

Abstract
In the 1920s, Otto Warburg observed the phenomenon of altered glucose metabolism in cancer cells. Although the initial hypothesis suggested that the alteration resulted from mitochondrial damage, multiple studies of the subject revealed a precise, multistage process rather than a random pattern. The phenomenon of aerobic glycolysis emerges not only from mitochondrial abnormalities common in cancer cells, but also results from metabolic reprogramming beneficial for their sustenance. The Warburg effect enables metabolic adaptation of cancer cells to grow and proliferate, simultaneously enabling their survival in hypoxic conditions. Altered glucose metabolism of cancer cells includes, inter alia, qualitative and quantitative changes within glucose transporters, enzymes of the glycolytic pathway, such as hexokinases and pyruvate kinase, hypoxia-inducible factor, monocarboxylate transporters, and lactate dehydrogenase. This review summarizes the current state of knowledge regarding inhibitors of cancer glucose metabolism with a focus on their clinical potential. The altered metabolic phenotype of cancer cells allows for targeting of specific mechanisms, which might improve conventional methods in anti-cancer therapy. However, several problems such as drug bioavailability, specificity, toxicity, the plasticity of cancer cells, and heterogeneity of cells in tumors have to be overcome when designing therapies based on compounds targeted in cancer cell energy metabolism.
Keywords: aerobic glycolysis, glucose transporters, pyruvate kinase, lactate dehydrogenase, monocarboxylate transporters, hypoxia-inducible factor, inhibitors, anti-cancer therapy
Introduction
In the year 2000, Hanahan and Weinberg published a well-known review: Hallmarks of cancer. In it, they organized the complexity of cancer biology into six major attributes: self-sufficiency in growth signals, insensitivity to anti-growth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis.1 Ten years later, they published an updated review and added two new hallmarks: evading the immune system and reprogrammed energy metabolism.2 Although alterations in cancer metabolism were shown by Otto Warburg in the 1920s, reprogrammed cancer metabolism has become a hot topic only in the past decade. An abnormal dependence on glycolysis as the sole source of ATP creation instead of oxidative phosphorylation, even in the presence of oxygen, is commonly called “the Warburg effect.” Changes in metabolism observed in many types of cancer provide a selective advantage during tumor initiation and progression.3 Despite the early hypothesis, altered glucose metabolism is not a coincidence or the effect of nonfunctional mitochondria, but the result of expedient changes. Mitochondrial abnormalities, such as disturbance in number, structure, or function of the organelle, are common in almost all types of cancer4; however, the universality of specific metabolism alteration points to the deterministic nature of the Warburg effect’s occurrence. These changes lead to aerobic glycolysis, allowing cancer cells to grow, proliferate, spread, and invade other tissues. Significant changes in cancer cell energy metabolism include (1) overexpression of glucose transporters leading to increased glucose uptake; (2) overexpression of enzymes of the glycolytic pathway leading to increased activity of glucose metabolism; (3) overexpression of PKM gene and increased amount of pyruvate kinase isoform M2, which allows redirection of glucose to the pentose phosphate pathway; (4) overexpression of lactate dehydrogenase which allows maintaining the NAD+/NADH ratio; (5) overexpression of monocarboxylate transporters, which orchestrate the trafficking of the lactate; and (6) increased transcriptional activity of hypoxia-inducible factor which up-regulates glucose metabolism.5,6 The metabolic differences between cancer and normal cells are shown in Figure 1.
Figure 1.

Comparison of glucose metabolism in a normal and cancer cell and potential inhibitors of glucose metabolism l. The majority of normal cells utilize glucose as a substrate in the glycolytic pathway. Then pyruvate, a product in this process, enters the mitochondria and after being converted into acetylo-CoA enters the tricarboxylic acid cycle. In cancer cells, overexpression of GLUT transporters causes increased glucose uptake compared to normal cells. Increased expression of glycolytic enzymes is also observed. M2 isoform of pyruvate kinase shows reduced catalytic activity which allows the directing of part of the created G-6-P to the pentose phosphate pathway. The PKM2 isoform also stimulates HIF-1 into increased transcriptional activity by binding to the transactivation domain of HIF1-α subunit. The activation of HIF results in increased expression of the key elements of glucose metabolism, for example, GLUT transporters, enzymes of the glycolytic pathway, and lactate dehydrogenase. Most of the pyruvate created in cancer cells is converted into lactate by lactate dehydrogenase. Lactate is transported outside of cells by the monocarboxylate transporters, whose amount is elevated in cancer cells.
The intensive growth and proliferation of cancer cells require constant energy supply and macromolecules synthesis. Cancer cells need significantly more glucose than normal cells, but increased numbers of GLUT transporters enable increased glucose uptake. Overexpression of the glycolytic pathway enzymes has a positive impact on the effectiveness of energy production. Simultaneously, the reduced catalytic activity of pyruvate kinase M2 (PKM2) allows to direct part of the created G-6-P to the pentose phosphate pathway, which provides certain essential elements, for example, NADPH or pentoses necessary for biomolecule synthesis.5,6 Moreover, PKM2 binds with HIF-1α and increases the transcriptional activity of the hypoxia-inducible factor which results in the overexpression of genes under the control of the factor, among other things, the critical elements of glucose metabolism. Increased lactate production contributes to angiogenesis, maintains a low pH of the cancer cell microenvironment, and maintains the NAD+/NADH ratio. Proper NAD+/NADH ratio is crucial for redox homeostasis, metabolism, cellular bioenergetics, genomic stability, gene expression, mitochondrial homeostasis, and adaptive stress responses.7 The NAD+ homeostasis is maintained by the biosynthesis, consumption, and recycling in different cellular compartments, including the cytosol, the mitochondria, and the nucleus. Continuous replenishment of NAD increases the proliferation and survival of rapidly dividing cancer cells because elevated NAD levels enhance glycolysis via glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and lactate dehydrogenase (LDH) that require NAD as a co-enzyme.7 To keep the intracellular balance of pH, the lactate needs to be efficiently transported outside of the cancer cells which is enabled by an increased number of monocarboxylate transporters.5,6
All these changes result in phenotypic distinctions between normal and cancer cells that might allow specific targeting of cancer cells for anti-cancer therapy. This can be achieved by inhibiting the key regulatory factors of the reprogrammed mechanism of glucose metabolism. In this review, we present the current state of knowledge regarding inhibitors of cancer glucose metabolism and discuss the reasons for discrepancies between expectations toward their efficiency and the reality of their clinical utility.
Inhibition of Glucose Transporters
The first stage of glucose metabolism is glucose transport through the cell membrane. As a result of metabolic reprogramming of cancer cells, expression of the GLUT1 noticeably increases.8–11 An increased number of glucose transporters and the resulting higher glucose uptake is a universal phenomenon observed in many types of cancers, including diffuse large B-cell lymphoma, colorectal cancer, hepatocellular carcinoma, larynx cancer, gastrointestinal stromal tumor, thyroid cancer, pancreatic cancer, renal cell carcinoma, prostate cancer, lung cancer, and sarcomas.12 Overexpression of GLUT1 may also correlate with the increased potential for metastasis and poor prognosis.13–15
The WZB117 compound (2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate) is a well-known anti-cancer drug prototype whose mechanism of action is based on inhibiting the glucose transporter. It inhibits sugar transport by reversibly binding at the exofacial sugar binding site.16 In vitro studies showed that WZB117 in human colon cancer cells (HT-29) could overcome the cells’ resistance to 5-Fluorouracil, the most commonly used drug in colon cancer therapy. The usage of WZB117 resulted in increased sensitivity to 5-fluorouracil, decreased glucose uptake, and a significant reduction of the cell’s proliferation.17
Another study on human breast cancer cell lines (MDA-MB231 and MCF-7) showed that inhibiting GLUT1 with WZB117 sensitizes the cells to radiation and even re-sensitizes radioresistant cells.18 Moreover, a study with an adriamycin-resistant breast cancer cell line (MCF-7) showed that WZB117 treatment partially restores the effects of adriamycin.19 It has also been shown that combining WZB117 with biguanides like metformin resulted in broad anti-cancer activities that allowed targeting cancers characterized by intratumoral heterogeneity.20 The compound study using a nude mouse model showed a reduction of tumors (non-small cell lung cancer and breast cancer) and weight loss, mainly in fat tissue, and fluctuation in the number of lymphocytes and platelets remaining within the normal range. The primary purpose of using WZB117 in an animal model was to check whether it might cause hyperglycemia—which it does, but not persistently.21 Another animal study showed that WZB117 administration inhibited tumor formation after implanting cancer stem cells without causing significant side effects in host animals.22 The high efficiency of WZB117 both in vitro and in vivo and its low toxicity leads to the conclusion that this compound might be a promising anti-cancer drug, if not as monotherapy, then as an enhancer of conventional therapy methods.
