

Abstract
A series of new asymmetric ureas, urethanes, and other derivatives of the framework structure have been synthesized by the reactions of adamantan-1-yl isocyanate generated in situ by the thermolysis of carbamothioates with nitrogen-containing nucleophiles and alcohols.
Keywords: isocyanates, urethanes, ureas, carbamothioates, thermolysis, biological activity
INTRODUCTION
Isocyanates are strategically important substrates in the synthesis of amides [1–3], polyurethanes [4–6], urethanes [7] and ureas, including those with biological activity [8–11]. Adamantane-containing isocyanates are initial substrates in the synthesis of asymmetric ureas, which act as effective inhibitors of human soluble epoxide hydrolase (human sEH) [12–15], an enzyme playing an important in the metabolism of epoxidated fatty acids. Urethanes derived from adamantan-1-yl isocyanate can act as potential acetyl- and butyrylcholinesterase inhibitors [16] and exhibit antimicrobial activity [17]. Some information is available of the potential antitubercular activity of adamantane-containing urethanes [18] and unsymmetrical ureas [19–22].
The practical value of derivatives of adamantan-1-yl isocyanate consists of their use as starting materials in supramolecular chemistry [23] and as catalysts in enantioselective processes. For example, (S)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-7-yl (adamantan-1-yl)carbamate has shown high efficiency in the synthesis of Remdesivir, one of the antiviral drugs used to treat COVID-19 [24].
Isocyanates are commonly prepared by phosgenation of amines or by Curtius rearrangement from acyl azides [25]. Most of the methods for the preparation of adamantan-1-yl isocyanate include the aforementioned treatment of 1-aminoadamantane hydrochloride with phosgene [26, 27] or triphosgene [28] and the implementation of Curtius rearrangement [29], including with the preliminary preparation of 1-azidoadamantane from adamantane-1-carboxylic acid [13, 30] or its acid chloride [31–35]. Alternative synthetic approaches to adamantan-1-yl isocyanate involve the cleavage of 1,3-dehydroadamantane in sulfuric acid in the presence of sodium cyanate [36], the reaction of 1-bromoadamantane with the silver salt of nitrocyanamide [37], and thermolysis of 2-(adamantan-1-yl)-5-methyl-1,3-oxothiolane [38].
Even though isocyanates are widely used in organic synthesis, this class of compounds has a few important disadvantages such as high toxicity and hydrolytic instability. To overcome these disadvantages, a lot of syntheses of urethanes, ureas, and other compounds were proposed, which combine in situ generation of isocyanates and their subsequent reactions with nucleophilic agents. The reported precursors of isocyanates include N-alkoxyphenylcarbamates [39], hydroxamic acids [40, 41], amides [42, 43], 1,1'-(1,2-phenylene)bisureas [44], and Cbz amines [45]. We have not found examples of in situ generation of adamantan-1-yl isocyanate in the literature.
RESULTS AND DISCUSSION
In this work, we propose a method for the synthesis of a wide range of unsymmetrical ureas, urethanes, and other derivatives, which involves in situ generation of adamantan-1-yl isocyanate. Since S-alkyl carbamothioates can be considered as adducts of mercaptans to isocyanates [46–48]. N-Adamantylated S-alkyl carbamothioates can act as a synthetic equivalent of adamantan-1-yl isocyanate [49], because S-alkyl (adamantan-1-yl)carbamothioates contain a bulky cage fragment at the nitrogen atom, which decreases the thermal stability of thiourethanes.
As the main starting substrate we chose S-ethyl (adamantan-1-yl)carbamothioate (1), readily available by the reaction of adamantan-1-ol or its nitrate with ethyl thiocyanate in sulfuric acid [50–52]. S-Methyl (adamantan-1-yl)carbamothioate, too, converts into adamantan-1-yl isocyanate, but, unlike carbamothioate 1, it thermolyses under more rigid conditions. As nucleophiles we employed alcohols and aliphatic, aromatic, and heterocyclic nitrogen-containing compounds.
Compound 1 reacted with alcohols to form N-(adamantan-1-yl)urethanes (Scheme 1). The reactions were performed in an excess of the nucleophilic reagent in the absence of a solvent. The reaction involved the intermediate formation of adamantan-1-yl isocyanate (A) and the attendant release of ethanethiol. The formation of adamantan-1-yl isocyanate was also confirmed by the observation of the corresponding peak in the gas chromatogram of the starting carbamothioate 1 analyzed at the injector temperature of 250°C. The yields of products 2–8 were 44–75%. The products of the reactions of compound 1 with aminoalcohols were isolated as hydrochlorides 5–8.
Scheme.

1.
The 1H NMR spectra of N-(adamantan-1-yl)carbamates 2–8 display the NH proton signals as singlets at 4.6–6.9 ppm. The signal of the quaternary carbon atom of the urethane fragment in the 13C NMR spectrum is observed at 156–160 ppm.
