

Abstract
Alcoholic liver disease (ALD) due to chronic alcohol consumption is a significant global disease burden and a leading cause of mortality. Alcohol abuse induces a myriad of aberrant changes in hepatocytes at both the cellular and molecular level. Although the disease spectrum of ALD is widely recognized, the precise triggers for disease progression are still to be fully elucidated. Oxidative stress, mitochondrial dysfunction, gut dysbiosis and altered immune system response plays an important role in disease pathogenesis, triggering the activation of inflammatory pathways and apoptosis. Despite many recent clinical studies treatment options for ALD are limited, especially at the alcoholic hepatitis stage. We have therefore reviewed some of the key pathways involved in the pathogenesis of ALD and highlighted current trials for treating patients.
Keywords: Liver, Alcohol, Oxidative stress, Inflammation, Gut microbiome, Mitochondria
Core Tip: Alcoholic liver disease (ALD) causes significant global disease burden inducing both cellular and molecular modifications in hepatocytes. Although the spectrum of disease is widely recognized, the precise disease pathogenesis is yet to be fully elucidated. In this review we summarize some of the key pathways responsible for the pathogenesis of ALD and highlight current available treatments.
INTRODUCTION
Alcoholic liver disease (ALD) is one of the most prevalent chronic liver diseases and causes significant mortality globally. Chronic alcohol consumption has been implicated in multiple medical conditions including cancer, diabetes, cardiovascular disease, liver and pancreatic disorders. However, no new treatment options have been developed for many years. At present, abstinence remains the most important treatment for ALD, however, there is a need to develop effective treatment options associated with alcohol misuse.
The spectrum of ALD is widely recognized ranging from simple liver steatosis; which can occur in up to 90% of heavy drinkers[1] to alcoholic hepatitis (AH), which develops in 10% to 35% of heavy drinkers[2]. AH can ultimately progress to fibrosis, where hepatic stellate cell (HSC) activation, collagen synthesis and accumulation of extracellular matrix proteins occurs due to the formation of protein adducts. Cirrhosis and lastly hepatocellular cancer are the final stages. Fibrosis/cirrhosis can cause hepatocyte inactivation, and is associated with abnormal DNA repair, damage to mitochondrial function and oxygen utilization disorders[3]. This can lead to hepatocellular failure and portal hypertension which ultimately requires a liver transplantation.
The molecular and biochemical mechanisms underlying the pathogenesis of ALD as well as the precise triggers for disease progression are not completely understood. Recent evidence suggests disease progression is thought to involve several pathological stages such as mitochondrial dysfunction, oxidative stress, altered methionine metabolism, iron dysregulation, gut dysbiosis, activation of inflammatory pathways and decreased production of antioxidants (Figure 1)[4]. Currently, no effective treatment for ALD exists due to the incomplete understanding of hepatic biochemical alterations and pathogenic mechanisms responsible for disease progression.
Figure 1.
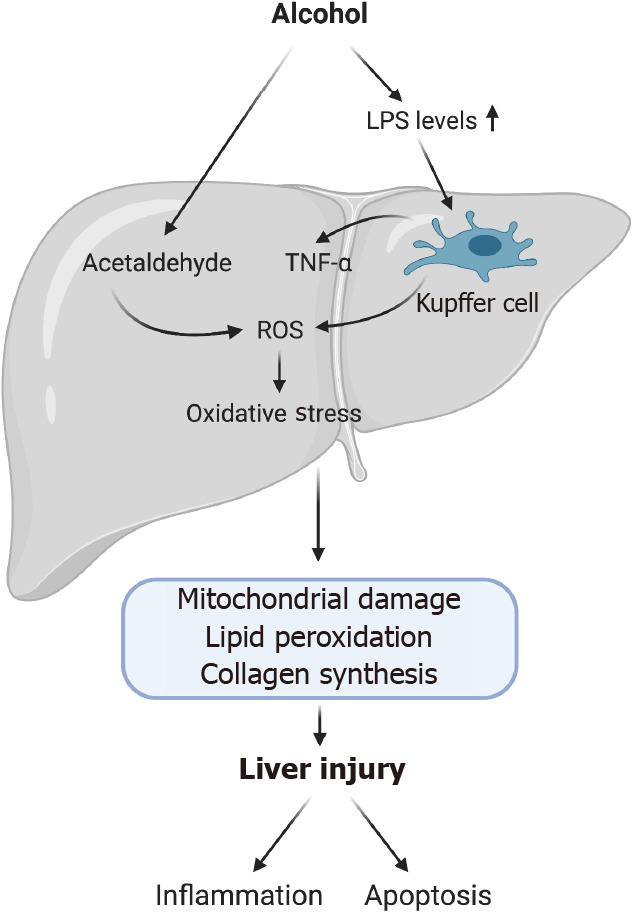
Alcohol-related induction of oxidative stress and liver injury. Alcohol misuse leads to loss of tight junctions in the gut increasing its permeability. This causes translocation of lipopolysaccharide into the liver activating toll-like receptor 4 on Kupffer cells (KCs). Activation of KCs can cause reactive oxygen species (ROS) production and pro-inflammatory cytokines such as tumor necrosis factor-α. ROS production also occurs due to the metabolism of alcohol. ROS production and inflammatory cytokines leads to inflammation and recruitment of inflammatory cells as well as activation of apoptotic pathways. (Figure created with BioRender.com). LPS: Lipopolysaccharide; ROS: Reactive oxygen species; TNF-α: Tumor necrosis factor-α.
EPIDEMIOLOGY AND PREVALENCE
Alcohol misuse is a leading cause of liver disease worldwide. ALD is a global disease burden and results in approximately 3 million deaths per year[5]. In 2016, 5.3% of all deaths were caused by the harmful effects of alcohol worldwide[5]. Alcohol accounts for 5.1% of total disease burden worldwide, measured in disability-adjusted life years[5]. Total alcohol consumption per capita increased in 2005 from 5.5 L to 6.4 L in 2010 and was sustained at 6.4 L in 2016[5]. Europe has the highest per capita alcohol consumption and disability-adjusted life years[5], as well as binge drinking particularly, in France and England[6].
Although the overall prevalence of ALD has remained stable from 2001-2016 at between 8.1%-8.8%, the proportion of ALD with stage 3 fibrosis and above has increased from 2.2%-6.6%[7], such that in the United States chronic liver disease and cirrhosis is the 12th leading cause of death[7]. In the United States, the number of adults listed as waiting for liver transplants also increased by 63% from 2007 to 2017[7], where approximately one third of all liver transplants are due to alcohol-related disease[8]. Due to the high-risk drinking rates, the number of deaths due to alcohol-related liver disease in the United States is projected to increase by 84% from 2019-2040[9]. In the United Kingdom, the number of deaths from ALD was reported at 5964 deaths in 2020 compared to 4954 deaths in 2019, increasing by 20%[10]. From 2001 the number of deaths due to ALD has increased by 72% from 2001-2020[10].
There are several factors which effect the development of ALD. There are significant differences in the amounts of alcohol consumed between males and females. It is well documented that men consume more alcohol than women, leading to a higher prevalence of alcohol related liver disease[11]. It is estimated 237 million men and 46 million women have alcohol use disorders[5]. Although men often consume more alcohol, women are more susceptible to the toxic effects of alcohol and have a higher risk of advanced liver disease[11], due to higher blood alcohol concentrations despite equal dosing between genders[11]. Therefore, female gender is an important risk factor for progression of ALD. Genomic data has also discovered that a variation in the PNPLA3 gene is associated with increased hepatic fat content, increasing the risk of both ALD and non-alcoholic fatty liver disease, which has the highest frequency in Hispanics[12].
DISEASE SPECTRUM
Sustained excessive alcohol consumption produces a vast range of hepatic lesions. The first stage is liver steatosis or alcoholic fatty liver, which can occur in up to 90% of heavy drinkers, emerging as early as 3 to 7 d after initial excessive alcohol consumption[1]. Steatosis is often asymptomatic with normal or only slightly increased liver enzymes levels. The deposition of microscopic fat droplets occurs initially in the centrilobular zone then spreads towards the periportal region of hepatocytes[13]. Steatosis, although reversible and originally thought to be a benign state, is a now a priming phase for AH, which develops in 10% to 35% of heavy drinkers and is a more severe stage of ALD. AH is characterized by hepatocyte ballooning, the formation of Mallory-Denk bodies, infiltration of white blood cells, Kupffer cell (KC) activation, and collagen deposition via once dormant HSCs[14], the latter playing an important role in fibrosis leading to cirrhosis. At this stage hepatocyte inactivation, abnormal DNA repair, damage to mitochondrial structures, oxygen utilization disorders, and the accumulation of extracellular matrix proteins occurs[3]. Continuation of hepatic scarring and the spread of collagen (bridging fibrosis) throughout the liver can lead to cirrhosis and in some cases hepatocellular cancer.
