

Abstract
The Hap2,3,4,5p transcription complex is required for expression of many mitochondrial proteins that function in electron transport and the tricarboxylic acid (TCA) cycle. We show that as the cells’ respiratory function is reduced or eliminated, the expression of four TCA cycle genes, CIT1, ACO1, IDH1, and IDH2, switches from HAP control to control by three genes, RTG1, RTG2, and RTG3. The expression of four additional TCA cycle genes downstream of IDH1 and IDH2 is independent of the RTG genes. We have previously shown that the RTG genes control the retrograde pathway, defined as a change in the expression of a subset of nuclear genes, e.g., the glyoxylate cycle CIT2 gene, in response to changes in the functional state of mitochondria. We show that the cis-acting sequence controlling RTG-dependent expression of CIT1 includes an R box element, GTCAC, located 70 bp upstream of the Hap2,3,4,5p binding site in the CIT1 upstream activation sequence. The R box is a binding site for Rtg1p-Rtg3p, a heterodimeric, basic helix-loop-helix/leucine zipper transcription factor complex. We propose that in cells with compromised mitochondrial function, the RTG genes take control of the expression of genes leading to the synthesis of α-ketoglutarate to ensure that sufficient glutamate is available for biosynthetic processes and that increased flux of the glyoxylate cycle, via elevated CIT2 expression, provides a supply of metabolites entering the TCA cycle sufficient to support anabolic pathways. Glutamate is a potent repressor of RTG-dependent expression of genes encoding both mitochondrial and nonmitochondrial proteins, suggesting that it is a specific feedback regulator of the RTG system.
Cells reconfigure their pattern of gene expression to accommodate changes in nutrient availability. Often, this is seen as an induction of genes that enable cells to utilize certain nutrients or as a repression of those genes when such nutrients are no longer available. Cells also reconfigure patterns of gene expression in response to different stress conditions, for instance, heat shock or osmotic stresses, as a mechanism of protection against those environmental insults. In cells of the budding yeast Saccharomyces cerevisiae, dramatic changes in gene expression are observed when cells switch from fermentative to oxidative metabolism, as in the diauxic shift, when cells growing on glucose—a repressing carbon source—begin to deplete the glucose from the medium (8). The expression of many nucleus-encoded mitochondrial proteins, such as components of the electron transport chain and enzymes of the tricarboxylic acid (TCA) cycle, become derepressed during the diauxic shift. The derepression of many of these proteins requires either Hap1p, an oxygen-sensing transcriptional activator (6, 29), the heteromeric Hap2,3,4,5p transcriptional complex (10, 11, 24, 26, 27, 30, 43), or both of these trans-acting regulators. Thus, the HAP system represents an important mechanism for global control of expression of key components of respiratory metabolism.
Yeast cells also modulate the expression of nuclear genes in response to mitochondrial dysfunctions (28). This interorganelle signaling pathway, called retrograde regulation, can be thought of as a stress response whose function is to accommodate various cellular activities to the changes in the mitochondrial state (39). The best-studied example of the retrograde response is that of the CIT2 gene, whose expression is sensitive to conditions or mutations that compromise mitochondrial functions, such as inhibition of respiration, loss of TCA cycle activity, or loss of mitochondrial DNA (3, 18, 19). Recently, we found a new cytosolic d-lactate dehydrogenase activity encoded by a previously uncharacterized gene, YEL071w, now named DLD3, that shows a similar retrograde response as CIT2, namely, increased expression in cells with dysfunctional mitochondria (4). CIT2 encodes a peroxisomal isoform of citrate synthase (CS2) that functions in the glyoxylate cycle; CS2 shares 75% sequence similarity with the mitochondrial citrate synthase (CS1) encoded by the CIT1 gene, suggesting that CIT1 and CIT2 arose by a duplication of some ancestral citrate synthase gene. In cells with compromised mitochondrial functions, for example, those without mitochondrial DNA ([rho0] petites), CIT2 expression is elevated by as much as 30- to 40-fold (19). Physiological studies suggest that this increase in CIT2 expression facilitates a more efficient utilization of carbon via the transfer of metabolites from the glyoxylate cycle to the TCA cycle (40). In contrast to CIT2, the level of CIT1 expression is unaffected by the loss of mitochondrial DNA (18).
The expression of CIT2 and DLD3 is dependent under all conditions tested on three genes, RTG1, RTG2, and RTG3. RTG1 and RTG3 encode basic helix-loop-helix/leucine zipper (bHLH/Zip) transcription factors that bind as a heterodimer to activate transcription to a novel DNA target site, GTCAC, called an R box (14). CIT2 and DLD3 contain two R boxes in their 5′ noncoding regions, both of which are required for maximal gene expression. RTG2 encodes a novel cytoplasmic protein that has an amino-terminal ATP binding domain similar to the hsp70/actin/sugar kinase superfamily of ATP binding proteins (2). Rtg2p also shares some sequence similarity with bacterial polyphosphatases and enzymes that hydrolyze guanosine tetra- and pentaphosphate (16). Although the precise function of Rtg2p is unknown, genetic data suggest that it acts upstream of Rtg1p and Rtg3p in the control of gene expression (36). The CIT2-DLD3 retrograde response appears to be controlled by an Rtg2p-dependent redistribution of the Rtg1p-Rtg3p transcriptional complex from a predominantly cytoplasmic location in cells with robust mitochondrial function to a nuclear location in cells whose mitochondrial functions have been compromised, such as in [rho0] petites (38).
None of the RTG genes are essential, nor are they required for growth of cells on some nonfermentable carbons sources. Although the RTG genes are required for both basal and retrograde expression of CIT2 and DLD3, two unexpected phenotypes of the rtg mutants were observed: an inability of cells to grow on acetate, and a growth requirement for glutamate and aspartate on minimal glucose medium (18). These phenotypes are characteristic of cells with blocks in both the TCA and glyoxylate cycles. The inability to grow on acetate is a common phenotype of cells lacking TCA cycle enzymes (23), and cit1 cit2 and aco1 mutants are glutamate auxotrophs (12, 15). These observations suggest a potential defect in the TCA cycle in rtg mutant cells. Biochemical experiments have suggested that rtg mutant cells may have multiple and cumulative lesions in the TCA cycle that impair the cells’ ability to grow on acetate medium (40). Consistent with this conclusion, it has been shown that RTG2 is required for expression of the ACO1 gene in glucose-repressed cells (41).