Another candidate for an anti-cancer drug targeting GLUT1 is STF-31. In 2011, Chan and co-workers conducted a study in which they showed that STF-31 treatment inhibits growth and glucose uptake in renal cell carcinomas with the loss of the von Hippel–Lindau tumor suppressor gene.23 A recent experimental re-evaluation of STF-31 has shown that this compound acts as nicotinamide phosphoribosyltransferase (NAMPT) or GLUT1 inhibitor, which depends on concentration and specific cellular context.24 It suggests that generally, STF-31 might be an effective inhibitor in the case of VHL-mutated cancers that are mainly dependent on the GLUT1 transporter.
Anti-cancer properties of resveratrol have been known for over two decades now. This compound belongs to the group of polyphenols and can be found in red grapes. In 2001, Park and co-workers conducted a study based on human myelocytic cell lines (U937 and HL-60), in which they proved inhibition of the glucose uptake.25 Subsequent research with the same cell lines confirmed this effect.26 They suggested that resveratrol binds to an endofacial site of GLUT1 and inhibits glucose transport through the plasma membrane. Another study of ovarian cancer cells proved that resveratrol suppresses intracellular trafficking of GLUT1 to the plasma membrane.27 Regardless of the exact mechanism of inhibition, resveratrol has shown anti-tumor activity at all stages of carcinogenesis in several types of cancer. However, despite the excellent efficiency of the compound, the application of resveratrol is a significant challenge due to low bioavailability.28
Another approach to glucose transporters inhibition is the use of glucose analogs. Although 2-deoxy-D-glucose (2DG) shows an effective competitive inhibiting effect with GLUT1 and hexokinase, clinical trials of this compound have been terminated because of non-specific cytotoxicity.29 Using a different analog of glucose, non-radioactive 2-deoxy-2-fluoro-D-glucose (19FDG), enabled overcoming the limitations of 2DG. A study on the impact of 19FDG combined with doxorubicin on the human cervical cancer line (HeLa) showed a decreased number of viable cells and decreased production of lactate. This effect was also confirmed in vivo using positron emission tomography of nude mice with tumors induced by breast cancer cells injection (MDA MB- 231).30 The results of this study suggest that 19FDG could be a promising enhancer of conventional methods for anti-cancer therapy.
Inhibition of Enzymes of the Glycolytic Pathway and Their Isoforms
After glucose enters a cell, it may be directed to the glycolytic pathway. As the result of metabolic rearrangements, most cancer cells prefer aerobic glycolysis which can be associated with the changes in expression or activity of enzymes involved in this process. Hexokinase (HK) is the first enzyme involved in this pathway, and at the same time, it is the first glycolysis checkpoint. The dominant isoform of hexokinase found in cancer cells is HKII. It may play a key role in the growth of some cancer types like glioblastoma multiforme since the knockdown of its expression leads to restored oxidative metabolism and increased sensitivity to radiation and temozolomide.31 Moreover, a study performed by Gu and co-workers proved that a high level of HKII is associated with therapy resistance, shorter progression-free survival, and overall survival in aggressive lymphoma.32
Lonidamine is a dechlorinate derivative of indazole-3-carboxylic acid. For a long time, it has been known as a compound inhibiting aerobic glycolysis by inhibiting mitochondria-bound HKII. Moreover, it was proven to modify the permeability of membranes, sensitizing cells to drugs and, in effect, allowing the usage of smaller drug doses. Combining lonidamine with anthracyclines resulted in efficient cytotoxic effects in the case of breast cancer,33 melanoma, and Ehrlich ascites carcinoma.34 The combination of lonidamine and diazepam in phase II study with glioblastoma multiforme patients resulted in the stabilization of tumor progression but has not shown significant therapeutic effects.35 The compound was also used in phases II and III of clinical trials for benign prostatic hyperplasia patients but resulted in severe hepatic adverse effects.29 In later studies, it was shown that lonidamine inhibits not only HKII, but also pyruvate and monocarboxylate transporters, targeting multiple sites of glucose metabolism at once.36 Although most of the lonidamine trials with patients were terminated, a broad range of effects in glucose metabolism caused by this compound points to the conclusion that it is worth re-evaluating in combination with other chemotherapeutics.
Another compound targeting HKII is a pyruvate analog 3-bromopyruvate (3-BP). Its alkylating activity leads to the alkylation of cysteine residues. Recent studies show that 3-bromopyruvate plays a significant role in the anti-cancer effects of many other compounds. For example, in a study on in vivo and in vitro models of hepatocellular carcinomas, 3-BP has been shown to increase the efficacy of sorafenib.37 This finding might be crucial since patients with a higher HKII expression level tend to show poor overall survival, even under sorafenib treatment. Another study based on human metastatic prostate cancer cell lines (DU-145 and PC3) proved that 3-bromopyruvate has the potential for a reduction of metastasis.38 However, 3-bromopyruvate has a high chemical activity and may non-specifically affect other cellular components; moreover, it cannot cross the blood–brain barrier which makes that compound ineffective in treating brain tumors and has no persistent effect on tumor tissue because of its higher permeability compared to normal tissue.39
Pachymic acid (PA) is a natural steroid extracted from the fungus Wolfiporia cocos. It is a compound known for its various medical properties, that is, anti-inflammatory, anti-aging, and insulin-like effects. Another effect of PA is the inhibition of HKII, causing dissociation of the enzyme from the mitochondria, releasing cytochrome, generating reactive oxygen species, and ATP depletion which induces mitochondrial apoptosis. The main anti-cancer activity of the pachymic acid is possibly based on ROS production stimulation, which leads to apoptotic death of cancer cells.40
Another essential enzyme of the glycolysis pathway is pyruvate kinase. It is involved in the last stage of glycolysis and serves as the last checkpoint of this process. The main products of the PKM gene are two isoforms: PKM1 and PKM2. For a long time, it was believed that cancer cells’ metabolism is characterized by the shift from expressing PKM1 isoform to PKM2. It was shown that PKM2 has a lower catalytic activity which favors promoting metabolic changes, giving cancer cells significant advantages.41 As it was indicated in a few studies and summarized in the review done by S. Mazurek, the modest activity of dimeric form of PKM2 allows accumulation of upstream intermediates among others, G-6-P, which can be directed to other metabolic pathways. PKM2 isoform overexpression enables gate-keeping function, forcing competition for other isoforms in this point of glycolysis.42 However, a study by Bluemlein and co-workers, performed on 25 human cancers, showed no evidence of isoform shift or distinction of dominating form in a proliferating tissue.43 Re-evaluation of RNA data of human tumors confirmed an elevated level of isoform PKM2 but as a result of general overexpression of the PKM gene rather than isoform switch.44 Examination of PKM2’s role in glucose metabolism with a knockout of the PKM gene via the CRISPR/Cas9 system showed that both isoforms favor aerobic glycolysis, but PKM2 is more efficient for the Warburg effect.45 Due to PKM2 isoform activity, cancer cells do not produce enough ATP, forcing them to rely on other ATP sources such as insufficient oxidative phosphorylation and glutaminolysis. Therefore, the PKM2 isoform has become an attractive target in the search for anti-cancer agents. However, this task remains challenging because of the similarity of PKM2 and PKM1 isoforms. Hsieh et al. presented a newly developed synthetic organic compound 1, which irreversibly inhibits the PKM2. It binds to Cys317 and Cys326 residues of the isoform, suppressing tumor growth in both human cancer cell lines (breast and cervical cancer) and xenografts without causing acute toxicity.46 Compound 1 requires further evaluation but may inspire new anti-cancer therapy strategies.
Another new inhibitor for the PKM2 is benserazide, an aromatic L-amino acid used for the treatment of Parkinson’s disease. A study based on human melanoma cells showed that benserazide binds directly to the PKM2, blocking its activity, and leading to the inhibition of aerobic glycolysis and restoration of oxidative phosphorylation. Despite the similarity of PKM1 and PKM2 isoforms, benserazide inhibited the PKM2 exclusively, and inhibition of tumor growth was observed both in vitro and in vivo.47 Using benserazide in anti-cancer therapy seems promising, especially considering that this compound is already in clinical use and can cross the blood–brain barrier, which may be useful in brain tumor treatment.
