

Abstract
N6‐methyladenosine (m6A), the newest and most prevalent layer of internal epigenetic modification in eukaryotic mRNA, has been demonstrated to play a critical role in cancer biology. Increasing evidence has highlighted that the interaction between cancer stem cells (CSCs) and the tumor immune microenvironment (TIME) is the root cause of tumorigenesis, metastasis, therapy resistance, and recurrence. In recent studies, the m6A modification has been tightly linked to this CSC‐TIME interplay, participating in the regulation of CSCs and TIME remolding. Interestingly, the m6A modification has also been identified as a novel decisive factor in the efficacy of immunotherapies—particularly anti‐PD‐1/PD‐L1 monotherapies—by changing the plasticity of the TIME. Given the functional importance of the m6A modification in the crosstalk between CSCs and the TIME, targeting m6A regulators will open new avenues to overcome therapeutic resistance, especially for immune checkpoint‐based immunotherapy. In the present review, we summarize the current landscape of m6A modifications in CSCs and the TIME, and also prospect the underling role of m6A modifications at the crossroads of CSCs and the TIME for the first time. Additionally, to provide the possibility of modulating m6A modifications as an emerging therapeutic strategy, we also explore the burgeoning inhibitors and technologies targeting m6A regulators. Lastly, considering recent advances in m6A‐seq technologies and cancer drug development, we propose the future directions of m6A modification in clinical applications, which may not only help to improve individualized monitoring and therapy but also provide enhanced and durable responses in patients with insensitive tumors.
Keywords: cancer stem cells, immune checkpoint, immunotherapy, m6A modification, metastasis, recurrence, therapeutic resistance, tumor immune microenvironment, PD‐1, PD‐L1
In this review, we evaluate the current landscape of m6A modification in the lethal teamwork of CSCs and the TIME for the first time. In addition, we summarize the existing inhibitors targeting m6A regulators and discuss the potential of modulating m6A modification as a therapeutic strategy. Moreover, we propose the future directions of using m6A modification in cancer diagnosis and treatment, as well as providing an overview of the emerging approaches to m6A‐targeted immunotherapy. We believe that this review will provide novel insight into the role of m6A modification in cancer‐specific basic and translational medicine.
1. BACKGROUND
Since RNA modifications were first reported in 1951,1, 2 more than 150 types have been discovered owing to advances in high‐throughput sequencing technology.3 Of these, N 6‐methyladenosine (m6A) is recognized as the most essential and widespread type of modification in eukaryotic mRNAs and noncoding RNAs.4 A lack of advanced sequencing technologies has prevented any major breakthroughs in this field over past decades; however, with the successive discoveries of m6A regulatory components, the importance and function of the m6A modification has gradually been revealed. High‐throughput sequencing technologies have identified that m6A modification sites are often enriched in the coding sequence, 3’ untranslated region (3’UTR), and in the vicinity of stop codons.1, 5, 6 As with DNA and protein modifications, RNA m6A modification is a dynamic, reversible, and multilayered process that alters target gene expression based on the three types of m6A regulators (methyltransferases, demethylases, and binding proteins).7, 8 To date, numerous studies have demonstrated its involvement in various physiological and pathological processes, most notably in tumorigenesis.9
Increasing evidence suggests that the m6A modification plays a nonnegligible role in the evolution and progression of multiple tumors. It is noteworthy that a number of studies have found that m6A is engaged in the maintenance and modulation of the stemness property of cancer stem cells (CSCs).10, 11, 12 CSCs are a small subpopulation of tumor cells that possess self‐renewal and clonal tumor initiation potential, and are often regarded as one of the sources of tumor relapse, metastasis, and therapeutic resistance.13 Meanwhile, the tumor immune microenvironment (TIME), the lethal synthesis partner of CSCs, unites CSCs to form an ecological system for the acceleration of tumor progression.14, 15, 16 From the perspective of the seed and soil hypothesis, CSCs can be likened to the most tenacious intrinsic seed, while the TIME tends to represent the fertile soil conductive to the growth and survival of CSCs, which collude to promote a more malignant tumor phenotype with stronger metastatic and invasive capacity.17, 18 Theoretically, CSCs are able to remodel the TIME but are also inversely affected by signals originating from it.15, 19 Notably, it has also been reported that both CSCs and the TIME are intimately associated with resistance to the most promising immunotherapy, in particular immune checkpoint blockade therapy, and disruption of the CSC‐TIME interplay is a critical step in reducing resistance propensity and enhancing antitumor activity.20, 21, 22, 23 Intriguingly, m6A modification not only actively participates in the remodeling of TIME processes but also appears to be involved in the crosstalk between CSCs and the TIME during various immune responses and hypoxia‐related reactions.24, 25, 26, 27 It is reasonable to infer that m6A may be a pivotal factor in this deadly teamwork.
To date, the function of the m6A modification in the interaction between CSCs and the TIME remains under active investigation. In the present review, we provide a novel perspective on the role of the m6A regulatory network with respect to CSCs and the TIME. Additionally, we discuss the relevant molecular mechanisms and potential therapeutic strategies based on m6A modification, providing an overview of targeting the crosstalk between CSCs and the TIME for cancer therapies.
2. THE REGULATORY COMPONENTS OF RNA m6A MODIFICATION
Similar to DNA and protein modifications, RNA m6A modification is a dynamic, reversible, and multilayered process that is determined by m6A‐specialized methyltransferases (writers), demethylases (erasers), and binding proteins (readers)28 (Figure 1). Owing to significant developments in the field of epitranscriptomics, a large number of writers, erasers, and readers have been identified.
FIGURE 1.
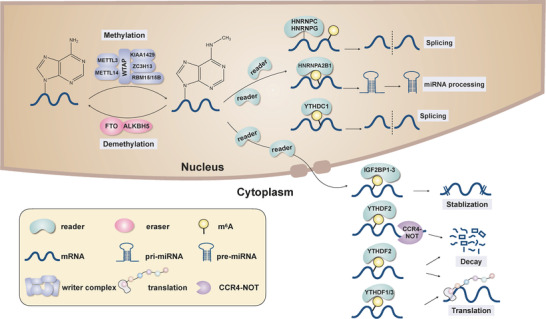
The cellular m6A machinery. m6A modification is a dynamic, reversible, and multilayered process. The writer complex has been identified as METTL3, METTL14, WATP, RBM15/15B, KIAA1429, and ZC3H13, which adds m6A methylation on target RNAs. The two erasers, FTO and ALKBH5, remove the m6A methylation from target RNAs. m6A is recognized by diverse readers, mainly HNRNPC, HNRNPG, HNRNBPA2B1, YTHDC1/2, and YTHDF1/2/3, which mediate various posttranscriptional processes including mRNA export, splicing, stabilization, decay, and translation, in addition to miRNA processing.
2.1. m6A methyltransferases/writers
m6A writers are capable of adding m6A to RNA, which is regarded as the installation of the m6A modification, and often form a multisubunit complex to exert their effects, which includes methyltransferase‐like 3 (METTL13), methyltransferase‐like 14 (METTL14), wilms tumor 1‐associated protein (WTAP), vir‐like m6A methyltransferase associated (VIRMA/KIAA1429), RNA‐binding motif protein 15/15B (RBM15/15B), and zinc finger CCCH‐type containing 13 (ZC3H13). METTL3 and METTL14 are the core subunits of this methyltransferase complex, with METTL3 playing a leading enzyme‐catalyzing role and METTL14 being less enzymatically efficient. In addition, METTL14 also functions to stabilize METTL3 and recognize the target RNA.29 Despite lacking catalytic function, WTAP facilitates the localization of the METTL3/14‐METTL14 heterodimer to nuclear speckles.30 The function of ZC3H13 is to maintain and enhance the nuclear localization of the writer complex.31 KIAA1429 and RBM15/15B are responsible for ensuring that this complex is recruited in a specific region to exert catalytic action.32, 33, 34 In addition to this classical writer complex, other m6A methyltransferases have been consecutively discovered: methyltransferase‐like 5 (METTL5), methyltransferase‐like 16 (METTL16), zinc finger CCHC‐type containing 4 (ZCCHC4), phosphorylated CTD interacting factor 1 (PCIF1), and NOP2/Sun RNA methyltransferase 2 (NSun2). PCIF1 performs m6A methylation on 2‐O‐methylated adenine located at the 5’ end of mRNAs.35 With the exception of PCIF1, these writers are involved in the m6A modification of noncoding RNAs. In particular, METTL5 and ZCCHC4 are responsible for adding m6A on 18S and 28S ribosomal RNAs.36, 37 METTL16 is specifically engaged in the m6A modification of U6 small nuclear RNAs and also regulates the expression of MAT2A.38 Nsun2 actively participates in the regulation of m6A modification of tRNAs.39 Further research will reveal the more detailed functions of these writers.
HIGHLIGHTS
The teamwork of cancer stem cells (CSCs) and the tumor immune microenvironment (TIME) is the root cause of tumor progression;
The m6A modification is participated in the CSC−TIME interplay, and the interaction between m6A and CSC−TIME was overviewed for the first time;
This will provide novel insight into the role of m6A in cancer‐specific basic and translational medicine.