Here we report that expression of the genes encoding the first three steps of the TCA cycle leading to the synthesis of α-ketoglutarate, CIT1, ACO1, IDH1, and IDH2, switches between a dependence on the HAP genes in cells with robust mitochondrial function to the RTG genes in cells whose mitochondrial respiratory capacity has been reduced or eliminated. The remaining TCA cycle genes tested—all of which encode enzymes catalyzing steps downstream of isocitrate dehydrogenase—have no dependence on the RTG genes for their expression in either derepressed [rho+] (respiratory competent) wild-type cells or [rho0] petites. We analyze the control of CIT1 expression in detail and show that it contains a functional R box in the 5′ flanking region of the gene that is required for Rtg1p-Rtg3p-dependent expression. We propose that the RTG control of genes encoding the first three enzymes of the TCA cycle leading to the synthesis of α-ketoglutarate is to ensure that sufficient glutamate is made for biosynthetic processes in cells with reduced respiratory capacity. Finally, we show that glutamate is a potent repressor of RTG-dependent gene expression, suggesting an important feedback regulation of glutamate synthesis. Like the HAP genes, which are responsible for a global control of gene expression in derepressed, respiratory competent cells, the RTG genes represent a major control pathway of gene expression in cells with reduced or compromised mitochondrial function.
MATERIALS AND METHODS
Growth media and growth conditions.
Yeast strains were grown at 30°C in YP medium (1% yeast extract, 2% Bacto Peptone) with 2% raffinose (YPR) or 2 or 5% glucose (YPD or YP5%D, respectively); in YNB medium (0.67% yeast nitrogen base) supplemented with 1% Casamino Acids, leucine (30 mg/liter), lysine (30 mg/liter), and 2 or 5% dextrose (YNBcasD or YNBcas5%D), 2% raffinose (YNBcasR), 2% potassium acetate (YNBcasAce, pH adjusted to 5.5), or 2% glycerol (YNBcasGly); or in minimal YNB medium (0.67% yeast nitrogen base without Casamino Acids) supplemented with leucine (30 mg/liter), lysine (30 mg/liter), uracil (20 mg/liter), and 2 or 5% glucose (YNBD or YNB5%D) or 2% raffinose (YNBR), with or without sodium glutamate (concentrations indicated in the text and figures).
Strains.
Yeast strains used in this study were PSY142 (MATα leu2 lys2 ura3 [rho+]) and its derivatives constructed as follows. To construct the rtg1Δ derivative, a 674-bp HindIII-SstI fragment of RTG1 was replaced with a 1.2-kb XhoI-HindIII fragment of the URA3 gene (18). Ura− derivatives were obtained by selection with 5-fluro-orotic acid. An rtg2Δ derivative was constructed by replacing a SalI-XbaI fragment of RTG2 with a 2.2-kb fragment of the LEU2 gene, thus deleting codons 23 to 573 of RTG2 (37). To construct an rtg3Δ derivative, codons 175 to 340 of RTG3 were replaced with a 1.6-kb fragment of the LEU2 gene (14). [rho0] derivatives of these strains were generated by several passages of [rho+] cells in YPD medium supplemented with 25 μg of ethidium bromide per ml. The hap2Δ deletion was obtained by replacing bp +44 to +686 of the HAP2 gene with the kanMX4 cassette (42). The hap2Δ deletion was introduced into the wild-type and Δrtg1, Δrtg2, and Δrtg3 mutant strains. The cit1Δ and cit2Δ deletion mutants were described previously (18, 19, 40).
Plasmid constructs.
The DNA region from −806 to +9 of CIT1 was amplified by PCR. The forward primer contained a 5′ EcoRI restriction site and reverse primer contained a 5′ HindIII restriction site. The CIT1 amplified product was inserted into the EcoRI-HindIII site of YIp356 to produce pCIT1-LacZ. pCIT1-LacZ was linearized by digestion with KpnI and integrated into the genomic CIT1 locus by standard yeast transformation procedures (32). The pACT1-LacZ plasmid was constructed similarly by inserting the PCR-amplified DNA region of ACT1 from −667 to +9 into the HindIII-EcoRI site of YIp356R. The pACT1-LacZ plasmid was linearized by digestion with NcoI and integrated into the URA3 locus. The pCIT2-LacZ and pDLD3-LacZ reporter gene constructs were described previously (4, 18). To make an R box mutant construct of pCIT1-LacZ, an internal pair of primers, 5′-gtacACGCGTTTTTTTCCGCCGCAG-3′ and 5′-gtacACGCGTCGCCTTTTAGCACAAAAATG-3′, (lowercase letters indicate plasmid sequence), was used to introduce two point mutations into the R box, changing GTCAC to GACGC. To construct pCIT1(UAS [upstream activation sequence])-CYC1-LacZ and pCIT1(R)(UAS)-CYC1-LacZ, a pair of primers, 5′-gactaagcttTGTATTTACCTTGCATTT-3′ and 5′-gactctcgagGGAAAAGCTCCAAAGGG-3′, (underlining indicates restriction sites), was used to amplify the −400 to −260 region of CIT1 in pCIT1-LacZ and pCIT1(R)-LacZ, respectively. The resulting amplified products were cleaved by HindIII and XhoI and inserted into the multiple cloning region of pWCJ100, which contains the minimal promoter of CYC1 fused to lacI-lacZ coding sequence. Plasmid pWCJ100 is a centromere-based vector derived from pKM270. pACT1-CIT1 was constructed by fusing the ACT1 region from −457 to −152 to the CIT1 region from −170 to +1990 in plasmid YIp352. pACT1-CIT1 was linearized by digestion with ApaI and integrated into the URA3 locus.
Electrophoretic mobility shift assay (EMSA).
Whole-cell extracts were prepared as described previously (18) except that [rho0] cells were used. Cells were grown in YNB5%D medium containing the necessary nutritional supplements and including 0.01% glutamic acid. A 140-bp DNA sequence from −400 to −260 of the CIT1 gene amplified by PCR was used as a probe. The resulting PCR product was gel purified and end labeled with [γ-32P]ATP by using T4 polynucleotide kinase. The reaction mixture contained 25 mM Tris-HCl (pH 7.5), 5 mM MgCl, 0.125 mM EDTA, 300 mM KCl, 10% glycerol, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 μg of aprotinin per ml, 0.5 μg of leupeptin per ml, 1 ng of 32P-labeled probe, 1 μg of salmon sperm DNA, and 40 μg of cell extract in a final volume of 20 μl. After 20 min of incubation at room temperature, the reaction mixture was applied to a 4% acrylamide gel (40:1 in 0.5× Tris-borate-EDTA buffer) prerun for 30 min and then run for 2.3 h at 4°C. The gel was dried and exposed to X-ray film.
Yeast transformation and β-galactosidase assays.
Yeast cells were transformed as described by Chen et al. (5). Transformants carrying the desired plasmids were selected on YNBcasD plates. Liquid precultures were inoculated with a pool of several independent transformants and grown in YNBcasD medium. Cells were collected by centrifugation and diluted into YNBcas5%D or into YNBcasR medium and grown overnight to an optical density at 600 nm of ∼2.0. Cells were collected by centrifugation, diluted into corresponding fresh medium, and collected at an optical density at 600 nm of ∼0.8 after 9 h growth at 30°C. For glutamate repression analysis, YNB5%D and YNBR media were used. The preparation of cell extracts and β-galactosidase assays were carried out as described by Rose et al. (33). For each plasmid-strain combination, assays were conducted in triplicate and independent experiments were carried out two to three times.
