

Abstract
Although the classic activities of p53 including induction of cell-cycle arrest, senescence, and apoptosis are well accepted as critical barriers to cancer development, accumulating evidence suggests that loss of these classic activities is not sufficient to abrogate the tumor suppression activity of p53. Numerous studies suggest that metabolic regulation contributes to tumor suppression, but the mechanisms by which it does so are not completely understood. Cancer cells rewire cellular metabolism to meet the energetic and substrate demands of tumor development. It is well established that p53 suppresses glycolysis and promotes mitochondrial oxidative phosphorylation through a number of downstream targets against the Warburg effect. The role of p53-mediated metabolic regulation in tumor suppression is complexed by its function to promote both cell survival and cell death under different physiological settings. Indeed, p53 can regulate both pro-oxidant and antioxidant target genes for complete opposite effects. In this review, we will summarize the roles of p53 in the regulation of glucose, lipid, amino acid, nucleotide, iron metabolism, and ROS production. We will highlight the mechanisms underlying p53-mediated ferroptosis, AKT/mTOR signaling as well as autophagy and discuss the complexity of p53-metabolic regulation in tumor development.
Keywords: p53, metabolism, transcriptional activation, tumor suppression, ferroptosis
Introduction
First described in 1979, tumor protein p53 (TP53, or p53) has been under intense scrutiny for more than 40 years. A search of the PubMed database using the keyword “p53” currently generates a list of more than 100,000 entries. Most of these studies focus on the role of p53 in cancer. However, p53 has other, less well-characterized biological functions, including critical contributions to development, stem cell biology, and non-neoplastic disorders [1–3]. p53 can be induced by extra- or intracellular stress (e.g., DNA damage, oncogene activation, ribosomal or telomere-associated stresses, and nutrient deprivation) to orchestrate the responses of numerous downstream signaling pathways. While p53 primarily functions as a transcription factor (TF), recent work has revealed several diverse roles, including those within the cell cytoplasm that are unrelated to gene transcription [4]. p53 can achieve multiple cellular effects, including cell cycle arrest, DNA repair, senescence, apoptosis, and ferroptosis [5, 6]. From an overall perspective, induction of p53 has been linked to improved fitness of host cells and the host organism as a whole.
Metabolism, including both anabolism and catabolism, are critical processes found in all living systems. For mammals, balanced systemic and cellular metabolism provides indispensable support for physiological homeostasis and health. By contrast, dysregulated metabolism can result in diverse diseases, including neoplasia [7–9]. In 2005, several groups provided the first evidence documenting the role of p53 in regulating metabolism [10–12]. After that, burgeoning researches referring to the roles of p53 in metabolic regulation have been following up. To date, all known functions of p53 have been linked to its ability to regulate one or more critical metabolic pathways. For its mostly noted relationship with cancer, there are more and more papers pointing out that p53 positively or negatively influences cancer initiation and development by reprogramming cancer cell metabolism. In a classic study, p53 3KR (lysine to arginine mutation, K→R) knock-in mice were constructed, in which p533KR mutant was deficient in cell-cycle arrest, senescence, and apoptosis [13]. Surprisingly, these mice did not develop early-onset tumors as did with the p53 knockout (KO) mice. Further study revealed that p533KR mice remained capable of p53-mediated metabolic regulation, including the ability to modulate energy metabolism and control reactive oxygen species (ROS). This work highlighted the importance of p53-mediated regulation of cellular metabolism as a component of its anti-tumor function. However, in certain circumstances, p53 can also promote cancer development by regulating cancer metabolism. In this review, we will summarize our current understanding of the main metabolic targets of wild type (WT) p53.We will mainly focus on cancer metabolism, while we also refer to other physiological or pathological contexts. We would also like to discuss the role of mutant p53 (mtp53) in regulating cell metabolism. We will conclude with a discussion of mechanisms underlying p53-mediated modulation of metabolism, notably as it relates to the pathogenesis of neoplastic disease. We also refer our readers to several excellent reviews of this field [14–18].
p53 regulates numerous and diverse metabolic pathways
The six nutrients that are essential to life are carbohydrates, fats, proteins, minerals, vitamins, and water. p53 is involved in pathways that regulate the metabolism of the first four of them. p53 also regulates nucleic acid biosynthesis and controls the production of ROS. In this section, we will review our current understanding of the roles of p53 in modulating the anabolism and/or catabolism of each of these critical biomolecules.
Glycolysis and gluconeogenesis
Glucose is the central molecule in energy and carbon metabolism. Once uptaken by the cell, glucose first undergoes a multi-enzymatic degradation process in the cytoplasm called glycolysis, in which glucose is converted to pyruvate ready for thorough breakdown to produce large amounts of ATP within the mitochondria of healthy cells. In cancer cells, however, glycolysis is often amplified and accompanied by the conversion of pyruvate to lactate which is then exported, but not importing pyruvate into mitochondria for ATP production (i.e., the Warburg effect). This pathway benefits the cancer cells, as it provides them with a means to meet their enormous demand for antioxidants and materials of anabolism derived from the intermediate products of the glycolytic pathway [7].
Under most circumstances, p53 can inhibit glycolysis at multiple steps (Figure 1 and Table 1). Among these, p53 reduces glucose uptake via direct suppression of the transcription of glucose transporters glucose transporter 1 (GLUT1), GLUT4, and GLUT12 [19–21]; this is accompanied by indirect suppression of GLUT1 and GLUT3 via the downregulation of their activators, paraoxonase 2 (PON2) and nuclear factor-κB (NF-κB), respectively [22, 23]. Moreover, under conditions of hypoxia, p53 activates Ras-related associated with diabetes (RRAD) to inhibit the translocation of GLUT1 to the plasma membrane in lung cancer cells [24]. The insulin receptor can also be subject to p53-mediated transcriptional repression; this may have a profound impact on insulin-mediated glucose uptake by skeletal muscle, liver, and adipose tissue [25]. Furthermore, glucose starvation activates p53 and induces the transcription of the long noncoding RNA (lncRNA) known as TRINGS (TP53-regulated inhibitor of necrosis under glucose starvation [26]). TRINGS suppresses the STRAP-GSK3β-NF-κB pathway to protect the cell from glucose starvation-caused cell necrosis. Once glucose has been transported into cells, p53 inhibits several glycolytic enzymes, including hexokinase 1 (HK1), HK2, glucose-6-phosphate isomerase (GPI), phosphoglucomutase (PGM), and β-enolase via transcriptional suppression or induction of specific microRNAs (miRNAs) [27–30]. Phosphofructokinase-1 (PFK-1) is the rate-limiting enzyme in glycolysis; p53 can limit PFK1 activity via its capacity to activate TP53-inducible glycolysis and apoptosis regulator (TIGAR) or suppress transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3). Inhibition of PFK1 can channel glycolytic carbon into the pentose phosphate pathway (PPP) to generate ribose 5-phosphate (R-5-P, material for synthesis of nucleotide) and NADPH (reducing agent for ROS control) [31, 32]. Interestingly, p53 may also inhibit the PPP via its capacity to limit the expression of PFKFB4 or binding to repress glucose-6-phosphate dehydrogenase (G6PDH), the enzyme that catalyzes the first reaction in the PPP using glucose 6-phosphate as a substrate [33, 34]. These conflicting results might be understood in the light of context-dependent p53 functions associated with the regulation of the PPP. Specifically, parkinson disease 2 (PARK2, or Parkin), which is a direct target of p53, suppresses glycolysis via direct inhibition of pyruvate kinase isoform M2 (PKM2) and hypoxia-inducible factor 1α (HIF1α) [35, 36]. Accumulation of pyruvate, the end-product of glycolysis, will ultimately inhibit glycolysis. As such, excess pyruvate is converted to lactate and then exported from the cell by the monocarboxylate transporter (MCT); p53 suppresses MCT1 expression, thereby impeding pyruvate export and glycolysis [37]. In addition to its role in regulating the activity of glycolytic enzymes, p53 promotes functional crosstalk with the master regulators of this pathway, including HIF and c-Myc [38]. In skeletal and cardiac cells, p53 also supports glycolysis by promoting the expression of PGM [39]. Moreover, p53 target, TIGAR, can bind HK2 to improve its anti-ROS activity [40]. Taken together, these findings reflect the complexity of p53’s roles in metabolism regulation, which will be reiterated below.
Figure 1. p53 regulates glucose, lipid, amino acid, nucleotide, and iron metabolism.
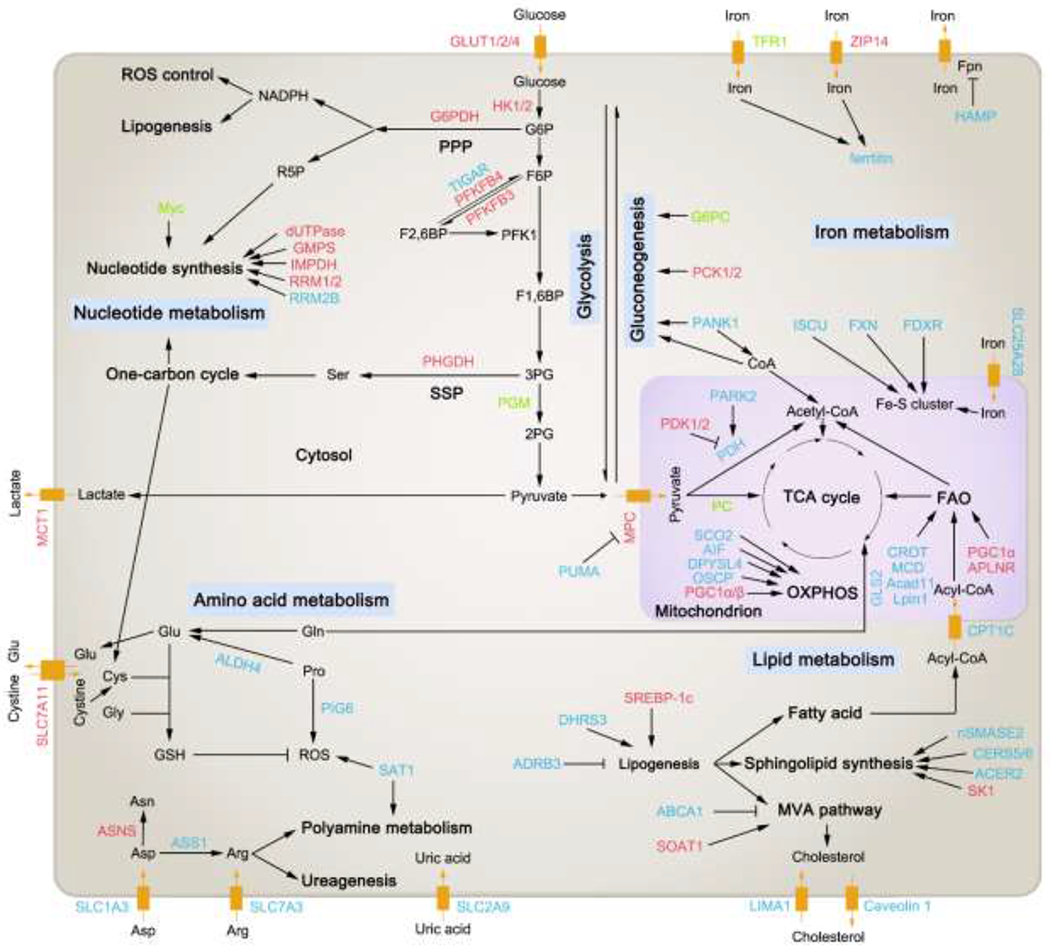
p53 participates in the regulation of metabolism of diverse biomolecules (including glucose, lipid, amino acid, nucleotide, and iron) in a transcription factor (TF)-dependent or –independent way. Major target genes regulated by p53 in these metabolic pathways are shown in this figure. For the full names of them, please refer to Table 1. Those proteins that are modulated positively by p53 (directly or indirectly), like promoting their expressions or activities, are depicted in blue. Oppositely, those proteins that are modulated negatively by p53 (directly or indirectly), like suppressing their expressions or activities, are depicted in red. If some protein can either be promoted or suppressed by p53 in different circumstances, it is depicted in green. Black arrows indicate positive effects. Black perpendicular bars indicate negative effects. G6P, glucose-6-phosphate; PPP, pentose phosphate pathway; F6P, fructose-6-phosphate; F2,6BP, fructose-2,6-bisphosphate; PFK1, phosphofructokinase 1; F1,6BP, fructose-1,6-bisphosphate; 3PG, 3-phosphoglycerate; SSP, serine synthesis pathway; 2PG, 2-phosphoglycerate; Fpn, ferroportin; Ser, serine; Glu, glutamate; Gln, glutamine; Cys, cysteine; Gly, glycine; Pro, proline; Asp, asparate; Asn, asparagine; Arg, arginine; GSH, glutathione; TCA cycle, tricarboxylic acid cycle; OXPHOS, oxidative phosphorylation; MVA pathway, mevalonate pathway.
Table 1.
Basic information about the metabolic target genes of wild type p53.
| Functional category of target gene | Target gene | Full name of target gene | Positively (+) or negatively (−) regulated by p53 | How does p53 regulate it | Metabolic effect of p53 by regulating this target gene | Promote (+) or suppress (−) cancer when p53 regulates this target gene (NA, not applicable in the orignal reference) | Other metabolic role of this target gene | Reference |
|---|---|---|---|---|---|---|---|---|
| Glucose metabolism | GLUT1/4 | Glucose transporter 1/4 | − | Suppress transcription | Inhibit glucose uptake and glycolysis | − | [19, 21, 22, 24] | |
| PON2 | Paraoxonase 2 | − | Suppress transcription | Inhibit glucose uptake and glycolysis | − | [22] | ||
| RRAD | Ras-related associated with diabetes | + | Promote transcription | Inhibit GLUT1 translocation to the plasma membrane to import glucose, thus inhibit glycolysis | − | [24] | ||
| Lipid metabolism | GLUT3 | Glucose transporter 3 | − | Inhibit nuclear factor-κB (NF-κB), which activates GLUT3 | Inhibit glucose uptake and glycolysis | − | [23] | |
| GLUT12 (SLC2A12) | Glucose transporter 12 (solute carrier family 2 member 12) | − | Suppress transcription | Inhibit glucose uptake and glycolysis | − | [20] | ||
| INSR (IR) | Insulin receptor | − | Suppress transcription | Inhibit glucose uptake and glycolysis | − | [25] | ||
| HK1 and GPI | HK1: hexokinase 1; GPI: glucose phosphate isomerase | − | Induce miR-34a to inhibit translation | Inhibit glycolysis | − | [27] | ||
| HK2 | Hexokinase 2 | +/− | Activate TIGAR to promote its activity or induce miR-34a and miR-143 to inhibit its translation | Lower ROS or inhibit glycolysis | − | ROS control | [27, 28, 40] | |
| PGM | Phosphoglycerate mutase | +/− | Promote transcription (in muscle) or suppress transcription (in fibroblast) or induce miR-34a to inhibit its translation | Promote or inhibit glycolysis | +/− | [27, 29, 39] | ||
| ENO3 | β-enolase | − | Suppress transcription | Inhibit glycolysis and lactate production | NA | [30] | ||
| TIGAR | TP53-induced glycolysis and apoptosis regulator | + | Promote transcription | Inhibit glycolysis to promote PPP or promote HK2 activity to lower ROS | + | ROS control | [31, 40] | |
| PFKFB3 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | − | Suppress transcription | Inhibit glycolysis but promote PPP | − | [32] | ||
| PFKFB4 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 | − | Suppress transcription | Promote glycolysis but inhibit PPP | − | [33] | ||
| G6PDH | Glucose-6-phosphate dehydrogenase | − | Bind to inhibit its activity | Inhibit PPP | − | [34] | ||
| MCT1 (SLC16A1) | Monocarboxylate transporter 1 (solute carrier family 16 member 1) | − | Suppress transcription | Inhibit lactate export and glycolysis | − | [37] | ||
| G6PC | Glucose-6-Phosphatase Catalytic Subunit | +/− | Promote expression or inhibit expression through activation of SIRT6 | Promote or suppress glyconeogenesis | −/+ | [41, 43] | ||
| PCK1 | Phosphoenolpyruvate carboxykinase 1 | − | Inhibit expression through activation of SIRT6 | Suppress glyconeogenesis | + | [43] | ||
| PCK2, GK, AQP3/9, and GOT1 | PCK2: phosphoenolpyruvate carboxykinase 2; AQP3/9: aquaporin 3/9; GOT1: glutamic-oxaloacetic transaminase 1 | + | Promote expression | Promote glyconeogenesis | − | [41] | ||
| GYS2 | Glycogen synthase 2 | − | Suppress transcription | Inhibit glycogen synthesis | + | [44] | ||
| FUCA1 | Alpha-L-fucosidase 1 | + | Promote transcription | Promote chemotherapy-induced apoptosis | − | [47] | ||
| PDK1 | Pyruvate dehydrogenase kinase 1 | − | Induce miR-34a to inhibit translation | Promote TCA cycle | − | [27] | ||
| PDK2 | Pyruvate dehydrogenase kinase 2 | − | Suppress transcription | Promote TCA cycle | − | [48] | ||
| PARK2 (Parkin) | Parkinson disease 2 | +/− | Promote transcription or bind to suppress its activity | (1) Promote TCA cycle (by enhance the expression of PDHA1--a component of PDH complex); (2) inhibit glycolysis (by suppress PKM2 and HIF1α); (3) positively regulate PTEN to lower ROS and repress the PI3K/AKT pathway; (4) promote OXPHOS with PINK1; (5) promote mitophagy to suppress ferroptosis | − | ROS control, autophagy, and ferroptosis | [35, 36, 49, 57, 234 , 259, 280] | |
| GLS2 | Glutaminase 2 | + | Promote transcription | Promote TCA cycle and ferroptosis | − | Amino acid metabolism | [51, 52] | |
| ME1/2 | Malic enzyme 1/2 | − | Suppress transcription | Inhibit NADPH synthesis and lipogenesis, suppress glutaminolysis, promote senescence | − | [53] | ||
| PC | Pyruvate carboxylase | +/− | Promote or suppress transcription | (1) Promote TCA cycle in PDAC; (2) Inhibit TCA cycle and OXPHO in pancreatic β cell and impair GSIS (glucose-stimulated insulin secretion), leading to glucose intolerance | − | [54, 69] | ||
| IDH1 | Isocitrate dehydrogenase 1 | + | Promote transcription | Promote TCA cycle and α-ketoglutarate generation in PDAC | − | [54] | ||
| TET2 | Ten-eleven translocation 2 | +/− | Support its function by accumulation of αKG or promote its autophagic degradation | Promote PDAC differentiation or decrease cancer therapeutic resistance | − | [54, 366] | ||
| SCO2 | Synthesis of cytochrome c oxidase 2 | + | Promote transcription | Promote formation of the cytochrome c oxidase complex (complex IV) and enhance OXPHOS | − | [55] | ||
| AIF | Apoptosis-inducing factor | + | Promote transcription | Promote stability of the mitochondrial complex I and enhance OXPHOS | − | [56] | ||
| DPYSL4 | Dihydropyrimidinase-like 4 | + | Promote transcription | Promote mitochondrial supercomplexes activity and enhance OXPHOS | − | [58] | ||
| OSCP | Oligomycin sensitivity-conferring protein | + | Bind to promote its activity | Promotes assembly of F1F0-ATP synthase and enhance OXPHOS | NA | [59] | ||
| RelA | RELA protooncogene, NF-κB subunit | − | Bind to suppress its activity | Promote mitochondrial gene expression and OXPHOS | − | [60] | ||
| TFAM | Transcription factor A, mitochondrial | + | Promote transcription or bind to promote its activity | Promote mitochondrial DNA replication and function to enhance OXPHOS | − | [61, 62] | ||
| POLG | Mitochondrial DNA polymerase subunit γ | + | Bind to promote its activity | Promote mitochondrial genome stability and enhance OXPHOS | NA | [63] | ||
| HmtSSB | Human single-stranded DNA-binding protein, mitochondrial | + | Bind to promote its activity | Promote mitochondrial genome stability and enhance OXPHOS | NA | [64] | ||
| Tom20, Tim23, and mtHsp60/70 | Tom20: translocase of outer membrane 20; Tim23: translocase of inner membrane 23; mtHsp60/70: mitochondrial heat shock protein 60/70 | + | Promote transcription | Promote mitochondrial normal function and enhance OXPHOS | − | [65] | ||
| MNF2 | Mitofusin 2 | + | Promote transcription | Promote mitochondrial fusion and enhance OXPHOS | − | [67] | ||
| apoB and apobec1 | apoB: apolipoprotein B; apobec1: apolipoprotein B mRNA editing enzyme catalytic subunit 1 | + | Promote transcription | Promote APOB biogenesis and lipid transportation | NA | [72] | ||
| PLTP, Abca12, and Cel | PLTP: phospholipid transfer protein; Abca12: ATP binding cassette subfamily A member 12; Cel: carboxyl ester lipase | + | Promote transcription | Promote lipid transport in circulation | NA | [73] | ||
| CYP19 | Aromatase | + | Promote transcription | Inihibit lipid accumulation and avoid fat | NA | [74] | ||
| SIRT1 | NAD-dependent protein deacetylase sirtuin-1 | + | Bind Foxo3a to promote its transcription | Inhibit lipid accumulation | NA | [75, 76] | ||
| DHRS3 | Dehydrogenase/reductase 3 | + | Promote transcription | Promote lipid droplet formation | NA | [77] | ||
| NPC1L1, TNF, and CCL2 | NPC1L1: NPC1 like intracellular cholesterol transporter 1; TNF: tumor necrosis factor; CCL2: C-C motif chemokine ligand 2 | + | Promote transcription | Promote fat accumulation | NA | [78] | ||
| SREBP-1c | Sterol regulatory element-binding protein-1c | − | Suppress transcription | Inhibit lipogenesis | − | [79] | ||
| OPN | Osteopontin | + | Promote transcription | Inhibit de novo lipogenesis | NA | [80] | ||
| ADRB3 | Adrenoceptor beta 3 | + | Promote transcription | Promote lipolysis | + | [81] | ||
| PANK1 | Pantothenate kinase 1 | + | Promote transcription | Promote CoA synthesis to facilitate β-oxidation and gluconeogenesis | NA | Glueose metabolism | [42] | |
| CROT | Carnitine O-octanoyltransferase | + | Promote transcription | Promote transport of medium length acyl chains to the mitochondria to facilitate FAO | NA | [73, 82] | ||
| MCD | Malonyl-CoA decarboxylase | + | Promote transcription | Promote import long chain fatty acid into mitochondria to facilitate FAO | − | [83] | ||
| Acad11 | Acyl-CoA dehydrogenase family member 11 | + | Promote transcription | Promote FAO | + | [84] | ||
| LPIN1 | Lpin1 | + | Promote transcription | Promote FAO | − | [85] | ||
| CPT1C | Carnitine palmitoyltransferase 1C | + | Promote transcription | Promote transport of long-chain fatty acids into mitochondria to facilitate FAO | + | [86] | ||
| PGC1α/β | Peroxisome proliferator-activated receptor γ coactivator 1α/β | − | Suppress transcription | (1) Inhibit mitochondria biogenesis to promote premature aging upon telomere attrition (2) PGC-1α directly binds p53 to promotes cell survival upon metabolic stress (3) p53 inhibits FAO by downregulating PGC1α | NA | [70, 71, 94, 322] | ||
| APLN and APLNR | APLN: Apelin; APLNR: Apelin receptor | − | Suppress transcription | Inhibit FAO | NA | [94] | ||
| SK1 | Sphingosine kinase 1 | − | Suppress transcription | Promote synthesis of proapoptotic ceramides and sphingosine, while inhibit synthesis of the anti-apoptotic S1P | − | [88] | ||
| CERS6 | Ceramide synthase 6 | + | Promote transcription | Promote synthesis of proapoptotic ceramide | − | [89] | ||
| nSMASE2 | Neutral sphingomyelinase 2 | + | Promote transcription | Promote synthesis of proapoptotic ceramide | − | [90] | ||
| ACER2 | Alkaline ceramidase 2 | + | Promote transcription | Inhibit ceramide but promote sphingosine and S1P synthesis | −/+ | [91] | ||
| CERS5 | Ceramide synthase 5 | + | Promote transcription | Promote synthesis of C16:0-ceramide to promote autophagy and mitochondrial respiration, and thus enhancing chemoresistance to chemotherapy | + | [92] | ||
| CDS1/2 | CDP-diacylglycerol synthase 1/2 | + | Promote transcription | Promote cardiolipin de novo synthesis | NA | [93] | ||
| Hmgcll1 | 3-hydroxymethyl-3-methylglutaryl-coA lyase like 1 | + | Promote transcription | Promote ketone bodies generation and energy production | − | [84] | ||
| LIMA1 | LIM domain and actin binding 1 | + | Promote transcription | Promote cholestrol uptake | − | [95, 96] | ||
| CAV | Caveolin 1 | + | Promote transcription | Promote cell cholesterol efflux | +/− | [97] | ||
| SHP | Small heterodimer partner | + | Promote transcription | Inhibit bile acid synthesis and intestinal lipid absorption | NA | [98] | ||
| ABCC3 and CYP2B6 | ABCC3: ATP binding cassette subfamily C member 3; CYP2B6: cytochrome P450 family 2 subfamily B member 6 | + | Promote transcription | Promote bile acid disposition and improve cholestasis | NA | [99] | ||
| ABCA1 | ATP binding cassette subfamily A member 1 | + | Promote transcription | Inhibit mevalonate pathway | − | [101] | ||
| HMGCR, MVK, FDPS, and FDFT1 | HMGCR: 3-hydroxy-3-methylglutaryl-CoA reductase; MVK: mevalonate kinase; FDPS: farnesyl diphosphate synthase; FDFT1: farnesyl-diphosphate farnesyltransferase 1 | + | Promote transcription | Promote mevalonate pathway | NA | [102] | ||
| SOAT1 | Sterol O-acyltransferase 1 | − | Suppress transcription | Inhibit mevalonate pathway | − | [103] | ||
| SCD1 | Stearoyl-CoA desaturase | − | Suppress transcription | Inhibit fatty acid desaturation | − | Cros stalk with AKT | [105] | |
| Sema3E | Semaphorin 3E | + | Promote transcription | Promote adipose tissue inflammation and insulin resistance | NA | [106, 107] | ||
| PRDM16 | PR/SET domain 16 | + | Promote transcription | Promote brown adipose tissue differentiation and thermogenesis | NA | [108] | ||