Inhibition of Lactate Dehydrogenase A
Another characteristic feature of many types of cancer is the high expression of lactate dehydrogenase A (LDHA). The primary function of this enzyme in physiological metabolism is converting lactate to pyruvate and back to maintain the NAD+/NADH ratio. In cancer cells, it plays a role in proliferation, invasion, metastasis, angiogenesis, and immune escape. LDHA gene knockdown resulted in growth inhibition and induction of apoptosis.48
The inhibition of LDHA can be achieved in different ways. One of the most commonly used compounds is galloflavin, a synthetic molecule binding to free LDHA. It shows good permeability and no significant effect on properly functioning cells. Treating cells with galloflavins showed effectiveness in studies on breast cancer cells and hepatocellular carcinoma cell lines.48 It was also shown that using galloflavin can sensitize Burkitt’s lymphoma cells to cisplatin without affecting normal lymphocytes.49 Rupiani et al. identified more soluble galloflavin mimetic—urolithin M6 (UM6), a metabolite produced by gut microbiota. The compound successfully imitated galloflavin’s mechanism of action.50 This study showed that identifying mimetics of galloflavin or modifying the original molecule could enable overcoming its poor physicochemical properties.
Another well-known inhibitor is FX11. It is a gossypol analog inhibiting LDHA by competing with NADH. The study on a panel of 15 patient-derived xenografts showed that FX11 inhibited LDHA but only in tumors containing TP53 mutated cells.51 This discovery does not exclude the possibility of using FX11 in therapy since most human cancers contain mutations of TP53.
Inhibition of the Monocarboxylate Transporters
One of the main characteristics of cancer cells is the production of large amounts of lactates due to the Warburg effect. Lactate accumulation could lead to the acidification of cells and, eventually, their death. Therefore, cancer cells must maintain lactate at an optimal level by transporting acids outside via monocarboxylate transporters (MCTs).52 Dominant transporters involved in maintaining the lactate level in cancer cells are MCT1 and MCT4. They are also involved in the processes of angiogenesis and tumor growth. In conclusion, inhibiting those transporters can have anti-cancer effects.
AR-C155858 and AZD3965 belong to a group of potent, selective MCT1 inhibitors. A study on human breast tumor cancer cells treated with AR-C155858 in the xenograft model showed no significant effects on tumor growth (only resulted in slight, statistically insignificant, increased lactate level).53 Another study performed on the same cancer line showed that both AR-C155858 and AZD3965 could cause slowly reversible inhibition of MCT1.54 Application of AZD3965 in human lymphoma and colon carcinoma cell lines resulted in lactate accumulation and increased mitochondrial metabolism. A combination of AZD3965 and drug-targeting mitochondrial metabolism such as metformin resulted in anti-cancer effects confirmed in vivo in xenograft models of human lymphoma.55 At this moment, the AZD3965 compound is in the first phase of clinical trials of lymphoma, Burkitt’s lymphoma, diffuse large B-cell lymphoma, and solid tumors (NCT01791595). Another potent MCT1 inhibitor is BAY-8002. In the study of diffuse large B-cell lymphoma cell lines and solid tumor models, the application of BAY-8002 resulted in a significant increase of accumulated lactate. Inhibition of the lactate trafficking results in pH change and indirectly reduces glycolysis, raising anti-tumor activity by affecting tumor growth.56
The group of compounds inhibiting both MCT1 and MCT4 transporters is represented by syrosingopine derived from reserpine, a natural alkaloid. A study performed on breast cancer cell lines (MDA-MB-453MDA-MD-453, SkBr3), leukemia cell lines (K562, HL60), colon carcinoma cell line (HCT116), and cervical cancer (HeLa) showed that the application of syrosingopine results in inhibition of MCT1 and MCT4 with 60-fold higher effect on MCT4. Moreover, syrosingopine enhanced the effect of metformin in breast cancer cell lines, which resulted in a loss of NAD+ regeneration capacity, inhibition of glycolysis, ATP depletion, and, eventually, cell death.57 Promising dual inhibitory effects are also shown by compounds based on N,N-dialkyl cyanocinnamic acids. The results of studies of human colon adenocarcinoma cells WiDr and human breast cancer cells MDA-MB-231 treated with N,N-dialkyl cyanocinnamic acids homolog showed equal inhibition of both transporters in vitro as well as in vivo in tumor xenograft models.58
Inhibition of Hypoxia-Inducible Factor (HIF-1)
In general, hypoxia can be defined as an effect emerging from deficient oxygen supply in the context of physiological demand. The state of insufficient O2 levels is an inseparable element of many cancer types. Worsen oxygenation can be caused by adverse diffusion geometry, disturbed circulation, and structural abnormalities of newly formatted blood vessels. Hypoxia is present in tumors when the O2 partial pressure falls below a critical value, which causes a progressive decrease in O2 consumption and ATP production rates in cells.59 Hypoxia causes profound changes in cancer cells’ proteomes due to the stimulation or inhibition of specific gene expression.
The hypoxia-inducible factors are transcription factors playing an important role in maintaining the Warburg effect. There are three isoforms, among which the role of HIF-3 remains the least known, but HIF-1 and HIF-2 share similar regulation characteristics.60 Hypoxic conditions stimulate the activation of this factor, but it can also be activated through mutation in the von Hippel–Lindau suppressor or through activation of Akt kinase which stimulates HIF-1 activation via Akt/PI3K/mTOR signal transduction pathway. Overexpression of the PKM2 isoform also stimulates HIF-1 into increased transcriptional activity by binding to the transactivation domain of the HIF1-α subunit. HIF-1 targets genes coding key elements of glucose metabolism such as glucose transporters, hexokinases, phosphofructokinase, or lactate dehydrogenase.61 Because of the complexity of HIF-1 involvement in various pathways and mechanisms, targeting it as a potential anti-cancer therapy element suggests it is an option worth further exploration.
Transcriptional activity of hypoxia-inducible factors requires dimerization of subunits α and β. The formation of the HIF-2α/HIF-β complex can be inhibited by small, synthetic molecules PT2385 and PT2399. Preclinical trials showed that treating human renal carcinoma cell lines with PT2385 and PT2399 results in inhibition of the HIF-2α and HIF2, leading to growth and proliferation suppression.62
Another possible approach for the reduction of HIF activity is inhibiting its chaperone protein. The proper conformation of HIF-1 requires the presence of heat shock protein 90 (HSP90). Application of ganetespib, a small molecule, in pancreatic ductal adenocarcinoma cells resulted in the inhibition of HSP90, which led to the withdrawal of proliferative, angiogenic, and anti-apoptotic effects of HIF-1 overexpression. Moreover, treating cells with ganetespib resulted in increased sensitivity to chemoradiotherapy. The anti-cancer effects of the compound were also confirmed in vivo using human pancreatic cancer xenografts.63
Another compound inhibiting the interaction of HSP90 and HIF-1 is thymoquinone. It is a natural phytochemical compound whose anti-angiogenic effects have only recently been linked to its inhibiting HIF-1 characteristics. Treating human renal cancer cell lines with thymoquinone resulted in the degradation of HIF-1α protein, decreased expression of genes under the control of this factor, and lower glucose, lactate, and ATP levels.64 Another element required for HIF-1 transcriptional activity is binding with the p300 cofactor. Inhibiting HIF can be achieved by interference with this binding. Thus, factor inhibiting HIF-1 (FIH-1) has properties allowing the disruption of HIF/p300 binding.65
Bortezomib, sold under the brand name Velcade, is a substance enhancing HIF-1 inhibition via FIH-1.65 Bortezomib is used in multiple myeloma therapy and in clinical trials evaluating its effectiveness in the treatment of other types of cancer, such as breast cancer (NCT00028639), kidney cancer (NCT00025376), and lung cancer (NCT00064012, NCT01833143). YC-1 (3-(5′-Hydroxymethyl-2′-furyl)-1-benzyl indazole) also shows inhibition via the FIH-1 effect. A recent study showed that a combination of YC-1, a hydrophobic compound, with the anti-cancer drug irinotecan, which is a hydrophile, in one amphiphilic molecule, resulted in the creation of a 5.7-fold more effective anti-cancer drug.66 This study also shows that overcoming hypoxia conditions can be crucial for other anti-cancer drugs’ efficacy.