2.2. m6A demethylases/erasers
Erasers ensure that the m6A modification is a dynamic and reversible process. Currently, there are two predominant m6A demethylases, fat mass and obesity associated protein (FTO) and AlkB homolog 5 (ALKBH5), which belong to the ALKB family of dependent dioxygenases and collaborate to balance the m6A levels in the transcriptome by abrogating m6A modification of RNAs.40, 41, 42 However, these two erasers work in different ways, with FTO sequentially converting m6A to N 6‐hydroxymethyladenosine (hm6A), N 6‐formyladenosine (f6A), and adenosine, while ALKBH5 removes m6A in a straightforward manner.42, 43 To a large extent, these erasers select target RNAs to perform demethylation based on the structure and conformation elicited by m6A.44 Additionally, recent studies have shown that AlkB homolog 3 (ALKBH3) is also an emerging eraser of m6A modifications but acts preferentially on tRNAs.45
2.3. m6A binding proteins/readers
The mechanism by which m6A truly exerts its biological effects is through the recruitment of relevant binding proteins (readers). At present, the YT521‐B homology (YTH) domain family proteins (YTH domain‐containing proteins 1 [YTHDC1], YTH domain‐containing proteins 2 [YTHDC2], YTH N6‐methyladenosine RNA binding protein 1 [YTHDF1], YTH N6‐methyladenosine RNA binding protein 2 [YTHDF2], and YTH N6‐methyladenosine RNA binding protein 3 [YTHDF3]) are the most well‐studied m6A readers and can be classified into three types based on location: nuclear YTHDC1, nucleocytoplasmic YTHDC2, and cytoplasmic YTHDF1, YTHDF2, and YTHDF3.46, 47, 48 The main functions of YTHDC1 include RNA splicing, X‐chromosome silencing mediation, and facilitation of m6A‐modified RNA export from the nucleus to the cytoplasm.46, 49 YTHDC2 is tightly linked to both translation and RNA degradation.50 YTHDF1 directly promotes the translation of m6A‐modified RNAs in cooperation with the translation machinery.51 The ability of YTHDF2 to trigger the decay of target RNAs is achieved by the direct recruitment of CCR4‐NOT adenosine complexes.52 Interestingly, YTHDF3 serves as a synergistic role, assisting YTHDF1 in enhancing translation and YTHDF2 in causing RNA degradation.53 However, recent study revealed that YTHDF2 may play a double‐faceted role in RNA stabilization, not only inducing RNA degradation, but also stabilizing some RNA transcripts.54 In addition to the YTH domain family, other m6A‐binding protein families have also been identified, such as the insulin‐like growth factor 2 mRNA‐binding proteins (IGF2BP1, IGF2BP2, and IGF2BP3) and the heterogeneous nuclear ribonucleoprotein (HNRNP) family (HNRNPC, HNRNPG, and HNRPA2B1). IGF2BP1‐3 are involved in the maintenance and enhancement of target RNA stability and storage by interacting with the typical consensus GG(m6A)CU sequence.55 Unlike the YTH domain family, HNRNPC and HNRNPG are recognized as indirect readers since they do not bind to m6A modification sites, but preferentially bind to emerging sites generated by changes in RNA structure caused by m6A modification. This process, in which m6A modification regulates RNA‐structure‐based accessibility of m6A readers to alter target RNA biological function, is known as the m6A switch mechanism. It is through this m6A switch mechanisms that HNRNPC and HNRNPG affect the abundance and splicing of target RNAs.56, 57 In addition to being an indirect m6A reader, HNRNPA2B1 also actively modulates microRNA (miRNA) processing.48 Furthermore, other novel m6A readers have been discovered, including leucine‐rich pentatricopeptide repeat containing (LRPPRC), fragile X mental retardation protein (FMRP), eukaryotic initiation factor 3 (eIF3), and ATP binding cassette subfamily F member 1 (ABCF1).58, 59, 60, 61 These novel readers can also recognize m6A modifications to influence the fate of RNAs.
2.4. m6A modification in CSCs
A growing body of evidence shows that CSCs are inextricably linked to tumor initiation and development62, 63; therefore, it is believed that targeting CSC‐relevant mechanisms is the key foundation of anticancer treatment. Recently, it has been demonstrated that m6A modification contributes greatly to the pluripotency and differentiation of mammalian stem cells.64, 65 In particular, many representative pluripotent genes, such as nanog homeobox (NANOG), octamer binding transcription factor 3/4 (OCT3/4), kruppel like factor 4 (KLF4), and SRY‐Box transcription factor 2 (SOX2), have been found to possess a large number of m6A modifications in their corresponding RNA transcripts.66, 67 Moreover, recent studies have shown that aberrant m6A deposition is also closely related to CSCs, which has been confirmed in a variety of malignancies.10, 11, 68 In the subsequent sections, we elaborate on the role of m6A modification in CSCs in different organ systems (Figure 2, Table 1).
FIGURE 2.
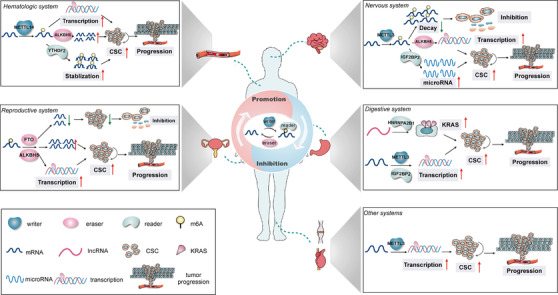
The relationship between m6A‐related regulators and various CSCs in different organ systems. The m6A‐related regulators are involved in various CSCs of different organ systems, including the nervous, digestive, reproductive, and hematologic systems. These regulators play a dual role in the modulation of various CSCs, promoting or inhibiting the stemness characteristics of CSCs by regulating the decay, splicing, stabilization, and translation of target RNAs.
TABLE 1.
The relationship between m6A regulators and various CSCs
| Cancer types | M6A regulators | Molecular axis | Function | Model system | Reference |
|---|---|---|---|---|---|
| GBM | METTL3 | METTL3/SOX2 | Promoting stemness of GSCs | In vivo | 71 |
| METTL14 | METTL3/METTL14/ADAM19 | Inhibiting stemness of GSCs | In vitro and in vivo | 12 | |
| ALKBH5 | ALKBH5/FOXM1 | Promoting stemness of GSCs | In vitro and in vivo | 11 | |
| FTO | Promoting stemness of GSCs | In vivo | 12 | ||
| IGF2BP2 | IGF2BP2/Let‐7family | Promoting stemness of GSCs | In vitro and in vivo | 73 | |
| HMGA1 | Promoting stemness of GSCs | In vivo | 74 | ||
| YTHDF2 | YTHDF2/MYC/IGFBP2 | Promoting stemness of GSCs | In vitro and in vivo | 75 | |
| PC | HNRNPA2B1 | HNRNPA2B1/UCA1/KRAS | Promoting stemness of pancreatic CSCs | In vitro | 76 |
| IGF2BP2 | IGF2BP2/DANCR | Promoting stemness of pancreatic CSCs | In vitro and in vivo | 77 | |
| CRC | METTL3 | METTL3/SOX2 | Promoting stemness of colorectal CSCs | In vitro and in vivo | 78 |
| YTHDF1 | YTHDF1/FZD9/WNT6/Wnt//β‐cantenin pathway | Promoting stemness of colorectal CSCs | In vitro and in vivo | 79 | |
| HCC | YTHDF2 | YTHDF2/OCT4 | Promoting stemness of liver CSCs | In vitro and in vivo | 80 |
| IGF2BP2 | IGF2BP2/ROS production | 81 | |||
| BC | ALKBH5 | ALKBH5/NANOG | Promoting stemness of BCSCs | In vitro and in vivo | 10 |
| OC | FTO | FTO/PDEC1/PDE4B/cAMP pathway | Inhibiting stemness of OCSCs | In vitro and in vivo | 85 |
| EC | ALKBH5 | ALKBH5/SOX2 | Promoting stemness of ECSCs | In vitro and in vivo | 25 |
| AML | METTL14 | SPI1/METTL14/MYB/MYC | Promoting stemness of LSCs | In vitro and in vivo | 68 |
| ALKBH5 | ALKBH5/TACC3 | Promoting stemness of LSCs | In vitro and in vivo | 88 | |
| FTO | Promoting stemness of LSCs | In vitro and in vivo | 89, 90 | ||
| IGF2BP1 | IGF2BP1/ALDH1A1/HOXB4/MYB | Promoting stemness of LSCs | In vitro and in vivo | 91 | |
| YTHDF2 | Promoting stemness of LSCs | In vitro and in vivo | 92 | ||
| OS | METTL3 | Pluripotency of stem cells/Wnt pathway | Promoting stemness of OSCs | In vitro | 94 |
| BCa | METTL3 | METTL3/AFF4/SOX2/MYC | Promoting stemness of BCa stem cells | In vitro | 95 |
| cSCC | METTL3 | METTL3/ΔNp6 | Promoting stemness of cSCC stem cells | In vitro | 96 |
m6A, N6‐methyladenosine; GBM, glioblastoma; PC, pancreatic cancer; CRC, colorectal cancer; HCC, hepatocellular carcinoma; BC, breast cancer; OC, ovarian cancer; EC, endometrial cancer; AML, acute myeloid leukemia; OS, osteosarcoma; BCa, bladder cancer; cSCC, cutaneous squamous cell carcinoma; GSCs, glioblastoma stem cells; CSCs, caner stem cells; BCSCs, breast cancer stem cells; OCSCs, ovarian cancer stem cells; ECSCs, endometrial cancer stem cells; LSCs, leukemia stem cells; OSCs, osteosarcoma cancer stem cells.
2.5. Nervous system
Glioblastoma (GBM) is the most prevalent and aggressive type of malignant nervous system tumors, with a high relapse potential and unfavorable prognosis.69, 70 The presence of glioblastoma stem cells (GSCs) is one of the root causes of this phenomenon and the primary concern related to GBM treatment strategies. Recent studies have demonstrated that m6A regulatory components, such as METTL3, ALKBH5, FTO, YTHDF2, and IGF2BP2, are involved in the modulation of GSCs.11, 12, 71, 72, 73, 74, 75 First, the m6A reader METTL3, a key mediator of GSCs, promotes the growth and self‐renewal of GSCs by stabilizing SOX2 mRNA.71 Downregulation of METTL3 has been shown to suppress the stemness features of GSCs and attenuate GBM invasiveness. Furthermore, high m6A modification levels enable reprogramming of GBM cells and transform non‐GSCs into GSCs. An in‐depth study uncovered that METTL3 protects serine and arginine rich splicing factors (SRSFs) mRNAs from nonsense‐mediated mRNA decay, which facilitates GBM development and progression72; however, with regard to the function of METTL3 in GBM, the opposite notion has been proposed.12 It was noted that overexpression of METTL3 inhibits the growth, self‐renewal, and frequency of GSCs by increasing the m6A abundance in the target mRNAs (eg, ADAM metallopeptidase domain 19 [ADAM19]), subsequently decreasing their expression. Further research is needed to explore these contradictory conclusions.