| Elovl3 | Elongation of very long chain fatty acids protein 3 | + | Promote transcription | Promote thermogenesis | NA | [109] | ||
| Amino acid metabolism | SLC1A3 | Solute carrier family 1 member 3 | + | Promote transcription | Promote aspartate uptake and cancer cell adaptation to glutamine deprivation | + | Amino acid metabolism | [113] |
| SLC7A3 | Solute carrier family 7 member 3 | + | Promote transcription | Promote arginine uptake and cancer cell adaptation to glutamine deprivation | + | Amino acid metabolism | [114] | |
| SLC7A11 | Solute carrier family 7 member 11 | − | Suppress transcription | Inihibit cystine uptake and promote ferroptosis | − | Amino acid metabolism | [115, 116, 228] | |
| p21 | CDKN1A, cyclin dependent kinase inhibitor 1A | + | Promote transcription | (1) Promote cell cycle arrest and cancer cell survival under serine/glutamine/cystine deprivation (2) stablilize NRF2 | +/− | Amino acid metabolism and ROS control | [120, 121, 172, 232] | |
| MDM2 | Mouse double minute 2 homolog | + | Promote transcription | (1) Promote serine metabolism and redox homeostasis; (2) promote adipocyte differentiation | + | Amino acid metabolism and ROS control | [110, 122] | |
| PHGDH | Phosphoglycerate dehydrogenase | − | Suppress transcription | Inhibit serine synthesis | − | Amino acid metabolism | [123] | |
| ASS1 | Argininosuccinate synthase 1 | + | Promote transcription | Promote arginine biosynthesis and inhibit AKT | + | Amino acid metabolism and cross talk with AKT | [124] | |
| ASNS | Asparagine synthetase | − | Suppress transcription | Suppress asparagine synthesis and promote senescence and cell cycle arrest | − | Crosstalk with AMPK | [127] | |
| PW1 | PEG3, paternally expressed gene 3 | + | Promote expression | Inhibit muscle differention and promote muscle atrophy and cachexia | + | Amino acid metabolism | [129] | |
| Ammonia metabolism | SAT1 | Spermidine/spermine N1-acetyltransferase 1 | + | Promote transcription | Promote ALOX15 activity and ferroptosis | − | Ferroptosis | [130] |
| CPS1, OTC, and ARG 1 | CPS1: carbamoylphosphate synthase 1; OTC: ornithine carbamoyltransferase; ARG1 : Arginase 1 | − | Suppress transcription | Suppresse ureagenesis and ammonia elimination | − | Amino acid metabolism | [131] | |
| Nucleotide metabolism | dUTPase | DUT, deoxyuridine triphosphatase | − | Suppress transcription | Inhibit dTTP biogenesis | − | [133] | |
| GMPS | Guanine monophosphate synthase | − | Suppress transcription | Inhibit GMP biogenesis | − | [134, 135] | ||
| IMPDH | Inosine monophosphate dehydrogenase | − | Induce miR-34a to inhibit its translation | Inhibit GTP biogenesis | − | [136] | ||
| RRM1/2 | Ribonucleotide reductase catalytic subunit M1/2 | − | Suppress expression by inhibiting mTOR | Inhibit nucleotide synthesis | − | [137] | ||
| p53R2 (RRM2B) | Ribonucleotide reductase regulatory TP53 inducible subunit M2B | + | Promote transcription | (1) Promote dNTP synthesis; (2) bind catalase to lower ROS | −/+ | ROS control | [138, 139, 195] | |
| ADORA2B | Adenosine A2b receptor | + | Promote transcription | Promote extracellular adenosine sensing and apoptosis | − | [144] | ||
| Iron metabolism | TFR1 | Transferrin receptor 1 | −/+ | Inhibit or promote expression posttranscriptionally | Induce cell cycle arrest by reducing intracellular iron level or promote ferroptosis by increasing intracellular iron level | − | [148, 152] | |
| ferritin | ferritin | + | Promote expression posttranscriptionally | Induce cell cycle arrest by reducing the labile iron level | − | [148] | ||
| ZIP14 (SLC39A14) | Zrt- and Irt-like protein 14 (solute carrier family 39 member 14) | − | Bind to promote its ubiquitin-mediated degradation | Inhibit iron uptake | − | [149, 150] | ||
| HAMP | Hepcidin antimicrobial peptide | + | Promote transcription | Reduce serum iron and induce iron sequestration in the reticuloendothelial macrophages | − | [151] | ||
| ISCU | Iron-sulfur cluster assembly enzyme | + | Promote transcription | Promote Fe-S cluster biogenesis to reduce labile iron level and ROS | − | ROS control | [153] | |
| FXN | Frataxin | + | Promote transcription | Promote Fe-S cluster biogenesis to reduce labile iron level and ROS | − | [154, 155] | ||
| FDXR | Ferredoxin reductase | + | Promote transcription | Promote Fe-S cluster biogenesis to reduce labile iron level and ROS | − | ROS control | [156, 157] | |
| SLC25A28 | Solute carrier family 25 member 28 | + | Binds to promote its activity | Promote abnormal accumulation of mitochondrial iron and ferroptosis | NA | [158] | ||
| ROS control | NRF2 | NFE2L2, nuclear factor, erythroid 2 like 2 | +/− | Promote its stabilization by inducing p21 or suppress its transcription by inhibiting Sp1 | Lower or enhance ROS | +/− | [172, 176, 183, 339] | |
| SESN1 (PA26) | Sestrin 1 (p53 activated gene 26) | + | Promote transcription | Lower ROS | − | Crosstalk with AMPK | [173, 175, 193, 273] | |
| SESN2 (Hi95) | Sestrin 2 (hypoxia induced gene 95) | + | Promote transcription | Lower ROS | −/+ | Crosstalk with AMPK | [174, 175, 193, 273] | |
| PML | PML nuclear body scaffold | + | Promote transcription | Function as a ROS sensor to activate p53 | − | [184–186] | ||
| COX-2 | PTGS2, prostaglandin-endoperoxide synthase 2 | −/+ | Suppress or promote expression | Enhance or lower ROS | −/+ | [187, 188] | ||
| NOS2 | Nitric oxide synthase 2 | − | Suppress transcription | Lower ROS | − | [187] | ||
| NOS3 (eNOS) | Nitric oxide synthase 3 (Endothelial NOS) | +/− | Promote or suppress transcription | (1) Protect cardiomyocytes from ischemia/ref low-induced death; (2) Protect organism from dietary obesity | NA | [21, 189] | ||
| NOX4 | NADPH oxidase 4 | − | Suppress expression | Lower ROS and cell metastasis | − | [190] | ||
| Mieap (SPATA18) | Mitochondria-eating protein (spermatogenesis associated 18) | + | Promote transcription | Promote repair or degradation of unhealthy mitochondria and lower ROS | NA | OXP HOS | [66] | |
| CAT | Catalase | +/− | Promote transcription or suppress activity | Lower or enhance ROS | NA | [191, 195] | ||
| GPX1 | Glutathione peroxidase 1 | + | Promote transcription | Lower ROS (may enhance ROS and apoptosis in specific circumstances) | − | [192] | ||
| MnSOD (SOD2) | Manganese superoxide dismutase (superoxide dismutase 2) | +/− | Promote or inhibit transcription or directly bind to suppress its activity or destablize protein | Lower or enhance ROS | − | [192, 196–198] | ||
| HO-1 (HMOX1) | Heme oxygenase-1 | +/− | Promote transcription or destablize protein | Lower or enhance ROS | NA | [194, 198, 199] | ||
| ALDH4 | ALDH4A1, aldehyde dehydrogenase 4 family member A1 | + | Promote transcription | Lower ROS | NA | Amino acid metabolism | [126] | |
| SLC2A9 (GLUT9) | Solute carrier family 2 member 9 (glucose transporter 9) | + | Promote transcription | Promote transport uric acid into cell to lower ROS | − | Uric acid metabolism | [132] | |
| TP53INP1 | Tp53-inducible nuclear protein 1 | + | Promote transcription | Lower ROS or promote autophagy-dependent cell death | − | Autophagy | [200, 263] | |
| PUMA | p53 up-regulated modulator of apoptosis | + | Promote transcription | Ehance ROS or suppress mitochondrial pyruvate uptake by inactivating MPC, thus suppressing OXPHOS | −/+ | OXP HOS | [68, 201, 202] | |
| BAX | BCL2-associated X protein | + | Promote transcription | Ehance ROS | − | [203, 204] | ||
| Noxa | PMAIP1: phorbol-12-myristate-13-acetate-induced protein 1 | + | Promote transcription | Ehance ROS | − | [205, 206] | ||
| NCF2/p67p hox | Neutrophil cytosolic factor 2 | + | Promote transcription | Ehance ROS | + | [208] | ||
| p66Shc | SHC1: SHC adaptor protein 1 | + | Promote transcription | Ehance ROS | − | [209] | ||
| PIG1 (Galectin-7/LGALS7) | p53-induced gene 1 (lectin, galactoside-binding, soluble, 7) | + | Promote transcription | Enhance ROS | − | [210, 211] | ||
| PIG3 (TP53I3) | p53-induced gene 3 (tumor protein p53 inducible protein 3) | + | Promote transcription | Enhance ROS | − | [195, 210] | ||
| PIG6 (PRODH/POX) | p53-induced gene 6 (proline dehydrogenase 1/proline oxidase) | + | Promote transcription | Catalyze proline dehydrogenation and enhance ROS | − | Amino acid metabolism | [125, 210] | |
| NQO1 and GST-α1 | NQO1: NAD(P)H quinone dehydrogenase 1; GST-α1: glutathione S-transferase α1 | − | Suppress transcription by inhibiting NRF2 | Ehance ROS | − | [115] | ||
| DRP1 | DNM1L: dynamin 1 like | + | Bind to promote its activity | Promote mitochondria fragmentation and dysfunction, and enhance mitochondrial ROS | NA | [212] | ||
| Opa1 | OPA1 mitochondrial dynamin like GTPase | + | Bind prohibitin 1 (PHB1) to release Opa1 | Promote mitochondria fragmentation (enhance ROS) and apoptosis | − | [213] | ||
| cyclophilin D (CypD) | PPIF: peptidylprolyl isomerase F | + | Bind to promote its activity | Promote mitochondrial permeability transition pore (PTP) opening (enhance ROS) and necrosis | − | [214] | ||
| CTSQ | cathepsin Q | + | Promote transcription | Promote necrosis along with ROS | − | [215] | ||
| Ferroptosis | CBS | Cystathionine β-synthase | − | Suppress expression posttranscriptionally | Promote ferroptosis | − | Amino acid metabolism | [119] |
| DPP4 | Dipeptidyl peptidase 4 | − | Bind to inhibit its activity | Inhibit ferroptosis | + | ROS control | [233] | |
| Autophagy | ATG2B/4A/4C/7/10, UVRAG, and VMP1(TME M49) | ATG2B/4A/4C/7/10: autophagy related 2B/4A/4C/7/10 ;UVRAG: UV radiation resistance associated; VMP1(TMEM4 9): vacuole membrane protein 1 (transmembrane protein 49) | + | Promote transcription | Promote autophagy and cell death | − | [239] | |
| ULK1/2 | Unc-51 like autophagy activating kinase 1/2 | + | Promote transcription | Promote autophagy and cell death | − | [239, 241] | ||
| DRAM | Damage-regulated autophagy modulator | + | Promote transcription | Promote autophagy and cell death | − | [243] | ||
| CTSD | Cathepsin D | + | Promote transcription | Promote autophagy and cell death | − | [244–247] | ||
| ISG20L1 (AEN) | Interferon stimulated exonuclease gene 20kDa-like 1 (apoptosis enhancing nuclease) | + | Promote transcription | Promote autophagy and cell death | − | [248] | ||
| Dapk1 | Death associated protein kinase 1 | + | Promote transcription | Promote autophagy and cell death | − | [249] | ||
| PIG8 (EI24) | p53-induced gene 8 (EI24 autophagy associated transmembrane protein) | + | Promote transcription | Promote autophagy and cell death | − | [250] | ||
| TGM2 | Transglutaminase 2 | + | Promote transcription | Promote autophagy | − | [251] | ||
| BNIP3 | BCL2 interacting protein 3 | +/− | Promote or inhibit transcription | Promote mitophagy or suppress hypoxia-induced apoptosis | + | [253, 254] | ||
| BNIP3L (NIX) | BCL2 interacting protein 3 like (NIP-3-like protein X) | + | Promote transcription | Promote mitophagy and cell death | − | [255, 256] | ||
| HMGB1 | High mobility group box 1 | − | Bind to inhibit its activity | Inhibit autophagy | + | [257] | ||
| LC3 | MAP1LC3A, microtubule associated protein 1 light chain 3 alpha | −/+ | Inhibit expression posttranscriptionally or induce TP53INP 1 to bind and promote its activity | Inhibit or promote autophagy | +/− | [262, 263] | ||
| RB1CC1 (FIP200) | RB1 inducible coiled-coil 1 (FAK family kinase-interacting protein of 200 KDa) | − | Bind to inhibit its activity | Inhibit autophagy | NA | [264] | ||
| Crosstalk with metabolic sensors | AMPKβ1/2 and TSC2 | AMPKβ1/2: protein kinase AMP-activated non-catalytic subunit α2/β2; TSC2: tuberous sclerosis complex 2 | + | Promote transcription | Activate AMPK but repress mTOR pathway | − | [271] | |
| LKB1 | Liver kinase B1 | + | Promote transcription | Activate AMPK pathway | − | [272] | ||
| IGF1R | Insulin like growth factor 1 receptor | − | Suppress transcription | Repress AKT pathway | − | [275, 276] | ||
| IGFBP3 | Insulin like growth factor binding protein 3 | + | Promote transcription | Repress AKT pathway | − | [277] | ||
| IGFBP1 | Insulin like growth factor binding protein 1 | + | Promote transcription | Inhibit apoptosis | + | [278] | ||
| PTEN | Phosphatase and tensin homolog | + | Promote transcription | Repress AKT and mTOR pathway | − | [271, 279] | ||
| PLK1 | Polo-like kinase 1 | − | Suppress transcription | Repress AKT pathway | − | [281–283] | ||
| PLK2 | Polo-like kinase 2 | + | Promote transcription | Repress mTOR pathway | − | [284, 285] | ||
| REDD1 | DDIT4: DNA damage inducible transcript 4 | + | Promote transcription | Repress mTOR pathway | − | [286–289] | ||
| PHLDA1/3 | Pleckstrin homology like domain family A member 1/3 | + | Promote transcription | Repress AKT pathway | − | [290–292] | ||
| SIVA | SIVA1: SIVA1 apoptosis inducing factor | + | Promote transcription | Promote mTOR pathway | + | [293–295] | ||
| Drug metabolism | CYPs, SULTs, and MRPs | CYPs: cytochromes P450 family; SULTs: sulfotransferase family; MRPs: multidrug resistance-associated protein family | + | Promote transcription | Promote metabolism of drugs, carcinogens, and pollutants | NA | [392–396] |
Gluconeogenesis is the process used by cells to synthesize glucose, part of which are inverse reactions compared with glycolysis. Theoretically, p53-mediated inhibition of glycolysis as discussed above could benefit the process of gluconeogenesis. Additionally, p53 has been found to induce several genes encoding enzymes that participate in gluconeogenesis, including glucose-6-phosphatase, catalytic subunit (G6PC), phosphoenolpyruvate carboxykinase 2 (PCK2), glycerol kinase (GK), aquaporin 3 (AQP3), AQP9, and glutamic-oxaloacetic transaminase 1 (GOT1), to enhance hepatic glucose production [41] (Figure 1 and Table 1). p53 also activates pantothenate kinase 1 (PANK1) to increase intracellular levels of coenzyme A (CoA) and promote gluconeogenesis [42]. However, p53 has also been reported to inhibit G6PC and PCK1 via activation of sirtuin 6 (SIRT6), thereby suppressing gluconeogenesis [43]. In hepatitis B virus (HBV)-associated hepatocellular carcinoma (HCC), p53 suppresses the synthesis of glycogen synthase 2 (GYS2), thereby reducing glycogen levels [44]. Interestingly, glucose level can in turn alter p53 activity [45, 46]. In addition to glucose metabolism, p53 also has an impact on cellular glycosylation process as it can activate the glycosidase, alpha-L-fucosidase 1 (FUCA1) to promote chemotherapy-induced cellular apoptosis [47].
The tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS)
TCA cycle and followed oxidative phosphorylation can thoroughly breakdown of biomolecules to produce energy in the form of ATP. In most cases, p53 promotes both of these processes (Figure 1 and Table 1). The conversion of pyruvate to acetyl-CoA by pyruvate dehydrogenase (PDH) is a critical step for pyruvate to enter the TCA cycle. Pyruvate conversion is promoted by p53; p53 can suppress pyruvate dehydrogenase kinase 1 and 2 (PDK1 and PDK2), which are the enzymes that phosphorylate and inactivate PDH [27, 48]. In addition, p53’s target Parkin can elevate the expression of a PDH component—PDHA1 [49]. The amino acid, glutamine, has anaplerotic properties and can be introduced into the TCA cycle, most notably when the supply of glucose is limited [50]. p53 amplifies this process by upregulating the expression of glutaminase 2 (GLS2) to promote glutaminolysis [51, 52]. However, p53 can also repress malic enzyme 1 (ME1) and ME2 to suppress glutaminolysis [53]. In pancreatic ductal adenocarcinoma (PDAC), p53 activated the TCA cycle by inducing pyruvate carboxylase (PC) and isocitrate dehydrogenase 1 (IDH1), resulting in the accumulation of α-ketoglutarate (αKG), which can be used as a substrate by chromatin-modifying enzymes, including ten-eleven translocation 2 (TET2). Signaling via the p53/αKG axis increases the level of chromatin 5-hydroxymethylcytosine (5hmC) and induces tumor-cell differentiation and growth suppression [54]. With respect to OXPHOS, p53 promotes the transcription of several components of the respiratory chain complexes (RCCs), including synthesis of cytochrome oxidase 2 (SCO2) and apoptosis-inducing factor (AIF) [55, 56]. p53’s target, Parkin, cooperates with PTEN induced kinase 1 (PINK1) to promote the translation of some RCC mRNAs [57]. Dihydropyrimidinase-like 4 (DPYSL4), another p53 target, binds mitochondrial supercomplexes to foster OXPHOS [58]. p53 also undergoes translocation into the mitochondria where it binds to oligomycin sensitivity-conferring protein (OSCP), which facilitates the assembly of F1 F0 -ATP synthase [59]. On the other hand, p53 binds NF-κB subunit RelA (also called p65) to suppress its mitochondrial translocation, which abolishes RelA-mediated OXPHOS inhibition [60]. Since TCA cycle and OXPHOS both happen in the mitochondria, increasing the copy number of mitochondria, maintaining its structural integrity and genomic stability, and repairing or removing damaged mitochondria will benefit the OXPHOS to proceed. Indeed, p53 activates a batch of target genes to achieve these outcomes [61–67].
Similar to the opposing roles identified for p53 in glycolysis, recent evidence suggests that p53 can also inhibit both the TCA cycle and OXPHOS (Figure 1 and Table 1). In HCC, WT p53 has an oncogenic role by inducing the expression of p53-upregulated modulator of apoptosis (PUMA) to inhibit pyruvate-driven OXPHOS [68], thereby promoting oncogenesis. Mechanistically, acting as a pro-apoptosis protein in most cases, in HCC, PUMA can bind and suppress mitochondrial pyruvate carrier (MPC). This interaction disrupts mitochondrial pyruvate uptake and represses OXPHOS. High levels of PUMA detected in patients with HCC correlate with a poor prognosis. Unlike in PDAC, where p53 induces PC expression [54], in pancreatic β-cells, activation of p53 results in the downregulation of PC, thereby impairing mitochondrial metabolism [69]. Upon telomere dysfunction, p53 is activated to repress peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC1α/β), which ultimately leads to the disruption of mitochondrial biogenesis and function [70]. Of note, PGC1α can also bind p53 and modulate its metabolic functions under nutrient stress to promote cell survival [71]. If starvation is prolonged, PGC1α will undergo degradation by ring finger protein 2 (RNF2), and p53 shifts to promote cell apoptosis. Thus, p53 and PGC1α compose a feedback regulatory loop that serves to promote a switch in p53 function under conditions of nutrient stress.
Lipid metabolism
Lipids are important for the cell to maintain membrane structures, provide energy, and transduce signals. Most normal cells (except for some specialized cell types, such as adipocytes and hepatocytes) exhibit low levels of de novo lipogenesis, as the demand for lipid is satisfied primarily via its absorption from the peripheral circulation. By contrast, tumor cells have dramatically higher lipid requirements and require amplification of de novo lipogenesis [8]. In this light, p53 has been found to play versatile roles in regulating lipid metabolism (Figure 1 and Table 1).
At the systemic level, p53 in liver cells regulates several genes (i.e., apolipoprotein B, apoB; apolipoprotein B mRNA editing enzyme catalytic subunit 1, apobec1; phospholipid transfer protein, PLTP; ATP binding cassette subfamily A member 12, Abca12; and carboxyl ester lipase, Cel) to influence systemic lipid transport and homeostasis, which may relate to atherosclerosis development [72, 73]. p53 also inhibits lipid accumulation in liver cells by regulating the levels of aromatase and SIRT1 [74–76]. Compared with WT mice, p53 KO mice exhibit marked obesity and hepatic lipid accumulation after feeding with a high-fat diet (HFD) [74]. Under nutrient stress, p53 induces SIRT1 in a forkhead box o3a (Foxo3a) dependent manner to impede fat storage [75, 76]. However, there are opposite observations that p53 can promote lipid accumulation [77, 78]. In one study, p53 induced the expression of dehydrogenase/reductase 3 (DHRS3), which promotes lipid droplet formation [77]. In another study, the p53 single nucleotide polymorphism (SNP) variant, p53(P72R), was associated with increased fat accumulation by regulating tumor necrosis factor (TNF) and NPC1 like intracellular cholesterol transporter 1 (NPC1L1) [78].
At the sub-cellular level, p53 mainly suppresses lipogenesis and promotes lipolysis and fatty acid oxidation (FAO) (Figure 1 and Table 1). Sterol regulatory element-binding protein-1c (SREBP-1c) is a master TF that controls the expression of a range of lipogenic enzymes. In adipocytes, activation of p53 leads to a decrease in SREBP-1c and impaired lipogenesis [79]. In liver cells, p53 activates osteopontin (OPN), thereby suppressing aging-associated cellular senescence and triglycerides (TG) synthesis [80]. Moreover, p53-mediated suppression of ME also hinders lipogenesis [53], and p53 transactivates the gene encoding beta-3-adrenergic receptor (ADRB3) to promote lipolysis [81]. Noticeably, a p53 R178C mutant (equivalent to human R181C) has a stronger effect on ADRB3. The R178C mice are lean with less body fat than do their WT counterparts.
Complete degradation of fatty acids via β-oxidation and OXPHOS results in more energy production than can be obtained from similar concentrations of glucose alone (Figure 1 and Table 1). CoA is a necessary co-factor for fatty acid oxidative degradation. p53 activates PANK1 to boost the synthesis of CoA, thereby promoting fatty acid β-oxidation [42]. p53 also facilitates the transport of fatty acids with different lengths into the mitochondria for degradation via activation of both carnitine O-octanoyltransferase (CROT) and malonyl-CoA decarboxylase (MCD) [73, 82, 83]. Of note, MCD is induced by p53 in response to fasting via a ribosome protein (RP)/ Mouse double minute 2 homolog (MDM2)/p53/MCD-dependent pathway; this serves to promote FAO and to ameliorate hepatosteatosis [83]. Additionally, p53 directly enhances β-oxidation by transactivating acyl-CoA dehydrogenase family member 11 (Acad11), which is important for p53 pro-survival function when the cell encounters glucose starvation [84]. Similarly, the p53 target gene, Lpin1 (LPIN1), also promotes FAO and cell survival under conditions of nutritional stress [85]. Since tumor cell growth requires de novo lipogenesis, p53’s above lipogenesis-inhibitory, lipolysis and lipid oxidation-promoting roles all serve to limit tumor growth. However, p53-mediated FAO can be utilized as a pro-survival mechanism in response to nutrient stresses [84, 85]. In this case, p53 may provide important advantages with respect to tumor cell survival, as the tumor microenvironment (TME) is often nutrient-deficient. Indeed, in a brain cancer model, one study revealed that the tumor cells relied on FAO activated by the p53 target gene, carnitine palmitoyltransferase 1C (CPT1C), to resist hypoxia and glucose deprivation. Decreased expression of CPT1C ultimately delayed tumor growth [86].
