
- Th1
T-helper 1
- Th2
T-helper 2
- Th17
T-helper 17
- Tmems
memory T-cells
- Treg
regulatory T-cells
Footnotes
Disclaimer: The authors declare no conflicts of interest.
REFERENCES
- 1.Raigani S, Karimian N, Huang V, et al. Metabolic and lipidomic profiling of steatotic human livers during ex situ normothermic machine perfusion guides resuscitation strategies. PLoS One. 2020;15(1):e0228011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minor T, von Horn C, Paul A. Role of temperature in reconditioning and evaluation of cold preserved kidney and liver grafts. Curr Opin Organ Transplant. 2017;22(3):267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garonzik-Wang JM, Lonze BE, Ruck JM, et al. Mitochondrial membrane potential and delayed graft function following kidney transplantation. Am J Transplant. 2019;19(2):585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran DT, Esckilsen S, Mulligan J, et al. Impact of mitochondrial permeability on endothelial cell immunogenicity in transplantation. Transplantation. 2018;102(6):935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wedel J, Stack MP, Seto T, et al. T cell-specific adaptor protein regulates mitochondrial function and CD4(+) T regulatory cell activity in vivo following transplantation. J Immunol. 2019;203(8):2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Kumar A, Carmeliet P. Metabolic pathways fueling the endothelial cell drive. Annu Rev Physiol. 2019;81:483–503. [DOI] [PubMed] [Google Scholar]
- 8.Duvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15(3):155–162. [DOI] [PubMed] [Google Scholar]
- 10.Le Bourgeois T, Strauss L, Aksoylar HI, et al. Targeting T cell metabolism for improvement of cancer immunotherapy. Front Oncol. 2018;8:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Delgoffe GM, Meyer CF, et al. Anergic T cells are metabolically anergic. J Immunol. 2009;183(10):6095–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hume DA, Radik JL, Ferber E, et al. Aerobic glycolysis and lymphocyte transformation. Biochem J. 1978;174(3):703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krauss S, Brand MD, Buttgereit F. Signaling takes a breath--new quantitative perspectives on bioenergetics and signal transduction. Immunity. 2001;15(4):497–502. [DOI] [PubMed] [Google Scholar]
- 15.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CH, Curtis JD, Maggi LB Jr, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174(8):4670–4677. [DOI] [PubMed] [Google Scholar]
- 20.Cham CM, Driessens G, O’Keefe JP, et al. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38(9):2438–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerriets VA, Kishton RJ, Nichols AG, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125(1):194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769–777. [DOI] [PubMed] [Google Scholar]
- 24.Sena LA, Li S, Jairaman A, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailis W, Shyer JA, Zhao J, et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature. 2019;571(7765):403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. [DOI] [PubMed] [Google Scholar]
- 27.Carr EL, Kelman A, Wu GS, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185(2):1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kidani Y, Elsaesser H, Hock MB, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. 2013;14(5):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berod L, Friedrich C, Nandan A, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nature Med. 2014;20(11):1327–1333. [DOI] [PubMed] [Google Scholar]
- 30.Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. 2014;259(1):88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi LZ, Wang R, Huang G, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanimine N, Germana SK, Fan M, et al. Differential effects of 2-deoxy-D-glucose on in vitro expanded human regulatory T cell subsets. PLoS One. 2019;14(6):e0217761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eleftheriadis T, Pissas G, Karioti A, et al. Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. J Basic Clin Physiol Pharmacol. 2013;24(4):271–276. [DOI] [PubMed] [Google Scholar]
- 35.De Rosa V, Galgani M, Porcellini A, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. 2015;16(11):1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beier UH, Angelin A, Akimova T, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J. 2015;29(6):2315–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. 2017;17(12):774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinberg SE, Singer BD, Steinert EM, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565(7740):495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu Z, Ye J, Dean JW, et al. Requirement of mitochondrial transcription factor A in tissue-resident regulatory T cell maintenance and function. Cell Rep. 2019;28(1):159–171.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schenk U, Frascoli M, Proietti M, et al. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal. 2011;4(162):ra12. [DOI] [PubMed] [Google Scholar]