RNA isolation and Northern blot analysis.
Total yeast RNA was isolated from 50- to 200-ml logarithmic-phase cultures, fractionated on 1.2% agarose gels, transferred to Nytran Plus, and hybridized at 65°C with probes specific for transcripts of the CIT2 and ACT1 genes as previously described (14). The region from +263 to +1344 of CIT1 was purified by cleaving a plasmid containing that sequence with ClaI and StuI and used as probe for CIT1 transcripts. The remaining probes were PCR amplified from selected coding regions of the genes of interest, using genomic DNA as a template. The PCR products were gel purified by using a GenecleanII kit from Bio 101 (Vista, Calif.). The regions amplified were −380 to +2855 of ACO1, −54 to +1237 of IDH1, −53 to +1279 of IDH2, −59 to +3226 of KGD1, −126 to +2171 of SDH1, and −63 to +1660 of FUM1. Hybridization signals were quantified with a Molecular Dynamics PhosphorImager.
RESULTS
Alternative dependence of expression of a CIT1-lacZ reporter gene on HAP2 and RTG1, RTG2, and RTG3.
To examine the role of the RTG genes in TCA cycle gene expression, we first investigated the regulation of CIT1 expression by using a reporter gene construct in which 806 bp of the 5′ flanking region of CIT1 was fused to the Escherichia coli lacZ gene. This construct was integrated into the CIT1 locus of wild-type PSY142 [rho+] and [rho0] cells and into a hap2Δ and rtg1Δ, rtg2Δ, or rtg3Δ mutant derivatives of these strains. In agreement with the findings of Rosenkrantz et al. (34), the CIT1-lacZ reporter gene construct fully recapitulated the known glucose repression of the native CIT1 gene, with more than a 15-fold decrease in β-galactosidase activity in extracts from [rho+] cells grown on glucose compared with extracts from cells grown on raffinose, a nonrepressing carbon source (Fig. 1A and B). Reporter gene activity in [rho+] rtgΔ cells was inhibited only modestly (25 to 50%) when those cells were grown on raffinose but was very strongly inhibited when those cells were grown on glucose. In derepressed [rho+] cells, the hap2Δ mutation nearly abolished CIT1 reporter gene expression; however, in glucose-repressed [rho+] cells, significantly more reporter gene activity remained in the hap2Δ background. Finally, in [rho+] cells grown on either raffinose or glucose, reporter gene activity was essentially eliminated in the hap2Δ rtgΔ double mutants. These results are fully consistent with the known requirement of the Hap2,3,4,5p complex for derepressed expression of the CIT1 gene (34).
FIG. 1.

Alternative dependence of CIT1 expression on RTG1, RTG2, RTG3, and HAP2. β-Galactosidase assays were carried out to determine the activity of a CIT1-lacZ reporter gene in wild-type (WT) PSY142 [rho+] and [rho0] cells and various mutant derivatives of these strains as indicated. The bp −806 to +9 region of the CIT1 gene was fused to the coding region of the E. coli lacZ gene, and the resulting construct was integrated at the chromosomal CIT1 locus. For each strain grown on either raffinose (YPR) or glucose (YP5%D) medium, four independent transformants were pooled from mid-log-phase cultures and β-galactosidase assays on whole-cell extracts were carried out in triplicate as described in Materials and Methods.
In contrast to these results, reporter gene expression was completely dependent on the RTG genes in [rho0] petite cells, whether those petites were grown on raffinose or glucose (Fig. 1C and D). Although expression in [rho0] cells still showed some dependence on HAP2 when cells were grown on raffinose, HAP2 was not required for expression in glucose-grown petite cells. Finally, as in [rho+] cells, expression of the CIT1-lacZ reporter gene was eliminated in the rtgΔ hap2Δ double mutants. We conclude from these results that the CIT1-lacZ expression is largely dependent on the HAP system in cells with robust mitochondrial respiratory function but switches to a synergistic dependence on the RTG and HAP systems when respiratory function is reduced, as in glucose-repressed [rho+] cells, or eliminated, as in [rho0] petites. In the most severe case of reduced mitochondrial function—[rho0] cells grown on glucose—CIT1 expression is independent of the HAP system.
Glutamate auxotrophy.
Previous studies showed that rtg mutant [rho+] cells grown on minimal glucose medium were glutamate auxotrophs (18). Glutamate auxotrophy could arise from a block in the synthesis of α-ketoglutarate, the direct precursor of glutamic acid. The finding that CIT1-lacZ expression was largely independent of the RTG genes in [rho+] cells grown on raffinose, but was strongly dependent on those genes in cells grown on glucose, raised the possibility that rtg mutant [rho+] cells do not require glutamate for growth on minimal raffinose medium. We therefore compared the glutamate requirements for growth of wild-type [rho+] and [rho0] cells and their rtgΔ mutant derivatives on minimal raffinose or glucose medium. Figure 2A and Fig. 2B show that the rtgΔ mutant [rho0] cells, in which CIT1-lacZ expression was strongly dependent on the RTG genes, were glutamate auxotrophs when grown on minimal raffinose medium. By contrast, the various rtgΔ [rho+] mutant cells, in which CIT1-lacZ expression is largely independent of the RTG genes, were glutamate prototrophs on minimal raffinose medium. On minimal glucose medium, both the [rho0] and [rho+] rtgΔ mutant strains were glutamate auxotrophs (Fig. 2C and D), again consistent with the nearly complete dependence of CIT1-lacZ expression in those cells on the RTG genes.
FIG. 2.

Glutamate auxotrophy of rtg mutant cells coincides with RTG-dependent gene expression. Wild-type (WT) PSY142 [rho+] and [rho0] cells and rtg1Δ, rtg2Δ or rtg3Δ mutant derivatives of those strains were streaked on YNBR or YNBD medium with or without 0.02% glutamate. (A) YNBR plus glutamate; (B) YNBR alone; (C) YNBD plus glutamate; (D) YNBD. Only the [rho0] mutant derivatives are glutamate auxotrophs in derepressed cells, whereas both [rho+] and [rho0] mutant derivatives are glutamate auxotrophs in glucose-repressed cells.
The CIT1 UAS contains a functional R box.