However, p53 can stimulate the synthesis of some types of lipids (Figure 1 and Table 1). Sphingolipids are an important type of lipid within the cell. Diverse metabolites associated with sphingolipid metabolism serve to regulate cell survival or apoptosis. For example, ceramide and sphingosine mediate antiproliferative responses, including cell cycle arrest, senescence, and apoptosis, while sphingosine-1-phosphate (S1P) to prevent apoptosis and promote angiogenesis and metastasis [87]. p53 can upregulate the levels of ceramide and/or sphingosine via induction of ceramide synthase 6 (CERS6) and neutral sphingomyelinase 2 (nSMASE2) and suppression of sphingosine kinase 1 (SK1) transcription [88–90]. Inhibition of SK1 also results in decreased levels of S1P [90]. These effects contribute to p53-mediated tumor-suppressive functions. However, p53 can also downregulate ceramide expression via induction of alkaline ceramidase 2 (ACER2), which results in the upregulation of both sphingosine and S1P [91]. Expression of ACER2 may have a dual role with respect to cell fate. Specifically, low levels of p53 induce moderate expression of ACER2 and thus promote cell survival via increased S1P and decreased ceramide levels, respectively. By contrast, high levels of p53 lead to robust expression of ACER2; this results in cell death due to the accumulation of sphingosine. The chemotherapeutic agents, oxaliplatin and 5-fluorouracil activate p53 in colon cancer, which ultimately serves to transactivate CerS5, resulting in elevated C16:0-ceramide levels. High levels of C16:0-ceramide impair cancer cell sensitivity to chemotherapeutic agents due to activation of autophagy and mitochondrial respiration [92]. Besides sphingolipid, p53 synergizes with SIRT6 to stimulate the synthesis of cardiolipin (CL) via activation of CDP-diacylglycerol synthase 1 and 2 (CDS1 and CDS2) [93]. p53 also promotes the generation of ketone bodies via the upregulation of 3-hydroxymethyl-3-methylglutaryl-coA lyase like (Hmgcll1), which promotes cell survival under conditions of nutrient starvation [84]. In some situations, p53 can also inhibit FAO. For example, p53-mediated suppression of both PGC1α and apelin receptor (APLNR) signaling pathways in the myocardium reduces the rate of FAO, thereby revealing a critical, context-dependent function of p53 in FAO regulation [94].
Another important function for p53 is regulating cholesterol metabolism and the mevalonate (MVA) pathway (MVP) (Figure 1 and Table 1). p53 may promote cholesterol uptake by inducing the expression of LIM domain and actin binding 1 (LIMA1), a newly identified regulator of cholesterol absorption [95, 96]. p53 also facilitates cellular cholesterol efflux via caveolin 1 (CAV) [97]. By activating a series of target genes, p53 regulates bile acid synthesis and disposition, and prevents cholestasis [98, 99]. The MVP is critical for the biosynthesis of isoprenoids, including cholesterol, and is tightly linked to neoplasia [100]. p53 either promotes or inhibits the MVP in different settings. In liver cancer, p53 transactivates ATP binding cassette subfamily A member 1 (ABCA1) to block the maturation of SREBP-2, resulting in inhibition of the MVP [101]. This effect is important for p53-mediated suppression of liver tumorigenesis; p53-null mice may develop liver cancer due to increased activity of the MVP. However, in human glioblastoma, p53 activates a cohort of MVP associated genes to favor this pathway [102]. p53 mutant state may also influence its effect on the MVP. In pancreatic cancer, WT p53 inhibits the expression of sterol O-acyltransferase 1 (SOAT1), thereby suppressing the MVP, while mtp53 elevates its expression to enhance the MVP [103]. In breast cancer, mtp53 also boosts the MVP and promotes the progression of cancer via SREBP, resulting in a highly disorganized mammary tissue structure [104].
p53 also participates in lipid metabolism in other ways. By repressing the expression of stearoyl-CoA desaturase 1 (SCD1), p53 shifts mono-unsaturated phospholipids to more saturated phospholipid species. This activity represses the oncogenic AKT (also called protein kinase B, PKB) pathway and impedes tumor growth [105]. p53 elevates semaphorin 3E (Sema3E) transcription to promote adipose tissue inflammation, which is involved in insulin resistance and obesity [106, 107]. By contrast, p53 promotes thermogenesis and the differentiation of brown adipose tissue (BAT) via activation of PR/SET domain 16 (PRDM16) and elongation of very long chain fatty acids protein 3 (Elovl3), which has an anti-obesity benefit [108, 109]. MDM2, a target and major negative regulator of p53, was found to regulate the initiation of adipogenesis in a CREB-dependent manner [110].
Amino acid metabolism
Cancer cells have an increased demand for some amino acids, most notably glutamine and serine [111, 112]. As such, the restriction of certain types of amino acids may dramatically impede cancer growth. p53 is involved in several pathways that regulate amino acid metabolism (Figure 1 and Table 1). p53 controls the expression of several amino acid transporters [113–116]. When the concentration of glutamine in peripheral circulation becomes limiting, p53 activates solute carrier family 1 member 3 (SLC1A3) to increase aspartate import; this helps cancer cells to circumvent the glutamine shortage by using aspartate to maintain energy production and for glutamine/nucleotide biosynthesis [113]. Similarly, p53 also activates SLC7A3 to enhance arginine uptake during glutamine deprivation [114]. Upregulated levels of arginine promote mammalian target of rapamycin complex 1 (mTORC1)-dependent cell growth. Taken together, these studies reveal p53-mediated pro-survival mechanisms that emerge in the setting of nutrient stress in both neoplastic and non-neoplastic cells. By contrast, p53 represses the expression of the cystine/glutamate transporter, SLC7A11, which reduces the intracellular concentration of the amino acid, cysteine [115, 116]. This will lower the antioxidant glutathione (GSH) biosynthesis from cysteine and confer cell enhanced susceptibility to ferroptosis (see below p53 regulates ferroptosis section). The transsulfuration pathway is critical for de novo cysteine biosynthesis [117]. Cystathionine β-synthase (CBS) catalyzes the conversion of homocysteine to cystathionine to promote the transsulfuration pathway and ferroptosis resistance [118]. p53 inhibits the expression of CBS via a p53/ELAVL1/linc00336/miR-6852/CBS axis to sensitize cell to ferroptosis (see below p53 regulates ferroptosis section) [119]. Upon serine starvation, p53 enhances its transactivation of p21 to cause cell cycle arrest [120]. This response redirects serine from nucleotide production toward GSH biosynthesis pathways in order to combat ROS, thereby promoting cancer cell survival. Similarly, the p53-p21 axis may also protect cancer cells from glutamine starvation [121]. The p53 target gene, MDM2, binds directly to activating transcription factor 3 and 4 (ATF3 and ATF4) under conditions of ROS-mediated stress to activate the serine synthesis pathway (SSP) [122]. However, in another study, p53 could inhibit de novo serine biosynthesis by suppressing phosphoglycerate dehydrogenase (PHGDH), a key enzyme in the SSP, thereby inducing cellular apoptosis [123]. In response to genotoxic stress, p53 activates argininosuccinate synthase 1 (ASS1) to increase arginine level, which suppresses AKT activation and protects the cells from genotoxicity-caused apoptosis [124]. As discussed above, p53 induces GLS2 to promote the conversion of glutamine to glutamate, thereby fueling the TCA cycle [52]. Interestingly, p53 is activated by protein phosphatase 2A (PP2A) to support cell survival under conditions of glutamine starvation [121]. p53 also mediates proline oxidation via upregulation of proline dehydrogenase 1 (PRODH; or p53-induced gene 6, PIG6), a proline oxidase that is required for the production of ROS and is a critical factor underlying cellular apoptosis [125]. Another p53 target, the enzyme aldehyde dehydrogenase 4 family member A1 (ALDH4A1 or ALDH4), catalyzes the conversion of proline to glutamate [126]. Recently, p53 was found to repress asparagine synthesis via transcriptional suppression of asparagine synthetase (ASNS); this led to the inhibition of lymphoma and colon cancer growth [127]. Interestingly, asparagine and aspartate can differentially regulate p53 activity by binding with liver kinase B1 (LKB1). Asparagine inhibits, but aspartate promotes p53 activity via the LKB1/AMPK/p53 regulatory axis.
Cachexia is a symptom correlating with poor prognosis in cancer patients [128]. One critical hallmark of cachexia is muscle wasting characterized by imbalance of proteolysis and protein synthesis. Toward this end, p53 transactivates paternally expressed gene 3 (PEG3, or PW1) to block myogenesis, thereby amplifying cachexia in response to the tumor load [129]. Ammonia metabolism (including polyamine metabolism and ureagenesis) is linked to amino acid metabolism. As such, p53 influences polyamine metabolism by inducing spermidine/spermine N1-acetyltransferase 1 (SAT1), thereby contributing to p53-mediated ferroptosis (see below p53 regulates ferroptosis section) [130]. p53 also suppresses ureagenesis and the elimination of ammonia via inhibition of urea cycle genes, including carbamoylphosphate synthase 1 (CPS1), ornithine carbamoyltransferase (OTC), and arginase 1 (ARG1), which slows down cell growth and results in tumor suppression [131]. On the other hand, p53 promotes uric acid uptake into the cell by inducing its transporter SLC2A9 (GLUT9) to reduce ROS production to inhibit tumorigenesis [132].
Nucleotide metabolism
In support of rapid proliferation, cancer cell enhances the rate of nucleotide production. p53-mediated cell cycle arrest results in reduced demand for this process. Meanwhile, p53 can also directly or indirectly limit nucleotide biogenesis (Figure 1 and Table 1). p53 inhibits the synthesis of dTTP and GMP by repressing deoxyuridine triphosphatase (dUTPase) and guanine monophosphate synthase (GMPS), respectively [133, 134]. Noteworthily, upon genotoxic stress, GMPS can strengthen ubiquitin specific protease 7 (USP7)-mediated p53 stabilization [135]. p53 also indirectly suppresses GTP production via the induction of miR-34a, a miRNA that disrupts the translation of inosine monophosphate dehydrogenase (IMPDH), a critical enzyme in GTP biosynthesis [136]. Through inhibition of mTORC1, p53 suppresses the expression of ribonucleotide reductase subunit 1 (RRM1) and 2 (RRM2), which leads to the diminished generation of all dNTPs [137]. These p53-mediated inhibitory roles and their impact on nucleotide synthesis impair the mitotic process in cancer cells. However, p53 can also promote nucleotide production, thereby facilitating the repair of damaged DNA and genome stability (Figure 1 and Table 1). The PPP and one-carbon cycle that generate biomolecules (such as R-5-P, purines, and pyrimidines) for nucleotide synthesis are both modulated by the actions of p53. Moreover, p53 activates p53R2 (RRM2B) to enhance the ribonucleotide reductase activity resulting from DNA damage [138, 139]. Additionally, p53 also maintains the integrity of the mitochondrial genome, as discussed above [61–64]. These results suggest that p53 regulates nucleotide production in a context-dependent fashion. Additionally, p53 may have a more global impact on nucleotide synthesis by regulating Myc, the master regulator of nucleotide metabolism [140–143]. It is also important to note that p53 induces the expression of membrane adenosine receptor adenosine A2b receptor (ADORA2B) to facilitate monitoring of extracellular adenosine levels [144]. Elevated levels of extracellular adenosine can be detected in the immediate microenvironments of numerous solid tumors. Ligand-engaged ADORA2B can activate the downstream apoptosis pathway, thereby contributing to p53-mediated tumor suppression. An interesting fact is, ATP/ADP can directly regulate binding interactions between p53 and its DNA targets[145] or indirectly influence p53 stabilization and activity via AMP-activated protein kinase (AMPK) or the mechanistic target of rapamycin (mTOR) pathways (see below Crosstalk between p53 and metabolic sensors section). Moreover, when pyrimidine biosynthesis is suppressed by inhibition of dihydroorotate dehydrogenase (DHODH), p53 is activated to induce cell apoptosis [146].
Iron metabolism
Iron is a necessary mineral for cell survival, most notably for cancer cells [147]. Cancer cells often reprogram iron metabolism pathways to facilitate the accumulation of cellular iron stores to promote cell growth and metastasis. p53 regulates iron metabolism mainly via its capacity to reduce the iron levels maintained in cells (Figure 1 and Table 1). p53 inhibits iron uptake via post-transcriptional suppression of the iron transporters transferrin receptor 1 (TFR1) and Zrt- and Irt-like protein 14 (ZIP14) [148, 149]. It is worth noting that p53-mediated inhibition of ZIP14 also influences the transport of other metal ions, including manganese, zinc, and cadmium [150]. By contrast, p53 can also activate hepcidin (or hepcidin antimicrobial peptide, HAMP) which serves to sequester iron within the reticuloendothelial macrophages, leading to decreased iron levels in plasma [151]. Both p53-mediated actions limit iron availability in cancer cells, thereby limiting cancer cell proliferation. However, p53 also upregulates TFR1 in acute ischemic stroke (AIS) patients via a p53/lncRNA PVT1/miR-214/TFR1 axis [152]. This results in amplified iron import and ferroptosis (see below p53 regulates ferroptosis section). Noticeably, miR-214 can inhibit the expression of p53; as such, this axis represents a reciprocally-regulating feedback loop.
Not all intracellular iron is available for use. Most intracellular iron is chelated to one or more storage proteins, like iron–sulfur clusters (ISC) and ferritin (Figure 1 and Table 1). p53 transactivates iron-sulfur cluster assembly enzyme (ISCU), frataxin (FXN), and ferredoxin reductase (FDXR) to promote the biosynthesis of ISCs as well as post-transcriptional induction of ferritin formation [148, 153–157]. These actions reduce the level of available iron and can result in cell cycle arrest and inhibition of cancer cell growth. In hepatic stellate cells, treatment with inducers of ferroptosis results in activation of bromodomain-containing 7 (BRD7); BRD7 binds p53 and facilitates its translocation to the mitochondria, where it forms a complex with SLC25A28 to cause abnormal accumulation of redox-active iron and cell ferroptosis (see below p53 regulates ferroptosis section) [158]. Interestingly, p53 expression and activity are also influenced by the cellular iron level and also by some p53 targets that are associated with iron turnover pathways [159]. Treatment with iron chelators results in a decrease in the available stores of intracellular iron; this promotes HIF1α-mediated p53 activation and cell cycle arrest [160–162]. By contrast, excess iron results in heme-dependent downregulation of p53 [163]. However, in macrophage, iron overload will activate p53 to induce macrophage M1 polarization [164]. Moreover, p53 target genes FDXR and FXN both regulate p53 expression, but in different ways [165, 166]. Taken together, these feedback mechanisms reveal the delicate balance maintained between p53 and intracellular iron levels.
ROS control
Reactive oxygen species (ROS) production is inevitable during cell life cycles, which has both beneficial and harmful effects on cell survival. Moderate level of ROS promotes intracellular signaling and cell proliferation, and generates inflammation as a host defense response to pathogens. However, excess ROS may induce DNA damage, genome instability, and cell death [167]. ROS also exhibits dual (promotive or suppressive) roles in cancer development [168]. Intracellular ROS are mainly generated by mitochondrial activities and a batch of metabolic enzymes, including nitric oxide synthases (NOSs), arachidonate lipoxygenases (ALOXs), NADPH oxidases (NOXs), cyclooxygenases (COXs), and cytochrome P450 family (CYPs) [169]. Correspondingly, the cell has developed a multitude of anti-oxidant mechanisms that serve to limit the intracellular accumulation of ROS [170]. The dysregulation of either ROS production system or the anti-ROS system will cause a deregulated ROS level. The strict control of ROS level (redox metabolism or ROS metabolism) not only benefits cellular homeostasis, but also protects the host organism from a diverse set of disorders, most notably cancer. p53 functions to lower or enhance ROS levels according to distinct cellular contexts (Figure 2 and Table 1).
Figure 2. p53 regulates ROS metabolism.
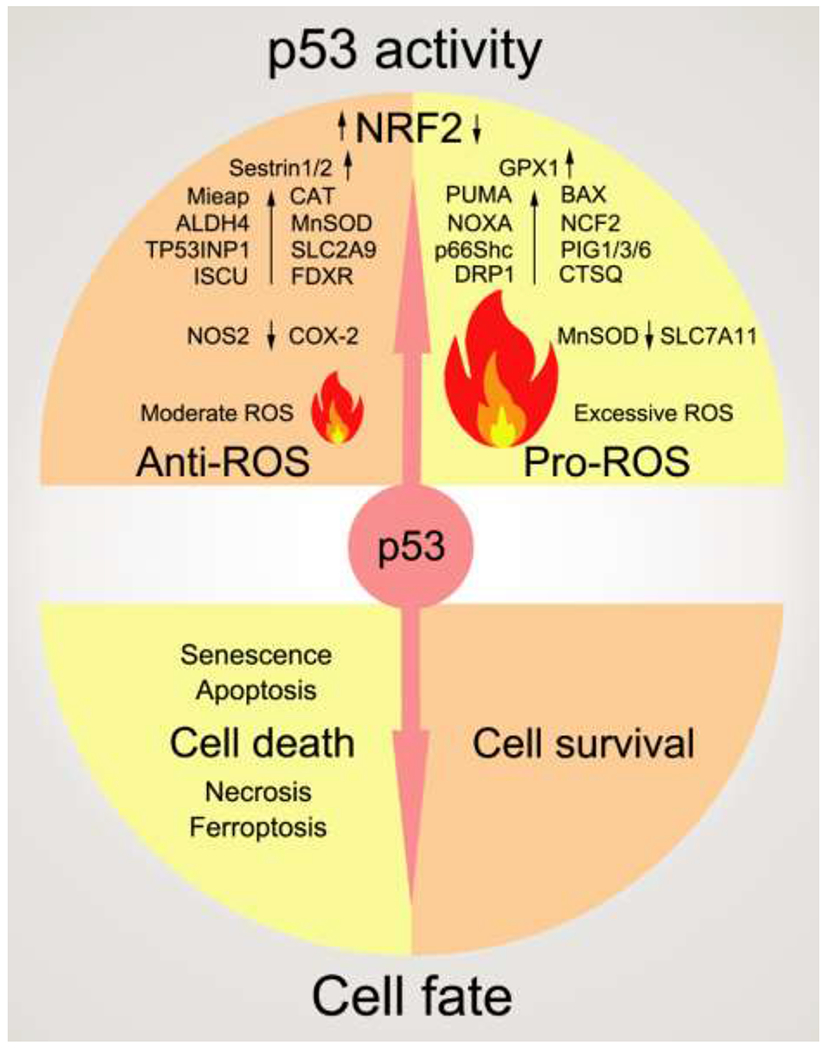
In different situations, p53 either enhances or lowers the ROS level, which may cause different cell fates (cell death or survival). Major target genes regulated by p53 in ROS control are shown in this figure. For the full names of them, please refer to Table 1. Black arrows pointing up indicate a positive effect of p53 on this protein. Oppositely, black arrows pointing down indicate a negative effect of p53 on this protein.
Nuclear factor, erythroid 2 like 2 (NFE2L2, also called NRF2) is a master TF that regulates redox metabolism via the transactivation of a cohort of antioxidant proteins [171]. p53 stabilizes NRF2 by activating p21 and Sestrins (Sestrin1 and Sestrin2, or SESN1/2), which block NRF2 major negative regulator Kelch-like ECH-associated protein 1 (Keap1) to degrade NRF2 [172–175]. However, p53 may also suppress NRF2 transcription by blocking the binding of the TF, Sp1, to the Nrf2 promoter [176]. By contrast, NRF2 suppresses p53 expression and activity via direct induction of MDM2 or inhibition of thioredoxin (TXN) interacting protein (TXNIP), which protects p53 from both ubiquitin-dependent and ubiquitin-independent degradation [177–182]. These results provide only a sense of the complexity surrounding the roles of p53 in the regulation of ROS [183]. PML nuclear body scaffold (PML), a direct target of p53, acts as a ROS sensor to activate p53 upon oxidative stress [184–186]. p53 directly reduces ROS levels by suppressing COX-2 and NOS2; both are enzymes that can stimulate ROS production [187]. Nevertheless, findings from one report indicate that p53 can also activate COX-2 [188]. In the setting of myocardial infarction, p53 is recruited to the promoter and induces expression of the enzyme, NOS3; this serves to protect cardiac cells from undergoing apoptosis [189]. However, endothelial p53 is reported to inhibit NOS3 and GLUT1 to protect organism from dietary obesity [21]. In human lung and breast epithelial cells, WT p53 suppresses TGF-β-mediated NADPH oxidase 4 (NOX4) expression to lower ROS level and cell metastasis. Interestingly, mtp53 has opposite effect as WT p53 in the same setting to stimulate ROS production and cancer cell metastasis [190]. As discussed previously, p53 target gene, TIGAR, reduces ROS level by inhibiting PFK1 or promoting HK2 [31, 40]. p53-activated mitochondria-eating protein (Mieap) helps to maintain mitochondrial health and stability, thereby limiting the production of ROS [66]. p53 can also activate various other antioxidant proteins, including ISCU, FDXR, peroxiredoxins, catalase, glutathione peroxidase 1 (GPX1), manganese superoxide dismutase (MnSOD), and heme oxygenase-1 (HO-1, or HMOX1) [153, 156, 191–194], which may logically reduce ROS levels. However, imbalanced induction of catalase, GPX1, and MnSOD may also serve to increase ROS and lead to cell apoptosis [192]. On the other hand, p53 also has a negative influence on the activity or expression of catalase and MnSOD [195–198]. Under homeostatic conditions, p53-activated p53R2 binds to catalase and enhances its capacity to combat ROS. However, p53 can also induce the expression of PIG3 to inhibit catalase activity and cause cell apoptosis under oxidative state [195]. Treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA) promotes p53-mediated suppression of MnSOD via the actions of the TF, Spl. TPA also promotes translocation of p53 to the mitochondria where it can bind to and repress MnSOD activity, thereby inhibiting its anti-ROS activity and promoting apoptosis [196, 197]. Similarly, p53 also destabilizes HO-1 protein in embryonic stem cells [198]. Moreover, in fibroblasts, HO-1 inhibits p53 to promote reprogramming [199]. These results suggest a complex interplay between p53 and HO-1 in different settings. As we introduced, p53-mediated serine synthesis (by inducing MDM2), proline degradation (via activating ALDH4), and uric acid uptake (through increasing SLC2A9) all serve to inhibit the effects of ROS [122, 126, 132]. Another p53 target gene, TP53-inducible nuclear protein 1 (TP53INP1), also suppresses ROS [200]. TP53INP1-deficient mice have a higher incidence of lymphoma associated with oxidative stress and exhibit shorter survival times.
Since excess ROS is harmful to cell survival and may result in cancer, p53 can switch from reducing ROS level to promote it when ROS stress is too acute (Figure 2 and Table 1). This facilitates p53-mediated pro-apoptotic functions that are critical components of its anti-cancer activity. In fact, PUMA, BCL2-associated X protein (BAX), and phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, or NOXA)—three major pro-apoptosis target genes of p53—execute their functions partly by triggering ROS generation [201–207]. Neutrophil cytosolic factor 2 (NCF2/p67phox) is the cytosolic subunit of the NADPH oxidase enzyme complex. p53 binds to the promoter of NCF2, thereby activating gene transcription and amplifying NOX2-generated ROS [208]. Likewise, the p53 target protein, SHC adaptor protein 1 (SHC1, or p66Shc), cooperates with p53 to upregulate cellular ROS and oxidant-mediated DNA damage. p66Shc gene-deleted mice exhibit enhanced resistance to oxidative stress and a longer life span [209]. Some p53-induced genes (PIGs) also assist p53 to increase ROS [210]. In urothelial cancer, PIG1 sensitizes cancer cells to cis-diamminedichloroplatinum (CDDP) by the accumulation of ROS [211]. PIG3, as we introduced before, binds to inhibit catalase, thereby enhancing ROS upon genotoxic stress [195]. In p53 WT cells, PIG6 catalyzes proline oxidation to augment ROS levels [125]. p53 also antagonizes the function of NRF2 via suppression of a cohort of its target genes, including xCT (SLC7A11), NAD(P)H quinone dehydrogenase 1 (NQO1), and glutathione S-transferase α1 (GST-α1), ultimately resulting in the accumulation of ROS [115]. Specially, p53-mediated suppression of SLC7A11 reduces the levels of intracellular GSH, thereby rendering cells more susceptible to ferroptosis [116] (see below p53 regulates ferroptosis section). Mitochondria localized p53 can promote ROS production in transcription-independent ways [212, 213]. In the case of Huntington’s disease, dynamin 1 like (DNM1L, Drp1) binds p53 to translocate it to mitochondria, where p53 promotes Drp1-mediated mitochondrial fragmentation and dysfunction [212]. This process results in ROS accumulation and neuronal cell death. Similarly, mitochondrial p53 binds directly to prohibitin 1 (PHB1) to release OPA1 mitochondrial dynamin like GTPase (Opa1) in cisplatin-sensitive gynecologic cancers. The mitochondrial metallopeptidase, Oma1, mediates the processing of L-Opa1 and induction of mitochondrial fragmentation, increased levels of ROS, and cancer cell apoptosis [213]. More directly, mitochondrial p53 can bind directly to cyclophilin D (CypD) to open mitochondrial permeability transition pore (PTP), which dramatically generates ROS to triggers cellular necrosis [214]. Of note, p53-activated cathepsin Q (CTSQ) can also cooperate with ROS to induce necrosis in response to DNA damage [215]. p53 also has manifold functions in regulating cell membrane lipid ROS and associated ferroptotic cell death (see below p53 regulates ferroptosis section). It deserves to be mentioned that ROS can modify all ten cysteines within the p53 protein, thereby influencing its structure and function [216]. GSH-mediated S-glutathionylation of p53 cysteine residues can also have an impact on its function [216].
Taken together, under different cellular contexts, p53 reduces ROS to promote cell survival or facilitates ROS generation to cause cell death to avoid more serious cell damages (Figure 2 and Table 1). Noteworthily, p53 has a reciprocal and complicated relationship with hypoxia and HIF pathway, about which we recommend two reviews for your references [217, 218].
p53 regulates ferroptosis
Ferroptosis, a newly identified regulated cell death (RCD) type, was first reported in 2012 [219]. Burgeoning researches in this field have revealed the potential roles of ferroptosis in development, immune system regulation, ischemia-reperfusion injury, and tumor suppression [220, 221]. In 2018, the Nomenclature Committee on Cell Death (NCCD) defined ferroptosis as “a form of RCD initiated by oxidative perturbations of the intracellular microenvironment that is under constitutive control by GPX4 and can be inhibited by iron chelators and lipophilic antioxidants” [222]. The three core components of ferroptosis are iron, lipid, and ROS. Metabolic dysregulation of any one of them may influence ferroptotic cell death. Since p53 participates in the regulation of the metabolism of all three of these elements, it is reasonable to hypothesize that p53 may play a critical role in modulating ferroptosis (Figure 3 and Table 1).