In addition, the eraser ALKBH5 is highly overexpressed in GSCs, predicting an unfavorable prognosis in patients with GBM. ALKBH5‐mediated demethylation of the transcription factor forkhead box M1 (FOXM1) results in elevated corresponding nascent transcripts and the subsequently detectable increased expression of mRNA and protein, which ultimately boosts GSC function and GBM germination. Moreover, knockdown of ALKBH5 has been shown to be effective in reducing the proliferative capacity of GSCs and weakening their stemness features.11 Furthermore, pharmacological inhibition of eraser FTO impedes GBM growth and tumor initiation while also prolonging the life expectancy of GSC‐engrafted mice.12
Moreover, two m6A binding proteins have been reported to participate in the regulation of GSCs. IGF2BP2 specifically binds to the let‐7 miRNA recognition sites of target transcripts to protect against let‐7 miRNA‐based splicing and silencing of these target genes, which is thought to increase the expression levels of corresponding mRNA and protein levels and subsequently induce and preserve GSCs specificity.73 Another study revealed the relationship between IGF2BP2 and GSCs in mesenchymal GBM, where IGF2BP2, DExH‐Box helicase 9 (DHX9), and HIF1A antisense RNA 2 (HIF1A‐AS2) can directly interact to stimulate the expression of target genes (high mobility group A1 [HMGA1]), eventually driving the GBM phenotype and enabling GSCs to acclimatize to hypoxic conditions.74 Similarly, YTHDF2 serves as an oncogenic trigger in GBM hierarchy, being upregulated in GSCs and supporting their stemness. This reader stabilizes transcripts of MYC and VEGFA in an m6A modification‐dependent manner, both of which subsequently interact with the downstream effector IGF2BP2 to establish a strong axis in GBM. The YTHDF2‐MYC‐IGF2BP2 axis may be a potential novel therapeutic target for GBM treatment.75
2.6. Digestive system
At present, it is understood that m6A regulators act as essential modulators of CSCs in three digestive malignancies: pancreatic cancer (PC), colorectal cancer (CRC), and hepatocellular carcinoma (HCC). First, HNRNPA2B1 has been reported to synergize with a long noncoding RNA urothelial cancer associated 1 (UCA1) to enhance KRAS expression and activity, ultimately facilitating CSCs properties and tumor growth in PC.76 In addition, IGF2BP2 stabilizes the long noncoding differentiation antagonizing nonprotein coding RNA (DANCR) in an m6A modification‐based manner, which work in concert to contribute to the stemness properties and progression in PC.77 Moreover, METTL3 collaborates with IGF2BP2 to increase m6A enrichment of SOX2 transcripts as well as extend their lifespan, maintaining and motivating the stemness of CRC cells.78 Researchers have also found that a high expression level of YTHDF1 promotes the tumorigenicity of CRC cells both in vivo and in vitro, while silencing YTHDF1 gives rise to a corresponding downregulation of classical CSC markers (CD44, CD133, OCT4, aldehyde dehydrogenase 1 [ALDH1], and leucine‐rich repeat‐containing G‐protein coupled receptor 5 [LGR5]), a smaller clonosphere, and slower tumor formation. Theoretically, suppression of YTHDF1 decreases frizzled class receptor 9 (FZD9) and Wnt family member 6 (WNT6) levels, ultimately limiting the stem cell‐related Wnt/β‐catenin signaling pathway.79 Several m6A regulators have been identified as playing pivotal roles in HCC CSCs regulation. YTHDF2 functions to regulate m6A methylation levels in the 5’UTR of OCT4 mRNA and influence the protein translation of OCT4 mRNA; therefore, the activity of CSCs can be enhanced or weakened by altering the expression level of YTHDF2. In vitro experiments have demonstrated that the loss of YTHDF2 lowers tumor burden and the likelihood of lung metastasis in HCC.80 Furthermore, another m6A binding protein IGF2BP2 enables to improve reactive oxygen species (ROS) production and induce genomic instability in HCC CSCs.81
2.7. Reproductive system
Breast cancer (BC) is the leading killer in women, with unparalleled morbidity and mortality rates.82 Similar to other tumors, aberrant m6A modification also promotes the strength of breast cancer stem cells (BCSCs).10 Upregulation of ALKBH5 increases m6A demethylation of NANOG, NANOG mRNA stability, and protein expression, ultimately boosting the BCSCs phenotype in BC. In vitro experiments have demonstrated that silencing ALKBH5 suppresses tumor formation and dramatically diminishes the proportion and function of BCSCs.10 Moreover, both ovarian and endometrial cancer (OC and EC) are serious diseases in women.83, 84 In OC, FTO plays a tumor suppressor role and impairs ovarian cancer stem cells (OCSCs) function when overexpressed. Owing to its demethylase activity, FTO destabilizes the mRNAs of two phosphodiesterase genes (phosphodiesterase 1C/4B [PDEC1 and PDE4B]) to strengthen cyclic adenosine monophosphate (cAMP) signaling and dampen the stemness characteristics of OCSCs.85 Similarly, endometrial cancer stem cells (ECSCs) are also controlled by ALKBH5, the high expression of which mediates the SOX2 level through its enhanced demethylation capacity, triggering ECSCs initiation and stemness states.25
2.8. Hematologic system
Leukemia stem cells (LSCs), featured by an unparalleled self‐renewal and growth capacity, are believed to be the initial trigger of the emergence and development of this hematologic malignancy, as well as problems related to treatment resistance and recurrence.86, 87 Researchers have successively unraveled the connection between m6A modification and LSCs, elucidating that m6A regulation is indispensable for LSCs growth.
METTL14, an essential element of the m6A writer complex, has been found to be highly expressed in both normal hematopoietic stem cells (HSCs) and acute myeloid leukemia (AML) cells; however, its expression decreases when these cells begin to differentiate. MYB and MYC, playing significant roles in the differentiation and self‐renewal of AML cells, are the direct targets of METTL14. This m6A writer targets the posttranscriptional regulation of MYB and MYC based on m6A modification, while its own expression is controlled by SFI1 centrin binding protein (SFI1). This biological process constructed a novel signaling axis (SPI1‐METTL14‐MYB/MYC) in the AML that determines the fate and activity of LSCs.68 Notably, the small molecule inhibitor of METTL3 was effective to decrease the AML stem cells growth and propagating, also enabled to prolong the survival of multiple AML patients‐derived‐xenografts mouses models.88 ALKBH5 is also aberrantly elevated in AML, which often predicts poor survival in AML patients. Inhibition of ALKBH5 impairs the growth and self‐renewal of LSCs yet has little effect on normal HSCs. Mechanistically, it has been discovered that ALKBH5 directly modifies Transforming acidic coiled‐coil 3 (TACC3) in an m6A posttranscriptional manner to ensure its transcripts half‐life and high expression. Moreover, it was highlighted that ALKBH5 is a specific target for eradicating LSCs in AML.89 With respect to another identified m6A eraser FTO, both its pharmacological inhibition and knockdown can inhibit LSCs population growth and function, which also increases the toxic effect of T cells on LSCs.90, 91 The m6A binding proteins, IGF2BP1 and YTHDF2, have also been associated with LSCs. IGF2BP1 orchestrates LSCs phenotype and tumor‐initial property. Suppression of IGF2BP1, genetically and pharmacologically, decreases LSCs proliferation, increases LSCs differentiation, induces programmed death of LSCs, and enhances the sensitivity of LSCs to chemotherapy. The potential and underlying cause may be the ability of IGF2BP1 to directly modulate crucial regulators and stemness markers of HSCs, including Aldehyde dehydrogenase 1 family member A1 (ALDH1A1), Homeobox B4 (HOXB4), and MYB.92 Similarly, YTHDF2 is also an active participant in the transformation of AML, and inactivation of YTHDF2 prolongs the half‐life of transcripts with m6A modifications, which are mostly associated with LSCs. The absence of YTHDF2 renders LSCs more sensitive to TNF‐induced apoptotic signals. Interestingly, silencing of YTHDF2 promotes HSCs to a certain extent, but does not allow hematopoiesis to become out of control.93
3. OTHER SYSTEMS
Unsurprisingly, in other systems, recent literature has also uncovered the presence of m6A modifications in the regulation of various CSCs. Since related studies are still at a relatively early stage, we have included these different systems into the same section for elaboration.
METTL3 is perceived as an essential performer of m6A modifications in osteosarcoma (OS), bladder cancer (BCa), and cutaneous squamous cell carcinoma (cSCC), mediating the stemness properties of CSCs and tumor progression.94, 95, 96, 97 The expression of METTL3 is elevated in osteosarcoma stem cells (OSCs), causing the increased m6A modifications in OSCs as compared with non‐OSCs, which may be one of the principal reasons why OS is prone to chemoresistance and metastasis. Moreover, bioinformatics analysis has suggested that these variations in m6A enrichment are likely related to pluripotency of stem cells and the Wnt pathway.95 In BCa, the emergence and self‐renewal of CSCs is limited when METTL3 is depleted. Mechanistically, METTL3 controls the expression of AF4/FMR2 family member 4 (AFF4), and AFF4 upregulates the essential stemness genes SOX2 and MYC by binding to their promoter regions.96 METTL3 deficiency has been proven to alter the expression of typical differentiation and undifferentiation markers (K10 and K14) in cSCC stem cells. Depletion of METTL3 dramatically restricts ΔNp63 expression, which consequently leads to poorer growth and tumorigenicity of cSCC stem cells.97 In addition, YTHDF1 has also been demonstrated to help tumor cells adapt to hypoxic conditions in lung cancer, while hypoxia‐related molecular events are known to facilitate and support CSCs development. This phenomenon suggests that YTHDF1 may govern CSCs in an indirect manner in lung cancer.24, 98
In summary, it can be clearly observed that m6A modification plays a key role in the emergence and development of CSCs in a variety of malignancies. The presence of CSCs is often considered the root source of tumor propagation and relapse, which in turn are also responsible for chemoradiotherapy resistance. Furthermore, chemotherapy drug transport and metabolism are also mediated by m6A modification, which is the first step in determining drug effectiveness against CSCs.99 Therefore, it is reasonable to assume that the biological process of m6A modification may be pivotal in eliminating CSCs and destroying tumors, providing a novel and potential therapeutic avenue for several cancers.