A 10-bp DNA segment of CIT1 from −367 to −358 has been shown to be important for glucose-repressed expression of the gene (34, 35). Notably, that 10-bp segment starts with GTCAC, an R box binding site for the Rtg1p-Rtg3p heterodimeric complex (14). To determine whether this R box confers the RTG-dependent expression of CIT1, we first constructed a CIT1-lacZ reporter gene in which a 140-bp fragment from −400 to −260 of the upstream region of CIT1 containing both the R box site and the Hap2,3,4,5p binding site, CCAAT, was fused to a CYC1 transcriptional start site linked to lacZ (Fig. 3A). The resultant CIT1 UAS-CYC1-lacZ construct in a centromere plasmid was transformed into [rho+] and [rho0] cells and into rtg3Δ, hap2Δ, and rtg3Δ hap2Δ double-mutant derivatives of those strains. This reporter gene mimicked the profile of expression of the −806 bp CIT1-lacZ reporter gene: it was subject to glucose repression; expression was dependent on RTG3 in [rho+] and [rho0] cells grown on glucose medium; and expression was dependent both on HAP2 and on RTG3 in [rho0] cells grown on raffinose medium (Fig. 3B).
FIG. 3.

A functional R box in the CIT1 UAS. (A) Diagram of a CIT1 UAS-CYC1-lacZ construct in which a 140-bp fragment of the upstream region of CIT1 from bp −400 to −260 was fused to the transcriptional start site of the CYC1 gene and fused to the reading frame of the E. coli lacZ gene. Positions of the putative Rtg1p-Rtg3p R box binding site, GTCAC, and the Hap2,3,4,5p binding site, ATTGG, are indicated. (B) Wild-type (WT) PSY142 [rho+] and [rho0] cells and rtg3Δ, hap2Δ, and rtg3Δ hap2Δ derivatives were transformed with the CIT1 UAS-CYC1-lacZ construct in a centromeric plasmid. Pools of 10 transformants of each were grown to mid-log phase on YNBcasR or YNBcas5%D medium, and β-galactosidase activity was determined in cell-free extracts. (C) Two mutations were introduced into the R box in the CIT1 UAS-CYC1-lacZ construct as indicated in boldface and described in Materials and Methods. This construct was placed into a centromeric plasmid to yield pCIT1(R)(UAS)-CYC1-LacZ. (D) PSY142 [rho+] and [rho0] cells were transformed either with pCIT1(UAS)-CYC1-LacZ, containing the wild-type R box, or with pCIT1(R)(UAS)-CYC1-LacZ, containing the mutant R box construct. Ten transformants of each were pooled and grown to mid-log phase in YNBcasR or YNBcas5%D medium, and β-galactosidase activities were measured in cell-free extracts. Standard errors from triplicate assays are <10%.
To determine whether the R box site is important for RTG1,3-dependent expression, two mutations were introduced into the R box of the CIT1 UAS-CYC1-lacZ wild-type reporter construct (Fig. 3C), and the effects of those mutations were analyzed in [rho+] and [rho0] cells grown on raffinose or glucose medium. In [rho+] cells grown on raffinose, the R box mutations reduced reporter gene expression less than threefold (Fig. 3D), whereas expression was reduced more than sixfold in [rho0] cells grown on raffinose, and nearly ninefold in [rho0] cells grown on glucose. These results suggest that the R box in the CIT1 UAS is an important cis-acting element for expression of CIT1, particularly in glucose-repressed [rho0] cells in which mitochondrial function is most severely affected. These results are consistent with the differential dependence of CIT1 expression on RTG3 in [rho+] and [rho0] cells grown on repressing versus nonrepressing carbon sources.
To determine whether the Rtg1p-Rtg3p complex binds to the CIT1 UAS, the 140-bp DNA fragment containing the R box was used as a probe in EMSAs with extracts from wild-type [rho+] and rtgΔ mutant strains. With extracts from wild-type cells, two bands were detected by EMSA (Fig. 4, lane 2). However, when extracts from rtg1Δ or rtg3Δ strains were used, the fainter, lower band disappeared (Fig. 4, lanes 3 and 5). The faster-migrating band was still present when an extract from a rtg2Δ strain was used (Fig. 4, lane 4). This result is consistent with those of previous EMSAs with a CIT2 UASr probe showing that Rtg2p is not required for the binding of the Rtg1p-Rtg3p complex to R box sites (14, 18) and that Rtg2p is a cytoplasmic protein (38). Collectively, these data strongly suggest that the Rtg1p-Rtg3p complex regulates CIT1 expression through interaction with the CIT1 R box.
FIG. 4.
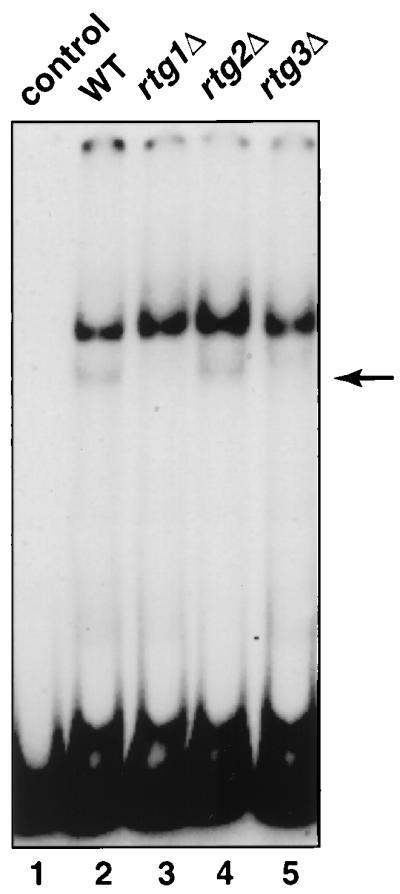
The Rtg1p-Rtg3p complex binds to the CIT1 UAS. Whole-cell extracts were prepared from wild-type (wt) [rho+] PSY142 cells and from rtg1Δ, rtg2Δ, and rtg3Δ derivatives grown on YNB5%D medium supplemented with 0.01% glutamate. EMSA was carried out with a [γ-32P]ATP-labeled 140-bp DNA probe of the CIT1 UAS as described in Materials and Methods. The control lane 1 is the probe alone. The arrow indicates the gel-retarded band whose presence is dependent on Rtg1p and Rtg3p.
Glutamate represses RTG gene functions.
Glutamate is a negative regulator of CIT1 expression in glucose-repressed cells (15, 35). ACO1, whose expression requires Rtg2p in glucose-repressed cells (41), is also subject to glutamate repression (12). These observations raise the possibility that glutamate repression of CIT1 expression is correlated with CIT1’s dependence on the RTG genes in glucose-repressed or respiratory deficient cells. To test this, we examined the effect of addition of glutamate to the growth medium on the expression of the −806 CIT1-lacZ reporter gene in [rho+] and [rho0] cells grown in medium with raffinose or glucose as the carbon source. In [rho+] cells grown on raffinose, the addition of 0.01 or 0.2% glutamate to the medium resulted in only a partial inhibition of reporter gene expression (Fig. 5A), comparable to the inhibitory effect of the rtgΔ mutations in derepressed [rho+] cells (Fig. 1). In contrast to these results, glutamate was a potent inhibitor of reporter gene expression in [rho+] and [rho0] cells grown on glucose and was slightly less effective as an inhibitor in [rho0] cells grown on raffinose. Similar patterns of repression by glutamate were observed when we used a CIT1 reporter gene containing the 140-bp CIT1 UAS (data not shown). These findings support the conclusion that glutamate is a repressor of RTG-dependent CIT1 expression.