The most common approach to blocking HIF-1 activity is inhibition at the expression level. Such a mechanism was observed in a study on melittin. This 26-amino acid peptide is the main component of honeybee venom whose original role is to activate nociceptor cells. Treating human liver cancer cells and, later, tumor xenografts with melittin decreased expression of HIF-1α whereby the HIF-1α/Akt pathway was diminished, which resulted in suppressed growth, proliferation, migration, invasion, and vasculogenic mimicry formation.67 Another compound of natural origin that has HIF expression inhibiting effect is actinolactomycin (ALM) extracted from Streptomyces flavoretus. The studies of human hepatoma cells (Hep3B) and human prostate cancer cells (PC3) treated with ALM revealed growth inhibition as a result of HIF-1α translation and mTOR signaling pathway suppression. These effects have been confirmed in both in vitro and in vivo models. Moreover, using low concentrations of ALM resulted in an enhanced effect of Everolimus, an inhibitor of mTOR already used in anti-cancer therapy.68 The combination of ALM and Everolimus enables a lower drug dose and more efficient therapy for solid tumor patients.68 Zebularine, the analog of cytidine belonging to a group of DNA methyltransferases, also shows a downregulation effect on HIF-1α expression. Using this compound in the study of the human colorectal cancer line (HCT116) and tumor xenografts resulted in the degradation of HIF-1α protein and the reduction of angiogenesis. Targeting HIF-1α with zebularine could increase the efficacy of chemotherapy used in colorectal cancer therapy.69
Clinical Perspectives
As a hallmark of cancer metabolism, aerobic glycolysis seems to be a promising drug target of anti-cancer therapy. However, despite significant efforts that have been put into developing metabolism-based drugs, we still lack effective cancer therapy. Out of the many compounds which proved to have high potential in in vitro studies, very few have been used in clinical trials (Table 1). Moreover, the results of clinical trials often showed a significant discrepancy between our expectations toward the efficiency of some drugs and their actual clinical utility. Several problems have to be taken into consideration when designing therapies based on compounds targeted in cancer cell energy metabolism. They are summarized as follows.
Table 1.
Inhibitors of Selected Points of Glucose Metabolism in Cancer Cells.
| Compound | Chemical formula | Type of substance | Mechanism of action | Cells/animal models in preclinical studies | Clinical development | References | |
|---|---|---|---|---|---|---|---|
| Inhibitors of glucose transporters | WZB117 | 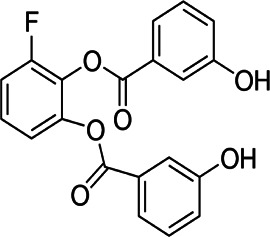 |
Hydoxy-benzoate | Binding at the exofacial sugar binding site | Colon, breast lung cancer cells | Preclinical | 18,19,21,23 |
| STF-31 | 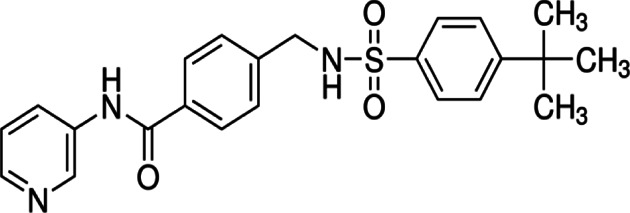 |
Benzamide | NAPMT or GLUT1 inhibitor | Renal cancer cells | Preclinical | 24,25 | |
| Resveratrol | 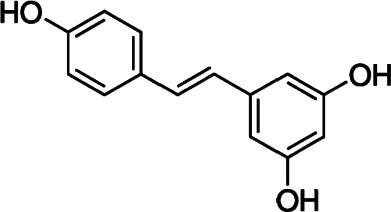 |
Polyphenol | Suppresses intracellular trafficking of GLUT1 to the plasma membrane | Myelotic, ovarian cancer cells | Phase I (colon, rectal cancer) | 26–28 | |
| 19FDG |  |
Glucose analog | Competitive inhibition | Cervical cancer cells, cervical cancer xenografts | Preclinical | 31 | |
| Inhibitors of glycolytic enzymes | Lonidamine |  |
Dechlorinate derivative of indazole-3-carboxylic acid | Inhibition of mitochondria-bound HKII | Glioblastoma multiforme cells | Phase III (benign prostatic hyperplasia) | 30,34,35 |
| 3-bromopyruvate | 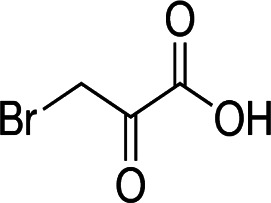 |
Pyruvate analog | HKII inhibition by alkylation of cysteine residues | Hepatocellular, metastatic prostate cancer cells | Preclinical, two case studies | 37,38,65 | |
| Pachymic acid | 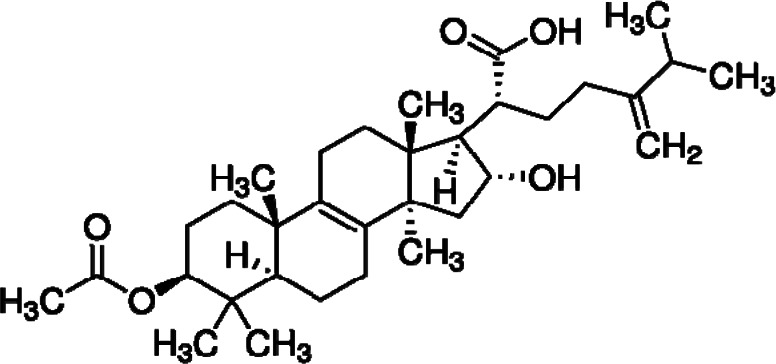 |
Triterpenoid | HKII inhibition | Breast, lung cancer cells | Preclinical | 39 | |
| Benserazide |  |
Aromatic L-amino acid decarboxylase | PKM2 inhibition by direct bind | Melanoma cancer cells | Phase IV, used in Parkinson’s disease | 45 | |
| Inhibitors of LDHA | Galloflavin |  |
Synthetic molecule | Binding to free LDHA | Breast, hepatocellular cancer, Burkitt’s lymphoma cells | Preclinical | 61,62 |
| Urolithin M6 | 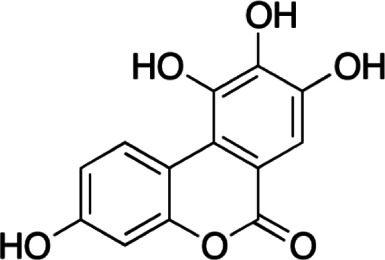 |
Natural galloflavin mimetic | Binding to free LDHA | Adipocytes, hepatocytes | Preclinical | 63 | |
| FX11 |  |
Gossypol analog | NADH competitive | 15 patient-derived mouse xenograft models | Preclinical | 62 | |
| Inhibitors of MCTs | AR-C155858 | 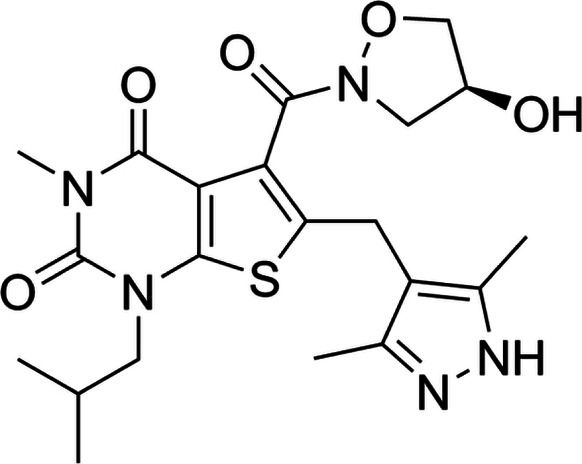 |
Pyrrole pyrimidine derivative | Selective MCT1 inhibitor | Breast cancer cells, tumor xenografts | Preclinical | 55 |
| AZD3965 | 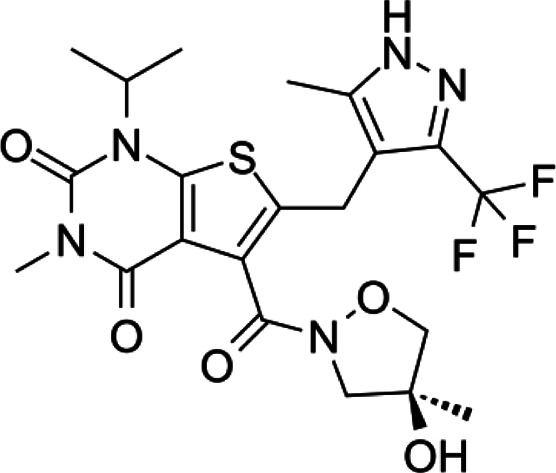 |
Pyrrole pyrimidine derivative | Selective MCT1 inhibitor | Breast, colon cancer cells, lymphoma xenografts | Phase I (solid tumors, DLBCL) | 57 | |
| BAY-8002 | 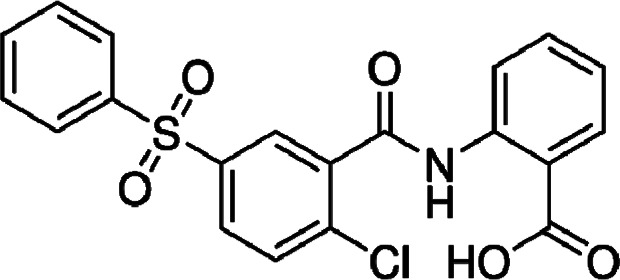 |
Synthetic compound | Selective MCT1 inhibitor | Diffuse large B-cell lymphoma cells | Preclinical | 58 | |
| Syrosingopine | 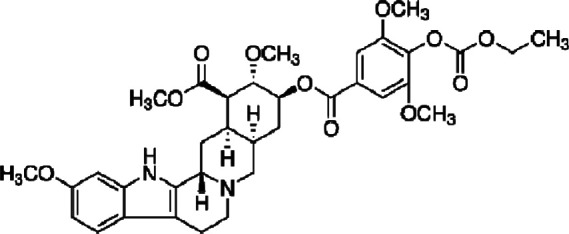 |
Reserpine derivative | Dual MCT1/MCT4 inhibitor | Breast, colon, cervical cancer, leukemia cells | Preclinical | 59 | |
| N,N-dialkyl cyanocinnamic acids homologs | 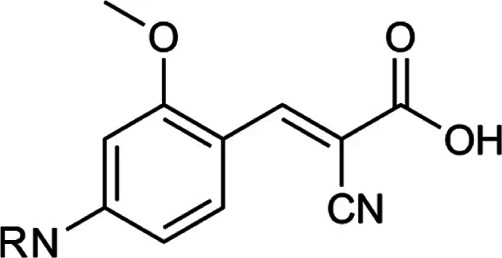 |
N,N-dialkyl cyanocinnamic acids homologs | Dual MCT1/MCT4 inhibitor | Breast, colon cancer cells | Preclinical | 60 | |
| Inhibitors of HIF | PT2385 and PT2399 | 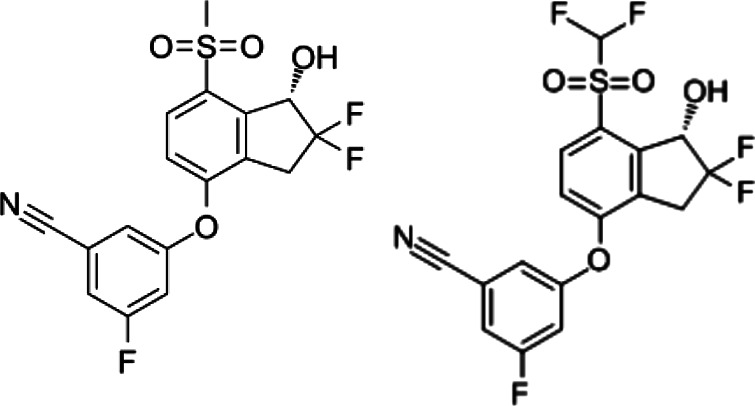 |
Synthetic molecules | Inhibition of HIFα/HIFβ dimerization | Renal cancer cells | Preclinical | 47 |
| Ganetespib | 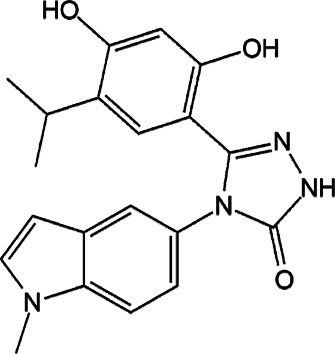 |
Synthetic molecule | Inhibition via inhibition of HSP90 | Pancreatic ductal adenocarcinoma, pancreatic cancer xenografts | Phase II (breast cancer, NSCLC) | 48 | |
| Thymoquinone | 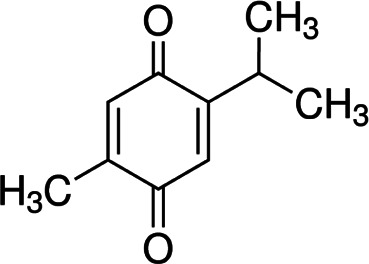 |