Although the pathogenesis of ALD is yet to be fully understood, it is thought to include multiple interplaying factors and pathways including the production of toxic ethanol metabolites, oxidative stress, innate and adaptive immune activation, fibrogenesis and cell death. Upon activation of these pathways tissue damage can occur leading to the progression of the disease. This review will focus on mechanisms involved in inflammation that predominantly occur at the AH stage.
ALCOHOL METABOLISM
Within the liver, alcohol dehydrogenase and cytochrome p450 2E1 (CYP2E1) are the main oxidative pathways of alcohol metabolism. Another minor pathway of alcohol metabolism in the liver is via the peroxisomal enzyme catalase[15]. A small proportion of ethanol may also be metabolized by non-oxidative pathways such as by interaction with fatty acids, generating fatty acid ethyl esters[16]. Alcohol dehydrogenase oxidatively metabolizes alcohol to acetaldehyde, a highly reactive and toxic by product that contributes to tissue damage. This conversion reaction requires the cofactor nicotinamide adenine dinucleotide (NAD+), creating reduced NAD+ in the process. Due to the toxic nature of acetaldehyde, it is further oxidized to acetate, catalyzed by the enzyme mitochondrial aldehyde dehydrogenase-2[17]. Increased conversion of NAD+ results in leakage of electrons and reactive oxygen species (ROS) production. The toxic metabolites produced during alcohol metabolism as well as increased ROS can also trigger endoplasmic reticulum (ER) stress. The second major pathway is via the microsomal ethanol-oxidizing system and involves CYP2E1, which is involved in ethanol oxidation to acetaldehyde[17]. The activity of CYP2E1 is induced by alcohol, increasing its hepatocellular content causing accumulation of CYP2E1[18]. Electron leakage from the CYP2E1 pathway, leads to ROS generation, including hydroxyethyl, superoxide anion and hydroxyl radicals[19]. ROS can also form lipid peroxides and DNA adducts such as N2-ethyldeoxyguanosine, which has been detected both in the livers of alcohol-fed rats as well as leukocytes in ALD patients[20].
AUTOPHAGY, MITOPHAGY AND INFLAMMASOMES
Alcohol metabolism increases ROS production and ER stress leading to calcium depletion, glycosylation and lipid overloading, triggering the unfolded protein response (UPR)[21]. The UPR can restore ER homeostasis by attenuating translation of proteins, increasing folding capacity and degrading unfolded proteins. However, a prolonged UPR causes inflammation, fat accumulation, mitochondrial stress and apoptosis via direct activation apoptosis signal-regulating kinase 1[22], nuclear factor-κB (NF-κB), c-Jun N-terminal kinases and P38[23]. Alcohol induced ER stress involving an impaired UPR was first identified in a model of intragastric alcohol fed mice[24].
Alcohol can induce autophagy, a self-degradative process which occurs by the action of lysosomes and can be selective only for damaged mitochondria (mitophagy). Evidence suggests that autophagy in ALD can have inhibitory effects on inflammation and steatosis as well as the ability to remove lipid droplets, Mallory-Denk bodies and damaged mitochondria[25]. Whilst tumor necrosis factor (TNF)-α induces autophagy, generation of ROS can lead to inhibition of TNF-α induced autophagy through activation of NF-κB[26]. This suggests dysfunction of autophagy is associated with ALD pathogenesis[27,28]. Multiple animal models have observed autophagy as a protective response in ALD, as well as confirming ameliorative effects in ALD. Acute ethanol feeding (6 g/kg bodyweight) increased autophagy as measured by autophagosome numbers, however, chronic ethanol feeding (5.2% ethanol by volume) inhibited hepatic autophagy[29], suggesting that this protective mechanism is lost with longer term alcohol consumption.
Mitophagy can also be induced as a protective response to both acute and chronic alcohol consumption due to accumulation of ROS or loss of mitochondrial membrane potential via elimination of dysfunctional mitochondria. The process of mitophagy depends on induction of autophagy and priming of damaged mitochondria for recognition and is mediated by phosphatase and tensin homolog-induced putative kinase 1-Parkin signalling pathway or Nip3-like protein X[30]. The duration of alcohol exposure can affect the mitophagy process[30]. In rats, binge-models of alcohol consumption have been shown to increase mitophagy, decreasing alcohol-induced hepatoxicity[30,31]. Acute ethanol consumption also increases transcription factor EB, a master regulator of lysosomal biogenesis, however, chronic ethanol exposure decreased transcription factor EB[29]. Accumulation of damaged mitochondria occurs in chronic ethanol models which releases mitochondrial damage-associated molecular patterns (DAMPs), which in turn promote inflammation and fibrogenesis contributing to accelerated disease state[30]. Mitochondrial DNA (a mitochondrial DAMP), can bind to toll-like receptor (TLR)-9 activating HSCs and fibrogenesis[31]. Therefore, targeting mitophagy may be a potential therapeutic for ALD.
Inflammasomes
Oxidative stress in response to alcohol metabolism can damage hepatocytes, releasing endogenous DAMPs. Recognition of DAMPs can induce inflammation by release of proinflammatory cytokines, immune cell localization and stimulation of the inflammasome[32]. Inflammasomes are expressed in hepatic cells and are multi-protein complex’s containing a nucleotide-binding oligomerization domain-like receptor (NLR). Inflammasome activation is thought to be a two-step process. Inflammasome sensor molecules can trigger the assembly of inflammasomes, including NLR molecules, for example NOD-, LRR- and pyrin domain-containing 3 (NLRP3)[33]. Assembly is initiated by TLR and pathogen-associated molecular pattern (PAMP)/DAMP signaling which results in the NLR forming complexes with pro-caspase 1 with or without an adaptor molecule, apoptosis-associated speck like CARD-domain containing protein (ASC)[4,33]. Inflammasome assembly initiates cleavage of procaspase-1 to its active form caspase-1. Activated caspase-1 then promotes the secretion and activation of pro-inflammatory cytokines via cleavage of pro-interleukin (IL)-1β and pro-IL-18 into their active forms IL-1β and IL-18[33]. IL-1β plays an important role in the infiltration of immune cells and IL-18 is important for the production interferon-gamma (IFN-γ)[33]. Inflammasome activation also leads to a pathway of cell death called pyroptosis.
NLRP3 can be activated by a variety of stimuli including bacterial toxins, mitochondrial dysfunction and production of ROS. Interestingly, increased levels of IL-1β, ASC and NLRP3 have been documented in the livers of ethanol fed mice, whereas elevated mRNA expression of IL-1β, IL-18 and caspase-1 has been documented in the liver of ALD patients which correlated with liver lesions[34] and has also been associated with the development of liver fibrosis. Blocking IL-1β activity strongly decreases liver inflammation and damage[35]. Higher levels of serum IL-1 has also been documented in patients with AH in comparison to healthy controls[36]. Caspase-1 knockout mice have also been shown to be protected from fibrosis as well as treatment with IL-1 receptor antagonist has been shown to attenuate steatosis and liver injury when administered 2 wk post-ethanol feeding[36]. Decreased inflammation, steatosis and IL-1β expression has been associated in NLRP3 deficiency. Research has also shown mice deficient in NLRP3 have protection against ethanol-induced inflammation including attenuation of steatosis and liver injury[37]. Previous research has also shown inflammasome components such as NLRP3 and ASC are present in HSCs and are required for the development of liver fibrosis by inducing changes including upregulation of transforming growth factor-β and collagen[38]. Therefore, this demonstrates the importance of IL-1β signaling, inflammasome components and activation in ALD.
INNATE AND ADAPTIVE IMMUNITY
Gut permeability
Increases in gut permeability due to alcohol has been confirmed in both clinical and experimental studies[39,40]. Increased gut permeability enables the entrance of PAMPs, such as lipopolysaccharide (LPS) in the portal circulation[41]. LPS is one of the exogenous ligands for TLR4, a pattern recognition receptor found on KCs. LPS interaction with TLR4 initiates downstream signaling via TIR-domain-containing adapter-inducing IFN-β and IFN regulatory factor 3, leading to production of proinflammatory cytokines proinflammatory cytokines such as TNF-α and IL-1β (Figure 2), the former can activate the extrinsic pathway of apoptosis via the TNF receptor 1 and TNF receptor 2 signaling[42]. High levels of these death receptors, including Fas, are expressed in all liver cells, therefore, the extrinsic pathway is the main apoptotic pathway in hepatocytes, such that hepatocyte apoptosis has been correlated with severity of disease in AH[43]. Serum TNF-α and IL-6 are also increased after exposure to alcohol and LPS[44], with TNF-α correlating with liver injury and mortality. However, chronic ethanol exposure to TNF-α knockout mice does not cause alcohol-associated inflammation and liver injury[45], therefore, mechanisms to reduce TNF-α may be an important tool in preventing inflammation.
Figure 2.