FIG. 5.

Glutamate is a repressor of RTG-dependent gene expression. (A) PSY 142 [rho+] and [rho0] strains with an integrated copy of the −806 CIT1-lacZ reporter gene and an rtg1Δ derivative of those strains were grown in YNBR or YNB5%D medium as controls. Parallel cultures contained glutamate in the medium at the indicated concentrations. Whole-cell extracts were prepared, and β-galactosidase activities were determined. Data are expressed as β-galactosidase activities normalized to the value for the wild-type (WT) grown in the absence of glutamate. (B) Same as panel A except that wild-type [rho+] and [rho0] strains lacked the CIT1 reporter gene and instead were transformed with centromere-based plasmids containing a CIT2-lacZ reporter gene (18) or a DLD3 reporter gene in which the bp −500 region of DLD3 was fused to lacZ (4).
The RTG genes are required for the expression the CIT2 and DLD3 genes in all strains and carbon sources that we have tested. If glutamate is a general negative regulator of RTG-dependent gene expression, then CIT2 and DLD3 expression should also be sensitive to glutamate repression independent of carbon source and the functional state of mitochondria. To test this, CIT2-LacZ and DLD3-lacZ reporter genes were introduced into [rho+] and [rho0] cells, which were then grown on either raffinose or glucose. Expression of both CIT2-lacZ and DLD3-lacZ was repressed by glutamate in the medium, and the extent of repression was independent of carbon source or the functional state of mitochondria (Fig. 5B). As a control, lacZ expression was placed under the control of the constitutive ACT1 promoter, but expression of that fusion gene was not repressed by glutamate (data not shown). We conclude from these experiments that glutamate is a general repressor of RTG-dependent gene expression.
Constitutive expression of CIT1 cannot rescue the phenotypes of RTG deletion mutants.
Although the RTG genes are essential for CIT2 expression, a cit2Δ mutant, unlike an rtgΔ mutant, is not a glutamate auxotroph, whereas the cit1Δ cit2Δ double mutant requires glutamate for growth (15). Given the finding that CIT1 expression becomes dependent on the RTG genes in glucose-repressed or respiratory activity-deficient cells, we examined whether constitutive expression of CIT1 would rescue the glutamate auxotrophy of rtgΔ mutant cells or their inability to grow on acetate medium. To these ends, we constructed a fusion gene in which CIT1 expression was placed under the control of the constitutive promoter of the ACT1 gene. The resulting plasmid, pACT1-CIT1, was transformed into a cit1Δ [rho+] strain and into rtgΔ mutant derivatives of the wild-type [rho+] strain. Controls included cells transformed with the vector alone, as well as a cit2Δ mutant strain. We then determined the ability of these strains to grow on YNBcasAce medium and on YNBD medium with or without glutamate supplementation. The constitutive expression of CIT1 from pACT1-CIT1 was not able to restore glutamate auxotrophy to any of the rtgΔ mutant strains (Fig. 6A and B). Although all of these strains are respiratory competent, since they were able to grow on medium with glycerol as the sole carbon source (Fig. 6C), the ACT1-CIT1 fusion gene restored growth to the cit1Δ mutant cells only when cells were grown on acetate medium (Fig. 6D). These data suggest that first, CIT1 could be expressed from the ACT1-CIT1 fusion gene sufficiently to restore acetate growth to a cit1Δ mutant, and second, the reduction in CIT1 and CIT2 expression in rtgΔ mutant cells is not solely responsible for their glutamate auxotrophy or inability of those cells to grow on acetate medium. Thus, other defects in the TCA cycle are likely to exist in these mutants.
FIG. 6.

Constitutive expression of CIT1 fails to rescue the glutamate auxotrophy and acetate− phenotypes of rtg mutant cells. Wild-type (WT) PSY142 [rho+] cells and cit1Δ, cit2Δ, rtg1Δ, rtg2Δ, or rtg3Δ derivatives were transformed with a centromere-based plasmid, pACT1-CIT1, in which CIT1 expression was placed under the control of the constitutive ACT1 promoter. As controls, these strains were also transformed with pRS416 alone. Cells were streaked on YNBD–0.02% glutamate (A), YNBD (B), YNBcasGly (C) or YNBcasAce (D) medium and grown for 2.5 to 3.5 days at 30°.
Expression of CIT1, ACO1, IDH1, and IDH2 requires the RTG genes in cells with dysfunctional mitochondria.
To investigate further the relation between the RTG genes and expression of TCA cycle genes, we carried out Northern blot analysis to examine the relative expression of eight genes encoding proteins that function in the TCA cycle: CIT1, ACO1, IDH1, IDH2, KGD1, SDH1, FUM1, and MDH1. The transcript levels of these genes, normalized to the level of ACT1 mRNA, were compared in [rho+] and [rho0] wild-type cells and in rtg1Δ, rtg2Δ, and rtg3Δ mutant derivatives of these strains grown in YPR medium. As a control, we also probed for CIT2 transcripts, whose behavior has previously been well characterized in those strains. Only the CIT2 gene showed a strong dependence on the RTG genes for expression in both [rho+] and [rho0] cells (Fig. 7). However, four of the TCA cycle genes, CIT1, ACO1, IDH1, and IDH2, which encode proteins catalyzing the first three steps of the TCA cycle from oxaloacetate to α-ketoglutarate, showed a dramatic dependence on the RTG genes for their expression in [rho0] cells, but not in [rho+] cells. In sharp contrast to the elevated expression of CIT2 in [rho0] cells, the transcript levels of these first four TCA cycle genes were about the same in [rho0] and [rho+] cells. Expression of the remaining TCA cycle genes tested, KGD1, SDH1, FUM1, and MDH1, was essentially unaffected by the rtgΔ mutations, and their expression in the wild-type [rho0] strain was significantly less than in [rho+] cells. Two transcripts that behaved identically were detected with the SDH1 probe. The origin of these two transcripts has not been investigated further. The HAP genes are known to be required for expression of KGD1, SDH1, FUM1, and MDH1 in derepressed [rho+] cells (7, 12, 22, 31). Their reduced expression in derepressed [rho0] cells provides the first indication to our knowledge that HAP functionality is reduced in derepressed, respiratory activity-deficient cells. From these experiments, we conclude that the expression of genes encoding proteins that function in the first three steps of the TCA cycle are under dual HAP-RTG control.