Phytochemical | Inhibiting HSP90/HIF interaction | Renal cancer cells | Preclinical | 49 | |
| YC-1 |  |
Indazole | Inhibition via HIF/FIH binding enhancement | Pulmonary adenocarcinoma cells | Preclinical | 50 | |
| Melittin |  |
Peptide | Inhibition of HIF-1α expression | Liver cancer cells, tumor xenografts | Preclinical | 51 | |
| Actinolactomycin | 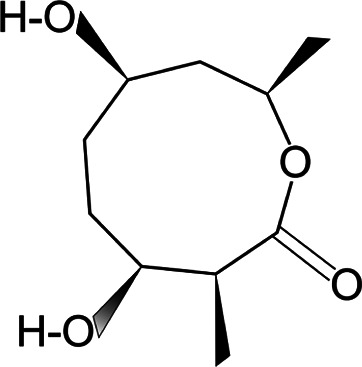 |
2-oxonano-noidal antibiotic | Inhibition of HIF-1α translation | Hepatoma, prostate cancer cells, tumor xenografts | Preclinical | 52 | |
| Zebularine |  |
DNA methyltransferase | Downregulating HIF-1α expression | Colorectal cancer cells, tumor xenografts | Preclinical | 53 |
FDG, fluorodeoxyglucose; GLUT, glucose transporter; HIF-1, hypoxia-inducible factor; HKII, hexokinase II; LDHA, lactate dehydrogenase A; MCT1, monocarboxylate transporter 1; NADH, nicotinamide adenine dinucleotide; PKM2, pyruvate kinase isozyme M2.
Drug Administration and Bioavailability
The 3-bromopyruvate is an excellent example of the difficulties associated with the search for effective drugs. 3BP showed great effectiveness in studies using cell lines and tumor xenograft animal models,70 raising hopes for developing a fast, powerful agent for cancer treatment. The 3BP has not yet undergone formal clinical trials but it was used in two single-case trials. In 2012, Ko and co-workers reported on the usage of 3BP in the treatment of a 16-year-old patient diagnosed with fibrolamellar hepatocellular carcinoma in the terminal phase.71 The authors suggested that 3BP treatment did not cause significant toxicity to the patient and let him survive for two more years with a higher quality of life than expected. Finally, the patient died not from cancer, but due to liver function overload, unable to eliminate the dead cancer cell debris formed after 3BP treatment.71 In another report, in 2014, El Sayed and co-workers used 3BP to treat a 28-year-old patient diagnosed with stage IV metastatic melanoma.72 Intravenous infusion of 3BP caused a burning venous sensation but there were no severe toxic effects. Unfortunately, in this case, the patient died a few months after starting the 3BP treatment due to complications related to the advanced stage of the tumor.72 It is important to mention that 3BP was also used as part of alternative therapy. In Germany, in 2016, three patients undergoing alternative cancer treatment died shortly after the infusion of 3BP, and non-medical practitioners had been charged with involuntary manslaughter.73
At present, it is known that free, unformulated 3BP could not be used for patient treatment. Apart from discounting side effects, such as a burning venous sensation accompanying the intravenous 3BP infusion, we have to be aware of many other obstacles, for example, the inactivation of 3BP by serum proteins and high GSH level. Moreover, despite 3BP’s effectiveness in killing glioblastoma cells, its utility in brain cancer treatment is hindered due to its inability to cross the blood–brain barrier. However, some of these obstacles can be overcome. Formulating 3BP with liposomal carriers or suitable nanoparticle carriers could potentially enable 3BP to cross the blood–brain barrier and prevent its inactivation in blood.39 Mixing 3BP with citric acid might result in better persistence of the compound in tumor tissue, and combining 3BP with a GHS depleting agent might overcome the resistance in GHS-rich tumors.39
Low bioavailability and lack of early response are common characteristics of other compounds targeting glucose metabolism. Resveratrol, which suppresses intracellular trafficking of GLUT1 to the plasma membrane, is a potentially effective compound in anti-cancer therapy. Resveratrol in phase I clinical trial for healthy volunteers was proven safe and had no significant side effects (NCT 00098969) but, unfortunately, it showed low bioavailability and poor solubility.74 To overcome these obstacles, researchers prepared synthesized resveratrol derivatives such as methoxylated, hydroxylated, and halogenated forms of the compound which exhibited favorable therapeutic potential.74 Poor physicochemical properties might also be overcome by finding mimetics of specific compounds. For example, galloflavin, which has poor solubility, could be replaced by its mimetic—urolithin M6. This drug still needs clinical evaluation but shows better properties and good effectiveness in inhibiting LDHA.50
Specificity and Toxicity
Unwanted toxicity and side effects are considerable challenges in developing drugs that target cancer cell metabolism. Toxic and side effects are mainly caused by inhibiting metabolic enzymes not only in cancer but also in normal cells—especially those that display aerobic glycolysis, such as cells of the immune system.75 Similar to tumors, primed T cells use aerobic glycolysis to maintain their high proliferative rate. The conversion of glucose into lactate in the presence of oxygen is a characteristic feature of the metabolic switch of naive into effector T cells. The results of some studies demonstrated that inhibition of LDH might lead to a decrease in T cells of IFN-γ production.76 2-Deoxyglucose (2DG) is an effective inhibitor of HK used to shut down glycolysis in cancer cells. Despite its efficiency, 2DG has also been shown to impair the metabolism of T cells which results in decreased secretion of cytokines and reduces T cell anti-tumor function.75
The increase of glucose uptake is an essential hallmark of cancer cells. Since upregulation of GLUT1 is a common feature of cancer cells, this protein is considered a promising target in anti-cancer therapy. However, GLUT1 facilitates the basal uptake of glucose in most normal cell types. Thus, even if the results of in vitro studies show excellent effectiveness of GLUT1 inhibition, we need to consider the potential undesirable impact of blocking GLUT1 in cells or tissues that need it for physiological glucose homeostasis. So far, there are no clinical trials for glucose transporters.