The inflammatory response during alcoholic liver disease. Excessive consumption of alcohol causes lipopolysaccharide release from the gut activating toll-like receptor 4 on Kupffer cells (KCs). Pattern recognition receptors become activated by pathogen-associated molecular patterns/damage-associated molecular patterns which induces inflammation via release of proinflammatory cytokines and inflammasome activation. Interleukin (IL)-18 production from KCs causes activation of natural killer cells. Toll-like receptor stimulation in hepatic stellate cells results in the expression of IL-6, transforming growth factor-β1 and tumor necrosis factor-α. (Figure created with BioRender.com). DAMPs: Damage-associated molecular patterns; FFA: Free fatty acids; PAMPs: Pathogen-associated molecular patterns; IL: Interleukin; LPS: Lipopolysaccharide; NF-κB: Nuclear factor-κB; NK: Natural killer; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; PRR: Pattern recognition receptor; ROS: Reactive oxygen species; TGF-β: Transforming growth factor β; TLR4: Toll-like receptor 4; TNF-α: Tumor necrosis factor-α.
Natural killer cells
LPS and HSCs can directly interact with immune cells such as natural killer (NK) cells, natural killer T (NKT) cells and T cells leading to disease progression[46]. NK cells can kill activated HSCs via TNF-α related apoptosis, however, HSCs isolated from ethanol-fed mice showed reduced sensitivity to NK cell killing[46]. Alcohol consumption also enhances splenic NK cell apoptosis and blocks NK cell release from the bone marrow, as well as accelerating progression to fibrosis due to reduced NK cell activity[46]. Gut-derived bacteria such as LPS may influence the activation of hepatic NKT cells leading to induction of HSCs and apoptosis, further exacerbating liver injury. Patients with ALD have shown a decreased number of circulating NK cells along with reduced cytotoxic activity resulting in decreased anti-viral, anti-fibrotic, and anti-tumor effects which can contribute to accelerated progression of disease state[47]. However, NK/ NKT cells may inhibit fibrosis through deletion of activated HSCs and production of IFN-γ[48]. On the other hand, activation of NKT cells also promotes liver fibrosis via enhancing hepatocellular damage and promoting HSC activation[48]. Therefore, a balance is required between inhibitory and stimulatory effects for liver health.
Neutrophil activation
Neutrophils can be recruited in response to liver injury to form neutrophil extracellular traps (NETs), which generate ROS and undertake phagocytosis. Acute alcohol consumption leads to neutrophil imbalance in the liver releasing spontaneous NETs[49]. The scavenging ability of macrophages to eliminate NETs diminishes resulting in persistent inflammation via hepatocyte damage[49]. During AH, the infiltration of neutrophils is believed to occur via the activation of KCs, which recruit cytokines and chemokines including IL-8 and IL-17. In mouse models, blockade of inflammatory mediators such as IL-8 and IL-17, which are necessary for neutrophil infiltration, can ameliorate liver disease[50,51], which supports neutrophil dysfunction in disease progression. Patients with ALD have a decreased baseline function of neutrophils in the liver[14], which may provide an explanation for high rates of bacterial, fungal and viral infection as well as organ failure and mortality. Neutrophil dysfunction has been shown to be reversed in patients with AH following endotoxin removal[52]. In AH patients, extensive modification of albumin occurs, further activating neutrophil infiltration causing inflammation and oxidative stress[46,49]. Monocyte chemoattractant protein-1 also known as C–C motif chemokine ligand 2 is involved in proinflammatory cytokine activation and its levels have been found to be correlated with neutrophil infiltration and disease severity[46]. Therefore, neutrophils have been implicated in disease pathogenesis and a balance between anti-bacterial and anti-inflammatory functions is important for ALD patients.
Adaptive immunity
The adaptive immune response has also been implicated in pathogenesis of ALD (Figure 3). Early studies in both animals and humans have shown excessive alcohol consumption reduces peripheral T cell numbers, disrupts the balance between phenotypes, impairs function and promotes apoptosis[53]. Alcohol consumption can cause lymphopenia as well as disrupt the balance between T cell phenotypes, causing a shift from naïve populations to memory cells, experimentally and clinically[54-56]. Cytotoxic CD8+ T cells are greatly reduced, and this reduction was shown to correlate with stage of fibrosis and Child-Pugh (CTP) score, impairing cytotoxic functions leading to immune incompetence[56]. Decreased numbers of regulatory T cells are also associated with immune activation and increased inflammatory cytokines in AH[57].
Figure 3.

Innate and adaptive immune response to alcohol exposure. Kupffer cells, hepatic stellate cells and natural killer cells are components of the innate immune system which becomes activated following chronic alcohol consumption. This leads to the release of inflammatory cytokines, causing further recruitment of inflammatory cells. The adaptive immune system also becomes activated releasing inflammatory mediators as well as antibody generation to protein and Malondialdehyde-acetaldehyde adducts. Both immune response mechanisms eventually become dysregulated over time with alcohol consumption. (Figure created with BioRender.com). HSC: Hepatic stellate cell; IFN-γ: Interferon-γ; IL: Interleukin; KC: Kupffer cell; LPS: Lipopolysaccharide; NK: Natural killer; NKT: Natural killer T; ROS: Reactive oxygen species; TGF-β: Transforming growth factor β; TLR4: Toll-like receptor 4; TNF-α: Tumor necrosis factor-α.
More recently increasing phenotypes of T cells have been implicated in ALD. Chemokines such as CCL5, a chemoattractant for immune cells such as T lymphocytes, has been found to be upregulated in the liver[58]. Various proteins are expressed on the cell surface such as T-cell receptor, which recognize antigens and elicit a response. Infiltration and activation of both CD4+ and CD8+ T cells have been found to be increased in the livers of patients with ALD[58,59]. Until recently it had not been defined whether the increased activation of T cells in the livers of ALD patients were caused by bystander activation or due to an antigen-specific response[58,59]. Protein adducts derived from alcohol metabolism and lipid peroxidation have been identified in the liver of patients which act as neoantigens. These neoantigens are presented to CD4+ T cells by antigen presenting cells inducing proliferation[58,59]. As well as antigen-specific activation, bystander activation of T cells can occur induced in the absence of via cytokines, DAMPs and PAMPs[59]. This infiltration of T cells has been found to be correlated with inflammation and necrosis in ALD as well as regeneration. Therefore, both antigen-specific and bystander activation may contribute to the progression of ALD but also provide a beneficial role, however, this requires further research.
Alterations in regulatory cells may also provide an explanation for disease pathogenesis, as oxidative stress is known to lower regulatory T cell populations in the liver[60]. Th17 cells have been identified in the livers of ALD patients and are critical for defense of bacterial infections[46]. These cells produce IL-17 and promote neutrophil infiltration. Mucosa-associated invariant T cells (MAIT), an innate-like subset of T cells which inhibit bacterial infection, have been found to be reduced in ALD patients, consequentially increasing bacterial infection. Transcription factors (RORC/ RORγt, ZBTB16/PLZF and Eomes) which control the differentiation of MAIT cells were lower in AH patients compared to heathy controls[61]. Therefore, MAIT cell function may provide an important therapeutic approach for the treatment of ALD.
During ALD a loss of peripheral B cells also occurs, as well as increased amounts of circulating immunoglobulin[53]. B cell numbers are documented to be lower in heavy drinkers (90 to 249 drinks/mo) compared to moderate (30 to 89 drinks/mo) to light drinkers (< 9 drinks/mo) as well as a loss or circulating B cells in patients consuming 164.9 to 400 g of alcohol/day on average. The differentiation of progenitor B cells can be affected by ethanol exposure via down-regulation of transcription factors (early B cell factor and Pax5) and cytokine receptors (IL-7R α)[62] and thus, alcohol use can affect subpopulations of B cells (B-1a, B-1b, B2-B)[53]. Exposure to 100 mmol/L of alcohol in vitro blocks the expression of transcription factors which has been shown to impair B cell differentiation[62]. Alcoholics cannot respond adequately to antigens which is likely due to a reduction in high-affinity antibody-producing B-2B subset[53]. Further, this decrease in B-2B subsets is typically associated with a decrease in the number of B-1a cells as well as a relative increase in the percentage of B-1b cells, important for T cell independent responses[53]. Although B cells numbers appear to be reduced in alcoholics, during cirrhosis, circulating levels of immunoglobulins (IgA, IgG, and IgE) may be increased against liver antigens. It has also been reported that IgG antibodies against CYP2E1 have developed in both alcohol fed rats and patients with advanced ALD[63]. The role of B cells in ALD requires further research.