3.1. m6A modification in the TIME
With the recognition of cancer as a heterogeneous disease, the TIME can no longer be separated from tumorigenesis and progression.100 The TIME is a complex and systematic structure, analogous to fertile soil for tumor growth, featured by various immune cellular compositions, regulatory‐protein expression, and inflammatory cytokines.18, 21 Most importantly, the TIME status is the leading cause of the differential responses and outcomes in cancer patients receiving the same treatment, especially for multiple immunotherapies.21, 101 Therefore, explaining the diversity and complexity of the TIME is an indispensable step in enhancing the predictive capacity and clinical guidance of immunotherapy, which will benefit countless patients. It is becoming apparent that TIME remodeling includes various biological processes, such as immune cell infiltration, immune checkpoint protein expression, and cytokine production, in which the m6A modifications function as a critical mediator (Figure 3, Table 2).
FIGURE 3.
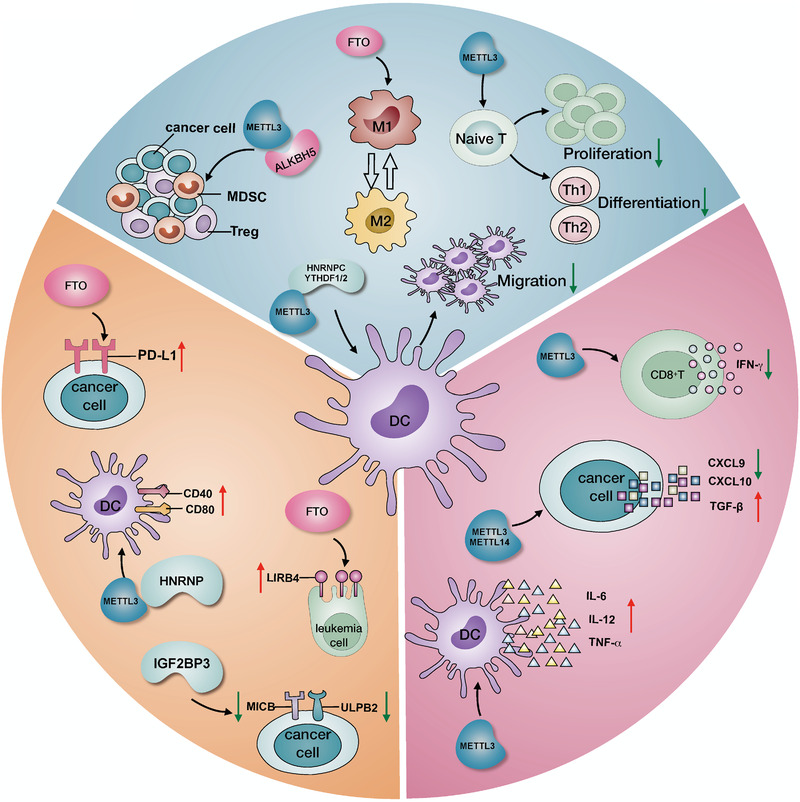
The m6A‐mediated TIME remodeling includes multiple aspects. m6A‐related regulators actively reprogram the TIME in various ways. For immune cells, these regulators are able to influence MDSC and Treg infiltration, M1 and M2 polarization, DC migration and function, and naïve T cell proliferation and differentiation. For immune checkpoints, PD‐L1, LIRB4, CD40, and CD80 are upregulated by certain m6A regulators, while MICB and ULPB2 are downregulated. For cytokine production, the secretion of IL6, IL12, TNF‐α, and TGF‐β is increased, but that of CXCL9, CXCL10, and IFN‐γ production is decreased.
TABLE 2.
m6A‐mediated TIME remodeling in various aspects
| TIME compositions | M6A regulators | Molecular mechanisms | Functions | Reference | |
|---|---|---|---|---|---|
| Immune cells | DC | METTL3, HNRNPC | Regulating CD40 and CD80 expression | DC migration and function | 27, 102 |
| YTHDF1, YTHDF2 | Regulating lysosomal protease expression | CCR7‐mediated DC migration and DC‐based immunec responses | 103 | ||
| Macrophage | FTO | Regulating STAT1 and PPAR‐γ mRNA degradation | M1 and M2 macrophages polarization | 105 | |
| Naïve T cell | METTL3 | Regulating SOCS family mRNA degradation | naïve T cell proliferation and differentiation | 106 | |
| Treg | METTL3 | Regulating SOCS family mRNA degradation | Tregs stability and suppressive function | 107 | |
| ALKBH5 | Regulating MCT4 expression | Tregs infiltration | 26 | ||
| MDSC | METTL3 | MDSCs infiltration | 108 | ||
| ALKBH5 | Regulating MCT4 expression | MDSCs infiltration | 26 | ||
| Immune checkpoints | PD‐L1 | FTO | Regulating IFN‐γ pathway | PD‐L1 expression | 110 |
| LIRB4 | FTO | Regulating mRNA degradation | LIRB4 expression | 88 | |
| ULPB2, MICB | IGF2BP3 | ULPB2 and MICB expression and migration | 111 | ||
| CD40, CD80 | METTL3, HNRNPC | CD40 and CD80 expression | 27, 102 | ||
| Cytokines | IL6, IL12, and TNF‐α | METTL3 | Regulating NF‐κB signaling | Production of IL6, IL12, and TNF‐α | 27 |
| IFN‐γ, CXCL9, and CXCL10 | METTL3, METTL14 | Regulating IFN‐γ‐STAT1‐IRF1 signal pathway | Production of IFN‐γ, CXCL9, and CXCL10 | 113 | |
| TGF‐β1 | METTL3 | Regulating TGF‐β1 mRNAs degradation and translation elongation | Production and activation of TGF‐β1 | 115 |
m6A, N 6‐methyladenosine; TIME, tumor immune microenvironment; DC, dendritic cell; CCR7, C‐C motif chemokine receptor 7; STAT1, signal transducer and activator of transcription 1; PPAR, peroxisome proliferators‐activated receptor; SOCS, suppressor of cytokine signaling; Treg, regulatory T cells; MCT4, monocarboxylate transporter 4; MDSC, myeloid‐derived suppressor cell; IFN‐γ, interferon gamma; LIRB4, leukocyte immunoglobulin like receptor B4; LIRB4, leukocyte immunoglobulin like receptor B4; MICB, MHC class I polypeptide‐related sequence B; IL‐6, interleukin 6; IL‐12, interleukin 12; TNF‐α, tumor necrosis factor alpha; NK‐κB, nuclear factor kappa B; CXCL9, C‐X‐C motif chemokine ligand 9; CXCL10, C‐X‐C motif chemokine ligand 10; TFG‐β1, transforming growth factor beta 1.