FIG. 7.

Northern blot analysis of TCA cycle gene expression. Total RNA was prepared from PSY142 [rho+] and [rho0] wild-type (WT) strains and their rtg1Δ, rtg2Δ, and rtg3Δ derivatives grown to mid-log phase on YPR medium. Blots were probed for each of the indicated genes as described in Materials and Methods. RNA loads were normalized to the level of transcripts of the ACT1 gene. CIT2 transcripts were also analyzed as a control.
DISCUSSION
In an analysis of the cis-acting elements controlling expression of the mitochondrial citrate synthase gene, CIT1, Rosenkrantz et al. (34, 35) concluded that there were distinct upstream activation regions required for glucose-repressed and derepressed expression of the gene. In particular, the region from −367 to −348 was necessary for glucose-repressed expression, whereas the region from −291 and −273 was necessary for expression in derepressed cells. The latter is clearly identified as the HAP control region: it contains a consensus Hap2,3,4,5p binding site whose integrity is necessary for CIT1 expression in derepressed cells, consistent with the known functionality of the Hap complex in cells with robust respiratory activity.
In the present study, we have shown that expression of CIT1 becomes increasingly dependent on the RTG genes as mitochondrial respiratory function is reduced. The upstream region of CIT1 identified by Rosenkrantz et al. (34, 35) as being necessary for glucose-repressed expression contains a single R box, GTCAC, which we have shown for the CIT2 and DLD3 genes is a binding site for the Rtg1p-Rtg3p, bHLH/Zip complex (4, 14). CIT2 and DLD3 each contain two closely spaced R boxes (in inverted orientation), both of which are necessary for maximal gene expression. Mutation of the single R box in the upstream region of CIT1 shows that the R box is most important for CIT1 expression in glucose-repressed cells, consistent with CIT1’s strong dependence on the RTG genes for its expression under glucose-repressed conditions. Together with the EMSAs showing that the Rtg1p-Rtg3p bind to a DNA probe containing the CIT1 R box, these findings provide strong support for the conclusion that the RTG genes play an important role in the regulation of CIT1 expression under conditions where HAP-dependent expression decreases.
We extended this dual HAP-RTG control of gene expression to include ACO1, IDH1, and IDH2. Velot et al. (41) reported that ACO1 expression required RTG2 specifically in glucose-repressed cells. Our studies confirm this finding and extend the control of ACO1 expression as well as expression of the above-mentioned genes to include RTG1 and RTG3. It was essential to test whether expression required all three of the RTG genes, because we do not yet know whether RTG2 affects gene expression in pathways not involving RTG1 or RTG3. We have inspected the 5′ flanking regions of ACO1, IDH1, and IDH2 to see whether they contain R box sites. ACO1 has two R boxes at −772 and −450. IDH1 contains three R boxes at −355, −476, and −576, but the only consensus R box for IDH2 evident from the database is one far upstream at −768. The functionality of these putative Rtg1p-Rtg3p binding sites and whether potential degenerate R box sites can serve as binding sites for Rtg1p-Rtg3p will have to be determined on a gene-by-gene basis. Finally, we found that expression of all of the other TCA cycle genes tested that encode proteins catalyzing steps of the cycle downstream of IDH1 and IDH2 was independent of the RTG genes in respiratory activity-deficient cells. Collectively, our findings show that the expression of genes encoding the first three steps of the TCA cycle from oxaloacetate to α-ketoglutarate come under increasing control of the RTG regulatory pathway as mitochondrial respiratory capacity is reduced. In this way, expression of those genes can be maintained to compensate for the loss of HAP gene control.
To evaluate the effects of the functional state of mitochondria on TCA cycle gene expression, it is worth emphasizing that the combination of strains and growth conditions that we chose have provided cells with a graded range of mitochondrial functions, from those with robust respiratory activity in derepressed [rho+] cells to those with severely debilitated mitochondrial function in glucose-repressed [rho0] cells. The striking observation was that the dependence of CIT1 expression on the HAP and RTG genes followed this graded range of mitochondrial function: at the extremes, expression was largely HAP or RTG dependent, whereas in the middle, expression showed a synergistic dependence on the two systems. Although we do not know the precise details of how the control of expression of these TCA cycle genes is handed off from largely HAP-dependent expression in derepressed [rho+] cells to RTG-dependent expression in glucose-repressed [rho0] petites, it is not likely to be as a result of modulation of the level of the RTG gene products, since they are expressed constitutively in all cases that we have examined (14, 36, 37). Indeed, the RTG pathway appears to be controlled by an Rtg2p-dependent regulation of the nuclear localization of Rtg1p and Rtg3p (38). There is evidence that some of the components of the HAP complex are induced in cells growing on nonfermentable carbon sources, but how HAP activity is tied to the functional state of mitochondria is less clear.
The retrograde response versus RTG control of TCA cycle gene expression.
Our previous analysis of the CIT2 and DLD3 genes showed that their expression required the RTG genes, independent of the cell’s respiratory state or the carbon source in the growth medium (4, 18, 19). A characteristic feature of CIT2 and DLD3 expression is their retrograde response, namely, a sharply elevated level of expression in cells with dysfunctional mitochondria. Based on the present findings, we can define two new patterns of gene regulation related to the mitochondrial state that apply to TCA cycle genes. The first is a dual dependence on the HAP and RTG genes, with an increasing reliance on the RTG genes for expression in cells whose mitochondrial respiratory function is reduced or eliminated. The overall levels of expression for those genes, CIT1, ACO1, IDH1, and IDH2, in contrast to retrograde response genes, are roughly the same in derepressed [rho+] and [rho0] cells. This combinatorial control between the HAP and RTG genes represents a heretofore unrecognized strategy by which cells regulate gene expression in response to carbon source and to changes in the functional state of mitochondria. The second pattern of control was evident from our Northern blot data showing that expression of TCA cycle genes functioning downstream of α-ketoglutarate, KGD1, SDH1, FUM1, and MDH1, are all down-regulated in [rho0] petites.
We can consider three general pathways used by yeast cells to modulate expression of genes related to mitochondrial oxidative metabolism. The first is responsive to carbon source and is controlled by the Hap2,3,4,5p complex (10, 11, 24, 26, 27, 30, 43). Second are the oxygen-sensing pathways exemplified by control via the positive regulator, Hap1p (6, 29), and the negative regulator, Rox1p, a heme-dependent repressor of hypoxic genes (20, 21, 44). Hap1p is a transcriptional activator that, together with heme, responds to oxygen levels to regulate expression of an assortment of genes that function in electron transport, oxidative stress, and heme, sterol, and unsaturated fatty acid biosynthesis (reviewed by Kwast et al. [17]). The RTG genes can now be included as an additional pathway of gene regulation that monitors the functional state of mitochondria. As mitochondrial respiratory functions are compromised, the RTG system takes over responsibility for expression of some, otherwise HAP-dependent genes as described here and elevates the expression of other genes such as CIT2 (19) to compensate for the mitochondrial defects.