DIAGNOSIS
Diagnosis of ALD is challenging as many patients present as asymptomatic. An ALD diagnosis is commonly made on a combination of clinical laboratory abnormalities, imaging and a history of alcohol abuse. Laboratory blood tests are used to identify abnormal aspartate aminotransferase (AST), alanine aminotransferase (ALT) level, gamma-glutamyl transpeptidase (GGT), mean corpuscular volume (MCV), carbohydrate-deficient transferrin (CDT) levels, albumin, prothrombin time (PT) (international normalized ratio), bilirubin and platelet counts. These blood tests are useful to suggest alcohol misuse but are inadequate at predicting alcohol use on their own or the disease severity[64]. Historically GGT was used alone as a marker for ALD, although elevated GGT alone has low sensitivity and specificity for alcohol abuse and may be limited by a high rate for false positives[64]. An AST/ALT ration above 2 is regarded as an indicator of ALD, although it is used less frequently to predict chronic alcohol abuse due to low sensitivity[64]. CDT is used as a biomarker for chronic ethanol intake (> 60 g ethanol/d) and has a higher specificity (sensitivity 46%-73%, specificity 70%) than conventional markers such as GGT and MCV. However, no individual biomarker alone provides suitable sensitivity and specificity for ALD diagnosis, therefore, a combination of these biomarkers, imaging and in some cases a biopsy can provide an improved diagnosis.
CLINICAL STAGING OF DISEASE SEVERITY
There are various algorithms used to assess the severity of liver disease as well as predicating survival and treatment options (Table 1). The first models developed were the CTP score and the model for end-stage liver disease (MELD) score. The CTP score identifies patients as class A, B or C determined by serum levels of bilirubin and albumin, prothrombin time, ascites, and encephalopathy[65]. These measures are scored 1-3, with 3 being the most severe. A CTP defined as Class A (5-6 points) indicates a 100% 1-year survival and 85% 2-year survival. Class B (7-9 points) has an 80% 1-year survival and 60% 2-year survival. Class C (10-15 points) has a 45% 1-year survival and a 35% 2-year survival[66]. The MELD score is determined by total bilirubin, creatinine, and international normalized ratio (INR) levels, and is a widely useful tool for evaluation of liver transplantation in patients[67]. MELD is calculated by the formula 9.57 × loge (creatinine) + 3.78 × loge (total bilirubin) + 11.2 × loge (INR) + 6.43[66]. Although these scores are useful in predicting mortality, they are less useful in assessing prognosis and treatment options. Research by Sheth et al[68] has shown that 30-d mortality predictions from the MELD scoring were 86% sensitive and 82% specific for MELD scores greater than 11[68].
Table 1.
Models for clinical staging in alcoholic liver disease
| Model |
Stratification |
Bilirubin |
Albumin |
Prothrombin Time |
Ascites |
Encephalopathy |
INR |
Creatinine |
White blood cell count |
Serum urea |
Age |
| Child-Pugh[65,66] | Severe: ≥ 10 | + | + | + | + | + | |||||
| Model for End-Stage Liver disease[66] | Severe: ≥ 21 | + | + | + | |||||||
| Maddrey Discriminant Function score[66] | Severe: ≥ 32 | + | + | ||||||||
| The Glasgow Alcoholic Hepatitis Score[66] | Poor prognosis: ≥ 9 | + | + | + | + | + | |||||
| Lille Model[66,69] | ≥ 0.45: Nonresponse. < 0.45: Response | + | + | + | + | + |
INR: International normalized ratio.
The Maddrey Discriminant Function score (MDF) and The Glasgow AH Score (GAHS) were developed to determine disease severity and treatment options in AH patients. The MDF score assesses serum bilirubin and prothrombin time via the equation DF = {4.6 x [PT (sec) - control PT (sec)]} + (serum bilirubin) and classifies disease as either severe (MDF > 32) or non-severe (MDF < 32) and patients who fall in the severe category are most likely to benefit from steroid treatment[66]. The diagnostic sensitivity and specificity of the MDF scoring system was 86% and 48% when the DF was greater than 32[68]. The GAHS was developed to predict outcomes and to initiate therapy in AH patients. The GAHS includes age, white blood cell count, serum urea, bilirubin, and PT. Each variable is given a score and a final combined score between 5-12 is obtained. Patients with a GAHS above 9 have a poorer prognostic outcome. A study has shown patients with an MDF > 32 and a GAHS > 9 who were treated with steroids has a higher survival rate than those without treatment (59% compared to 38%)[67]. The Lille model is another prognostic model developed to identify response to corticosteroids in severe AH patients after 7 d of treatment[69]. Lille Model Score is defined as [exp(-R)]/[1 + exp(-R)]. Where r = 3.19 – 0.101*(age, years) + 0.147*(albumin day 0, g/L) + 0.0165* (evolution in bilirubin level, μmol/L) - 0.206*(renal insufficiency) - 0.0065*(bilirubin day 0, μmol/L) - 0.0096*(PT, seconds)[66]. It is useful for predicting short-term survival due to its high sensitivity and specificity and is able to identify patients at high risk of death at 6 mo[69].
THERAPEUTIC INTERVENTION
The consequences of excessive alcohol consumption causes significant morbidity and mortality with 704300 projected deaths due to alcohol-related liver disease in the United States between 2019-2040[9]. However, with a lack of new therapeutic options, abstinence is still regarded as the most important treatment, as well as treatments such as nutritional therapy, pharmacological therapy, combination therapy and transplantation (Table 2).
Table 2.
Current available treatments for alcoholic liver disease
| Intervention |
Objective |
Treatment method |
Treatment effects |
| Abstinence | Stop drinking | Abstinence combined with disulfiram, naltrexone or acamprosate | Improve overall survival at all stages[70]. Acamprosate has been shown to be effective in reducing withdrawal symptoms[77] |
| Nutritional Therapy | Replenish nutrition | 1.5 g protein and 35 to 49 kcal per kg of body weight. Supplementation with vitamins | Nutritional support showed improved hepatic encephalopathy and reduced infections in AH patients[80] |
| Corticosteroid | Anti-inflammatory | 40 mg daily for 28 d, then 20 mg daily for 7 d, and 10 mg daily for 7 d | Short-term histological improvement has been documented, however, no improvement in long term survival[83] |
| Pentoxifylline | Anti-cytokine | 400 mg orally three times a day for 4 wk | Reduction in the levels of cytokines and lower mortality rate[86] |
| Infliximab | Anti-cytokine | Not confirmed. 5 mg/kg studied | Further studies required. Treatment has shown to predispose patients to higher rate of infections as well as higher likelihood of mortality[93] |
| Liver transplantation | Surgery | Healthy ‘donor’ liver transplanted. 6 mo abstinence required | Transplantation has been shown to improve in quality of life[95,96] |
| N-acetylcysteine | Antioxidant | Not confirmed | In animal models NAC has been shown to prevent relapse and improve injury[111]. Further research is required |
| SAM | Antioxidant | Not confirmed | SAM therapy has improved survival and delayed the need for transplants however other studies have not found evidence to support or refute its use[102,103] |
| Silymarin | Herbal | Eurosil 85® 420 mg a day | Treatment with Silymarin reduced mortality and improved 4-yr overall survival in cirrhotic patients as well as improving liver function[119] |
| Betaine | Nutrient | Not confirmed | Animal models have shown betaine supplementation can attenuate alcoholic fatty liver[106-108,120,121] |
AH: Alcoholic hepatitis; NAC: N-acetylcysteine; SAM: S-adenosyl methionine.
Abstinence
Abstinence is the most important treatment for patients with ALD[70]. Abstinence from alcohol improves overall survival and prognosis as well as preventing further disease progression[71]. A reduction in portal pressure, decreased progression to cirrhosis and an improvement in survival has been shown after a period of abstinence[70,72]. However, relapse rates are as high as 67%-81% in alcoholics[73]. Several drugs such as disulfiram, naltrexone and acamprosate have been trialed to sustain abstinence and treat alcohol addiction. However, disulfiram has little evidence at improving abstinence[74]. Naltrexone, an opioid antagonist aimed to control alcohol cravings lowers the risk of relapse, although, it also has been shown to cause hepatocellular injury[71,75]. Acamprosate is used to minimize withdrawal symptoms when abstaining from alcohol[76]. It has been shown to reduce withdrawal in 15 controlled trials[77].
Nutritional therapy
Malnutrition is often correlated with disease severity in ALD patients[78]. Alcoholics suffer from deficiencies in several vitamins and minerals, including vitamin A, vitamin D, thiamine, folate, pyridoxine, and zinc[78]. Supplementation with zinc has shown to improve and prevent liver disease, as well as block mechanisms of liver injury including ‘leaky gut’, oxidative stress and apoptosis in animal models[79]. The recommended amount for ALD patients is 1.5 g of protein/kg body weight[2]. Supplementation with micronutrients may be necessary if an individual develops deficiencies. Nutritional support in AH has been reported to improve hepatic encephalopathy and reduce infections[80]. A reverse in both energy and protein deficits has been shown reduce morbidity and mortality in patients with acute AH and cirrhosis[81].
Steroid and anti-cytokine therapy
Steroids serve as the primary treatment for severe AH[82]. Treatment with glucocorticoids have decreased proinflammatory cytokines as well as inhibiting neutrophil activation[83]. Glucocorticoid therapy in AH patients showed short-term histological improvement and 28-d survival, however, long-term survival (beyond 1 year) was not improved[83].
