

Abstract
Protein-lipid binding interactions play a key role in the regulation of peripheral membrane protein function. Liposome-binding assays are a simple and affordable means of screening for specific protein-lipid interactions. Liposomes are prepared by mixing phospholipid combinations of interest before drying and rehydration. Sonication of the lipid mixture produces small unilamellar vesicles (SUVs) which are incubated with a protein of interest to allow for any binding to occur. Liposomes and liposome-protein complexes are floated on a sucrose gradient by centrifugation to separate them from unbound protein. Bound protein levels are easily determined by SDS-PAGE and Western blotting. This approach provides a reliable means of assaying novel protein-lipid interactions in vitro. Here we use liposome floatation to show the binding of the SNARE-activating protein Sec18 (mammalian NSF) to phosphatidic acid.
Keywords: Liposome, Phospholipids, Membrane trafficking, Membrane fusion, Sec18, NSF, Phosphatidic acid, SNARE
1. Introduction
SNARE-mediated membrane fusion is a vital process required for maintaining eukaryotic cell homeostasis. The process occurs through a series of well-defined stages that carry out compartment contact, membrane bilayer fusion, and luminal mixing events [1]. Specific lipids have been shown to serve in an important regulatory capacity to facilitate the membrane fusion across a breadth of systems. The importance of protein-lipid interactions has been underscored through the use of cell-free in vitro systems where isolated organelles can be purified to examine the separate phases of membrane fusion in isolation. For instance, the yeast vacuole homotypic fusion system has shown that specific phosphoinositides (PI), phosphatidic acid (PA), diacylglycerol (DAG), and ergosterol are essential for vacuole fusion throughout the different stages of membrane fusion [2–16]. Separately, in vitro-reconstituted proteoliposome studies have shown that phosphatidylcholine (PC), phosphatidylethanolamine (PE), PA, DAG, and phosphatidylinositol 3-phosphate (PI3P) are the required minimal lipid components for successful compartment fusion [17]. Many of these lipids are known to bind directly to proteins involved in fusion, and these lipid-protein interactions serve to regulate protein function and localization throughout the different stages of fusion.
With the ongoing emergence of novel fusion-related protein-lipid interactions, it is important to be able to determine specific lipid-binding characteristics of relevant proteins in vitro. This can be done using several previously described assays including lipid-conjugated bead pull-downs, liposome-binding assays, surface plasmon resonance (SPR), and microscale thermophoresis (MST) [18–21]. Each of these approaches is effective in certain contexts but has limitations in others. SPR and MST, for example, provide quantitative binding measurements, but are costly and generally require user training. Lipid-conjugated bead pull-downs can identify novel protein-lipid interactions from crude cell lysate, but utilize lipids in a non-native state. Ultimately acquiring data from multiple assays will give the most comprehensive look at any protein-lipid interaction tested. However, it is important to have a reliable screening method to determine specific protein-lipid binding before investing time and money into multiple experiments. Liposome-binding assays use lipids in a physiologically relevant membrane-like bilayer and require a relatively short amount of time to complete. These factors make them an ideal initial approach for probing specific protein-lipid binding.
Here we describe an approach for assaying protein-lipid interactions using purified recombinant proteins and small unilamellar vesicles (SUVs). A protein is incubated with SUVs prepared using a variety of lipid compositions, and samples are loaded onto sucrose gradients and centrifuged at high speed. Liposomes and any bound protein float to the top of the gradient while unbound protein remains in the bottom layers. Protein-lipid binding levels are easily probed by SDS-PAGE and Western blotting of the top fraction of the sucrose gradient. The liposome-binding assay protocol included here has been used by our lab to detect specific protein-lipid interactions for many different proteins and lipids of interest. Here we specifically describe the binding of the SNARE-activating protein Sec18 to PA [15].
2. Materials
All reagents should be prepared using ultrapure water. Liposome-binding buffer can be made in advance and stored at room temperature. Liposome-loading and -stacking buffers should be made fresh before each experiment. The 5× SDS loading sample buffer can be made in advance and stored at −20 °C.
