

Abstract
Kir5.1 (encoded by Kcnj16 gene) is an inwardly rectifying K+ (Kir) channel highly expressed in the aldosterone-sensitive distal nephron of the kidney where it forms a functional channel with Kir4.1. Kir4.1/Kir5.1 channels are responsible for setting the transepithelial voltage in the distal nephron and collecting ducts and are thereby major determinants of fluid and electrolyte distribution. These channels contribute renal blood pressure control and have been implicated in salt-sensitive hypertension. However, mechanisms pertaining to the impact of Kir4.1/Kir5.1-mediated K+ transport on the renin-angiotensin-aldosterone system (RAAS) remain unclear. Herein, we utilized a knockout of Kcnj16 in the Dahl salt-sensitive rat (SSKcnj16−/−) to investigate the relationship between Kir5.1 and RAAS balance and function in the sensitivity of blood pressure to the dietary Na+/K+ ratio. The knockout of Kcnj16 caused substantial elevations in plasma RAAS hormones (aldosterone and angiotensin peptides) and altered the RAAS response to changing the dietary Na+/K+ ratio. Blocking aldosterone with spironolactone caused rapid mortality in SSKcnj16−/− rats. Supplementation of the diet with high K+ was protective against mortality resulting from aldosterone-mediated mechanisms. Captopril and losartan treatment had no effect on the survival of SSKcnj16−/− rats. However, neither of these drugs prevented mortality of SSKcnj16−/− rats when switched to high Na+ diet. These studies revealed that the knockout of Kcnj16 markedly altered RAAS regulation and function, suggesting Kir5.1 as a key regulator of the RAAS, particularly when exposed to changes in dietary sodium and potassium content.
Keywords: RAAS, salt-sensitive hypertension, Kcnj16, Kcnj10, hypokalemia, ENaC, HSD11β2
Introduction
In the kidney, Na+ and K+ transport in the late distal convoluted tubule (DCT2), connecting tubule (CNT), and cortical collecting duct (CCD), which collectively comprise the aldosterone-sensitive distal nephron (ASDN), is a critical determinant of the pressure-natriuresis relationship responsible for the long-term control of arterial pressure [1, 2]. Basolaterally expressed inwardly rectifying K+ (Kir) channels are critical for K+ transport in these segments [3–5]. The Kir4.1/Kir5.1 heterotetramer is the predominant basolateral K+ channel in the ASDN and sets the basolateral membrane potential, determines the driving force for ion transport, and senses and controls plasma K+ levels [6–8]. We previously reported that the Kir4.1/Kir5.1 heteromeric channel is required for the kidney to maintain numerous homeostatic mechanisms, including acid/base balance, whole body K+ homeostasis, and blood pressure control. In particular, we found that a high salt (HS) diet caused elevated blood pressure accompanied by increased expression of Kir5.1 in the ASDN of the Dahl salt-sensitive rat, an established model of salt-sensitive (SS) hypertension. Our previous studies revealed that a knockout of Kcnj16 in the Dahl salt-sensitive rat (SSKcnj16−/−) eliminated renal Kir4.1/Kir5.1 channel expression and function in the kidney and resulted in hypotension, salt wasting tubulopathy, and hypokalemia, which was exacerbated and led to mortality within a few days when SSKcnj16−/− rats were fed a high salt diet [9]. Kir5.1 has been proposed to be involved in the responsiveness of blood pressure to diet, and dysfunction of this channel may contribute to the pathophysiology of salt-sensitive hypertension. Although it is clear that Kir4.1/Kir5.1 channels regulate ion transport and influence blood pressure in the ASDN [5, 10], little progress has been made in defining the mechanism and extent of these channels’ effect on the renin-angiotensin-aldosterone system (RAAS).
The RAAS is a hormonal homeostatic system responsible for sustaining blood pressure in spite of inevitable fluctuations in plasma electrolytes resulting from variable dietary intake. The main effector peptides of the RAAS, angiotensin II (Ang II) and aldosterone, respond to deviations in plasma Na+ and K+, respectively, and act on the kidney to maintain blood pressure homeostasis. RAAS dysfunction or dysregulation is associated with numerous cardiorenal pathologies, and overactivity of the RAAS is associated with the development of hypertension and progression of chronic kidney disease (CKD). The capacity for RAAS hormones to alter renal ion transport is key to its effects on blood pressure. In the ASDN, the Na+-Cl− co-transporter (NCC) and the epithelial Na+ channel (ENaC) are both responsive to aldosterone signaling and have been established as significant contributors to the development of SS hypertension [11, 12]. We and others previously reported that NCC and ENaC were inappropriately activated in Dahl SS rats fed a HS diet, and inhibition of these Na+ transporters was sufficient to prevent the development of SS hypertension [11, 13–17]. However, modulation of Na+ transport by basolateral Kir4.1/Kir5.1 channels in the pathophysiology of SS hypertension has not been fully elucidated. The mechanism of Kir4.1/Kir5.1 channels’ involvement in salt-sensitive hypertension and the capacity of these channels to influence the RAAS requires investigation and is the main goal of this study. Despite their overlapping functionality and a shared site of action in the ASDN, there is thus far little evidence directly implicating renal Kir4.1/Kir5.1 channels in RAAS homeostasis or function. We hypothesize that Kir5.1 subunit exerts its control over blood pressure both through its established role in determining fluid and electrolyte homeostasis and via an additional regulatory role in RAAS modulation. We utilized a rat model with a knockout of Kcnj16 on the Dahl SS rat background to investigate the capacity of Kir5.1 to influence RAAS signaling in the sensitivity of blood pressure to the dietary Na+/K+ ratio.
Methods
Animals.
The genetic background used for the studies is the re-derived Rapp Dahl SS rat (SS/JrHsdMcwi; referred to as SSWT), which has been inbred for over 50 generations at the Medical College of Wisconsin (MCW). This strain is a proven model for the study of salt-sensitive hypertension [11, 18–20]. A Kcnj16 knockout rat model (SSKcnj16−/−) on the Dahl SS rat background was created at the MCW Gene Editing Rat Resource Center using zinc finger nuclease technology to induce an 18 base-pair deletion in the second transmembrane domain (TM2) of the Kir5.1 protein, resulting in the knockout of Kir5.1 subunit as previously described [9, 21]. Animals were housed in a temperature and humidity-controlled facility and maintained on a standard 12/12 dark/light cycle with water and food provided ad libitum. SSKcnj16−/− and SSWT rats were fed a purified AIN-76A diet (# 113755, Dyets, Inc., Bethlehem, PA) containing 0.4% NaCl and 0.36% K+ from wean unless otherwise noted (LS LK; 0.4% NaCl represents normal salt content but referenced here as a LS for simplicity in comparison to high salt (HS) diet). For experiments investigating the impact of altering dietary sodium and potassium intake the following diet compositions are used and specified in the text: HS LK (# 113756), HS HK (# 113522) and LS KH (# 113521) all form Dyets, Inc., Bethlehem, PA. All rats except SSKcnj16−/− rats on a HS LK diet (this diet results in rapid mortality of SSKcnj16−/− rats [9] requiring samples to be taken for measurement after 24 hours) were fed the specified diet for a 5 week duration before measurements were taken at approximately 12 weeks of age. Numbers of male and female rats included in all experimental groups are indicated in all figure legends.
Quantification of RAAS hormones.