Pentoxifylline, a phosphodiesterase inhibitor, is an anti-TNF-α agent. Pentoxifylline has been trialed in 101 patients with severe AH[84,85]. In-hospital mortality was 40% lower in those patients who were treated with pentoxifylline as well as reducing the likelihood of hepatorenal syndrome (HRS). 50% of deaths in the pentoxifylline treatment group were due to HRS, compared to 92% in the placebo group. Pentoxifylline also exhibited a higher 6-mo survival and a reduced incidence of HRS in patients with severe AH[86].
The Steroids or Pentoxifylline for AH trial (double-blind, randomized control trial) has evaluated the effects of both treatment with prednisolone and/or pentoxifylline[87-89]. 1103 patients underwent randomization with 1053 suitable for primary end point analysis. The primary endpoint was mortality at 28 d[89]. Results showed primary end point mortality at 28 d was 17% in the placebo–placebo group, 14% in the prednisolone–placebo group, 19% in the pentoxifylline–placebo group, and 13% in the prednisolone–pentoxifylline group, showing that pentoxifylline did not improve patients overall survival[89]. Although not significant, the steroid group showed a trend toward reduced 28-d mortality[89]. However, in those patients who received steroid treatment the rate of serious infection was nearly doubled[89,90].
Another anti-cytokine therapy used in the treatment of ALD is infliximab, a monoclonal chimeric anti-TNF antibody. In a primary randomized study using infliximab, 20 AH patients were given either 5 mg/kg of infliximab as well as 40 mg/d of prednisone or prednisone alone[91]. The results indicated there was no change in overall mortality, however, combination therapy decreased cytokine levels[84]. In France, a clinical trial studied prednisolone (40 mg/d for 4 wk) treatment compared to prednisolone with infliximab (10 mg/kg, at study entry, 2 wk and 4 wk after entry) in 36 patients[92]. Unfortunately the trial was stopped early due to mortality and infection[84], and therefore, this study has received criticism for the dose of infliximab in the trial as this predisposed patients to infections[93]. These trials suggest that anti-cytokine treatment in ALD is associated with an increase likelihood of severe infections and mortality. Canakinumab, a licensed monoclonal antibody inhibitor of IL-1 is currently being studied to treat ALD, as IL-1 has the ability to mediate disease progression in ALD[58].
Liver transplantation
Liver transplantation is a common treatment for end stage chronic liver disease; however, it remains controversial due to the increasing demand for donor organs as well as concerns of relapse from abstinence. Prior to surgery patients must abstain from alcohol for a fixed period of 6 mo[94]. Studies have shown patients who receive transplants have a better quality of life[95,96], however, less than 20% of patients whom have an end-stage liver disease receive surgery[97].
Antioxidants
Oxidative stress is a major contributor to the pathogenesis of chronic liver disease; therefore, antioxidant therapy has been considered to be beneficial in the treatment of ALD. Antioxidant agents able to mediate ROS include vitamins E and C, N-acetylcysteine (NAC) as well as S-adenosyl methionine (SAM), and betaine.
SAM operates to synthesize glutathione, the primary cellular antioxidant[98,99]. Patients with AH and cirrhosis have decreased hepatic SAM levels[100]. In animal models, SAM supplementation can reverse liver injury and mitochondrial damage caused by alcohol[101]. However, no significant difference between SAM supplementation and placebo groups has been reported[102,103]. Betaine, a nutrient involved in the formation of glutathione, is effective in protecting against damage from chronic alcohol consumption[104,105]. Supplementation with dietary betaine in animal studies has shown to ameliorate effects of oxidative stress[106-109]. In rats, NAC was able to reduce ethanol seeking behavior by 77%[110] as well as inhibiting ethanol intake by up to 70%[111]. In 174 patients with severe AH, combination therapy with NAC and prednisolone, compared to prednisolone only increased 1-mo survival in patients with AH, although 6-mo survival did not improve[112]. There are several trials underway investigating treatment options for ALD. A current clinical trial is assessing the effects of SAM and choline treatment for 24 wk against a placebo (trial number NCT 03938662). Choline can help the liver undergo glucose metabolism as well as repairing the cell membrane[113]. As damaged livers cannot produce SAM sufficiently, administration of choline and SAM may be a beneficial treatment in patients with ALD.
Fecal bacteria transplants
Chronic alcohol consumption leads to bacterial overgrowth promoting gut dysbiosis which correlates to disease severity. In cirrhosis patients Bacteroidetes and Firmicutes phyla were found to be decreased[114], however, Proteobacteria, Fusobacteria and Actinobacteria phyla were increased[114]. Cirrhosis is also characterized by reduced beneficial autochthonous bacteria such as Lachnospiraceae, Ruminococcaceae, and Clostridiales XIV as well increased pathogenic bacteria such as Enterococcaeae, Staphylococcaceae and Enterobacteriaceae[115]. The significance of bacteria in liver disease has been demonstrated when ethanol-fed, germ-free mice developed severe inflammation and necrosis when supplemented with gut microbiota from AH patients[116]. Furthermore, subsequent transfer of gut microbiota from patients without ALD, led to less inflammation and liver injury[117] implicating the importance of healthy bacteria. More recently administration of probiotics containing beneficial bacteria such as Lactobacillus and Bifidobacterium to patients with ALD has improved liver damage and function, including reduction of ALT, AST and bilirubin[118]. Modulation of the gut microbiota with Profermin®, a disease specific food for special medical purposes, has been hypothesized to reduce disease progression (trial number NCT03863730). These studies provide strong evidence that fecal bacteria transplants/probiotic administration may prove an effective mode for the treatment of ALD.
CONCLUSION
Previous research has uncovered many elements in the pathogenesis of ALD, however, the precise triggers and biochemical alterations are yet to be fully understood. Oxidative stress can impair proliferation and alter the immune system leading to bacterial overgrowth and an increased risk of infection. Poor treatments options are available for patients with ALD which have not transformed for several years. Treatment options rely on abstinence, steroids, nutritional therapy and lastly liver transplantation. Current new therapies are aimed at reducing pro-inflammatory signals as well as treating the gut-liver axis. This highlights a need for new therapeutic intervention and advancements in the understanding of the mechanisms involved in disease pathogenesis.
Footnotes
Conflict-of-interest statement: There is no conflict of interest.
Manuscript source: Invited manuscript
Peer-review started: April 30, 2021
First decision: July 8, 2021
Article in press: August 12, 2021
Specialty type: Biochemistry and molecular biology
Country/Territory of origin: United Kingdom
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Naserian S S-Editor: Fan JR L-Editor: A P-Editor: Yuan YY
Contributor Information
Lucy Petagine, Center for Nutraceuticals, School of Life Sciences, University of Westminster, London W1W 6UW, United Kingdom.
Mohammed Gulrez Zariwala, Center for Nutraceuticals, School of Life Sciences, University of Westminster, London W1W 6UW, United Kingdom.
Vinood B Patel, Center for Nutraceuticals, School of Life Sciences, University of Westminster, London W1W 6UW, United Kingdom. v.b.patel@westminster.ac.uk.
References
- 1.Fleming KA, McGee JO. Alcohol induced liver disease. J Clin Pathol. 1984;37:721–733. doi: 10.1136/jcp.37.7.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ. Treatment of alcoholic liver disease. Therap Adv Gastroenterol. 2011;4:63–81. doi: 10.1177/1756283X10378925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong LZ, Chandimali N, Han YH, Lee DH, Kim JS, Kim SU, Kim TD, Jeong DK, Sun HN, Lee DS, Kwon T. Pathogenesis, Early Diagnosis, and Therapeutic Management of Alcoholic Liver Disease. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20112712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagy LE. The Role of Innate Immunity in Alcoholic Liver Disease. Alcohol Res. 2015;37:237–250. doi: 10.35946/arcr.v37.2.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO Global status report on alcohol and health 2018. [cited 10 March 2021]. Available from: https://apps.who.int/iris/bitstream/handle/10665/274603/9789241565639-eng.pdf?ua=1 .
- 6.Mellinger JL. Epidemiology of Alcohol Use and Alcoholic Liver Disease. Clin Liver Dis (Hoboken) 2019;13:136–139. doi: 10.1002/cld.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang K, Hirode G, Singal AK, Sundaram V, Wong RJ. Alcoholic Liver Disease Epidemiology in the United States: A Retrospective Analysis of 3 US Databases. Am J Gastroenterol. 2020;115:96–104. doi: 10.14309/ajg.0000000000000380. [DOI] [PubMed] [Google Scholar]
- 8.Parker R, Holt A. Transplanting Patients with Alcohol-related Liver Disease in the National Health System: New Rules and Decisions. Alcohol Alcohol. 2018;53:145–150. doi: 10.1093/alcalc/agx103. [DOI] [PubMed] [Google Scholar]
- 9.Julien J, Ayer T, Bethea ED, Tapper EB, Chhatwal J. Projected prevalence and mortality associated with alcohol-related liver disease in the USA, 2019-40: a modelling study. Lancet Public Health. 2020;5:e316–e323. doi: 10.1016/S2468-2667(20)30062-1. [DOI] [PubMed] [Google Scholar]
- 10.ONS Quarterly alcohol-specific deaths in England and Wales - Office for National Statistics. 2021. [cited 10 March 2021]. Available from: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/datasets/quarterlyalcoholspecificdeathsinenglandandwales .