Normal proliferating cells have similar metabolic requirements to cancer cells, which raises questions about whether changes in expression of GLUTs or certain glycolytic enzyme activity in cancers are sufficient to provide a therapeutic window to let us effectively target cancer cells without causing severe toxic effects. Although early clinical testing of 2-DG caused a response in patients, the use of this drug was limited by the toxicity associated with hypoglycemia symptoms. In recent clinical trials, 2-DG has been used at lower doses, but those are insufficient to inhibit disease progression.77,78 In effect, at tolerable doses, there was no clinical effect.
The solution to these problems could be engineering systems for restricted delivery of drugs at the tumor site. Local administration is a promising strategy for targeted drug delivery in specific cancers. Local delivery of therapeutics using nanocarriers, implants, aerosols, and hydrogels can bypass physiological barriers, acting as a promising strategy in treating locally accessible tumors.79
Plasticity of Cancer Cells
It has been shown that cancer cells can overcome the inhibition of specific pathways. Although the Warburg effect is the most significant characteristic of tumor metabolism, it is not the only source of energy for cancer cells that are able to use other energy sources to maintain proliferation, such as glutamine, amino acids, and fatty acids. Moreover, metabolic plasticity enables cancer cells to switch their metabolism phenotypes between glycolysis and oxidative phosphorylation during tumorigenesis and metastasis.80 Cancer cells, due to the activity of the PKM2 isoform instead of the PKM1 and reduced production of ATP, have to rely on glutaminolysis in terms of energy production.6 Glutaminolysis may compensate for inhibited glycolytic flux, and glycolysis can compensate for decreased glutaminolysis. The relationships between those two pathways points out the need for targeting them at the same time. In mouse model with Myc-induced hepatocellular carcinoma, deletion of the GLS1 gene whose product takes part in the glutaminolysis and deletion of the gene coding for hexokinase II resulted in inhibition of tumor formation, decreased level of TCA and PPP intermediates, and disturbance in the energy balance.81
Thus, considering tumor metabolic plasticity, single agents are unlikely to become an effective anti-cancer therapy. Targeting simultaneously two or more metabolic pathways would be more effective in blocking the relapse and development of resistances. The studies using the mouse cancer model proved that a triple metabolic blockage by targeting glycolysis, glutaminolysis, and de novo synthesis of fatty acids might be a feasible and effective strategy to combat malignant tumors.82.
Heterogeneity of Cells in Tumors
Cancer is a dynamic disease, and tumors consist of a diverse collection of cells with differential metabolic requirements and levels of sensitivity to treatment. Heterogeneity is the main reason for their resistance and accurate assessment of tumor cells’ metabolic preference is essential for developing effective therapies.83 Nowadays, it is well-accepted that there are specific cell subpopulations in tumors, such as slow-growing cells, dormant cells, and cancer stem cells that rely on oxidative phosphorylation. Moreover, tumors’ intricate architecture and nutrient conditions are affected by other cell types such as cancer-associated fibroblasts (CAFs), dysfunctional blood vessels, and immune cells. Thus, heterogeneity of cells in tumors and tumor cells’ plasticity are the reason for metabolism-targeted drugs possibly useless as monotherapy, and targeting the multitude of potential metabolic strategies that tumors can activate will be necessary for effective therapy.
Recently, metabolic alterations have been shown to play a role in the sensitization of cancer cells to widely used first-line chemotherapeutics. This suggests that metabolic pathways are essential mediators of resistance toward anti-cancer agents.84
Metabolism-targeting drugs are tested with other anti-cancer therapies. Pyruvate mimetic compound dichloroacetate (DCA) stimulates mitochondrial function by inhibiting regulatory pyruvate dehydrogenase kinases (PDK) at the expense of glycolysis to reverse the Warburg effect and block the growth advantage of tumor cells. Ongoing clinical studies evaluate the effects of DCA versus placebo in combination with cisplatin and radiation treatment in patients with stage III-IV squamous cell carcinoma of the head and neck (SCCHN) (NCT01386632).85
An inhibitor of HIF-2 PT2385 has been tested on healthy volunteers (NCT02553356) for safety and tolerability and patients with advanced clear cell renal carcinoma (NCT02293980) in multiple doses to identify the recommended phase II dose of PT2385.86 PT2385 is also tested in combination with nivolumab and cabozantinib. PT2385 tablets are tested in treating patients with recurrent glioblastoma as monotherapy (NCT03216499). The monocarboxylate transporters inhibitor AZD3965 is tested in patients with advanced cancers (NCT01791595).
Anti-cancer strategies based on the Warburg effect involve not only the use of drug compounds but also some dietary changes. Ketogenic diets that rely on cancer’s dependence on glycolysis even in the presence of oxygen are well-known adjuvant treatment strategies for cancer patients. Ketogenic diets rich in fat and poor in carbohydrates reduce the systemic amount of glucose that the cancer cells can utilize. The results of many preclinical studies suggest that ketogenic diets slow tumor growth, prolong the survival rate, and sensitize cancer cells to traditional chemo- or radiotherapies. Unfortunately, ketogenic diets also show pro-tumor effects or severe side effects in certain cancer models.87 It is suggested that the efficacy of ketogenic diets could be affected by cancer type or even subtype, as well as the genetic background. Thus, molecular context can determine a ketogenic diet’s impact on cancer cells.87
Despite the many clinical studies on ketogenic diets in cancer patients, clear evidence demonstrating an advantageous effect and anti-tumor efficiency is still lacking. Römer et al. have recently performed a systematic search of databases to find all the studies analyzing the effectiveness of a ketogenic diet in cancer patients as a sole or complementary therapy.88 However, they did not find conclusive evidence for its anti-tumor effects or improved overall survival. The reason was a low quality and high heterogeneity of most studies.88 Majority of studies were case reports or pilot studies focusing only on tolerability and safety of ketogenic diets. Despite the preliminary nature of these studies, consistent findings include a moderate reduction of blood glucose levels, induction of ketosis, as well as improvement in quality of life (for a review see Ref. 87). Recently, Seyfried at al.89 have reported the first case of confirmed IDH1-mutant glioblastoma successfully treated with ketogenic metabolic therapy and surgical debulking without chemo- or radiotherapy. The authors suggest that the very long-term survival of the patient could be due in part to a therapeutic metabolic synergy between ketogenic diet and the IDH1 mutation that simultaneously target the glycolysis and glutaminolysis pathways that are important for tumor growth.89 However, more consistent clinical evidence from large patient groups with comparable methodology and dietary protocols is necessary before a ketogenic diet can be considered effective and recommended to cancer patients as a treatment.