The characterization of RAAS components was performed by quantification of the steady-state angiotensin peptide and aldosterone levels in equilibrated heparinized plasma samples by Attoquant Diagnostics (Vienna, Austria). Stable isotope-labeled internal standards for both classical and alternative renin-angiotensin system (RAS) branch constituents (Ang I (1-10), Ang II (1-8), Ang III (2-8), Ang 1-7, Ang IV (3-8), and Ang 1-5) were added to stabilized plasma samples at a concentration of 200 pg/mL and subjected to liquid chromatography tandem-mass spectrometry (LC-MS/MS)-based angiotensin quantification as described previously [22–25]. It was reported that similar outcomes are achieved using protease inhibitor-stabilized and equilibrated samples for angiotensin peptide quantification, thus both methods are representative of equilibrium hormone levels [25]. This technique was used to evaluate RAAS hormones in plasma samples from SSWT and SSKcnj16−/− rats fed each of four experimental diets: 1) LS LK (standard diet: 0.4% NaCl, 0.36% K+); 2) LS HK (0.4% NaCl, 1.41% K+); 3) HS LK (4% NaCl, 0.36% K+); and 4) HS HK (4% NaCl, 1.41% K+). These analyses included six angiotensin metabolites (Ang I (1-10), Ang II (1-8), Ang III (2-8), Ang IV (3-8), Ang 1-7, and Ang 1-5), aldosterone levels, and ratios of different derivatives to give surrogate measurements of angiotensin-converting enzyme (ACE) activity (Ang II/Ang I), plasma renin activity (PRA, Ang I + Ang II), and the adrenal response to Ang II (AA2-Ratio).
Administration of inhibitors targeting RAAS and analysis of electrolytes.
To evaluate functional and physiological repercussions of altered RAAS signaling in SSKcnj16−/− rats, we administered pharmacological inhibitors targeting aldosterone and angiotensin signaling in vivo. Spironolactone, a specific mineralocorticoid receptor (MR) antagonist, was used to block the action of aldosterone. Rats were given once daily IP injections of 50 mg/kg spironolactone for up to 10 days. This dose was chosen for maximal antagonism of MR in the ASDN while still safe and well tolerated in rats [26]. To prepare injections, spironolactone (S3378; Sigma-Aldrich) was dissolved in DMSO and ethanol at a 1:5 ratio and vehicle injections were performed (N=4) indicating no alteration to plasma electrolytes in control animals. A similar procedure was repeated to administer the angiotensin converting enzyme inhibitor (ACEi) captopril (C4042; Sigma-Aldrich), and angiotensin receptor blocker (ARB) losartan (PHR1602; Sigma-Aldrich), which were also given once daily IP (50 and 30 mg/kg/day, respectively in 0.9% saline). SSWT rats were used as a control strain in comparison with the effects of RAAS modulation in the SSKcnj16−/− strain.
To evaluate changes in renal ion transport, urine and blood electrolyte composition were assessed in SSWT and SSKcnj16−/− rats before and after administration of corresponding drugs. Rats were individually housed in metabolic cages to allow 24-hour urine collection at multiple time points. Control urine was collected before beginning any treatment. After the initial 24-hour control urine collection, rats received a daily dose of spironolactone or captopril/ losartan administered by IP injection. Standard 24-hour urine collection was repeated after the first administration and at the end of the protocol. Arterial blood samples were collected from the abdominal aorta under isoflurane anesthesia, and cortical renal tissue was harvested after flushing the blood from the kidneys. Electrolyte concentrations, pH, creatinine, and hemoglobin (ctHb) measurements from whole blood and 24-hour urine samples were determined by ABL800 FLEX blood gas analyzer (Radiometer, Copenhagen, Denmark) in SSWT and SSKcnj16−/− rats (both LS LK and LS HK diets) before and after injections. For comparison, arterial blood was also analyzed from untreated SSKcnj16−/− rats and SSWT control rats.
Histological and biochemical analyses.
To harvest kidneys for immunohistochemistry (IHC), rats were anesthetized with isoflurane and kidneys were flushed with PBS via aortic catheterization at 3 ml/min per kidney until blanched. Kidneys were formalin fixed (10% normal buffered formalin), paraffin embedded, sectioned (4 μm), and mounted on slides as previously described [9]. Hydroxysteroid 11-β dehydrogenase 2 (HSD11β2) was visualized with HSD11β2 antibody (1:100; Abcam ab203132) and streptavidin-biotin immunohistochemistry. Images were created using Nikon Eclipse Ni-E microscope (20X/0.50 and 40X/0.75 objectives). For Western blot analysis, kidney cortical lysates were prepared as previously described [9]. Kidney tissue samples (15-25 mg) were pulse sonicated in Laemmli buffer with a protease inhibitor cocktail (Roche, Basel Switzerland) to achieve a final protein concentration of 25 mg/ml. The resulting supernatant was subjected to SDS-PAGE, transferred onto nitrocellulose membrane (Millipore, Bedford MA) and probed with antibodies to α-ENaC (1:1000, Stressmarq Biosciences Inc, Vancouver, cat# SPC-403D) or HSD11β2 (1:1000; Abcam ab203132), and visualized by enhanced chemiluminescence (Amersham Biosciences Inc, Piscataway, NJ). Antibody to β-actin was used as control.
Statistical methods.
Blood and urine measurements are presented in tables as mean ± SEM. RAAS hormone measurements are presented in boxplots with individual data points displayed. Bounds of boxes represent SEM, whiskers span three standard deviations, median is denoted by a horizontal line within each box, and mean is denoted by a square within each box. For statistical analysis of data, equal variance and normality (Shapiro-Wilk) tests were performed followed by two-way ANOVA to determine significant effects resulting from the knockout and dietary treatments. For data comparisons before and after drug administration, one-way repeated measures ANOVAs were used. Data which failed the normality test was analyzed using the Kruskal-Wallis one-way ANOVA on ranks followed by post hoc tests. P-values of less than 0.05 were considered significant.
Study approval.
All animal studies were conducted at Medical College of Wisconsin and protocols were approved by the MCW Institutional Animal Care and Use Committees (IACUC) and were performed in accordance with the standards set forth by the NIH Guide for the Care and Use of Laboratory Animals.
Results
SSKcnj16−/− rats exhibit broad elevations in plasma RAAS hormones.
To evaluate whether Kir5.1 impacts overall RAAS balance, we used a highly sensitive mass spectrometry-based method to quantify equilibrium plasma hormone levels in SSWT and SSKcnj16−/− rats on a standard diet (LS LK; 0.4% NaCl, 0.36% K+). Angiotensin metabolites (Ang I, Ang II, Ang III, Ang IV, Ang 1-7, Ang 1-5) and aldosterone were dramatically elevated in SSKcnj16−/− compared to SSWT rats (Figures 1 and 2C). Plasma levels of Ang II and aldosterone were increased by an order of magnitude in the knockout (371 vs. 3674 and 183 vs. 1878 pM Ang II and aldosterone in SSWT and SSKcnj16−/− rats, respectively). Clinical metrics and surrogate measures of enzyme activity were computed based on RAAS hormone quantification. As renin is the rate-limiting enzyme of the RAAS, plasma renin activity (PRA) is used clinically to estimate RAAS activation in patients and identify cases of essential hypertension. PRA, represented by the total quantity of Ang I and Ang II, was significantly elevated in SSKcnj16−/− rats compared to SSWT rats (Figure 2B). Angiotensin converting enzyme (ACE) activity, represented by the Ang II to Ang I ratio, was also elevated in the Kcnj16 (Kir5.1) knockout (Figure 2A). However, the adrenal response to Ang II was not altered in the knockout strain (AA2 ratio; can be used clinically to detect primary aldosteronism and represented by the aldosterone to Ang II ratio; Figure 2D). There were no sex differences in measurements of angiotensin metabolites (Figure S3), aldosterone (Figure S4A), or surrogate functional parameters (PRA, ACE activity, and AA2 ratio; Figures S4B–D) in SSKcnj16−/− rats.