Figure 3. p53 regulates ferroptosis.
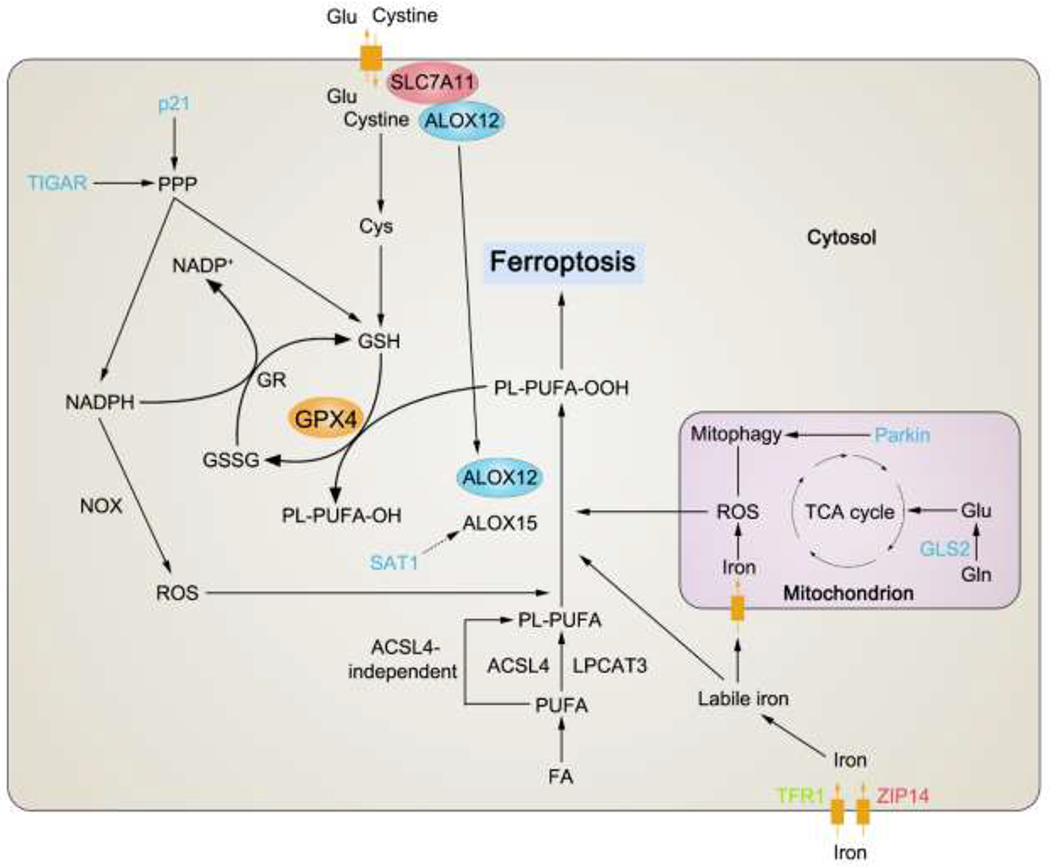
p53 mediates (either promote or inhibit) ferroptosis via distinct mechanisms. Major target genes regulated by p53 in ferroptosis modulation are shown in this figure. For the full names of them, please refer to Table 1. Those proteins that are modulated positively by p53 (directly or indirectly), like promoting their expressions or activities, are depicted in blue. Oppositely, those proteins that are modulated negatively by p53 (directly or indirectly), like suppressing their expressions or activities, are depicted in red. If some protein can either be promoted or suppressed by p53 in different circumstances, it is depicted in green. Black arrows indicate positive effects. Black perpendicular bars indicate negative effects. ALOX12, arachidonate 12-lipoxygenase, 12S type; ALOX15, arachidonate 15-lipoxygenase; GPX4, glutathione peroxidase 4; GR, glutathione reductase; GSSG, oxidized glutathione; FA, fatty acid; PUFA, polyunsaturated fatty acid; ACSL4, acyl-CoA synthetase long chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase-3; PL-PUFA, polyunsaturated fatty acid-containing phospholipid; PL-PUFA-OOH, polyunsaturated fatty acid-containing phospholipid hydroperoxides; PL-PUFA-OH, polyunsaturated fatty acid-containing phospholipid alcohol.
The first evidence documenting a role for p53 in the regulation of ferroptosis was published in 2015 [116]. In this study, p53 was shown to promote ferroptosis via its capacity to inhibit the import of cystine into target cells. Mechanistically, p53 was found to suppress the transcription of SLC7A11, which is a core subunit of the cystine/glutamate antiporter, xCT. Cysteine (cystine is its oxidized dimeric form) is the necessary material for the biosynthesis of GSH, an antioxidant used by GPX4 to inhibit ferroptosis [223]. Suppression of SLC7A11 by p53 reduces the intracellular levels of GSH and sensitizes cells to ferroptosis. Interestingly, the p533KR mutant form of the protein described earlier retains the capacity to repress SLC7A11 and as such, it has the capacity to induce ferroptosis [116]. However, p534KR (3K→R as discussed above, with the addition of a K98R mutation) and an African-specific human SNP, p53(P47S), are unable to promote ferroptosis to inhibit tumor growth [224–227]. These results highlight the contribution of p53-mediated regulation of ferroptosis to its tumor suppression function. Monoubiquitination of lysine (K) 120 of histone H2B (H2Bub1) identified at the SLC7A11 promoter epigenetically activates its transcription. p53 can also silence SLC7A11 by recruiting USP7 to its promoter, thereby promoting H2Bub1 deubiquitination [228]. p53 mediated SLC7A11 suppression can also function independently of cystine transport. The lipoxygenase, ALOX12, oxidizes membrane polyunsaturated fatty acids (PUFAs) to induce ferroptosis. SLC7A11 can bind to and sequester ALOX12, thereby impairing its enzymatic activity [229]. Reduction of SLC7A11 mediated by p53 promotes the release of ALOX12 to exert its pro-ferroptosis function. ALOX15 is another member of the ALOX family that mediates ferroptosis [230]. p53 can enhance ALOX15 expression by activating SAT1 [130]. Furthermore, p53-mediated inhibition of SSP resulting from the suppression of PHGDH may also serve to limit GSH generation and may promote ferroptotic cell death [123]. Glutaminolysis also plays a role in supporting ferroptosis [231]. p53 activates glutaminolysis by inducing GLS2, thereby amplifying ferroptosis [51, 52]. p53 also activates ferroptosis via its capacity to modulate iron metabolism. The p53 target gene, FXDR, modulates ISL3- and erastin-(two typical small molecules for inducing ferroptosis) induced ferroptosis via its impact on available intracellular iron levels [165]. As discussed above, p53 also influences intracellular iron levels via its interactions with SLC25A28 or by activating lncRNA PVT1 to trigger ferroptosis [152, 158]. Additionally, p53 also regulates ferroptosis markers prostaglandin-endoperoxide synthase 2 (PTGS2, or COX-2) and CBS via the Ras/Raf/ERK cascade or the p53/ELAVL1/linc00336/miR-6852/CBS axis, respectively [119, 188].
Under certain contexts, p53 can also protect cells from undergoing ferroptosis (Figure 3 and Table 1). As noted earlier, p53-activated p21 permits cells to adapt to nutrient deprivation by preserving GSH to defend against ROS-mediated damage [120]; this pathway may also serve to help the cell survive ferroptosis since GSH can be used by GPX4 to inhibit ferroptosis. A group depleted cellular cysteine by erastin-2 to induce ferroptosis [232]. They observed that nutlin-3 stabilized p53 could delay the onset of ferroptosis. This protective role of p53 was again due to its activation of p21 to preserve GSH level. In colorectal cancer, p53 binds to relocate dipeptidyl peptidase 4 (DPP4) in the nucleus, which disrupts the DPP4-NOX1 complex, thereby hindering membrane lipid peroxidation and ferroptosis [233]. Mitochondrial activity is critical for the induction of ferroptosis [234]. The p53 target, Parkin, inhibits erastin-induced ferroptosis via its role in promoting mitophagy [35, 36, 49, 234].
In summary, despite several notable exceptions, p53 is generally involved in metabolic regulatory functions that promote cellular ferroptosis. This property may be among its most definitive weapons against cancer.
p53 regulates autophagy
Autophagy, from an etymologic point of view, means “self-eating” [235]. Indeed, autophagy is a cellular catabolic process to degrade proteins, organelles, and membranes for reuse in response to metabolic stresses. Autophagy is actually an ensemble of many types of cargo-selective degradation processes [236]. The roles of autophagy in the setting of cancer remain complex. While autophagy can support cancer development by providing energy and building blocks that support survival and proliferation, and by limiting the extent of tumor necrosis and inflammation, autophagic elimination of damaged organelles and proteins may also serve to inhibit cancer initiation and development. Excessive autophagy may result in death of cancer cell [237, 238].
Nuclear p53 typically promotes autophagy (Figure 4 and Table 1). In 2013, a group used high-throughput sequencing (HTS) to systematically study global p53 transcriptional networks upon DNA damage [239]. They identified a suite of autophagy-associated genes, including autophagy related 2B/4A/4C/7/10 (ATG2B/4A/4C/7/10), Unc-51 like autophagy activating kinase 1 and 2 (ULK1 and ULK2), UV radiation resistance associated (UVRAG), and vacuole membrane protein 1 (VMP1), that were regulated by p53. The authors concluded that p53-activated autophagy had no specific impact on cell cycle arrest, but instead, it synergized with p53-induced apoptosis and tumor-suppression in response to DNA damage [240]. For example, treatment with either camptothecin (CPT) or etoposide (Eto) resulted in DNA damage accompanied by autophagy that was mediated by p53-induced upregulation of ULK1 and ULK2 [241]. This autophagy process contributes to CPT- and Eto-caused cell death. It is worth noting that ATG7 has a feedback role in regulating p53 target selection during nutrient withdrawal [242]. By binding to p53, ATG7 promotes the expression of the cell cycle arrest regulator, p21, but not any of the pro-apoptosis targets; this is similar to the impact of PGC1α in this setting [71]. The p53 target, damage-regulated autophagy modulator (DRAM), is also activated in response to DNA damage [243]. DRAM downregulation was observed in a panel of human cancer cells. Interestingly, while induction of DRAM alone has only minimal impact on cell death, this response can enhance p53-mediated apoptosis by inducing autophagy. Cathepsin D (CTSD) promotes autophagy in different cell types [244, 245]. p53 can activate cathepsin D to induce autophagy, thereby contributing to p53-mediated tumor suppression and chemosensitivity [246, 247]. In addition to these targets, p53 also activates interferon stimulated exonuclease gene 20kDa-like 1 (ISG20L1), death associated protein kinase 1 (Dapk1), EI24 autophagy associated transmembrane protein (EI24, or PIG8), and transglutaminase 2 (TGM2) to facilitate autophagy and tumor suppression [248–251]. Additionally, spermine was demonstrated to activate p53 by increasing activity of p-p53 and acetyl-p53, causing induction of autophagy in HT1080 cells [252]. Mitophagy, a type of selective autophagy that eliminates mitochondria, is also modulated by the actions of p53. In radioresistant cancer cells, p53 raises BCL2 interacting protein 3 (BNIP3) levels to induce mitophagy, thereby clearing the abnormal mitochondria to maintain OXPHOS and inhibiting glycolysis [253]. However, in hypoxia stress, p53 switches to suppress BNIP3 to protect the cells from BNIP3-caused autophagic cell death [254]. Nevertheless, p53 activates BCL2 interacting protein 3 like (BNIP3L) under hypoxia or anticancer drug KP46 treatment to initiate mitophagy to suppress cancer [255, 256]. Additionally, Parkin, a p53 target gene, modulates mitophagy to avoid ferroptosis [234]. Nuclear p53 also has an autophagy-inhibitory function. Cytoplasmic high mobility group box 1 (HMGB1) binds to Beclin 1 to enhance autophagic flux [257]. Nuclear p53 binds directly to HMGB1 and sequesters it in the nucleus, thereby limiting its capacity to induce autophagy [258]. Reciprocally, HMGB1 also sequesters p53 in the nucleus and thus weakens cytoplasmic p53-mediated apoptosis.
Figure 4. p53 regulates autophagy.
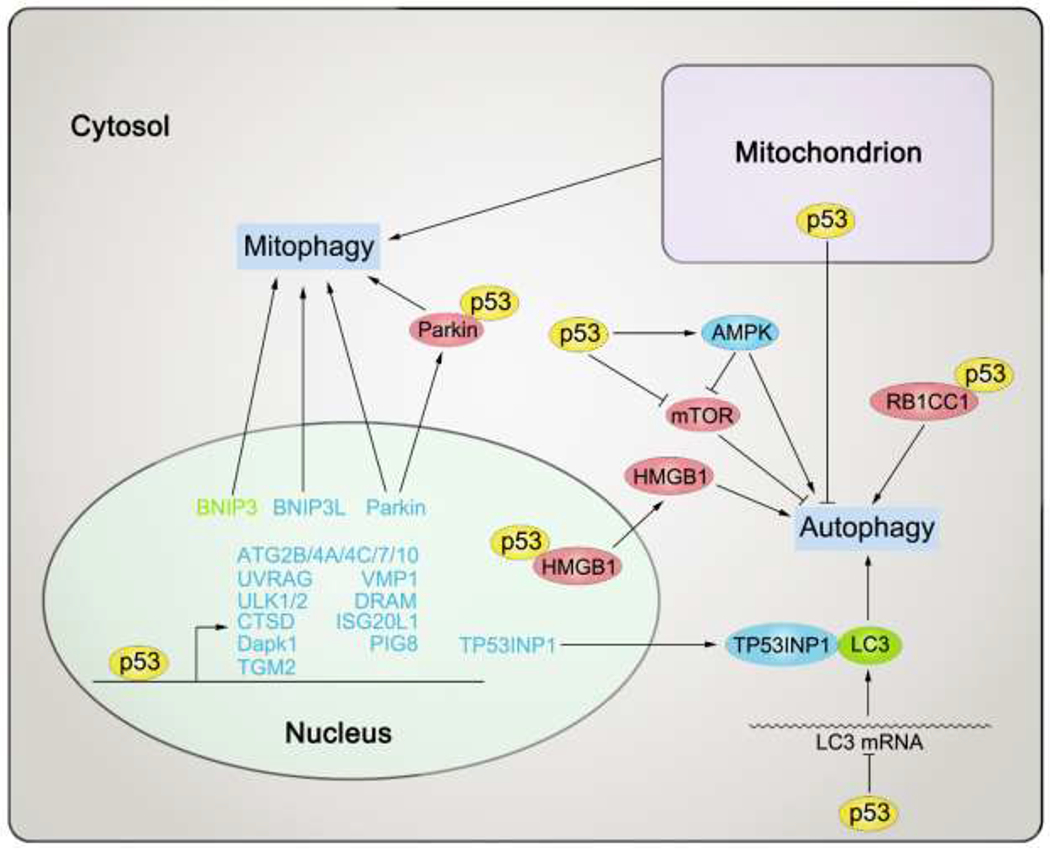
Nuclear and cytoplasmic p53 both regulate autophagy. Major target genes regulated by p53 in autophagy modulation are shown in this figure. For the full names of them, please refer to Table 1. Those proteins that are modulated positively by p53 (directly or indirectly), like promoting their expressions or activities, are depicted in blue. Oppositely, those proteins that are modulated negatively by p53 (directly or indirectly), like suppressing their expressions or activities, are depicted in red. If some protein can either be promoted or suppressed by p53 in different circumstances, it is depicted in green. Black arrows indicate positive effects. Black perpendicular bars indicate negative effects.
By contrast, cytoplasmic and/or mitochondrial p53 can often suppress autophagy [4] (Figure 4 and Table 1). In mouse heart and pancreatic islet β cells, cytosolic p53 binds directly to Parkin, thereby impairing Parkin-mediated mitophagy. In type I diabetes, inhibition of p53 restores mitochondria function and insulin secretion in β cell [259, 260]. Microtubule associated protein 1 light chain 3 alpha (MAP1LC3A, or LC3) is a positive regulator of autophagosome formation and is also an autophagy marker [261]. In colorectal cancer cell line, HCT116, prolonged nutrient starvation treatment results in post-transcriptional downregulation of LC3 mediated by cytoplasmic p53, thereby reducing the rate of cellular autophagy [262]. Although the extent of autophagy is decreased by p53, a limited but sustained autophagic flux is beneficial for cancer cells for survival in the setting of chronic starvation. However, results from another study revealed that the p53 target, TP53INP1, binds to both LC3 and ATG8 family proteins to promote autophagic cell death and tumor suppression [263]. Cytoplasmic p53 also interacts with RB1 inducible coiled-coil 1 (RB1CC1, or FIP200) to inhibit autophagy [264]. A K382R mtp53 loses the capacity for this interaction and thus the ability to suppress autophagy.
p53 can also affect autophagy by regulating AMPK and mTOR pathways, which are both master regulators of autophagy [265–267] (see below crosstalk between p53 and metabolic sensors section). In summary, p53 mainly promotes autophagy, resulting in cell death and tumor inhibition. However, in some settings, the actions of p53 serve to inhibit autophagy and promote cell survival.
Crosstalk between p53 and metabolic sensors
Accurate perception of the intracellular nutrient, metabolite, and energy status is critical for appropriate regulation of cell metabolism. Two well-defined central metabolic sensors are AMPK and mTOR [268]. AMPK mainly senses the glucose and energy state and can be activated by a decrease in the ATP/ADP ratio to induce catabolism while suppressing anabolism to produce energy [267, 268]. By contrast, mTOR is activated in environments enriched in nutrients and energy, including growth factors, a high ATP/ADP ratio, oxygen, and certain amino acids [268, 269]. Activation of mTOR promotes anabolism and represses catabolism, and as such, it antagonizes the actions of AMPK. Another important signaling pathway in metabolism is the PI3K-AKT signaling axis, which not only bridges upstream growth factor signals to mTOR, but also has multiple roles in metabolic sensing and regulation that are independent of mTOR [270]. p53 regulates metabolism via complex crosstalk mechanisms involving AMPK, AKT, and mTOR pathways (Figure 5 and Table 1).
Figure 5. Crosstalk between p53 and major metabolic sensors.
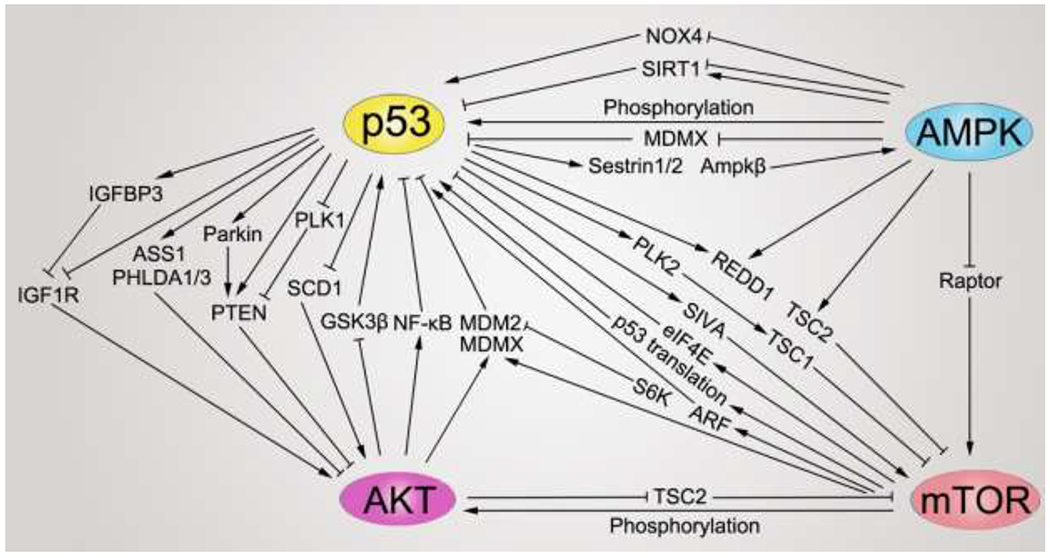
p53 has complicated interactions with major metabolic sensors: AMPK, AKT, and mTOR. Main mechanisms by which p53 interplays with AMPK, AKT, and mTOR are shown in this figure. For the full names of the p53 target genes, please refer to Table 1. Black arrows indicate positive effects. Black perpendicular bars indicate negative effects. NOX4, NADPH oxidase 4; SIRT1, sirtuin 1; MDMX, MDM4 regulator of p53; Raptor, regulatory associated protein of mTOR complex 1; eIF4E, eukaryotic translation initiation factor 4E; ARF, also called CDKN2A, cyclin dependent kinase inhibitor 2A; S6K, ribosomal protein S6 kinase B1; GSK3β, glycogen synthase kinase 3 β; NF-κB, nuclear factor κB subunit 1.
p53 directly transactivates AMPK subunit, AMPKβ1/2, to promote the AMPK pathway [271]. The kinase, LKB1, is an upstream activator of AMPK. p53 binds to the LKB1 promoter to induce its expression which indirectly results in AMPK activation [272]. p53 also controls LKB1/AMPK signaling via regulation of aspartate-asparagine homeostasis [127]. Similarly, AMPK activator Sestrins are also p53 targets [273]. Induction of Sestrin1/2 by p53 activates AMPK but suppresses mTOR. Insulin like growth factor 1 (IGF1) is an extracellular signaling molecule that binds to the IGF1 receptor (IGF1R) and also activates the AKT pathway [274]. p53 suppresses IGF1R transcription to block IGF1/AKT signaling [275, 276]. IGF binding proteins (IGFBPs) interact with and inhibit the actions of IGF1, and induction of IGFBP3 by p53 inhibits IGF1/AKT signaling and cell growth [277]. Interestingly, IGFBP1 is also a p53 target, although it exerts pro-survival function in hepatic cells by binding and deactivating BCL2 antagonist/killer 1 (BAK) to protect cells from apoptosis [278]. By repressing SCD1, p53 alters the levels of phosphatidylinositol phosphates (PIPs) in the cytoplasmic membrane, which attenuates the activation of AKT [105]. Phosphatase and tensin homolog (PTEN) is another important tumor suppressor that dephosphorylates PI(3,4,5)P3 to inactivate AKT. p53-mediated activation of PTEN expression results in suppression of the AKT/mTOR pathway [271, 279]. p53 target, Parkin, maintains mitochondrial function to lower ROS level, which protects PTEN from AMPK-mediated S-nitrosylation and degradation [280]. Polo-like kinase 1 (PLK1) is an activator of AKT by phosphorylating PTEN to abrogate its inhibitory effect on the AKT pathway. p53 suppresses PLK1 transcription, thereby supporting PTEN-mediated inhibition of AKT [281–283]. By contrast, PLK2 is activated by p53 and binds to tuberous sclerosis complex 1 and 2 (TSC 1 and TSC2), thereby enhancing the inhibition of mTOR [284, 285]. Meanwhile, TSC2 itself is a p53 target [271]. ASS1 is an intrinsic AKT repressor that functions by inhibiting AKT phosphorylation and activation. ASS1 expression is directly induced by p53 in the setting of genotoxic stress [124]. Hypoxia has a repressive effect on mTOR that dependends on TSC1/2 complex and DNA damage inducible transcript 4 (DDIT4, or REDD1), which is also a p53 target gene [286, 287]. In a quite recent study, researchers identified mouse p53 K136 (corresponding to human p53 K139) acetylation is critical for p53-mediated REDD1 and Sestrin1/2 activation, and the following mTOR inhibition [288]. In contrast to results from the aforementioned p534KR variant, mouse p535KR (4K→R as discussed above, with the addition of a K136R mutation) cannot suppress mTOR pathway signaling and exhibits early development of tumors. This research indicates that mTOR repression by p53 can contribute independently to p53-mediated tumor suppression regardless of its impact on cell cycle arrest, senescence, apoptosis, and ferroptosis. Interestingly, REDD1 exerts feedback inhibition on mTORC1-dependent p53 translation [289]. Pleckstrin homology like domain family A member 1 and 3 (PHLDA1 and PHLDA3) compete with AKT to bind to its activator, PIP3, thereby impeding AKT activation. p53 induces the expression of PHLDA1/3 to repress AKT [290–292]. To sum up, major influences of p53 on metabolic sensors are activating AMPK, while inhibiting the AKT and mTOR pathway. However, one group has reported that the p53 target gene, SIVA1 apoptosis inducing factor (SIVA), can enhance mTOR activity specifically in non–small cell lung cancer (NSCLC) [293]. p53-activated SIVA amplifies p53-induced apoptosis. However, SIVA can also promote tumor growth via stimulation of the mTOR pathway and cancer metabolism; high levels of SIVA have been associated with poor outcome in NSCLC patients [293]. SIVA can also reversely decrease p53 stabilization by promoting p53/MDM2 interactions or cyclin dependent kinase inhibitor 2A (CDKN2A, or ARF, a p53 activator) degradation [294, 295].
