

Abstract
Bioorthogonal chemistry represents a class of high-yielding chemical reactions that proceed rapidly and selectively in biological environments without side reactions towards endogenous functional groups. Rooted in the principles of physical organic chemistry, bioorthogonal reactions are intrinsically selective transformations not commonly found in biology. Key reactions include native chemical ligation and the Staudinger ligation, copper-catalysed azide–alkyne cycloaddition, strain-promoted [3 + 2] reactions, tetrazine ligation, metal-catalysed coupling reactions, oxime and hydrazone ligations as well as photoinducible bioorthogonal reactions. Bioorthogonal chemistry has significant overlap with the broader field of ‘click chemistry’ — high-yielding reactions that are wide in scope and simple to perform, as recently exemplified by sulfuryl fluoride exchange chemistry. The underlying mechanisms of these transformations and their optimal conditions are described in this Primer, followed by discussion of how bioorthogonal chemistry has become essential to the fields of biomedical imaging, medicinal chemistry, protein synthesis, polymer science, materials science and surface science. The applications of bioorthogonal chemistry are diverse and include genetic code expansion and metabolic engineering, drug target identification, antibody–drug conjugation and drug delivery. This Primer describes standards for reproducibility and data deposition, outlines how current limitations are driving new research directions and discusses new opportunities for applying bioorthogonal chemistry to emerging problems in biology and biomedicine.
Bioorthogonal chemistry encompasses a class of high-yielding rapid and selective chemical reactions that proceed in biological environments, with little or no reactivity towards endogenous functional groups. Rooted in the principles of physical organic chemistry and classic organic reactivity, bioorthogonal reactions are intrinsically selective transformations not commonly found in biology. Chemical tools to study biological processes with molecular detail are foundational to modern science. The advent of recombinant protein expression enabled tracking protein dynamics in living systems using fluorescent proteins and antibodies1,2. These tools are often essential for studying intricate protein systems, but can be limited by their large size. Common genetic tags such as fluorescent proteins can disrupt protein function and trafficking, and for these same reasons they cannot be easily translated to non-protein biomolecules such as glycans, lipids and nucleic acids. An expanded set of biomolecules can be tagged via bioorthogonal chemistry. The classes of reactions include native chemical ligation and the Staudinger ligation, copper-catalysed azide–alkyne cycloaddition, strain-promoted [3 + 2] reactions, tetrazine ligation, metal-catalysed coupling reactions, oxime and hydrazone ligations as well as photoinducible bioorthogonal reactions (Fig. 1). Bioorthogonal reactions are intrinsically chemoselective, and therefore do not require proximity effects to achieve site-selective labelling. Also, bioorthogonal chemistry must readily proceed in aqueous environments at biocompatible pH and temperature, and be non-toxic under these conditions. Another consideration is the reaction rate3, as it is generally advantageous for reactions to proceed rapidly at the low concentrations required for many biological experiments. The size of reaction partners is an intrinsically important parameter as the native function of many targets can be sensitive to bulky chemical groups.
Fig. 1 |. Different classes of bioorthogonal reactions.
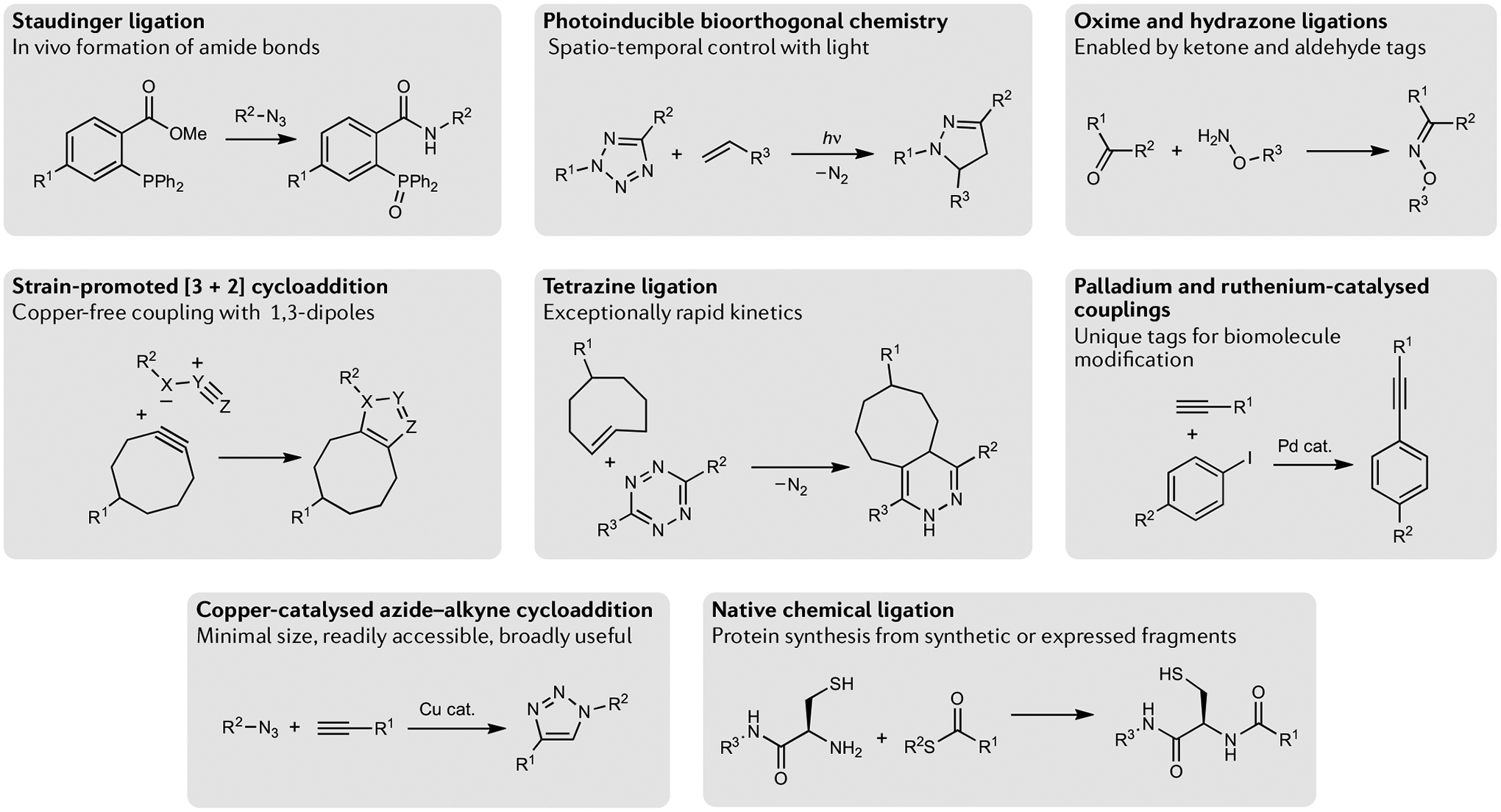
The broad range of bioorthogonal reactions with their associated reactants, key reagents, products and key feature(s) are highlighted here.
Chemoselective.
A chemical reaction that is selective for a certain functional group even in the presence of differing functional groups. Reaction partners in bioorthogonal chemistry are chemoselective for each other, even in biological settings.
Bioorthogonal chemistry enables probing biological systems through selective covalent bond formations that minimally disrupt the system(s) being studied. These reactions have been applied broadly both in cellular systems as well as in living animals; the growing list of applications includes probe construction, biomedical imaging, medicinal chemistry, polymer science and materials science as well as surface science. Similar to the broadening of applications enabled by bioorthogonal chemistry, the toolbox of chemical reactions has grown to meet the diverse needs of the chemical biology community.
Click chemistry.
Bioorthogonal chemistry has significant overlap with the broader field of click chemistry, which is defined by high-yielding and modular reactions that are wide in scope, simple to perform and generate only inert by-products4,5. Click chemistry also encompasses reactions that are not strictly bioorthogonal (as defined above) as these include selective reactions of functional groups commonly found in biological molecules. For example, alkylations and conjugate addition reactions of cysteines are classic approaches to achieving residue-selective protein modification, and the ‘thiol–ene’ reaction between unactivated alkenes and free thiols has become a broadly used tool across a range of applications, including organic synthesis, polymer science and materials science6.
Click chemistry.
A concept, coined by K. B. Sharpless and colleagues, describing bond-forming reactions that are thermodynamically driven, highly selective and reliable, and proceed in water without toxic by-products. Click reactions are often, but not strictly, bioorthogonal.
Scope.
Within the scope of this Primer are transformations where both reaction partners are not commonly found in biology and are the sources for chemoselectivity. This type of approach is complementary to strategies in which genetically programmed molecular recognition is responsible for selectivity, where effective chemistry is often still required, but the biological system is an active player and, thus, is at least somewhat perturbed by the modification(s). Here, examples include the use of reversible covalent reactions to modify specific protein sequences such as with genetically encoded tetracysteine motifs7. Another approach uses enzymatic modifications of proteins with encoded peptide sequences as a method of creating covalent bonds to exogenous molecules with high selectivity, enabling site-selective modifications at the carboxy terminus, amino terminus or internal positions8. Glycosyltransferases have been used for selective chemoenzymatic modification of glycans9. Directed evolution has been used to develop fusion proteins for covalent self-labelling with probe molecules10. These genetically programmed transformations represent powerful approaches to covalent bond formation, but they differ from bioorthogonal approaches where chemoselectivity derives directly from the reaction partners.
In this Primer, the scope of bioorthogonal chemistries examined includes the early developments such as native chemical ligation, oxime ligation and the Staudinger ligation, which all exemplified how reaction design and mechanistic insight can guide the development of new chemical tools to study biology. With improved rates and minimally sized reactants, the copper-catalysed cycloaddition between azides and alkynes to form triazoles has been transformative for numerous scientific disciplines including bioorthogonal chemistry. Using ring strain to enhance reactivity is a classic concept in organic chemistry that has proven especially powerful in the area of bioorthogonal chemistry. Initially demonstrated with azides, the [3 + 2] cycloadditions of cyclooctyne derivatives with 1,3-dipoles have emerged as broadly important bioorthogonal reactions. Inverse electron-demand Diels–Alder (IEDDA) reactions of strained alkenes with tetrazines are also facilitated by the release of ring strain, and the reactions of trans-cyclooctene (TCO) represent the fastest bioorthogonal reactions developed to date. Spatio-temporal control over bioorthogonal chemistry can be achieved through the application of light through photoinduced processes that liberate reactive nitrile imine dipolarophiles or cycloalkynes. Metal-catalysed cross-coupling and ruthenium-mediated olefin metathesis have also emerged as complementary tools that can function in complex biological settings.
Here, we describe the range of reactions that encompass the bioorthogonal chemistry toolbox (Experimentation) and highlight the broad range of applications that implement this chemistry from site-specific protein labelling both in vitro and in vivo, as well as polymers and materials sciences (Applications). We discuss how handling these highly reactive reagents should be treated to ensure their use across diverse sets of applications (Reproducibility and data deposition). Finally, we examine the current limitations of bioorthogonal chemistry, the exciting areas of development in this research area (Limitations and optimizations) and what to anticipate in the next decade (Outlook).
Experimentation
The underlying mechanisms of key reactions in the bioorthogonal chemistry toolbox and their ideal operating conditions are discussed in this section, including native chemical ligation, oxime and hydrazone ligations, the Staudinger ligation, copper-catalysed azide–alkyne cycloaddition, strain-promoted [3 + 2] reactions, tetrazine ligation, metal-catalysed coupling reactions and photoinducible bioorthogonal reactions.
Native chemical ligation.
For chemical protein synthesis and bioconjugation, a significant body of work has focused on selective reactivity at the protein N terminus. Grounded in classic work by Wieland and pioneered by Kent, Dawson and Muir, native chemical ligation involves the ligation of a peptide bearing a C-terminal thioester with a second peptide with an N-terminal cysteine11–14 (Fig. 2a). An initial thiol–thioester exchange reaction at the N terminus is key to the chemoselectivity of native chemical ligation, providing a transient thioester intermediate that undergoes an intramolecular S,N-acyl transfer to afford a native peptide bond to cysteine. As thioester exchange is the rate-determining step, native chemical ligation can be accelerated by nucleophilic catalysts including thiols or imidazole12,15. Subsequent to ligation, desulfurization reactions can serve to convert the internal cysteine into alanine16,17. Using selenocysteine and peptide selenoesters has enabled accelerating these ligation reaction rates, and selenocysteines can function as alanine surrogates in complex target synthesis as deselenization reactions can proceed in the presence of unprotected cysteine and methionine residues18,19. Expressed protein ligation is another variant of native chemical ligation where the fragment containing the C-terminal thioester is created from a recombinant protein fused to an intein20. These methods are important tools for the synthesis and semi-synthesis of proteins that are too large to make by automated peptide synthesis alone and also provide a way to create proteins bearing site-specific post-translational modifications (for example, glycoproteins) and unnatural amino acids (UAAs).
Fig. 2 |. The native chemical ligation and oxime and hydrazine ligations.
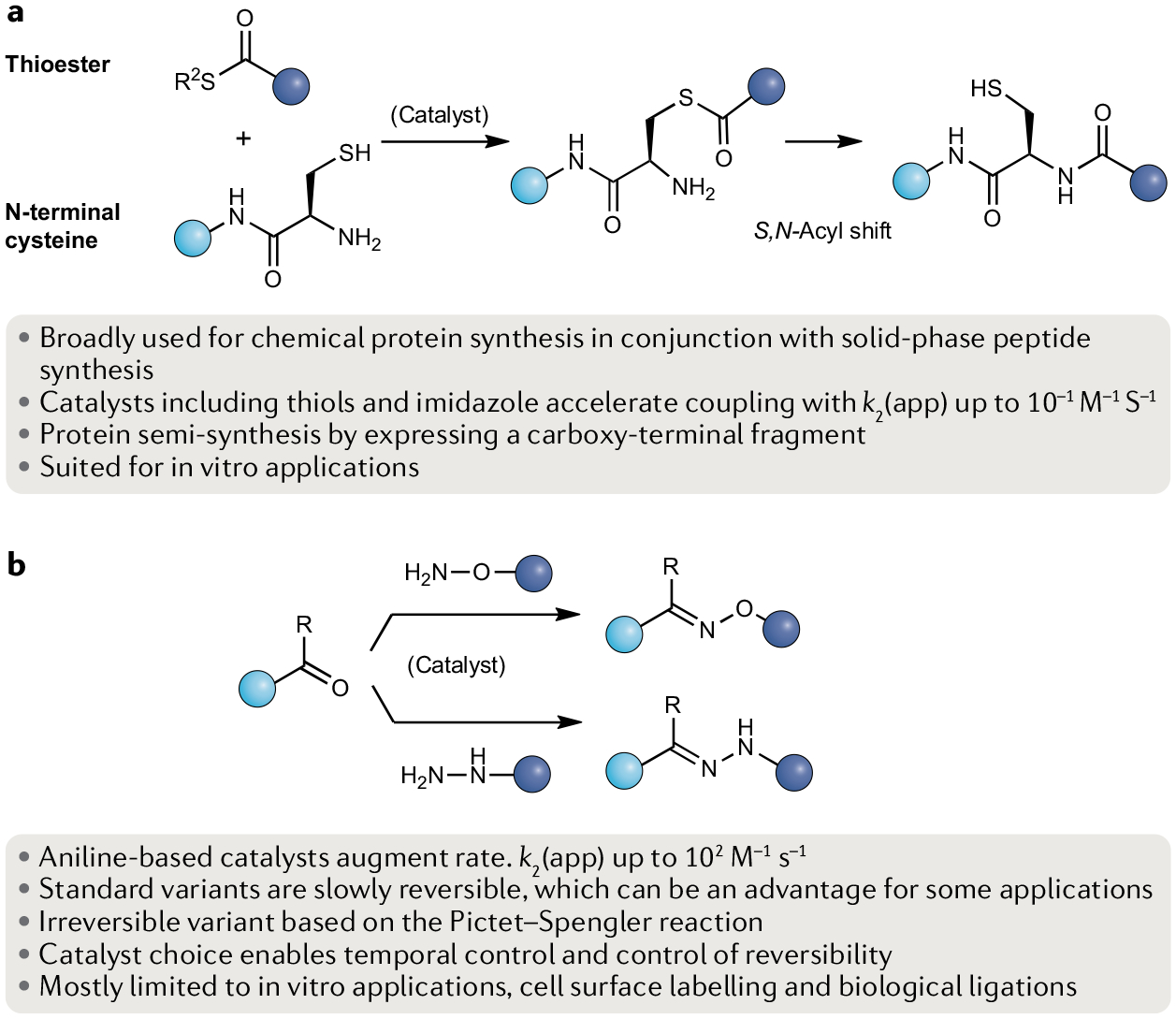
a | Native chemical ligation is enabled by a catalysed reaction between a thioester and an amino-terminal cysteine residue to afford a native amide bond through a thioester intermediate that undergoes an S,N-acyl shift. b | Using aniline-based catalysts, the oxime and hydrazone ligations occur between a carbonyl group with a hydroxylamine or a hydrazine, respectively. k2, second-order rate constant.
Nucleophilic catalysts.
Electron-rich additives that increase the reaction rate for certain polar bioorthogonal chemistries. in general, the most effective catalysts target the rate-limiting step for a given transformation. Catalysts must also adhere to the same strict requirements as bioorthogonal reagents (non-toxic, chemoselective and so on) if used in a biological context.
Post-translational modifications.
Chemical transformations that occur on reactive side chains (such as lysine, serine and cysteine) of proteins. The identity of the modification can drastically affect the function and activity of the target protein. The modifications can be installed enzymatically or can occur spontaneously in solution.
Numerous techniques complementary to native chemical ligation for the formation of amide bonds have been developed, including serine–threonine ligation, which involves a C-terminal o-formylphenolate reacting with an N-terminal serine or threonine21,22. Also, the α-ketoacid–hydroxylamine (KAHA) ligation involves a C-terminal ketoacid group reacting with N-terminal hydroxylamine functionality and the KAT ligation between O-carbamoylhydroxylamines and potassium acyltrifluoroborates23,24. Other approaches for the selective modification at a protein’s N terminus include the direct condensation of N-terminal serine25, cysteine26 and tryptophan27 side-chain nucleophiles with aldehydes to form cyclic products. Inspired by luciferin biosynthesis, N-terminal cysteine can also rapidly react with 2-cyanobenzothiazole derivatives in vitro or on the surface of live cells28. Transamination reactions with pyridoxal-5′-phosphate29 or Rapoport’s salt30 convert the N terminus into a ketone or aldehyde capable of oxime formation. One-step N-terminal conjugation can be also achieved with 2-pyridinecarboxaldehyde derivatives to form a cyclic imidazolidinone product31. In combination with solid-phase peptide synthesis, native chemical ligation methods and variants that form native amide bonds have found extensive applications in the chemical synthesis and semi-synthesis of peptides and proteins12,13.
Solid-phase peptide synthesis.
Amino acids are iteratively coupled from the carboxy terminus to the amino terminus on a solid support. Protecting group strategies ensure that only one amide bond is formed at a time, without oligomerization or cross-reactivity with reactive side chains. After cleavage from the solid support, peptides are typically purified through high-performance liquid chromatography.
Oxime/hydrazone ligation chemistry.
The formation of oximes and hydrazones from the condensation of carbonyls with hydrazines and alkoxylamines, respectively, represents some of the oldest reactions applied to biological ligation applications. Carbonyl groups were some of the first extrinsic functional groups explored as reacting partners for bioorthogonal applications32 (Fig. 2b). As products of condensation, hydrazones and oximes display higher stability towards hydrolysis than imines owing to the stabilizing effect of the heteroatom adjacent to the sp2 nitrogen atom; with oximes being more stable to hydrolysis than hydrazones33. Under physiological conditions, and especially in acidic environments (pH 5–7), however, these ligation reactions can be reversible, a feature that can be tuned for the release of desired cargo34–36. At neutral pH, these reactions proceed with a second-order rate constant (k2) of 0.01 M−1 2 s−1, slower than most commonly employed bioorthogonal cycloaddition reactions37,38. Initial findings showed improved reaction rates with lower pH conditions (pH 4–6) or the use of aniline as a catalyst, through reactive Schiff base formation39–43. Improvements in reaction rates were further achieved at neutral pH through application of substituted anilines such as 5-methoxyanthranilic, 3,5-diaminobenzoic and 2-aminobenzenephosphonic acids, with enhancements of reaction rates of up to 40 times compared with the original aniline catalysts44,45. Amine buffer systems have also been recently shown to promote condensation with fast rates46.
Schiff base.
A subclass of imine compounds characterized by a carbon–nitrogen double bond, with a general formula of R1R2C=NR3, where R3 is not a hydrogen atom. They often arise from the condensation reaction between an amine and a carbonyl, and are classified as secondary ketimines or aldimines.
The Pictet–Spengler ligation is a more hydro-lytically stable variation of oxime ligation between indolyl-substituted nucleophiles and aldehydes27, and variations using hydrazine nucleophiles have also been reported47. Carbonyls have been traditionally incorporated into biomolecules via oxidative cleavage of vicinal diols in sugar moieties using sodium periodate, allowing facile modification of glycoproteins through sialic acid oxidation48,49. Oxidative cleavage of 1,2-amino alcohols, including N-terminal serine and threonine, is an analogous strategy50. Nucleic acids can also be functionalized through sodium periodate oxidation of 3′-ribonucleotides to yield dialdehydes51. The use of carbonyl-bearing UAAs and nucleic acids also allows for the site-selective incorporation of modification sites52–54. Comparatively, fewer methods have been developed for the incorporation of alkoxyamine and hydrazine nucleophiles, mostly limited to synthetic nucleic acids, through the use of phosphoramidites to modify 5′-end ribonucleotides and appropriately functionalized nucleobases. Although improvements to reaction rates and hydrolytic stability have been achieved, these ligations are best suited to in vitro, cell surface labelling, and to biological ligation applications owing to their innate reversibility and potential cross-reactivity with carbonyl-bearing metabolites27,47–54. However, the recent development of new catalysts and more selective reacting partners has allowed for more specialized applications with reaction rates as fast as 0.1 M−1 s−1 (REFS44,45).