- 11.Durazzo M, Belci P, Collo A, Prandi V, Pistone E, Martorana M, Gambino R, Bo S. Gender specific medicine in liver diseases: a point of view. World J Gastroenterol. 2014;20:2127–2135. doi: 10.3748/wjg.v20.i9.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mavrelis PG, Ammon HV, Gleysteen JJ, Komorowski RA, Charaf UK. Hepatic free fatty acids in alcoholic liver disease and morbid obesity. Hepatology. 1983;3:226–231. doi: 10.1002/hep.1840030215. [DOI] [PubMed] [Google Scholar]
- 14.Celli R, Zhang X. Pathology of Alcoholic Liver Disease. J Clin Transl Hepatol. 2014;2:103–109. doi: 10.14218/JCTH.2014.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teschke R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines. 2018;6 doi: 10.3390/biomedicines6040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zelner I, Matlow JN, Natekar A, Koren G. Synthesis of fatty acid ethyl esters in mammalian tissues after ethanol exposure: a systematic review of the literature. Drug Metab Rev. 2013;45:277–299. doi: 10.3109/03602532.2013.795584. [DOI] [PubMed] [Google Scholar]
- 17.Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–685. doi: 10.1016/j.cld.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dilger K, Metzler J, Bode JC, Klotz U. CYP2E1 activity in patients with alcoholic liver disease. J Hepatol. 1997;27:1009–1014. doi: 10.1016/s0168-8278(97)80144-4. [DOI] [PubMed] [Google Scholar]
- 19.Osna NA, Donohue TM Jr, Kharbanda KK. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017;38:147–161. doi: 10.35946/arcr.v38.2.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev. 2010;3:178–185. doi: 10.4161/oxim.3.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int. 2012;2012:216450. doi: 10.1155/2012/216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibusuki R, Uto H, Oda K, Ohshige A, Tabu K, Mawatari S, Kumagai K, Kanmura S, Tamai T, Moriuchi A, Tsubouchi H, Ido A. Human neutrophil peptide-1 promotes alcohol-induced hepatic fibrosis and hepatocyte apoptosis. PLoS One. 2017;12:e0174913. doi: 10.1371/journal.pone.0174913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee YJ, Shukla SD. Histone H3 phosphorylation at serine 10 and serine 28 is mediated by p38 MAPK in rat hepatocytes exposed to ethanol and acetaldehyde. Eur J Pharmacol. 2007;573:29–38. doi: 10.1016/j.ejphar.2007.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 25.Czaja MJ, Ding WX, Donohue TM Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6:24. doi: 10.1186/s40169-017-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song X, Yin S, Huo Y, Liang M, Fan L, Ye M, Hu H. Glycycoumarin ameliorates alcohol-induced hepatotoxicity via activation of Nrf2 and autophagy. Free Radic Biol Med. 2015;89:135–146. doi: 10.1016/j.freeradbiomed.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Cederbaum AI. Autophagy Protects against CYP2E1/Chronic Ethanol-Induced Hepatotoxicity. Biomolecules. 2015;5:2659–2674. doi: 10.3390/biom5042659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr. Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcohol Clin Exp Res. 2015;39:2354–2363. doi: 10.1111/acer.12904. [DOI] [PubMed] [Google Scholar]
- 30.Ma X, McKeen T, Zhang J, Ding WX. Role and Mechanisms of Mitophagy in Liver Diseases. Cells. 2020;9 doi: 10.3390/cells9040837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemasters JJ, Zhong Z. Mitophagy in hepatocytes: Types, initiators and role in adaptive ethanol metabolism☆. Liver Res. 2018;2:125–132. doi: 10.1016/j.livres.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rotimi VO, Akindutire D. Clostridium difficile in the normal adult faecal flora. Afr J Med Med Sci. 1986;15:73–77. [PubMed] [Google Scholar]
- 33.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voican CS, Njiké-Nakseu M, Boujedidi H, Barri-Ova N, Bouchet-Delbos L, Agostini H, Maitre S, Prévot S, Cassard-Doulcier AM, Naveau S, Perlemuter G. Alcohol withdrawal alleviates adipose tissue inflammation in patients with alcoholic liver disease. Liver Int. 2015;35:967–978. doi: 10.1111/liv.12575. [DOI] [PubMed] [Google Scholar]
- 35.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2016;64:955–965. doi: 10.1002/hep.28456. [DOI] [PubMed] [Google Scholar]
- 37.Petrasek J, Iracheta-Vellve A, Saha B, Satishchandran A, Kodys K, Fitzgerald KA, Kurt-Jones EA, Szabo G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J Leukoc Biol. 2015;98:249–256. doi: 10.1189/jlb.3AB1214-590R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA, Mehal WZ. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1248–G1257. doi: 10.1152/ajpgi.90223.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, Banan A, Fields JZ. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riva A, Patel V, Kurioka A, Jeffery HC, Wright G, Tarff S, Shawcross D, Ryan JM, Evans A, Azarian S, Bajaj JS, Fagan A, Mehta K, Lopez C, Simonova M, Katzarov K, Hadzhiolova T, Pavlova S, Wendon JA, Oo YH, Klenerman P, Williams R, Chokshi S. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut. 2018;67:918–930. doi: 10.1136/gutjnl-2017-314458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neuman MG, Maor Y, Nanau RM, Melzer E, Mell H, Opris M, Cohen L, Malnick S. Alcoholic Liver Disease: Role of Cytokines. Biomolecules. 2015;5:2023–2034. doi: 10.3390/biom5032023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michelena J, Altamirano J, Abraldes JG, Affò S, Morales-Ibanez O, Sancho-Bru P, Dominguez M, García-Pagán JC, Fernández J, Arroyo V, Ginès P, Louvet A, Mathurin P, Mehal WZ, Caballería J, Bataller R. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology. 2015;62:762–772. doi: 10.1002/hep.27779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawaratani H, Tsujimoto T, Douhara A, Takaya H, Moriya K, Namisaki T, Noguchi R, Yoshiji H, Fujimoto M, Fukui H. The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm. 2013;2013:495156. doi: 10.1155/2013/495156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, Tan HY, Wang N, Feng Y, Wang X. Recent Insights Into the Role of Immune Cells in Alcoholic Liver Disease. Front Immunol. 2019;10:1328. doi: 10.3389/fimmu.2019.01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laso FJ, Almeida J, Torres E, Vaquero JM, Marcos M, Orfao A. Chronic alcohol consumption is associated with an increased cytotoxic profile of circulating lymphocytes that may be related with the development of liver injury. Alcohol Clin Exp Res. 2010;34:876–885. doi: 10.1111/j.1530-0277.2010.01160.x. [DOI] [PubMed] [Google Scholar]
- 48.Tian Z, Chen Y, Gao B. Natural killer cells in liver disease. Hepatology. 2013;57:1654–1662. doi: 10.1002/hep.26115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu K, Wang FS, Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol Immunol. 2021;18:38–44. doi: 10.1038/s41423-020-00560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology. 2016;150:1704–1709. doi: 10.1053/j.gastro.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, Feng D, Bertola A, Wang H, Kunos G, Gao B. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology. 2015;62:1070–1085. doi: 10.1002/hep.27921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mookerjee RP, Stadlbauer V, Lidder S, Wright GA, Hodges SJ, Davies NA, Jalan R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology. 2007;46:831–840. doi: 10.1002/hep.21737. [DOI] [PubMed] [Google Scholar]
- 53.Pasala S, Barr T, Messaoudi I. Impact of Alcohol Abuse on the Adaptive Immune System. Alcohol Res. 2015;37:185–197. doi: 10.35946/arcr.v37.2.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song K, Coleman RA, Zhu X, Alber C, Ballas ZK, Waldschmidt TJ, Cook RT. Chronic ethanol consumption by mice results in activated splenic T cells. J Leukoc Biol. 2002;72:1109–1116. [PubMed] [Google Scholar]
- 55.Zhang H, Meadows GG. Chronic alcohol consumption in mice increases the proportion of peripheral memory T cells by homeostatic proliferation. J Leukoc Biol. 2005;78:1070–1080. doi: 10.1189/jlb.0605317. [DOI] [PubMed] [Google Scholar]