2.1. Liposome Preparation
Phosphatidylcholine (PC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine); phosphatidylethanolamine (PE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine); phosphatidylserine (PS, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine); phosphatidic acid (PA, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate); diacylglycerol (DAG, 1-palmitoyl-2-oleoyl-sn-glycerol).
Phospholipid stock solutions: Lipids dissolved in CHCl3 or CHCl3:MeOH:H2O for phosphoinositides (1:2:0.8) and stored under nitrogen at −20 °C (see Note 1).
Small glass test tubes (10 × 75 mm).
Hamilton glass syringe (100 μL).
Vacuum concentrator.
Liposome-binding buffer: 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4. Add 800 mL water to a glass beaker. Weigh 8 g NaCl, 0.2 g KCl, 1.44 g NaHPO4, and 0.24 g KH2PO4 and dissolve in the water. Adjust pH of the solution to 7.4 using HCl. Make up to 1 L with water (see Note 2).
Temperature-controlled water bath.
Water bath sonicator.
2.2. Liposome-Binding Assay
Sec18: His6-Sec18 can be expressed as a fusion protein in E. coli and purified using Ni-NTA and size-exclusion chromatography [15].
Standard Eppendorf tubes (1.7 mL).
Liposome-binding buffer.
Liposome-loading buffer: Liposome-binding buffer, 1.65 M sucrose. Weigh 5.65 g sucrose and add to small graduated cylinder. Add liposome-binding buffer to 10 mL final volume (see Note 3).
Liposome-stacking buffer: Liposome-binding buffer, 0.75 M sucrose. Weigh 2.57 g sucrose and add to small graduated cylinder. Add liposome-binding buffer to 10 mL final volume (see Note 3).
5× SDS-loading sample buffer: 250 mM Tris pH 6.8, 10% sodium dodecyl sulfate (SDS), 30% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue. Add 25 mL water to a glass beaker. Weigh 1.51 g Tris and 5.0 g SDS and dissolve in water. Add 15 mL glycerol and 2.5 mL β-mercaptoethanol and stir until homogenous. Adjust pH to 6.8 with HCl. Weigh 0.01 g bromophenol blue and dissolve in solution. Make up to 50 mL with water. Store 1 mL aliquots at −20 °C.
Temperature-controlled water bath.
Ultracentrifuge with swinging bucket rotor and thin-wall ultracentrifuge tubes (see Note 4).
3. Methods
3.1. Liposome Preparation
Mix phospholipids from stocks to produce a mixture with the desired lipid mole percentages. The total lipid mass used should be 2.6 μmoles. When using lipids in addition to PC and PE to the lipid mixture, add the appropriate amount of methanol to adjust the final solvent to CHCl3:MeOH (1:2) (see Note 1).
Dry lipid mixture under a stream of nitrogen until chloroform completely evaporates. The dried lipids should leave a film at the bottom of the test tube (see Note 5).
Place tube in a vacuum concentrator and further dry lipids for 1 h: medium speed, room temperature (see Note 6).
Add 2.6 mL liposome-binding buffer to the tube of dried lipids. Allow lipids to hydrate for 1 h with periodic vortexing. The lipid film should resuspend into the buffer and be completely removed from the sides of the tube (see Note 7).
Place tube with mixture in a water bath sonicator and sonicate until the suspension clarifies (see Notes 8 and 9).
3.2. Liposome-Binding Assay
Each liposome composition being tested should be set up in its own fresh tube to avoid cross contamination of lipids. The number of total samples assayed at one time should not exceed the number of tubes that can be spun by the rotor being used so all can be centrifuged simultaneously.
Add 150 μL liposome solution (SUVs) to a standard Eppendorf tube.
Add 2 μg purified recombinant Sec18 to the tube. Gently vortex on a low setting to mix (see Note 10).
Incubate the sample solution at temperature for 10 min (see Note 11).
Add 630 μL liposome-loading buffer to the sample. Gently pipette the solution up and down to mix (see Note 12).
Add the full sample volume to the bottom of an ultracentrifuge tube. Make sure that no residual sample sticks to the sides of the tube.