Phosphine-based transformations.
The Staudinger ligation — a reaction between triarylphosphines and organic azides — exploits the mild electrophilicity of organic azides and their propensity to react with mild nucleophiles55,56. Phosphine–azide chemistry was first reported by Staudinger in the context of azide reduction57. The reaction produced iminophosphorane intermediates that hydrolysed in water to give amine and phosphine oxide products. The relative simplicity of the Staudinger reaction — and the virtual absence of triarylphosphines and azides in biological settings — provided an ideal platform for biocompatible reaction development. Bertozzi and co-workers built upon this foundation to create a, now iconic, bioorthogonal transformation58. A methyl ester was installed on the triarylphosphine core, which served as an electrophilic trap for the iminophosphorane intermediate, circumventing hydrolysis and driving formation of a ligated adduct. This variant of the Staudinger reaction was termed the Staudinger ligation (or the Staudinger–Bertozzi ligation59). The Staudinger ligation was the first bioorthogonal transformation to be widely used in cells and living systems60 (Fig. 3a). The two reactants — organic azides and phosphines — are both bioavailable and biocompatible. Azides also rank among the smallest bioorthogonal functional groups, making them a go-to choice for many applications. Additionally, phosphines can be outfitted with various probes for detection in biological environments and have been used to image, retrieve and profile biomolecules56. Initially used for cell surface glycan labelling58, the Staudinger ligation has since been used to target proteins61–63, nucleic acids64 and other biomolecules65–67. Fluorogenic phosphines and azides68,69 as well as more water-soluble probes have enabled additional studies70,71. The Staudinger ligation is one of the few bioorthogonal reactions that can be used in rodent models72,73. Although versatile, the ligation still ranks among the slowest bioorthogonal chemistries74 (Fig. 3a), which inspired the pursuit of more rapid reactions and drove many early advances in bioorthogonal cycloadditions as described below.
Fig. 3 |. The Staudinger ligation types and the copper-catalysed azide–alkyne reaction.
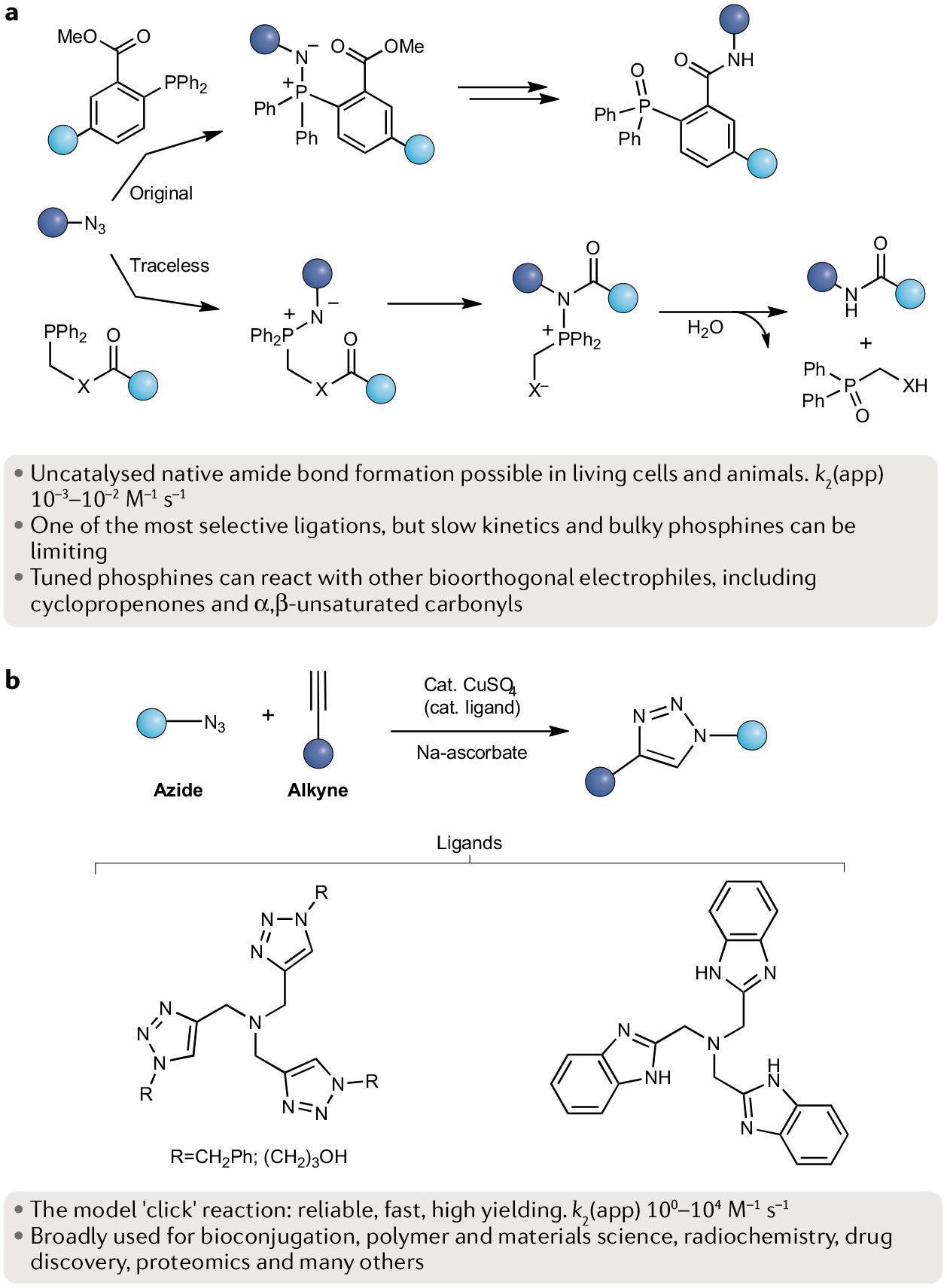
a | Both the original and traceless Staudinger ligations enable native amide bond formation between an azide and a carbonyl group using triaryl phosphines to afford the product and a phosphine oxide as a by-product. b | The model ‘click reaction’ between an azide and an alkyne enabled by use of a copper-based catalysis route to afford a triazole product. Ligand examples are provided. k2, second-order rate constant.
The Staudinger ligation also spurred the development of other phosphine-based transformations. The most well known feature phosphines with alternative electrophilic traps (for example, thioesters)75,76. Some of these variants react with azides to form amide bonds with concomitant release of the phosphine probe. Such ‘traceless’ versions of the ligation were developed contemporaneously by Raines and Bertozzi77,78 and have seen application in peptide and protein synthesis56,79,80. Alternative phosphine nucleophiles, including phosphite81 and phosphonite82 probes, have been used to functionalize peptides and proteins. These reagents react with azides to form covalent adducts under physiological conditions.
Bioorthogonal phosphines have also been used for ligation chemistries with electrophiles other than azides. Recent examples include Michael-type reactions with α,β-unsaturated amides83,84 and cyclopropenone derivatives85–88.
Azide–alkyne cycloaddition.
The azide and terminal alkyne functional groups are very small and lack the ability to associate with other molecules by anything more stable than weak dipolar interactions. Accordingly, they often minimally perturb the functional properties of the molecules to which they are attached. Azides and alkynes are largely unreactive with most other molecules in biological systems, and so can move through cells and organisms intact, only to be revealed when provided with the right conditions or catalysts for their reaction.
The Huisgen 1,3-dipolar cycloaddition of organic azides and alkynes, giving 1,2,3-triazoles, is usually much too slow to be of use in biological applications. The introduction of copper(I) catalysts, reported independently by the groups of Meldal89 and Sharpless90, transformed the process into one of wide use by accelerating triazole formation up to 100 million-fold (Fig. 3b). Copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) enables the exclusive formation of 1,4-disubstituted triazoles, which are good mimics of native peptide-based trans-amide connectors, in terms of size and geometry, and are far more stable than amides to thermal and chemical cleavage. The CuAAC reaction is modular, compatible with aqueous conditions, usually free of by-products and user-friendly, all characteristics that enable successful click reactions4. Its mechanism is stepwise, proceeding through an easily formed binuclear copper acetylide intermediate91,92. A wide range of catalysts have been employed, including simple copper(I) salts90, small-molecule copper-binding ligands92,93 and heterogeneous, polymeric and nano-structured materials of different kinds94–96. The development of CuAAC has driven many applications in the realms of organic synthesis, polymer functionalization, drug discovery and chemical biology.
Although a few examples exist of terminal alkyne biosynthetic pathways in microorganisms and fungi97,98, naturally occurring azides are exceedingly rare. Proteins, lipids, oligonucleotides and cell surface moieties can be labelled with azides or alkynes through benign chemical transformations or via metabolic incorporation of labelled metabolites, allowing their conjugation to an orthogonally labelled partner. The use of CuAAC for bioorthogonal bioconjugation was largely enabled by the development of water-soluble copper-binding ligands, which allowed users to address the most striking limitation of CuAAC for biology: the copper-mediated formation of reactive oxygen species99–101. These ligands stabilize the required copper(I) oxidation state and provide access to coordination sites on the metal, accelerating the reaction while minimizing copper-mediated oxidative stress. Ligands with notable characteristics such as the ability to scavenge reactive oxygen species99–101 or to enhance local concentrations of reactive partners by combining substrate and copper-binding moieties102–107 have further allowed CuAAC to be adapted for more demanding biological applications such as the labelling of living cellular surfaces108, labelling tagged enzymes or other biomolecules in complex cellular mixtures109, fast click reactions inside cellular compartments110,111 and versatile dye labelling of a wide variety of cellular structures for super-resolution expansion microscopy112.
Reactive oxygen species.
Highly reactive forms of oxygen involved in diverse cellular signalling processes, and tightly regulated in cells. For bioorthogonal chemistry, reactive oxygen species arise from the oxidation of copper(i) to copper(ii) in water, which generates damaging superoxide or hydroxyl radicals. An accumulation of reactive oxygen species damages nucleic acids, proteins and lipids, and is cytotoxic.
A few limitations remain in the application of CuAAC to bioconjugation. First, copper is highly regulated in cells, and tightly bound by a network of interacting proteins and chaperones; thus, the best copper-binding ligands for CuAAC cannot retain the metal ion when encountering endogenous copper-binding proteins113. In addition, thiols — abundant in the form of protein cysteines and intracellular glutathione — bind copper tightly, and thereby inactivate catalysts and trigger copper-mediated oxidative stress114. A systematic evaluation of copper-mediated toxicities in various human cell lines illustrates the practical consequences of these phenomena114. Copper-free variants of CuAAC, detailed below, were developed in part to address these limitations, but suffer from reduced reaction rates, susceptibility to unproductive side reactions or use of sterically larger connecting groups that can compromise function. Thus, fully biocompatible accelerating ligands for CuAAC, able to rapidly catalyse intracellular reactions without inducing biological stress and exchanging copper with endogenous biomolecules, await discovery.
Strain-promoted [3 + 2] cycloadditions.
Strained cyclooctynes were introduced as an alternative to CuAAC to circumvent copper catalysts in various applications115. Cyclooctynes are the most commonly used substrates for strain-promoted azide–alkyne cycloadditions (SPAAC; as shown in Fig. 4a), with rates that are rapid enough to be used in living systems without the need for a catalyst116,117. Many different cyclooctynes have been developed specifically for use in SPAAC reactions, including a difluorinated cyclooctyne (DIFO)116,118,119 (Fig. 4a). However, early versions of cyclooctynes (OCT; Fig. 4a) suffered from slow reaction rates and poor aqueous solubility. These limitations spurred the development of alternative scaffolds that enabled rates as fast as 1 M−1 s−1, including bicyclic derivatives (for example, bicyclo[2.1.0]nonyne (BCN))120 (Fig. 4a), benzannulated cyclooctynes (for example, biarylazacyclooctynone (BARAC) and dibenzocyclooctyne (DIBO or DBCO))121–124 (Fig. 4a) and more hydrophilic variants125–127. More highly strained cycloheptynes have also been used for SPAAC reactions to achieve further rate acceleration (for example, 3,3,6,6-tetramethylthiaheptyne (TMTH))128–130 (Fig. 4a). Fluorogenic cyclooctynes serve to both conjugate and introduce reaction-dependent fluorescence read-outs for SPAAC reactions131,132. Distortion/interaction modelling has also been a useful tool for tuning the reactivity of strained alkyne systems133–135. Overall, there are many options for using SPAAC chemistry to report on different biological events or to connect components, making this bioorthogonal chemistry a popular choice for in situ and in vivo applications.
Fig. 4 |. Cycloaddition-based bioorthogonal chemical reaction types.
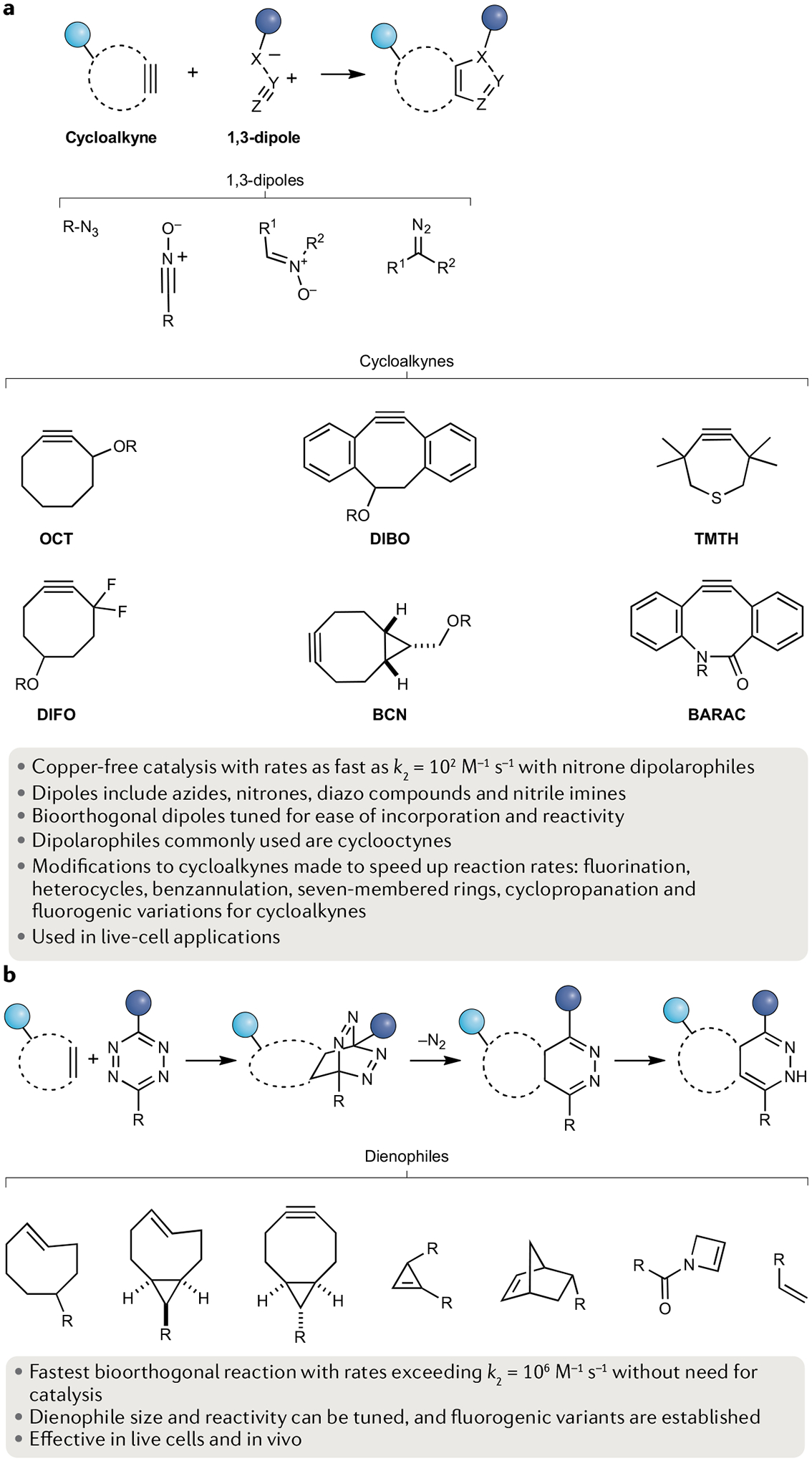
a | Strained alkynes enable copper-free catalysis of cycloaddition to dipoles such as azides. b | The fastest biorthogonal reaction involves the inverse-electron demand [4 + 2] addition between a tetrazine and a dienophile. BARAC, biarylazacyclooctynone; BCN, bicyclo[2.1.0]nonyne; DIBO, dibenzocyclooctyne; DIFO, difluorinated cyclooctyne; k2, second-order rate constant; OCT, cyclooctyne; TMTH, 3,3,6,6-tetramethylthiaheptyne.
Although SPAAC reactions are both versatile and robust, dipoles other than azide have been found to react with faster rates in the analogous cycloaddition reactions, which can be critical for the success of bioorthogonal reactions in living systems. Different dipoles also offer additional sites for stereoelectronic tuning of their reactivity. As an example, strain-promoted alkyne–nitrone cycloaddition reactions involve nitrone dipoles that are faster and have three sites of substitution available for reaction tuning136–139. Other dipoles that react with cyclooctynes include diazo compounds and nitrile oxides140–143. Strain-promoted sydnone–alkyne cycloadditions are also versatile and rapid alternatives to SPAAC reactions, which have been applied in positron-emission tomography imaging using 18F-labelled antibodies, where fast reaction rates are required144–146. The strain-driven quadricyclane (QC) ligation involving nickel bis(dithiolene) derivatives offers another unique tool for bioorthogonal bond creation147. Taken together, these examples illustrate the large and growing number of possibilities for fast and efficient bioorthogonal chemistry utilizing strained alkynes with different dipoles as reaction partners.
Inverse electron-demand Diels–Alder reactions.
The bioorthogonal tetrazine ligation refers to the reaction between a 1,2,4,5-tetrazine and an alkene or alkyne dienophile via a sequence of [4 + 2]/retro [4 + 2] cycloaddition to provide a dihydropyridazine or pyridazine conjugate (Fig. 4b). Tetrazine ligation is an IEDDA reaction that is notable for rapid kinetics without the need for catalysis, which yields nitrogen gas as the only by-product. Tetrazines initially found utility in organic synthesis148,149 and were noted for their rapid kinetics in reactions with strained dienophiles150.
Dienophile.
An alkene or alkyne that reacts with a conjugated diene in [4 + 2] cycloadditions. Diels–Alder cycloadditions are enabled by electron-poor dienophiles and electron-rich dienes. Conversely, inverse electron-demand Diels–Alder reactions occur between electron-rich dienophiles and electron-poor dienes.
In 2008, the bioorthogonal reactions of tetrazines with strained alkenes were first described151–153, including the reactions with derivatives of TCO and norbornene by the groups of Fox and Weissleder, respectively151,152. There has since been a growing diversity of chemical richness for both tetrazines and their dienophile reaction partners for the tuning rate, stability and design of ‘minimal’ reporter molecules. As an IEDDA reaction, the tetrazine ligation is accelerated by electron-withdrawing groups on the tetrazine.
Tetrazines are most often prepared through condensation of Pinner salts or nitriles with hydrazine and facilitated by Lewis acidic, thiol or sulfur catalysts154–156. Milder protocols have been developed for directly introducing intact tetrazine groups via palladium-catalysed coupling reactions of aryl halides or aryl boronic acids157,158. Carboxylic esters also serve as a handle for the preparation of unsymmetrical and monosubstituted tetrazines159.
A range of dienophiles have been developed for tetrazine ligation. TCO can be prepared by flow photochemistry160,161, and variants have been designed that combine with tetrazines in the most rapid bioorthogonal reactions known — with k2 = 104–106 M−1 s−1 under aqueous conditions162,163 — with applications that extend to radiochemistry, in vivo chemistry, cell imaging and click to release chemistry. Highly strained cyclooctynes such as BCN also react rapidly (k2 = 102–103 M−1 s−1) with tetrazines, and produce aromatic conjugates that can simplify workflows where defined stereochemistry is needed164. Cyclopropenes (k2 = 1–104 M−1 s−1) are the smallest class of ring-strained dienophiles, offering a complementary approach when size and stability are important, such as for genetic encoding and metabolic incorporation165,166. Norbornene retains good kinetics at a low cost and has proven an advantage for scale-up and materials applications. Small dienophiles such as cyclobutenes, azetidines, unstrained olefins and isonitriles also react efficiently with tetrazines but typically require large excess of one reagent owing to slower kinetics167–171. Similarly, triazines can serve as surrogates for tetrazines that offer advantages of smaller size and greater stability172.
Another useful feature of tetrazine ligation is that many tetrazine–fluorophore conjugates are fluorogenic. Resonant energy transfer to the tetrazine reduces fluorescence, but upon reaction with a dienophile the fluorescence is ‘switched’ on with up to an 11,000-fold increase in fluorescence intensity173–175. Fluorogenic tetrazine–fluorophores have found particularly strong use in the field of live-cell microscopy, and the area is actively growing with a focus on improving the signal to noise ratio that can result from background tetrazine reactivity in the intracellular environment176,177. External cues for temporal control of the tetrazine ligation have been developed for photoactivatable cyclooctynes or redox activation of dihydrotetrazine to tetrazines using electrochemistry, photocatalysts or enzymes178–180. This toolkit of dienes and dienophiles has enabled an exciting range of applications as further detailed throughout the applications section below.
Photoinducible bioorthogonal chemistry.
In contrast to thermal processes, light-triggered reactions often display higher yields and selectivity without the need for transition metal-mediated catalysis. The first example of using light to initiate selective reactions efficiently in biological systems is a photoinduced 1,3-dipolar cycloaddition reaction between a 2,5-diaryltetrazole and an alkene, often referred to as photoclick chemistry181 (Fig. 5a). This chemistry was based on the seminal work by Huisgen, who reported outstanding reactivity of the transient nitrile imine dipole generated photochemically from 2,5-diphenyltetrazoles in benzene182. Milder conditions using a handheld low-powered UV lamp183 facilitated the rapid development of tetrazole photoclick chemistry for bioorthogonal protein modifications in vitro184 and inside bacterial cells185. Mechanistically, the reaction proceeds through a photoinduced cycloreversion to exude N2 and generate a highly reactive nitrile imine followed by [3 + 2] cycloaddition with an alkene dipolarophile. A prominent feature of tetrazole photoclick chemistry is that the pyrazoline adducts are fluorescent, making the reaction fluorogenic184.