AMPK, AKT, and mTOR can also regulate the level and activity of p53 (Figure 5). AMPK is activated in response to glucose deprivation and phosphorylates p53 to induce its activation [10]. Activated p53 results in cell cycle and metabolic arrest which promotes cell survival in response to stress. AMPK also phosphorylates MDM4 regulator of p53 (MDMX), thereby eliminating its capacity to inhibit p53 [296]. In liver cancer cells, AMPK promotes p53 acetylation and activation via inhibition of SIRT1, which deacetylates and inhibits p53 [297]. However, AMPK may also enhance SIRT1 activity via upregulation of cellular NAD+ levels, which may in turn serve to suppress p53 [298]. Moreover, in diabetes, AMPK abrogates NOX4-dependent p53 activation and apoptosis of glomerular epithelial cells (podocytes) [299]. Eukaryotic translation initiation factor 4E (eIF4E) is a major downstream effector of mTOR, which inhibits p53 transactivation function and p53-mediated cellular apoptosis [300]. However, constitutive activation of mTOR due to loss of TSC1 or TSC2 ultimately amplifies p53 levels via upregulation of p53 translation or by promoting ARF-mediated stabilization of p53 [301, 302]. Under genotoxic stress, mTOR can activate p53 via the mTOR/S6K1/MDM2/p53 pathway [303]. Besides affecting p53 level and activity via the mTOR pathway, AKT regulates p53 in other ways. AKT phosphorylates MDM2 at Ser186 to enhance MDM2-mediated p53 ubiquitination and degradation [304]. In HCC, up-regulation of the eukaryotic elongation factor 1A2 (EEF1A2) increases MDMX protein stability in PI3K/AKT/USP2a and PI3K/AKT/mTOR ways, leading to inactivated p53 and restraint growth of HCC [305]. In HTLV-1-transformed cells, AKT is activated and results in p53 inhibition partly via concomitant activation of the TF, NF-κB [306]. AKT can also suppress p53-mediated apoptosis by suppressing glycogen synthase kinase 3β (GSK3β). thereby blocking Tip60-catalyzed acetylation of p53 at Lys120 and induction of PUMA [307].
To summarize, p53 and metabolic sensors are linked to one another via a highly intertwined network. A three parts model for metabolite sensing and signaling consists of metabolic sensor, transducer, and effector [268]. By crosstalk with AMPK, AKT, and mTOR, p53 plays dual roles in this system. On the one hand, modulated by the metabolic sensors, p53 acts as a metabolic transducer that transits upstream metabolic signals toward a variety of downstream effectors. On the other hand, p53 in turn enhances or suppresses the effects of metabolic sensors, just like a metabolic super-sensor. The main effects of p53 are to enhance catabolism and limit anabolism. The interplay between these network components serves to coordinate the rapid and accurate regulation of cell metabolism and are closely related to cancer initiation and development.
The role of p53 mutants in modulating cell metabolism
p53 is mutated in more than half of cancer patients. More than 1500 different mutations have been found across the p53 protein (http://p53.iarc.fr/), among which those that happen at six hotspot codons (175, 245, 248, 249, 273, and 282) account for about 28% of the total p53 mutations [308, 309]. Mtp53 can exhibit loss of function (LOF), gain of function (GOF), or dominant negative effect (DNE) compared to WT p53, which may confer them oncogenic functions to greatly influence cancer initiation and development [308, 309]. A large part of these effects are due to the metabolism-regulatory roles of mtp53 [310–312] (Figure 6).
Figure 6. Mutant p53 regulates cell metabolism.
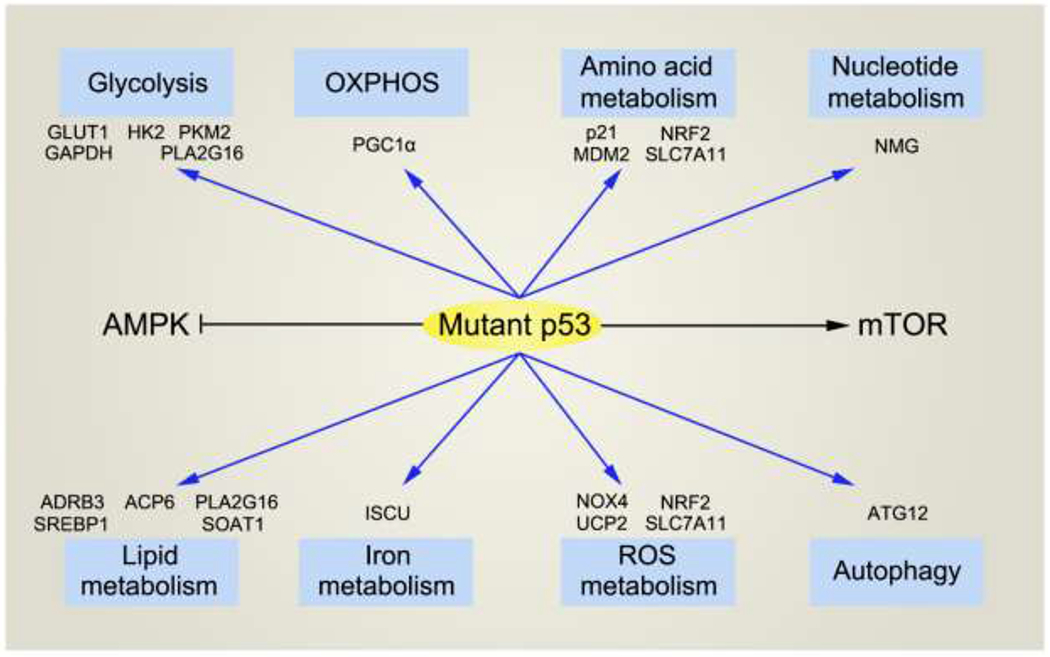
Mtp53 is involved in the regulation of all critical metabolic pathways that WT p53 participates in. Major target genes regulated by mtp53 are shown in this figure. Blue arrows indicate regulatory effects (including both positive and negative effects). Black arrow indicates positive effects. Black perpendicular bar indicates negative effects. PKM2, pyruvate kinase isoform M2; PLA2G16, phospholipase A2, group XVI; ACP6, lysophosphatidic acid phosphatase type 6.
Mtp53 modulates glycolysis and OXPHOS in various ways (Figure 6). While WT p53 suppresses GLUT1 expression and activity in several ways as we introduced before [19, 22, 24]. mtp53 switches to promote GLUT1 translocation to the plasma membrane in a RhoA/ROCK/GLUT1 signalling pathway [313]. This will boosts the function of GLUT1 to enhance glycolysis and tumorigenesis. Similarly, mtp53 also transactivates HK2 and PLA2G16 or prevents GAPDH nuclear translocation to support glycolysis [314–316]. Mtp53 gains novel function to bind and inhibit AMPK, resulting in increased glycolysis and lipogenesis [317]. Analogously, mtp53 activates mTOR/PKM2 axis to enhance glycolysis and chemoresistance in cancer cells [318]. In human cervix cancer cells, mtp53 R248Q promotes glycolysis but inhibits OXPHOS under both normoxia and hypoxia [319]. Interestingly, in a mesenchymal stem cell (MSC)-based cancer model, mtp53 augments both glycolysis and OXPHOS [320]. In one family with the Li–Fraumeni syndrome, mtp53 R181C promotes the biogenesis and activity of mitochondria in human myoblast [321]. In another study, mtp53 with a R72 SNP also increases cancer cell OXPHOS by modulating PGC1α function [322]. Additionally, mtp53 obtains the ability to transactivate mitochondrial citrate transporter SLC25A1 in a Foxo1-dependent manner, which may also affect cancer cell glycolysis and OXPHOS [323]. It is worth noting that the roles of distinct mtp53 variants in adjusting glycolysis and OXPHOS are also different [324].
Mtp53 can also regulate lipid, amino acid, nucleotide, and iron metabolism (Figure 6). In mouse adipocyte, mtp53 R178C (equivalent to human R181C) has a stronger lipolysis activity than WT p53 by inducing ADRB3 [81]. In high-grade serous ovarian cancer (HGSOC), mtp53 augments the level of lysophosphatidic acid (LPA), an oncogenic lipid, by downregulating the LPA-degrading enzyme lysophosphatidic acid phosphatase type 6 (ACP6), resulting in enhanced adhesion and metastasis in HGSOC [325]. Like binding KLF5 to activate PLA2G16 transcription [315]. mtp53 R172H (equivalent to human R175H) cooperates E26 transformation-specific 2 (ETS2) to induce PLA2G16 transcription, which supports phospholipid metabolism [326]. As we discussed above, mtp53 also promotes lipogenesis by repressing AMPK [317]. In addition, mtp53 boosts MVP via SREBP and SOAT1 [103, 104]. Interestingly, MVP can has feedback effects on mtp53 [327–329]. For example, MVP can stabilize mtp53 via MVP-DNAJA1 and MVP-RhoA axises [328, 329]. Upon glutamine deprivation, mtp53 can activate p21 to promote cancer cell survival [330]. Similarly, under serine starvation, mtp53 R248W (but not R175H) can lower ROS and enhance SSP to support cancer cell survival by inducing p21 and MDM2 [331]. Unlike WT p53, which suppresses SLC7A11 in diverse ways, mtp53 inhibits SLC7A11 expression by binding NRF2 to repress NRF2-mediated transcription of SLC7A11. Targeting SLC7A11–glutathione axis may provide an effective way to treat cancer with accumulated mtp53 [332]. On the other hand, mtp53 can cooperate with NRF2 to transactivate some proteasome genes, leading to a global influence on protein homeostasis [333]. For nucleotide metabolism, mtp53 and ETS2 form a complex to activate a batch of nucleotide metabolism genes (NMG) to support fast proliferation of cancer cells [334]. Different p53 mutants also maintain differential ability to modulate cellular iron metabolism [335]. Among them, mtp53 R175H loses the ability to induce ISCU expression [153]. In addition, mtp53 R270H (equivalent to human R273H) is suppressed by master iron metabolic regulator iron regulatory protein 2 (IRP2) [336].
Mtp53 also has a role in mediating cellular ROS level (Figure 6). Unlike their WT counterpart, which has dual roles in regulating ROS, mtp53 mainly functions to enhance intracellular ROS to boost cancer survival and development [337]. Under oxidative stress, mtp53 R273H can bind to inhibit NRF2, resulting in reduced expression of phase 2 detoxifying enzymes (including NQO1 and HO-1) and increased ROS level [338]. In another study, upon binding NRF2, mtp53 R280K selectively activates or represses NRF2 target genes, like TXN or HO-1 respectively, to promote breast cancer cell survival and migration [339]. As we mentioned above, mtp53 can reverse the effect of WT p53 to sustain TGF-β-mediated NOX4 expression, ROS production, and cancer cell metastasis [190]. Mtp53 also supports ROS increase by blocking SESN1/AMPK/PGC-1α/UCP2 anti-oxidative axis [340]. On the contrary, in human melanoma cells, mtp53 R175H can utilize exogenous pyruvate to scavenge diphenylene iodonium (DPI) and glucose depletion-caused H2O2 production to prevent cell apoptosis [341].
Just like WT p53, since mtp53 can regulate lipid, iron, and ROS metabolism, it is not hard to speculate that mtp53 may be also involved in the regulation of cell ferroptosis. All the effects of mtp53 to affect lipid, iron, and ROS metabolism may contribute to its ferroptosis-regulatory role. For example, accumulated mtp53 in cancer cell has a stronger inhibitory effect on SLC7A11 than WT p53, which causes a more ferroptosis-senstive state in that cell [6, 332]. Actually, several colorectal cancer cell lines with mtp53 (CACO2, DLD1, and SW837) show enhanced sensitivity to cell death caused by erastin than the cells with WT TP53 (HCT116 and SW48) [233]. Whether there are other ways that mtp53 can mediate cancer cell ferroptosis awaits further investigation.
Mtp53 also regulates autophagy (Figure 6) [342, 343]. In one study, researchers found that when the transfected mtp53 in p53 KO HCT116 colon carcinoma cells localized in the cytoplasm, they could effectively suppress autophagy, but the nucleus-localized mtp53 failed to do so [344]. In another study, mtp53 was proved to inhibit autophagy via various ways, including forming complex with p50 subunit of NF-κB to repress ATG12 expression, stimulating mTOR, and impeding AMPK signaling. The activation of mTOR by mtp53 rendered cancer cells augmented sensitivity to mTOR inhibition [345]. Similarly, mtp53 R273H inhibits autophagy in lung cancer cells, causing these cells more sensitive to proteasome inhibitor [346]. In the opposite direction, certain autophagy types can degrade mtp53 to promote cancer cell death [347, 348].
Taken together, mtp53 is involved in the regulation of all major metabolic pathways that WT p53 participates in, although may has opposite effects. Some of mtp53’s metabolism-regulatory functions result from its ability to adjust the activities of metabolic sensors (like AMPK or mTOR, see Figure 6) [317, 318, 340, 345]. Though most of these novel functions of mtp53 are tumor-promoting, they also open new windows for cancer treatment. Further study is needed to illustrate the mechanisms underlying metabolic regulation by mtp53 and find out novel therapeutic methods to target mtp53 and associated metabolic pathways.
A refined model of p53-mediated tumor suppression activity
p53 gene originates from about 800 million years ago and is highly conserved across evolution [349–351]. The long-term conservation of p53 and its functional network suggests that these features are critical in providing support for multicellular life. Research carried out over the past 40 years on the biology and chemistry of p53 has revealed two major and interlinked functions that include its involvement in the response to environmental perturbations and its capacity to modulate various aspects of tumor biology. However, for most of the organisms that harbor the p53 gene, there is little chance to get cancer due to a limited life span. By contrast, environmental stresses are everywhere and may emerge at any time, both in somatic and germ cells. Therefore, the most fundamental function of p53 in most species and at most times may be to help cells to withstand and to recover from environmental stress, and to maintain genomic stability and homeostasis. p53 has evolved many powerful mechanisms, including transcriptional or post-transcriptional modulation of a vast array of targets that facilitate its role in preventing one or more types of stress. As such, p53 may be considered to be a pluripotent “guardian of the cell”. With this perspective, the role of p53 in tumor biology may represent a specific application of its power to limit the impact of environmental or intracellular stress.
In the first decade after its discovery, p53 was regarded as an oncogene [352]. In 1989, p53 was demonstrated to be a tumor suppressor [353–355]. The following years’ researches consolidated the tumor-suppressive role of p53. However, in recent years (especially after 2010), more and more studies have come out to claim a tumor-supportive role of WT p53 in some circumstances [26, 31, 68, 84, 86, 92, 113, 114, 120, 232, 293]. Most of these cases are due to the metabolic regulatory roles of p53. The extreme complexity of cell metabolism (especially cancer cell metabolism), coupled with the diverse effects of p53, make it is impossible to uniquely classify p53 as either an oncogene or a tumor suppressor. We have emphasized the fact that the role of p53 in cancer is highly context-dependent. Actually, to judge every cancer-related gene as an oncogene or a tumor suppressor must depend on a specific context or background. However, current findings suggest that we can categorize all p53-mediated effects into two simple categories: pro-survival and pro-death (Figure 7). Under some conditions (e.g., the insult is not too acute), p53 induces cellular responses that are focused on protecting the cell from damage or demise. For example, when cells confront mild DNA damage, p53 promotes cell cycle arrest, thereby avoiding further DNA damage and creating an opportunity for genome repair. In cases in which cellular energy is limited, p53 activates glucose- and lipid-based OXPHOS to supply ATP. This pro-survival strategy is economically efficient because repairing a cell typically requires substantially less time, energy, and raw materials than would be needed for its full disposal and replacement. Under other circumstances (e.g., the stress is too severe to resist), activation of p53 leads to cell demise in different ways, including senescence, apoptosis, necrosis, and ferroptosis. On the surface of it, this pro-death choice looks cruel. In fact, elimination of those severely damaged cells is beneficial for the other healthy cells. This is a sacrifice of small amounts of cells to exchange for higher fitness of the local cell community or the entire host organism. As such, both pro-survival and pro-death activities may contribute to p53-mediated tumor-suppressive functions.
Figure 7. A simplified model of the relationship between metabolic regulation roles of p53 and cell fates.
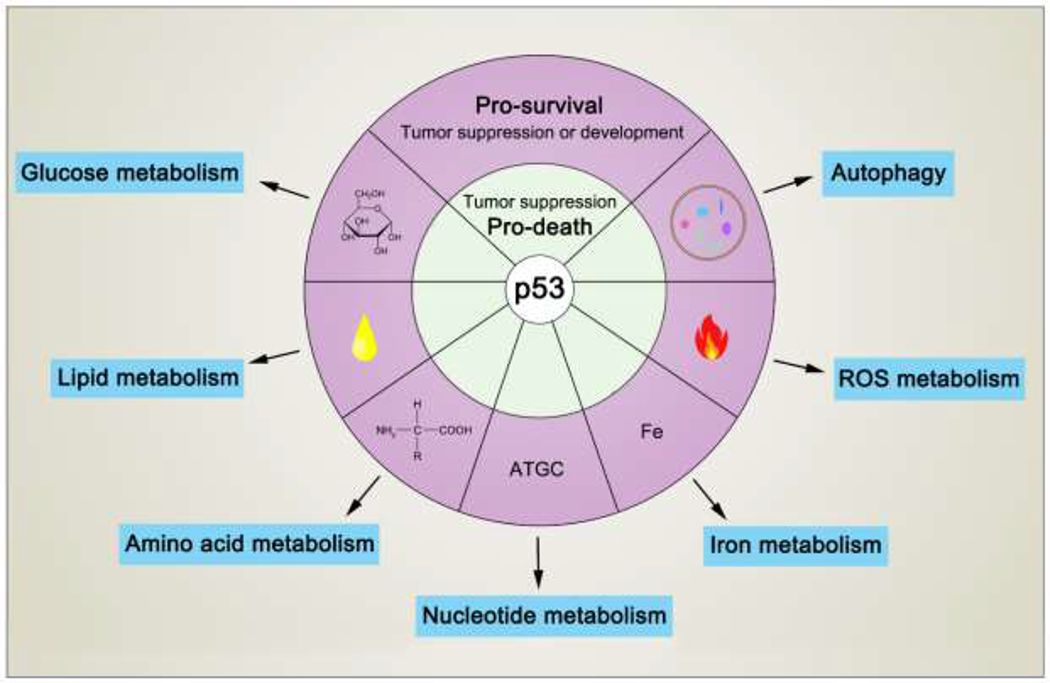
Major metabolic processes regulated by p53 are shown in this figure. p53-mediated metabolic changes will lead to two main outcomes: cell survival (may suppress tumor or promote tumor development) or cell death (will suppress tumor).
For the pro-survival mode of p53 to suppress cancer, there are at least two situations. First, some stresses have a risk of causing a neoplastic transformation of normal cells, such as DNA damage or excessive accumulation of ROS. While p53 limits these stresses to promote cell survival, it is at the same time also reducing the chance of neoplastic transformation. Second, some p53-induced metabolic adaptations serve to antagonize the metabolic needs of existing tumor cells, including FAO and OXPHOS promotion, and AKT/mTOR inhibition. However, although tumor cell has specialized metabolism hallmarks, it also shares many similarities with their non-neoplastic counterparts. For example, the accumulation of excessive amounts of ROS can promote death of both tumor and non-tumor cells. Nutrient deprivation is another adversity that is frequently encountered by tumor cells. As such, p53-mediated pro-survival effects may also be beneficial for cancer cells to survive diverse stresses. For example, p53 activates TIGAR to limit ROS-mediated damage to tumor cells and can also induce the expression of p21 to protect tumor cells from the negative sequelae of serine starvation [31, 40, 120]. In this context, some p53 targets that have been characterized as tumor-suppressive (like p21) or neutral (like SLC7A3) at most times, however, may act as an accomplice to the tumor cells. Another possibility is that many of p53’s metabolic targets themselves are mainly oncogenes, like MDM2, TIGAR, and SIVA. When p53 activates these genes to promote normal cell survival, they may also facilitate tumor cell development. p53-mediated pro-survival mechanism may even impede the efficacy of some tumor therapeutics (like metformin treatment) to kill tumor cell [356].
For the pro-death mode of p53 to suppress cancer, this maybe a stronger and safer way than the prosurvival mode. When p53 switches on this mode, it drives tumor cells into irreversible cell death processes and finally eliminates them once for all. Nonetheless, it is also critical to recognize that various pro-death pathways function with different efficacies. Once researchers took p53-mediated cell cycle arrest, senescence, and apoptosis as its final trump card to fight cancer. However, a series of experiments later excluded the necessity of them by which p53 suppresses tumor [13, 357, 358]. Findings linking p53 to mechanisms underlying ferroptosis have shed new light on this paradox [116]. Though just coined for less than ten years, ferroptosis has shown highly relevant to health and disease, especially cancer [220, 359–361]. Particularly, ferroptosis is the only cell death type that results from the dysregulation and imbalance of several core metabolic pathways (lipid, ROS, iron metabolism, and autophagy [362]) that are closely related to cancer initiation and development. As discussed above, p53 is involved in the regulation of all key pathways involved in ferroptosis (see above p53 regulates ferroptosis section). Results from studies carried out with p534KR mice provide solid support for the contributions of ferroptosis to p53-mediated tumor suppression [224, 288]. The multiplicity of p53’s roles in ferroptosis makes this cell death module outstand as potentially the most important mechanism that makes p53 one of the most vital tumor suppressors. However, there are still many questions remaining with respect to basic mechanisms of ferroptosis, the precise modes of action mediated by p53 in the regulation of ferroptosis, and the status of ferroptosis in the p53-mediated tumor-suppressive network. It is worthy of paying more attention and effort to clarify these critical issues.
Concluding remarks and further perspectives
p53 is a versatile protein with multiple roles in promoting physiological and pathological regulation. The cellular processes regulated by p53 belong to several major classes, like cell cycle arrest, DNA repair, angiogenesis, metastasis, senescence, apoptosis, and ferroptosis. In nature as a hub to receive and integrate upstream signals and orchestrate numerous downstream actions to deal with different types of stresses, p53 is highly valued mostly because of its great power to suppress cancer, although it is not yet clear which of the aforementioned p53-mediated biological processes contributes most profoundly to its tumor-suppressive function. Many recent studies highlight p53-mediated regulation of cellular metabolism as a fundamental mechanism to control cancer.
Metabolism is the basis of life. For cancer cells, due to the dramatically enhanced material and energy demand, a highly active metabolic state becomes a necessity. In the meantime, there is also an increased need to reduce the damage caused by these high-speed metabolic processes or a harsh cancer microenvironment. Both requirements together reshape a highly reprogrammed cancer metabolism mode, which is absolutely critical for tumorigenesis and tumor development [7–9]. Going through the reported p53 (both direct and indirect) targets to date, you will find that a large portion of them are functioning in the cellular metabolic networks. Most of these metabolism-associated p53 targets are related to cancer development. In this review, we introduced the roles of p53 in the regulation of glucose, lipid, amino acid, nucleotide, iron, and ROS metabolism. Then we mentioned two metabolic processes—ferroptosis and autophagy—that are controlled by p53. Next, we described the complex crosstalk between p53 and the major metabolic sensing pathways (i.e., AMPK, AKT, and mTOR). Further, we also discussed the role of mtp53 in modulating cell metabolism. Finally, we considered current ideas focused on p53-mediated tumor suppression and used our assessment of the role of p53 in cellular metabolism to suggest a refined model for this controversial but vital issue. Unfortunately, limited by the space, we cannot reasonably cover all published research on topics related to this field. In this section, we will consider several important points that were not fully discussed in the main part of this review.
First, p53 mainly functions as a TF in the nucleus where it serves to directly activate or inhibit the expression of its target genes. However, it is critical to recognize that p53 has many functions that are unrelated to its role as a TF. For example, cytoplasmic p53 (especially mitochondrial p53) participates in the regulation of glycolysis, apoptosis, ROS control, and autophagy via their interactions with other proteins [4, 34]. Many of the cytoplasmic functions of p53 are in direct opposition to its nuclear functions [4]. Even when in the nucleus, p53-mediated actions may not always relate to its role as a TF [228, 233, 258]. Additionally, many of the direct targets of p53 are themselves among the regulators of gene expression, including miRNA, lncRNA, and enzymes that promote epigenetic modifications [363–366], which may have a broad effect on gene expression. So that when we claim a protein as a metabolic target of p53, there is a caveat, that p53 may not directly affect the expression of this protein as a TF. Experimental validation will be needed in order to identify all direct transcriptional targets of p53.
Second, given the complexity of the p53-regulated metabolic network, it is not at all clear how this system might be effectively coordinated. There are two aspects to this question. One is that how p53 is induced or activated upon diverse metabolic stresses. After decades of study, many upstream activators of p53 have been identified [367, 368]. However, there may be other not-yet-identified regulators that can transduce stress signals to induce p53. A second important question focuses on how exactly p53 chooses its downstream target in response to a given metabolic state or stress. This remains one of the core challenges in the p53 field. The nature, strength, and duration of a given stress or stresses may be a critical determinant of the influence and outcomes of p53-mediated activation. One concept that is currently widely accepted suggests that if the stress is mild, transient, and easy to resist, p53 will be programmed in pro-survival mode, and will facilitate cell cycle arrest, DNA repair, reduced ROS levels, and increased energy production. By contrast, if stress is severe, prolonged, and difficult to tolerate, p53 will shift to a pro-death mode and will facilitate induction of senescence, apoptosis, and ferroptosis. At the molecular level, cell type, p53 expression level, localization, post-translational modifications, availability of binding partners, and the genetic or epigenetic status of p53 target genes will all contribute to p53 target selectivity [4, 71, 224, 242, 369]. Furthermore, positive or negative feedback regulatory mechanisms, notably those that link p53 with other master metabolic mediators including AMPK, AKT, mTOR, NRF2, HIF, and Myc, play important roles in promoting specific p53 functions [44, 71, 127, 135, 152, 165, 166, 183, 199, 218, 242, 289, 294, 295, 367, 370]. It is worth noting that baseline and stress-induced p53 may promote different functions with respect to cell metabolism [371]. In the future, more accurate and integrated mechanisms for p53-mediated metabolic regulation will be identified.
Third, a highly relevant issue to the second topic is which of these p53 functions are primary contributors to its tumor-suppressive role. This issue was discussed at length above (see A refined model of p53-mediated tumor suppression activity section). Here we would like to conclude that while cancer suppression may involve both pro-survival and pro-death functions of p53, the final effect of pro-survival choice is rather context-dependent. As such, we suggest that the pro-death (especially the pro-ferroptosis) mode may be among the more definitive mechanisms underlying p53-mediated tumor suppression (Figure 7).