3.2. Immune cells
The TIME is broadly populated with various immune cells, and their composition, distribution, and function largely determine the TIME characteristics.21 Dendritic cell (DC)‐directed antigen presentation is the initial step in the activation of antitumor effects of specific immunity, while m6A modification influences DCs in many ways. METTL3, HNRNPC, YTHDF1, and YTHDF2 affect the maturation and phenotype of DCs, further disrupting their key immune functions. On one hand, both METTL3 and HNRNPC regulate DC maturation and function by altering the costimulatory molecular (CD40 and CD80) expression.27, 102 METTL3 has been found to promote translation of CD40 and CD80 mRNAs. In addition, loss of METTL3 in DCs decreases the transcription of TLR4 signaling adaptor Tirap, resulting in a diminished ability to activate T cells.27 On the other hand, YTHDF1 and YTHDF2 manipulate DC migration and function through completely different mechanisms. In DCs, YTHDF1 directly binds to lysosomal protease transcripts marked by m6A and promotes the translation of lysosomal cathepsins, rendering DCs incapable of neoantigen‐presentation and cross‐priming.103 CCR7‐mediated DC migration and DC‐based immune response pathways can also involve the presence of m6A modifications. The lnc‐RNA double PHD fingers 3 (Dpf3), which plays a pivotal role in these pathways, directly impedes hypoxia‐inducible factor 1‐alpha (HIF‐1α) activity and HIF‐1α‐dependent glycolytic metabolism, culminating in the inhibition of DC migration and inflammatory responses. Silencing of YTHDF2 further exacerbates this CCR7‐induced DC migration process and completely disables DCs by alleviating m6A modification‐based RNA degradation of lnc‐Dpf3.104 In addition to DCs, macrophage polarization is also fine‐tuned by m6A modification. Depletion of the m6A demethylase FTO not only inactivates the NF‐κB signaling pathway but also restricts the polarization of M1 and M2 macrophages owing to the accelerated degradation of signal transducer and activator of transcription 1 (STAT1) and peroxisome proliferators‐activated receptor‐γ (PPAR‐γ) mRNA marked by m6A.105 Furthermore, m6A methylation‐mediated mRNA decay disturbs homeostasis and activates naïve T cells and Tregs.106, 107 Downregulation of METTL3 results in a considerable decline in m6A‐dependent mRNA degradation of the suppressor of cytokine signaling (SOCS) family, which encode many STAT family repressor proteins. Consequently, these inhibitor proteins, such as rate‐limiting enzymes, restrain naïve T cell proliferation and differentiation mediated by IL‐7 signaling and also decrease IL‐2 signaling, disturbing the stability and suppressive function of Tregs.106, 107 There is no doubt that CD8+ T cells are also influenced by m6A regulators. In vivo experiments, both METTL3 and METTL14‐deficient tumors render higher infiltration of CD8+ T cells and enhanced secretion ability of cytokines than the controls.108 Meanwhile, YTHDF1‐knock out mice exhibit increased cross‐priming of CD8+ T cells by DCs as compared with WT mice.103 Interestingly, patients with low YTHDF1 expression have higher proportion of CD8+ T cells in the TIME.103
Moreover, the expression level of METTL3 is positively correlated with myeloid‐derived suppressor cells (MDSCs) infiltration in the TIME, which work together to create an immunosuppressive environment. Both METTL3 and MDSCs are independent factors for reduced survival in cervical cancer patients.109 Similarly, the m6A eraser ALKBH5 indirectly manipulates the splicing and expression of the target gene monocarboxylate transporter 4 (MCT4), a crucial lactate transporter, the expression of which is subject to m6A demethylation by ALKBH5. Inhibition of ALKBH5 by CRISPR or pharmacological molecules reduces MCT4 expression, leading to a dramatic decrease in lactate content of the TIME. A lower lactate concentration is accompanied by a smaller proportion of suppressive immune cells (Tregs and MDSCs) in the TIME, which notably harbor ALKBH5 deletions or mutations and typically predict sensitive responses and favorable efficacy of anti‐PD‐1 treatment in melanoma patients.26 Moreover, Li et al constructed a comprehensive m6A regulator‐based risk signature that implicates a strong relationship between m6A regulators and immune cell infiltration in the TIME. Based on bioinformatics analysis, they also prompted m6A may collaborate with the PI3K‐AKT‐mTOR signaling pathway to reprogram the TIME.110
3.3. Immune checkpoints
Conceivably, some critical immune checkpoints are also supervised by m6A regulators. In colon cancer cells, FTO overexpression causes a corresponding boost in PD‐L1 protein expression in an IFN‐γ‐dependent manner.111 In addition, FTO actively upregulates immune checkpoint LIRB4 in AML cells by reducing YTHDF2‐induced mRNA degradation. Blockade of FTO is an effective way to sensitize AML cells to T‐cell cytotoxicity by targeting leukocyte immunoglobulin like receptor B4 (LIRB4), which alleviates tumor immune evasion in AML to some degree.90 Further, the m6A binding protein IGF2BP3 also takes part in the modulation of tumor immune evasion, showing a remarkably higher expression level in tumor cells than in normal tissue. Powerfully oncogenic IGF2BP3 not only downregulates the expression of stress‐induced ligands (UL16 binding protein 2 [ULPB2] and MHC class I polypeptide‐related sequence B [MICB]) but also prohibits these proteins from trafficking to the cell surface. Moreover, NK cells fail to recognize and annihilate these ingenious tumor cells via the natural killer group 2 member D (NKG2D) receptor.112 Furthermore, both METTL3 and HNRNPC mediate the expression of the costimulatory molecules CD40 and CD80 in DCs. METTL3‐deficient DCs demonstrate lower CD40 and CD80 translation levels than METTL3‐wild type DCs. It has also been shown that METLL3 promotes CD40 and CD80 expression in DCs by increasing the translation efficiency of the corresponding mRNAs.27
Anti‐PD‐1/L1 treatment is perceived to be the most promising method of annihilating and eradicating malignancies,113 and researchers have gradually unveiled the roles of certain m6A regulators in anti‐PD‐1/L1 therapy. Intriguingly, the efficacy of anti‐PD‐1 is considerably augmented by simultaneous depletion of METTL3 and METTL14 in both colorectal cancer and melanoma. Mechanistically, the depletion of METTL3 and METTL14 may increase the infiltration and function of CD8+T cells.108 Similarly, the m6A eraser ALKBH5 indirectly affects responses to anti‐PD‐1 immunotherapy by creating a suppressive TIME. Conceivably, harboring ALKBH5 deletions or mutations typically predicts sensitive responses and a favorable efficacy of anti‐PD‐1 treatment in melanoma patients.26 In parallel, knockdown of FTO results in increased PD‐1 expression, which reverses melanoma resistance to anti‐PD‐1 therapy in preclinical experiments.114 Moreover, YTHDF1‐deficient mice are featured by more CD8+ T cells in TIME, tending to display preferable efficacy profiles and outcomes for PD‐L1 checkpoint blockade.103 Taken together, this evidence demonstrates that all aspects of m6A regulators are competent in modulating the immune responses to anti‐PD‐1/L1 treatment.
3.4. Cytokines
It has been reported that the cytokine production process is also controlled by m6A regulators‐mediated signaling pathways. METLL3 enhances NF‐κB signaling by controlling the levels of downstream effector molecules. MELLT3‐knockout DCs exhibit a dramatic decrease in production of the cytokines IL6, IL12, and TNF‐α following lipopolysaccharides (LPS) stimulation. Mechanistically, METTL3 deficiency causes a lower translation level of these cytokine mRNAs.27 Also, the type IFN‐β production raised by human cytomegalovirus or double‐stranded DNA was influenced by METTL14 and ALKBH5. Inhibiting the METTL14 genetic expression decreased the production and accumulation of type IFN‐β in the above process; however, inhibition of ALKBH5 obtained the opposite result.115 Previous studies have uncovered that these cytokines are positively involved in the accumulation and response of T cells and the subsequent antitumor effect.116, 117, 118, 119 Importantly, the combined targeting of IL6 with PD‐1/PD‐L1 and blocking of TNF and PD‐1 enables to increase the efficacy of PD‐1/PD‐L1 treatment, which significantly reduce tumor progression.117, 118, 120 Moreover, it has also been found that FTO is engaged in NF‐κB signaling, and inhibition of FTO leads to inactivation of this pathway.105 In colorectal cancer and melanoma, simultaneous depletion of METTL3 and METTL14 induces mass production of cytokines, including IFN‐γ, CXCL9, and CXCL10. The underlying mechanism is that suppression of METTL3 and METLL14 contributes to the stabilization of STAT1 and interferon regulatory factor 1 (IRF1) mRNAs, which orchestrate the IFN‐γ‐STAT1‐IRF1 signaling pathway in the TIME.108 Most strikingly, METTL3 has also been found to mediate the expression and secretion of TGF‐β1, which further modulates the TGF‐β‐induced epithelial‐mesenchymal transition (EMT) process of tumors. It has been suggested that the METTL3 expression level is negatively correlated with TGF‐β1 mRNA decay and translation elongation. In METTL3‐knockdown cancer cells, the half‐life, secretion, and activation of TGF‐β are inhibited, which contributes to the suppression of the biological process of EMT.121
To summarize, the m6A modification has shown tremendous potential in remodeling the TIME in various dimensions, including the function of multiple immune cells, cytokine secretion, and expression of immune checkpoint proteins. Therefore, it is believed that only the tip of the iceberg related to the role and mechanisms of the m6A modification in the reprograming of the TIME has been uncovered to date. Elucidation of the manner by which m6A modification remodels the TIME is not only essential for perceiving tumor biology but also provides a novel opportunity to explore the potential of this RNA regulation‐based therapy in cancer.
3.5. m6A modification: an underlying bridging between CSCs and the TIME
CSCs are notorious for being the root cause of tumor recurrence and resistance, which render current treatments ineffective in a vast number of patients. Moreover, the TIME is also a primary determinant of the efficacy of various therapies, especially immunotherapy. It has become apparent that CSCs and the TIME are never mutually exclusive in tumors but establish a deadly teamwork to facilitate tumor development and progression, serving as putative catalysts for each other. In the crosstalk between the CSC‐TIME, both intercellular contact and noncontact interactions occur. Taking the noncontact interactions as an example, tumor cells produce various cytokines to stimulate the expansion of suppressive immune cells, while these immune cells also secrete soluble factors to enhance the CSCs plasticity and the EMT process.122
Due to the rapid and unrestrained proliferation of tumor cells, hypoxia is a pervasive and prominent feature of the TIME. Hypoxia is indispensable for CSCs maintenance but also supports the acquisition of stemness characteristics in tumors. Strikingly, it has been reported that certain m6A regulators collaborate with HIF‐1α and HIF‐2α to promote the CSCs phenotype in various tumors.10, 25 Exposure to a hypoxic microenvironment profoundly stimulates the expression of HIFs and ALKBH5 in BC cells, which ultimately advances BCSCs stemness features and enrichment. During this biological process, HIFs may act as upstream regulators of ALKBH5‐mediated demethylation targeting the pluripotent gene NANOG, since changing the expression level of HIFs alters the activity of ALKBH5 accordingly. Dual regulation by HIFs and ALKBH5 gives rise to higher expression and lower degradation levels of target gene NANOG, ultimately increasing the percentage of BCSCs in BC.10 The same applies to ECSCs, in which suppression of HIFs also markedly decreases the protein expression level of ALKBH5 and thus diminishes its demethylation capacity. HIFs and ALKBH5 form a subtle collaboration to mediate the level of SOX2, which is the trigger for ECSCs initiation and development. This HIFs‐ALKBH5‐SOX2 axis endeavors to maintain the ECSCs phenotype and function in the TIME.25 These findings indicate that the m6A modification functions as a connector in the process of hypoxia‐induced stemness in ECSCs.
Meanwhile, the costimulatory and adhesion molecules also actively participate in the contact interaction in the CSC‐TIME connection, for instance, PD‐L1 signaling acts as an essential role to by promoting immune evasion and CSC growth. They work together to create a hypoxic and immunosuppressive environment that inhibits antitumor effects and promotes tumor progression and metastasis.122 The interaction between CSCs and the TIME remains under active investigation; however, some clues have been found that m6A modifications may play an important role in this interaction. m6A modification not only influences CSCs and the TIME but also mediates communication between them, and dissecting the detailed role of m6A modification will have profound implications in the discovery of novel related targets for tumor therapy.