Fig. 5 |. Light-activated click chemistry and metal-catalysed coupling reactions.
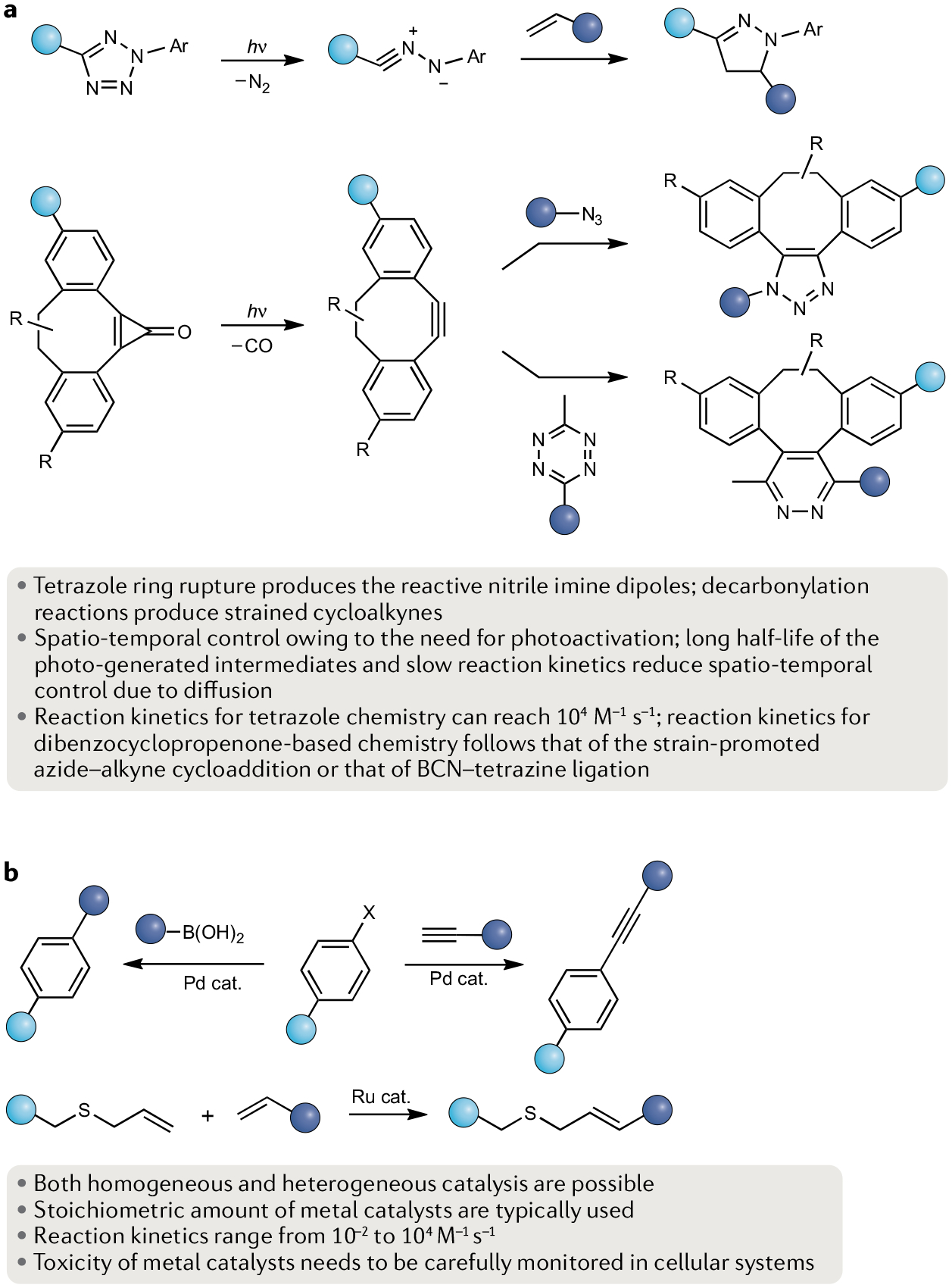
a | Two examples of photoclick chemistry: a photoinduced tetrazole-alkene 1,3-dipolar cycloaddition reaction to generate a pyrazoline adduct (top) and a photo-triggered alkyne-azide cycloaddition reaction (bottom). The cycloalkyne is masked in the dibenzocyclopropenone form. b | Two examples of palladium-catalysed reactions: Suzuki–Miyaura cross-coupling and copper-free Sonogashira cross-coupling (top left and right, respectively), and ruthenium-mediated olefin cross-metathesis (bottom) involving the use of allyl chalcogen-based privileged substrates. BCN, bicyclo[2.1.0]nonyne.
Photoclick chemistry.
Click chemistry in which reactions that are initiated using light as an external stimuli. Photoclick reactions can use light sources ranging from short-wavelength to near-IR light and allow for spatial and temporal control of reactions.
2,5-Diaryltetrazoles can be prepared from cycloadditions of azides with nitriles, condensations of phenylsulfonyl hydrazide with arene diazonium salts or copper-catalysed regioselective N-arylation of 5-aryl-2H-tetrazoles with either phenyl iodonium salts or aryl boronic acids186. As the rate of cycloaddition is dictated primarily by the HOMO (dipole)–LUMO (dipolarophile) energy gap, electron-rich tetrazoles187 and electron-deficient alkenes such as acrylamide188 and fumarate189 are privileged substrates for tetrazole photoclick chemistry. Because of these electronic requirements, tetrazole photoclick chemistry is mutually exclusive to the tetrazine ligation190. Tetrazoles bearing the steric shielding groups show improved reaction selectivity191. For enhanced spatio-temporal control, photoactivation of tetrazoles can be achieved using a 405-nm laser192 and a near-IR femtosecond laser193 by modifying tetrazole structures as well as combining a near-IR laser with nanoparticles capable of two-photon upconversion194.
HOMO.
(Highest occupied molecular orbital). A molecule’s highest energy molecular orbital containing an electron pair.
LUMO.
(Lowest unoccupied molecular orbital). A molecule’s lowest energy molecular orbital not containing an electron. The energies of HOMO and LUMO are related to the reactivity of the molecule and the energy difference between the HOMO and LUMO is termed the HOMO–LUMO gap.
Two-photon upconversion.
A molecule is excited from the ground state (S0) to the second excited singlet state (S2) by simultaneous absorption of two photons, via a virtual state. A photon with frequency greater than those of the absorbed photons is emitted upon relaxation from the excited state, that is, two-photon upconversion.
For increased stability in biological systems, strained alkenes such as 3,3-disubstituted cyclopropene195, spiro[2.3]hex-1-ene (REF.196) and water-soluble azaspiro[2.3]hex-1-ene (REF.197) have been developed for tetrazole photoclick chemistry with robust reaction kinetics (k2 = 102–104 M−1 s−1). The alkene-based chemical reporters, along with the optimized tetrazole reagents, have allowed bioorthogonal labelling of membrane proteins191, DNA198, RNA199 and glycans200. The fluorogenic property has been exploited in the design of photoactivatable fluorescent probes for cellular proteins201 and the detection of oncometabolites202.
The spatio-temporal control afforded by photoinduction can be infused with SPAAC. For example, a dibenzocyclopropenone undergoes decarbonylation upon photoirradiation at 350 nm to generate dibenzocyclooctyne, which then reacts rapidly with azides to form the cycloadducts203 (Fig. 5a). The utility of this dibenzocyclopropenone-based photoclick chemistry was demonstrated through successful glycan labelling203 as well as surface immobilization204. More recently, a cyclopropenone‐caged dibenzoannulated bicyclo[6.1.0] nonyne probe was applied in conjugation with tetrazine ligation using irradiation at 365 nm (REF.179). Spirocyclic aminocyclopropenes can be protected with either light or enzyme labile groups and uncaged for controlled reactions with tetrazines205. The photoclick chemistry toolbox also includes hetero-Diels–Alder reactions based on 2-naphthoquinone-3-methide (REF.206), o-quinodimethane207 and 9,10-phenanthrenequinone208, light-induced tetrazine ligation180 and diarylsydnone–alkene photoligation209.
Palladium-catalysed and ruthenium-catalysed bioorthogonal reactions.
The power of palladium-catalysed cross-coupling reactions has been harnessed for biomolecular functionalization in vitro and in living cells (Fig. 5b). Early efforts uncovered conditions with limited reaction efficiency and biocompatibility210–213, including high catalyst loading, high temperature and the presence of organic co-solvent, but further developments have led to the discoveries of novel catalyst systems and additives that enable efficient palladium-mediated cross-coupling reactions with a broader substrate scope and utility in biological systems214,215.
In homogeneous catalysis, the palladium–2-amino-4,6-dihydroxypyrimidine catalyst allowed fast and high-yielding Suzuki–Miyaura cross-coupling between proteins bearing p-iodobenzylcysteine and various aryl and vinyl-boronic acids in phosphate buffer216. The palladium–2-dimethylamino-4,6-dihydroxypyrimidine catalyst enabled copper-free Sonogashira cross-coupling between homopropargylglycine-encoded proteins and aryl iodides in aqueous media as well as inside Escherichia coli217. The aryl–palladium(II) complexes from decarboxylative palladation of substituted benzoic acid derivatives enabled functionalization of a styrene-modified protein with biotin and a cyanine dye via a Heck-type mechanism218. In heterogeneous catalysis, polystyrene microspheres encapsulating Pd0 allowed cytosolic uptake of the palladium catalyst and subsequent in situ synthesis of a fluorescent dye via Suzuki–Miyaura cross-coupling inside HeLa cells219. The main advantage of using the palladium-encapsulated microspheres is that the palladium catalysts stay inside cells to perform intracellular chemistry for an extended period without posing a toxicity risk.
Homogeneous catalysis.
The catalyst and reaction mixture are in the same phase.
Heterogeneous catalysis.
The catalyst and reaction mixture are in a different phase.
Obtaining kinetic parameters for these cross-coupling reactions is challenging owing to the complex reaction mechanisms involved and the excess amount of palladium catalysts used. In cases where stable palladium catalysts were used, the apparent k2 values were determined to be 0.011 M−1 s−1 for Suzuki–Miyaura cross-coupling220 and 5.2 M−1 s−1 for copper-free Sonogashira cross-coupling221. By placing an alkyne reporter in the middle of a selected protein sequence environment to facilitate recruitment of the palladium complex, faster reactions were observed for Sonogashira cross-coupling with apparent k2 values as high as 1.3 × 104 M−1 s−1 (REF.222).
Palladium-mediated cross-coupling reactions have been used for modifying proteins with fluorophores218,223, polyethylene glycol224 and carbohydrates224 in vitro, on the surface of E. coli225, inside bacterial cells217 and on the mammalian cell surface220–222, as well as for modifying DNA226. As palladium is an abiotic transition metal, the toxicity225 and cell permeability of palladium complexes need to be carefully monitored in cellular applications. Also, non-specific sequestration of palladium catalysts by biomolecules227 may further decrease the cross-coupling reaction efficiency.
Ruthenium-mediated cross-metathesis of olefins has also been adopted for bioorthogonal chemistry (Fig. 5b). As ruthenium alkylidenes are highly reactive and prone to decomposition in protic solvents, the use of allyl chalcogen-based privileged substrates for accelerated cross-metathesis228, MgCl2 as a competitive, hard Lewis acid and tert-butanol as co-solvent has proven crucial. This unique reactivity was harnessed for modifying proteins bearing allyl chalcogen tags such as S-allylcysteine229 and Se-allyl selenocysteine230 in vitro using the Hoveyda–Grubbs second-generation catalyst.Kinetic studies231 showed that Se-allyl-selenocysteine is ten times more reactive towards allyl alcohol than S-allyl-cysteine, with an apparent k2 value of 0.3 M−1 s−1. The development of a genetically encoded allylsulfide reporter, S-allylhomocysteine, further enhanced the utility of this chemistry232. It remains to be seen whether this reaction is sufficiently robust for cellular applications.
Lewis acid.
A chemical species that can accept a pair of non-bonding electrons.
Safety.
Bioorthogonal reagents are designed to react quickly. Accordingly, the energetic properties of any bioorthogonal reagent that is purchased or prepared should be carefully considered prior to use. Safety analysis is especially important for nitrogen-rich compounds including azides, diazo compounds, tetrazines and tetrazoles, or compounds with other high-energy functional groups (for example, alkynes and nitro groups). For reagents that are synthesized, the safety of all synthetic intermediates should be critically evaluated even for compounds already known in the literature. Some common starting materials for bioorthogonal chemistry also have documented safety concerns. For example, sodium azide (used to prepare reagents for CuAAC and SPAAC) is incompatible with acids and halogenated solvents such as dichloromethane233, and anhydrous hydrazine (often used to prepare tetrazines) is highly toxic, energetic and not available for purchase in all countries234.
For nitrogen-rich bioorthogonal reagents, it is generally observed that the safety profile improves with an increasing carbon to nitrogen ratio. One rule of thumb is to avoid compounds that do not have at least six carbons (or other atoms of about the same size) per high-energy functional group (such as azide, diazo, nitro and so on)4. However, there are exceptions235, and therefore testing safety properties is recommended for any compound with a high-energy functional group. Differential scanning calorimetry (DSC) represents a simple and informative initial measurement of energetic safety that requires only milligrams of material. In DSC, heat flow associated with phase transitions is measured as a function of time and temperature. To obtain accurate data, DSC analyses of energetic compounds must be performed in sealed pans rated for pressure. Energetic compounds display an exothermic decomposition that can be analysed for onset temperature, decomposition energy and rate of decomposition. Based on these DSC data, the Yoshida correlation can also be used to inform whether to further test for explosivity or impact sensitivity of the material236. Therefore, DSC provides a good starting point for understanding the overall energetics of a material237, and to help guide the reaction scale and safety precautions (for example, blast shields) that should be employed238.
Differential scanning calorimetry.
(DSC). A technique that measures heat flow rates to determine phase transitions and quantitative heats of decomposition of a compound of interest. DSC requires only milligram quantities of a sample and provides a rapid measurement of the thermal properties of a compound.
Yoshida correlation.
The impact sensitivity and explosive propagation properties of compounds can be derived from differential scanning calorimetry data.
Applications
Bioorthogonal chemistry is rich in chemical diversity and offers a palette of tools for many applications. Live-cell imaging has been revolutionized by bioorthogonal reactions and these chemistries have been essential for genetic code expansion and detecting chemical reporters following their metabolic incorporation into target biomolecules239. Activity-based protein profiling (ABPP) utilizes advancements in reaction and probe design for understanding protein function in complex biological systems240. Applications of bioorthogonal chemistry in polymer and materials science and surface chemistry have expanded significantly, including fabrication of three-dimensional (3D) networks, hydrogels for live cells and drug-delivery strategies241. Advances in reaction chemistry have not only been used for bond-forming reactions but also bond-breaking ‘decaging’ reactions for release or activation of biomolecules or small molecules242. Together, the tools available for bioorthogonal chemistry have provided a diverse set of methods that are impacting multidisciplinary research and are expanding to medicinal applications.
Metabolic engineering.
Metabolic engineering is a tactic used to install non-natural functional groups into target biomolecules using the cell’s own enzymatic machinery. Metabolites outfitted with bioorthogonal functional groups (chemical reporters) can hijack biosynthetic pathways and be incorporated into target biomolecules239. Once installed, the reporters are detected via bioorthogonal ligation with complementary reagents243–245 (Fig. 6a). This two-step approach (the chemical reporter strategy) has been routinely used to image and profile proteins61, glycans246,247 and other biomolecules248,249 in cells and other environments250–252 (Fig. 6b). Importantly, the chemical reporter strategy provides a mechanism to examine structures that are difficult to study via genetic manipulation, such as lipids67,253, small-molecule cofactors254 and other secondary metabolites255.
Fig. 6 |. Applications for labelling different molecule types in cells.
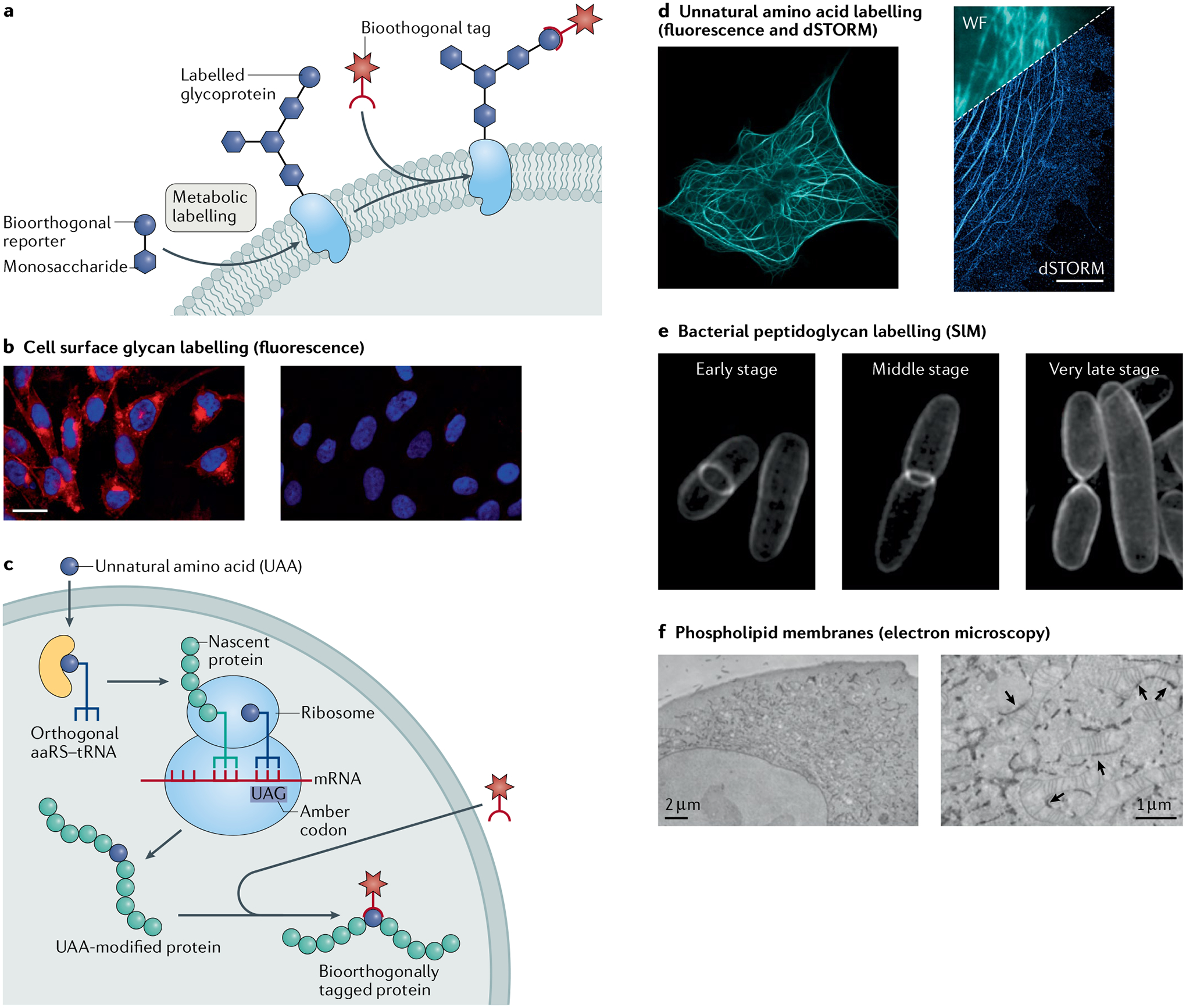
a | Model for metabolic engineering for cell labelling and imaging. b | Fluorescence microscopy of CHO cells incubated in the presence (left) or absence (right) of peracetylated N-azidoacetylmannosamine (Ac4ManNAz) and labelled with a fluorophore by the Staudinger ligation. c | Model for genetic code expansion as a strategy for cell labelling and imaging. d | Fluorescence and direct stochastic optical reconstruction microscopy (dSTORM) super-resolution images of COS-7 cells where microtubule-associated protein was encoded with an unnatural trans-cyclooctene (TCO) amino acid and tetrazine ligation was used to attach a microscopy dye. e | Structured illumination microscopy (SIM) images of Escherichia coli, where N-azidoacetyl-muramic acid (NAM) was metabolically incorporated into the bacterial peptidoglycan and fluorophore-labelled by copper(I)-catalysed azide–alkyne cycloaddition (CuAAC). f | Electron microscopy images of HeLa cells, where azido-choline was metabolically incorporated, and cyclooctyne/azide click chemistry was used to conjugate electron microscopy imaging agents. The arrows indicate sites of endoplasmic reticulum–mitochondria contacts. aaRS, aminoacyl-tRNA synthetase; WF, widefield image. Images in panel b adapted with permission from REF.252, ACS. Images in panel d reprinted from REF.176, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Images in panel e reprinted from REF.286, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Images in panel f reprinted from REF.296, Springer Nature Limited.
The most widely used chemical reporters are the azide and alkyne. These motifs are among the smallest and most stable bioorthogonal functional groups, and they are tolerated by a wide variety of cellular enzymes256,257. Azides and alkynes can also be readily appended to biosynthetic precursors for metabolic labelling258. Typical detection chemistries include CuAAC (primarily in the context of fixed or lysed samples) or SPAAC (in the case of live cells)259,260. Other bioorthogonal functional groups can be employed for metabolic engineering, although smaller functional groups are the most useful. Ketones261 and cyclopropenes165,262 are included in this group. Importantly, these motifs (and their ligation chemistries) are orthogonal to typical azide/alkyne reactions, enabling multicomponent detection.