Fourth, besides directly mediating cancer cell metabolism, p53 has also been found to modulate TME, especially regulating the immune cells in it [372]. Cancer immunology is a key topic both in cancer research and treatment [373, 374]. Like in cancer cells or other normal cells, metabolism within immune cells is also vital for its development and function, especially in immune cells of TME [375, 376]. Numerous researches have established a solid regulatory role for p53 in various immunological processes [377–381]. Given the pivotal role of p53 in regulating cancer cell metabolism, it is reasonable to speculate that p53 may also modulate metabolism of cancer-associated immune cells to affect their anti-tumor functions. Some evidence has emerged to support this idea. In cytolytic T cells, p53 KO strengthened glycolysis, which might boost the augmented tumor suppressive activity of T cells [382, 383]. ROS and HO-1, a p53-regulated metabolite or p53 target gene respectively, both have feedback effect to engage p53 to mediate macrophage activation [164, 384]. Moreover, p53-regulated metabolism in cancer cell can alter the biochemical features of TME (like cancer acidity caused by enhanced glycolysis), which may affect immune cell function [385]. Another clue is that ferroptosis in T cell will affect its activity in virus or parasite infection [386]. It will be interesting to investigate the significance of ferroptosis-regulatory roles of p53 in cancer-related immune cell functions. In summary, this field about p53-mediated metabolic regulation in cancer immunology is a promising direction for further scientific and clinical exploration.
Fifth, in this review, we focused on p53-mediated regulation of cancer-associated metabolism. We also recognize that p53-regulated actions may have an impact on other metabolic disorders, including obesity, diabetes, and liver and cardiovascular diseases, as well as processes associated with stem cell biology and aging [18, 387–389]. p53 even has a role in the exercise metabolism [390]. Another fact is, although p53 undergoes activation in response to nutrient deprivation, it also participates in the response to nutrient excess [391]. What should be kept in mind is that p53 may have distinct effects in systemic metabolism versus cellular metabolism [18]. In addition, p53 not only regulates the synthesis and degradation of endogenous biomolecules, it is also involved in the metabolic modulation of drugs, dietary carcinogens, and environmental pollutants [392–396]. The roles of p53 in these fields are not as fully studied as in cancer metabolism. However, the importance of these subjects should not be underestimated. More attention should be focused on the functions of p53 in these settings.
Sixth, we mainly discussed the metabolic functions of the full-length WT and mutant p53. However, there are many additional p53 variants, including p53 isoforms and polymorphisms that play interesting roles in regulating metabolism. Moreover, other p53 family members, including p63 and p73, exert critical roles in metabolic regulation. Readers are referred to several good reviews that focus specifically on these topics [17, 371, 397–399].
Finally, given the importance and wide-reaching effects of p53 on numerous aspects of metabolism, future research will be needed to explore how these findings might be used to develop treatments for cancer and/or other metabolic diseases [400–402]. There are several obstacles to overcome. p53 has many other functions besides metabolism modulation. For metabolic regulation, p53 can affect diverse pathways. Even when focused on a single pathway, p53 may have opposing roles, depending on the specific context. As such, it is not clear how unique p53-mediated metabolic effects might be achieved. One potential avenue for exploration might focus on combinations of targeting upstream p53 regulators or downstream p53 effectors while targeting p53. For cancer cells, it is not yet clear whether one should focus on stimulation of p53-mediated pro-survival or pro-death pathways, as the responses are highly case- and context-dependent. It is important to recognize that p53-mediated pro-survival activities may promote cancer development. This issue should be taken very seriously when considering cancer treatments that activate p53. Moreover, since mtp53 also participates in the metabolic regulation to affect cancer development, finding a means to eliminate the negative effects of mtp53 or to restore WT functions in mtp53 may ultimately be a practical, albeit challenging method to defeat cancer [403, 404]. If we consider other p53 variants (i.e., isoforms or SNP) and other p53 family members (p63 and p73), this issue becomes even more complicated. In the meantime, additional research into the potential adverse effects of p53 activation is certainly warranted [405–408].
In summary, we have reviewed our current understanding of the interrelationships between p53, cancer metabolism, and tumor suppression. We highlighted ferroptosis as a novel and potentially fundamental mechanism utilized by p53 to suppress cancer. Given the ongoing and rapid development in all of these fields, we anticipate that additional interesting and important findings that link p53 to cancer metabolism, ferroptosis, and tumor suppression will emerge in the near future.
Acknowledgements
This work was supported by the National Cancer Institute of the National Institutes of Health under Award 5R01CA216884, 5R01CA254970, 5R01CA085533 and 5R01CA224272 to W.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
The authors declare that there are no conflicts of interest.
Refrences
- [1].Brady CA, Attardi LD: p53 at a glance. Journal of cell science 2010, 123(Pt 15):2527–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bowen ME, Attardi LD: The role of p53 in developmental syndromes. Journal of molecular cell biology 2019, 11(3):200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jain AK, Barton MC: p53: emerging roles in stem cells, development and beyond. Development 2018, 145(8). [DOI] [PubMed] [Google Scholar]
- [4].Green DR, Kroemer G: Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458(7242):1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kruse JP, Gu W: Modes of p53 regulation. Cell 2009, 137(4):609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu J, Zhang C, Wang J, Hu W, Feng Z: The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. International journal of molecular sciences 2020, 21(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ward PS, Thompson CB: Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell 2012, 21(3):297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pavlova NN, Thompson CB: The Emerging Hallmarks of Cancer Metabolism. Cell metabolism 2016, 23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vander Heiden MG, DeBerardinis RJ: Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168(4):657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB: AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular cell 2005, 18(3):283–293. [DOI] [PubMed] [Google Scholar]
- [11].Feng Z, Zhang H, Levine AJ, Jin S: The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of the United States of America 2005, 102(23):8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM: The antioxidant function of the p53 tumor suppressor. Nature medicine 2005, 11(12):1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W: Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149(6):1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vousden KH, Prives C: Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137(3):413–431. [DOI] [PubMed] [Google Scholar]
- [15].Kastenhuber ER, Lowe SW: Putting p53 in Context. Cell 2017, 170(6):1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kruiswijk F, Labuschagne CF, Vousden KH: p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nature reviews Molecular cell biology 2015, 16(7):393–405. [DOI] [PubMed] [Google Scholar]
- [17].Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH: Metabolic regulation by p53 family members. Cell metabolism 2013, 18(5):617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L: Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Molecular metabolism 2020, 33:2–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E: The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer research 2004, 64(7):2627–2633. [DOI] [PubMed] [Google Scholar]
- [20].Zawacka-Pankau J, Grinkevich VV, Hunten S, Nikulenkov F, Gluch A, Li H, Enge M, Kel A, Selivanova G: Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: targeting Warburg effect to fight cancer. The Journal of biological chemistry 2011, 286(48):41600–41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yokoyama M, Okada S, Nakagomi A, Moriya J, Shimizu I, Nojima A, Yoshida Y, Ichimiya H, Kamimura N, Kobayashi Y et al. : Inhibition of Endothelial p53 Improves Metabolic Abnormalities Related to Dietary Obesity. Cell reports 2014, 7(5):1691–1703. [DOI] [PubMed] [Google Scholar]
- [22].Nagarajan A, Dogra SK, Sun L, Gandotra N, Ho T, Cai G, Cline G, Kumar P, Cowles RA, Wajapeyee N: Paraoxonase 2 Facilitates Pancreatic Cancer Growth and Metastasis by Stimulating GLUT1-Mediated Glucose Transport. Molecular cell 2017, 67(4):685–701 e686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kawauchi K, Araki K, Tobiume K, Tanaka N: p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nature cell biology 2008, 10(5):611–618. [DOI] [PubMed] [Google Scholar]
- [24].Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, Chan CS, Hu W, Feng Z: Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget 2014, 5(14):5535–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Webster NJ, Resnik JL, Reichart DB, Strauss B, Haas M, Seely BL: Repression of the insulin receptor promoter by the tumor suppressor gene product p53: a possible mechanism for receptor overexpression in breast cancer. Cancer research 1996, 56(12):2781–2788. [PubMed] [Google Scholar]
- [26].Khan MR, Xiang SX, Song ZY, Wu M: The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. Embo Journal 2017, 36(23):3483–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim HR, Roe JS, Lee JE, Cho EJ, Youn HD: p53 regulates glucose metabolism by miR-34a. Biochemical and biophysical research communications 2013, 437(2):225–231. [DOI] [PubMed] [Google Scholar]
- [28].Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ et al. : Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell reports 2014, 8(5):1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D: Glycolytic enzymes can modulate cellular life span. Cancer research 2005, 65(1):177–185. [PubMed] [Google Scholar]
- [30].Huang J, Du J, Lin W, Long Z, Zhang N, Huang X, Xie Y, Liu L, Ma W: Regulation of lactate production through p53/beta-enolase axis contributes to statin-associated muscle symptoms. EBioMedicine 2019, 45:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH: TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126(1):107–120. [DOI] [PubMed] [Google Scholar]
- [32].Franklin DA, He Y, Leslie PL, Tikunov AP, Fenger N, Macdonald JM, Zhang Y: p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Scientific reports 2016, 6:38067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ros S, Floter J, Kaymak I, Da Costa C, Houddane A, Dubuis S, Griffiths B, Mitter R, Walz S, Blake S et al. : 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017, 36(23):3287–3299. [DOI] [PubMed] [Google Scholar]
- [34].Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X: p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature cell biology 2011, 13(3):310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu K, Li F, Han H, Chen Y, Mao Z, Luo J, Zhao Y, Zheng B, Gu W, Zhao W: Parkin Regulates the Activity of Pyruvate Kinase M2. The Journal of biological chemistry 2016, 291(19):10307–10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, Chen J, Tomsky K, Xie H, Khella CA et al. : Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nature communications 2017, 8(1):1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Boidot R, Vegran F, Meulle A, Le Breton A, Dessy C, Sonveaux P, Lizard-Nacol S, Feron O: Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer research 2012, 72(4):939–948. [DOI] [PubMed] [Google Scholar]
- [38].Gomes AS, Ramos H, Soares J, Saraiva L: p53 and glucose metabolism: an orchestra to be directed in cancer therapy. Pharmacological research 2018, 131:75–86. [DOI] [PubMed] [Google Scholar]
- [39].Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS Jr., Chien KR, Gualberto A: p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 1999, 10(5):295–306. [PubMed] [Google Scholar]
- [40].Cheung EC, Ludwig RL, Vousden KH: Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proceedings of the National Academy of Sciences of the United States of America 2012, 109(50):20491–20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Goldstein I, Yizhak K, Madar S, Goldfinger N, Ruppin E, Rotter V: p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer & metabolism 2013, 1(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang SJ, Yu G, Jiang L, Li T, Lin Q, Tang Y, Gu W: p53-Dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell cycle 2013, 12(5):753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C, Gu B, Li Z, Wang L, Yang Y et al. : Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(29):10684–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen SL, Zhang CZ, Liu LL, Lu SX, Pan YH, Wang CH, He YF, Lin CS, Yang X, Xie D et al. : A GYS2/p53 Negative Feedback Loop Restricts Tumor Growth in HBV-Related Hepatocellular Carcinoma. Cancer research 2019, 79(3):534–545. [DOI] [PubMed] [Google Scholar]
- [45].Garufi A, D’Orazi G: High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity. J Exp Clin Canc Res 2014, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schisano B, Tripathi G, McGee K, McTernan PG, Ceriello A: Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54(5):1219–1226. [DOI] [PubMed] [Google Scholar]
- [47].Baudot AD, Crighton D, O’Prey J, Somers J, Sierra Gonzalez P, Ryan KM: p53 directly regulates the glycosidase FUCA1 to promote chemotherapy-induced cell death. Cell cycle 2016, 15(17):2299–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Contractor T, Harris CR: p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer research 2012, 72(2):560–567. [DOI] [PubMed] [Google Scholar]
- [49].Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z: Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proceedings of the National Academy of Sciences of the United States of America 2011, 108(39):16259–16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M et al. : Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Molecular cell 2014, 56(3):414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z: Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(16):7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y et al. : Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(16):7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jiang P, Du W, Mancuso A, Wellen KE, Yang X: Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493(7434):689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Morris JP, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, Baslan T, Marinkovic ZS, Sanchez-Rivera FJ, Leach SD et al. : alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573(7775):595-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM: p53 regulates mitochondrial respiration. Science 2006, 312(5780):1650–1653. [DOI] [PubMed] [Google Scholar]
- [56].Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, Rotter V: Regulation of AIF expression by p53. Cell death and differentiation 2006, 13(12):2140–2149. [DOI] [PubMed] [Google Scholar]
- [57].Gehrke S, Wu ZH, Klinkenberg M, Sun YP, Auburger G, Guo S, Lu BW: PINK1 and Parkin Control Localized Translation of Respiratory Chain Component mRNAs on Mitochondria Outer Membrane. Cell metabolism 2015, 21(1):95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nagano H, Hashimoto N, Nakayama A, Suzuki S, Miyabayashi Y, Yamato A, Higuchi S, Fujimoto M, Sakuma I, Beppu M et al. : p53-inducible DPYSL4 associates with mitochondrial supercomplexes and regulates energy metabolism in adipocytes and cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2018, 115(33):8370–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bergeaud M, Mathieu L, Guillaume A, Moll UM, Mignotte B, Le Floch N, Vayssiere JL, Rincheval V: Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F(1)F0-ATP synthase. Cell cycle 2013, 12(17):2781–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Johnson RF, Witzel II, Perkins ND: p53-Dependent Regulation of Mitochondrial Energy Production by the RelA Subunit of NF-kappa B. Cancer research 2011, 71(16):5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wen S, Gao J, Zhang L, Zhou H, Fang D, Feng S: p53 increase mitochondrial copy number via up-regulation of mitochondrial transcription factor A in colorectal cancer. Oncotarget 2016, 7(46):75981–75995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Itoh H, Kang D, Kohno K: P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer research 2003, 63(13):3729–3734. [PubMed] [Google Scholar]
- [63].Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, Huang P: Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. The EMBO journal 2005, 24(19):3482–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wong TS, Rajagopalan S, Townsley FM, Freund SM, Petrovich M, Loakes D, Fersht AR: Physical and functional interactions between human mitochondrial single-stranded DNA-binding protein and tumour suppressor p53. Nucleic acids research 2009, 37(2):568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Saleem A, Iqbal S, Zhang Y, Hood DA: Effect of p53 on mitochondrial morphology, import, and assembly in skeletal muscle. American journal of physiology Cell physiology 2015, 308(4):C319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kitamura N, Nakamura Y, Miyamoto Y, Miyamoto T, Kabu K, Yoshida M, Futamura M, Ichinose S, Arakawa H: Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PloS one 2011, 6(1):e16060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang WL, Cheng XF, Lu JJ, Wei JF, Fu GH, Zhu F, Jia CK, Zhou L, Xie HY, Zheng SS: Mitofusin-2 is a novel direct target of p53. Biochemical and biophysical research communications 2010, 400(4):587–592. [DOI] [PubMed] [Google Scholar]
- [68].Kim J, Yu L, Chen W, Xu Y, Wu M, Todorova D, Tang Q, Feng B, Jiang L, He J et al. : Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer cell 2019, 35(2):191–203 e198. [DOI] [PubMed] [Google Scholar]
- [69].Li X, Cheng KKY, Liu Z, Yang JK, Wang B, Jiang X, Zhou Y, Hallenborg P, Hoo RLC, Lam KSL et al. : The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic beta-cells. Nature communications 2016, 7:11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C et al. : Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470(7334):359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sen N, Satija YK, Das S: PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Molecular cell 2011, 44(4):621–634. [DOI] [PubMed] [Google Scholar]
- [72].Ashur-Fabian O, Har-Zahav A, Shaish A, Wiener Amram H, Margalit O, Weizer-Stern O, Dominissini D, Harats D, Amariglio N, Rechavi G: apoB and apobec1, two genes key to lipid metabolism, are transcriptionally regulated by p53. Cell cycle 2010, 9(18):3761–3770. [PubMed] [Google Scholar]
- [73].Goldstein I, Ezra O, Rivlin N, Molchadsky A, Madar S, Goldfinger N, Rotter V: p53, a novel regulator of lipid metabolism pathways. Journal of hepatology 2012, 56(3):656–662. [DOI] [PubMed] [Google Scholar]
- [74].Wang X, Zhao X, Gao X, Mei Y, Wu M: A new role of p53 in regulating lipid metabolism. Journal of molecular cell biology 2013, 5(2):147–150. [DOI] [PubMed] [Google Scholar]
- [75].Nemoto S, Fergusson MM, Finkel T: Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306(5704):2105–2108. [DOI] [PubMed] [Google Scholar]
- [76].Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L: Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429(6993):771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Deisenroth C, Itahana Y, Tollini L, Jin A, Zhang Y: p53-Inducible DHRS3 is an endoplasmic reticulum protein associated with lipid droplet accumulation. The Journal of biological chemistry 2011, 286(32):28343–28356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kung CP, Leu JI, Basu S, Khaku S, Anokye-Danso F, Liu Q, George DL, Ahima RS, Murphy ME: The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell reports 2016, 14(10):2413–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y et al. : p53 Activation in adipocytes of obese mice. The Journal of biological chemistry 2003, 278(28):25395–25400. [DOI] [PubMed] [Google Scholar]
- [80].Gomez-Santos B, Saenz de Urturi D, Nunez-Garcia M, Gonzalez-Romero F, Buque X, Aurrekoetxea I, Gutierrez de Juan V, Gonzalez-Rellan MJ, Garcia-Monzon C, Gonzalez-Rodriguez A et al. : Liver osteopontin is required to prevent the progression of age-related nonalcoholic fatty liver disease. Aging cell 2020, 19(8):e13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kang JG, Lago CU, Lee JE, Park JH, Donnelly MP, Starost MF, Liu C, Kwon J, Noguchi AC, Ge K et al. : A Mouse Homolog of a Human TP53 Germline Mutation Reveals a Lipolytic Activity of p53. Cell reports 2020, 30(3):783–792 e785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hage-Sleiman R, Bahmad H, Kobeissy H, Dakdouk Z, Kobeissy F, Dbaibo G: Genomic alterations during p53-dependent apoptosis induced by gamma-irradiation of Molt-4 leukemia cells. PloS one 2017, 12(12):e0190221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liu Y, He Y, Jin A, Tikunov AP, Zhou L, Tollini LA, Leslie P, Kim TH, Li LO, Coleman RA et al. : Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(23):E2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jiang D, LaGory EL, Kenzelmann Broz D, Bieging KT, Brady CA, Link N, Abrams JM, Giaccia AJ, Attardi LD: Analysis of p53 transactivation domain mutants reveals Acad11 as a metabolic target important for p53 pro-survival function. Cell reports 2015, 10(7):1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S: ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Molecular cell 2011, 44(3):491–501. [DOI] [PubMed] [Google Scholar]
- [86].Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, Tsuchihara K, Bungard D, Berger SL, Jones RG et al. : Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell death and differentiation 2013, 20(4):659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ponnusamy S, Meyers-Needham M, Senkal CE, Saddoughi SA, Sentelle D, Selvam SP, Salas A, Ogretmen B: Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future oncology 2010, 6(10):1603–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Heffernan-Stroud LA, Helke KL, Jenkins RW, De Costa AM, Hannun YA, Obeid LM: Defining a role for sphingosine kinase 1 in p53-dependent tumors. Oncogene 2012, 31(9):1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Fekry B, Jeffries KA, Esmaeilniakooshkghazi A, Ogretmen B, Krupenko SA, Krupenko NI: CerS6 Is a Novel Transcriptional Target of p53 Protein Activated by Non-genotoxic Stress. The Journal of biological chemistry 2016, 291(32):16586–16596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shamseddine AA, Clarke CJ, Carroll B, Airola MV, Mohammed S, Rella A, Obeid LM, Hannun YA: P53-dependent upregulation of neutral sphingomyelinase-2: role in doxorubicin-induced growth arrest. Cell death & disease 2015, 6:e1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Xu R, Garcia-Barros M, Wen S, Li F, Lin CL, Hannun YA, Obeid LM, Mao C: Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2. Cell death and differentiation 2018, 25(5):841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Brachtendorf S, Wanger RA, Birod K, Thomas D, Trautmann S, Wegner MS, Fuhrmann DC, Brune B, Geisslinger G, Grosch S: Chemosensitivity of human colon cancer cells is influenced by a p53-dependent enhancement of ceramide synthase 5 and induction of autophagy. Biochimica et biophysica acta Molecular and cell biology of lipids 2018, 1863(10):1214–1227. [DOI] [PubMed] [Google Scholar]
- [93].Li M, Hou T, Gao T, Lu X, Yang Q, Zhu Q, Li Z, Liu C, Mu G, Liu G et al. : p53 cooperates with SIRT6 to regulate cardiolipin de novo biosynthesis. Cell death & disease 2018, 9(10):941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Saleme B, Das SK, Zhang Y, Boukouris AE, Lorenzana Carrillo MA, Jovel J, Wagg CS, Lopaschuk GD, Michelakis ED, Sutendra G: p53-Mediated Repression of the PGC1A (PPARG Coactivator 1alpha) and APLNR (Apelin Receptor) Signaling Pathways Limits Fatty Acid Oxidation Energetics: Implications for Cardio-oncology. Journal of the American Heart Association 2020, 9(15):e017247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ohashi T, Idogawa M, Sasaki Y, Tokino T: p53 mediates the suppression of cancer cell invasion by inducing LIMA1/EPLIN. Cancer letters 2017, 390:58–66. [DOI] [PubMed] [Google Scholar]
- [96].Zhang YY, Fu ZY, Wei J, Qi W, Baituola G, Luo J, Meng YJ, Guo SY, Yin H, Jiang SY et al. : A LIMA1 variant promotes low plasma LDL cholesterol and decreases intestinal cholesterol absorption. Science 2018, 360(6393):1087–1092. [DOI] [PubMed] [Google Scholar]
- [97].Bist A, Fielding CJ, Fielding PE: p53 regulates caveolin gene transcription, cell cholesterol, and growth by a novel mechanism. Biochemistry 2000, 39(8):1966–1972. [DOI] [PubMed] [Google Scholar]
- [98].Kim DH, Lee JW: Tumor suppressor p53 regulates bile acid homeostasis via small heterodimer partner. Proceedings of the National Academy of Sciences of the United States of America 2011, 108(30):12266–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chen P, Li D, Chen Y, Sun J, Fu K, Guan L, Zhang H, Jiang Y, Li X, Zeng X et al. : p53-mediated regulation of bile acid disposition attenuates cholic acid-induced cholestasis in mice. British journal of pharmacology 2017, 174(23):4345–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ: The interplay between cell signalling and the mevalonate pathway in cancer. Nature reviews Cancer 2016, 16(11):718–731. [DOI] [PubMed] [Google Scholar]
- [101].Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JPt, Tschaharganeh DF, Kastenhuber ER, Barsotti AM, Culp-Hill R et al. : p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176(3):564–580 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Laezza C, D’Alessandro A, Di Croce L, Picardi P, Ciaglia E, Pisanti S, Malfitano AM, Comegna M, Faraonio R, Gazzerro P et al. : p53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell death & disease 2015, 6:e1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Oni TE, Biffi G, Baker LA, Hao Y, Tonelli C, Somerville TDD, Deschenes A, Belleau P, Hwang CI, Sanchez-Rivera FJ et al. : SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. The Journal of experimental medicine 2020, 217(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A et al. : Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148(1–2):244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Rueda-Rincon N, Bloch K, Derua R, Vyas R, Harms A, Hankemeier T, Khan NA, Dehairs J, Bagadi M, Binda MM et al. : p53 attenuates AKT signaling by modulating membrane phospholipid composition. Oncotarget 2015, 6(25):21240–21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Shimizu I, Yoshida Y, Katsuno T, Tateno K, Okada S, Moriya J, Yokoyama M, Nojima A, Ito T, Zechner R et al. : p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell metabolism 2012, 15(1):51–64. [DOI] [PubMed] [Google Scholar]
- [107].Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y, Minamino T: Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell metabolism 2013, 18(4):491–504. [DOI] [PubMed] [Google Scholar]
- [108].Molchadsky A, Ezra O, Amendola PG, Krantz D, Kogan-Sakin I, Buganim Y, Rivlin N, Goldfinger N, Folgiero V, Falcioni R et al. : p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell death and differentiation 2013, 20(5):774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kon N, Wang D, Li T, Jiang L, Qiang L, Gu W: Inhibition of Mdmx (Mdm4) in vivo induces anti-obesity effects. Oncotarget 2018, 9(7):7282–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hallenborg P, Feddersen S, Francoz S, Murano I, Sundekilde U, Petersen RK, Akimov V, Olson MV, Lozano G, Cinti S et al. : Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation. Cell death and differentiation 2012, 19(8):1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wise DR, Thompson CB: Glutamine addiction: a new therapeutic target in cancer. Trends in biochemical sciences 2010, 35(8):427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Yang M, Vousden KH: Serine and one-carbon metabolism in cancer. Nature reviews Cancer 2016, 16(10):650–662. [DOI] [PubMed] [Google Scholar]
- [113].Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD, Vousden KH: A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell metabolism 2018, 28(5):721–736 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lowman XH, Hanse EA, Yang Y, Ishak Gabra MB, Tran TQ, Li H, Kong M: p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell reports 2019, 26(11):3051–3060 e3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Faraonio R, Vergara P, Di Marzo D, Pierantoni MG, Napolitano M, Russo T, Cimino F: p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. The Journal of biological chemistry 2006, 281(52):39776–39784. [DOI] [PubMed] [Google Scholar]