Carefully layer 3.2 mL liposome-stacking buffer onto the sample layer in the ultracentrifuge tube (see Notes 13 and 14).
Carefully layer liposome-binding buffer on to the stacking buffer layer. Add binding buffer to within 3–4 mm of the top of the tube (see Note 14).
Spin all sample tubes in an ultracentrifuge at 200,000 × g for 90 min at 4 °C.
Harvest 200 μL of suspension from the top interface between the liposome-binding buffer and liposome-stacking buffer. Additionally, harvest the bottom 100 μL of suspension in the liposome-loading buffer layer to assay for unbound protein. Add the samples to clean Eppendorf tubes (see Notes 15 and 16).
Add 1 mL liposome-binding buffer to the harvested liposome suspension (top interface). Spin tube in a refrigerated tabletop centrifuge to pellet liposomes (16,000 × g, 10 min, 4 °C). Carefully remove top 1.1 ml buffer and discard. To remaining 100 μL sample add 20 μL 5× SDS-loading sample buffer. Add 20 μL 5× SDS-loading sample buffer to unbound protein sample (bottom of tube suspension).
Incubate samples at 95 °C for 5 min. Load 10 μL of each sample and resolve by SDS-PAGE and Western blotting. Figure 1 shows example blots of His6-Sec18 lipid binding (9) (see Notes 17 and 18).
Fig. 1.
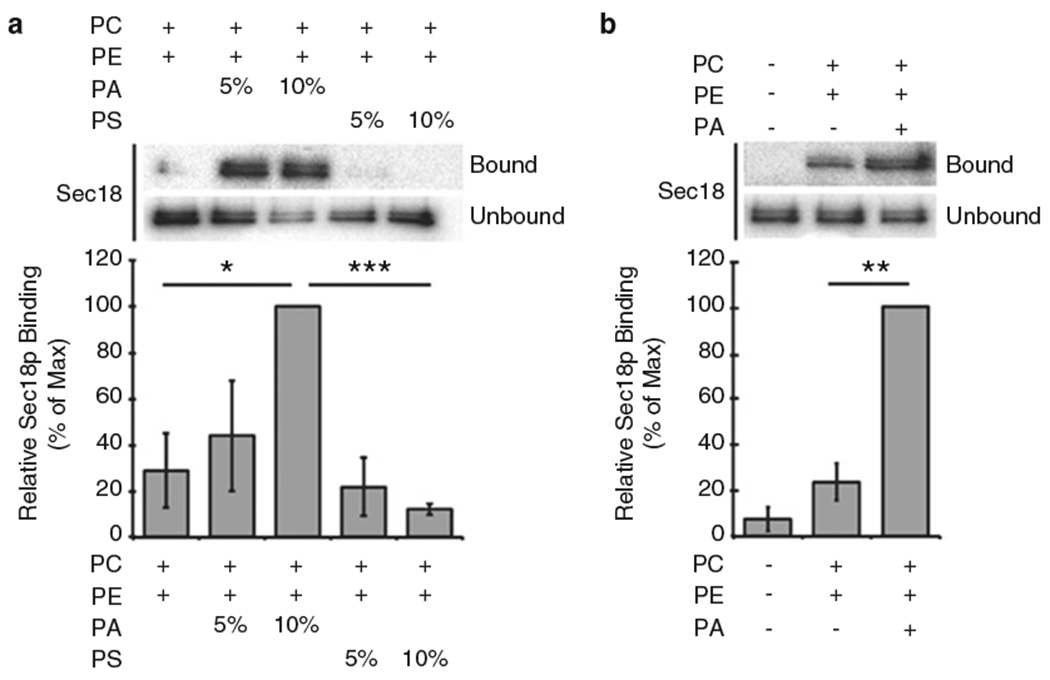
Sec18 preferentially binds to liposomes containing PA. Recombinant His6-Sec18 (2 μg) was incubated with liposomes of the indicated compositions for 10 min at 30 °C. Liposomes were isolated by centrifugation and washed before bound proteins were resolved by SDS-PAGE. Bar graphs show average normalized densitometry values measured for three separate experiments. Binding was observed in liposomes with increasing concentrations of PA or PS (a) and with no liposomes (b). *p < 0.05; **p < 0.001; ***p < 0.0001. (Reproduced from [15] with permission from John Wiley and Sons)
Acknowledgments
This work was supported in part by NIH grant GM101132 to RAF.