Metabolic labelling strategies — although straightforward — are not often selective for a single target. The metabolite employed can be incorporated into multiple biomolecules, depending on the enzymatic pathways employed, because some metabolites are common to multiple biosynthetic pathways. Additional modifications to probes and pathways are necessary to label specific cell populations or individual biomolecules263–265. For example, cell-selective labelling can be achieved using targeted delivery strategies or caged metabolites that are released in response to specific enzymes. Caged probes have been primarily used to label cancer cell targets264,266, although other cell and tissue types can be labelled. Biosynthetic enzymes can also be engineered to selectively process non-natural metabolites267–269, and are the underlying strategy for site-specific protein tagging/genetic code expansion technologies with bioorthogonal amino acids.
Genetic code expansion.
Metabolic engineering approaches, such as residue-specific incorporation of UAAs bearing bioorthogonal side chains, allow globally modifying proteins and targeting proteomes of entire cells or organisms270, but genetic code expansion approaches enable the site-specific genetic encoding of UAAs to modify proteins with medicinal chemistry-like precision271,272. Genetic code expansion relies on the use of an orthogonal aminoacyl-tRNA synthetase (aaRS)–tRNA pair to direct co-translational UAA incorporation in response to an amber stop codon introduced at a chosen position in a gene of interest. Orthogonality refers to mutual and exclusive recognition of an orthogonal aaRS–tRNA pair for charging the UAA in the host of choice without interfering with endogenous aaRS, tRNAs and natural amino acids. In the past 20 years, numerous orthogonal aaRS–tRNA pairs have been developed for incorporation of UAAs with bioorthogonal functionalities271 to site-specifically modify and label proteins in bacteria, yeast, mammalian cells and multicellular model organisms (Fig. 6c). This strategy offers a modular approach for installing diverse probes with a single genetic system and removes limitations that the translational machinery may place on the size of probes. The most common and useful bioorthogonal UAAs that can be encoded include keto, azide, terminal alkyne, strained alkyne/alkene and tetrazine functionalities and have been used for site-specific protein conjugation through oxime ligation, Staudinger ligation, CuAAC, SPAAC, photoclick and IEDDA reactions271,273. Many of these approaches have been used for generating in vitro defined protein conjugates such as PEGylated and biotinylated proteins, antibody–drug conjugates (ADCs) and proteins labelled with spectroscopic probes such as fluorophores and spin labels. An important recent advance has been the encoding of IEDDA reaction partners274, such as strained alkenes/alkynes164,275,276 and tetrazines179,277,278 that permit rapid and selective labelling in live cells (both on the cell surface and intracellularly; shown in Fig. 6d) and have allowed researchers to visualize and image proteins in their native environment176,275,279–281 and to monitor protein turnover282. Site-specific incorporation of two distinct bioorthogonal UAAs into the same protein using further engineered translational machineries has enabled selective dual protein labelling for applications such as Förster resonance energy transfer studies on protein folding283,284. Furthermore, the exquisite selectivity and fast rate (Fig. 4b) of the IEDDA reaction between a site-specifically incorporated cyclooctyne-bearing UAA and a tetrazine–inhibitor conjugate has been used to selectively inhibit a target enzyme within live cells285. Introducing a photoisomerizable linker into tetrazine–inhibitor conjugate has allowed reversible switching of enzyme activity with light in living mammalian cells285.
Cell imaging.
Bioorthogonal chemistry has proven a powerful and versatile approach for imaging various processes in living cells. Strategies for cell imaging that allow for direct attachment of reporter molecules to an expanded set of important biomolecules are free of conventional restraints imposed by genetically fused tags, and benefit from a researcher’s choice of chemistry for their particular application(s). Many bioorthogonal reactions have been used for cell imaging, but some are more well suited than others. For example, CuAAC and SPAAC have been used in bacteria to image peptidoglycan remodelling during cell division286 (Fig. 6e), and SPAAC has been used to image differential expression of different glycosylated proteins within cells and transparent model organisms such as zebrafish246 and Caenorhabditis elegans287. Strain-promoted alkyne–nitrone cycloaddition reactions have been used to image external growth factor receptors136, and for duplex labelling to image different bacterial species simultaneously139. In combination with genetic code expansion to site-specifically incorporate strained dienophile-bearing UAAs, the tetrazine ligation has enabled labelling and imaging of cell surface and cytoskeletal proteins by super-resolution microscopy176,279,288,289 (Fig. 6f). Furthermore, the tetrazine ligation has been used to image and enrich diverse proteomes in bacteria290, mammalian cells290, Drosophila melanogaster291 and the mouse brain292 in a temporally and spatially resolved fashion. Aside from being spatially permissive and applicable to different types of biomolecule, bioorthogonal chemistry strategies enable the use of newer and advanced imaging agents.
A suite of organic fluorophores covering the entire visible spectrum can be chosen for super-resolution microscopy techniques. In addition, some bioorthogonal reagents and fluorophore pairs are capable of turn-on fluorescence to enhance the signal to noise ratio132,174,201,293–295. Depending on the application, the turn-on approach has enabled ‘no-wash’ imaging strategies175. The groups of Bertozzi and Tsien combined bioorthogonal chemistry with electron microscopy in creative ways to perform super-resolution imaging of non-protein biomolecules. Selected dyes could be used to image cells by fluorescence, and then these cells were fixed and the dyes used to photooxidize the electron microscopy stain diaminobenzidine (DAB) to create contrast for electron microscopy imaging296 (Fig. 6d). This approach led to unprecedented resolution in images of subcellular localization of DNA, RNA, lipids and metabolites. Another complementary technique is bioorthogonal Raman spectroscopy, where spectroscopically bioorthogonal Raman tags, such as alkynes, azides and nitriles, are directly engineered into biological targets in live cells297. Taken together, these bioorthogonal chemistry approaches provide a wide variety of possibilities for molecule-specific imaging of cells with different imaging modalities.
Pharmaceutical applications.
Bioorthogonal chemistry has found widespread use in drug discovery programmes, including applications in target identification (for example, phenotypic screening), target engagement and selectivity profiling of lead compounds298. An immensely valuable method to evaluate these directions has been ABPP, which assesses protein functionality in complex physiological systems299–301. As compared with assessing mRNA or total protein levels, ABPP differentiates active proteins from inactive proteins resulting from zymogens, post-translationally modified and/or inhibitor-bound forms. This method is dependent on an activity-based probe that consists of a chemical ‘warhead’ (for example, an electrophile), designed to covalently react in an activity-dependent manner, attached via a linker to a reporter group that allows for downstream analysis of labelled proteins by in-gel fluorescence, western blot and/or mass spectrometry-based proteomics that are attached to each other with a linker299,300. Relevant applications of ABPP include identification of activity in different cancer cell lines302, characterization of aberrant enzymatic activity in aggressive cancer cells303 and illumination of previously uncharacterized protein targets302–305.
Zymogens.
inactive forms of an enzyme. The enzyme takes its active form following a natural biochemical process such as cleavage, hydrolysis or post-translational modification.
When evaluating reversible small molecules as ligands across the proteome, a probe can be designed for photoaffinity labelling301,306,307. In addition to a protein binding element, photoaffinity probes possess a photo-reactive group, such as benzophenone, phenyl azide or diazirine. Upon irradiation with UV light, these groups form reactive species, each by a unique chemical mechanism, to generate covalent bonds with proximal amino acid residues306,307. Similar to ABPP, a reporter group is necessary for identification. Photoaffinity labelling has been useful for finding protein targets in phenotypic screens306, as well as off-target interactions that can lead to undesired toxicological outcomes, and discovering novel protein–protein interaction partners as exemplified by the interaction of IFITM3 with γ-secretase308,309.
Common tagging groups for activity-based and photoaffinity probes include biotin and various fluorophores; however, the incorporation of these large reporter groups can negatively impact cellular permeability and localization. Towards this end, less perturbative CuAAC chemistry handles (for example, azide or alkyne) have been incorporated to improve the physiochemical properties of the probes240,310,311. Copper-free clickable handles have also been utilized in activity-based probes to increase experimental throughput and avoid copper removal steps that can result in protein loss312. These bioorthogonal probes have been useful for screening compound libraries in the development of selective inhibitors305,313 and for assessing target engagement and proteome selectivity for small-molecule drug candidates314–316.
Bioorthogonal chemistry handles also grant modularity during a proteomic workflow for both ABPP and photoaffinity labelling (Fig. 7a). For example, cleavable linkers and/or isotopic reporters can be incorporated to assess sites of engagement and/or perform quantitative analysis317,318. A variant of ABPP employs a generally reactive warhead, such as cysteine-reactive iodoacetamide appended to an alkyne handle, to discover novel ligandable hotspots in the proteome319,320. These ligands can provide useful handles for developing novel small-molecule inhibitors or for other therapeutic modalities such as targeted protein degradation321.
Fig. 7 |. Examples of biorthogonal chemistry applications in vitro and in vivo.
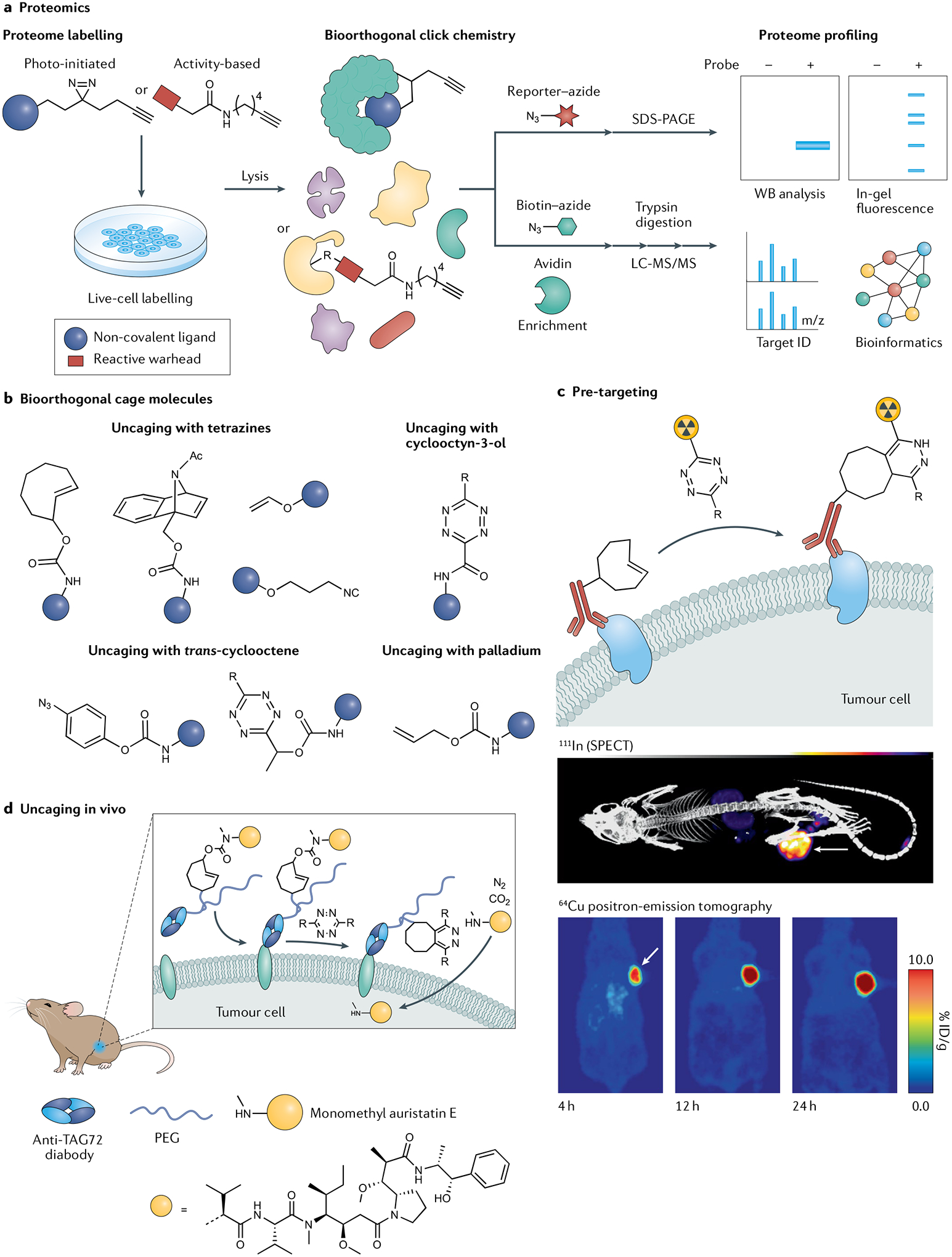
a | Enrichment strategies for proteome labelling enabled by the copper-catalysed azide–alkyne reaction. b | Examples of bioorthogonal cage molecules. c | The tetrazine ligation as a strategy for pre-targeted radiochemical imaging of cancer. d | Uncaging small-molecule cargo has been applied at tumour sites in animal models. LC-MS/MS, liquid chromatography with tandem mass spectrometry; SPECT, single-photon emission computed tomography; WB, western blot. Middle image in panel c originally published in REF.351, JNM. Rossin, R., Läppchen, T., Van Den Bosch, S. M., Laforest, R. & Robillard, M. S. Diels–Alder reaction for tumor pretargeting: in vivo chemistry can boost tumor radiation dose compared with directly labeled antibody. J. Nucl. Med. 54, 1989–1995 (2013). © SNMMI. Bottom image in panel c adapted with permission from REF.354, ACS. Panel d adapted from REF.357, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Targeted protein degradation.
A technique used for targeting specific proteins for degradation within a cell. Commonly, hetero-bifunctional small-molecule compounds are used for targeting the protein of interest to an E3 ubiquitin ligase protein. This facilitates the polyubiquination and subsequent degradation of the targeted protein.
An additional area of interest in drug discovery that has benefited from bioorthogonal chemistry is the development of ADCs. ADCs combine a potent small-molecule drug and a highly selective antibody for delivery to an antigen-specific cell type322. This approach has been utilized primarily in oncology indications to avoid toxic effects to normal tissue cells by targeting the cytotoxic molecule directly to cancerous tissues. Bioorthogonal chemistry has played a pivotal role in linking the small molecule to the antibody322. Traditionally, this linkage occurs via acylation of lysine residues by N-hydroxysuccinimide esters or alkylation of cysteines by maleimide; however, heterogeneous labelling of the antibodies has led to the exploration of alternative chemistries, including the development of site-selective small-molecule conjugation strategies that exploit the incorporation of UAAs, followed by bioorthogonal conjugation reactions such as SPAAC or oxime condensation323.
Bioorthogonal uncaging strategies.
Successes in the rapidly evolving field of bioorthogonal conjugation inspired the development of bioorthogonal cleavage approaches to release or activate small molecules and biomolecules242 (Fig. 7b). Most cleavage reactions were derived from their bioorthogonal bond-forming counterparts, typically resulting in conjugation intermediates designed to decompose and release a payload. By initially targeting and later releasing a bioactive molecule, bioorthogonal uncaging represents a way to achieve spatio-temporal control over cargo delivery in biological systems. In addition to the parameters that are key for successful bioorthogonal conjugation (reactivity and stability), the release rate and the nature of the group being released are important factors when choosing a cleavage reaction.
The first examples used the Staudinger ligation and Staudinger reaction to achieve, respectively, the elimination of a carbamate from the part originating from the triphenylphosophine324 or an azide to amine reduction71,325, optionally followed by further release of a self-immolative linker to liberate the payload. Similarly, cycloaddition of TCO with organic azide affords an unstable product that rearranges to liberate the corresponding amine326. The well-known SPAAC between cyclooctyne and azide has not yet been transformed into a dissociative reaction, but the conjugation between cyclooctyne and sydnones has led to efficiently cleavable iminosydnones144. Also, hydroxyl-functionalized cyclooctyne was used to release an amide-linked payload from a tetrazine following cycloaddition and intramolecular cyclization327. The IEDDA between tetrazines and dienophiles has proven to be a fertile ground for the development of several tetrazine-triggered cleavage reactions, such as the cleavage of allylic-substituted TCO (IEDDA pyridazine elimination)328, vinyl ethers329–331, 3-isocyanopropyls332 and carbamate-linked benzonorbornadienes333. From this series, allylic-substituted TCO has the highest reactivity328, mirroring the reactivity differences between the click conjugation reactions. Recently, click conjugation reactivity was increased by three orders of magnitude by the development of a new IEDDA pyridazine elimination mechanism where the roles are reversed, with highly reactive TCO triggering payload release from a tetrazine linker334. Isonitriles were shown to cleave this tetrazine linker through a slightly different mechanism335. Finally, the well-known fluoride-mediated removal of the tert-butyldimethylsilyl (TBS) protecting group in organic synthesis was recently reworked into an effective bioorthogonal cleavage reaction by using imaging agent-derived Phe-BF3 as the fluoride source336.
Self-immolative linker.
A class of linker that, when exposed to a certain trigger, is designed to break the payload connecting bonds via an intramolecular process.
Bradley and co-workers have used transition metals as triggers for bioorthogonal cleavage reactions219. The amine-protecting allyloxycarbonyl (alloc) group can be catalytically removed in physiological conditions by ruthenium and palladium complexes337,338. Pd0 nanoparticles and palladium complexes encapsulated in nanoparticles afforded selective alloc removal in cells and mice219. Likewise, the amine-protecting propargyl group has been catalytically removed by palladium338, copper339 and gold340 complexes as well as metallic palladium341. Hydroxyl groups were caged with an allene moiety and uncaged with palladium catalysts342. Although the use of metals or metal complexes may pose challenges in living systems in view of long-term stability and biocompatibility, their catalytic nature offers compelling opportunities, for example in prodrug activation.
Prodrug.
A pharmacologically inactive precursor compound that is converted into an active drug through in vivo chemical modification achieved via metabolic/enzymatic processes. Prodrugs are employed to improve pharmacokinetic properties (absorption, distribution, metabolism and elimination) and pharmacodynamics properties (selectivity, reduction of adverse effects) of the active drug molecule.
In vivo chemistry.
A central development in bioorthogonal chemistry has been the enormous reaction rate increase as new transformations are developed. As the reactivity increased, so did the application scope, leading to today’s use in a wide range of fields. Higher reactivity enables equimolar reagent stoichiometry, lower concentrations and shorter reaction times, and therefore allows demanding applications such as those found in medicine.
The Staudinger ligation and SPAAC were successfully used for the detection of metabolically engineered tissues in mice343, but the reaction kinetics (k2 ~ 10−3–101 M−1 s−1) required a high dose of secondary reagent (such as triarylphosphines and cyclooctynes, respectively) to achieve detectable binding, limiting the application scope. Nevertheless, there are examples of medical applications, such as SPAAC-mediated clearing of azide-containing warfarin344 and cardiac homing of endogenous stem cells345.
The rate constant for the tetrazine ligation (k2 ~ 105 M−1 s−1) is on a par with biomolecule association rates, opening up applications where chemistry substitutes for, or works in conjunction with, biology, at the same timescale and the same low concentrations. It was shown to be fast enough to be used for pre-targeted radioimmunoimaging, which involved treating tumour-bearing mice with TCO-tagged monoclonal antibody followed a few days later by administration of an equimolar amount of a fast-clearing radiolabelled tetrazine probe and its reaction with the tumour-bound monoclonal antibody346–348 (Fig. 7c). This approach has since been markedly improved by the development of tags with improved reactivity and stability349, pharmacokinetics350, the introduction of bioorthogonal monoclonal antibody clearing agents351,352 and the development of a wide range of probes for imaging353,354 and for radiotherapy355,356, together affording strongly increased tumour to normal tissue ratios. As a result, the tetrazine ligation is now an established method for companion diagnostic imaging of monoclonal antibodies and pre-targeted radioimmunotherapy in live animals.
Mirroring its parent reaction, the high reactivity of the IEDDA pyridazine elimination reaction has led to widespread utility328, applications of which include unmasking of TCO-containing ADCs357,358 (Fig. 7d), prodrugs359, proteins360 and peptide antigens361. Contrary to the imaging and radiotherapy applications, in cleavage applications the tetrazine is typically not radiolabelled and can be dosed in excess to achieve complete activation yields, as demonstrated for the activation of a tumour-bound ADC by a tetrazine administered intravenously in a second step357,358. Other approaches centre on the pre-administration of the tetrazine362,363, such as the intratumoural injection of a tetrazine followed by intravenous administration of a TCO prodrug, local capture and drug activation359. The IEDDA between liposome-encapsulated tetrazine and vinyl ether prodrug afforded an enhanced permeability and retention-mediated drug release in tumours364, and the iminosydnone release reaction was used to trigger fragmentation of and payload release from a tumour-bound micelle344. Likewise, nanoparticle-bound drugs were efficiently activated inside cancer cells in vivo using the desilylation reaction336. Finally, nanoparticle-encapsulated palladium complexes have been successfully used for on-tumour prodrug activation of alloc-caged doxorubicin as well as its micelle-encapsulated analogue365,366.
Most applications of in vivo chemistry have been mainly performed in mice. Bioorthogonal uncaging using the TCO–tetrazine pyridazine elimination has recently entered phase I clinical evaluation as a method of breast cancer chemotherapy367. Future clinical studies are expected to be fuelled by very promising results in several of the imaging, radiotherapy and drug delivery studies.
Polymer and materials science.
Bioorthogonal reactions have emerged as enabling tools in the areas of polymer science, materials science and surface science. In the field of polymer modification, CuAAC has been impactful with applications extending to the synthesis and functionalization of essentially every polymer class, including adhesives, block and multiblock copolymers, brush and comb copolymers as well as star, hyperbranched and dendronized polymers281,368–371. Bioorthogonal chemistry can also be used to create high molecular weight polymers through polymer–polymer couplings372. The tetrazine ligation has been used to investigate defects in polymer network formation373. Bioorthogonal reactions have also been used as tools for the construction of monodisperse macromolecules including dendrimers374 and sequence-specific polymers375. Here, the sequential application of two mutually orthogonal reactions can be used to produce well-defined macromolecules. For example, CuAAC in combination with sulfuryl fluoride exchange (SuFEx) chemistry (BOX 1) has been applied to the preparation of sequence-specific polymers376, and SPAAC in combination with tetrazine ligation has been used to produce dendrimer scaffolds attached to antibodies that can be used to amplify the signal in pre-targeted positron-emission tomography imaging377.