- 41.Li YH, Xu F, Thome R, et al. Mdivi-1, a mitochondrial fission inhibitor, modulates T helper cells and suppresses the development of experimental autoimmune encephalomyelitis. J Neuroinflammation. 2019;16(1):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buck MD, O’Sullivan D, Klein Geltink RI, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166(1):63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masopust D, Kaech SM, Wherry EJ, et al. The role of programming in memory T-cell development. Curr Opin Immunol. 2004;16(2):217–225. [DOI] [PubMed] [Google Scholar]
- 44.van der Windt GJW, Everts B, Chang C-H, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fraser KA, Schenkel JM, Jameson SC, et al. Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity. 2013;39(1):171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Sullivan D, van der Windt GJW, Huang SC-C, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41(1):75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Windt GJW, O’Sullivan D, Everts B, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A. 2013;110(35):14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gubser PM, Bantug GR, Razik L, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14(10):1064–1072. [DOI] [PubMed] [Google Scholar]
- 49.Borges da Silva H, Beura LK, Wang H, et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature. 2018;559(7713):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ledderose C, Liu K, Kondo Y, et al. Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J Clin Invest. 2018;128(8):3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dang EV, Barbi J, Yang HY, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arterbery AS, Osafo-Addo A, Avitzur Y, et al. Production of proinflammatory cytokines by monocytes in liver-transplanted recipients with de novo autoimmune hepatitis is enhanced and induces TH1-like regulatory T cells. J Immunol. 2016;196(10):4040–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kitz A, de Marcken M, Gautron AS, et al. AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO reports. 2016;17(8):1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JH, Elly C, Park Y, et al. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity. 2015;42(6):1062–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michalek RD, Gerriets VA, Nichols AG, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U S A. 2011;108(45):18348–18353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang K, Shrestha S, Zeng H, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. 2013;39(6):1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sinclair LV, Rolf J, Emslie E, et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han JM, Jeong SJ, Park MC, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149(2):410–424. [DOI] [PubMed] [Google Scholar]
- 60.Shrestha S, Yang K, Guy C, et al. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16(2):178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bantug GR, Fischer M, Grählert J, et al. Mitochondria-endoplasmic reticulum contact sites function as immunometabolic hubs that orchestrate the rapid recall response of memory CD8(+) T cells. Immunity. 2018;48(3):542–555.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blagih J, Coulombe F, Vincent EE, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42(1):41–54. [DOI] [PubMed] [Google Scholar]
- 64.Watson CJ, Dark JH. Organ transplantation: historical perspective and current practice. Br J Anaesth. 2012;108 Suppl 1:i29–i42. [DOI] [PubMed] [Google Scholar]
- 65.Fisher JD, Acharya AP, Little SR. Micro and nanoparticle drug delivery systems for preventing allotransplant rejection. Clin Immunol. 2015;160(1):24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105(12):4743–4748. [DOI] [PubMed] [Google Scholar]
- 67.Hester J, Schiopu A, Nadig SN, et al. Low-dose rapamycin treatment increases the ability of human regulatory T cells to inhibit transplant arteriosclerosis in vivo. Am J Transplant. 2012;12(8):2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buchholz M, Schatz A, Wagner M, et al. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25(15):3714–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walczak-Drzewiecka A, Ratajewski M, Wagner W, et al. HIF-1alpha is upregulated in activated mast cells by a process that involves calcineurin and NFAT. J Immunol. 2008;181(3):1665–1672. [DOI] [PubMed] [Google Scholar]
- 70.Liu Z, Hou J, Chen J, et al. Deletion of CD98 heavy chain in T cells results in cardiac allograft acceptance by increasing regulatory T cells. Transplantation. 2012;93(11):1116–1124. [DOI] [PubMed] [Google Scholar]
- 71.Paolicchi E, Gemignani F, Krstic-Demonacos M, et al. Targeting hypoxic response for cancer therapy. Oncotarget. 2016;7(12):13464–13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brook MO, Wood KJ, Jones ND. The impact of memory T cells on rejection and the induction of tolerance. Transplantation. 2006;82(1):1–9. [DOI] [PubMed] [Google Scholar]
- 73.Alegre ML. Danger, metabolism and T cell memory. Am J Transplant. 2018;18(10):2375. [Google Scholar]
- 74.Yin Y, Choi SC, Xu Z, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee CF, Lo YC, Cheng CH, et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep. 2015;13(4):760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fontaine E Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front Endocrinol (Lausanne). 2018;9:753. [DOI] [PMC free article] [PubMed] [Google Scholar]