Conclusions
Metabolic reprogramming is a common characteristic of cancer cells; therefore, targeting key metabolism regulators seems to be a very good strategy in anti-cancer therapy. Over the years, many small chemical compounds have been tested for the inhibition of the glycolytic pathway components but only a few entered clinical trials. Taking cancer’s heterogeneity and metabolic plasticity into account, it is very unlikely that metabolic-targeted drugs can be used as monotherapy. A major problem encountered in strategies to inhibit metabolic pathways is cancer’s capability for obtaining the necessary metabolites from other sources. In effect, a better strategy is to use two or more drugs targeted at different metabolic pathways. Moreover, considering that inhibitors of glucose metabolism can increase the sensitivity of cancer cells to some chemotherapeutics, it seems more effective to use metabolic inhibitors in combination with other targeted therapies, chemotherapy or radiotherapy. Moreover, combining current and future metabolism-based drugs with targeted delivery systems can help avoid toxic effects and further advance their use in cancer treatment. Finally, for successful anti-cancer therapy, metabolic profiling of tumors in patients is necessary. Specific biomarkers indicating metabolic vulnerabilities of different types of cancers and a better knowledge of the interactions between cancer cells and their environment will help design more effective anti-cancer therapies.
Appendix 1
- 2DG
2-deoxy-D-glucose
- 3-BP
3-bromopyruvate
- AKT
v-akt murine thymoma viral oncogene homolog 1 (protein kinase)
- ALM
actinolactomycin
- FIH-1
factor inhibiting HIF-1
- GSH
glutathione
- HIF-1
hypoxia-inducible factor 1
- HKII
hexokinase II
- HSP90
heat shock protein 90
- LDHA
lactate dehydrogenase A
- MCT
monocarboxylate transporter
- mTOR
mammalian target of rapamycin (protein kinase)
- NAD+/NADH
oxidized and reduced nicotinamide adenine dinucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- PI3K
phosphoinositide 3-kinase
- PKM2
pyruvate kinase M
- VHL
von Hippel–Lindau protein
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD
Anna Krzeslak https://orcid.org/0000-0002-4982-7901
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D1, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 4.Seyfried TN, Arismendi-Morillo G, Mukherjee P, Chinopoulos C. On the origin of ATP synthesis in cancer. iScience. 2020;23(11):101761. doi: 10.1016/j.isci.2020.101761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaupel P, Schmidberger H, Mayer A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. 2019;95(7):912–919. doi: 10.1080/09553002.2019.1589653 [DOI] [PubMed] [Google Scholar]
- 6.Tekade RK, Sun X. The Warburg effect and glucose-derived cancer theranostics. Drug Discov Today. 2017;22(11):1637–1653. [DOI] [PubMed] [Google Scholar]
- 7.Xie N, Zhang L, Gao W, et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 2020;5:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krześlak A, Jóźwiak P, Forma E, et al. Diagnostic value of glucose transporter 1 and 3 (GLUT1 and GLUT3) mRNA level in postmenopausal women with urinary bladder cancer. Przeglad Menopauzalny. 2012;11:178–182. [Google Scholar]
- 9.Krzeslak A, Wojcik-Krowiranda K, Forma E, et al. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol Oncol Res. 2012;18:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao ZX, Lu LW, Qiu J, et al. Glucose transporter-1 as an independent prognostic marker for cancer: a meta-analysis. Oncotarget. 2017;9(2):2728–2738. doi: 10.18632/oncotarget.18964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu M, Yongzhi H, Chen S, Luo X, Lin Y, Zhou Y, et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget. 2017;8(26):43356–43367. doi: 10.18632/oncotarget.17445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jóźwiak P, Krześlak A, Bryś M, Lipińska A. Glucose-dependent glucose transporter 1 expression and its impact on viability of thyroid cancer cells. Oncol Rep. 2015;33:913–920. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Ye C, Chen C, et al. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: A systematic review and meta-analysis. Oncotarget. 2017;8(10):16875–16886. doi: 10.18632/oncotarget.15171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, Xie Z, Li B. The clinicopathologic impacts and prognostic significance of GLUT1 expression in patients with lung cancer: A meta-analysis. Gene. 2019;689:76–83. doi: 10.1016/j.gene.2018.12.006 [DOI] [PubMed] [Google Scholar]
- 16.Ojelabi OA, Lloyd KP, Simon AH, De Zutter JK, Carruthers A. WZB117 (2-Fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J Biol Chem. 2016;291(52):26762–26772. doi: 10.1074/jbc.M116.759175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu W, Fang Y, Wang XT, Liu J, Dan X, Sun LL. Overcoming 5-Fu resistance of colon cells through inhibition of Glut1 by the specific inhibitor WZB117. Asian Pac J Cancer Prev. 2014;15:7037–7041. doi: 10.7314/apjcp.2014.15.17.7037 [DOI] [PubMed] [Google Scholar]
- 18.Zhao F, Ming J, Zhou Y, Fan L. Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother Pharmacol. 2016;77(5):963–972. doi: 10.1007/s00280-016-3007-9 [DOI] [PubMed] [Google Scholar]
- 19.Chen Q1, Meng YQ, Xu XF, Gu J. Blockade of GLUT1 by WZB117 resensitizes breast cancer cells to adriamycin. Anti Canc Drugs. 2017;28(8):880–887. doi: 10.1097/CAD.0000000000000529 [DOI] [PubMed] [Google Scholar]
- 20.Yakisich JS, Azad N, Kaushik V, Iyer AKV. The biguanides metformin and buformin in combination with 2-Deoxy-glucose or WZB-117 inhibit the viability of highly resistant human lung cancer cells. Stem Cells Int. 2019;2019:6254269. doi: 10.1155/2019/6254269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11(8):1672–1682. doi: 10.1158/1535-7163.MCT-12-0131 [DOI] [PubMed] [Google Scholar]
- 22.Shibuya K, Okada M, Suzuki S, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. 2015;6(2):651–661. doi: 10.18632/oncotarget.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan DA, Sutphin PD, Nguyen P, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70. doi: 10.1126/scitranslmed.3002394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraus D, Reckenbeil J, Veit N, et al. Targeting glucose transport and the NAD pathway in tumor cells with STF-31: A re-evaluation. Cell Oncol. 2018;41(5):485–494. doi: 10.1007/s13402-018-0385-5 [DOI] [PubMed] [Google Scholar]
- 25.Park JB. Inhibition of glucose and dehydroascorbic acid uptakes by resveratrol in human transformed myelocytic cells. J Nat Prod. 2001;64(3):381–384. [DOI] [PubMed] [Google Scholar]
- 26.Salas M, Obando P, Ojeda L, et al. Resolution of the direct interaction with and inhibition of the human GLUT1 hexose transporter by resveratrol from its effect on glucose accumulation. Am J Physiol Cell Physiol. 2013;305(1):C90–C99. doi: 10.1152/ajpcell.00387.2012 [DOI] [PubMed] [Google Scholar]
- 27.Gwak H, Haegeman G, Tsang BK, Song YS. Cancer-specific interruption of glucose metabolism by resveratrol is mediated through inhibition of Akt/GLUT1 axis in ovarian cancer cells. Mol Carcinog. 2015;54(12):1529–1540. doi: 10.1002/mc.22227 [DOI] [PubMed] [Google Scholar]
- 28.Salehi B, Mishra AP, Nigam M, et al. Resveratrol: A double-edged sword in health benefits. Biomedicines. 2018;6(3):91. doi: 10.3390/biomedicines6030091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front Pharmacol. 2011;2:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niccoli S, Boreham DR, Phenix CP, Lees SJ. Non-radioactive 2-deoxy-2-fluoro-D-glucose inhibits glucose uptake in xenograft tumors and sensitizes HeLa cells to doxorubicin in vitro. PLoS One. 2017;12(11):e0187584. doi: 10.1371/journal.pone.0187584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf A, Agnihotri S, Micallef J, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208(2):313–326. doi: 10.1084/jem.20101470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu JJ, Singh A, Xue K, et al. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget. 2017;9(3):4020–4033. doi: 10.18632/oncotarget.23425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Cosimo S, Ferretti G, Papaldo P, Carlini P, Fabi A, Cognetti F. Lonidamine: Efficacy and safety in clinical trials for the treatment of solid tumors. Drugs Today. 2003;39(3):157–174. [DOI] [PubMed] [Google Scholar]