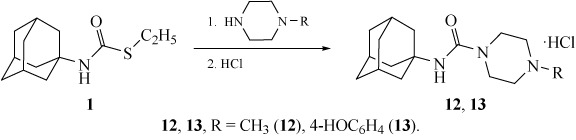
The reactions of carbamothioate 1 with amines gave substituted N-(adamantan-1-yl)ureas 9–13 (Schemes 2 and 3). Products 12 and 13 were isolated as hydrochlorides. The reactions were performed in the absence of a solvent under heating in an excess of the amine (products 9, 12, and 13) or by fusion (products 10 and 11).
Scheme.

2.
Scheme.
3.
The 1H NMR spectra of ureas 9–13 contain proton signals of the NH group attached to the adamantanyl radical as singlets at 6.6–7.2 ppm. The signal of the quaternary carbon atom of the C=O group in the 13C NMR spectrum is observed at 159–161 ppm.
Heating of carbamothioate 1 in an excess of hydrazine hydrate resulted in the isolation of 4-(adamantan-1-yl)semicarbazide (14). The acylsemicarbazide, an intermediate product of the reaction with acetylhydrazine, underwent spontaneous cyclization to form 2-(adamantan-1-yl)amino-5-methyl-1,3,4-oxadiazole (15) (Scheme 4).
Scheme.

4.
In the 1H NMR spectrum of 1,3,4-oxadiazole 15, the methyl protons appear as a singlet at 2.51 ppm. The NH proton gives a singlet at 5.62 ppm. In the 13C NMR spectrum, no quaternary carbon signal was observed in the region of 160 ppm.
The reaction of carbamothioate 1 with primary amides formed N-adamantanyl-N'-acylureas 16 and 17 (Scheme 5). The structure of the synthesized compounds was confirmed by the NMR spectra. The 1H NMR spectra show NH proton signals as singlets at 7.0–9.0 ppm.
Scheme.

5.
EXPERIMENTAL
The IR spectra were run on a Shimadzu IR Affinity-1 spectrometer in KBr pellets. The 1H and 13C NMR spectra were obtained on a JEOL NMR-ECX400 spectrometer at 400 and 100 MHz, respectively, in DMSO-d6, using residual proton and carbon signals of the solvent as internal references. The melting points were measured in capillaries on an SRS OptiMelt MPA 100 apparatus and are uncorrected. The elemental analyses were obtained in a EuroVector 3000 EA analyzer, using L-cystine as reference.
N-(Adamantan-1-yl) S-ethyl carbamothioate (1) was prepared as described in [50].
2-Hydroxyethyl (adamant-1-yl)carbamate (2). A mixture of 2 g (0.0084 mol) of carbamothioate 1 and 5 mL (0.09 mol) of ethylene glycol was heated under reflux for 1 h. The hot reaction mixture was poured into water. The product precipitated as a brown oil which gradually solidified and was separated by decantation and recrystallized. Yield 1.16 g (58%). Colorless crystals, mp 76–78°C (benzene–heptane) (77–78°C [49]). C13H21NO3.
2-Bromoethyl (adamant-1-yl)carbamate (3). A mixture of 2 g (0.0084 mol) of carbamothioate 1 and 5 mL (0.07 mol) of 2-bromoethanol was heated at 140°C for 1 h. Excess 2- bromoethanol was removed in a vacuum. The residue was purified by vacuum distillation, and the fraction at 155–157°C (9 mmHg) was collected. Yield 1.54 g (61%). Colorless oil. nD20 1.5650 [49]. C13H20BrNO2.
Benzyl (adamant-1-yl)carbamate (4). A mixture of 2 g (0.0084 mol) of carbamothioate 1 and 3 mL (0.029 mol) of benzyl alcohol was heated at 170°C for 1 h. Excess benzyl alcohol was removed in a vacuum. The residue was distilled at 203–205°C (2 mmHg) and crystallized from pentane. Yield 1.31 g (55%). Colorless crystals, mp 29–31°C. IR spectrum, ν, cm–1: 3340, 1710, 1505, 1285, 1050, 735, 695. 1H NMR spectrum, δ, ppm: 1.65–2.00 m (15H, CHAd), 4.97 s (2H, CH2), 5.89 s (1H, NH), 7.34–7.39 m (5Harom). 13C NMR spectrum, δ, ppm: 28.6 (CH), 38.2 (CH2), 41.6 (CH2), 50.2 (C), 68.6 (CH2), 127.3 (CH), 128.4 (CH), 129.6 (CH), 136.4 (C), 159.3 (C). Found, %: C 75.65; H 8.21; N 4.83. C18H23NO2. Calculated, %: C 75.76; H 8.12; N 4.91.