PD‐L1 signaling is also an essential bridge between CSCs and the TIME. Immune cells produce cytokines to promote PD‐L1 expression in CSCs, while CSCs with a higher PD‐L1 expression induce immune evasion in the TIME.123 The m6A regulator FTO was identified to participate in this process. FTO enables to upregulate the PD‐L1 expression in colon cancer cells and subsequently promotes immune escape in the TIME.111
Integrins family members are known to participate in the signaling transduction between intracellular and extracellular matrix within the TIME and also play a significant role in cancer stemness, progression, and drug resistance.124, 125 Previous studies have elucidated that integrin‐α6 is concentrated on CSCs and maintains the stemness characteristics of CSCs.126 Moreover, recent studies have suggested that the m6A writer MELLT3 upregulates the expression level of integrin‐α6 in BCa cells, facilitating a more malignant phenotype with greater migration and invasion abilities.127 Therefore, it is reasonable to speculate that m6A modification may also determine the expression level of integrins, which are involved in prosurvival signaling and TIME reprogramming, thus facilitating the stemness features and function of cancer cells.
Taken together, m6A regulators are frequently present during the interaction between CSCs and the TIME. Even though the direct regulatory function of m6A modifications in this interaction has not yet been uncovered, there are many clues indicating that it plays an important role in regulating a variety of key molecules in this process. The specific communication mechanisms remain elusive, warranting further research to explore the role of m6A modification in the CSC‐TIME interplay.
4. THERAPEUTIC POTENTIAL OF TARGETING m6A REGULATORS
Based on existing research, it is clear that m6A regulators play pivotal roles in carcinogenesis, demonstrating great potential in cancer treatment. To date, several relevant studies have explored the therapeutic value of certain m6A regulators. FTO inhibitors are the most well‐researched candidates targeting m6A modification in cancer therapy, with several FTO inhibitors that increase the m6A abundance on RNAs having already been successfully identified. First, FB23 and FE23‐2, other types of small‐molecule FTO inhibitors, are able to bind directly to the FTO active pocket, dealing a fatal blow to the proliferation of AML cells.128 In addition, another two FTO inhibitors, CS1 and CS2, have been found to limit the growth and function of LSCs, sensitize them to T‐cell cytotoxicity, and decrease their immune evasion ability.90 Another MA2 has also been shown to successfully suppress the phenotype of GSCs and impair tumor progression.12 Noticeably, entacapone has been identified as a potential novel FTO inhibitor in metabolic diseases based on the structure virtual screening FDA‐approved drugs. In addition, IGF2BP1 inhibitors have also demonstrated favorable antitumor effects in several malignancies including leukemia, melanoma, and ovarian cancer.92, 129 Moreover, a small‐molecule ALKBH5 inhibitor, ALK‐04, has been shown to cause a considerable increase in the efficacy of anti‐PD‐1 therapy both in vivo and in vitro.26 Further, the simultaneous suppression of METTL3 and METTL14 has also been demonstrated to augment the efficacy of anti‐PD‐1 therapy in colorectal cancer and melanoma, owing to the higher infiltration of CD8+ T cells and massive cytokine release in the TIME.108 Additionally, the METTL3 pharmacological inhibitor (STM2457) was also capable of effectively preventing AML growth and improving survival in AML models in preclinical experiments.88 Meanwhile, in NSCLC, the resistance to gefitinib caused by METTL3‐mediated autophagy process was reversed by β‐elemene.130 Together, these studies indicate that m6A regulator inhibitors will be useful in cancer treatment.
On the other hand, a bidirectional dCasRx m6A modification editing platform has been constructed, which is composed of nucleus dCasRx and either a reader (METTL3) or an eraser (ALKBH5). This editing platform is capable of regulating the methylation status at specific m6A sites in HEK293T and GBS cells, ultimately affecting the expression of target genes and cancer cell proliferation.131 This remarkable emerging technology provides a solid foundation for the clinical targeting of m6A modification in cancer treatment. Furthermore, some preclinical research also found that depletion of some m6A regulators can sensitize tumor cells to chemotherapies, including breast cancer and NK/T cell lymphoma.132, 133 Nevertheless, further detailed studies are required to realize its potential. Finally, it is notable that m6A expression profiles have great potential to differentiate the immune characteristics of patients with tumors, which is likely to accurately guide the application of immunotherapy in clinical.134, 135
5. DISCUSSION AND PERSPECTIVE
Despite still being in the immature stage of exploration, numerous studies have indicated that m6A modification and the corresponding regulators orchestrate a range of critical pathological processes in tumorigenesis and development by regulating the epitranscriptome. Notably, m6A modification actively participates in the development of CSCs in various tumors, determining their fate and functions to influence tumor progression. Both genetic and pharmacological inhibition of m6A regulators enable the suppression of CSCs self‐renewal and growth, thus limiting tumor formation and progression. Moreover, m6A modification also remodels the TIME in various aspects, including immune cell regulation, cytokine production, and immune checkpoint expression. Targeting m6A regulators is an effective method for sensitizing immune responses in anti‐PD‐1/L1 immunotherapy both in vitro and in vivo, which further profoundly indicates a novel strategy to compensate for the limitations of immunotherapy. The m6A modification is a dual regulator in both CSCs and TIME, but its direct function in this interaction has not yet been proposed. However, some clues indicate that it is involved in the deadly teamwork of the CSCs‐TIME. m6A regulators collaborate with hypoxic factors, integrins, and PD‐L1 to influence interactions between the CSC and TIME, ultimately promoting the tumor development and progression. It is clear that the CSC‐TIME interplay has an essential function in the therapeutic resistance and unfavorable survival. We believe more relevant research will further reveal the regulate function of m6A modification in such crosstalk, which will also be informative for cancer eradication and therapy.
As rapid advancements are made in m6A sequencing and detection methods and continuous refinement of m6A‐based drugs development is performed, it is promising that m6A modification will open a new opportunity for tumor diagnosis and treatment.136, 137, 138, 139 Given the central role of m6A modification in tumorigenesis and development, it is reasonable to speculate that m6A modification possesses significant value in the clinical diagnosis and treatment of cancer. First, m6A regulators are promising biomarkers for distinguishing benign from malignant tumors and predicting metastasis, therapy resistance, and recurrence, which may be helpful in early diagnosis and individualized monitoring. In addition to m6A regulators, the global m6A profile based on blood or tissue may also be a reliable choice as a cancer biomarker for diagnosis, classification, and prognosis, warranting more advanced m6A‐seq technologies. Furthermore, it is noteworthy that m6A modification is closely related to the most promising immunotherapies, in particular anti‐PD‐1/PD‐L1 monotherapies, and its associated regulators hold great promise for the screening of appropriate immunotherapy candidates to achieve precision medicine. Moreover, the combination of m6A‐related biomarkers and other classical biomarkers is likely to spark new ideas for better clinical guidelines.
With regard to cancer treatment, m6A modification has demonstrated tremendous potential in various aspects. First, as more studies explore and validate the specificity and side effects of m6A regulatory inhibitors and editing platforms, the two m6A‐related therapies may function as emerging targeted treatments for tumor eradication. Second, combining m6A‐based and mainstream treatments is also an attractive and promising blueprint for the future. These m6A‐targeted regimens may compensate for the limitations and deficiencies of other current therapies to some extent. Nevertheless, the timing and sequence of combined regimens are critical during cancer treatment, and more research is needed to investigate and validate optimal decisions with a view to maximizing patient benefit. In addition, it is clear that aberrant m6A deposition level and m6A regulators expression play pivotal roles in therapeutic resistance mechanisms, such as chemoradiotherapy resistance and immunotherapy unresponsiveness. This may provide a new opportunity to patients with advanced drug‐resistant cancer for whom no medication is currently available, filling the significant gap still remaining in the current field of cancer therapeutics.
6. CONCLUSION
In conclusion, we have overviewed the landscape of m6A modifications in the cross‐linkage between CSC and TME for the first time, which may bring the possibility of m6A modifications as a new therapeutic target for tumor treatment. In addition, we also attempted to point out the direction of m6A modifications in the future clinical applications, which can be suggestive for individual therapy and improvement in efficacy of current treatments. Lastly, only the tip of the iceberg has been uncovered regarding the mechanisms related to m6A modification in the crosstalk between CSCs and the TIME, and deeper large‐scale studies are warranted for further exploration with a view to opening a new therapeutic avenue in cancer.
AUTHOR CONTRIBUTIONS
NS and JH designed this study and provided funding support. CQZ, YJL, and ZHZ drafted the manuscript and completed the figures. YJL, GCZ, and PW collected the references and completed the tables. All the authors reviewed and approved the final manuscript.
CONSENT FOR PUBLICATION
Consent for publication of this paper has been obtained from the authors.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
All authors would like to thank Dr Shasha Liu and Dr Xiaoxiao Lu for their kind help in the picture drawing. This work was supported by the CAMS Innovation Fund for Medical Sciences (2017‐I2M‐1‐005, 2016‐I2M‐1‐001), the National Natural Science Foundation of China (81802299, 81502514), the Fundamental Research Funds for the Central Universities (3332018070), and the National Key Basic Research Development Plan (2018YFC1312105).
Zhang Z, Zhang C, Luo Y, et al. RNA N 6‐methyladenosine modification in the lethal teamwork of cancer stem cells and the tumor immune microenvironment: Current landscape and therapeutic potential. Clin Transl Med. 2021;11:e525. 10.1002/ctm2.525
Zhihui Zhang, Chaoqi Zhang, and Yuejun Luo contributed equally.
Contributor Information
Nan Sun, Email: sunnan@vip.126.com.
Jie He, Email: prof.jiehe@gmail.com.