- 56.Matos LC, Batista P, Monteiro N, Ribeiro J, Cipriano MA, Henriques P, Girão F, Carvalho A. Lymphocyte subsets in alcoholic liver disease. World J Hepatol. 2013;5:46–55. doi: 10.4254/wjh.v5.i2.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Almeida J, Polvorosa MA, Gonzalez-Quintela A, Marcos M, Pastor I, Hernandez Cerceño ML, Orfao A, Laso FJ. Decreased peripheral blood CD4+/CD25+ regulatory T cells in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2013;37:1361–1369. doi: 10.1111/acer.12095. [DOI] [PubMed] [Google Scholar]
- 58.Wang H, Mehal W, Nagy LE, Rotman Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell Mol Immunol. 2021;18:73–91. doi: 10.1038/s41423-020-00579-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70:249–259. doi: 10.1016/j.jhep.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. 2007;46:1519–1529. doi: 10.1002/hep.21823. [DOI] [PubMed] [Google Scholar]
- 61.Gao B, Ma J, Xiang X. MAIT cells: a novel therapeutic target for alcoholic liver disease? Gut. 2018;67:784–786. doi: 10.1136/gutjnl-2017-315284. [DOI] [PubMed] [Google Scholar]
- 62.Wang H, Zhou H, Mahler S, Chervenak R, Wolcott M. Alcohol affects the late differentiation of progenitor B cells. Alcohol Alcohol. 2011;46:26–32. doi: 10.1093/alcalc/agq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vidali M, Stewart SF, Rolla R, Daly AK, Chen Y, Mottaran E, Jones DE, Leathart JB, Day CP, Albano E. Genetic and epigenetic factors in autoimmune reactions toward cytochrome P4502E1 in alcoholic liver disease. Hepatology. 2003;37:410–419. doi: 10.1053/jhep.2003.50049. [DOI] [PubMed] [Google Scholar]
- 64.Litten RZ, Bradley AM, Moss HB. Alcohol biomarkers in applied settings: recent advances and future research opportunities. Alcohol Clin Exp Res. 2010;34:955–967. doi: 10.1111/j.1530-0277.2010.01170.x. [DOI] [PubMed] [Google Scholar]
- 65.Angermayr B, Cejna M, Karnel F, Gschwantler M, Koenig F, Pidlich J, Mendel H, Pichler L, Wichlas M, Kreil A, Schmid M, Ferlitsch A, Lipinski E, Brunner H, Lammer J, Ferenci P, Gangl A, Peck-Radosavljevic M. Child-Pugh versus MELD score in predicting survival in patients undergoing transjugular intrahepatic portosystemic shunt. Gut. 2003;52:879–885. doi: 10.1136/gut.52.6.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rahimi E, Pan JJ. Prognostic models for alcoholic hepatitis. Biomark Res. 2015;3:20. doi: 10.1186/s40364-015-0046-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Forrest EH, Morris AJ, Stewart S, Phillips M, Oo YH, Fisher NC, Haydon G, O'Grady J, Day CP. The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids. Gut. 2007;56:1743–1746. doi: 10.1136/gut.2006.099226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheth M, Riggs M, Patel T. Utility of the Mayo End-Stage Liver Disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol. 2002;2:2. doi: 10.1186/1471-230X-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Louvet A, Naveau S, Abdelnour M, Ramond MJ, Diaz E, Fartoux L, Dharancy S, Texier F, Hollebecque A, Serfaty L, Boleslawski E, Deltenre P, Canva V, Pruvot FR, Mathurin P. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology. 2007;45:1348–1354. doi: 10.1002/hep.21607. [DOI] [PubMed] [Google Scholar]
- 70.Pessione F, Ramond MJ, Peters L, Pham BN, Batel P, Rueff B, Valla DC. Five-year survival predictive factors in patients with excessive alcohol intake and cirrhosis. Effect of alcoholic hepatitis, smoking and abstinence. Liver Int. 2003;23:45–53. doi: 10.1034/j.1600-0676.2003.01804.x. [DOI] [PubMed] [Google Scholar]
- 71.European Association for the Study of Liver. EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol. 2012;57:399–420. doi: 10.1016/j.jhep.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 72.Brunt PW, Kew MC, Scheuer PJ, Sherlock S. Studies in alcoholic liver disease in Britain. I. Clinical and pathological patterns related to natural history. Gut. 1974;15:52–58. doi: 10.1136/gut.15.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller WR, Walters ST, Bennett ME. How effective is alcoholism treatment in the United States? J Stud Alcohol. 2001;62:211–220. doi: 10.15288/jsa.2001.62.211. [DOI] [PubMed] [Google Scholar]
- 74.Cheng HY, McGuinness LA, Elbers RG, MacArthur GJ, Taylor A, McAleenan A, Dawson S, López-López JA, Higgins JPT, Cowlishaw S, Lingford-Hughes A, Hickman M, Kessler D. Treatment interventions to maintain abstinence from alcohol in primary care: systematic review and network meta-analysis. BMJ. 2020;371:m3934. doi: 10.1136/bmj.m3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Srisurapanont M, Jarusuraisin N. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev. 2000:CD001867. doi: 10.1002/14651858.CD001867. [DOI] [PubMed] [Google Scholar]
- 76.Palmer AJ, Neeser K, Weiss C, Brandt A, Comte S, Fox M. The long-term cost-effectiveness of improving alcohol abstinence with adjuvant acamprosate. Alcohol Alcohol. 2000;35:478–492. doi: 10.1093/alcalc/35.5.478. [DOI] [PubMed] [Google Scholar]
- 77.Boothby LA, Doering PL. Acamprosate for the treatment of alcohol dependence. Clin Ther. 2005;27:695–714. doi: 10.1016/j.clinthera.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 78.Halsted CH. Nutrition and alcoholic liver disease. Semin Liver Dis. 2004;24:289–304. doi: 10.1055/s-2004-832941. [DOI] [PubMed] [Google Scholar]
- 79.Mohammad MK, Zhou Z, Cave M, Barve A, McClain CJ. Zinc and liver disease. Nutr Clin Pract. 2012;27:8–20. doi: 10.1177/0884533611433534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crabb DW, Im GY, Szabo G, Mellinger JL, Lucey MR. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology. 2020;71:306–333. doi: 10.1002/hep.30866. [DOI] [PubMed] [Google Scholar]
- 81.Kamran U, Towey J, Khanna A, Chauhan A, Rajoriya N, Holt A. Nutrition in alcohol-related liver disease: Physiopathology and management. World J Gastroenterol. 2020;26:2916–2930. doi: 10.3748/wjg.v26.i22.2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mathurin P, O'Grady J, Carithers RL, Phillips M, Louvet A, Mendenhall CL, Ramond MJ, Naveau S, Maddrey WC, Morgan TR. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: meta-analysis of individual patient data. Gut. 2011;60:255–260. doi: 10.1136/gut.2010.224097. [DOI] [PubMed] [Google Scholar]
- 83.Menachery J, Duseja A. Treatment of decompensated alcoholic liver disease. Int J Hepatol. 2011;2011:219238. doi: 10.4061/2011/219238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105:14–32; quiz 33. doi: 10.1038/ajg.2009.593. [DOI] [PubMed] [Google Scholar]
- 85.McCullough AJ, O'Shea RS, Dasarathy S. Diagnosis and management of alcoholic liver disease. J Dig Dis. 2011;12:257–262. doi: 10.1111/j.1751-2980.2010.00470.x. [DOI] [PubMed] [Google Scholar]
- 86.Lenz K, Buder R, Kapun L, Voglmayr M. Treatment and management of ascites and hepatorenal syndrome: an update. Therap Adv Gastroenterol. 2015;8:83–100. doi: 10.1177/1756283X14564673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Forrest E, Mellor J, Stanton L, Bowers M, Ryder P, Austin A, Day C, Gleeson D, O'Grady J, Masson S, McCune A, Patch D, Richardson P, Roderick P, Ryder S, Wright M, Thursz M. Steroids or pentoxifylline for alcoholic hepatitis (STOPAH): study protocol for a randomised controlled trial. Trials. 2013;14:262. doi: 10.1186/1745-6215-14-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Forrest EH, Atkinson SR, Richardson P, Masson S, Ryder S, Thursz MR, Allison M STOPAH trial Management Group. Application of prognostic scores in the STOPAH trial: Discriminant function is no longer the optimal scoring system in alcoholic hepatitis. J Hepatol. 2018;68:511–518. doi: 10.1016/j.jhep.2017.11.017. [DOI] [PubMed] [Google Scholar]
- 89.Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, Downs N, Gleeson D, MacGilchrist A, Grant A, Hood S, Masson S, McCune A, Mellor J, O'Grady J, Patch D, Ratcliffe I, Roderick P, Stanton L, Vergis N, Wright M, Ryder S, Forrest EH STOPAH Trial. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 2015;372:1619–1628. doi: 10.1056/NEJMoa1412278. [DOI] [PubMed] [Google Scholar]