- [116].Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W: Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520(7545):57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Sbodio JI, Snyder SH, Paul BD: Regulators of the transsulfuration pathway. British journal of pharmacology 2019, 176(4):583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Liu N, Lin X, Huang C: Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. British journal of cancer 2020, 122(2):279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, Shi Y, Chen L, Xiao D, Yu F et al. : Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell death and differentiation 2019, 26(11):2329–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH: Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493(7433):542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M: The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Molecular cell 2013, 50(2):200–211. [DOI] [PubMed] [Google Scholar]
- [122].Riscal R, Schrepfer E, Arena G, Cisse MY, Bellvert F, Heuillet M, Rambow F, Bonneil E, Sabourdy F, Vincent C et al. : Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Molecular cell 2016, 62(6):890–902. [DOI] [PubMed] [Google Scholar]
- [123].Ou Y, Wang SJ, Jiang L, Zheng B, Gu W: p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. The Journal of biological chemistry 2015, 290(1):457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Miyamoto T, Lo PHY, Saichi N, Ueda K, Hirata M, Tanikawa C, Matsuda K: Argininosuccinate synthase 1 is an intrinsic Akt repressor transactivated by p53. Science advances 2017, 3(5):e1603204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM: Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer research 2001, 61(5):1810–1815. [PubMed] [Google Scholar]
- [126].Yoon KA, Nakamura Y, Arakawa H: Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. Journal of human genetics 2004, 49(3):134–140. [DOI] [PubMed] [Google Scholar]
- [127].Deng L, Yao P, Li L, Ji F, Zhao S, Xu C, Lan X, Jiang P: p53-mediated control of aspartate-asparagine homeostasis dictates LKB1 activity and modulates cell survival. Nature communications 2020, 11(1):1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ: Cancer cachexia: understanding the molecular basis. Nature reviews Cancer 2014, 14(11):754–762. [DOI] [PubMed] [Google Scholar]
- [129].Schwarzkopf M, Coletti D, Sassoon D, Marazzi G: Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes & development 2006, 20(24):3440–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Ou Y, Wang SJ, Li D, Chu B, Gu W: Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proceedings of the National Academy of Sciences of the United States of America 2016, 113(44):E6806–E6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Li L, Mao Y, Zhao L, Li L, Wu J, Zhao M, Du W, Yu L, Jiang P: p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567(7747):253–256. [DOI] [PubMed] [Google Scholar]
- [132].Itahana Y, Han R, Barbier S, Lei Z, Rozen S, Itahana K: The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene 2015, 34(14):1799–1810. [DOI] [PubMed] [Google Scholar]
- [133].Wilson PM, Fazzone W, LaBonte MJ, Lenz HJ, Ladner RD: Regulation of human dUTPase gene expression and p53-mediated transcriptional repression in response to oxaliplatin-induced DNA damage. Nucleic acids research 2009, 37(1):78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Holzer K, Drucker E, Roessler S, Dauch D, Heinzmann F, Waldburger N, Eiteneuer EM, Herpel E, Breuhahn K, Zender L et al. : Proteomic Analysis Reveals GMP Synthetase as p53 Repression Target in Liver Cancer. The American journal of pathology 2017, 187(2):228–235. [DOI] [PubMed] [Google Scholar]
- [135].Reddy BA, van der Knaap JA, Bot AGM, Mohd-Sarip A, Dekkers DHW, Timmermans MA, Martens JWM, Demmers JAA, Verrijzer CP: Nucleotide Biosynthetic Enzyme GMP Synthase Is a TRIM21-Controlled Relay of p53 Stabilization. Molecular cell 2014, 53(3):458–470. [DOI] [PubMed] [Google Scholar]
- [136].Kim HR, Roe JS, Lee JE, Hwang IY, Cho EJ, Youn HD: A p53-inducible microRNA-34a downregulates Ras signaling by targeting IMPDH. Biochemical and biophysical research communications 2012, 418(4):682–688. [DOI] [PubMed] [Google Scholar]
- [137].He Z, Hu X, Liu W, Dorrance A, Garzon R, Houghton PJ, Shen C: P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget 2017, 8(25):41422–41431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y: A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404(6773):42–49. [DOI] [PubMed] [Google Scholar]
- [139].Nakano K, Balint E, Ashcroft M, Vousden KH: A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene 2000, 19(37):4283–4289. [DOI] [PubMed] [Google Scholar]
- [140].Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV: MYC, Metabolism, and Cancer. Cancer Discov 2015, 5(10):1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, Dang CV: Global regulation of nucleotide biosynthetic genes by c-Myc. PloS one 2008, 3(7):e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Ho JSL, Ma WL, Mao DYL, Benchimol S: p53-dependent transcriptional repression of c-myc is required for G(1) cell cycle arrest. Mol Cell Biol 2005, 25(17):7423–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Sachdeva M, Zhu SM, Wu FT, Wu HL, Walia V, Kumar S, Elble R, Watabe K, Mo YY: p53 represses c-Myc through induction of the tumor suppressor miR-145. Proceedings of the National Academy of Sciences of the United States of America 2009, 106(9):3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Long JS, Crighton D, O’Prey J, Mackay G, Zheng L, Palmer TM, Gottlieb E, Ryan KM: Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Molecular cell 2013, 50(3):394–406. [DOI] [PubMed] [Google Scholar]
- [145].Okorokov AL, Milner J: An ATP/ADP-dependent molecular switch regulates the stability of p53-DNA complexes. Mol Cell Biol 1999, 19(11):7501–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG: Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(29):12828–12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Torti SV, Torti FM: Iron and cancer: more ore to be mined. Nature reviews Cancer 2013, 13(5):342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM: Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. The Journal of biological chemistry 2008, 283(49):33911–33918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Zhao N, Zhang AS, Wortham AM, Jue S, Knutson MD, Enns CA: The Tumor Suppressor, P53, Decreases the Metal Transporter, ZIP14. Nutrients 2017, 9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Aydemir TB, Cousins RJ: The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). The Journal of nutrition 2018, 148(2):174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Weizer-Stern O, Adamsky K, Margalit O, Ashur-Fabian O, Givol D, Amariglio N, Rechavi G: Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. British journal of haematology 2007, 138(2):253–262. [DOI] [PubMed] [Google Scholar]
- [152].Lu J, Xu F, Lu H: LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life sciences 2020, 260:118305. [DOI] [PubMed] [Google Scholar]
- [153].Funauchi Y, Tanikawa C, Yi Lo PH, Mori J, Daigo Y, Takano A, Miyagi Y, Okawa A, Nakamura Y, Matsuda K: Regulation of iron homeostasis by the p53-ISCU pathway. Scientific reports 2015, 5:16497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Shimizu R, Lan NN, Tai TT, Adachi Y, Kawazoe A, Mu A, Taketani S: p53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene 2014, 551(1):79–85. [DOI] [PubMed] [Google Scholar]
- [155].Sawamoto M, Imai T, Umeda M, Fukuda K, Kataoka T, Taketani S: The p53-dependent expression of frataxin controls 5-aminolevulinic acid-induced accumulation of protoporphyrin IX and photo-damage in cancerous cells. Photochemistry and photobiology 2013, 89(1):163–172. [DOI] [PubMed] [Google Scholar]
- [156].Liu G, Chen X: The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene 2002, 21(47):7195–7204. [DOI] [PubMed] [Google Scholar]
- [157].Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW, Vogelstein B: Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nature medicine 2001, 7(10):1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Zhang Z, Guo M, Shen M, Kong D, Zhang F, Shao J, Tan S, Wang S, Chen A, Cao P et al. : The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox biology 2020, 36:101619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Zhang J, Chen X: p53 tumor suppressor and iron homeostasis. The FEBS journal 2019, 286(4):620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Saletta F, Suryo Rahmanto Y, Noulsri E, Richardson DR: Iron chelator-mediated alterations in gene expression: identification of novel iron-regulated molecules that are molecular targets of hypoxia-inducible factor-1 alpha and p53. Molecular pharmacology 2010, 77(3):443–458. [DOI] [PubMed] [Google Scholar]
- [161].An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM: Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 1998, 392(6674):405–408. [DOI] [PubMed] [Google Scholar]
- [162].Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS: Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). The Journal of clinical investigation 2007, 117(7):1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X et al. : Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell reports 2014, 7(1):180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, Gong JP, Liu ZJ: Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med-Us 2018, 7(8):4012–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhang Y, Qian Y, Zhang J, Yan W, Jung YS, Chen M, Huang E, Lloyd K, Duan Y, Wang J et al. : Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes & development 2017, 31(12):1243–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Palomo GM, Cerrato T, Gargini R, Diaz-Nido J: Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Human molecular genetics 2011, 20(14):2807–2822. [DOI] [PubMed] [Google Scholar]
- [167].Ray PD, Huang BW, Tsuji Y: Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling 2012, 24(5):981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Chio IIC, Tuveson DA: ROS in Cancer: The Burning Question. Trends in molecular medicine 2017, 23(5):411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Holmstrom KM, Finkel T: Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature reviews Molecular cell biology 2014, 15(6):411–421. [DOI] [PubMed] [Google Scholar]
- [170].Sarsour EH, Kumar MG, Chaudhuri L, Kalen AL, Goswami PC: Redox control of the cell cycle in health and disease. Antioxidants & redox signaling 2009, 11(12):2985–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kaspar JW, Niture SK, Jaiswal AK: Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free radical biology & medicine 2009, 47(9):1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD: Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Molecular cell 2009, 34(6):663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, Kley N: PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999, 18(1):127–137. [DOI] [PubMed] [Google Scholar]
- [174].Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P et al. : Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002, 21(39):6017–6031. [DOI] [PubMed] [Google Scholar]
- [175].Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG: Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell metabolism 2013, 17(1):73–84. [DOI] [PubMed] [Google Scholar]
- [176].Tung MC, Lin PL, Wang YC, He TY, Lee MC, Yeh SD, Chen CY, Lee H: Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6(39):41692–41705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW, Kwak MK: Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Arch Biochem Biophys 2011, 507(2):356–364. [DOI] [PubMed] [Google Scholar]
- [178].Asher G, Lotem J, Sachs L, Kahana C, Shaul Y: Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQ01. Proceedings of the National Academy of Sciences of the United States of America 2002, 99(20):13125–13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Asher G, Tsvetkov P, Kahana C, Shaul Y: A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes & development 2005, 19(3):316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Jung H, Kim MJ, Kim DO, Kim WS, Yoon SJ, Park YJ, Yoon SR, Kim TD, Suh HW, Yun S et al. : TXNIP Maintains the Hematopoietic Cell Pool by Switching the Function of p53 under Oxidative Stress. Cell metabolism 2013, 18(1):75–85. [DOI] [PubMed] [Google Scholar]
- [181].He XQ, Ma Q: Redox Regulation by Nuclear Factor Erythroid 2-Related Factor 2: Gatekeeping for the Basal and Diabetes-Induced Expression of Thioredoxin-Interacting Protein. Molecular pharmacology 2012, 82(5):887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Hou YH, Wang YT, He Q, Li LY, Xie H, Zhao Y, Zhao J: Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res 2018, 336:32–39. [DOI] [PubMed] [Google Scholar]
- [183].Rotblat B, Melino G, Knight RA: NRF2 and p53: Januses in cancer? Oncotarget 2012, 3(11):1272–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, Lowe SW: PML is a direct p53 target that modulates p53 effector functions. Molecular cell 2004, 13(4):523–535. [DOI] [PubMed] [Google Scholar]
- [185].Niwa-Kawakita M, Ferhi O, Soilihi H, Le Bras M, Lallemand-Breitenbach V, de The H: PML is a ROS sensor activating p53 upon oxidative stress. The Journal of experimental medicine 2017, 214(11):3197–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Niwa-Kawakita M, Wu HC, The H, Lallemand-Breitenbach V: PML nuclear bodies, membrane-less domains acting as ROS sensors? Seminars in cell & developmental biology 2018, 80:29–34. [DOI] [PubMed] [Google Scholar]
- [187].Gallo O, Schiavone N, Papucci L, Sardi I, Magnelli L, Franchi A, Masini E, Capaccioli S: Down-regulation of nitric oxide synthase-2 and cyclooxygenase-2 pathways by p53 in squamous cell carcinoma. The American journal of pathology 2003, 163(2):723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Han JA, Kim JI, Ongusaha PP, Hwang DH, Ballou LR, Mahale A, Aaronson SA, Lee SW: P53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. The EMBO journal 2002, 21(21):5635–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Gogna R, Madan E, Khan M, Pati U, Kuppusamy P: p53’s choice of myocardial death or survival: Oxygen protects infarct myocardium by recruiting p53 on NOS3 promoter through regulation of p53-Lys(118) acetylation. EMBO molecular medicine 2013, 5(11):1662–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Boudreau HE, Casterline BW, Burke DJ, Leto TL: Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. British journal of cancer 2014, 110(10):2569–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].O’Connor JC, Wallace DM, O’Brien CJ, Cotter TG: A novel antioxidant function for the tumor-suppressor gene p53 in the retinal ganglion cell. Investigative ophthalmology & visual science 2008, 49(10):4237–4244. [DOI] [PubMed] [Google Scholar]
- [192].Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, Ichimiya M, Sengupta S, Mechanic L, Okamura S et al. : p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer research 2004, 64(7):2350–2356. [DOI] [PubMed] [Google Scholar]
- [193].Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM: Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304(5670):596–600. [DOI] [PubMed] [Google Scholar]
- [194].Meiller A, Alvarez S, Drane P, Lallemand C, Blanchard B, Tovey M, May E: p53-dependent stimulation of redox-related genes in the lymphoid organs of gamma-irradiated mice - identification of Haeme-oxygenase 1 as a direct p53 target gene. Nucleic acids research 2007, 35(20):6924–6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Kang MY, Kim HB, Piao C, Lee KH, Hyun JW, Chang IY, You HJ: The critical role of catalase in prooxidant and antioxidant function of p53. Cell death and differentiation 2013, 20(1):117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Dhar SK, Xu Y, Chen Y, St Clair DK: Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. The Journal of biological chemistry 2006, 281(31):21698–21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Zhao Y, Chaiswing L, Velez JM, Batinic-Haberle I, Colburn NH, Oberley TD, St Clair DK: p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer research 2005, 65(9):3745–3750. [DOI] [PubMed] [Google Scholar]
- [198].Toro A, Anselmino N, Solari C, Francia M, Oses C, Sanchis P, Bizzotto J, Echegaray CV, Petrone MV, Levi V et al. : Novel Interplay between p53 and HO-1 in Embryonic Stem Cells. Cells-Basel 2021, 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Stepniewski J, Pacholczak T, Skrzypczyk A, Ciesla M, Szade A, Szade K, Bidanel R, Langrzyk A, Grochowski R, Vandermeeren F et al. : Heme oxygenase-1 affects generation and spontaneous cardiac differentiation of induced pluripotent stem cells. Iubmb Life 2018, 70(2):129–142. [DOI] [PubMed] [Google Scholar]
- [200].Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, Barelier S, Vasseur S, Spoto RP, Pebusque MJ et al. : Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer research 2009, 69(1):219–226. [DOI] [PubMed] [Google Scholar]
- [201].Nakano K, Vousden KH: PUMA, a novel proapoptotic gene, is induced by p53. Molecular cell 2001, 7(3):683–694. [DOI] [PubMed] [Google Scholar]
- [202].Liu Z, Lu H, Shi H, Du Y, Yu J, Gu S, Chen X, Liu KJ, Hu CA: PUMA overexpression induces reactive oxygen species generation and proteasome-mediated stathmin degradation in colorectal cancer cells. Cancer research 2005, 65(5):1647–1654. [DOI] [PubMed] [Google Scholar]
- [203].Miyashita T, Reed JC: Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80(2):293–299. [DOI] [PubMed] [Google Scholar]
- [204].Jiang J, Huang Z, Zhao Q, Feng W, Belikova NA, Kagan VE: Interplay between bax, reactive oxygen species production, and cardiolipin oxidation during apoptosis. Biochemical and biophysical research communications 2008, 368(1):145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N: Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288(5468):1053–1058. [DOI] [PubMed] [Google Scholar]
- [206].He H, Zang LH, Feng YS, Chen LX, Kang N, Tashiro S, Onodera S, Qiu F, Ikejima T: Physalin A induces apoptosis via p53-Noxa-mediated ROS generation, and autophagy plays a protective role against apoptosis through p38-NF-kappaB survival pathway in A375-S2 cells. Journal of ethnopharmacology 2013, 148(2):544–555. [DOI] [PubMed] [Google Scholar]
- [207].Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G: Mechanisms of cytochrome c release from mitochondria. Cell death and differentiation 2006, 13(9):1423–1433. [DOI] [PubMed] [Google Scholar]
- [208].Italiano D, Lena AM, Melino G, Candi E: Identification of NCF2/p67phox as a novel p53 target gene. Cell cycle 2012, 11(24):4589–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M et al. : A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21(24):3872–3878. [DOI] [PubMed] [Google Scholar]
- [210].Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B: A model for p53-induced apoptosis. Nature 1997, 389(6648):300–305. [DOI] [PubMed] [Google Scholar]
- [211].Matsui Y, Ueda S, Watanabe J, Kuwabara I, Ogawa O, Nishiyama H: Sensitizing effect of galectin-7 in urothelial cancer to cisplatin through the accumulation of intracellular reactive oxygen species. Cancer research 2007, 67(3):1212–1220. [DOI] [PubMed] [Google Scholar]
- [212].Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X: Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. The Journal of clinical investigation 2013, 123(12):5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kong B, Wang Q, Fung E, Xue K, Tsang BK: p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. The Journal of biological chemistry 2014, 289(39):27134–27145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM: p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149(7):1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Tu HC, Ren D, Wang GX, Chen DY, Westergard TD, Kim H, Sasagawa S, Hsieh JJ, Cheng EH: The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proceedings of the National Academy of Sciences of the United States of America 2009, 106(4):1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Eriksson SE, Ceder S, Bykov VJN, Wiman KG: p53 as a hub in cellular redox regulation and therapeutic target in cancer. Journal of molecular cell biology 2019, 11(4):330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Sermeus A, Michiels C: Reciprocal influence of the p53 and the hypoxic pathways. Cell death & disease 2011, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Obacz J, Pastorekova S, Vojtesek B, Hrstka R: Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol Cancer 2013, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al. : Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE et al. : Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171(2):273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Dixon SJ, Stockwell BR: The Hallmarks of Ferroptosis. Annu Rev Canc Biol 2019, 3:35–54. [Google Scholar]
- [222].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW et al. : Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death and differentiation 2018, 25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al. : Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156(1–2):317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W: Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell reports 2016, 17(2):366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK et al. : An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes & development 2016, 30(8):918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Singh KS, Leu JI, Barnoud T, Vonteddu P, Gnanapradeepan K, Lin C, Liu Q, Barton JC, Kossenkov AV, George DL et al. : African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nature communications 2020, 11(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Leu JI, Murphy ME, George DL: Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proceedings of the National Academy of Sciences of the United States of America 2019, 116(17):8390–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, He Q, Zhao S, Zhang GA, Wang Y et al. : Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO reports 2019, 20(7):e47563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, Gu W: ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nature cell biology 2019, 21(5):579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Shintoku R, Takigawa Y, Yamada K, Kubota C, Yoshimoto Y, Takeuchi T, Koshiishi I, Torii S: Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer science 2017, 108(11):2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Gao M, Monian P, Quadri N, Ramasamy R, Jiang X: Glutaminolysis and Transferrin Regulate Ferroptosis. Molecular cell 2015, 59(2):298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, Dixon SJ: p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell reports 2018, 22(3):569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR et al. : The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell reports 2017, 20(7):1692–1704. [DOI] [PubMed] [Google Scholar]
- [234].Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, Jiang X: Role of Mitochondria in Ferroptosis. Molecular cell 2019, 73(2):354–363 e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Yorimitsu T, Klionsky DJ: Autophagy: molecular machinery for self-eating. Cell death and differentiation 2005, 12 Suppl 2:1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Reggiori F, Komatsu M, Finley K, Simonsen A: Selective types of autophagy. International journal of cell biology 2012, 2012:156272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Mathew R, Karantza-Wadsworth V, White E: Role of autophagy in cancer. Nature reviews Cancer 2007, 7(12):961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].White E: Deconvoluting the context-dependent role for autophagy in cancer. Nature reviews Cancer 2012, 12(6):401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD: Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes & development 2013, 27(9):1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Kenzelmann Broz D, Attardi LD: TRP53 activates a global autophagy program to promote tumor suppression. Autophagy 2013, 9(9):1440–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Gao W, Shen Z, Shang L, Wang X: Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell death and differentiation 2011, 18(10):1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T: Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336(6078):225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM: DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126(1):121–134. [DOI] [PubMed] [Google Scholar]
- [244].Tatti M, Motta M, Di Bartolomeo S, Scarpa S, Cianfanelli V, Cecconi F, Salvioli R: Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Human molecular genetics 2012, 21(23):5159–5173. [DOI] [PubMed] [Google Scholar]
- [245].Zheng W, Chen QP, Wang C, Yao D, Zhu L, Pan Y, Zhang JH, Bai Y, Shao CL: Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol Carcinogen 2020, 59(6):651–660. [DOI] [PubMed] [Google Scholar]
- [246].Wu GS, Saftig P, Peters C, El-Deiry WS: Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 1998, 16(17):2177–2183. [DOI] [PubMed] [Google Scholar]
- [247].Ikeguchi M, Sakatani T, Ueta T, Fukuda K, Oka S, Hisamitsu K, Yamaguchi K, Tsujitani S, Kaibara N: Correlation between cathepsin D expression and p53 protein nuclear accumulation in oesophageal squamous cell carcinoma. J Clin Pathol 2002, 55(2):121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Eby KG, Rosenbluth JM, Mays DJ, Marshall CB, Barton CE, Sinha S, Johnson KN, Tang LJ, Pietenpol JA: ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer 2010, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Martoriati A, Doumont G, Alcalay M, Bellefroid E, Pelicci PG, Marine JC: dapk1, encoding an activator of a p19(ARF)-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 2005, 24(8):1461–1466. [DOI] [PubMed] [Google Scholar]
- [250].Lehar SM, Naeht M, Jacks T, Vater CA, Chittenden T, Guild BC: Identification and cloning of EI24, a gene induced by p53 in etoposide-treated cells. Oncogene 1996, 12(6):1181–1187. [PubMed] [Google Scholar]
- [251].Yeo SY, Itahana Y, Guo AK, Han R, Iwamoto K, Nguyen HT, Bao Y, Kleiber K, Wu YJ, Bay BH et al. : Transglutaminase 2 contributes to a TP53-induced autophagy program to prevent oncogenic transformation. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Chae YB, Kim MM: Activation of p53 by spermine mediates induction of autophagy in HT1080 cells. Int J Biol Macromol 2014, 63:56–63. [DOI] [PubMed] [Google Scholar]
- [253].Chang HW, Kim MR, Lee HJ, Lee HM, Kim GC, Lee YS, Nam HY, Lee M, Jang HJ, Lee KE et al. : p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 2019, 38(19):3729–3742. [DOI] [PubMed] [Google Scholar]
- [254].Feng X, Liu X, Zhang W, Xiao WH: p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. Embo Journal 2011, 30(16):3397–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Fei PW, Wang WG, Kim SH, Wang SL, Burns TF, Sax JK, Buzzai M, Dicker DT, McKenna WG, Bernhard EJ et al. : Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer cell 2004, 6(6):597–609. [DOI] [PubMed] [Google Scholar]
- [256].Wilfinger N, Austin S, Scheiber-Mojdekhar B, Berger W, Reipert S, Praschberger M, Paur J, Trondl R, Keppler BK, Zielinski CC et al. : Novel p53-dependent anticancer strategy by targeting iron signaling and BNIP3L-induced mitophagy. Oncotarget 2016, 7(2):1242–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Tang DL, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ et al. : Endogenous HMGB1 regulates autophagy. J Cell Biol 2010, 190(5):881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ, Li LY et al. : p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer research 2012, 72(8):1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Ikeda K, Ueyama T, Ogata T et al. : Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(8):3116–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S: Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nature communications 2013, 4. [DOI] [PubMed] [Google Scholar]
- [261].Tanida I, Ueno T, Kominami E: LC3 conjugation system in mammalian autophagy. Int J Biochem Cell B 2004, 36(12):2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M: p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(43):18511–18516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, Carrier A, Iovanna JL, Dusetti NJ: TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell death and differentiation 2012, 19(9):1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Morselli E, Shen SS, Ruckenstuhl C, Bauer MA, Marino G, Galluzzi L, Criollo A, Michaud M, Maiuri MC, Chano T et al. : p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell cycle 2011, 10(16):2763–2769. [DOI] [PubMed] [Google Scholar]