Box 1 |. Sulfuryl fluoride exchange chemistry.
Sulfuryl fluoride exchange (SuFEx) chemistry represents a class of biocompatible click reactions described in 2014 by Sharpless and colleagues, who identified sulfuryl fluorides (RSO2F) and related species as having balanced reactivity well matched to bioorthogonal applications5. Sulfur(VI) fluorides are compatible with water and are potently activated by acid-mediated stabilization of fluoride as a leaving group in aqueous media, the H···F interaction being the strongest known hydrogen bond400. Sulfuryl fluorides and related nitrogen-containing groups (such as sulfuramidimidoyl fluorides)401 are thereby proving to be quite selective in biomolecular labelling promoted by initial non-covalent interactions. In the formation and modification of polymeric materials, activation of the S–F bond by silyl reagents has proven especially useful. Thus, the combination of building blocks containing S–F and Si–O bonds, when addressed by the right nucleophilic catalysts, gives very high yields of S–O linkages, thermodynamically driven by the concomitant formation of ultra-stable Si–F by-products402. The term ‘exchange’ in the SuFEx moniker is meant to emphasize the fact that both the departing fluoride and the entering alkoxide/aryloxide must be appropriately activated in order for the reaction to proceed, and that thermodynamic factors governing the swap of S–F for S–O bonds are more balanced than is typical for other bioorthogonal ligations. As the most recently recognized click reaction, SuFEx chemistry is only now becoming recognized as an especially useful tool for numerous biological applications403, and relevant reagents for SuFEx chemistry are becoming increasingly available404,405.
Sequence-specific polymers.
Macromolecules that are monodisperse with defined monomer sequences or block sequences. This requires controlled, sequential addition of subunits using highly efficient bond-forming chemical reactions. Naturally occurring examples of sequence-specific macromolecules include DNA, RNA and protein.
Multivalency is a ubiquitous phenomenon in biology involving the simultaneous binding of multiple copies of a ligand displayed on one surface to complementary binders or receptors on another surface. Bioorthogonal chemistry has been used broadly to enable the multivalent display of designer ligands based on naturally occurring biomacromolecules. For example, CuAAC has been used to label the outer and inner surfaces of viral particles378,379, and has been a widely used tool for the creation of multivalent glycomaterials, glycopolymers and glycosurfaces380. Bioorthogonal reactions are also important to the functionalization of surfaces as well as nanoparticles381. CuAAC emerged as an early tool for electrode and surface functionalization382, and, more recently, SPAAC, tetrazine ligation and other bioorthogonal reactions have become important tools for the selective attachment of proteins and other biomolecules to surfaces383. Beyond surface modification, bioorthogonal chemistry also enables the creation of 3D networks, with applications that include the development of hydrogel supports for cell culture applications241. Here, copper-free methods including SPAAC and tetrazine ligation are proving especially useful for the assembly of 3D scaffolds that display biological ligands capable of supporting tissue-like structures384,385.
Materials construction (and deconstruction) in response to external stimulus is a field of increasing importance in various applications, and bioorthogonal reactions allow for creative applications of molecular connectivity in tailored circumstances or environments. Examples include photoinducible ligations, which have proven highly useful for the activation and patterning of surfaces and 3D materials, such as for biomolecular attachment in 3D scaffolds386,387. The tetrazole photoclick chemistry has also been employed in materials science for preparation of photodegradable supramolecular hydrogels388, grafting polymers onto silicon and cellulose surfaces389, and synthesis of polymeric networks390. Thiol–ene and thiol–yne chemistry has so far been the technique of choice for light-based 3D printing, such as for interesting hydrogel materials391. The inherent reversibility of properly designed thiol–ene connections has also been exploited for controllable disassembly392. Similarly, the rapid kinetics of tetrazine ligation can be used for further advantage in materials assembly through interfacial processes that can be used for attaching ultrasound microbubbles to cell surfaces393, as a method for modulating cell–cell adhesion394 and as a method for the creation of molecularly patterned fibres395, core-shell particles396 and hydrogel channels397 or shapes398 capable of directing cell behaviour in three dimensions.
Reproducibility and data deposition
The characterization of new bioorthogonal reagents and small-molecule conjugates follows the standards of the field of organic chemistry. At a minimum, new compounds are generally characterized by 1H and 13C NMR spectroscopy as well as elemental analysis and/or high-resolution mass spectrometry. Additional characterization by NMR (probing other nuclei, 2D NMR experiments as well as measuring the nuclear Overhauser effect), Fourier transform infrared spectroscopy, chromatography (for example, high-performance liquid chromatography or gas chromatography) and X-ray crystallography are also routinely performed to support structural assignments. In general, copies of spectral data along with spectral peak listings are submitted for peer review and made available as supporting information by the journal at the time of publication. Additionally, X-ray crystallographic data are typically deposited in the Cambridge Structural Database. Where possible, kinetic rate constants should be measured under purely aqueous conditions to enable rate comparisons across different reaction types and, where applicable, the effect of the catalyst concentration on the rate should also be indicated. One of the hallmarks of bioorthogonal chemistry is that reactions are reliable and successful under a wide range of conditions. However, the high reactivity associated with the most reactive bioorthogonal substrates can also require additional considerations to verify reagent quality prior to use. It is generally advisable to store reagents under refrigeration and to periodically validate reagent quality by spectroscopic (such as NMR) or chromatographic (such as high-performance liquid chromatography) analysis.
Additionally, reagent stability should be evaluated under physiological conditions. Valuable methods include studying the reagent in the biological milieu of interest (for example, blood, serum, cell lysate) as well as in the presence of glutathione at 10 mM, which approximates the thiol’s intracellular concentration. In addition to rigorous analytical chemical characterization, bioorthogonal reactions should be characterized on labelled biomolecules whenever possible. Mass spectrometry as well as quantification by high-performance liquid chromatography or gel electrophoresis should be routinely used for deducing reaction conversions and yields. To validate bioorthogonal reaction efficiency in live cells, it is generally useful to quantify labelling using fluorescent probes and in-gel fluorescence. Mass spectrometry can be used to validate overexpressed protein targets in bacteria. In mammalian cells, it is useful to validate specificity through labelling of recombinant fluorescent proteins with fluorescent probes, where co-localization of fluorescence from the protein and probe can be used to provide a measure of the specificity and efficacy of labelling.
Limitations and optimizations
The impact of bioorthogonal chemistry on biology and biomedical research has been facilitated by the increasing availability of commercial reagents to the wider scientific community, and a growing number of companies now specialize in reagents for bioorthogonal and click chemistry. An increased variety of bioorthogonal reagents in formats that are ready to use for bioconjugation and applications including fluorescent imaging, radiochemistry and proteomics would be beneficial for researchers using these tools. There is a continued need to develop improved reagents that display rapid kinetics while exhibiting high stability towards extended incubation in the cellular or in vivo environment. There are opportunities for synthetic chemists to further impact the scientific community by creating new and improved bioorthogonal reagents.
The expanding catalogue of bioorthogonal reactions has brought the associated challenge of deciding how to choose a bioorthogonal reaction for a given application. As pointed out above, different reactions in the bioorthogonal toolbox have associated strengths and limitations, and it is advisable for practitioners to begin by prioritizing the characteristics that are most important for the application. With small and readily available reaction partners and fast rates under catalytic conditions, CuAAC has been applied across a remarkable range of applications in vitro and in extracellular and cell surface settings, but, as noted above, is typically less well suited for intracellular labelling applications. For intracellular and in vivo applications where fast reactivity is required, the kinetics of tetrazine ligation can enable fast labelling even at sub-micromolar concentrations. For tetrazine ligation, a consideration is that both reaction partners are relatively bulky, especially for TCO dienophiles, which are also the most reactive. Cycloalkyne [3 + 2] cycloadditions also function efficiently in the intracellular environment, with the advantage of using azides and other small dipoles as one reaction partner. However, the kinetics are generally less rapid than tetrazine ligation and require concentrations in the high micromolar to millimolar range. Native chemical ligation serves as the standard for the conjugation of large peptidic or protein fragments using native amide linkages but is relegated to in vitro applications. The Staudinger ligation offers an alternative for the formation of amide bonds in cellular or in vivo settings, but can be limited by bulky reagents and relatively slow kinetics. Oxime, palladium-catalysed and ruthenium-catalysed reactions offer unique new functional group handles in bioorthogonal chemistry applications. With some exceptions, these tools are generally best suited for applications in vitro and in the extracellular environment. Photoclick chemistry has proven to be a powerful tool for exercising spatio-temporal control over bioorthogonal reactivity in varied settings, including live-cell applications. Continued efforts to reduce the size of photoinducible groups and red-shift activation wavelengths will enable a wider range of applications. Finally, the development of cleavage reactions with increased reactivity and release kinetics would provide researchers with a wider set of uncaging tools.
Outlook
Bioorthogonal chemistry has emerged as an enabling method for the construction of molecules with complexity and in contexts that were previously unthinkable. The impact of bioorthogonal reactivity has been far-reaching with applications spanning protein synthesis, drug discovery, in vivo chemistry and polymer science. Moving forwards, bioorthogonal chemistry will continue to develop into a tool capable of delivering new drugs, creating new materials and answering biological questions that cannot be addressed by modern molecular biology tools (such as GFP and CRISPR–Cas9) or small-molecule ligands.
Despite tremendous progress in the development and application of bioorthogonal chemistry, there is much work to be done. Although all bioorthogonal groups are much lower in molecular weight than directly expressed protein tags, bioorthogonal linkers still represent a perturbation to the biological system under study, and bulky derivatives may not be accepted by the enzymatic machinery used to incorporate a tag into a biological target. There is still a need for new reagents that are small, safe, stable and capable of rapid kinetics, with high value for reagents that work efficiently inside living cells and animals. Initial efforts to minimize the size and improve the physiological properties of bioorthogonal groups have been successful, yet few classes of bioorthogonal reactions can produce native biological bonds such as amides. New classes of reactions capable of producing native covalent bonds with rapid kinetics in living systems would be valuable tools for chemical biology. The continued advancement of ‘orthogonal bioorthogonal’ reactions399, which enable the simultaneous use of multiple bioorthogonal reactant pairs in living systems, offers another level of control over the biological system under study and is valuable. Furthermore, bioorthogonal covalent reactions that can be reversed using benign triggers would be ideal tools for studying and manipulating biological systems.
Biological imaging has been advanced through the development of methods that couple bioorthogonal chemistry to imaging in living systems. Innovations in the development of fluorogenic bioorthogonal reactions as well as in vivo radiochemistry have emerged as powerful imaging tools from the cellular to the whole animal level346–356. Complementary approaches are needed that will continue to push the limits of detection by ameliorating non-specific interactions or side reactions that can decrease signal to noise ratios, and thereby enable new frontiers in imaging, including the live-cell imaging of low abundance biomolecular targets (such as endogenous proteins), and in the development of in vivo approaches for diagnosing, monitoring and staging human diseases in the clinic. These developments will benefit from ongoing efforts in synthesizing sophisticated dyes and fluorophores with custom-designed physical properties to illuminate individual biomolecules in complex biological systems.
Chemical techniques for site-specific attachment of bioorthogonal handles onto biomacromolecules have made tremendous advancements in recent years. Recombinant methods to install uniquely reactive peptide tags have simplified site-specific incorporation with broad substrate scope onto protein targets. But these methods do suffer from incomplete conversions and require high reagent concentrations, which results in limited in vivo use. Genetic code expansion has also been instrumental for installing bioorthogonal handles with amino acid precision devoid of extra peptide tags. However, extensive re-engineering of machinery is often necessary to accommodate new substrates. Simple methods for quantitative, non-invasive, highly pure, site-specific attachment of small molecules to proteins in a living system with broad substrate scope are highly desirable, and represent an idealized protein conjugation method to strive for. Substantial effort has been focused on protein conjugation strategies; expanding these strategies to other biomacromolecules and complex metabolites remains largely unexplored and a critical area for newer methodology.
Photoclick chemistry has also provided a powerful method for the study of cellular processes with spatio-temporal precision. Moving forwards, the control offered by photoinducible bioorthogonal chemistry may enable real-time imaging of individual biological molecule dynamics and provide a tool for elucidating the biomolecule function in signalling pathways. The development of photoinducible bioorthogonal chemistry based on tissue-penetrant near-IR light180 may offer the ability to induce rapid reactivity in vivo, providing a new level of precision for studying pathogenesis in living animals.
From the intersection of organic chemistry and biology, bioorthogonal chemistry has emerged as an essential way to construct biological molecules and to study them in their native environment. With ever-increasing momentum, bioorthogonal chemistry is enabling research in biology and biomedicine and is influencing scientists across the spectrum of disciplines through a broad range of applications.
Acknowledgements
J.A.P. acknowledges support over the years from the Alfred P. Sloan Foundation, the Camille & Henry Dreyfus Foundation and the National Institutes of Health (NIH) (R01 GM126226). J.M.F. acknowledges support from the NIH (R01 GM132460), National Science Foundation (NSF) (DMR 1809612 and 2011824) and Pfizer. Q.L. acknowledges support from the NIH (R35 GM130307) and NSF (CHE-1904558). K.L acknowledges support from the German Science Foundation DFG through programmes SFB1035 and SPP1623.
Footnotes
Competing interests
W.L. and C.W.a.E. are employees of Pfizer Inc. M.S.R. is an employee and shareholder of Tagworks Pharmaceuticals. S.L.S., D.A.B., R.H., S.S.N., M.X., M.G.F., K.L., Q.L., J.P.P., J.A.P. and J.M.F. declare no competing interests.
References
- 1.Nienhaus GU The green fluorescent protein: a key tool to study chemical processes in living cells. Angew. Chem. Int. Ed 47, 8992–8994 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Winter G & Milstein C Man-made antibodies. Nature 349, 293–299 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Lang K & Chin JW Bioorthogonal reactions for labeling proteins. ACS Chem. Biol 9, 16–20 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Kolb HC, Finn MG & Sharpless KB Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed 40, 2004–2021 (2001). [DOI] [PubMed] [Google Scholar]; This seminal review article outlines the concept and requirements of click chemistry.
- 5.Dong J, Krasnova L, Finn MG & Sharpless KB Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew. Chem. Int. Ed 53, 9430–9448 (2014). [DOI] [PubMed] [Google Scholar]; This article presents an initial description of SuFEx chemistry as an efficient biocompatible click chemistry tool.
- 6.Hoyle CE & Bowman CN Thiol–ene click chemistry. Angew. Chem. Int. Ed 49, 1540–1573 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Griffin BA, Adams SR & Tsien RY Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Park K-Y, Suazo KF & Distefano MD Recent progress in enzymatic protein labelling techniques and their applications. Chem. Soc. Rev 47, 9106–9136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopez Aguilar A et al. Tools for studying glycans: recent advances in chemoenzymatic glycan labeling. ACS Chem. Biol 12, 611–621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liss V, Barlag B, Nietschke M & Hensel M Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci. Rep 5, 17740 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson P, Muir T, Clark-Lewis I & Kent S Synthesis of proteins by native chemical ligation. Science 266, 776–779 (1994). [DOI] [PubMed] [Google Scholar]; This article presents an initial description of native chemical ligation, which enabled the use of fully deprotected peptides through bioorthogonal reaction of a thioester motif with an N-terminal cysteine residue.
- 12.Agouridas V et al. Native chemical ligation and extended methods: mechanisms, catalysis, scope, and limitations. Chem. Rev 119, 7328–7443 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Kent SBH Total chemical synthesis of proteins. Chem. Soc. Rev 38, 338–351 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Wieland T, Bokelmann E, Bauer L, Lang HU & Lau H Über Peptidsynthesen. 8. Mitteilung Bildung von S-haltigen Peptiden durch intramolekulare Wanderung von Aminoacylresten. Justus Liebigs Ann. Chem 583, 129–149 (1953). [Google Scholar]
- 15.Dawson PE, Churchill MJ, Ghadiri MR & Kent SBH Modulation of reactivity in native chemical ligation through the use of thiol additives. J. Am. Chem. Soc 119, 4325–4329 (1997). [Google Scholar]
- 16.Yan LZ & Dawson PE Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. J. Am. Chem. Soc 123, 526–533 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Wan Q & Danishefsky SJ Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew. Chem. Int. Ed 46, 9248–9252 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Metanis N, Keinan E & Dawson PE Traceless ligation of cysteine peptides using selective deselenization. Angew. Chem. Int. Ed 49, 7049–7053 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulkarni SS, Sayers J, Premdjee B & Payne RJ Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nat. Rev. Chem 2, 0122 (2018). [Google Scholar]
- 20.Muir TW, Sondhi D & Cole PA Expressed protein ligation: a general method for protein engineering. Proc. Natl Acad. Sci. USA 95, 6705–6710 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents the first description of expressed protein ligation — the application of native chemical ligation to ligate small synthetic sequences to much larger recombinant protein fragments.
- 21.Tam JP & Miao Z Stereospecific pseudoproline ligation of N-terminal serine, threonine, or cysteine-containing unprotected peptides. J. Am. Chem. Soc 121, 9013–9022 (1999). [Google Scholar]
- 22.Zhang Y, Xu C, Lam HY, Lee CL & Li X Protein chemical synthesis by serine and threonine ligation. Proc. Natl Acad. Sci. USA 110, 6657–6662 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bode JW, Fox RM & Baucom KD Chemoselective amide ligations by decarboxylative condensations of N-alkylhydroxylamines and α-ketoacids. Angew. Chem. Int. Ed 45, 1248–1252 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Noda H, Erős G & Bode JW Rapid ligations with equimolar reactants in water with the potassium acyltrifluoroborate (KAT) amide formation. J. Am. Chem. Soc 136, 5611–5614 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Geoghegan KF & Stroh JG Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjugate Chem. 3, 138–146 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Zhang L & Tam JP Thiazolidine formation as a general and site-specific conjugation method for synthetic peptides and proteins. Anal. Biochem 233, 87–93 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Agarwal P, van der Weijden J, Sletten EM, Rabuka D & Bertozzi CR A Pictet–Spengler ligation for protein chemical modification. Proc. Natl Acad. Sci. USA 110, 46–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren H et al. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed 48, 9658–9662 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS & Francis MB N-terminal protein modification through a biomimetic transamination reaction. Angew. Chem. Int. Ed 45, 5307–5311 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Witus LS et al. Site-specific protein transamination using N-methylpyridinium-4-carboxaldehyde. J. Am. Chem. Soc 135, 17223–17229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacDonald JI, Munch HK, Moore T & Francis MB One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat. Chem. Biol 11, 326–331 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Rideout D Self-assembling cytotoxins. Science 233, 561–563 (1986). [DOI] [PubMed] [Google Scholar]
- 33.Kalia J & Raines RT Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed 47, 7523–7526 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brudno Y et al. Refilling drug delivery depots through the blood. Proc. Natl Acad. Sci. USA 111, 12722–12727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deygen IM et al. Novel prodrug of doxorubicin modified by stearoylspermine encapsulated into PEG-chitosan-stabilized liposomes. Langmuir 32, 10861–10869 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Matson JB & Stupp SI Drug release from hydrazone-containing peptide amphiphiles. ChemComm 47, 7962–7964 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kölmel DK & Kool ET Oximes and hydrazones in bioconjugation: mechanism and catalysis. Chem. Rev 117, 10358–10376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saito F, Noda H & Bode JW Critical evaluation and rate constants of chemoselective ligation reactions for stoichiometric conjugations in water. ACS Chem. Biol 10, 1026–1033 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Gaertner HF et al. Construction of protein analogs by site-specific condensation of unprotected fragments. Bioconjugate Chem. 3, 262–268 (1992). [DOI] [PubMed] [Google Scholar]
- 40.Jencks WP Studies on the mechanism of oxime and semicarbazone formation1. J. Am. Chem. Soc 81, 475–481 (1959). [Google Scholar]
- 41.Sander EG & Jencks WP Equilibria for additions to the carbonyl group. J. Am. Chem. Soc 90, 6154–6162 (1968). [Google Scholar]
- 42.Dirksen A, Hackeng TM & Dawson PE Nucleophilic catalysis of oxime ligation. Angew. Chem. Int. Ed 45, 7581–7584 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Dirksen A, Dirksen S, Hackeng TM & Dawson PE Nucleophilic catalysis of hydrazone formation and transimination: implications for dynamic covalent chemistry. J. Am. Chem. Soc 128, 15602–15603 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Crisalli P & Kool ET Importance of ortho proton donors in catalysis of hydrazone formation. Org. Lett 15, 1646–1649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crisalli P & Kool ET Water-soluble organocatalysts for hydrazone and oxime formation. J. Org. Chem 78, 1184–1189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larsen D et al. Exceptionally rapid oxime and hydrazone formation promoted by catalytic amine buffers with low toxicity. Chem. Sci 9, 5252–5259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agarwal P et al. Hydrazino-Pictet–Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjugate Chem. 24, 846–851 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Sudalai A, Khenkin A & Neumann R Sodium periodate mediated oxidative transformations in organic synthesis. Org. Biomol. Chem 13, 4374–4394 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Zeng Y, Ramya TNC, Dirksen A, Dawson PE & Paulson JC High-efficiency labeling of sialylated glycoproteins on living cells. Nat. Methods 6, 207–209 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chelius D & Shaler TA Capture of peptides with N-terminal serine and threonine: a sequence-specific chemical method for peptide mixture simplification. Bioconjugate Chem. 14, 205–211 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Hansske F, Sprinzl M & Cramer F Reaction of the ribose moiety of adenosine and AMP with periodate and carboxylic acid hydrazides. Bioorg. Chem 3, 367–376 (1974). [Google Scholar]
- 52.Haney CM & Horne WS Oxime side-chain cross-links in an α-helical coiled-coil protein: structure, thermodynamics, and folding-templated synthesis of bicyclic species. Chem. Eur. J 19, 11342–11351 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Haney CM, Loch MT & Horne WS Promoting peptide α-helix formation with dynamic covalent oxime side-chain cross-links. ChemComm 47, 10915–10917 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Hardisty RE, Kawasaki F, Sahakyan AB & Balasubramanian S Selective chemical labeling of natural T modifications in DNA. J. Am. Chem. Soc 137, 9270–9272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Köhn M & Breinbauer R The Staudinger ligation — a gift to chemical biology. Angew. Chem. Int. Ed 43, 3106–3116 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Bednarek C, Wehl I, Jung N, Schepers U & Bräse S The Staudinger ligation. Chem. Rev 120, 4301–4354 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Staudinger H & Meyer J Über neue organische phosphorverbindungen III. phosphinmethylenderivate und phosphinimine. Helv. Chim. Acta 2, 635–646 (1919). [Google Scholar]
- 58.Saxon E & Bertozzi CR Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010 (2000). [DOI] [PubMed] [Google Scholar]; This article presents the first description of cell surface engineering using azido reporters and bioorthogonal chemistry.