Footnotes
PC and PE are used as bulk structural lipids in combination with any lipids to be tested in our assay. We use PC and PE at a ratio of 80:20 and liposomes containing only these lipids are used as a negative control. Additional lipids can be added to the liposomes generally up to about 40% of total lipid content while still forming a bilayer. In our lab, we replace a portion of the PC content with our lipids of interest and keep PE at a constant 20% total lipid content. PC is generally characterized as a bulk structural lipid that has minimal binding activity to most peripheral membrane proteins. PE is also generally considered a bulk, structural lipid, but contains a primary amine group which interacts with other phospholipid head groups including PA [22]. This interaction can lead to changes in the charge state of phosphates located on adjacent lipids which may have dramatic effects on protein binding to the membrane. For this reason, our lab keeps a constant level of PE present in all of our liposomes.
Alternate buffered saline solutions that are compatible with your protein of interest can be used in place of the binding buffer. We have also used HEPES-buffered saline in our lab at pH values ranging from 6.8 to 7.4. Additional salts such as MgCl2 and CaCl2 can be added if they are expected to play a role in the protein-lipid interaction being assayed. Any changes made to the composition of the binding buffer should be included in the loading and stacking buffers as well.
The concentrated sucrose solutions can first be made in polypropylene conical tubes to allow for vortexing. Once all the sugar has dissolved, the tube’s contents can be transferred to a graduated cylinder before being brought to volume. It is advisable to rinse the conical tube with a small amount of liposome-binding buffer and add this solution before bringing to volume to ensure that all sugar is transferred to the final solution. It may also be beneficial to make the liposome-loading buffer in advance and allow it to nutate at room temperature for at least 30 min. The concentration of sucrose in the loading buffer is very high and it is sometimes difficult to distinguish between bubbles that have formed from vortexing and sugar that remains undissolved. Nutation for an extended period of time will yield a uniform solution in which all of the sucrose has dissolved.
We use the SW-60 rotor with 11 × 60 mm thin-wall polypropylene tubes. Any tubes with a volume of at least 2 mL can be used with a compatible ultracentrifuge swinging bucket rotor. Other investigators have had success working on a smaller scale using the fixed-angle TLA-100 rotor.
It is important to keep the dried lipids near the bottom of the tube for the later hydration step. To do this, hold the tube at a slight angle and use a gentle stream of nitrogen to blow the lipid mixture down and off the sides toward the bottom. This will leave an oily film at the bottom of the tube free from most of the original solvent. If no additional solvent evaporates from the lipid mix, the sample has dried enough to move on to the next step. There is an additional drying step that will remove any residual solvent left after this step.
The vacuum concentrator used by our lab is able to spin glass test tubes at low speed without issue. If no concentrator is available for this setup, lipids can be dried in a vacuum desiccator for 12 h until all solvent is evaporated. We have prepared liposomes using both of these drying methods and observed no significant difference between them.
Hydration of lipids should be carried out at a temperature greater than the highest transition temperature (Tm) of the lipids contained in the mixture. This can be done easily by heating the buffer in a temperature-controlled water bath to an appropriate temperature before adding it to the tube. After addition of the buffer, the tube should be placed in the water bath to keep the mixture above the relevant Tm. Using an appropriate elevated temperature will also help to resuspend the entire lipid film while vortexing. We use parafilm to seal the tops of our tubes to allow vortexing without losing any suspension. Vortexing should be done on a high enough setting to completely agitate the full volume of the mixture.
Sonication should be performed at a temperature above the highest Tm of the lipids in the mixture. The suspension will usually clarify within about 10 min, but sonication times vary and can be longer. Once fully sonicated, the solution should be transparent but might still be somewhat hazy. Haziness is caused by large particles that remain in the suspension and cause light scattering. These can be removed from the final liposome suspension by performing a brief spin in a tabletop centrifuge to give a clear solution of SUVs. Liposome solutions should not be frozen and can be stored at 4 °C for 5–7 days.