Figure 1. Quantification of equilibrium angiotensin metabolites in SSKcnj16−/− and SSWT rats.
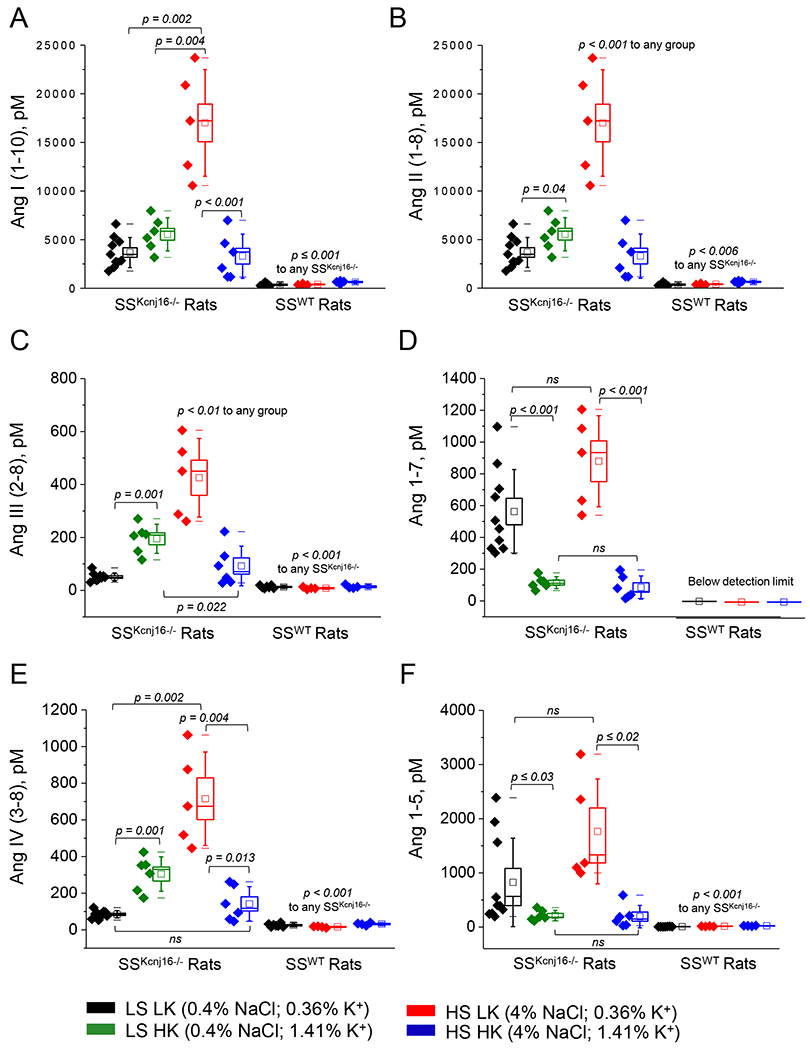
Ang I (1-10) (A), Ang II (1-8) (B), Ang III (2-8) (C), Ang 1-7 (D), Ang IV (3-8) (E), and Ang 1-5 (F) were quantified from SSKcnj16−/− (N=5-10 males) and SSWT (N=4-7 males) plasma samples collected from rats being fed one of 4 diets. In the boxplots, black data points denote rats fed a low salt, low potassium diet (LS LK, 0.4% NaCl and 0.36% K+); green indicates a low salt, high potassium diet (LS HK, 0.4% NaCl and 1.41% K+); red indicates a high salt, low potassium diet (HS LK, 4% NaCl and 0.36% K+); and blue indicates a high salt, high potassium diet (HS HK, 4% NaCl and 1.41% K+). Please see Figure S1 for expanded scale of angiotensin derivatives in SSWT rats. Measurements are shown in pM. Bounds of boxes represent SEM, whiskers span 3 standard deviations, median is denoted by a horizontal line, and mean is denoted by a square within each box. p-values are given for each graph.
Figure 2. Equilibrium aldosterone and surrogate RAAS measures in SSWT and SSKcnj16−/− rats.
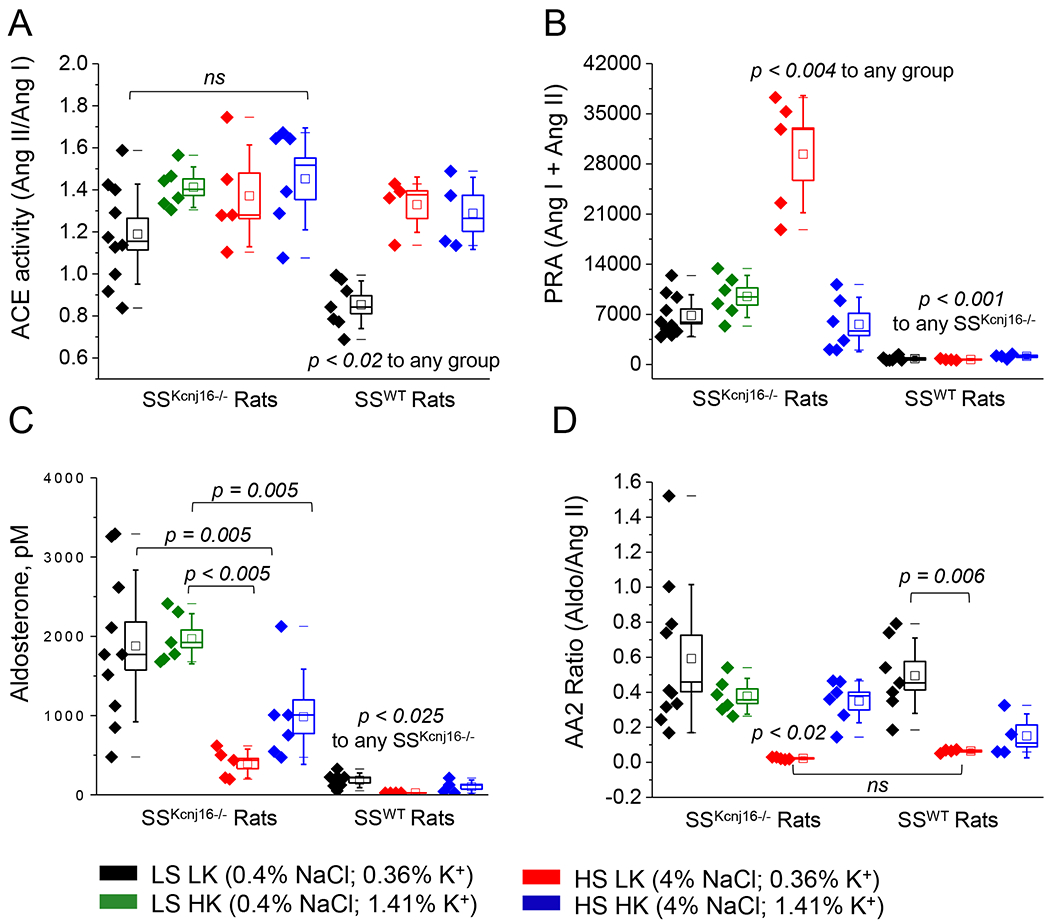
RAAS components were quantified using a mass spectrometry-based analysis of plasma samples collected from SSKcnj16−/− (N=5-10 male) and SSWT (N=4-7 male) rats fed one of 4 diets as described in Figure 1. (A) ACE activity indicated by the ratio of plasma Ang II to Ang I. (B) Plasma renin activity (PRA) represented by the combined content of Ang I and Ang II in pM. (C) Plasma aldosterone in pM (please see Figure S2 for expanded scale of aldosterone level in SSWT rats). (D) The ratio of aldosterone to Ang II (AA2 ratio). Bounds of boxes represent SEM, whiskers span 3 standard deviations, median is denoted by a horizontal line, and mean is denoted by a square within each box. p-values are given for each in the figure graph.