A central role for glutamate.
A number of previous studies had shown that glutamate is an inhibitor of expression of some TCA cycle genes as well as CIT2 (13, 15, 35, 41), but the regulatory targets affected by glutamate were unknown. It is now clear that glutamate inhibition occurs via RTG-dependent gene expression, which we suggest is a reflection of a negative feedback loop that regulates glutamate levels in cells with compromised mitochondrial function. In yeast, there are three known pathways for glutamate synthesis (1, 9, 25): two glutamic dehydrogenase isozymes encoded by GDH1 and GDH3 and glutamine synthetase encoded by GLT1. All three pathways use α-ketoglutarate as a common precursor of glutamate, which itself is a precursor for the synthesis of other amino acids and nucleotides. As illustrated in the model shown in Fig. 8, the RTG pathway could be activated to ensure that (i) there is sufficient synthesis of α-ketoglutarate for glutamate synthesis when the HAP system is downregulated, as in respiratory deficient cells, and (ii) anaplerotic pathways and gluconeogenesis are adequately maintained as a result of increased supply of intermediates to the TCA cycle, particularly succinate, from the glyoxylate cycle. The dramatic increase in CIT2 expression in respiratory activity-deficient cells could effectively increase the flux of carbon through the glyoxylate cycle to provide the net carbon needed for anabolic pathways.
FIG. 8.

Model for the role of the RTG genes in the control of expression of TCA and glyoxylate cycle genes. As mitochondrial function decreases (shaded triangle), the expression of CIT1, ACO1, IDH1, and IDH2 becomes increasingly dependent on the RTG genes and less dependent on the HAP genes. Glutamate is shown as a feedback regulator of RTG-dependent pathways of gene expression. α-KG, α-ketoglutarate.
Glutamate appears to be a key player in the RTG pathways. When supplemented in the growth medium, glutamate is a potent repressor of RTG-dependent gene expression, suggesting that if the RTG pathway indeed functions to maintain adequate levels of α-ketoglutarate for glutamate biosynthesis, it is subject to a strong glutamate feedback loop via transcriptional control, where expression of genes encoding the first three enzymes of the TCA cycle that lead to the synthesis of α-ketoglutarate are repressed by glutamate. Considering the findings to date, we are inclined to speculate that the intracellular level of glutamate or its direct precursor α-ketoglutarate might be a key signal for regulation of the RTG-dependent pathways.
ACKNOWLEDGMENTS
We thank A. Chelstowska for providing the pACT1-LacZ construct, and we thank C. Epstein for providing primers to generate probes for TCA cycle genes and for a critical reading of the manuscript. We also thank members of the Butow laboratory for helpful discussions.
This work was supported by grants from the National Institutes of Health (GM22525) and from The Robert A. Welch Foundation (I-0642).
Footnotes
This paper is dedicated to our friend and colleague, Paul Srere.
REFERENCES
- 1.Avendano A, DeLuna A, Olivera H, Valenzuela L, Gonzalez A. GDH3 encodes a glutamate dehydrogenase isozyme, a previously unrecognized route for glutmate biosynthesis in Saccharomyces cerevisiae. J Bacteriol. 1997;179:5594–5597. doi: 10.1128/jb.179.17.5594-5597.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin and hsp70 heat shock proteins. Proc Natl Acad Sci USA. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chelstowska A, Butow R A. RTG genes in yeast that function in communication between mitochondria and the nucleus are also required for expression of genes encoding peroxisomal proteins. J Biol Chem. 1995;270:18141–18146. doi: 10.1074/jbc.270.30.18141. [DOI] [PubMed] [Google Scholar]
- 4.Chelstowska, A., Z. Liu, Y. Jia, D. Amberg, and R. A. Butow. Yeast, in press. [DOI] [PubMed]
- 5.Chen D-C, Yang B-C, Kuo T-T. One-step transformation of yeast in stationary phase. Curr Genet. 1992;21:83–84. doi: 10.1007/BF00318659. [DOI] [PubMed] [Google Scholar]
- 6.Creusot E, Verdiere J, Gaisne M, Slonimski P P. CYP1 (HAP1) regulator of oxygen-dependent gene expression in yeast. I. Overall organization of the protein sequence displays several novel structural domains. J Mol Biol. 1988;204:263–276. doi: 10.1016/0022-2836(88)90574-8. [DOI] [PubMed] [Google Scholar]
- 7.Daignon-Fornier B, Valens M, Lemire B D, Bolotin-Fukuhara M. Structure and regulation of SDH3 the yeast gene encoding the cytochrome b560 subunit of respiratory complex II. J Biol Chem. 1994;269:15469–15472. [PubMed] [Google Scholar]
- 8.DeRisi J L, Iyer V R, Brown P O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 9.Folch J L, Antaramian A, Rodriguez L, Bravo A, Brunner A, Gonzalez A. Isolation and characterization of a Saccharomyces cerevisiae mutant with impaired glutamate synthase activity. J Bacteriol. 1989;171:6776–6781. doi: 10.1128/jb.171.12.6776-6781.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forsburg S L, Guarente L. Communication between mitochondria and the nucleus in regulation of cytochrome genes in the yeast Saccharomyces cerevisiae. Annu Rev Cell Biol. 1989;5:153–180. doi: 10.1146/annurev.cb.05.110189.001101. [DOI] [PubMed] [Google Scholar]
- 11.Forsburg S L, Guarente L. Identification and characterization of HAP4: a third component of the CCAAT-bound HAP2/HAP3 heterodimer. Genes Dev. 1989;3:1166–1178. doi: 10.1101/gad.3.8.1166. [DOI] [PubMed] [Google Scholar]
- 12.Gangloff S P, Marguet D, Lauquin G J M. Molecular cloning of the yeast mitochondrial aconitase gene (Aco1) and evidence of a synergistic regulation of expression by glucose plus glutamate. Mol Cell Biol. 1990;10:3551–3561. doi: 10.1128/mcb.10.7.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haselbeck R J, McAlister-Henn L. Function and expression of yeast mitochondrial NAD- and NADP-specific isocitrate dehydrogenases. J Biol Chem. 1993;268:12116–12112. [PubMed] [Google Scholar]