2-({[(Adamantan-1-yl)carbamoyl]oxy}ethyl)cyclohexylammonium chloride (5). A mixture of 3 g (0.0126 mol) of carbamothioate 1 and 5 g (0.035 mol) of 2-(cyclohexylamino)ethanol was heated at 200°C for 2 h and poured into water. The product was extracted with benzene (3 × 40 mL), the combined organic extracts were washed with 5% NaOH and water (3 × 100 mL), dried, and saturated with gaseous HCl. The solution was evaporated to 1/3 of the volume and cooled. The precipitate that formed was filtered off. Yield 3.35 g (75%). Colorless crystals, mp 207–209°C (benzene). IR spectrum, ν, cm–1: 3305, 1725, 1520, 1225, 1075. 1H NMR spectrum, δ, ppm: 1.25–2.20 m (25H, CHAd, CH), 3.26–3.28 m (2H, CH2), 3.60–3.68 m (1H, CH), 4.18–4.23 m (2H, CH2), 6.88 s (1H, NH), 9.17 s (2H, NH2+). 13C NMR spectrum, δ, ppm: 25.9 (CH2), 26.3 (CH2), 28.5 (CH), 30.4 (CH2), 39.2 (CH2), 39.7 (CH2), 46.5 (CH2), 50.4 (C), 56.2 (CH), 66.4 (CH2), 157.8 (C). Found, %: C 63.85; H 9.43; N 7.74. C19H33ClN2O2. Calculated, %: C 63.94; H 9.32; N 7.85.
2-({[(Adamantan-1-yl)carbamoyl]oxy}ethyl)phenylammonium chloride (6). A mixture of 2.4 g (0.01 mol) of carbamothioate 1 and 1.4 mL (0.01 mol) of 2-(phenylamino)ethanol was heated at 200°C for 2 h and poured into water. The product was extracted with benzene (3 × 30 mL), the combined organic extracts were washed with 5% NaOH and water (3 × 100 mL), dried, and saturated with gaseous HCl. The solution was evaporated to 1/3 of the volume and cooled. The precipitate that formed was filtered off. Yield 1.96 g (56%). Colorless crystals, mp 162–165°C (benzene). IR spectrum, ν, cm–1: 3345, 2600, 2430, 1700, 1600, 1520, 1285, 1020. 1H NMR spectrum, δ, ppm: 1.59–1.99 m (15H, CHAd), 3.40–3.44 m (2H, CH2), 4.11 t (2H, CH2, J 7.3 Hz), 6.77 s (1H, NH), 7.20–7.59 m (5Harom), 9.82 s (2H, NH2+). 13C NMR spectrum, δ, ppm: 28.4 (CH), 38.8 (CH2), 39.6 (CH2), 42.6 (CH2), 50.3 (C), 66.3 (CH2), 121.4 (CH), 123.4 (CH), 127.8 (CH), 139.8 (C), 158.0 (C). Found, %: C 64.94; H 7.67; N 8.09. C19H27ClN2O2. Calculated, %: C 65.04; H 7.76; N 7.98.
4-{[(Adamantan-1-yl)carbamoyl]oxy}quinuclidinium chloride (7). A mixture of 1.5 g (0.0063 mol) of carbamothioate 1 and 2 g (0.0157 mol) of quinuclidil-3-ol was heated at 200°C for 2 h and poured into water. The product was extracted with benzene (3 × 30 mL), the combined organic extracts were washed with 5% NaOH and water (3 × 100 mL), dried, and saturated with gaseous HCl. The solvent was evaporated, and the residue was recrystallized from acetonitrile. Yield 0.94 g (44%). Colorless crystals, mp 291–293°C (decomp.). IR spectrum, ν, cm–1: 3250, 2550, 2440, 1715, 1535, 1035. 1H NMR spectrum, δ, ppm: 1.60–2.07 m (19H, CHAd, CH), 2.23–2.26 m (1H, CH), 3.02–3.06 m (1H, CH), 3.18–3.26 m (4H, CH2), 3.61–3.66 m (1H, CH), 4.79–4.83 m (1H, CH), 6.82 s (1H, NH), 10.46 s (1H, NH+). 13C NMR spectrum, δ, ppm: 24.3 (CH2), 27.7 (CH2), 28.1 (CH), 28.4 (CH), 39.9 (CH2), 42.6 (CH2), 46.1 (CH2), 50.7 (C), 53.6 (CH2), 76.4 (CH), 157.0 (C). Found, %: C 63.34; H 8.67; N 8.13. C18H29ClN2O2. Calculated, %: C 63.42; H 8.58; N 8.22.