Although RAAS values were compared between SSWT and SSKcnj16−/− rats on an identical LS LK diet, it is important to note that these rats were exposed to vastly differing plasma K+ levels (Figure S7), as we previously showed that SSKcnj16−/− rats are hypokalemic on the LS LK diet [9]. To control for this discrepancy in plasma K+, we performed the same RAAS measurements in SSKcnj16−/− rats fed a LS HK diet (0.4% NaCl, 1.41% K+). Although plasma K+ was partially restored by dietary potassium supplementation, it was not enough to protect from hypokalemia, and corresponding plasma RAAS levels in SSKcnj16−/− rats on the LS HK diet did not become comparable to SSWT rats (Figure S5). Ang II became further elevated, but aldosterone was unaffected on the LS HK diet compared to the LS LK diet (Figures 1B and 2C). Overall, the substantial differences in RAAS balance observed in the SSKcnj16−/− rats persisted despite dietary K+ supplementation and augmentation of plasma K+.
Knockout of Kcnj16 altered the RAAS response to dietary Na+/K+ ratio.
Our previous work examined the effects of varying dietary Na+ and K+ content on blood pressure in SSWT and SSKcnj16−/− rats, and found that Kir5.1 was required for diet-specific blood pressure responses and salt-sensitive hypertension [9]. To determine whether the observed effect of Kir5.1 on blood pressure occurred at least partially through diet-specific RAAS changes, we measured the RAAS response to systematically varying Na+ and K+ intake in SSWT and SSKcnj16−/− rats. Plasma RAAS hormones were quantified in SSWT and SSKcnj16−/− rats being fed each of four diets consistent with our previous blood pressure study: 1) LS LK (standard diet: 0.4% NaCl, 0.36% K+); 2) LS HK (0.4% NaCl, 1.41% K+); 3) HS LK (4% NaCl, 0.36% K+); and 4) HS HK (4% NaCl, 1.41% K+) diets.
Effect of HS diet on plasma RAAS hormones.
We and others reported that the administration of a HS LK diet elicits a robust increase in blood pressure, which is characteristic of the salt-sensitive hypertension phenotype in SSWT rats [9]. Plasma RAAS analysis in SSWT rats showed that elevating dietary Na+ without altering K+ in the HS LK diet attenuated aldosterone but did not change Ang II levels compared to the standard LS LK diet (Figures 1, 2C, S1 and S2). Figures S1 and S2 show an expanded scale of the data from Figures 1 and 2C and represent a summary of angiotensin metabolite balance in SSWT rats. None of the angiotensin metabolites evaluated in SSWT rats were affected by the HS LK diet, except Ang 1-5 which was moderately elevated (Figure S1). As previously shown, the HS LK diet has vastly different physiological consequences in SSKcnj16−/− rats compared to SSWT rats. Specifically, the transition from a LS LK to a HS LK diet did not promote blood pressure elevation in these rats, but stimulated K+ wasting and exacerbated hypokalemia, which resulted in rapid mortality of all SSKcnj16−/− rats within a few days [9]. Plasma RAAS quantification in SSKcnj16−/− rats revealed that the switch to HS LK diet more than doubled Ang II (and all other angiotensin metabolites) and dramatically attenuated plasma aldosterone levels preceding HS-induced deaths (Figures 1B and 2C, respectively). In addition, HS LK treatment in SSWT increased ACE activity but did not alter PRA. Conversely, HS LK did not alter ACE2/ACE balance in SSKcnj16−/− rats but did elevate PRA. The HS LK diet decreased the AA2 ratio similarly in both SSWT and SSKcnj16−/− rats (Figure 2).
Capacity of dietary potassium to modulate the RAAS response on a HS diet.
To evaluate RAAS balance and the contribution of dietary K+ to modulate the renal response to Na+, plasma RAAS quantification was performed in SSWT and SSKcnj16−/− rats fed a HS HK diet. The analysis revealed that supplementing the HS diet with HK caused a modest elevation in plasma Ang II in SSWT rats, but significantly reduced Ang II to levels comparable to the LS LK diet in SSKcnj16−/− rats (Figures 1B and S1B). Accordingly, PRA, which was elevated in SSKcnj16−/− rats on a HS LK diet, returned to baseline levels with the addition of HK. The HS HK diet also augmented the AA2 ratio and plasma aldosterone in SSKcnj16−/− rats, partially restoring it to LS LK levels. The HS HK diet did not change ACE activity in SSKcnj16−/− rats. The addition of HK to HS did not alter plasma aldosterone, AA2 ratio, PRA, or ACE activity in SSWT rats (Figure 2).
Knockout of Kcnj16 altered balance between the renin-angiotensin system (RAS) axes.
To assess alterations in the balance between the classical and alternate RAS axes, plasma angiotensin effector peptides were grouped according to whether they primarily contribute to the classical (Ang II, Ang III, and Ang IV) or alternative (Ang 1-7 and Ang 1-5) RAS branch. We found that alternative branch peptides represented a greater proportion of the total angiotensin metabolites in SSKcnj16−/− rat plasma compared to SSWT plasma (Figure 3). This finding is consistent with the SSKcnj16−/− rat phenotype, which includes lower basal mean arterial pressure and reduced renal fibrosis [9]. Furthermore, the balance between the classical and alternative axes was markedly altered by Na+ and K+ intake in SSKcnj16−/− rats but remained relatively stable in SSWT rats regardless of diet (Figure 3).
Figure 3. Angiotensin peptides of the classical versus alternative RAS axis.
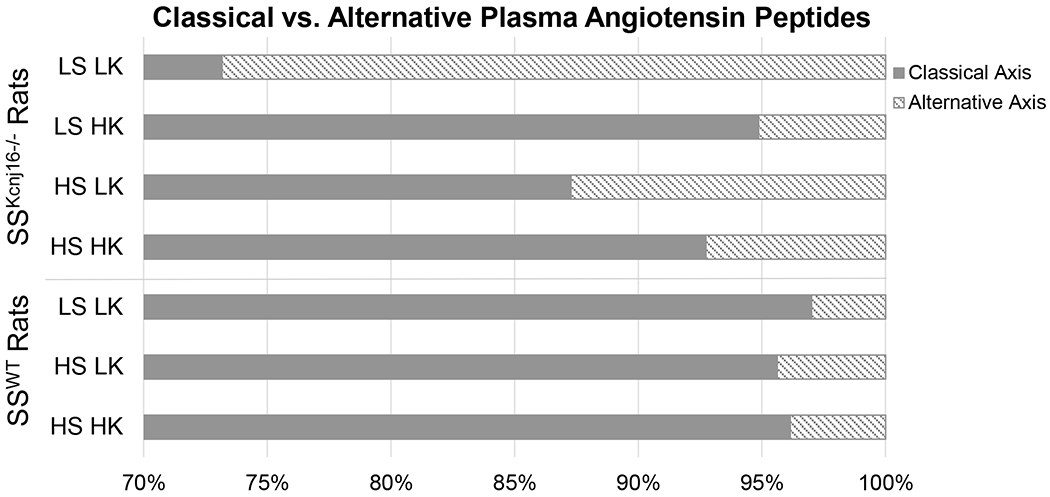
The proportion of classical axis related peptides (Ang II, Ang III, and Ang IV) and alternative axis related peptides (Ang 1-7 and Ang 1-5) represented in plasma from SSWT and SSKcnj16−/− rats. Proportions shown represent data compiled from raw angiotensin metabolite measurements depicted in Figure 1 (N=4-7 male SSWT and N=5-10 male SSKcnj16−/− rats). Diets are indicated on the y-axis and the percentage of plasma angiotensin metabolites designated to the classical RAS axis versus the alternative RAS axis is represented on the x-axis.