- 59.Shah L, Laughlin ST & Carrico IS Light-activated Staudinger–Bertozzi ligation within living animals. J. Am. Chem. Soc 138, 5186–5189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schilling CI, Jung N, Biskup M, Schepers U & Bräse S Bioconjugation via azide–Staudinger ligation: an overview. Chem. Soc. Rev 40, 4840–4871 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Kiick KL, Saxon E, Tirrell DA & Bertozzi CR Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl Acad. Sci. USA 99, 19–24 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ovaa H et al. Chemistry in living cells: detection of active proteasomes by a two-step labeling strategy. Angew. Chem. Int. Ed 42, 3626–3629 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Tsao M-L, Tian F & Schultz PG Selective Staudinger modification of proteins containing p-azidophenylalanine. ChemBioChem 6, 2147–2149 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Wang CCY, Seo TS, Li Z, Ruparel H & Ju J Site-specific fluorescent labeling of DNA using Staudinger ligation. Bioconjugate Chem. 14, 697–701 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Vocadlo DJ, Hang HC, Kim E-J, Hanover JA & Bertozzi CR A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc. Natl Acad. Sci. USA 100, 9116–9121 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin DD et al. Rapid detection, discovery, and identification of post-translationally myristoylated proteins during apoptosis using a bio-orthogonal azidomyristate analog. FASEB J. 22, 797–806 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hang HC, Wilson JP & Charron G Bioorthogonal chemical reporters for analyzing protein lipidation and lipid trafficking. Acc. Chem. Res 44, 699–708 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lemieux GA, de Graffenried CL & Bertozzi CR A fluorogenic dye activated by the Staudinger ligation. J. Am. Chem. Soc 125, 4708–4709 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Hangauer MJ & Bertozzi CR A FRET-based fluorogenic phosphine for live-cell imaging with the Staudinger ligation. Angew. Chem. Int. Ed 47, 2394–2397 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lukasak B, Morihiro K & Deiters A Aryl azides as phosphine-activated switches for small molecule function. Sci. Rep 9, 1–6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo J, Liu Q, Morihiro K & Deiters A Small-molecule control of protein function through Staudinger reduction. Nat. Chem 8, 1027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prescher JA, Dube DH & Bertozzi CR Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004). [DOI] [PubMed] [Google Scholar]; This article presents the first description of bioorthogonal chemistry in live mice.
- 73.Dube DH, Prescher JA, Quang CN & Bertozzi CR Probing mucin-type O-linked glycosylation in living animals. Proc. Natl Acad. Sci. USA 103, 4819–4824 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin FL, Hoyt HM, van Halbeek H, Bergman RG & Bertozzi CR Mechanistic investigation of the Staudinger ligation. J. Am. Chem. Soc 127, 2686–2695 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Ren G, Zheng Q & Wang H Aryl fluorosulfate trapped Staudinger reduction. Org. Lett 19, 1582–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tam A & Raines RT Protein engineering with the traceless Staudinger ligation. Methods Enzymol. 462, 25–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saxon E, Armstrong JI & Bertozzi CRA “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett 2, 2141–2143 (2000). [DOI] [PubMed] [Google Scholar]
- 78.Nilsson BL, Kiessling LL & Raines RT Staudinger ligation: a peptide from a thioester and azide. Org. Lett 2, 1939–1941 (2000). [DOI] [PubMed] [Google Scholar]
- 79.Kleineweischede R & Hackenberger CP Chemoselective peptide cyclization by traceless Staudinger ligation. Angew. Chem. Int. Ed 47, 5984–5988 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Merkx R, Rijkers DT, Kemmink J & Liskamp RM Chemoselective coupling of peptide fragments using the Staudinger ligation. Tetrahedron Lett. 44, 4515–4518 (2003). [Google Scholar]
- 81.Böhrsch V, Serwa R, Majkut P, Krause E & Hackenberger CP Site-specific functionalisation of proteins by a Staudinger-type reaction using unsymmetrical phosphites. ChemComm 46, 3176–3178 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Vallée MRJ et al. Staudinger-phosphonite reactions for the chemoselective transformation of azido-containing peptides and proteins. Org. Lett 13, 5440–5443 (2011). [DOI] [PubMed] [Google Scholar]
- 83.Lee YJ, Kurra Y & Liu WR Phospha-Michael addition as a new click reaction for protein functionalization. ChemBioChem 17, 456–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bos J & Muir TW A chemical probe for protein crotonylation. J. Am. Chem. Soc 140, 4757–4760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shih H-W & Prescher JA A bioorthogonal ligation of cyclopropenones mediated by triarylphosphines. J. Am. Chem. Soc 137, 10036–10039 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Row RD, Shih H-W, Alexander AT, Mehl RA & Prescher JA Cyclopropenones for metabolic targeting and sequential bioorthogonal labeling. J. Am. Chem. Soc 139, 7370–7375 (2017). [DOI] [PubMed] [Google Scholar]
- 87.Row RD & Prescher JA A cyclopropenethione–phosphine ligation for rapid biomolecule labeling. Org. Lett 20, 5614–5617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heiss TK & Prescher JA Cyclopropeniminium ions exhibit unique reactivity profiles with bioorthogonal phosphines. J. Org. Chem 84, 7443–7448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tornøe CW, Christensen C & Meldal M Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem 67, 3057–3064 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Rostovtsev VV, Green LG, Fokin VV & Sharpless KB A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed 41, 2596–2599 (2002). [DOI] [PubMed] [Google Scholar]; Together with Tornøe et al. (2002), these articles present the initial description of copper-catalysed azide–alkyne cycloaddition chemistry.
- 91.Himo F et al. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc 127, 210–216 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Presolski SI, Hong V, Cho S-H & Finn MG Tailored ligand acceleration of the Cu-catalyzed azide–alkyne cycloaddition reaction: practical and mechanistic implications. J. Am. Chem. Soc 132, 14570–14576 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chan TR, Hilgraf R, Sharpless KB & Fokin VV Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett 6, 2853–2855 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Lipshutz BH & Taft BR Heterogeneous copper-in-charcoal-catalyzed click chemistry. Angew. Chem. Int. Ed 45, 8235–8238 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Chen J et al. Enzyme-like click catalysis by a copper-containing single-chain nanoparticle. J. Am. Chem. Soc 140, 13695–13702 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Wang F et al. A biocompatible heterogeneous MOF–Cu catalyst for in vivo drug synthesis in targeted subcellular organelles. Angew. Chem. Int. Ed 58, 6987–6992 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Zhu X, Liu J & Zhang W De novo biosynthesis of terminal alkyne-labeled natural products. Nat. Chem. Biol 11, 115–120 (2015). [DOI] [PubMed] [Google Scholar]
- 98.Marchand JA et al. Discovery of a pathway for terminal-alkyne amino acid biosynthesis. Nature 567, 420–424 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Hong V, Presolski SI, Ma C & Finn MG Analysis and optimization of copper-catalyzed azide–alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed 48, 9879–9883 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hong V, Steinmetz NF, Manchester M & Finn MG Labeling live cells by copper-catalyzed alkyne–azide click chemistry. Bioconjugate Chem. 21, 1912–1916 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Soriano del Amo D et al. Biocompatible copper(I) catalysts for in vivo imaging of glycans. J. Am. Chem. Soc 132, 16893–16899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuang G-C, Michaels HA, Simmons JT, Clark RJ & Zhu L Chelation-assisted, copper(II)-acetate-accelerated azide–alkyne cycloaddition. J. Org. Chem 75, 6540–6548 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Besanceney-Webler C et al. Increasing the efficacy of bioorthogonal click reactions for bioconjugation: a comparative study. Angew. Chem. Int. Ed 50, 8051–8056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Uttamapinant C et al. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed 51, 5852–5856 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bevilacqua V et al. Copper-chelating azides for efficient click conjugation reactions in complex media. Angew. Chem. Int. Ed 53, 5872–5876 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Su Y, Li L, Wang H, Wang X & Zhang Z All-in-one azides: empowered click reaction for in vivo labeling and imaging of biomolecules. ChemComm 52, 2185–2188 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Inoue N, Onoda A & Hayashi T Site-specific modification of proteins through N-terminal azide labeling and a chelation-assisted CuAAC reaction. Bioconjugate Chem. 30, 2427–2434 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Li S et al. Copper-catalyzed click reaction on/in live cells. Chem. Sci 8, 2107–2114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Speers AE, Adam GC & Cravatt BF Activity-based protein profiling in vivo using a copper(I)-catalyzed azide–alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc 125, 4686–4687 (2003). [DOI] [PubMed] [Google Scholar]
- 110.Yang M et al. Biocompatible click chemistry enabled compartment-specific pH measurement inside E. coli. Nat. Commun 5, 4981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Clavadetscher J et al. Copper catalysis in living systems and in situ drug synthesis. Angew. Chem. Int. Ed 55, 15662–15666 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Sun DE et al. Click-ExM enables expansion microscopy for all biomolecules. Nat. Methods 18, 107–113 (2021). [DOI] [PubMed] [Google Scholar]
- 113.Morgan MT et al. Ratiometric two-photon microscopy reveals attomolar copper buffering in normal and Menkes mutant cells. Proc. Natl Acad. Sci. USA 116, 12167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kennedy DC et al. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc 133, 17993–18001 (2011). [DOI] [PubMed] [Google Scholar]
- 115.Agard NJ, Prescher JA & Bertozzi CR A strain-promoted [3 + 2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc 126, 15046–15047 (2004). [DOI] [PubMed] [Google Scholar]; This article presents the first use of strained molecules to facilitate rapid, catalyst-free bioorthogonal labelling.
- 116.Baskin JM et al. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl Acad. Sci. USA 104, 16793–16797 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dommerholt J, Rutjes FPJT & van Delft FL Strain-promoted 1,3-dipolar cycloaddition of cycloalkynes and organic azides. Top. Curr. Chem 374, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chupakhin EG & Krasavin MY Achievements in the synthesis of cyclooctynes for ring strain-promoted [3 + 2] azide–alkyne cycloaddition. Chem. Heterocycl. Compd 54, 483–501 (2018). [Google Scholar]
- 119.Codelli JA, Baskin JM, Agard NJ & Bertozzi CR Second-generation difluorinated cyclooctynes for copper-free click chemistry. J. Am. Chem. Soc 130, 11486–11493 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dommerholt J et al. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed 49, 9422–9425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Debets MF, van der Doelen CWJ, Rutjes FPJT & van Delft FL Azide: a unique dipole for metal-free bioorthogonal ligations. ChemBioChem 11, 1168–1184 (2010). [DOI] [PubMed] [Google Scholar]
- 122.Jewett JC, Sletten EM & Bertozzi CR Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc 132, 3688–3690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ning X, Guo J, Wolfert MA & Boons G-J Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast Huisgen cycloadditions. Angew. Chem. Int. Ed 47, 2253–2255 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nainar S et al. Temporal labeling of nascent RNA using photoclick chemistry in live cells. J. Am. Chem. Soc 139, 8090–8093 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Friscourt F et al. Polar dibenzocyclooctynes for selective labeling of extracellular glycoconjugates of living cells. J. Am. Chem. Soc 134, 5381–5389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sletten EM & Bertozzi CR A hydrophilic azacyclooctyne for Cu-free click chemistry. Org. Lett 10, 3097–3099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stöckmann H et al. Development and evaluation of new cyclooctynes for cell surface glycan imaging in cancer cells. Chem. Sci 2, 932–936 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Almeida GD, Townsend LC & Bertozzi CR Synthesis and reactivity of dibenzoselenacycloheptynes. Org. Lett 15, 3038–3041 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Almeida GD, Sletten EM, Nakamura H, Palaniappan KK & Bertozzi CR Thiacycloalkynes for copper-free click chemistry. Angew. Chem. Int. Ed 51, 2443–2447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weterings J et al. TMTHSI, a superior 7-membered ring alkyne containing reagent for strain-promoted azide–alkyne cycloaddition reactions. Chem. Sci 11, 9011–9016 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Friscourt F, Fahrni CJ & Boons G-J A fluorogenic probe for the catalyst-free detection of azide-tagged molecules. J. Am. Chem. Soc 134, 18809–18815 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jewett JC & Bertozzi CR Synthesis of a fluorogenic cyclooctyne activated by Cu-free click chemistry. Org. Lett 13, 5937–5939 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ess DH & Houk KN Distortion/interaction energy control of 1,3-dipolar cycloaddition reactivity. J. Am. Chem. Soc 129, 10646–10647 (2007). [DOI] [PubMed] [Google Scholar]
- 134.Liang Y, Mackey JL, Lopez SA, Liu F & Houk KN Control and design of mutual orthogonality in bioorthogonal cycloadditions. J. Am. Chem. Soc 134, 17904–17907 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Liu F, Liang Y & Houk KN Bioorthogonal cycloadditions: computational analysis with the distortion/interaction model and predictions of reactivities. Acc. Chem. Res 50, 2297–2308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McKay CS, Blake JA, Cheng J, Danielson DC & Pezacki JP Strain-promoted cycloadditions of cyclic nitrones with cyclooctynes for labeling human cancer cells. ChemComm 47, 10040–10042 (2011). [DOI] [PubMed] [Google Scholar]
- 137.McKay CS, Chigrinova M, Blake JA & Pezacki JP Kinetics studies of rapid strain-promoted [3 + 2]-cycloadditions of nitrones with biaryl-azacyclooctynone. Org. Biomol. Chem 10, 3066–3070 (2012). [DOI] [PubMed] [Google Scholar]
- 138.McKay CS, Moran J & Pezacki JP Nitrones as dipoles for rapid strain-promoted 1,3-dipolar cycloadditions with cyclooctynes. ChemComm 46, 931–933 (2010). [DOI] [PubMed] [Google Scholar]; This article presents the first description of strain-promoted bioorthogonal reactions of nitrones with strained cycloalkynes.
- 139.Sherratt AR et al. Dual strain-promoted alkyne–nitrone cycloadditions for simultaneous labeling of bacterial peptidoglycans. Bioconjugate Chem. 27, 1222–1226 (2016). [DOI] [PubMed] [Google Scholar]
- 140.Gutsmiedl K, Wirges CT, Ehmke V & Carell T Copper-free “click” modification of DNA via nitrile oxide–norbornene 1,3-dipolar cycloaddition. Org. Lett 11, 2405–2408 (2009). [DOI] [PubMed] [Google Scholar]
- 141.McGrath NA & Raines RT Diazo compounds as highly tunable reactants in 1,3-dipolar cycloaddition reactions with cycloalkynes. Chem. Sci 3, 3237–3240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moran J, McKay CS & Pezacki JP Strain-promoted 1,3-dipolar cycloadditions of diazo compounds with cyclooctynes. Can. J. Chem 89, 148–151 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Sanders BC et al. Metal-free sequential [3 + 2]-dipolar cycloadditions using cyclooctynes and 1,3-dipoles of different reactivity. J. Am. Chem. Soc 133, 949–957 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bernard S et al. Bioorthogonal click and release reaction of iminosydnones with cycloalkynes. Angew. Chem. Int. Ed 56, 15612–15616 (2017). [DOI] [PubMed] [Google Scholar]
- 145.Richard M et al. New fluorine-18 pretargeting PET imaging by bioorthogonal chlorosydnone–cycloalkyne click reaction. ChemComm 55, 10400–10403 (2019). [DOI] [PubMed] [Google Scholar]
- 146.Wallace S & Chin JW Strain-promoted sydnone bicyclo-[6.1.0]-nonyne cycloaddition. Chem. Sci 5, 1742–1744 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sletten EM & Bertozzi CR A bioorthogonal quadricyclane ligation. J. Am. Chem. Soc 133, 17570–17573 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carboni RA & Lindsey RV Reactions of tetrazines with unsaturated compounds. a new synthesis of pyridazines. J. Am. Chem. Soc 81, 4342–4346 (1959). [Google Scholar]
- 149.Zhang J, Shukla V & Boger DL Inverse electron demand Diels–Alder reactions of heterocyclic azadienes, 1-aza-1,3-butadienes, cyclopropenone ketals, and related systems. A retrospective. J. Org. Chem 84, 9397–9445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thalhammer F, Wallfahrer U & Sauer J Reaktivität einfacher offenkettiger und cyclischer dienophile bei Diels–Alder-reaktionen mit inversem elektronenbedarf. Tetrahedron Lett. 31, 6851–6854 (1990). [Google Scholar]
- 151.Blackman ML, Royzen M & Fox JM Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels–Alder reactivity. J. Am. Chem. Soc 130, 13518–13518 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents an initial report of tetrazine ligation with TCO and the first example of very rapid kinetics in bioorthogonal chemistry (k2 > 103 M−1 s−1).
- 152.Devaraj NK, Weissleder R & Hilderbrand SA Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjugate Chem. 19, 2297–2299 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents an initial report of tetrazine ligation with norbornene and the first example of tetrazine ligation in a live-cell application.
- 153.Pipkorn R et al. Inverse-electron-demand Diels–Alder reaction as a highly efficient chemoselective ligation procedure: synthesis and function of a BioShuttle for temozolomide transport into prostate cancer cells. J. Pept. Sci 15, 235–241 (2009). [DOI] [PubMed] [Google Scholar]
- 154.Mao W et al. Organocatalytic and scalable syntheses of unsymmetrical 1,2,4,5-tetrazines by thiol-containing promotors. Angew. Chem. Int. Ed 58, 1106–1109 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Qu Y, Sauvage F-X, Clavier G, Miomandre F & Audebert P Metal-free synthetic approach to 3-monosubstituted unsymmetrical 1,2,4,5-tetrazines useful for bioorthogonal reactions. Angew. Chem. Int. Ed 57, 12057–12061 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Yang J, Karver MR, Li W, Sahu S & Devaraj NK Metal-catalyzed one-pot synthesis of tetrazines directly from aliphatic nitriles and hydrazine. Angew. Chem. Int. Ed 51, 5222–5225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lambert WD et al. Installation of minimal tetrazines through silver-mediated Liebeskind–Srogl coupling with arylboronic acids. J. Am. Chem. Soc 141, 17068–17074 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wu H, Yang J, Šečkutė J & Devaraj NK In Situ synthesis of alkenyl tetrazines for highly fluorogenic bioorthogonal live-cell imaging probes. Angew. Chem 126, 5915–5919 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xie Y et al. Divergent synthesis of monosubstituted and unsymmetrical 3,6-disubstituted tetrazines from carboxylic ester precursors. Angew. Chem. Int. Ed 59, 16967–16973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pigga JE & Fox JM Flow photochemical syntheses of trans-cyclooctenes and trans-cycloheptenes driven by metal complexation. Isr. J. Chem 60, 207–218 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Royzen M, Yap GPA & Fox JM A photochemical synthesis of functionalized trans-cyclooctenes driven by metal complexation. J. Am. Chem. Soc 130, 3760–3761 (2008). [DOI] [PubMed] [Google Scholar]
- 162.Darko A et al. Conformationally strained trans-cyclooctene with improved stability and excellent reactivity in tetrazine ligation. Chem. Sci 5, 3770–3776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report describes ultrarapid bioorthogonal reactions with rates as fast as k2 = 3.3 × 106 M−1 s−1.
- 163.Taylor MT, Blackman ML, Dmitrenko O & Fox JM Design and synthesis of highly reactive dienophiles for the tetrazine–trans-cyclooctene ligation. J. Am. Chem. Soc 133, 9646–9649 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lang K et al. Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels–Alder reactions. J. Am. Chem. Soc 134, 10317–10320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents the first example of site-specific protein labelling via IEDDA in living cells with rapidly reacting cyclooctyne and TCO dienophiles.
- 165.Patterson DM, Nazarova LA, Xie B, Kamber DN & Prescher JA Functionalized cyclopropenes as bioorthogonal chemical reporters. J. Am. Chem. Soc 134, 18638–18643 (2012). [DOI] [PubMed] [Google Scholar]
- 166.Ramil CP et al. Spirohexene–tetrazine ligation enables bioorthogonal labeling of class B G protein-coupled receptors in live cells. J. Am. Chem. Soc 139, 13376–13386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Niederwieser A et al. Two-color glycan labeling of live cells by a combination of Diels–Alder and click chemistry. Angew. Chem. Int. Ed 52, 4265–4268 (2013). [DOI] [PubMed] [Google Scholar]
- 168.Rieder U & Luedtke NW Alkene–tetrazine ligation for imaging cellular. DNA. Angew. Chem. Int. Ed 53, 9168–9172 (2014). [DOI] [PubMed] [Google Scholar]
- 169.Stöckmann H, Neves AA, Stairs S, Brindle KM & Leeper FJ Exploring isonitrile-based click chemistry for ligation with biomolecules. Org. Biomol. Chem 9, 7303–7305 (2011). [DOI] [PubMed] [Google Scholar]
- 170.Engelsma SB et al. Acylazetine as a dienophile in bioorthogonal inverse electron-demand Diels–Alder ligation. Org. Lett 16, 2744–2747 (2014). [DOI] [PubMed] [Google Scholar]
- 171.Liu K et al. A genetically encoded cyclobutene probe for labelling of live cells. ChemComm 53, 10604–10607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kamber DN et al. 1,2,4-Triazines are versatile bioorthogonal reagents. J. Am. Chem. Soc 137, 8388–8391 (2015). [DOI] [PubMed] [Google Scholar]
- 173.Carlson JCT, Meimetis LG, Hilderbrand SA & Weissleder R BODIPY–tetrazine derivatives as superbright bioorthogonal turn-on probes. Angew. Chem. Int. Ed 52, 6917–6920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Devaraj NK, Hilderbrand S, Upadhyay R, Mazitschek R & Weissleder R Bioorthogonal turn-on probes for imaging small molecules inside living cells. Angew. Chem. Int. Ed 49, 2869–2872 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents an initial description of fluorogenic tetrazine–dye conjugates for live-cell imaging applications.