4-({[(Adamantan-1-yl)carbamoyl]oxy}methyl)quinuclidinium chloride (8). A mixture of 1.5 g (0.0063 mol) of carbamothioate 1 and 2 g (0.014 mol) of 3-(hydroxymethyl)quinuclidine [53] was heated at 200°C for 2 h and then poured into water. The product was extracted with benzene (3 × 30 mL), the combined organic extracts were washed with 5% NaOH and water (3 × 100 mL), dried, and saturated with gaseous HCl. The solvent was evaporated, and the residue was recrystallized from acetonitrile. Yield 1.44 g (65%). Colorless crystals, mp 151–153°C. IR spectrum, ν, cm–1: 3260, 2500, 2410, 1710, 1530, 1225, 1070. 1H NMR spectrum, δ, ppm: 1.52–2.19 m (21H, CHAd, CH), 2.78–3.02 m (6H, CH2), 3.95 d (2H, CH, J 6.9 Hz), 4.60 s (1H, NH), 9.20 s (1H, NH+). 13C NMR spectrum, δ, ppm: 27.7 (CH2), 28.4 (CH), 28.5 (CH2), 30.2 (CH), 36.4 (CH), 38.7 (CH2), 39.9 (CH2), 48.4 (CH2), 49.9 (CH2), 50.6 (C), 65.8 (CH2), 156.8 (C). Found, %: C 64.32; H 8.87; N 7.74. C19H31ClN2O2. Calculated, %: C 64.30; H 8.80; N 7.89.
1-(Adamantan-1-yl)-3-[2-(diethylamino)ethyl]urea (9). A mixture of 1.5 g (0.0063 mol) carbamothioate 1 and 3 mL (0.021 mol) of 2-(diethylamino)ethylamine was heated under reflux for 2 h and poured into water. The product was extracted with chloroform (3 × 50 mL), the combined organic extracts were washed with 5% NaOH and water (3 × 100 mL), dried, and the solvent was evaporated. The residue was recrystallized from benzene. Yield 1.25 g (68%). Colorless crystals, mp105–107°C (decomp.). IR spectrum, ν, cm–1: 3435, 3340, 1655, 1630, 1560. 1H NMR spectrum, δ, ppm: 1.05 t (3H, CH3, J 7.3 Hz), 1.67–2.02 m (15H, CHAd), 2.12–2.25 m (6H, CH2), 2.71–2.79 m (2H, CH2), 6.97 s (1H, NH), 8.75 s (1H, NH). 13C NMR spectrum, δ, ppm: 12.2 (CH3), 29.5 (CH), 36.2 (CH2), 39.5 (CH2), 42.9 (CH2), 47.7 (CH2), 50.9 (C), 55.3 (CH2), 159.8 (C). Found, %: C 69.19; H 10.78; N 14.50. C17H31N3O. Calculated, %: C 69.62; H 10.58; N 14.33.
1-(Adamantan-1-yl)-3-(2-methylquinolin-4-yl)urea (10). A mixture of 1.5 g (0.0063 mol) carbamothioate 1 and 1.5 g (0.0087 mol) of 4-amino-2-methylquinoline was fused at 130°C for 1 h. After the reaction had been complete, as evidenced by the solidification of the reaction mixture, water was added to the reaction flask, and the mixture was stirred until a precipitate formed. The precipitate was filtered off, washed with water, dried, and recrystallized. Yield 1.45 g (69%). Colorless crystals, mp 155–160°C (toluene). IR spectrum, ν, cm–1: 3410, 1705, 1630, 1610, 1550, 1275, 1220, 740. 1H NMR spectrum, δ, ppm: 1.65–2.12 m (15H, CHAd), 2.54 s (3H, CH3), 6.63 s (1H, NH), 7.47–8.23 m (5Harom), 8.71 s (1H, NH). 13C NMR spectrum, δ, ppm: 24.2 (CH3), 29.6 (CH), 36.6 (CH2), 43.9 (CH2), 50.4 (C), 112.4 (CH), 119.6 (C), 120.3 (CH), 126.4 (CH), 128.3 (CH), 130.1 (CH), 134.9 (C), 149.3 (C), 160.3 (C). Found, %: C 75.06; H 7.40; N 12.70. C21H25N3O. Calculated, %: C 75.22; H 7.46; N 12.54.
1-(Adamantan-1-yl)-3-(5-propyl-1,3,4-thiadiazol-2-yl)urea (11). A mixture of 1.5 g (0.0063 mol) of carbamothioate 1 and 0.9 g (0.0063 mol) of 2-amino-5-propyl-1,3,4-thiadiazole was fused at 140°C for 2 h. After the reaction had been complete, as evidenced by the solidification of the reaction mixture, water was added to the reaction flask, and the mixture was stirred until a precipitate formed. The precipitate was filtered off, washed with water, dried, and recrystallized. Yield 2.00 g (74%). Colorless crystals, mp 195–198°C (benzene–hexane). IR spectrum, ν, cm–1: 3490, 3310, 1720, 1690, 1600, 1530. 1H NMR spectrum, δ, ppm: 0.94 t (3H, CH3, J 7.5 Hz), 1.63–2.21 m (17H, CHAd, CH2), 2.78 t (2H, CH2, J 7.2 Hz), 6.95 s (1H, NH), 9.14 s (1H, NH). 13C NMR spectrum, δ, ppm: 13.8 (CH3), 19.3 (CH2), 29.6 (CH), 30.8 (CH2), 36.4 (CH2), 42.8 (CH2), 50.9 (C), 148.5 (C), 160.4 (C), 168.7 (C). Found, %: C 60.10; H 7.50; N 17.33. C16H24N4OS. Calculated, %: C 59.97; H 7.55; N 17.48.