- [265].Kim J, Kundu M, Viollet B, Guan KL: AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology 2011, 13(2):132–U171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Jung CH, Ro SH, Cao J, Otto NM, Kim DH: mTOR regulation of autophagy. Febs Lett 2010, 584(7):1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Mihaylova MM, Shaw RJ: The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology 2011, 13(9):1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Wang YP, Lei QY: Metabolite sensing and signaling in cell metabolism. Signal Transduct Tar 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Saxton RA, Sabatini DM: mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Hoxhaj G, Manning BD: The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nature Reviews Cancer 2020, 20(2):74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Feng ZH, Hu WW, de Stanchina E, Teresky AK, Jin SK, Lowe S, Levine AJ: The regulation of AMPK beta 1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer research 2007, 67(7):3043–3053. [DOI] [PubMed] [Google Scholar]
- [272].Co NN, Iglesias D, Celestino J, Kwan SY, Mok SC, Schmandt R, Lu KH: Loss of LKB1 in High-Grade Endometrial Carcinoma: LKB1 is a Novel Transcriptional Target of p53. Cancer-Am Cancer Soc 2014, 120(22):3457–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Budanov AV, Karin M: p53 target genes Sestrin1 and Sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134(3):451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Hakuno F, Takahashi SI: IGF1 receptor signaling pathways. J Mol Endocrinol 2018, 61(1):T69–T86. [DOI] [PubMed] [Google Scholar]
- [275].Werner H, Karnieli E, Rauscher FJ, LeRoith D: Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proceedings of the National Academy of Sciences of the United States of America 1996, 93(16):8318–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Ohlsson C, Kley N, Werner H, LeRoith D: p53 regulates insulin-like growth factor-I (IGF-I) receptor expression and IGF-I-induced tyrosine phosphorylation in an osteosarcoma cell line: interaction between p53 and Sp1. Endocrinology 1998, 139(3):1101–1107. [DOI] [PubMed] [Google Scholar]
- [277].Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N: Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 1995, 377(6550):646–649. [DOI] [PubMed] [Google Scholar]
- [278].Leu JIJ, George DL: Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes & development 2007, 21(23):3095–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW: Regulation of PTEN transcription by p53. Molecular cell 2001, 8(2):317–325. [DOI] [PubMed] [Google Scholar]
- [280].Gupta A, Anjomani-Virmouni S, Koundouros N, Dimitriadi M, Choo-Wing R, Valle A, Zheng YX, Chiu YH, Agnihotri S, Zadeh G et al. : PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. Molecular cell 2017, 65(6):999-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, Robertson P, Bourdon JC, Perkins N, Fuller-Pace F et al. : p53-dependent repression of polo-like kinase-1 (PLK1). Cell cycle 2010, 9(20):4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Li ZG, Li J, Bi PP, Lu Y, Burcham G, Elzey BD, Ratliff T, Konieczny SF, Ahmad N, Kuang SH et al. : Plk1 Phosphorylation of PTEN Causes a Tumor-Promoting Metabolic State. Mol Cell Biol 2014, 34(19):3642–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Ren Y, Bi CF, Zhao XH, Lwin T, Wang C, Yuan J, Silva AS, Shah BD, Fang B, Li T et al. : PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. Journal of Clinical Investigation 2018, 128(12):5517–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Matthew EM, Hart LS, Astrinidis A, Navaraj A, Dolloff NG, Dicker DT, Henske EP, El-Deiry WS: The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell cycle 2009, 8(24):4168–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Burns TF, Fei PW, Scata KA, Dicker DT, El-Deiry WS: Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (Taxol)-exposed cells. Mol Cell Biol 2003, 23(16):5556–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA: REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Molecular cell 2002, 10(5):995–1005. [DOI] [PubMed] [Google Scholar]
- [287].Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witter LA, Ellisen LW, Kaelin WG: Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes & development 2004, 18(23):2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Kon N, Ou Y, Wang SJ, Li H, Rustgi AK, Gu W: mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Genes & development 2021, 35(1–2):59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Vadysirisack DD, Baenke F, Ory B, Lei K, Ellisen LW: Feedback Control of p53 Translation by REDD1 and mTORC1 Limits the p53-Dependent DNA Damage Response. Mol Cell Biol 2011, 31(21):4356–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Chen Y, Takikawa M, Tsutsumi S, Yamaguchi Y, Okabe A, Shimada M, Kawase T, Sada A, Ezawa I, Takano Y et al. : PHLDA1, another PHLDA family protein that inhibits Akt. Cancer science 2018, 109(11):3532–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Kawase T, Ohki R, Shibata T, Tsutsumi S, Kamimura N, Inazawa J, Ohta T, Ichikawa H, Aburatani H, Tashiro F et al. : PH Domain-Only Protein PHLDA3 Is a p53-Regulated Repressor of Akt. Cell 2009, 136(3):535–550. [DOI] [PubMed] [Google Scholar]
- [292].Cheng J, Huang Y, Zhang XH, Yu Y, Wu SM, Jiao J, Tran L, Zhang WR, Liu R, Zhang LZ et al. : TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nature communications 2020, 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Van Nostrand JL, Brisac A, Mello SS, Jacobs SBR, Luong R, Attardi LD: The p53 Target Gene SIVA Enables Non-Small Cell Lung Cancer Development. Cancer Discov 2015, 5(6):622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Du W, Jiang P, Li N, Mei Y, Wang X, Wen L, Yang X, Wu M: Suppression of p53 activity by Siva1. Cell death and differentiation 2009, 16(11):1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Wang XW, Zha M, Zhao XC, Jiang P, Du WJ, Tam AYH, Mei YD, Wu M: Siva1 inhibits p53 function by acting as an ARF E3 ubiquitin ligase. Nature communications 2013, 4. [DOI] [PubMed] [Google Scholar]
- [296].He GF, Zhang YW, Lee JH, Zeng SX, Wang YYV, Luo ZJ, Dong XC, Viollet B, Wahl GM, Lu H: AMP-Activated Protein Kinase Induces p53 by Phosphorylating MDMX and Inhibiting Its Activity. Mol Cell Biol 2014, 34(2):148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Lee CW, Wong LLY, Tse EYT, Liu HF, Leong VYL, Lee JMF, Hardie DG, Ng IOL, Ching YP: AMPK Promotes p53 Acetylation via Phosphorylation and Inactivation of SIRT1 in Liver Cancer Cells. Cancer research 2012, 72(17):4394–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J: AMPK regulates energy expenditure by modulating NAD(+) metabolism and SIRT1 activity. Nature 2009, 458(7241):1056–U1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh-Choudhury G, Barnes JL, Abboud HE: AMP-activated Protein Kinase (AMPK) Negatively Regulates Nox4-dependent Activation of p53 and Epithelial Cell Apoptosis in Diabetes. Journal of Biological Chemistry 2010, 285(48):37503–37512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Mungamuri SK, Yang YH, Thor AD, Somasundaram K: Survival signaling by Notchl: Mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer research 2006, 66(9):4715–4724. [DOI] [PubMed] [Google Scholar]
- [301].Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, Guan KL: Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. Embo Journal 2007, 26(23):4812–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Miceli AP, Saporita AJ, Weber JD: Hypergrowth mTORC1 Signals Translationally Activate the ARF Tumor Suppressor Checkpoint. Mol Cell Biol 2012, 32(2):348–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Lai KP, Leong WF, Chau JFL, Jia DY, Zeng L, Liu HJ, He L, Hao AJ, Zhang HB, Meek D et al. : S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. Embo Journal 2010, 29(17):2994–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y: Akt enhances Mdm2-mediated ubiquitination and degradation of p53. Journal of Biological Chemistry 2002, 277(24):21843–21850. [DOI] [PubMed] [Google Scholar]
- [305].Pellegrino R, Calvisi DF, Neumann O, Kolluru V, Wesely J, Chen X, Wang CM, Wuestefeld T, Ladu S, Elgohary N et al. : EEF1A2 Inactivates p53 by Way of PI3K/AKT/mTOR-Dependent Stabilization of MDM4 in Hepatocellular Carcinoma. Hepatology 2014, 59(5):1886–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN: Activated AKT regulates NF-kappa B activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 2005, 24(44):6719–6728. [DOI] [PubMed] [Google Scholar]
- [307].Charvet C, Wissler M, Brauns-Schubert P, Wang SJ, Tang Y, Sigloch FC, Mellert H, Brandenburg M, Lindner SE, Breit B et al. : Phosphorylation of Tip60 by GSK-3 Determines the Induction of PUMA and Apoptosis by p53. Molecular cell 2011, 42(5):584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS: Why are there hotspot mutations in the TP53 gene in human cancers? Cell death and differentiation 2018, 25(1):154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Olivier M, Hollstein M, Hainaut P: TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Csh Perspect Biol 2010, 2(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Stein Y, Rotter V, Aloni-Grinstein R: Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. International journal of molecular sciences 2019, 20(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Liu J, Zhang C, Hu W, Feng Z: Tumor suppressor p53 and metabolism. Journal of molecular cell biology 2019, 11(4):284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Mantovani F, Collavin L, Del Sal G: Mutant p53 as a guardian of the cancer cell. Cell death and differentiation 2019, 26(2):199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Zhang C, Liu J, Liang YJ, Wu R, Zhao YH, Hong XH, Lin MH, Yu HY, Liu LX, Levine AJ et al. : Tumour-associated mutant p53 drives the Warburg effect. Nature communications 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Mathupala SP, Heese C, Pedersen PL: Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. The Journal of biological chemistry 1997, 272(36):22776–22780. [DOI] [PubMed] [Google Scholar]
- [315].Xia W, Bai HS, Deng Y, Yang Y: PLA2G16is a mutant p53/KLF5 transcriptional target and promotes glycolysis of pancreatic cancer. J Cell Mol Med 2020, 24(21):12642–12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Butera G, Pacchiana R, Mullappilly N, Margiotta M, Bruno S, Conti P, Riganti C, Donadelli M: Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Bba-Mol Cell Res 2018, 1865(12):1914–1923. [DOI] [PubMed] [Google Scholar]
- [317].Zhou G, Wang JP, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA et al. : Gain-of-Function Mutant p53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Molecular cell 2014, 54(6):960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Dando I, Cordani M, Donadelli M: Mutant p53 and mTOR/PKM2 regulation in cancer cells. Iubmb Life 2016, 68(9):722–726. [DOI] [PubMed] [Google Scholar]
- [319].Hernandez-Resendiz I, Gallardo-Perez JC, Lopez-Macay A, Robledo-Cadena DX, Garcia-Villa E, Gariglio P, Saavedra E, Moreno-Sanchez R, Rodriguez-Enriquez S: Mutant p53(R248Q) downregulates oxidative phosphorylation and upregulates glycolysis under normoxia and hypoxia in human cervix cancer cells. J Cell Physiol 2019, 234(5):5524–5536. [DOI] [PubMed] [Google Scholar]
- [320].Lonetto G, Koifman G, Silberman A, Attery A, Solomon H, Levin-Zaidman S, Goldfinger N, Porat Z, Erez A, Rotter V: Mutant p53-dependent mitochondrial metabolic alterations in a mesenchymal stem cell-based model of progressive malignancy. Cell death and differentiation 2019, 26(9):1566–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Wang PY, Ma WZ, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU et al. : Increased Oxidative Metabolism in the Li-Fraumeni Syndrome. New Engl J Med 2013, 368(11):1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J, Watters A, Chen Q, Kossenkov AV, Murphy ME: Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1 alpha. Genes & development 2018, 32(3–4):230–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Kolukula VK, Sahu G, Wellstein A, Rodriguez OC, Preet A, lacobazzi V, D’Orazi G, Albanese C, Palmieri F, Avantaggiati ML: SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5(5):1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Eriksson M, Ambroise G, Ouchida AT, Queiroz AL, Smith D, Gimenez-Cassina A, Iwanicki MP, Muller PA, Norberg E, Vakifahmetoglu-Norberg H: Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol Cell Biol 2017, 37(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Chryplewicz A, Tienda SM, Nahotko DA, Peters PN, Lengyel E, Eckert MA: Mutant p53 regulates LPA signaling through lysophosphatidic acid phosphatase type 6. Scientific reports 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Xiong SB, Tu HL, Kollareddy M, Pant V, Li Q, Zhang Y, Jackson JG, Suh YA, Elizondo-Fraire AC, Yang PR et al. : Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(30):11145–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Parrales A, Thoenen E, Iwakuma T: The interplay between mutant p53 and the mevalonate pathway. Cell death and differentiation 2018, 25(3):460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Parrales A, Ranjan A, Iyer SV, Padhye S, Weir SJ, Roy A, Iwakuma T: DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nature cell biology 2016, 18(11):1233-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Ingallina E, Sorrentino G, Bertolio R, Lisek K, Zannini A, Azzolin L, Severino LU, Scaini D, Mano M, Mantovani F et al. : Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nature cell biology 2018, 20(1):28-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Humpton TJ, Hock AK, Maddocks ODK, Vousden KH: p53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer & metabolism 2018, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Tran TQ, Lowman XH, Reid MA, Mendez-Dorantes C, Pan M, Yang Y, Kong M: Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 2017, 36(14):1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, Pearson HB, Fisher OM, Read M, Guerra GR et al. : Inhibiting the system x((C)over-bar)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nature communications 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, Rajkowska K, Gaweda-Walerych K, Ingallina E, Tonelli C et al. : Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nature cell biology 2016, 18(8):897-+. [DOI] [PubMed] [Google Scholar]
- [334].Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y et al. : Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nature communications 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Clarke SL, Thompson LR, Dandekar E, Srinivasan A, Montgomery MR: Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism. Nutrients 2019, 11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Zhang YH, Feng XL, Zhang J, Chen MY, Huang E, Chen XB: Iron regulatory protein 2 is a suppressor of mutant p53 in tumorigenesis. Oncogene 2019, 38(35):6256–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Cordani M, Butera G, Pacchiana R, Masetto F, Mullappilly N, Riganti C, Donadelli M: Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P et al. : Mutant p53(R273H) attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. Journal of cell science 2012, 125(22):5578–5586. [DOI] [PubMed] [Google Scholar]
- [339].Lisek K, Campaner E, Ciani Y, Walerych D, Del Sal G: Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9(29):20508–20523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Cordani M, Butera G, Dando I, Torrens-Mas M, Butturini E, Pacchiana R, Oppici E, Cavallini C, Gasperini S, Tamassia N et al. : Mutant p53 blocks SESN1/AMPK/PGC-1 alpha/UCP2 axis increasing mitochondrial O-2-center dot production in cancer cells. British journal of cancer 2018, 119(8):994–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Chavez-Perez VA, Strasberg-Rieber M, Rieber M: Metabolic utilization of exogenous pyruvate by mutant p53 (R175H) human melanoma cells promotes survival under glucose depletion. Cancer Biol Ther 2011, 12(7):647–656. [DOI] [PubMed] [Google Scholar]
- [342].Shi Y, Norberg E, Vakifahmetoglu-Norberg H: Mutant p53 as a Regulator and Target of Autophagy. Frontiers in oncology 2020, 10:607149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Cordani M, Butera G, Pacchiana R, Donadelli M: Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Bba-Rev Cancer 2017, 1867(1):19–28. [DOI] [PubMed] [Google Scholar]
- [344].Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T, Kroemer G: Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell cycle 2008, 7(19):3056–3061. [DOI] [PubMed] [Google Scholar]
- [345].Cordani M, Oppici E, Dando I, Butturini E, Pozza ED, Nadal-Serrano M, Oliver J, Roca P, Mariotto S, Cellini B et al. : Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol Oncol 2016, 10(7):1008–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Saini H, Hakeem I, Mukherjee S, Chowdhury S, Chowdhury R: Autophagy Regulated by Gain of Function Mutant p53 Enhances Proteasomal Inhibitor-Mediated Cell Death through Induction of ROS and ERK in Lung Cancer Cells. J Oncol 2019, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan LF, Ince TA, Kroemer G, Brugge JS et al. : Chaperone-mediated autophagy degrades mutant p53. Genes & development 2013, 27(15):1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Maan M, Pati U: CHIP promotes autophagy-mediated degradation of aggregating mutant p53 in hypoxic conditions. Febs Journal 2018, 285(17):3197–3214. [DOI] [PubMed] [Google Scholar]
- [349].Levine AJ: p53: 800 million years of evolution and 40 years of discovery. Nature Reviews Cancer 2020, 20(8):471–480. [DOI] [PubMed] [Google Scholar]
- [350].Belyi VA, Ak P, Markert E, Wang HJ, Hu WW, Puzio-Kuter A, Levine AJ: The Origins and Evolution of the p53 Family of Genes. Csh Perspect Biol 2010, 2(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Lane DP, Cheok CF, Brown C, Madhumalar A, Ghadessy FJ, Verma C: Mdm2 and p53 are highly conserved from placozoans to man. Cell cycle 2010, 9(3):540–547. [DOI] [PubMed] [Google Scholar]
- [352].Levine AJ, Oren M: The first 30 years of p53: growing ever more complex. Nature Reviews Cancer 2009, 9(10):749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, Vantuinen P, Ledbetter DH, Barker DF, Nakamura Y et al. : Chromosome-17 Deletions and P53 Gene-Mutations in Colorectal Carcinomas. Science 1989, 244(4901):217–221. [DOI] [PubMed] [Google Scholar]
- [354].Eliyahu D, Michalovitz D, Eliyahu S, Pinhasikimhi O, Oren M: Wild-Type P53 Can Inhibit Oncogene-Mediated Focus Formation. Proceedings of the National Academy of Sciences of the United States of America 1989, 86(22):8763–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Finlay CA, Hinds PW, Levine AJ: The P53 Proto-Oncogene Can Act as a Suppressor of Transformation. Cell 1989, 57(7):1083–1093. [DOI] [PubMed] [Google Scholar]
- [356].Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao FP, Viollet B, Thompson CB: Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer research 2007, 67(14):6745–6752. [DOI] [PubMed] [Google Scholar]
- [357].Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Broz DK, Basak S, Park EJ, McLaughlin ME et al. : Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 2011, 145(4):571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Valente LJ, Gray DHD, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A: p53 Efficiently Suppresses Tumor Development in the Complete Absence of Its Cell-Cycle Inhibitory and Proapoptotic Effectors p21, Puma, and Noxa. Cell reports 2013, 3(5):1339–1345. [DOI] [PubMed] [Google Scholar]
- [359].Stockwell BR, Jiang XJ, Gu W: Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol 2020, 30(6):478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [360].Tang DL, Chen X, Kang R, Kroemer G: Ferroptosis: molecular mechanisms and health implications. Cell Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Hassannia B, Vandenabeele P, Vanden Berghe T: Targeting Ferroptosis to Iron Out Cancer. Cancer cell 2019, 35(6):830–849. [DOI] [PubMed] [Google Scholar]
- [362].Liu J, Kuang FM, Kroemer G, Klionsky DJ, Kang R, Tang DL: Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem Biol 2020, 27(4):420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Chen S, Thorne RF, Zhang XD, Wu M, Liu L: Non-coding RNAs, guardians of the p53 galaxy. Seminars in cancer biology 2020. [DOI] [PubMed] [Google Scholar]
- [364].Hermeking H: MicroRNAs in the p53 network: micromanagement of tumour suppression. Nature reviews Cancer 2012, 12(9):613–626. [DOI] [PubMed] [Google Scholar]
- [365].Chaudhary R, Lal A: Long noncoding RNAs in the p53 network. Wiley interdisciplinary reviews RNA 2017, 8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [366].Zhang JX, Tan P, Guo L, Gong J, Ma JJ, Li J, Lee M, Fang SH, Jing J, Johnson G et al. : p53-dependent autophagic degradation of TET2 modulates cancer therapeutic resistance. Oncogene 2019, 38(11):1905–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [367].Hu W, Feng Z, Levine AJ: The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes & cancer 2012, 3(3–4):199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].Horn HF, Vousden KH: Coping with stress: multiple ways to activate p53. Oncogene 2007, 26(9):1306–1316. [DOI] [PubMed] [Google Scholar]
- [369].Liu Y, Tavana O, Gu W: p53 modifications: exquisite decorations of the powerful guardian. Journal of molecular cell biology 2019, 11(7):564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [370].Yu L, Yu TT, Young KH: Cross-talk between Myc and p53 in B-cell lymphomas. Chronic diseases and translational medicine 2019, 5(3):139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Hafsi H, Hainaut P: Redox Control and Interplay Between p53 Isoforms: Roles in the Regulation of Basal p53 Levels, Cell Fate, and Senescence. Antioxidants & redox signaling 2011, 15(6):1655–1667. [DOI] [PubMed] [Google Scholar]
- [372].Uehara I, Tanaka N: Role of p53 in the Regulation of the Inflammatory Tumor Microenvironment and Tumor Suppression. Cancers 2018, 10(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Finn OJ: Molecular origins of cancer - Cancer immunology. New Engl J Med 2008, 358(25):2704–2715. [DOI] [PubMed] [Google Scholar]
- [374].Chen DS, Mellman I: Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39(1):1–10. [DOI] [PubMed] [Google Scholar]
- [375].Biswas SK: Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43(3):435–449. [DOI] [PubMed] [Google Scholar]
- [376].Leone RD, Powell JD: Metabolism of immune cells in cancer. Nature Reviews Cancer 2020, 20(9):516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Munoz-Fontela C, Mandinova A, Aaronson SA, Lee SW: Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol 2016, 16(12):741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Miciak J, Bunz F: Long story short: p53 mediates innate immunity. Bba-Rev Cancer 2016, 1865(2):220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Braun MW, Iwakuma T: Regulation of cytotoxic T-cell responses by p53 in cancer. Transl Cancer Res 2016, 5(6):692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Blagih J, Buck MD, Vousden KH: p53, cancer and the immune response. Journal of cell science 2020, 133(5). [DOI] [PubMed] [Google Scholar]
- [381].Levine AJ: P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. International journal of molecular sciences 2020, 21(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [382].Banerjee A, Thyagarajan K, Chatterjee S, Chakraborty P, Kesarwani P, Soloshchenko M, Al-Hommrani M, Andrijauskaite K, Moxley K, Janakiraman H et al. : Lack of p53 Augments Antitumor Functions in Cytolytic T Cells. Cancer research 2016, 76(18):5229–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [383].Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, Huang SCC, van der Windt GJW, Blagih J, Qiu J et al. : Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153(6):1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [384].Nakamura K, Zhang M, Kageyama S, Ke B, Fujii T, Sosa RA, Reed EF, Datta N, Zarrinpar A, Busuttil RW et al. : Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. Journal of hepatology 2017, 67(6):1232–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [385].Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, Tagliabue E, Castelli C, Rivoltini L: Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Seminars in cancer biology 2017, 43:74–89. [DOI] [PubMed] [Google Scholar]
- [386].Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M: T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. Journal of Experimental Medicine 2015, 212(4):555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Labuschagne CF, Zani F, Vousden KH: Control of metabolism by p53 - Cancer and beyond. Biochimica et biophysica acta Reviews on cancer 2018, 1870(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [388].Spike BT, Wahl GM: p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes & cancer 2011, 2(4):404–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Rufini A, Tucci P, Celardo I, Melino G: Senescence and aging: the critical roles of p53. Oncogene 2013, 32(43):5129–5143. [DOI] [PubMed] [Google Scholar]
- [390].Bartlett JD, Close GL, Drust B, Morton JP: The Emerging Role of p53 in Exercise Metabolism. Sports Med 2014, 44(3):303–309. [DOI] [PubMed] [Google Scholar]
- [391].Humpton TJ, Vousden KH: Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harbor perspectives in medicine 2016, 6(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [392].Krais AM, Speksnijder EN, Melis JP, Singh R, Caldwell A, Gamboa da Costa G, Luijten M, Phillips DH, Arlt VM: Metabolic activation of 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine and DNA adduct formation depends on p53: Studies in Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice. International journal of cancer 2016, 138(4):976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [393].Wohak LE, Monien B, Phillips DH, Arlt VM: Impact of p53 function on the sulfotransferase-mediated bioactivation of the alkylated polycyclic aromatic hydrocarbon 1-hydroxymethylpyrene in vitro. Environmental and molecular mutagenesis 2019, 60(8):752–758. [DOI] [PubMed] [Google Scholar]
- [394].Krais AM, Speksnijder EN, Melis JP, Indra R, Moserova M, Godschalk RW, van Schooten FJ, Seidel A, Kopka K, Schmeiser HH et al. : The impact of p53 on DNA damage and metabolic activation of the environmental carcinogen benzo[a]pyrene: effects in Trp53(+/+), Trp53(+/-) and Trp53(-/-) mice. Archives of toxicology 2016, 90(4):839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [395].Sun J, Wen Y, Zhou Y, Jiang Y, Chen Y, Zhang H, Guan L, Yao X, Huang M, Bi H: p53 attenuates acetaminophen-induced hepatotoxicity by regulating drug-metabolizing enzymes and transporter expression. Cell death & disease 2018, 9(5):536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [396].Hu H, Yu T, Arpiainen S, Lang MA, Hakkola J, Abu-Bakar A: Tumour suppressor protein p53 regulates the stress activated bilirubin oxidase cytochrome P450 2A6. Toxicol Appl Pharm 2015, 289(1):30–39. [DOI] [PubMed] [Google Scholar]
- [397].Barnoud T, Parris JLD, Murphy ME: Common genetic variants in the TP53 pathway and their impact on cancer. Journal of molecular cell biology 2019, 11(7):578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [398].Anbarasan T, Bourdon JC: The Emerging Landscape of p53 Isoforms in Physiology, Cancer and Degenerative Diseases. International journal of molecular sciences 2019, 20(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [399].Napoli M, Flores ER: The p53 family orchestrates the regulation of metabolism: physiological regulation and implications for cancer therapy. British journal of cancer 2017, 116(2):149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Duffy MJ, Synnott NC, O’Grady S, Crown J: Targeting p53 for the treatment of cancer. Seminars in cancer biology 2020. [DOI] [PubMed] [Google Scholar]
- [401].Huang J: Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacology & therapeutics 2020:107720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [402].Levine AJ: Targeting Therapies for the p53 Protein in Cancer Treatments. Annu Rev Canc Biol 2019, 3:21–34. [Google Scholar]
- [403].Bykov VJN, Eriksson SE, Bianchi J, Wiman KG: Targeting mutant p53 for efficient cancer therapy. Nature Reviews Cancer 2018, 18(2):89–102. [DOI] [PubMed] [Google Scholar]
- [404].Sabapathy K, Lane DP: Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol 2018, 15(1):13–30. [DOI] [PubMed] [Google Scholar]
- [405].Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang PR, Manshouri T, Li Y et al. : p53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer cell 2012, 21(6):793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [406].Botchkarev VA, Komarova EA, Siebenhaar F, Botchkareva NV, Komarov PG, Maurer M, Gilchrest BA, Gudkov AV: p53 is essential for chemotherapy-induced hair loss. Cancer research 2000, 60(18):5002–5006. [PubMed] [Google Scholar]
- [407].Bertheau P, Espie M, Turpin E, Lehmann J, Plassa LF, Varna M, Janin A, de The H: TP53 status and response to chemotherapy in breast cancer. Pathobiology 2008, 75(2):132–139. [DOI] [PubMed] [Google Scholar]
- [408].Gudkov AV, Komarova EA: Dangerous habits of a security guard: the two faces of p53 as a drug target. Human molecular genetics 2007, 16:R67–R72. [DOI] [PubMed] [Google Scholar]