- 175.Meimetis LG, Carlson JC, Giedt RJ, Kohler RH & Weissleder R Ultrafluorogenic coumarin-tetrazine probes for real-time biological imaging. Angew. Chem. Int. Ed 53, 7531–7534 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Beliu G et al. Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy. Commun. Biol 2, 261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Werther P et al. Live-cell localization microscopy with a fluorogenic and self-blinking tetrazine probe. Angew. Chem. Int. Ed 59, 804–810 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ehret F, Wu H, Alexander SC & Devaraj NK Electrochemical control of rapid bioorthogonal tetrazine ligations for selective functionalization of microelectrodes. J. Am. Chem. Soc 137, 8876–8879 (2015). [DOI] [PubMed] [Google Scholar]
- 179.Mayer SV, Murnauer A, von Wrisberg M-K, Jokisch M-L & Lang K Photo-induced and rapid labeling of tetrazine-bearing proteins via cyclopropenone-caged bicyclononynes. Angew. Chem. Int. Ed 58, 15876–15882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang H et al. Rapid bioorthogonal chemistry turn-on through enzymatic or long wavelength photocatalytic activation of tetrazine ligation. J. Am. Chem. Soc 138, 5978–5983 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lim RK & Lin Q Photoinducible bioorthogonal chemistry: a spatiotemporally controllable tool to visualize and perturb proteins in live cells. Acc. Chem. Res 44, 828–839 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Clovis JS, Eckell A, Huisgen R & Sustmann R 1.3-Dipolare cycloadditionen, XXV. der nachweis des freien diphenylnitrilimins als zwischenstufe bei cycloadditionen. Chem. Ber 100, 60–70 (1967). [Google Scholar]
- 183.Wang Y, Rivera Vera CI & Lin Q Convenient synthesis of highly functionalized pyrazolines via mild, photoactivated 1,3-dipolar cycloaddition. Org. Lett 9, 4155–4158 (2007). [DOI] [PubMed] [Google Scholar]
- 184.Song W, Wang Y, Qu J, Madden MM & Lin Q A photoinducible 1,3-dipolar cycloaddition reaction for rapid, selective modification of tetrazole-containing proteins. Angew. Chem. Int. Ed 47, 2832–2835 (2008). [DOI] [PubMed] [Google Scholar]; This article presents the first report of tetrazole-based photoclick chemistry as a new bioorthogonal reaction for biological applications.
- 185.Song W, Wang Y, Qu J & Lin Q Selective functionalization of a genetically encoded alkene-containing protein via “photoclick chemistry” in bacterial cells. J. Am. Chem. Soc 130, 9654–9655 (2008). [DOI] [PubMed] [Google Scholar]
- 186.Kumar GS & Lin Q Light-triggered click chemistry. Chem. Rev 10.1021/acs.chemrev.0c00799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang Y, Song W, Hu WJ & Lin Q Fast alkene functionalization in vivo by photoclick chemistry: HOMO lifting of nitrile imine dipoles. Angew. Chem. Int. Ed 48, 5330–5333 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lee YJ et al. A genetically encoded acrylamide functionality. ACS Chem. Biol 8, 1664–1670 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang J et al. A biosynthetic route to photoclick chemistry on proteins. J. Am. Chem. Soc 132, 14812–14818 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kamber DN et al. Isomeric cyclopropenes exhibit unique bioorthogonal reactivities. J. Am. Chem. Soc 135, 13680–13683 (2013). [DOI] [PubMed] [Google Scholar]
- 191.An P, Lewandowski TM, Erbay TG, Liu P & Lin Q Sterically shielded, stabilized nitrile imine for rapid bioorthogonal protein labeling in live cells. J. Am. Chem. Soc 140, 4860–4868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report describes the exploitation of the steric shielding effect to improve bioorthogonality of the tetrazole photoclick chemistry in cellular systems.
- 192.An P, Yu Z & Lin Q Design and synthesis of laser-activatable tetrazoles for a fast and fluorogenic red-emitting 1,3-dipolar cycloaddition reaction. Org. Lett 15, 5496–5499 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yu Z, Ohulchanskyy TY, An P, Prasad PN & Lin Q Fluorogenic, two-photon-triggered photoclick chemistry in live mammalian cells. J. Am. Chem. Soc 135, 16766–16769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lederhose P et al. Near-infrared photoinduced coupling reactions assisted by upconversion nanoparticles. Angew. Chem. Int. Ed 55, 12195–12199 (2016). [DOI] [PubMed] [Google Scholar]
- 195.Yu Z, Pan Y, Wang Z, Wang J & Lin Q Genetically encoded cyclopropene directs rapid, photoclick-chemistry-mediated protein labeling in mammalian cells. Angew. Chem. Int. Ed 51, 10600–10604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yu Z & Lin Q Design of spiro[2.3]hex-1-ene, a genetically encodable double-strained alkene for superfast photoclick chemistry. J. Am. Chem. Soc 136, 4153–4156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report describes an unprecedented double-strained alkene for accelerated tetrazole photoclick chemistry and the effect of chloride ion on reaction kinetics.
- 197.An P, Wu HY, Lewandowski TM & Lin Q Hydrophilic azaspiroalkenes as robust bioorthogonal reporters. ChemComm 54, 14005–14008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Arndt S & Wagenknecht HA “Photoclick” postsynthetic modification of DNA. Angew. Chem. Int. Ed 53, 14580–14582 (2014). [DOI] [PubMed] [Google Scholar]
- 199.Holstein JM, Stummer D & Rentmeister A Enzymatic modification of 5′-capped RNA with a 4-vinylbenzyl group provides a platform for photo-click and inverse electron-demand Diels–Alder reaction. Chem. Sci 6, 1362–1369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schart VF et al. Triple orthogonal labeling of glycans by applying photoclick chemistry. ChemBioChem 20, 166–171 (2019). [DOI] [PubMed] [Google Scholar]
- 201.Yu Z, Ho LY & Lin Q Rapid, photoactivatable turn-on fluorescent probes based on an intramolecular photoclick reaction. J. Am. Chem. Soc 133, 11912–11915 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kulkarni RA et al. Photoinducible oncometabolite detection. ChemBioChem 20, 360–365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J & Popik VV Selective labeling of living cells by a photo-triggered click reaction. J. Am. Chem. Soc 131, 15769–15776 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes the first use of dibenzocyclopropenones as the photo-masked precursor of dibenzycyclooctyne and the subsequent click reactions with azides.
- 204.Orski SV et al. High density orthogonal surface immobilization via photoactivated copper-free click chemistry. J. Am. Chem. Soc 132, 11024–11026 (2010). [DOI] [PubMed] [Google Scholar]
- 205.Jiang T et al. Modular enzyme-and light-based activation of cyclopropene–tetrazine ligation. ChemBioChem 20, 2222–2226 (2019). [DOI] [PubMed] [Google Scholar]
- 206.Arumugam S & Popik VV Light-induced hetero-Diels–Alder cycloaddition: a facile and selective photoclick reaction. J. Am. Chem. Soc 133, 5573–5579 (2011). [DOI] [PubMed] [Google Scholar]
- 207.Feist F, Menzel JP, Weil T, Blinco JP & Barner-Kowollik C Visible light-induced ligation via o-quinodimethane thioethers. J. Am. Chem. Soc 140, 11848–11854 (2018). [DOI] [PubMed] [Google Scholar]
- 208.Li J et al. Visible light-initiated bioorthogonal photoclick cycloaddition. J. Am. Chem. Soc 140, 14542–14546 (2018). [DOI] [PubMed] [Google Scholar]
- 209.Zhang L et al. Discovery of fluorogenic diarylsydnone–alkene photoligation: conversion of ortho-dual-twisted diarylsydnones into planar pyrazolines. J. Am. Chem. Soc 140, 7390–7394 (2018). [DOI] [PubMed] [Google Scholar]
- 210.Ojida A, Tsutsumi H, Kasagi N & Hamachi I Suzuki coupling for protein modification. Tetrahedron Lett. 46, 3301–3305 (2005). [Google Scholar]
- 211.Kodama K et al. Regioselective carbon–carbon bond formation in proteins with palladium catalysis; new protein chemistry by organometallic chemistry. ChemBioChem 7, 134–139 (2006). [DOI] [PubMed] [Google Scholar]
- 212.Kodama K et al. Site-specific functionalization of proteins by organopalladium reactions. ChemBioChem 8, 232–238 (2007). [DOI] [PubMed] [Google Scholar]
- 213.Brustad E et al. A genetically encoded boronate-containing amino acid. Angew. Chem. Int. Ed 47, 8220–8223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Jbara M, Maity SK & Brik A Palladium in the chemical synthesis and modification of proteins. Angew. Chem. Int. Ed 56, 10644–10655 (2017). [DOI] [PubMed] [Google Scholar]
- 215.Isenegger PG & Davis BG Concepts of catalysis in site-selective protein modifications. J. Am. Chem. Soc 141, 8005–8013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Chalker JM, Wood CS & Davis BG A convenient catalyst for aqueous and protein Suzuki–Miyaura cross-coupling. J. Am. Chem. Soc 131, 16346–16347 (2009). [DOI] [PubMed] [Google Scholar]; This article describes the first example of palladium-catalysed cross-coupling reactions for selective protein modification.
- 217.Li N, Lim RK, Edwardraja S & Lin Q Copper-free Sonogashira cross-coupling for functionalization of alkyne-encoded proteins in aqueous medium and in bacterial cells. J. Am. Chem. Soc 133, 15316–15319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Simmons RL, Yu RT & Myers AG Storable arylpalladium(II) reagents for alkene labelling in aqueous media. J. Am. Chem. Soc 133, 15870–15873 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yusop RM, Unciti-Broceta A, Johansson EMV, Sánchez-Martín RM & Bradley M Palladium-mediated intracellular chemistry. Nat. Chem 3, 239–243 (2011). [DOI] [PubMed] [Google Scholar]; This article presents the first report of the effective use of palladium-based uncaging inside cells.
- 220.Ma X, Wang H & Chen W N-Heterocyclic carbene-stabilized palladium complexes as organometallic catalysts for bioorthogonal cross-coupling reactions. J. Org. Chem 79, 8652–8658 (2014). [DOI] [PubMed] [Google Scholar]
- 221.Li N, Ramil CP, Lim RKV & Lin Q A genetically encoded alkyne directs palladium-mediated protein labelling on live mammalian cell surface. ACS Chem. Biol 10, 379–384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lim RKV, Li N, Ramil CP & Lin Q Fast and sequence-specific palladium-mediated cross-coupling reaction identified from phage display. ACS Chem. Biol 9, 2139–2148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cheng G, Lim RKV, Li N & Lin Q Storable palladacycles for selective functionalization of alkyne-containing proteins. Chem. Commun 49, 6809–6811 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Dumas A et al. Self-liganded Suzuki–Miyaura coupling for site-selective protein PEGylation. Angew. Chem. Int. Ed 52, 3916–3921 (2013). [DOI] [PubMed] [Google Scholar]
- 225.Spicer CD, Triemer T & Davis BG Palladium-mediated cell-surface labelling. J. Am. Chem. Soc 134, 800–803 (2012). [DOI] [PubMed] [Google Scholar]
- 226.Lercher L, McGouran JF, Kessler BM, Schofield CJ & Davis BG DNA modification under mild conditions by Suzuki–Miyaura cross-coupling for the generation of functional probes. Angew. Chem. Int. Ed 52, 10553–10558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Spicer CD & Davis BG Palladium-mediated site-selective Suzuki–Miyaura protein modification at genetically encoded aryl halides. Chem. Commun 47, 1698–1700 (2011). [DOI] [PubMed] [Google Scholar]
- 228.Lin YA, Chalker JM & Davis BG Olefin cross-metathesis on proteins: investigation of allylic chalcogen effects and guiding principles in metathesis partner selection. J. Am. Chem. Soc 132, 16805–16811 (2010). [DOI] [PubMed] [Google Scholar]
- 229.Lin YA, Chalker JM, Floyd N, Bernardes GJL & Davis BG Allyl sulfides are privileged substrates in aqueous cross-metathesis: application to site-selective protein modification. J. Am. Chem. Soc 130, 9642–9643 (2008). [DOI] [PubMed] [Google Scholar]; This article describes the first example of selective ruthenium-mediated cross-metathesis of olefins with protein substrates.
- 230.Chalker JM, Lin YA, Boutureira O & Davis BG Enabling olefin metathesis on proteins: chemical methods for installation of S-allyl cysteine. Chem. Commun 25, 3714–3716 (2009). [DOI] [PubMed] [Google Scholar]
- 231.Lin YA et al. Rapid cross-metathesis for reversible protein modifications via chemical access to Se-allylselenocysteine in proteins. J. Am. Chem. Soc 135, 12156–12159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bhushan B et al. Genetic incorporation of olefin cross-metathesis reaction tags for protein modification. J. Am. Chem. Soc 140, 14599–14603 (2018). [DOI] [PubMed] [Google Scholar]
- 233.Hruby VJ, Boteju L & Li G in Chemical & Engineering News Vol. 71, 2 (American Chemical Society, Safety Letters, 1993). [Google Scholar]
- 234.Niemeier JK & Kjell DP Hydrazine and aqueous hydrazine solutions: evaluating safety in chemical processes. Org. Process. Res. Dev 17, 1580–1590 (2013). [Google Scholar]
- 235.Richardson MB et al. Synthesis and explosion hazards of 4-azido-l-phenylalanine. J. Org. Chem 83, 4525–4536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sperry JB et al. Thermal stability assessment of peptide coupling reagents commonly used in pharmaceutical manufacturing. Org. Process. Res. Dev 22, 1262–1275 (2018). [Google Scholar]; This study provides an excellent overview of the different techniques for analysing the energetics of materials.
- 237.Green SP et al. On the use of differential scanning calorimetry for thermal hazard assessment of new chemistry: avoiding explosive mistakes. Angew. Chem. Int. Ed 59, 15798–15802 (2020). [DOI] [PubMed] [Google Scholar]
- 238.Gordon AJ & Ford RA The Chemist’s Companion: A Handbook of Practical Data, Techniques, and References (Wiley, 1972). [Google Scholar]
- 239.Grammel M & Hang HC Chemical reporters for biological discovery. Nat. Chem. Biol 9, 475–484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Parker CG & Pratt MR Click chemistry in proteomic investigations. Cell 180, 605–632 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Madl CM & Heilshorn SC Bioorthogonal strategies for engineering extracellular matrices. Adv. Funct. Mater 28, 1706046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tu J, Xu M & Franzini RM Dissociative bioorthogonal reactions. ChemBioChem 20, 1615–1627 (2019). [DOI] [PubMed] [Google Scholar]
- 243.Lim RKV & Lin Q Bioorthogonal chemistry: recent progress and future directions. ChemComm 46, 1589–1600 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sletten EM & Bertozzi CR Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed 48, 6974–6998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.McKay CS & Finn MG Click chemistry in complex mixtures: bioorthogonal bioconjugation. Chem. Biol 21, 1075–1101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Laughlin ST, Baskin JM, Amacher SL & Bertozzi CR In vivo imaging of membrane-associated glycans in developing zebrafish. Science 320, 664–667 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Agarwal P, Beahm BJ, Shieh P & Bertozzi CR Systemic fluorescence imaging of zebrafish glycans with bioorthogonal chemistry. Angew. Chem. Int. Ed 54, 11504–11510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Salic A & Mitchison TJ A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl Acad. Sci. USA 105, 2415–2420 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Nainar S et al. Metabolic incorporation of azide functionality into cellular RNA. ChemBioChem 17, 2149–2152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Alvarez-Castelao B et al. Cell-type-specific metabolic labelling of nascent proteomes in vivo. Nat. Biotechnol 35, 1196–1201 (2017). [DOI] [PubMed] [Google Scholar]
- 251.Yuet KP et al. Cell-specific proteomic analysis in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 112, 2705–2710 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Chang PV, Prescher JA, Hangauer MJ & Bertozzi CR Imaging cell surface glycans with bioorthogonal chemical reporters. J. Am. Chem. Soc 129, 8400–8401 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Liang D et al. A real-time, click chemistry imaging approach reveals stimulus-specific subcellular locations of phospholipase D activity. Proc. Natl Acad. Sci. USA 116, 15453–15462 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jiang H, Kim JH, Frizzell KM, Kraus WL & Lin H Clickable NAD analogues for labeling substrate proteins of poly(ADP-ribose) polymerases. J. Am. Chem. Soc 132, 9363–9372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Cañeque T, Müller S & Rodriguez R Visualizing biologically active small molecules in cells using click chemistry. Nat. Rev. Chem 2, 202–215 (2018). [Google Scholar]
- 256.Tian Y & Lin Q Fitness factors for bioorthogonal chemical probes. ACS Chem. Biol 14, 2489–2496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Patterson DM, Nazarova LA & Prescher JA Finding the right (bioorthogonal) chemistry. ACS Chem. Biol 9, 592–605 (2014). [DOI] [PubMed] [Google Scholar]
- 258.Kim EJ Chemical reporters and their bioorthogonal reactions for labeling protein O-GlcNAcylation. Molecules 23, 2411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Thirumurugan P, Matosiuk D & Jozwiak K Click chemistry for drug development and diverse chemical–biology applications. Chem. Rev 113, 4905–4979 (2013). [DOI] [PubMed] [Google Scholar]
- 260.Zhang X & Zhang Y Applications of azide-based bioorthogonal click chemistry in glycobiology. Molecules 18, 7145–7159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Mahal LK, Yarema KJ & Bertozzi CR Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125–1128 (1997). [DOI] [PubMed] [Google Scholar]
- 262.Cole CM, Yang J, Šečkutė J & Devaraj NK Fluorescent live-cell imaging of metabolically incorporated unnatural cyclopropene-mannosamine derivatives. ChemBioChem 14, 205–208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Nguyen K et al. Cell-selective bioorthogonal metabolic labeling of RNA. J. Am. Chem. Soc 139, 2148–2151 (2017). [DOI] [PubMed] [Google Scholar]
- 264.Chang PV, Dube DH, Sletten EM & Bertozzi CR A strategy for the selective imaging of glycans using caged metabolic precursors. J. Am. Chem. Soc 132, 9516–9518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Xie R, Hong S & Chen X Cell-selective metabolic labeling of biomolecules with bioorthogonal functionalities. Curr. Opin. Chem. Biol 17, 747–752 (2013). [DOI] [PubMed] [Google Scholar]
- 266.Wang H et al. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Chem. Biol 13, 415–424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ngo JT et al. Cell-selective metabolic labeling of proteins. Nat. Chem. Biol 5, 715–717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Debets MF et al. Metabolic precision labeling enables selective probing of O-linked N-acetylgalactosamine glycosylation. Proc. Natl Acad. Sci. USA 117, 25293–25301 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Islam K et al. Defining efficient enzyme–cofactor pairs for bioorthogonal profiling of protein methylation. Proc. Natl Acad. Sci. USA 110, 16778–16783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Stone SE, Glenn WS, Hamblin GD & Tirrell DA Cell-selective proteomics for biological discovery. Curr. Opin. Chem. Biol 36, 50–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Lang K & Chin JW Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev 114, 4764–4806 (2014). [DOI] [PubMed] [Google Scholar]
- 272.Young DD & Schultz PG Playing with the molecules of life. ACS Chem. Biol 13, 854–870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Lee KJ, Kang D & Park HS Site-specific labeling of proteins using unnatural amino acids. Mol. Cell 42, 386–396 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mayer S & Lang K Tetrazines in inverse-electron-demand Diels–Alder cycloadditions and their use in biology. Synthesis 49, 830–848 (2017). [Google Scholar]
- 275.Lang K et al. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat. Chem 4, 298 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article present the first example of site-specific protein labelling via IEDDA in living cells.