- 14.Jia Y, Rothermel B, Thornton J, Butow R A. A basic helix-loop-helix zipper transcription complex functions in a signaling pathway from mitochondria to the nucleus. Mol Cell Biol. 1997;17:1110–1117. doi: 10.1128/mcb.17.3.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K S, Rosenkrantz M S, Guarente L. Saccharomyces cerevisiae contains two functional citrate synthase genes. Mol Cell Biol. 1986;6:1936–1942. doi: 10.1128/mcb.6.6.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koonin E V. Yeast protein controlling inter-organelle communication is related to bacterial phosphatases containing the Hsp70-type ATP-binding domain. Trends Biochem Sci. 1994;19:156–157. doi: 10.1016/0968-0004(94)90275-5. [DOI] [PubMed] [Google Scholar]
- 17.Kwast K E, Burke P V, Poyton R O. Oxygen sensing and the transcriptional regulation of oxygen-responsive genes in yeast. J Exp Biol. 1998;201:1177–1195. doi: 10.1242/jeb.201.8.1177. [DOI] [PubMed] [Google Scholar]
- 18.Liao X, Butow R A. RTG1 and RTG2: Two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72:61–71. doi: 10.1016/0092-8674(93)90050-z. [DOI] [PubMed] [Google Scholar]
- 19.Liao X, Small W C, Srere P A, Butow R A. Intramitochondrial functions regulate nonmitochondrial citrate synthase (CIT2) expression in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:38–46. doi: 10.1128/mcb.11.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowry C V, Zitomer R S. Oxygen regulation of anaerobic and aerobic genes mediated by a common factor in yeast. Proc Natl Acad Sci USA. 1984;81:6129–6133. doi: 10.1073/pnas.81.19.6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowry C V, Zitomer R S. ROX1 encodes a heme-induced repression factor regulating ANB1 and CYC7 of Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:4651–4658. doi: 10.1128/mcb.8.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McAlister-Henn L, Thompson L M. Isolation and expression of the gene encoding yeast mitochondrial malate dehydrogenase. J Bacteriol. 1987;169:5157–5166. doi: 10.1128/jb.169.11.5157-5166.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCammon M T. Mutants of Saccharomyces cerevisiae with defects in acetate metabolism: isolation and characterization of Acn− mutants. Genetics. 1996;144:57–69. doi: 10.1093/genetics/144.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNabb D S, Y. X, Guarente L. Cloning of yeast HAP5: a novel subunit of a heterotrimeric complex required for CCAAT binding. Genes Dev. 1995;9:47–58. doi: 10.1101/gad.9.1.47. [DOI] [PubMed] [Google Scholar]
- 25.Miller S M, Magasankik B. Role of NAD-linked glutamate dehydrogenase in nitrogen metabolism in Saccharomyces cerevisiae. J Bacteriol. 1990;172:4927–4935. doi: 10.1128/jb.172.9.4927-4935.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olesen J, Guarente L. The HAP2 subunit of yeast CCAAT transcriptional activator contains adjacent domains for subunit association and DNA recognition: model for the HAP2/3/4 complex. Genes Dev. 1990;4:1714–1729. doi: 10.1101/gad.4.10.1714. [DOI] [PubMed] [Google Scholar]
- 27.Olesen J T, Hahn S, Guarente L. Yeast HAP2 and HAP3 activators both bind to the CYC1 upstream activation site UAS2 in an interdependent manner. Cell. 1987;51:953–961. doi: 10.1016/0092-8674(87)90582-4. [DOI] [PubMed] [Google Scholar]
- 28.Parikh V S, Morgan M M, Scott R, Clements L S, Butow R A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science. 1987;235:576–580. doi: 10.1126/science.3027892. [DOI] [PubMed] [Google Scholar]
- 29.Pfeifer K, Prezant T, Guarente L. Yeast HAP1 activator binds to two upstream sites of different sequences. Cell. 1987;49:291–301. doi: 10.1016/0092-8674(87)90751-3. [DOI] [PubMed] [Google Scholar]
- 30.Pinkham J L, Olesen J T, Guarente L. Sequence and nuclear localization of the Saccharomyces cerevisiae HAP2 protein, a transcriptional activator. Mol Cell Biol. 1987;7:578–585. doi: 10.1128/mcb.7.2.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Repetto B, Tzagoloff A. Structure and regulation of KGD1, the structural gene for yeast alpha-ketoglutarate dehydrogenase. Mol Cell Biol. 1989;9:2695–2705. doi: 10.1128/mcb.9.6.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rose M D, Winston F, Heiter P. Methods in yeast genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 33.Rose M D, Winston F, Heiter P. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1990. [Google Scholar]
- 34.Rosenkrantz M, Kell C S, Pennell E A, Devenish L J. The HAP2,3,4 transcriptional activator is required for derepression of the yeast citrate synthase gene, CIT1. Mol Microbiol. 1994;13:119–131. doi: 10.1111/j.1365-2958.1994.tb00407.x. [DOI] [PubMed] [Google Scholar]
- 35.Rosenkrantz M, Kell C S, Pennell E A, Webster M, Devenish L J. Distinct upstream activation regions for glucose-repressed and derepressed expression of the yeast citrate synthase gene, CIT1. Curr Genet. 1994;25:185–195. doi: 10.1007/BF00357161. [DOI] [PubMed] [Google Scholar]
- 36.Rothermel B, Thornton J, Butow R A. Rtg3p, a basic helix-loop-helix/leucine zipper protein functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J Biol Chem. 1997;272:19801–19807. doi: 10.1074/jbc.272.32.19801. [DOI] [PubMed] [Google Scholar]
- 37.Rothermel B A, Shyjan A W, Etheredge J L, Butow R A. Transactivation by Rtg1p, a basic helix-loop-helix protein that functions in communication between mitochondria and the nucleus in yeast. J Biol Chem. 1995;49:29476–29482. doi: 10.1074/jbc.270.49.29476. [DOI] [PubMed] [Google Scholar]
- 38.Sekito, T., and R. A. Butow. 1999. Unpublished data.
- 39.Shyjan A W, Butow R A. Intracellular dialogue. Curr Biol. 1993;3:398–400. doi: 10.1016/0960-9822(93)90212-7. [DOI] [PubMed] [Google Scholar]
- 40.Small W C, Brodeur R D, Sandor A, Fedorova N, Li G, Butow R A, Srere P A. Enzymatic and metabolic studies on retrograde regulation mutants in yeast. Biochemistry. 1995;16:5569–5576. doi: 10.1021/bi00016a031. [DOI] [PubMed] [Google Scholar]
- 41.Velot C, Haviernik P, Lauquin G J M. The Saccharomyces cerevisiae RTG2 gene is a regulator of aconitase expression under catabolite repression conditions. Genetics. 1996;144:893–903. doi: 10.1093/genetics/144.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical pr PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 43.Xing Y, Fikes J D, Guarente L. Mutations in yeast HAP2/HAP3 define a hybrid CCAAT box binding domain. EMBO J. 1993;12:4647–655. doi: 10.1002/j.1460-2075.1993.tb06153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zitomer R S, Lowry C V. Regulation of gene expression by oxygen in Saccharomyces cerevisiae. Microbiol Rev. 1992;56:1–11. doi: 10.1128/mr.56.1.1-11.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]