It is also possible to perform this assay using liposomes prepared by extrusion. This process requires a mini extruder setup in which lipid suspensions are passed through a filter of a defined pore size to create a uniform suspension of large unilamellar vesicles (LUVs). LUVs have a significantly larger diameter (120–140 nm) than SUVs prepared by sonication (15–50 nm) and display different membrane curvature. If measuring curvature effects on protein-lipid interactions in the assay is of interest, it may be helpful to use liposomes prepared by both methods for comparison. If comparing only differences in binding relative to lipid composition of the membranes, all liposomes used should be prepared using the same method to maintain consistent curvature. In both cases, liposomes should not be frozen and can be stored at 4 °C for 5–7 days.
The mass of protein used may need to be altered to optimize the signal achieved. The volume of protein added should not exceed 20 μL. It may also be important to consider the composition of the purified recombinant protein’s storage buffer. Most standard storage buffers (phosphate, HEPES, etc.) should not dramatically interfere with a protein’s ability to bind lipids. We have used proteins stored in a buffer solution containing 10% glycerol in this assay without issue.
The optimal temperature to assay protein-lipid binding may vary by system. We incubate our assays at 30 °C, the optimal growth temperature for yeast. Choose a temperature most appropriate for the protein being tested. Keep in mind that the phospholipids being used may have physiologically relevant Tm values and liposome membrane fluidity may be dramatically altered depending on the temperature used for the experiment.
The liposome-loading buffer can also be added and mixed with the sample solution before incubation at temperature. We have obtained similar results using each approach.
. If using smaller tubes than those listed here (11 × 60 mm) the volume of stacking buffer added will be reduced. Add enough stacking buffer to bring the top of the layer near the top of the tube (within 8–10 mm). Leave enough space to add a 200 μL binding buffer layer to the top of the gradient.
Layering sucrose solutions when creating the density gradient can be difficult. Care must be taken to prevent mixing of the gradient’s layers. The most common cause of layer mixing is pipetting too quickly. We have had the most success making gradients by holding the tube at an angle while slowly pipetting the next sucrose solution down the side. It may also be beneficial to use a syringe to add each layer to the gradient. Syringes allow for slower addition of each solution and can dramatically reduce layer mixing. Labeling the top of each layer with a marker may help to locate each interface both after layering is complete and after the sample has been centrifuged. After each of these steps there should be a visible interface between layers.
The top interface of the sucrose gradient should be visible after the ultracentrifuge spin is complete. If the interface is not easily visible it is possible that the layers have mixed and proper liposome floatation will not have occurred. If there is difficulty locating the interface it may be beneficial to mark the tube prior to centrifugation as described in Note 10.
Unbound protein should sediment to the bottom of the tube and remain in the bottom loading buffer layer. The unbound fraction can be harvested by inserting a Hamilton syringe to the bottom of the tube and withdrawing 100 μL of sample. We generally run an experiment that contains no added liposomes to verify our protein is properly sedimenting to the bottom fraction in the tube during centrifugation (see Fig. 1b).
It is important to heat all samples before loading them into a gel to prevent precipitation of sample components. Samples may be stored at −20 °C but should be reheated and quickly centrifuged before gel loading.
If desired, relative binding levels of a protein of interest to liposomes of different lipid compositions can be measured and presented in a semiquantitative manner (see Fig. 1). To do this, densitometry values for each Western blot are measured using appropriate software (ImageJ, Image Lab, etc.) and normalized within each trial. Data can then be combined across multiple trials to analyze the significance of binding that is observed. It may be beneficial to include a loading control containing the total protein content loaded into each assay. Liposome-bound protein levels can then be expressed as a percentage of total protein.