REFERENCES
- 1.Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971‐3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rottman F, Shatkin AJ, Perry RP. Sequences containing methylated nucleotides at the 5' termini of messenger RNAs: possible implications for processing. Cell. 1974;3(3):197‐199. [DOI] [PubMed] [Google Scholar]
- 3.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machnicka MA, Milanowska K, Osman Oglou O, et al. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013;41(Database issue):D262‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149(7):1635‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature. 2012;485(7397):201‐206. [DOI] [PubMed] [Google Scholar]
- 7.Dai D, Wang H, Zhu L, Jin H, Wang X. N6‐methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9(2):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batista PJ. The RNA modification N(6)‐methyladenosine and its implications in human disease. Genom, Proteom Bioinform. 2017;15(3):154‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Weng H, Chen J. m(6)A modification in coding and non‐coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37(3):270‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF‐dependent and ALKBH5‐mediated m⁶A‐demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047‐2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Zhao BS, Zhou A, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem‐like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591‐606.e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui Q, Shi H, Ye P, et al. m(6)A RNA methylation regulates the self‐renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622‐2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.López de Andrés J, Griñán‐Lisón C, Jiménez G, Marchal JA. Cancer stem cell secretome in the tumor microenvironment: a key point for an effective personalized cancer treatment. J Hematol Oncol. 2020;13(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18(11):669‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lathia JD, Mack SC, Mulkearns‐Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011;8(5):482‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oskarsson T, Batlle E, Massagué J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14(3):306‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Zhang H, Jiang X, Qian C, Liu Z, Luo D. Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol Cancer. 2017;16(1):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song M, Ping Y, Zhang K, et al. Low‐dose IFNγ induces tumor cell stemness in tumor microenvironment of non‐small cell lung cancer. Cancer Res. 2019;79(14):3737‐3748. [DOI] [PubMed] [Google Scholar]
- 20.Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—a clinical update. Nat Rev Clin Oncol. 2020;17(4):204‐232. [DOI] [PubMed] [Google Scholar]
- 21.Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Codony‐Servat J, Rosell R. Cancer stem cells and immunoresistance: clinical implications and solutions. Transl Lung Cancer Res. 2015;4(6):689‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Codd AS, Kanaseki T, Torigo T, Tabi Z. Cancer stem cells as targets for immunotherapy. Immunology. 2018;153(3):304‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Fan S, Wu M, et al. YTHDF1 links hypoxia adaptation and non‐small cell lung cancer progression. Nat Commun. 2019;10(1):4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen G, Liu B, Yin S, et al. Hypoxia induces an endometrial cancer stem‐like cell phenotype via HIF‐dependent demethylation of SOX2 mRNA. Oncogenesis. 2020;9(9):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li N, Kang Y, Wang L, et al. ALKBH5 regulates anti‐PD‐1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117(33):20159‐20170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Hu X, Huang M, et al. Mettl3‐mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355(6330):1147‐1152. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Feng J, Xue Y, et al. Structural basis of N(6)‐adenosine methylation by the METTL3‐METTL14 complex. Nature. 2016;534(7608):575‐578. [DOI] [PubMed] [Google Scholar]
- 30.Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6‐methyladenosine methyltransferase. Cell Res. 2014;24(2):177‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen J, Lv R, Ma H, et al. Zc3h13 Regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self‐renewal. Mol Cell. 2018;69(6):1028‐1038.e1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue Y, Liu J, Cui X, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knuckles P, Lence T, Haussmann IU, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA‐binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32(5‐6):415‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patil DP, Chen CK, Pickering BF, et al. m(6)A RNA methylation promotes XIST‐mediated transcriptional repression. Nature. 2016;537(7620):369‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sendinc E, Valle‐Garcia D, Dhall A, et al. PCIF1 catalyzes m6Am mRNA methylation to regulate gene expression. Mol Cell. 2019;75(3):620‐630.e629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Tran N, Ernst FGM, Hawley BR, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47(15):7719‐7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma H, Wang X, Cai J, et al. N(6‐)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15(1):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169(5):824‐835.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Ma Y, Han W, et al. Proteinase‐activated receptor 2 promotes cancer cell migration through RNA methylation‐mediated repression of miR‐125b. J Biol Chem. 2015;290(44):26627‐26637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerken T, Girard CA, Tung YC, et al. The obesity‐associated FTO gene encodes a 2‐oxoglutarate‐dependent nucleic acid demethylase. Science. 2007;318(5855):1469‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang C, Klukovich R, Peng H, et al. ALKBH5‐dependent m6A demethylation controls splicing and stability of long 3'‐UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. 2018;115(2):E325‐e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei J, Liu F, Lu Z, et al. Differential m(6)A, m(6)A(m), and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973‐985.e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou S, Toh JD, Wong KH, Gao YG, Hong W, Woon EC. N(6)‐methyladenosine: a conformational marker that regulates the substrate specificity of human demethylases FTO and ALKBH5. Sci Rep. 2016;6:25677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueda Y, Ooshio I, Fusamae Y, et al. AlkB homolog 3‐mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7:42271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507‐519. [DOI] [PubMed] [Google Scholar]
- 47.Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m(6)A transcripts by the 3'→5' RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374‐387.e312. [DOI] [PubMed] [Google Scholar]
- 48.Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Ann Rev Cell Dev Biol. 2017;33:319‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roundtree IA, Luo GZ, Zhang Z, et al. YTHDC1 mediates nuclear export of N(6)‐methyladenosine methylated mRNAs. eLife. 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kretschmer J, Rao H, Hackert P, Sloan KE, Höbartner C, Bohnsack MT. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5'‐3' exoribonuclease XRN1. RNA (New York, NY). 2018;24(10):1339‐1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Zhao BS, Roundtree IA, et al. N(6)‐methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m(6)A‐containing RNA through direct recruitment of the CCR4‐NOT deadenylase complex. Nat Commun. 2016;7:12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)‐methyladenosine‐modified RNA. Cell Res. 2017;27(3):315‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dixit D, Prager BC, Gimple RC, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. 2021;11(2):480‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang H, Weng H, Sun W, et al. Recognition of RNA N(6)‐methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)‐methyladenosine‐dependent RNA structural switches regulate RNA‐protein interactions. Nature. 2015;518(7540):560‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou KI, Shi H, Lyu R, et al. Regulation of co‐transcriptional pre‐mRNA splicing by m(6)A through the low‐complexity protein hnRNPG. Mol Cell. 2019;76(1):70‐81.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arguello AE, DeLiberto AN, Kleiner RE. RNA chemical proteomics reveals the N(6)‐methyladenosine (m(6)A)‐regulated protein‐RNA interactome. J Am Chem Soc. 2017;139(48):17249‐17252. [DOI] [PubMed] [Google Scholar]
- 59.Edupuganti RR, Geiger S, Lindeboom RGH, et al. N(6)‐methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol. 2017;24(10):870‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang F, Kang Y, Wang M, et al. Fragile X mental retardation protein modulates the stability of its m6A‐marked messenger RNA targets. Hum Mol Genet. 2018;27(22):3936‐3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer KD, Patil DP, Zhou J, et al. 5' UTR m(6)A promotes cap‐independent translation. Cell. 2015;163(4):999‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105‐111. [DOI] [PubMed] [Google Scholar]
- 63.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275‐291. [DOI] [PubMed] [Google Scholar]
- 64.Bertero A, Brown S, Madrigal P, et al. The SMAD2/3 interactome reveals that TGFβ controls m(6)A mRNA methylation in pluripotency. Nature. 2018;555(7695):256‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao BS, He C. Fate by RNA methylation: m6A steers stem cell pluripotency. Genome Biol. 2015;16(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geula S, Moshitch‐Moshkovitz S, Dominissini D, et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347(6225):1002‐1006. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6‐methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22(2):191‐205.e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weller M, Wick W, Aldape K, et al. Glioma. Nat Rev Dis Primers. 2015;1:15017. [DOI] [PubMed] [Google Scholar]
- 70.Zhang C, Zhang Z, Li F, et al. Large‐scale analysis reveals the specific clinical and immune features of B7‐H3 in glioma. Oncoimmunology. 2018;7(11):e1461304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Visvanathan A, Patil V, Arora A, et al. Essential role of METTL3‐mediated m(6)A modification in glioma stem‐like cells maintenance and radioresistance. Oncogene. 2018;37(4):522‐533. [DOI] [PubMed] [Google Scholar]
- 72.Li F, Yi Y, Miao Y, et al. N(6)‐methyladenosine modulates nonsense‐mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79(22):5785‐5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Degrauwe N, Schlumpf TB, Janiszewska M, et al. The RNA binding protein IMP2 preserves glioblastoma stem cells by preventing let‐7 target gene silencing. Cell Rep. 2016;15(8):1634‐1647. [DOI] [PubMed] [Google Scholar]