In SSKcnj16−/− rats, angiotensin metabolites responded to dietary alterations distinctly according to whether the peptide belongs to the classical or alternative RAS branch. In the classical branch of the RAS, LS HK caused modest increases in Ang II, Ang III, and Ang IV, while the addition of HS in the HS HK diet caused these metabolites to return to control LS LK levels (Figure 1). In the alternate RAS branch, where Ang 1-7 is the main effector, Ang 1-7 and Ang 1-5 respond to LS HK with a modest decrease in the metabolite, which is sustained with the addition of high Na+ on the HS HK diet (Figure 1). Thus, unlike the classical RAS axis, the alternate axis is responsive to potassium intake but insensitive to sodium intake in SSKcnj16−/− rats.
Kcnj16 knockout increased renal ENaC and HSD11β2 expression.
To better understand the causes and effects of aberrant aldosterone signaling in SSKcnj16−/− rats, we assessed the expression of two proteins which are critically involved in the ASDN aldosterone signaling pathway: hydroxysteroid 11-beta dehydrogenase type 2 (HSD11β2) and ENaC. It is established that aldosterone binds to the mineralocorticoid receptor (MR) to exert its effects on sodium transport in the ASDN through the stimulation of activity and expression of ENaC and NCC. Although MR is widely expressed in the kidney, it has equal affinity for both glucocorticoids and mineralocorticoids. The ASDN is the major site for aldosterone signaling due to co-expression of MR with HSD11β2 specifically in this nephron segment. HSD11β2 is an enzyme complex that oxidizes and inactivates glucocorticoids preventing their binding to MR, accounting for the specificity for MR to bind aldosterone in the ASDN. Deficiency in HSD11β2 has also been implicated in salt-sensitive hypertension [27–29]. Western blot and IHC analysis revealed increased expression of HSD11β2 in the distal tubules of SSKcnj16−/− rats compared to SSWT controls (Figures 4A and C, respectively). Furthermore, downstream targets of aldosterone signaling were found to be elevated in SSKcnj16−/− rats compared to SSWT rats. Subunit expression of α-ENaC was significantly elevated in SSKcnj16−/− rats (Figure 4B) similar to increased expression of both total and phosphorylated form of NCC found previously and represent compensatory overexpression or sodium transporters in response to significant electrolyte wasting phenotype observed in SSKcnj16−/− rats [9].
Figure 4. ENaC and HSD11β2 expression in SSWT and SSKcnj16−/− rats.
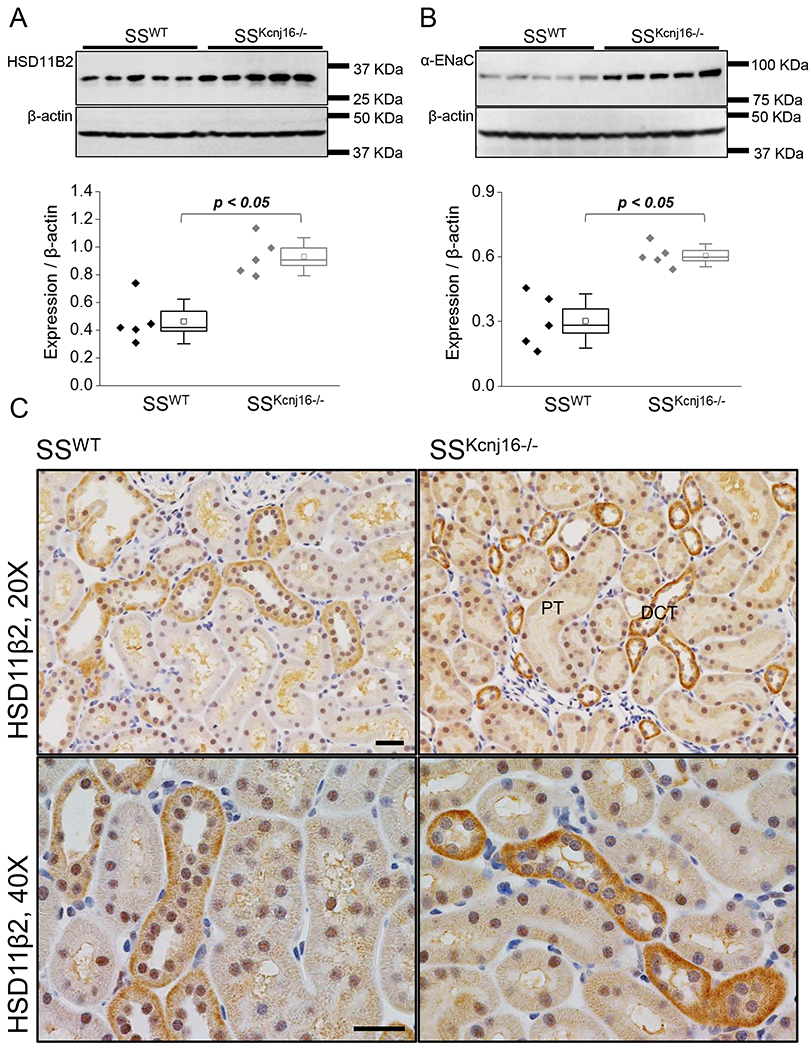
(A) Western blot analysis of HSD11β2 expression in kidney tissue from SSWT and SSKcnj16−/− rats on the standard LS LK diet (0.4% NaCl and 0.36% K+; N=5 males per group). (B) Western blot quantification of α-ENaC protein in kidney tissue from SSWT and SSKcnj16−/− rats on a LS LK diet (N=5 males per group). p-values are given for each graph. (C) Representative immunohistochemical staining of HSD11β2 protein in SSWT and SSKcnj16−/− rats at 20x and 40x magnification. Scale bars are 20 μm.
Targeted pharmacological RAAS inhibition revealed that amplified aldosterone signaling is required for survival of SSKcnj16−/− rats.