- 276.Plass T et al. Amino acids for Diels–Alder reactions in living cells. Angew. Chem. Int. Ed 51, 4166–4170 (2012). [DOI] [PubMed] [Google Scholar]
- 277.Jang HS, Jana S, Blizzard RJ, Meeuwsen JC & Mehl RA Access to faster eukaryotic cell labeling with encoded tetrazine amino acids. J. Am. Chem. Soc 142, 7245–7249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Blizzard RJ et al. Ideal bioorthogonal reactions using a site-specifically encoded tetrazine amino acid. J. Am. Chem. Soc 137, 10044–10047 (2015). [DOI] [PubMed] [Google Scholar]
- 279.Nikić I et al. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew. Chem. Int. Ed 53, 2245–2249 (2014). [DOI] [PubMed] [Google Scholar]
- 280.Peng T & Hang HC Site-specific bioorthogonal labeling for fluorescence imaging of intracellular proteins in living cells. J. Am. Chem. Soc 138, 14423–14433 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lahann J (ed.) Click Chemistry for Biotechnology and Materials Science 411 (Wiley, 2009). [Google Scholar]
- 282.Mideksa YG et al. Site-specific protein labeling with fluorophores as a tool to monitor protein turnover. ChemBioChem 21, 1861–1867 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Sachdeva A, Wang K, Elliott T & Chin JW Concerted, rapid, quantitative, and site-specific dual labeling of proteins. J. Am. Chem. Soc 136, 7785–7788 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wang K et al. Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat. Chem 6, 393–403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Tsai Y-H, Essig S, James JR, Lang K & Chin JW Selective, rapid and optically switchable regulation of protein function in live mammalian cells. Nat. Chem 7, 554–561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Liang H et al. Metabolic labelling of the carbohydrate core in bacterial peptidoglycan and its applications. Nat. Commun 8, 15015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Laughlin ST & Bertozzi CR In vivo imaging of Caenorhabditis elegans glycans. ACS Chem. Biol 4, 1068–1072 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Nikić I et al. Debugging eukaryotic genetic code expansion for site-specific click-PAINT super-resolution microscopy. Angew. Chem. Int. Ed 55, 16172–16176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Uttamapinant C et al. Genetic code expansion enables live-cell and super-resolution imaging of site-specifically labeled cellular proteins. J. Am. Chem. Soc 137, 4602–4605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Elliott TS, Bianco A, Townsley FM, Fried SD & Chin JW Tagging and enriching proteins enables cell-specific proteomics. Cell Chem. Biol 23, 805–815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Elliott TS et al. Proteome labeling and protein identification in specific tissues and at specific developmental stages in an animal. Nat. Biotechnol 32, 465–472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Krogager TP et al. Labeling and identifying cell-specific proteomes in the mouse brain. Nat. Biotechnol 36, 156–159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Shieh P, Hangauer MJ & Bertozzi CR Fluorogenic azidofluoresceins for biological imaging. J. Am. Chem. Soc 134, 17428–17431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Mao W et al. A general strategy to design highly fluorogenic far-red and near-infrared tetrazine bioorthogonal probes. Angew. Chem. Int. Ed 60, 2393–2397 (2021). [DOI] [PubMed] [Google Scholar]
- 295.Shieh P et al. CalFluors: a universal motif for fluorogenic azide probes across the visible spectrum. J. Am. Chem. Soc 137, 7145–7151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Ngo JT et al. Click-EM for imaging metabolically tagged nonprotein biomolecules. Nat. Chem. Biol 12, 459–465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Hong S, Lin L, Xiao M & Chen X Live-cell bioorthogonal Raman imaging. Curr. Opin. Chem. Biol 24, 91–96 (2015). [DOI] [PubMed] [Google Scholar]
- 298.Bunnage ME, Chekler ELP & Jones LH Target validation using chemical probes. Nat. Chem. Biol 9, 195–199 (2013). [DOI] [PubMed] [Google Scholar]
- 299.Cravatt BF, Wright AT & Kozarich JW Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem 77, 383–414 (2008). [DOI] [PubMed] [Google Scholar]
- 300.Cravatt BF, Hsu K-L & Weerapana E Activity-Based Protein Profiling Vol. 420 (Springer, 2019). [Google Scholar]
- 301.Geurink PP, Prely LM, van der Marel GA, Bischoff R & Overkleeft HS in Activity-Based Protein Profiling 85–113 (Springer, 2011). [DOI] [PubMed] [Google Scholar]; This book provides a summary of ABPP and photoaffinity labelling with relevant applications in natural product target discovery and microbial pathogenesis.
- 302.Jessani N, Liu Y, Humphrey M & Cravatt BF Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc. Natl Acad. Sci. USA 99, 10335–10340 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Nomura DK et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Barglow KT & Cravatt BF Activity-based protein profiling for the functional annotation of enzymes. Nat. Methods 4, 822–827 (2007). [DOI] [PubMed] [Google Scholar]
- 305.Nomura DK, Dix MM & Cravatt BF Activity-based protein profiling for biochemical pathway discovery in cancer. Nat. Rev. Cancer 10, 630–638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Smith E & Collins I Photoaffinity labeling in target-and binding-site identification. Future Med. Chem 7, 159–183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Preston GW & Wilson AJ Photo-induced covalent cross-linking for the analysis of biomolecular interactions. Chem. Soc. Rev 42, 3289–3301 (2013). [DOI] [PubMed] [Google Scholar]
- 308.Zuhl AM et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of β-secretase inhibitors. Nat. Commun 7, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Hur J-Y et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 586, 735–740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Speers AE, Adam GC & Cravatt BF Activity-based protein profiling in vivo using a copper (I)-catalyzed azide–alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc 125, 4686–4687 (2003). [DOI] [PubMed] [Google Scholar]
- 311.Speers AE & Cravatt BF Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol 11, 535–546 (2004). [DOI] [PubMed] [Google Scholar]
- 312.van der Linden WA et al. Two-step bioorthogonal activity-based proteasome profiling using copper-free click reagents: a comparative study. Bioorg. Med. Chem 20, 662–666 (2012). [DOI] [PubMed] [Google Scholar]
- 313.Jessani N & Cravatt BF The development and application of methods for activity-based protein profiling. Curr. Opin. Chem. Biol 8, 54–59 (2004). [DOI] [PubMed] [Google Scholar]
- 314.Van Esbroeck AC et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10–2474. Science 356, 1084–1087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Medina-Cleghorn D, Heslin A, Morris PJ, Mulvihill MM & Nomura DK Multidimensional profiling platforms reveal metabolic dysregulation caused by organophosphorus pesticides. ACS Chem. Biol 9, 423–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Huang Z et al. Global portrait of protein targets of metabolites of the neurotoxic compound BIA 10–2474. ACS Chem. Biol 14, 192–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Weerapana E, Speers AE & Cravatt BF Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP) — a general method for mapping sites of probe modification in proteomes. Nat. Protoc 2, 1414 (2007). [DOI] [PubMed] [Google Scholar]
- 318.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Roberts AM, Ward CC & Nomura DK Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr. Opin. Biotechnol 43, 25–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Vinogradova E et al. An activity-guided map of electrophile–cysteine interactions in primary human immune cells. Cell 10.2139/ssrn.3476689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Zhang X, Crowley VM, Wucherpfennig TG, Dix MM & Cravatt BF Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol 15, 737–746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Lambert JM & Morris CQ Antibody–drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv. Ther 34, 1015–1035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Agarwal P & Bertozzi CR Site-specific antibody–drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chem. 26, 176–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes key advancements in the ADC field with a focus on bioorthogonal chemistry and protein engineering.
- 324.Azoulay M, Tuffin G, Sallem W & Florent J-C A new drug-release method using the Staudinger ligation. Bioorg. Med. Chem. Lett 16, 3147–3149 (2006). [DOI] [PubMed] [Google Scholar]
- 325.Brakel RV, Vulders RC, Bokdam RJ, Grüll H & Robillard MS A doxorubicin prodrug activated by the Staudinger reaction. Bioconjugate Chem. 19, 714–718 (2008). [DOI] [PubMed] [Google Scholar]
- 326.Matikonda SS et al. Bioorthogonal prodrug activation driven by a strain-promoted 1,3-dipolar cycloaddition. Chem. Sci 6, 1212–1218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Zheng Y et al. Enrichment-triggered prodrug activation demonstrated through mitochondria-targeted delivery of doxorubicin and carbon monoxide. Nat. Chem 10, 787–794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Versteegen RM, Rossin R, ten Hoeve W, Janssen HM & Robillard MS Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem. Int. Ed 52, 14112–14116 (2013). [DOI] [PubMed] [Google Scholar]; This article presents an initial description of bioorthogonal uncaging of small molecules.
- 329.Wu H, Alexander SC, Jin S & Devaraj NK A bioorthogonal near-infrared fluorogenic probe for mRNA detection. J. Am. Chem. Soc 138, 11429–11432 (2016). [DOI] [PubMed] [Google Scholar]
- 330.Neumann K et al. Tetrazine-responsive self-immolative linkers. ChemBioChem 18, 91–95 (2017). [DOI] [PubMed] [Google Scholar]
- 331.Jiménez-Moreno E et al. Vinyl ether/tetrazine pair for the traceless release of alcohols in cells. Angew. Chem. Int. Ed 56, 243–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Tu J, Xu M, Parvez S, Peterson RT & Franzini RM Bioorthogonal removal of 3-isocyanopropyl groups enables the controlled release of fluorophores and drugs in vivo. J. Am. Chem. Soc 140, 8410–8414 (2018). [DOI] [PubMed] [Google Scholar]
- 333.Xu M, Galindo-Murillo R, Cheatham T & Franzini R Dissociative reactions of benzonorbornadienes with tetrazines: scope of leaving groups and mechanistic insights. Org. Biomol. Chem 15, 9855–9865 (2017). [DOI] [PubMed] [Google Scholar]
- 334.van Onzen AH et al. Bioorthogonal tetrazine carbamate cleavage by highly reactive trans-cyclooctene. J. Am. Chem. Soc 142, 10955–10963 (2019). [DOI] [PubMed] [Google Scholar]
- 335.Tu J et al. Isonitrile-responsive and bioorthogonally removable tetrazine protecting groups. Chem. Sci 11, 169–179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Wang Q et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426 (2020). [DOI] [PubMed] [Google Scholar]
- 337.Völker T, Dempwolff F, Graumann PL & Meggers E Progress towards bioorthogonal catalysis with organometallic compounds. Angew. Chem. Int. Ed 53, 10536–10540 (2014). [DOI] [PubMed] [Google Scholar]
- 338.Li J et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem 6, 352–361 (2014). [DOI] [PubMed] [Google Scholar]
- 339.Wang X et al. Copper-triggered bioorthogonal cleavage reactions for reversible protein and cell surface modifications. J. Am. Chem. Soc 141, 17133–17141 (2019). [DOI] [PubMed] [Google Scholar]
- 340.Pérez-López AM et al. Gold-triggered uncaging chemistry in living systems. Angew. Chem. Int. Ed 56, 12548–12552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Weiss JT et al. Extracellular palladium-catalysed dealkylation of 5-fluoro-1-propargyl-uracil as a bioorthogonally activated prodrug approach. Nat. Commun 5, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Wang J et al. Palladium-triggered chemical rescue of intracellular proteins via genetically encoded allene-caged tyrosine. J. Am. Chem. Soc 138, 15118–15121 (2016). [DOI] [PubMed] [Google Scholar]
- 343.Chang PV et al. Copper-free click chemistry in living animals. Proc. Natl Acad. Sci. USA 107, 1821–1826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article compares several copper-free chemistries for metabolic labelling in mice.
- 344.Ursuegui S, Recher M, Krężel W & Wagner A An in vivo strategy to counteract post-administration anticoagulant activity of azido-warfarin. Nat. Commun 8, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Li Z et al. Pretargeting and bioorthogonal click chemistry-mediated endogenous stem cell homing for heart repair. ACS Nano 12, 12193–12200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Rossin R et al. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem. Int. Ed 49, 3375–3378 (2010). [DOI] [PubMed] [Google Scholar]; This article presents an initial use of bioorthogonal chemistry for pre-targeted radiochemical imaging in live mice.
- 347.Zeglis BM et al. A pretargeted PET imaging strategy based on bioorthogonal Diels–Alder click chemistry. J. Nucl. Med 54, 1389–1396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Devaraj NK, Thurber GM, Keliher EJ, Marinelli B & Weissleder R Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl Acad. Sci. USA 109, 4762–4767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Rossin R et al. Highly reactive trans-cyclooctene tags with improved stability for Diels–Alder chemistry in living systems. Bioconjugate Chem. 24, 1210–1217 (2013). [DOI] [PubMed] [Google Scholar]
- 350.Rossin R, van Duijnhoven SM, Läppchen T, van den Bosch SM & Robillard MS Trans-cyclooctene tag with improved properties for tumor pretargeting with the Diels–Alder reaction. Mol. Pharm 11, 3090–3096 (2014). [DOI] [PubMed] [Google Scholar]
- 351.Rossin R, Läppchen T, Van Den Bosch SM, Laforest R & Robillard MS Diels–Alder reaction for tumor pretargeting: in vivo chemistry can boost tumor radiation dose compared with directly labeled antibody. J. Nucl. Med 54, 1989–1995 (2013). [DOI] [PubMed] [Google Scholar]
- 352.Meyer J-P et al. Bioorthogonal masking of circulating antibody–TCO groups using tetrazine-functionalized dextran polymers. Bioconjugate Chem. 29, 538–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Keinänen O et al. Pretargeting of internalizing trastuzumab and cetuximab with a 18F-tetrazine tracer in xenograft models. EJNMMI Res. 7, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Zeglis BM et al. Optimization of a pretargeted strategy for the PET imaging of colorectal carcinoma via the modulation of radioligand pharmacokinetics. Mol. Pharm 12, 3575–3587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Poty S et al. Leveraging bioorthogonal click chemistry to improve 225Ac-radioimmunotherapy of pancreatic ductal adenocarcinoma. Clin. Cancer Res 25, 868–880 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Rondon A et al. Pretargeted radioimmunotherapy and SPECT imaging of peritoneal carcinomatosis using bioorthogonal click chemistry: probe selection and first proof-of-concept. Theranostics 9, 6706 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents a therapeutic proof of concept of IEDDA-mediated pre-targeted radioimmunotherapy.
- 357.Rossin R et al. Chemically triggered drug release from an antibody–drug conjugate leads to potent antitumour activity in mice. Nat. Commun 9, 1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Rossin R et al. Triggered drug release from an antibody–drug conjugate using fast “click-to-release” chemistry in mice. Bioconjugate Chem. 27, 1697–1706 (2016). [DOI] [PubMed] [Google Scholar]
- 359.Mejia Oneto JM, Khan I, Seebald L & Royzen M In vivo bioorthogonal chemistry enables local hydrogel and systemic pro-drug to treat soft tissue sarcoma. ACS Cent. Sci 2, 476–482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Zhang G et al. Bioorthogonal chemical activation of kinases in living systems. ACS Cent. Sci 2, 325–331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.van der Gracht AM et al. Chemical control over T-cell activation in vivo using deprotection of trans-cyclooctene-modified epitopes. ACS Chem. Biol 13, 1569–1576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Li H, Conde J, Guerreiro A & Bernardes GJ Tetrazine carbon nanotubes for pretargeted in vivo ‘click-to-release’ bioorthogonal tumour imaging. Angew. Chem. Int. Ed 59, 16032 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Yao Q et al. Synergistic enzymatic and bioorthogonal reactions for selective prodrug activation in living systems. Nat. Commun 9, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Xie X et al. Bioorthogonal nanosystem for near-infrared fluorescence imaging and prodrug activation in mouse model. ACS Mater. Lett 1, 549–557 (2019). [Google Scholar]
- 365.Miller MA et al. Modular nanoparticulate prodrug design enables efficient treatment of solid tumors using bioorthogonal activation. ACS Nano 12, 12814–12826 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Miller MA et al. Nano-palladium is a cellular catalyst for in vivo chemistry. Nat. Commun 8, 1–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04106492 (2021). [DOI] [PubMed]
- 368.Díaz DD et al. Click chemistry in materials synthesis. 1. Adhesive polymers from copper-catalyzed azide–alkyne cycloaddition. J. Polym. Sci. A Polym. Chem 42, 4392–4403 (2004). [Google Scholar]
- 369.Arslan M, Acik G & Tasdelen MA The emerging applications of click chemistry reactions in the modification of industrial polymers. Polym. Chem 10, 3806–3821 (2019). [Google Scholar]
- 370.Qin A, Lam JWY & Tang BZ Click polymerization. Chem. Soc. Rev 39, 2522–2544 (2010). [DOI] [PubMed] [Google Scholar]
- 371.Binder WH & Sachsenhofer R ‘Click’ chemistry in polymer and material science: an update. Macromol. Rapid Commun 29, 952–981 (2008). [Google Scholar]
- 372.Hansell CF et al. Additive-free clicking for polymer functionalization and coupling by tetrazine–norbornene chemistry. J. Am. Chem. Soc 133, 13828–13831 (2011). [DOI] [PubMed] [Google Scholar]
- 373.Zhou H et al. Crossover experiments applied to network formation reactions: improved strategies for counting elastically inactive molecular defects in PEG gels and hyperbranched polymers. J. Am. Chem. Soc 136, 9464–9470 (2014). [DOI] [PubMed] [Google Scholar]
- 374.Arseneault M, Wafer C & Morin J-F Recent advances in click chemistry applied to dendrimer synthesis. Molecules 20, 9263–9294 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Martens S, Holloway JO & Du Prez FE Click and click-inspired chemistry for the design of sequence-controlled polymers. Macromol. Rapid Commun 38, 1700469 (2017). [DOI] [PubMed] [Google Scholar]
- 376.Yang C, Flynn JP & Niu J Facile synthesis of sequence-regulated synthetic polymers using orthogonal SuFEx and CuAAC click reactions. Angew. Chem. Int. Ed 57, 16194–16199 (2018). [DOI] [PubMed] [Google Scholar]
- 377.Cook BE, Membreno R & Zeglis BM Dendrimer scaffold for the amplification of in vivo pretargeting ligations. Bioconjugate Chem. 29, 2734–2740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Wang Q et al. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc 125, 3192–3193 (2003). [DOI] [PubMed] [Google Scholar]
- 379.Abedin MJ, Liepold L, Suci P, Young M & Douglas T Synthesis of a cross-linked branched polymer network in the interior of a protein cage. J. Am. Chem. Soc 131, 4346–4354 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Mellet CO, Méndez-Ardoy A & Fernández JMG in Click Chemistry in Glycoscience (eds Witczak ZJ & Bielski R) 143–182 (Wiley, 2013). [Google Scholar]
- 381.Haun JB, Devaraj NK, Hilderbrand SA, Lee H & Weissleder R Bioorthogonal chemistry amplifies nanoparticle binding and enhances the sensitivity of cell detection. Nat. Nanotechnol 5, 660–665 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Collman JP, Devaraj NK & Chidsey CED “Clicking” functionality onto electrode surfaces. Langmuir 20, 1051–1053 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Escorihuela J, Marcelis ATM & Zuilhof H Metal-free click chemistry reactions on surfaces. Adv. Mater. Interfaces 2, 1500135 (2015). [Google Scholar]
- 384.Azagarsamy MA & Anseth KS Bioorthogonal click chemistry: an indispensable tool to create multifaceted cell culture scaffolds. ACS Macro Lett. 2, 5–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Alge DL, Azagarsamy MA, Donohue DF & Anseth KS Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine–norbornene chemistry. Biomacromolecules 14, 949–953 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Brown TE & Anseth KS Spatiotemporal hydrogel biomaterials for regenerative medicine. Chem. Soc. Rev 46, 6532–6552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Selvanathan A et al. Photo-click chemistry strategies for spatiotemporal control of metal-free ligation, labeling, and surface derivatization. Pure Appl. Chem 85, 1499–1513 (2013). [Google Scholar]
- 388.He M, Li J, Tan S, Wang R & Zhang Y Photodegradable supramolecular hydrogels with fluorescence turn-on reporter for photomodulation of cellular microenvironments. J. Am. Chem. Soc 135, 18718–18721 (2013). [DOI] [PubMed] [Google Scholar]
- 389.Dietrich M et al. Photoclickable surfaces for profluorescent covalent polymer coatings. Adv. Funct. Mater 22, 304–312 (2012). [Google Scholar]
- 390.Hufendiek A, Carlmark A, Meier MAR & Barner-Kowollik C Fluorescent covalently cross-linked cellulose networks via light-induced ligation. ACS Macro Lett. 5, 139–143 (2016). [DOI] [PubMed] [Google Scholar]
- 391.Wallin TJ et al. Click chemistry stereolithography for soft robots that self-heal. J. Mater. Chem. B 5, 6249–6255 (2017). [DOI] [PubMed] [Google Scholar]
- 392.Diehl KL et al. Click and chemically triggered declick reactions through reversible amine and thiol coupling via a conjugate acceptor. Nat. Chem 8, 968–973 (2016). [Google Scholar]
- 393.Zlitni A, Janzen N, Foster FS & Valliant JF Catching bubbles: targeting ultrasound microbubbles using bioorthogonal inverse-electron-demand Diels–Alder reactions. Angew. Chem. Int. Ed 53, 6459–6463 (2014). [DOI] [PubMed] [Google Scholar]
- 394.Koo H et al. Bioorthogonal click chemistry-based synthetic cell glue. Small 11, 6458–6466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Liu S et al. Meter-long multiblock copolymer microfibers via interfacial bioorthogonal polymerization. Adv. Mater 27, 2783–2790 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Dicker KT et al. Core-shell patterning of synthetic hydrogels via interfacial bioorthogonal chemistry for spatial control of stem cell behavior. Chem. Sci 9, 5394–5404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Dicker KT et al. Spatial patterning of molecular cues and vascular cells in fully integrated hydrogel channels via interfacial bioorthogonal cross-linking. ACS Appl. Mater. Interfaces 11, 16402–16411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Truong VX, Ablett MP, Richardson SM, Hoyland JA & Dove AP Simultaneous orthogonal dual-click approach to tough, in-situ-forming hydrogels for cell encapsulation. J. Am. Chem. Soc 137, 1618–1622 (2015). [DOI] [PubMed] [Google Scholar]
- 399.Patterson DM & Prescher JA Orthogonal bioorthogonal chemistries. Curr. Opin. Chem. Biol 28, 141–149 (2015). [DOI] [PubMed] [Google Scholar]
- 400.Westrum EF & Pitzer KS Thermodynamics of the system KHF2–KF–HF, including heat capacities and entropies of KHF2 and KF. The nature of the hydrogen bond in KHF2. J. Am. Chem. Soc 71, 1940–1949 (1949). [Google Scholar]
- 401.Liu F et al. Biocompatible SuFEx click chemistry: thionyl tetrafluoride (SOF4)-derived connective hubs for bioconjugation to DNA and proteins. Angew. Chem. Int. Ed 58, 8029–8033 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Dong J, Sharpless KB, Kwisnek L, Oakdale JS & Fokin VV SuFEx-based synthesis of polysulfates. Angew. Chem. Int. Ed 53, 9466–9470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Jones LH Emerging utility of fluorosulfate chemical probes. ACS Med. Chem. Lett 9, 584–586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Guo T et al. A new portal to SuFEx click chemistry: a stable fluorosulfuryl imidazolium salt emerging as an “F−SO2+ ” donor of unprecedented reactivity, selectivity, and scope. Angew. Chem. Int. Ed 57, 2605–2610 (2018). [DOI] [PubMed] [Google Scholar]
- 405.Zhou H et al. Introduction of a crystalline, shelf-stable reagent for the synthesis of sulfur(VI) fluorides. Org. Lett 20, 812–815 (2018). [DOI] [PubMed] [Google Scholar]