References
- 1.Jahn R, Lang T, Südhof TC (2003) Membrane fusion. Cell 112:519–533 [DOI] [PubMed] [Google Scholar]
- 2.Boeddinghaus C, Merz AJ, Laage R, Ungermann C (2002) A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J Cell Biol 157:79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabrera M, Nordmann M, Perz A, Schmedt D, Gerondopoulos A, Barr F et al. (2014) The Mon1-Ccz1 GEF activates the Rab7 GTPase Ypt7 via a longin fold-Rab interface and association with PI-3-P-positive membranes. J Cell Sci 27(Pt 5):1043–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol 3:613–618 [DOI] [PubMed] [Google Scholar]
- 5.Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W (2004) Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol 167:1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jun Y, Fratti RA, Wickner W (2004) Diacylglycerol and its formation by Phospholipase C regulate Rab- and SNARE- dependent yeast vacuole fusion. J Biol Chem 279:53186–53195 [DOI] [PubMed] [Google Scholar]
- 7.Karunakaran S, Sasser T, Rajalekshmi S, Fratti RA (2012) SNAREs, HOPS, and regulatory lipids control the dynamics of vacuolar actin during homotypic fusion. J Cell Sci 14:1683–1692 [DOI] [PubMed] [Google Scholar]
- 8.Karunakaran S, Fratti R (2013) The lipid composition and physical properties of the yeast vacuole affect the Hemifusion-fusion transition. Traffic 14:650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato M, Wickner W (2001) Ergosterol is required for the Sec18/ATP-dependent priming step of homotypic vacuole fusion. EMBO J 20:4035–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawrence G, Brown CC, Flood BA, Karunakaran S, Cabrera M, Nordmann M et al. (2014) Dynamic association of the PI3P-interacting Mon1-Ccz1 GEF with vacuoles is controlled through its phosphorylation by the type-1 casein kinase Yck3. Mol Biol Cell 25:1608–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayer A, Scheglmann D, Dove S, Glatz A, Wickner W, Haas A (2000) Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol Biol Cell 11:807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miner GE, Starr ML, Hurst LR, Sparks RP, Padolina M, Fratti RA (2016) The central poly-basic region of the soluble SNARE (soluble N-Ethylmaleimide-sensitive factor attachment protein receptor) Vam7 affects binding to phosphatidylinositol 3-phosphate by the PX (Phox homology) domain. J Biol Chem 291:17651–17663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miner GE, Starr ML, Hurst LR, Fratti RA (2017) Deleting the DAG kinase Dgk1 augments yeast vacuole fusion through increased Ypt7 activity and altered membrane fluidity. Traffic 18:315–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasser T, Qiu QS, Karunakaran S, Padolina M, Reyes A, Flood B et al. (2012) Yeast lipin 1 orthologue pah1p regulates vacuole homeostasis and membrane fusion. J Biol Chem 287:2221–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starr ML, Hurst LR, Fratti RA (2016) Phosphatidic acid sequesters Sec18p from cis-SNARE complexes to inhibit priming. Traffic 17:1091–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stroupe C, Collins KM, Fratti RA, Wickner W (2006) Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J 25:1579–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mima J, Wickner W (2009) Complex lipid requirements for SNARE-and SNARE chaperone dependent membrane fusion. J Biol Chem 284:27114–27122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Vecchio K, Stahelin RV (2016) Using surface plasmon resonance to quantitatively assess lipid-protein interactions. Methods Mol Biol 1376:141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manifava M, Thuring JW, Lim ZY, Packman L, Holmes AB, Ktistakis NT (2001) Differential binding of traffic-related proteins to phosphatidic acid- or phosphatidylinositol (4,5)-bisphosphate-coupled affinity reagents. J Biol Chem 276:8987–8994 [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka K, Morimitsu Y, Uchida K, Schekman R (1998) Coat assembly directs v-SNARE concentration into synthetic COPII vesicles. Mol Cell 2:703–708 [DOI] [PubMed] [Google Scholar]
- 21.van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R (2012) Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem 287:16447–16453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kooijman EE, Tieleman DP, Testerink C, Munnik T, Rijkers DT, Burger KN et al. (2007) An electrostatic/hydrogen bond switch as the basis for the specific interaction of phosphatidic acid with proteins. J Biol Chem 282:11356–11364 [DOI] [PubMed] [Google Scholar]