To better understand the physiological implications of the aberrant RAAS signaling and differences between angiotensin and aldosterone pathways, we used spironolactone, captopril, and losartan in vivo to inhibit MR, ACE, and angiotensin receptor, respectively. Blocking aldosterone signaling with spironolactone caused mortality in all SSKcnj16−/− rats (Figure 5A) without significantly altering their low plasma K+ levels (Table 1). Survival of SSWT rats was unaffected by spironolactone treatment and plasma electrolyte measurements reveal a typical response to a potassium-sparing diuretic (Table 1 and Figure S5). As our previous studies have shown that potassium supplementation improved the cardio-renal phenotype and survival of SSKcnj16−/− rats [9], we predicted that increased dietary potassium intake would compensate for hypokalemia in SSKcnj16−/− rats and restore spironolactone-mediated potassium-sparing diuretic effects observed in SSWT rats. Potassium supplementation in SSKcnj16−/− rats dramatically changed their response to spironolactone, showing changes in plasma electrolytes that were more similar to SSWT rats than to SSKcnj16−/− rats on the standard LS LK diet (Table1 and Figure S5). Like SSWT rats, LS HK fed SSKcnj16−/− rats did not experience mortalities with spironolactone treatment. Table 1 summarizes plasma electrolytes in SSWT and SSKcnj16−/− rats (fed either LS LK or LS HK diets) injected with spironolactone. Changes in urinary electrolyte excretion following spironolactone administration are shown in Figure S6. In contrast to spironolactone, inhibiting Ang II signaling with captopril (ACEi) or losartan (ARB) had no effect on survival of LS LK fed SSKcnj16−/− rats (Figure 5B). Unlike the effects of K+ supplementation, neither captopril nor losartan were able to prevent SSKcnj16−/− rats’ mortalities when switched to the HS LK diet. Changes in urinary electrolyte excretion with daily captopril or losartan administration in SSWT and SSKcnj16−/− rats are shown in Table S1.
Figure 5. Survival of SSWT and SSKcnj16−/− rats following pharmacological RAAS modulation.
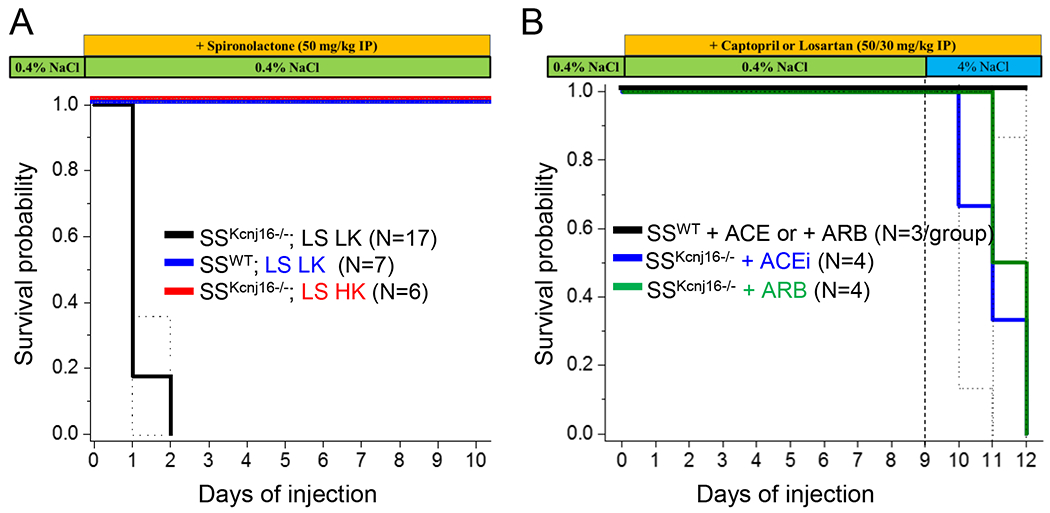
(A) Kaplan-Meier survival probability of SSWT rats on a LS LK diet (0.4% NaCl and 0.36% K+; N = 7), SSKcnj16−/− rats on a LS LK diet (0.4% NaCl and 0.36% K+; N = 17 total, 10 males and 7 females), and SSKcnj16−/− rats on a LS HK diet (0.4% NaCl and 1.41% K+; N = 6) were given IP injections of 50 mg/kg spironolactone daily for 10 days. (B) Kaplan-Meier survival probability of SSWT (N=3 males per ACEi and ARB treatment group) and SSKcnj16−/− rats (N=4 males per ACEi and ARB treatment group) injected with captopril or losartan (daily IP injections of 50 and 30 mg/kg, respectively) 8 days on a LS KL diet followed by 5 days on a HS LK diet.
Table 1.
Effect of spironolactone on plasma electrolytes.
| SSWT (LS LK) | SSKcnj16−/− (LS LK) | SSKcnj16−/− (LS HK) | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Measurement | − Drug (N = 14) | + Drug (N = 6) | − Drug (N = 12) | + Drug (N = 6) | − Drug (N = 10) | + Drug (N = 6) |
|
| ||||||
| K+ (mmol/L) | 4.3 ± 0.2 | 5.2 ± 0.3* | 2.6 ± 0.2 | 2.3 ± 0.3 | 2.8 ± 0.1 | 4.0 ± 0.2* |
| Na+ (mmol/L) | 140 ± 1 | 145 ± 2* | 139 ± 1 | 140 ± 2 | 142 ± 2 | 144 ± 1 |
| Ca++ (mmol/L) | 1.31 ± 0.04 | 1.20 ± 0.06 | 1.29 ± 0.03 | 0.86 ± 0.11* | 1.21 ± 0.07 | 1.25 ± 0.06 |
| Cl− (mmol/L) | 106 ± 1 | 108 ± 2 | 112 ± 1 | 110 ± 5 | 114 ± 1 | 118 ± 1 |
| pH | 7.25 ± 0.03 | 7.28 ± 0.05 | 7.11 ± 0.02 | 6.97 ± 0.05* | 7.19 ± 0.03 | 7.12 ± 0.02 |
| ctHb (g/dL) | 16.8 ± 0.5 | 15.9 ± 0.9 | 16.3 ± 0.5 | 16.3 ± 0.8 | 11.4 ± 0.5 | 14.6 ± 0.3* |
| Creatinine (mg/dL) | 0.34 ± 0.07 | 0.37 ± 0.11 | 0.35 ± 0.08 | 1.74 ± 0.11* | --- | 0.37 ± 0.04 |
Discussion
Basolateral Kir4.1/Kir5.1 channel is responsible for setting the resting membrane potential and the transepithelial voltage in the distal nephron and collecting ducts and are thereby major determinants of fluid and electrolyte distribution [4, 30]. Importantly, this channel have been predicted to contribute to renal control of blood pressure and have been implicated in salt-sensitive hypertension [9]. However, the specific mechanisms and extent of this regulatory role remains unclear. The impact of Kir4.1/Kir5.1 channel activity on blood pressure control has previously been explained by its ability to alter the kidneys’ pressure natriuresis response through its control over sodium reabsorption and fluid balance [10]. RAAS is well established for its role in regulating blood pressure, electrolyte homeostasis and fluid volume. We hypothesized that Kir5.1 exerts its control over blood pressure both through its established role in determining K+ homeostasis and via an additional regulatory role of RAAS signaling in ASDN. We utilized a knockout of Kcnj16 on the Dahl SS rat background to investigate the relationship between Kir5.1 and the RAAS in the sensitivity of blood pressure to the dietary Na+/K+ ratio.
Consistent with our hypothesis, the knockout of Kcnj16 (Kir5.1) resulted in extensive elevations in aldosterone and angiotensin metabolites in SSKcnj16−/− rats compared to SSWT rats (Figures 1 and 2), indicating the direct crosstalk between Kir5.1 function and RAAS balance. Although ACE activity and PRA were both elevated in SSKcnj16−/− rats, AA2 ratio remained unchanged, indicating that the knockout does not alter Ang II-dependent aldosterone production by the adrenal glands. Physiologically, such an excess in plasma Ang II and aldosterone should correspond with blood pressure elevations, but SSKcnj16−/− rats have chronically low blood pressure [9]. This clearly suggests that Kir5.1 is critical not only to maintain plasma RAAS balance, but also to mediate functional RAAS responses in the kidney required for appropriate blood pressure adjustments.