N-[(Adamantan-1-yl)carbamoyl]-1-methylpiperazinium chloride (12). A mixture of 1.5 g (0.0063 mol) of carbamothioate 1 and 3 mL (0.027 mol) of N-methylpiperazine was heated under reflux for 3 h and poured into water. The product was extracted with chloroform (3 × 30 mL), the combined organic fractions were washed with 5% NaOH and water (3 × 100 mL), dried, and saturated with gaseous HCl. The precipitate that formed was filtered off. Yield 1.43 g (73%). White powder, mp 217–220°C. IR spectrum, ν, cm–1: 3505, 2510, 1640, 1538, 1283. 1H NMR spectrum, δ, ppm: 1.60–1.99 m (15H, CHAd), 2.64 s (3H, CH3), 3.29–3.50 m (4H, CH2), 3.86–4.00 m (4H, CH2), 6.74 s (1H, NH), 8.86 s (1H, NH+). 13C NMR spectrum, δ, ppm: 29.7 (CH), 36.9 (CH2), 41.1 (CH2), 41.4 (CH3), 44.2 (CH2), 49.1 (CH2), 51.6 (C), 159.3 (C). Found, %: C 61.30; H 8.80; N 13.20. C16H28ClN3O. Calculated, %: C 61.15; H 8.92; N 13.39.
4-[(Adamantan-1-yl)carbamoyl]-1-(4-hydroxyphenyl)piperazinium chloride (13). A mixture of 1 g (0.0042 mol) of carbamothioate 1 and 2 g (0.011 mol) of N-(4-hydroxyphenyl)piperazine was fused at 150°C for 1 h and poured into water. After the reaction had been complete, as evidenced by the solidification of the reaction mixture, water was added to the reaction flask, and the product was extracted with chloroform (3 × 30 mL). The combined organic fractions were washed with 5% NaOH and water (3 × 100 mL), dried, and saturated with gaseous HCl. The precipitate that formed was filtered off and recrystallized. Yield 1.22 g (75%). Colorless crystals, mp 252–255°C (methanol). IR spectrum, ν, cm–1: 3400, 1618, 1540, 1150, 1000. 1H NMR spectrum, δ, ppm: 1.58–2.03 m (15H, CHAd), 3.34–3.52 m (4H, CH2), 3.86–4.04 (4H, CH2), 7.12 s (1H, NH), 7.26 d (2Harom, J 8.0 Hz), 7.41 d (2Harom, J 8.0 Hz), 8.94 s (1H, NH+). 13C NMR spectrum, δ, ppm: 29.7 (CH), 36.9 (CH2), 38.9 (CH2), 42.3 (CH2), 44.1 (CH2), 51.6 (C), 120.4 (CH), 125.8 (CH), 141.7 (C), 158.6 (C), 159.5 (C). Found, %: C 64.22; H 7.81; N 10.59. C21H30ClN3O2. Calculated, %: C 64.35; H 7.72; N 10.72.
4-(Adamantan-1-yl)semicarbazide (14). A mixture of 1.5 g (0.0063 mol) of carbamothioate 1 and 5 mL of hydrazine hydrate heated under reflux for 1.5 h and poured into water. The precipitate that formed was filtered off, dried, and recrystallized from propan-2-ol. Yield 1.14 g (87%). Colorless crystals, mp 268–270°C [54]. C11H19N3O.
(Adamantan-1-yl)-5-methyl-1,3,4-oxadiazol-2-amine (15). A mixture of 1.5 g (0.0063 mol) carbamothioate 1 and 2 g (0.027 mol) of acetylhydrazine was heated at 140°C for 1.5 h and poured into water. The precipitate that formed was filtered off, dried, and recrystallized from propan-2-ol. Yield 0.61 g (42%). Colorless crystals, mp 275–278°C (propan-2-ol). IR spectrum, ν, cm–1: 3330, 1617, 1540. 1H NMR spectrum, δ, ppm: 1.55–2.03 m (15H, CHAd), 2.51 s (3H, CH3), 5.62 s (1H, NH). 13C NMR spectrum, δ, ppm: 11.1 (CH3), 29.1 (CH), 35.8 (CH2), 40.7 (CH2), 52.9 (C), 150.2 (C), 164.9 (C). Found, %: C 67.00; H 8.30; N 17.86. C13H19N3O. Calculated, %: C 66.92; H 8.21; N 18.01.