- 90.Dao A, Rangnekar AS. Steroids for Severe Alcoholic Hepatitis: More Risk Than Reward? Clin Liver Dis (Hoboken) 2018;12:151–153. doi: 10.1002/cld.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spahr L, Rubbia-Brandt L, Frossard JL, Giostra E, Rougemont AL, Pugin J, Fischer M, Egger H, Hadengue A. Combination of steroids with infliximab or placebo in severe alcoholic hepatitis: a randomized controlled pilot study. J Hepatol. 2002;37:448–455. doi: 10.1016/s0168-8278(02)00230-1. [DOI] [PubMed] [Google Scholar]
- 92.Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, Davion T, Oberti F, Broët P, Emilie D Foie-Alcool group of the Association Française pour l'Etude du Foie. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39:1390–1397. doi: 10.1002/hep.20206. [DOI] [PubMed] [Google Scholar]
- 93.Mookerjee RP, Tilg H, Williams R, Jalan R. Infliximab and alcoholic hepatitis. Hepatology. 2004;40:499–500; author reply 500. doi: 10.1002/hep.20344. [DOI] [PubMed] [Google Scholar]
- 94.Lucey MR, Brown KA, Everson GT, Fung JJ, Gish R, Keeffe EB, Kneteman NM, Lake JR, Martin P, McDiarmid SV, Rakela J, Shiffman ML, So SK, Wiesner RH. Minimal criteria for placement of adults on the liver transplant waiting list: a report of a national conference organized by the American Society of Transplant Physicians and the American Association for the Study of Liver Diseases. Liver Transpl Surg. 1997;3:628–637. doi: 10.1002/lt.500030613. [DOI] [PubMed] [Google Scholar]
- 95.Singal AK, Bashar H, Anand BS, Jampana SC, Singal V, Kuo YF. Outcomes after liver transplantation for alcoholic hepatitis are similar to alcoholic cirrhosis: exploratory analysis from the UNOS database. Hepatology. 2012;55:1398–1405. doi: 10.1002/hep.25544. [DOI] [PubMed] [Google Scholar]
- 96.Singal AK, Chaha KS, Rasheed K, Anand BS. Liver transplantation in alcoholic liver disease current status and controversies. World J Gastroenterol. 2013;19:5953–5963. doi: 10.3748/wjg.v19.i36.5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lucey MR. Liver transplantation for alcoholic liver disease. Nat Rev Gastroenterol Hepatol. 2014;11:300–307. doi: 10.1038/nrgastro.2013.247. [DOI] [PubMed] [Google Scholar]
- 98.Cederbaum AI. Hepatoprotective effects of S-adenosyl-L-methionine against alcohol- and cytochrome P450 2E1-induced liver injury. World J Gastroenterol. 2010;16:1366–1376. doi: 10.3748/wjg.v16.i11.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–1542. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, Lu SC. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2004;28:173–181. doi: 10.1097/01.ALC.0000108654.77178.03. [DOI] [PubMed] [Google Scholar]
- 101.Lieber CS. S-Adenosyl-L-methionine and alcoholic liver disease in animal models: implications for early intervention in human beings. Alcohol. 2002;27:173–177. doi: 10.1016/s0741-8329(02)00230-6. [DOI] [PubMed] [Google Scholar]
- 102.Medici V, Virata MC, Peerson JM, Stabler SP, French SW, Gregory JF 3rd, Albanese A, Bowlus CL, Devaraj S, Panacek EA, Richards JR, Halsted CH. S-adenosyl-L-methionine treatment for alcoholic liver disease: a double-blinded, randomized, placebo-controlled trial. Alcohol Clin Exp Res. 2011;35:1960–1965. doi: 10.1111/j.1530-0277.2011.01547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Le MD, Enbom E, Traum PK, Medici V, Halsted CH, French SW. Alcoholic liver disease patients treated with S-adenosyl-L-methionine: an in-depth look at liver morphologic data comparing pre and post treatment liver biopsies. Exp Mol Pathol. 2013;95:187–191. doi: 10.1016/j.yexmp.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kharbanda KK, Todero SL, King AL, Osna NA, McVicker BL, Tuma DJ, Wisecarver JL, Bailey SM. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int J Hepatol. 2012;2012:962183. doi: 10.1155/2012/962183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kharbanda KK, Mailliard ME, Baldwin CR, Beckenhauer HC, Sorrell MF, Tuma DJ. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J Hepatol. 2007;46:314–321. doi: 10.1016/j.jhep.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 106.Kharbanda KK, Rogers DD 2nd, Mailliard ME, Siford GL, Barak AJ, Beckenhauer HC, Sorrell MF, Tuma DJ. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J Nutr. 2005;135:519–524. doi: 10.1093/jn/135.3.519. [DOI] [PubMed] [Google Scholar]
- 107.Alirezaei M, Jelodar G, Niknam P, Ghayemi Z, Nazifi S. Betaine prevents ethanol-induced oxidative stress and reduces total homocysteine in the rat cerebellum. J Physiol Biochem. 2011;67:605–612. doi: 10.1007/s13105-011-0107-1. [DOI] [PubMed] [Google Scholar]
- 108.Kim SJ, Jung YS, Kwon DY, Kim YC. Alleviation of acute ethanol-induced liver injury and impaired metabolomics of S-containing substances by betaine supplementation. Biochem Biophys Res Commun. 2008;368:893–898. doi: 10.1016/j.bbrc.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 109.Petagine L, Everitt H, Preedy V, Sherwood R, Patel V. Acute Alcohol Tissue Damage: Protective Properties of Betaine. J Ren Hepatic Disord. 2021;5:19–29. [Google Scholar]
- 110.Lebourgeois S, González-Marín MC, Jeanblanc J, Naassila M, Vilpoux C. Effect of N-acetylcysteine on motivation, seeking and relapse to ethanol self-administration. Addict Biol. 2018;23:643–652. doi: 10.1111/adb.12521. [DOI] [PubMed] [Google Scholar]
- 111.Quintanilla ME, Rivera-Meza M, Berríos-Cárcamo P, Salinas-Luypaert C, Herrera-Marschitz M, Israel Y. Beyond the "First Hit": Marked Inhibition by N-Acetyl Cysteine of Chronic Ethanol Intake But Not of Early Ethanol Intake. Parallel Effects on Ethanol-Induced Saccharin Motivation. Alcohol Clin Exp Res. 2016;40:1044–1051. doi: 10.1111/acer.13031. [DOI] [PubMed] [Google Scholar]
- 112.Nguyen-Khac E, Thevenot T, Piquet MA, Benferhat S, Goria O, Chatelain D, Tramier B, Dewaele F, Ghrib S, Rudler M, Carbonell N, Tossou H, Bental A, Bernard-Chabert B, Dupas JL AAH-NAC Study Group. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med. 2011;365:1781–1789. doi: 10.1056/NEJMoa1101214. [DOI] [PubMed] [Google Scholar]
- 113.Sanders LM, Zeisel SH. Choline: Dietary Requirements and Role in Brain Development. Nutr Today. 2007;42:181–186. doi: 10.1097/01.NT.0000286155.55343.fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwenger KJ, Clermont-Dejean N, Allard JP. The role of the gut microbiome in chronic liver disease: the clinical evidence revised. JHEP Rep. 2019;1:214–226. doi: 10.1016/j.jhepr.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, Noble NA, Unser AB, Daita K, Fisher AR, Sikaroodi M, Gillevet PM. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60:940–947. doi: 10.1016/j.jhep.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, Puchois V, Martin JC, Lepage P, Le Roy T, Lefèvre L, Langelier B, Cailleux F, González-Castro AM, Rabot S, Gaudin F, Agostini H, Prévot S, Berrebi D, Ciocan D, Jousse C, Naveau S, Gérard P, Perlemuter G. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65:830–839. doi: 10.1136/gutjnl-2015-310585. [DOI] [PubMed] [Google Scholar]
- 117.Sarin SK, Pande A, Schnabl B. Microbiome as a therapeutic target in alcohol-related liver disease. J Hepatol. 2019;70:260–272. doi: 10.1016/j.jhep.2018.10.019. [DOI] [PubMed] [Google Scholar]
- 118.Li F, Duan K, Wang C, McClain C, Feng W. Probiotics and Alcoholic Liver Disease: Treatment and Potential Mechanisms. Gastroenterol Res Pract. 2016;2016:5491465. doi: 10.1155/2016/5491465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gillessen A, Schmidt HH. Silymarin as Supportive Treatment in Liver Diseases: A Narrative Review. Adv Ther. 2020;37:1279–1301. doi: 10.1007/s12325-020-01251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Balkan J, Oztezcan S, Küçük M, Cevikbaş U, Koçak-Toker N, Uysal M. The effect of betaine treatment on triglyceride levels and oxidative stress in the liver of ethanol-treated guinea pigs. Exp Toxicol Pathol. 2004;55:505–509. doi: 10.1078/0940-2993-00347. [DOI] [PubMed] [Google Scholar]
- 121.Kharbanda KK, Todero SL, Ward BW, Cannella JJ 3rd, Tuma DJ. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol Cell Biochem. 2009;327:75–78. doi: 10.1007/s11010-009-0044-2. [DOI] [PubMed] [Google Scholar]