Although Ang II and aldosterone were similarly elevated in SSKcnj16−/− rats, their diet-specific responses and in vivo consequences were distinct. Increased aldosterone in SSKcnj16−/− rats was partially attenuated with increased Na+ intake (HS LK), but elevations in Ang II became further increased (Figure 2C and 1B, respectively). Increased dietary K+ is known to have beneficial effects in modulating salt-sensitive hypertension by altering renal Na+ handling. We previously reported that supplementing the HS challenge with HK (HS HK diet) attenuated but did not abolish salt-induced blood pressure elevations in SSWT rats, and completely prevented the salt-induced mortalities in SSKcnj16−/− rats [9]. Notably, SSKcnj16−/− rats showed no HS-induced blood pressure response, indicating a loss of the salt-sensitive phenotype. Furthermore, targeted pharmacological inhibition of Ang II and aldosterone revealed that the amplified aldosterone, but not Ang II signaling is critical for the survival of SSKcnj16−/− rats (Figure 5). Although specific mechanisms remain unclear, our combined results suggest that RAAS regulation by Kir4.1/Kir5.1 channels is mediated by aldosterone rather than angiotensin signaling in the kidney.
Our results suggest that Kir4.1/Kir5.1 channels’ critical role as potassium sensors in the distal nephron is required to dynamically alter the renal response to aldosterone dependent on present environmental conditions to maintain blood pressure and electrolyte homeostasis. Sometimes referred to as “the aldosterone paradox,” high plasma aldosterone can be induced by either hyperkalemia or hypovolemia, but to vastly different effects [31]. In hyperkalemic conditions, aldosterone signaling in the ASDN results in net K+ secretion to restore K+ homeostasis without affecting blood pressure, while in hypovolemia aldosterone causes Na+ retention, volume restoration and increases blood pressure. Herein, we found that without contribution from Kir5.1, aldosterone loses this flexibility, suggesting a crucial regulatory role of Kir4.1/Kir5.1 channels is required for the dynamic physiological effects of aldosterone. Although more work needs to be done to delineate a precise mechanism, this study, paired with the work of others, elucidates a promising potential mechanism. In states of hyperkalemia, high plasma K+ inhibits Kir4.1/Kir5.1 channels, depolarizing the basolateral membrane which results in less electroneutral NaCl reabsorption through NCC in the early DCT (DCT1) and consequently higher Na+ delivery to the ASDN. This, along with aldosterone, increases Na+ reabsorption through ENaC, driving K+ secretion through apical channels [32]. The outcome is K+ secretion without Na+ retention and thus, no increase in blood pressure. However, if high aldosterone is a result of hypervolemia, the opposite mechanism should occur. Increased Kir4.1/Kir5.1 activity hyperpolarizes the membrane NCC activation in DCT1, reducing Na+ delivery to the ASDN resulting in net Na+ retention without K+ secretion. In SSKcnj16−/− rats’ hypovolemic conditions, aldosterone should functionally increase blood pressure without K+ wasting. However, this cannot occur in SSKcnj16−/− rats because this mechanism is dependent on K+ sensing by basolateral Kir4.1/Kir5.1 channels. Accordingly, deletion of Kir5.1 has been shown to result in chronic depolarization of the basolateral membrane [33], which would eliminate aldosterone’s ability to respond dynamically to extracellular potassium, effectively forcing aldosterone to permanently function as if in hyperkalemic conditions. The resulting chronic K+ secretion without Na+ retention provides an elegant explanation for how such high plasma aldosterone, which is classically associated with hyperkalemia and increased blood pressure, can be paired with hypokalemia and low blood pressure in SSKcnj16−/− rats. Dysfunction of this mechanism may also contribute to the loss of the salt-sensitive phenotype in SSKcnj16−/− rats as well as aldosterone’s apparent insensitivity to dietary K+ modulation.
Although the results of this study indicate that Kir5.1 is likely most directly involved in aldosterone signaling, alterations in RAS balance and diet-specific responses of angiotensin peptides are likely very relevant to the SSKcnj16−/− phenotype. The overall function of the RAS is dependent on the balance between the ‘classical’ and ‘alternative’ axes, which are known to have opposing physiological effects. The classical axis produces vasoconstriction, inflammation, and increases in blood pressure, while the alternative axis causes vasodilation and is anti-inflammatory. We found that peptides of the alternative RAS branch (Ang 1-7 and Ang 1-5) were proportionally more represented in SSKcnj16−/− rats than in SSWT rats on all diets. This may contribute to low blood pressure in SSKcnj16−/− rats. Also, balance between the classical and alternative RAS was more affected by alterations in diet composition in SSKcnj16−/− rats compared to SSWT rats. Specifically, in SSKcnj16−/− rats, the alterative axis was responsive to dietary potassium but not sodium intake, which could partially explain their corresponding loss of the SS hypertensive phenotype.
Overall, we conclude that Kir5.1 is critical for specific RAAS responses to alterations in the dietary Na+/K+ ratio, for RAAS hormones to exert their effects on blood pressure, and for maintaining appropriate balance between the classical and alternative RAS axes. The genetic knockout of Kir5.1 caused an apparent uncoupling of the RAAS from renal ion transport in the ASDN, eliminating blood pressure responsiveness to Na+/K+ intake, resulting in the previously reported resolution of SS hypertensive phenotype. This study supports Kir4.1/Kir5.1’s involvement in the aldosterone paradox, as the knockout Kir5.1 resulted in loss of dynamic control of aldosterone function in the ASDN, effectively blinding aldosterone to environmental conditions, compromising its role in blood pressure homeostasis. We propose Kir5.1 as a key protein in regulating plasma RAAS hormones and facilitating physiological responses to RAAS signaling for the maintenance of homeostasis.
Supplementary Material
Clinical Perspectives:
Renal Kir5.1 containing channels play a role in blood pressure modulation in response to dietary salt. The renin-angiotensin-aldosterone system (RAAS) also has a prominent role in diet-specific blood pressure alterations. However very little is known about the capacity for these channels to modulate plasma RAAS hormones or the potential involvement of Kir5.1 in RAAS signaling in the kidney.
We found that genetic knockout of Kcnj16 to eliminate Kir5.1 channels resulted in profound elevations in plasma RAAS hormones and altered the response to dietary sodium and potassium.
Understanding the relationship between RAAS signaling and renal Kir5.1 channels in diet-specific blood pressure responses may elucidate novel therapeutic targets or strategies for a subset of hypertensive patients.
Acknowledgements
The authors would also like to thank Christine Duris (MCW Children’s Research Institute Imaging Core) for help with immunohistochemical analysis.
Funding Information
This research was supported by the National Institutes of Health grants R35 HL135749, P01 HL116264 (to A.S.), R56 DK121750 (to O.P.), F31 DK122647 (to A.D.M.), and R01 HL122358 (to M.R.H.), American Heart Association grants 16EIA26720006 (to A.S.) and 17SDG33660149 (to O.P.), Michael H. Keelan, Jr., MD, Research Foundation Grant and the Cardiovascular Center at the Medical College of Wisconsin (to O.P.), and Department of Veteran Affairs I01 BX004024 (to A.S.).