[(Adamantan-1-yl)carbamoyl]formamide (16). A mixture of 1.5 g (0.0063 mol) carbamothioate 1 and 5 mL (0.126 mol) of formamide was heated at 150°C for 1.5 h and poured into water. The product was extracted with chloroform (3 × 30 mL), the combined organic fractions were washed with 5% NaOH and water (3 × 100 mL), dried, and evaporated. The residue was purified by recrystallization. Yield 1.18 g (85%). Colorless crystals, mp 134–136°C (heptane–benzene). IR spectrum, ν, cm–1: 3180, 1680, 1545, 1310, 790. 1H NMR spectrum, δ, ppm: 1.65–2.17 m (15H, CHAd), 7.34 s (1H, NH), 7.87 s (1H, NH), 8.19 s (1H, COH), 8.22 s (1H, COH). 13C NMR spectrum, δ, ppm: 29.6 (CH), 37.0 (CH2), 43.4 (CH2), 50.8 (C), 160.3 (C), 167.4 (C). Found, %: C 64.93; H 8.24; N 12.70. C12H18N2O2. Calculated, %: C 64.84; H 8.16; N 12.60.
[(Adamantan-1-yl)carbamoyl]acetamide (17). A mixture of 1 g (0.0042 mol) of carbamothioate 1 and 3 g (0.051 mol) of acetamide was fused at 150°C for 2 h and poured into water. The precipitate that formed was filtered off, dried, and recrystallized from benzene. Yield 1.03 g (70%), mp 168–172°C. IR spectrum, ν, cm–1: 3280, 1690, 1625, 1550, 1290. 1H NMR spectrum, δ, ppm: 1.60–2.10 m (15H, CHAd), 1.99 s (3H, CH3), 8.33 s (1H, NH), 9.29 s (1H, NH). 13C NMR spectrum, δ, ppm: 24.3 (CH3), 29.6 (CH), 35.9 (CH2), 43.0 (CH2), 51.2 (C), 160.8 (C), 169.8 (C). Found, %: C 66.00; H 8.60; N 11.93. C13H20N2O2. Calculated, %: C 66.07; H 8.53; N 11.85.
CONCLUSIONS
A new method for the synthesis of unsymmetrical ureas, urethanes, and other derivatives, which involves initial thermolysis of S-ethyl (adamantan-1-yl)carbamothioate to form adamantan-1-yl isocyanate and its in situ reaction with alcohols and nitrogen-containing nucleophiles. The synthesized compounds contain privileged pharmacophoric fragments and can be considered as promising structures in terms of potential biological activity.
FUNDING
The work was financially supported by the Russian Science Foundation (project no. 21-73-20103). Spectral studied were funded by the Ministry for Education and Science of the Russian Federation in the framework of the design part of the State order no. 0778-2020-0005.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Schafer G., Matthey C., Bode J.W. Angew. Chem. Int. Ed. 2012;51:9173. doi: 10.1002/anie.201204481. [DOI] [PubMed] [Google Scholar]
- 2.Williams J.D., Kerr W.J., Leach S.G., Lindsay D.M. Angew. Chem. Int. Ed. 2018;57:12126. doi: 10.1002/anie.201807393. [DOI] [PubMed] [Google Scholar]
- 3.Pace V., de la Vega-Hernández K., Urban E., Langer T. Org. Lett. 2016;18:2750. doi: 10.1021/acs.orglett.6b01226. [DOI] [PubMed] [Google Scholar]
- 4.Golling F.E., Pires R., Hecking A., Weikard J., Richter F., Danielmeier K., Dijkstra D. Polymer Int. 2019;68:848. doi: 10.1002/pi.5665. [DOI] [Google Scholar]
- 5.Jurrat M., Pointer-Gleadhill B.J., Ball L.T., Chapman A., Adriaenssens L. J. Am. Chem. Soc. 2020;142:8136. doi: 10.1021/jacs.0c03520. [DOI] [PubMed] [Google Scholar]
- 6.Jia M., Hadjichristidis N., Gnanou Y., Feng X. Angew. Chem. Int. Ed. 2021;60:1593. doi: 10.1002/anie.202011902. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh A.K., Brindisi M. J. Med. Chem. 2015;58:2895. doi: 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akgul O., Singh S., Andring J.T., McKenna R., Selleri S., Carta F., Angeli A., Supuran C.T. Eur. J. Med. Chem. 2021;212:113035. doi: 10.1016/j.ejmech.2020.113035. [DOI] [PubMed] [Google Scholar]
- 9.Fedorova, V.A., Kadyrova, R.A., Slita, A.V., Muryleva, A.A., Petrova, P.R., Kovalskaya, A.V., Lobov, A.N., Zileeva, Z.R., Tsypyshev, D.O., Borisevich, S.S., Tsypysheva, I.P., Vakhitova, J.V., and Zarubaev, V.V., Nat. Prod. Res., 2019, p. 1. 10.1080/14786419.2019.1696791