- 74.Mineo M, Ricklefs F, Rooj AK, et al. The long non‐coding RNA HIF1A‐AS2 facilitates the maintenance of mesenchymal glioblastoma stem‐like cells in hypoxic niches. Cell Rep. 2016;15(11):2500‐2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dixit D, Prager BC, Gimple RC, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Y, Feng W, Gu S, et al. The UCA1/KRAS axis promotes human pancreatic ductal adenocarcinoma stem cell properties and tumor growth. Am J Cancer Res. 2019;9(3):496‐510. [PMC free article] [PubMed] [Google Scholar]
- 77.Hu X, Peng WX, Zhou H, et al. IGF2BP2 regulates DANCR by serving as an N6‐methyladenosine reader. Cell Death Different. 2020;27(6):1782‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li T, Hu PS, Zuo Z, et al. METTL3 facilitates tumor progression via an m(6)A‐IGF2BP2‐dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bai Y, Yang C, Wu R, et al. YTHDF1 regulates tumorigenicity and cancer stem cell‐like activity in human colorectal carcinoma. Front Oncol. 2019;9:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang C, Huang S, Zhuang H, et al. YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene. 2020;39(23):4507‐4518. [DOI] [PubMed] [Google Scholar]
- 81.Kessler SM, Laggai S, Barghash A, et al. IMP2/p62 induces genomic instability and an aggressive hepatocellular carcinoma phenotype. Cell Death Dis. 2015;6(10):e1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin. 2011;61(6):409‐418. [DOI] [PubMed] [Google Scholar]
- 83.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bell DW, Ellenson LH. Molecular genetics of endometrial carcinoma. Ann Rev Pathol. 2019;14:339‐367. [DOI] [PubMed] [Google Scholar]
- 85.Huang H, Wang Y, Kandpal M, et al. FTO‐dependent N (6)‐methyladenosine modifications inhibit ovarian cancer stem cell self‐renewal by blocking cAMP signaling. Cancer Res. 2020;80(16):3200‐3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13(11):470‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129(12):1577‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yankova E, Blackaby W, Albertella M, et al. Small‐molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593(7860):597‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shen C, Sheng Y, Zhu AC, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self‐renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27(1):64‐80.e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Su R, Dong L, Li Y, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38(1):79‐96.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van Der Werf I, Jamieson C. The Yin and Yang of RNA methylation: an imbalance of erasers enhances sensitivity to FTO demethylase small‐molecule targeting in leukemia stem cells. Cancer Cell. 2019;35(4):540‐541. [DOI] [PubMed] [Google Scholar]
- 92.Elcheva IA, Wood T, Chiarolanzio K, et al. RNA‐binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia. 2020;34(5):1354‐1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paris J, Morgan M, Campos J, et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25(1):137‐148.e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cheng M, Sheng L, Gao Q, et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF‐κB/MYC signaling network. Oncogene. 2019;38(19):3667‐3680. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y, Zeng L, Liang C, et al. Integrated analysis of transcriptome‐wide m(6)A methylome of osteosarcoma stem cells enriched by chemotherapy. Epigenomics. 2019;11(15):1693‐1715. [DOI] [PubMed] [Google Scholar]
- 96.Gao Q, Zheng J, Ni Z, et al. The m(6)A methylation‐regulated AFF4 promotes self‐renewal of bladder cancer stem cells. Stem cells Int. 2020;2020:8849218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou R, Gao Y, Lv D, Wang C, Wang D, Li Q. METTL3 mediated m(6)A modification plays an oncogenic role in cutaneous squamous cell carcinoma by regulating ΔNp63. Biochem Biophys Res Commun. 2019;515(2):310‐317. [DOI] [PubMed] [Google Scholar]
- 98.Foster JG, Wong SC, Sharp TV. The hypoxic tumor microenvironment: driving the tumorigenesis of non‐small‐cell lung cancer. Future Oncol (Lond, Engl). 2014;10(16):2659‐2674. [DOI] [PubMed] [Google Scholar]
- 99.Li B, Jiang J, Assaraf YG, Xiao H, Chen ZS, Huang C. Surmounting cancer drug resistance: new insights from the perspective of N(6)‐methyladenosine RNA modification. Drug Resist Updat. 2020;53:100720. [DOI] [PubMed] [Google Scholar]
- 100.Wang L, Zhu B, Zhang M, Wang X. Roles of immune microenvironment heterogeneity in therapy‐associated biomarkers in lung cancer. Semin Cell Dev Biol. 2017;64:90‐97. [DOI] [PubMed] [Google Scholar]
- 101.Zhang C, Zhang G, Sun N, et al. An individualized immune signature of pretreatment biopsies predicts pathological complete response to neoadjuvant chemoradiotherapy and outcomes in patients with esophageal squamous cell carcinoma. Signal Transduct Target Therap. 2020;5(1):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jia R, Li X, Yu C, Fan M, Guo J. The splicing factor hnRNP C regulates expression of co‐stimulatory molecules CD80 and CD40 in dendritic cells. Immunol Lett. 2013;153(1‐2):27‐32. [DOI] [PubMed] [Google Scholar]
- 103.Han D, Liu J, Chen C, et al. Anti‐tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu J, Zhang X, Chen K, et al. CCR7 chemokine receptor‐inducible lnc‐Dpf3 restrains dendritic cell migration by inhibiting HIF‐1α‐mediated glycolysis. Immunity. 2019;50(3):600‐615.e615. [DOI] [PubMed] [Google Scholar]
- 105.Gu X, Zhang Y, Li D, Cai H, Cai L, Xu Q. N6‐methyladenosine demethylase FTO promotes M1 and M2 macrophage activation. Cell Signal. 2020;69:109553. [DOI] [PubMed] [Google Scholar]
- 106.Li HB, Tong J, Zhu S, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL‐7/STAT5/SOCS pathways. Nature. 2017;548(7667):338‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tong J, Cao G, Zhang T, et al. m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018;28(2):253‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang L, Hui H, Agrawal K, et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti‐PD‐1 therapy. EMBO J. 2020;39(20):e104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ni HH, Zhang L, Huang H, Dai SQ, Li J. Connecting METTL3 and intratumoural CD33(+) MDSCs in predicting clinical outcome in cervical cancer. J Transl Med. 2020;18(1):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yi L, Wu G, Guo L, Zou X, Huang P. Comprehensive analysis of the PD‐L1 and immune infiltrates of m(6)A RNA methylation regulators in head and neck squamous cell carcinoma. Mol Therap Nucleic Acid. 2020;21:299‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsuruta N, Tsuchihashi K, Ohmura H, et al. RNA N6‐methyladenosine demethylase FTO regulates PD‐L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020;530(1):235‐239. [DOI] [PubMed] [Google Scholar]
- 112.Schmiedel D, Tai J, Yamin R, Berhani O, Bauman Y, Mandelboim O. The RNA binding protein IMP3 facilitates tumor immune escape by downregulating the stress‐induced ligands ULPB2 and MICB. eLife. 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen L, Han X. Anti‐PD‐1/PD‐L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125(9):3384‐3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang S, Wei J, Cui YH, et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti‐PD‐1 blockade. Nat Commun. 2019;10(1):2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I. RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev. 2018;32(23‐24):1472‐1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Garris CS, Arlauckas SP, Kohler RH, et al. Successful anti‐PD‐1 cancer immunotherapy requires T cell‐dendritic cell crosstalk involving the cytokines IFN‐γ and IL‐12. Immunity. 2018;49(6):1148‐1161.e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mace TA, Shakya R, Pitarresi JR, et al. IL‐6 and PD‐L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tsukamoto H, Fujieda K, Miyashita A, et al. Combined blockade of IL6 and PD‐1/PD‐L1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res. 2018;78(17):5011‐5022. [DOI] [PubMed] [Google Scholar]
- 119.Bertrand F, Rochotte J, Colacios C, et al. Blocking tumor necrosis factor α enhances CD8 T‐cell‐dependent immunity in experimental melanoma. Cancer Res. 2015;75(13):2619‐2628. [DOI] [PubMed] [Google Scholar]
- 120.Bertrand F, Montfort A, Marcheteau E, et al. TNFα blockade overcomes resistance to anti‐PD‐1 in experimental melanoma. Nat Commun. 2017;8(1):2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li J, Chen F, Peng Y, et al. N6‐methyladenosine regulates the expression and secretion of TGFβ1 to affect the epithelial‐mesenchymal transition of cancer cells. Cells. 2020;9(2):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor‐intrinsic PD‐L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front Oncol. 2018;8:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25(4):234‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dustin ML. Integrins and their role in immune cell adhesion. Cell. 2019;177(3):499‐501. [DOI] [PubMed] [Google Scholar]
- 126.Lathia JD, Gallagher J, Heddleston JM, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6(5):421‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jin H, Ying X, Que B, et al. N(6)‐methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang Y, Su R, Sheng Y, et al. Small‐molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35(4):677‐691.e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c‐Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10(5):818‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu S, Li Q, Li G, et al. The mechanism of m(6)A methyltransferase METTL3‐mediated autophagy in reversing gefitinib resistance in NSCLC cells by β‐elemene. Cell Death Dis. 2020;11(11):969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xia Z, Tang M, Zhang H, et al. Epitranscriptomic editing of the RNA N6‐methyladenosine modification by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res. 2021;49:7361‐7374. 2020:2020.2010.2027.356436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu X, Gonzalez G, Dai X, et al. Adenylate kinase 4 modulates the resistance of breast cancer cells to tamoxifen through an m(6)A‐based epitranscriptomic mechanism. Mol Ther. 2020;28(12):2593‐2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ma H, Shen L, Yang H, Gong H, Du X, Li J. m6A methyltransferase Wilms' tumor 1‐associated protein facilitates cell proliferation and cisplatin resistance in NK/T cell lymphoma by regulating dual‐specificity phosphatases 6 expression via m6A RNA methylation. IUBMB Life. 2021;73(1):108‐117. [DOI] [PubMed] [Google Scholar]
- 134.He X, Tan L, Ni J, Shen G. Expression pattern of m(6)A regulators is significantly correlated with malignancy and antitumor immune response of breast cancer. Cancer Gene Therap. 2021;28(3‐4):188‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. m(6)A regulator‐mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Niu Y, Wan A, Lin Z, Lu X, Wan G. N (6)‐Methyladenosine modification: a novel pharmacological target for anti‐cancer drug development. Acta Pharmaceut Sinica B. 2018;8(6):833‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Garcia‐Campos MA, Edelheit S, Toth U, et al. Deciphering the “m(6)A Code” via antibody‐independent quantitative profiling. Cell. 2019;178(3):731‐747.e716. [DOI] [PubMed] [Google Scholar]
- 139.Meyer KD. DART‐seq: an antibody‐free method for global m(6)A detection. Nat Meth. 2019;16(12):1275‐1280. [DOI] [PMC free article] [PubMed] [Google Scholar]