Abbreviations
- AA2 ratio
ratio aldosterone to angiotensin II
- ACE
angiotensin converting enzyme
- ACEi
angiotensin converting enzyme inhibitor
- ARB
angiotensin receptor blocker
- ASDN
aldosterone sensitive distal nephron
- CCD
cortical collecting duct
- CNT
connecting tubule
- DCT
distal convoluted tubule
- DCT1
early distal convoluted tubule
- DCT2
late distal convoluted tubule
- ENaC
epithelial sodium channel
- HS
high salt
- HSD11β2
11-β-hydroxysteroid dehydrogenase 2
- HS HK
high sodium, high potassium
- HS LK
high sodium, low potassium
- Kir
inward rectifier potassium channel
- LS HK
low sodium, high potassium
- LS LK
low sodium, low potassium
- MR
mineralocorticoid receptor
- NCC
sodium chloride cotransporter
- PRA
plasma renin activity
- RAAS
renin-angiotensin-aldosterone system
- RAS
renin angiotensin system
- SS
salt-sensitive
Footnotes
Declarations of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Staruschenko A (2012) Regulation of transport in the connecting tubule and cortical collecting duct. Compr. Physiol 2, 1541–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cowley AW Jr. (1992) Long-term control of arterial blood pressure. Physiol. Rev 72, 231–300 [DOI] [PubMed] [Google Scholar]
- 3.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH and Wang WH (2017) Potassium sensing by renal distal tubules requires Kir4.1. J. Am. Soc. Nephrol 28, 1814–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palygin O, Pochynyuk O and Staruschenko A (2017) Role and mechanisms of regulation of the basolateral Kir 4.1/Kir 5.1K(+) channels in the distal tubules. Acta Physiol. 219, 260–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palygin O, Pochynyuk O and Staruschenko A (2018) Distal tubule basolateral potassium channels: cellular and molecular mechanisms of regulation. Curr. Opin. Nephrol. Hypertens 27, 373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J and Paulais M (2008) Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am. J. Physiol. Renal Physiol 294, F1398–1407 [DOI] [PubMed] [Google Scholar]
- 7.Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A and Teulon J (2002) An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J. Physiol 538, 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sepulveda FV, Pablo Cid L, Teulon J and Niemeyer MI (2015) Molecular aspects of structure, gating, and physiology of pH-sensitive background K2P and Kir K+-transport channels. Physiol. Rev 95, 179–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, Geurts AM, Hodges MR and Staruschenko A (2017) Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight. 2, e92331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Staruschenko A (2018) Beneficial effects of high potassium: contribution of renal basolateral K+ channels. Hypertension. 71, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlov TS, Levchenko V, O’Connor PM, Ilatovskaya DV, Palygin O, Mori T, Mattson DL, Sorokin A, Lombard JH, Cowley AW Jr. and Staruschenko A (2013) Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J. Am. Soc. Nephrol 24, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pavlov TS and Staruschenko A (2017) Involvement of ENaC in the development of salt-sensitive hypertension. Am. J. Physiol. Renal Physiol 313, F135–F140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aoi W, Niisato N, Sawabe Y, Miyazaki H and Marunaka Y (2006) Aldosterone-induced abnormal regulation of ENaC and SGK1 in Dahl salt-sensitive rat. Biochem. Biophys. Res. Commun 341, 376–381 [DOI] [PubMed] [Google Scholar]
- 14.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S and Tomita K (2009) Aberrant ENaC activation in Dahl salt-sensitive rats. J. Hypertens 27, 1679–1689 [DOI] [PubMed] [Google Scholar]
- 15.Murphy SR, Dahly-Vernon AJ, Dunn KM, Chen CC, Ledbetter SR, Williams JM and Roman RJ (2012) Renoprotective effects of anti-TGF-beta antibody and antihypertensive therapies in Dahl S rats. Am. J. Physiol. Regul. Integr. Comp. Physiol 303, R57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou MS, Jaimes EA and Raij L (2006) Benazepril combined with either amlodipine or hydrochlorothiazide is more effective than monotherapy for blood pressure control and prevention of end-organ injury in hypertensive Dahl rats. J. Cardiovasc. Pharmacol 48, 857–861 [DOI] [PubMed] [Google Scholar]
- 17.Zhou MS, Schulman IH, Jaimes EA and Raij L (2008) Thiazide diuretics, endothelial function, and vascular oxidative stress. J. Hypertens 26, 494–500 [DOI] [PubMed] [Google Scholar]
- 18.Cowley AW Jr. (2006) The genetic dissection of essential hypertension. Nat. Rev. Genet 7, 829–840 [DOI] [PubMed] [Google Scholar]
- 19.Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C and Coffman TM (2019) Animal Models of Hypertension: A Scientific Statement From the American Heart Association. Hypertension. 73, e87–e120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rudemiller NP and Mattson DL (2015) Candidate genes for hypertension: insights from the Dahl S rat. Am. J. Physiol. Renal Physiol 309, F993–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puissant MM, Muere C, Levchenko V, Manis AD, Martino P, Forster HV, Palygin O, Staruschenko A and Hodges MR (2019) Genetic mutation of Kcnj16 identifies Kir5.1-containing channels as key regulators of acute and chronic pH homeostasis. FASEB J. 33, 5067–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antlanger M, Domenig O, Kovarik JJ, Kaltenecker CC, Kopecky C, Poglitsch M and Säemann MD (2017) Molecular remodeling of the renin-angiotensin system after kidney transplantation. J. Ren. Ang. Aldost. System. 18, 1470320317705232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilatovskaya DV, Levchenko V, Pavlov TS, Isaeva E, Klemens CA, Johnson J, Liu P, Kriegel AJ and Staruschenko A (2019) Salt-deficient diet exacerbates cystogenesis in ARPKD via epithelial sodium channel (ENaC). EBioMedicine. 40, 663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blass G, Klemens CA, Brands MW, Palygin O and Staruschenko A (2019) Postprandial Effects on ENaC-Mediated Sodium Absorption. Sci. Rep 9, 4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovarik JJ, Antlanger M, Domenig O, Kaltenecker CC, Hecking M, Haidinger M, Werzowa J, Kopecky C and Saemann MD (2016) Molecular regulation of the renin-angiotensin system in haemodialysis patients. Nephrol. Dial. Transpl 31, 851. [DOI] [PubMed] [Google Scholar]
- 26.Tokumura T, Muraoka A, Masutomi T and Machida Y (2005) Stability of spironolactone in rat plasma: strict temperature control of blood and plasma samples is required in rat pharmacokinetic studies. Biol. Pharmac. Bull 28, 1126–1128 [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Singh RJ, Usa K, Netzel BC and Liang M (2008) Renal medullary 11 beta-hydroxysteroid dehydrogenase type 1 in Dahl salt-sensitive hypertension. Physiol. Genom 36, 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullins LJ, Kenyon CJ, Bailey MA, Conway BR, Diaz ME and Mullins JJ (2015) Mineralocorticoid Excess or Glucocorticoid Insufficiency: Renal and Metabolic Phenotypes in a Rat Hsd11b2 Knockout Model. Hypertension. 66, 667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey MA, Craigie E, Livingstone DEW, Kotelevtsev YV, Al-Dujaili EAS, Kenyon CJ and Mullins JJ (2011) Hsd11b2 haploinsufficiency in mice causes salt sensitivity of blood pressure. Hypertension. 57, 515–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang WH (2016) Basolateral Kir4.1 activity in the distal convoluted tubule regulates K secretion by determining NaCl cotransporter activity. Curr. Opin. Nephrol. Hypert 25, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arroyo JP, Ronzaud C, Lagnaz D, Staub O and Gamba G (2011) Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology. 26, 115–123 [DOI] [PubMed] [Google Scholar]
- 32.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL and Ellison DH (2015) Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 21, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu P, Gao ZX, Su XT, Wang MX, Wang WH and Lin DH (2019) Kir4.1/Kir5.1 Activity Is Essential for Dietary Sodium Intake-Induced Modulation of Na-Cl Cotransporter. J. Am. Soc. Nephrol 30, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.