- 10.Lukin A., Kramer J., Hartmann M., Weizel L., Hernandez-Olmos V., Falahati K., Burghardt I., Kalinchenkova N., Bagnyukova D., Zhurilo N., Rautio J., Forsberg M., Ihalainen J., Auriola S., Leppänen J., Konstantinov I., Pogoryelov D., Proschak E., Dar’in D., Krasavin M. Bioorg. Chem. 2018;80:655. doi: 10.1016/j.bioorg.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Kodani S.D., Bhakta S., Hwang S.H., Pakhomova S., Newcomer M.E., Morisseau C., Hammock B.D. Bioorg. Med. Chem. Lett. 2018;28:762. doi: 10.1016/j.bmcl.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim I.-H., Morisseau C., Watanabe T., Hammock B.D. J. Med. Chem. 2004;47:2110. doi: 10.1021/jm030514j,. [DOI] [PubMed] [Google Scholar]
- 13.Burmistrov V.V., Danilov D.V., D’yachenko V.S., Rasskazova E.V., Butov G.M. Russ. J. Org. Chem. 2020;56:735. doi: 10.1134/S1070428020050024. [DOI] [Google Scholar]
- 14.Burmistrov V., Morisseau C., D’yachenko V., Karlov D., Butov G.M., Hammock B.D. Bioorg. Med. Chem. Lett. 2020;30:126908. doi: 10.1016/j.bmcl.2019.126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burmistrov V., Morisseau C., Danilov D., Harris T.R., Dalinger I., Vatsadze I., Shkineva T., Butov G.M., Hammock B.D. Bioorg. Med. Chem. Lett. 2015;25:5514. doi: 10.1016/j.bmcl.2015.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krátký M., Štěpánková Š., Vorčáková K., Vinšová J. Bioorg. Chem. 2018;80:668. doi: 10.1016/j.bioorg.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 17.Krátký M., Vinšová J. Bioorg. Med. Chem. 2016;24:1322. doi: 10.1016/j.bmc.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Krátký M., Jandourek O., Baranyai Z., Novotna E., Stolaríkova J., Bosze S., Vinsova J. Eur. J. Med. Chem. 2019;181:111578. doi: 10.1016/j.ejmech.2019.111578. [DOI] [PubMed] [Google Scholar]
- 19.North E.J., Scherman M.S., Bruhn D.F., Scarborough J.S., Maddox M.M., Jone V., Grzegorzewicz A., Yang L., Hess T., Morisseau C., Jackson M., McNeil M.R., Lee R.E. Bioorg. Med. Chem. 2013;21:2587. doi: 10.1016/j.bmc.2013.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherman M.S., North E.J., Jones V., Hess T.N., Grzegorzewicz A.E., Kasagamic T., Kim I.-H., Merzlikin O., Lenaerts A.J., Lee R.E., Jackson M., Morisseau C., McNeil M.R. Bioorg. Med. Chem. 2012;20:3255. doi: 10.1016/j.bmc.2012.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown J.R., North E.J., Hurdle J.G., Morisseau C., Scarborough J.S., Sun D., Korduláková J., Scherman M.S., Jones V., Grzegorzewicz A., Crew R.M., Jackson M., McNeil M.R., Lee R.E. Bioorg. Med. Chem. 2011;19:5585. doi: 10.1016/j.bmc.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alsayed S.S.R., Lun S., Payne A., Bishai W.R., Gunosewoyo H. Bioorg. Chem. 2021;106:104486. doi: 10.1016/j.bioorg.2020.104486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isenbugel K., Ritter H., Branscheid R., Kolb U. Macromol. Rapid Commun. 2010;31:2121. doi: 10.1002/marc.201000597. [DOI] [PubMed] [Google Scholar]
- 24.Wang M., Zhang Lu, Huo, X., Zhang Z., Yuan Q., Li P., Chen J., Zou Y., Wu Z., Zhang W. Angew. Chem. Int. Ed. 2020;59:20814. doi: 10.1002/anie.202011527. [DOI] [PubMed] [Google Scholar]
- 25.Ozaki S. Chem. Rev. 1972;72:457. doi: 10.1021/cr60279a002. [DOI] [Google Scholar]
- 26.Skelly P.D., Ray Jr. W.J., Timberlake J.W. J. Org. Chem. 1985;50:267. doi: 10.1021/jo00202a021. [DOI] [Google Scholar]
- 27.Serkova I.V., Proshina A.N., Ustinova A.K., Ledneva B.V., Fomina-Ageeva E.V., Ashba A.M., Bezuglov V.V., Bachurin S.O. Doklady Chem. 2018;478:9. doi: 10.1134/S0012500818010044. [DOI] [Google Scholar]
- 28.Palomero O.E., Jones R.A. Organometallics. 2019;38:2689. doi: 10.1021/acs.organomet.9b00336. [DOI] [Google Scholar]
- 29.Fortman G.C., Captain B., Hoff C.D. Organometallics. 2009;28:3587. doi: 10.1021/om900004k. [DOI] [Google Scholar]
- 30.Blazek V., Bregovic N., Mlinaric-Majerski K., Basaric N. Tetrahedron. 2011;67:3846. doi: 10.1016/j.tet.2011.03.096. [DOI] [Google Scholar]
- 31.Farooq O., Wang Q., Wu A.-H., Olah G.A. J. Org. Chem. 1990;55:4282. doi: 10.1021/jo00301a014. [DOI] [Google Scholar]
- 32.Butov G.M., Burmistrov V., Saad K.R. Modern Org. Chem. Res. 2017;2:124. doi: 10.22606/mocr.2017.23005. [DOI] [Google Scholar]
- 33.Burmistrov V., Morisseau C., Harris T.R., Butov G., Hammock B.D. Bioorg. Chem. 2018;76:510. doi: 10.1016/j.bioorg.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butov G.M., Burmistrov V.V., Pitushkin D.A. Russ. J. Org. Chem. 2017;53:673. doi: 10.1134/S1070428017050050. [DOI] [Google Scholar]
- 35.Davis M., Dahl J. Synth. Commun. 2008;38:1153. doi: 10.1080/00397910701865926. [DOI] [Google Scholar]
- 36.Butov G.M., Mokhov V.M., Burmistrov V.V., Saad K.R., Pitushkin D.A. Russ. J. Org. Chem. 2014;50:1276. doi: 10.1134/S1070428014090073. [DOI] [Google Scholar]
- 37.Boyer, J.H., Manimaran, T., and Wolford, L.T., J. Chem. Soc. Perkin Trans. 1, 1988, p. 2137. 10.1039/P19880002137
- 38.Shiryaev A.K., Kryslov I.Yu. Russ. J. Org. Chem. 2002;38:1382. doi: 10.1023/A:1021636704131. [DOI] [Google Scholar]
- 39.Derasp J.S., Barbera E.A., Seguin N.R., Brzezinski D.D., Beauchemin A.M. Org. Lett. 2020;22:7403. doi: 10.1021/acs.orglett.0c02782. [DOI] [PubMed] [Google Scholar]
- 40.Dube P., Nathel N.F.F., Vetelino M., Couturier M., Aboussafy C.L., Pichette S., Jorgensen M.L., Hardink M. Org. Lett. 2009;11:5622. doi: 10.1021/ol9023387. [DOI] [PubMed] [Google Scholar]
- 41.Yadav A.K., Srivastava V.P., Yadav L.D.S. RSC Adv. 2014;4:24498. doi: 10.1039/c4ra03805c. [DOI] [Google Scholar]
- 42.Yoshimura A., Luedtke M.W., Zhdankin V.V. J. Org. Chem. 2012;77:2087. doi: 10.1021/jo300007c. [DOI] [PubMed] [Google Scholar]
- 43.Bruffaerts J., von Wolff, N., Diskin-Posner Y., BenDavid Y., Milstein D. J. Am. Chem. Soc. 2019;141:16486. doi: 10.1021/jacs.9b08942. [DOI] [PubMed] [Google Scholar]
- 44.Saha D., Taily I.M., Naik S., Banerjee P. Chem. Commun. 2021;57:631. doi: 10.1039/D0CC07125K. [DOI] [PubMed] [Google Scholar]
- 45.Kimab H.-K., Lee A. Org. Biomol. Chem. 2016;14:7345. doi: 10.1039/C6OB01290F. [DOI] [PubMed] [Google Scholar]
- 46.Gilbert A.K., Zhao Y., Otteson C.E., Pluth M.D. J. Org. Chem. 2019;84:14469. doi: 10.1021/acs.joc.9b01873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torrico-Vallejos S., Erben M.F., Hey-Hawkins E., Della Védova C.O. Tetrahedron Lett. 2011;52:5352. doi: 10.1016/j.tetlet.2011.08.027. [DOI] [Google Scholar]
- 48.Abdelazeem A.H., Alqahtani A.M., Omar H.A., Bukhari S.N.A., Gouda A.M. J. Mol. Struct. 2020;1219:128567. doi: 10.1016/j.molstruc.2020.128567. [DOI] [Google Scholar]
- 49.Klimochkin Yu.N., Moiseev I.K., Vladyko G.V., Korobchenko L.V., Boreko E.I. Pharm. Chem. J. 1991;25:485. doi: 10.1007/BF00772005. [DOI] [Google Scholar]
- 50.Klimochkin Yu.N. Zh. Org. Khim. 1987;23:2026. [Google Scholar]
- 51.Klimochkin Yu.N., Moiseev I.K., Abramov O.V., Vladyko G.V., Korobchenko L.V., Boreko E.I. Pharm. Chem. J. 1991;25:489. doi: 10.1007/BF00772006. [DOI] [Google Scholar]
- 52.Klimochkin Yu.N., Ivleva E.A., Shiryaev V.A. Russ. J. Org. Chem. 2021;557:355. doi: 10.1134/S1070428021030052. [DOI] [Google Scholar]
- 53.Koikov L.N., Lisitsa E.A., Alekseeva N.A., Turchin K.F., Filipenko T.Ya. Chem. Heterocycl. Compd. 1992;28:1289. doi: 10.1007/BF00532080. [DOI] [Google Scholar]
- 54.Oliver J.E., Stokes J.B. J. Med. Chem. 1970;13:779. doi: 10.1021/jm00298a060. [DOI] [PubMed] [Google Scholar]