- 34.Huang Y, Sun G, Sun X, et al. The potential of lonidamine in combination with chemotherapy and physical therapy in cancer treatment. Cancers. 2020;12(11):3332. doi: 10.3390/cancers12113332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oudard S, Carpentier A, Banu E, et al. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neuro Oncol. 2003;63(1):81–86. [DOI] [PubMed] [Google Scholar]
- 36.Nancolas B, Guo L, Zhou R, et al. The anti-tumor agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem J. 2016;473(7):929–936. doi: 10.1042/BJ2015112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoo JJ, Yu SJ, Na J, et al. Hexokinase-II inhibition synergistically augments the anti-tumor efficacy of sorafenib in hepatocellular carcinoma. Int J Mol Sci. 2019;20(6):1292. doi: 10.3390/ijms20061292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pichla M, Sroka J, Pienkowska N, et al. Metastatic prostate cancer cells are highly sensitive to 3-bromopyruvic acid. Life Sci. 2019;227:212–223. doi: 10.1016/j.lfs.2019.03.066 [DOI] [PubMed] [Google Scholar]
- 39.EL Sayed SM. Enhancing anticancer effects, decreasing risks and solving practical problems facing 3-bromopyruvate in clinical oncology: 10 years of research experience. Int J Nanomedicine. 2018;13:4699–4709. doi: 10.2147/IJN.S170564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miao G, Han J, Zhang J, Wu Y, Tong G. Targeting pyruvate kinase M2 and hexokinase II, pachymic acid impairs glucose metabolism and induces mitochondrial apoptosis. Biol Pharm Bull. 2019;42(1):123–129. doi: 10.1248/bpb.b18-00730 [DOI] [PubMed] [Google Scholar]
- 41.Anastasiou D, Poulogiannis G, Asara JM, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–1283. doi: 10.1126/science.1211485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazurek S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43(7):969–980. doi: 10.1016/j.biocel.2010.02.005 [DOI] [PubMed] [Google Scholar]
- 43.Bluemlein K, Grüning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011;2(5):393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhan C, Yan L, Wang L, et al. Isoform switch of pyruvate kinase M1 indeed occurs but not to pyruvate kinase M2 in human tumorigenesis. PLoS One. 2015;10(3):e0118663. doi: 10.1371/journal.pone.0118663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Wang Y, Ruan Y, et al. PKM2 promotes reductive glutamine metabolism. Cancer Biol Med. 2018;15(4):389–399. doi: 10.20892/j.issn.2095-3941.2018.0122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh IS, Gopula B, Chou CC, et al. Development of novel irreversible pyruvate kinase M2 inhibitors. J Med Chem. 2019;62(18):8497–8510. doi: 10.1021/acs.jmedchem.9b00763 [DOI] [PubMed] [Google Scholar]
- 47.Zhou Y, Huang Z, Su J, et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int J Cancer. 2019;147:139–151. doi: 10.1002/ijc.32756 [DOI] [PubMed] [Google Scholar]
- 48.Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018;7(12):6124–6136. doi: 10.1002/cam4.1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manerba M, Di Ianni L, Govoni M, Roberti M, Recanatini M, Di Stefano G. LDH inhibition impacts on heat shock response and induces senescence of hepatocellular carcinoma cells. Eur J Pharm Sci. 2017;15:91–98. doi: 10.1016/j.ejps.2017.05.015 [DOI] [PubMed] [Google Scholar]
- 50.Rupiani S, Guidotti L, Manerba M, et al. Synthesis of natural urolithin M6, a galloflavin mimetic, as a potential inhibitor of lactate dehydrogenase A. Org Biomol Chem. 2016;14(46):10981–10987. doi: 10.1039/c6ob01977c [DOI] [PubMed] [Google Scholar]
- 51.Rajeshkumar NV, Dutta P, Yabuuchi S, et al. Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Res. 2015;75(16):3355–3364. doi: 10.1158/0008-5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol. 2019;9:1143. doi: 10.3389/fonc.2019.01143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guan X, Bryniarski MA, Morris ME. In vitro and in vivo efficacy of the monocarboxylate transporter 1 inhibitor AR-C155858 in the murine 4T1 breast cancer tumor model. AAPS J. 2018;21(1):3. doi: 10.1208/s12248-018-0261-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guan X, Rodriguez-Cruz V, Morris ME. Cellular uptake of MCT1 inhibitors AR-C155858 and AZD3965 and their effects on MCT-mediated transport of L-lactate in murine 4T1 breast tumor cancer cells. AAPS J. 2019;21(2):13. doi: 10.1208/s12248-018-0279-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beloueche-Babari M, Wantuch S, Casals Galobart T, et al. MCT1 inhibitor AZD3965 increases mitochondrial metabolism, facilitating combination therapy and noninvasive magnetic resonance spectroscopy. Cancer Res. 2017;77(21):5913–5924. doi: 10.1158/0008-5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quanz M, Bender E, Kopitz C, et al. Preclinical efficacy of the novel monocarboxylate transporter 1 inhibitor BAY-8002 and associated markers of resistance. Mol Cancer Ther. 2018;17(11):2285–2296. doi: 10.1158/1535-7163 [DOI] [PubMed] [Google Scholar]
- 57.Benjamin D, Robay D, Hindupur SK, et al. Dual inhibition of the lactate transporters MCT1 and MCT4 Is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018;25(11):3047–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonnalagadda S, Jonnalagadda SK, Ronayne CT, et al. Novel N,N-dialkyl cyanocinnamic acids as monocarboxylate transporter 1 and 4 inhibitors. Oncotarget. 2019;10(24):2355–2368. doi: 10.18632/oncotarget.26760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Höckel M, Vaupel P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93(4):266–276. doi: 10.1093/jnci/93.4.266 [DOI] [PubMed] [Google Scholar]
- 60.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15(6):1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soni S, Padwad YS. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017;56(4):503–515. doi: 10.1080/0284186X.2017.1301680 [DOI] [PubMed] [Google Scholar]
- 62.Martínez-Sáez O, Gajate Borau P, Alonso-Gordoa T, Molina-Cerrillo J, Grande E. Targeting HIF-2 α in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit Rev Oncol Hematol. 2017;111:117–123. doi: 10.1016/j.critrevonc.2017.01.013 [DOI] [PubMed] [Google Scholar]
- 63.Nagaraju GP, Zakka KM, Landry JC, Shaib WL, Lesinski GB, El-Rayes BF. Inhibition of HSP90 overcomes resistance to chemotherapy and radiotherapy in pancreatic cancer. Int J Cancer. 2019;145(6):1529–1537. doi: 10.1002/ijc.32227 [DOI] [PubMed] [Google Scholar]
- 64.Lee YM, Kim GH, Park EJ, et al. Thymoquinone selectively kills hypoxic renal cancer cells by suppressing HIF-1α-mediated glycolysis. Int J Mol Sci. 2019;20(5):1092. doi: 10.3390/ijms20051092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masoud GN, Li W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5(5):378–389. doi: 10.1016/j.apsb.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang B, Huang X, Wang H, Gou S. Promoting antitumor efficacy by suppressing hypoxia via nano self-assembly of two irinotecan-based dual drug conjugates having a HIF-1α inhibitor. J Mater Chem B. 2019;7(35):5352–5362. doi: 10.1039/c9tb00541b [DOI] [PubMed] [Google Scholar]
- 67.Chen Q, Lin W, Yin Z, et al. Melittin inhibits hypoxia-induced vasculogenic mimicry formation and epithelial-mesenchymal transition through suppression of HIF-1α/Akt pathway in liver cancer. Evid Based Complement Alternat Med. 2019;19:9602935. doi: 10.1155/2019/9602935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng J, Hu L, Yang Z, et al. 2-Oxonanonoidal antibiotic actinolactomycin inhibits cancer progression by suppressing HIF-1α. Cell. 2019;8(5):439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei TT, Lin YT, Tang SP, et al. Metabolic targeting of HIF-1α potentiates the therapeutic efficacy of oxaliplatin in colorectal cancer. Oncogene. 2019;39(2):414–427. doi: 10.1038/s41388-019-0999-8 [DOI] [PubMed] [Google Scholar]
- 70.Fan T, Sun G, Sun X, Zhao L, Zhong R, Peng Y. Tumor energy metabolism and potential of 3-Bromopyruvate as an inhibitor of aerobic glycolysis: Implications in tumor treatment. Cancers. 2019;11(3):317. doi: 10.3390/cancers11030317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr. 2012;44(1):163–170. doi: 10.1007/s10863-012-9417-4 [DOI] [PubMed] [Google Scholar]
- 72.El Sayed SM, Mohamed WG, Seddik MA, et al. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: a concise literature review and case study. Chin J Cancer. 2014;33(7):356–364. doi: 10.5732/cjc.013.10111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res. 2019;150:104511. doi: 10.1016/j.phrs.2019.104511 [DOI] [PubMed] [Google Scholar]
- 74.Salehi B, Mishra AP, Nigam M, et al. Resveratrol: A double-edged sword in health benefits. Biomedicines. 2018;6(3):91. doi: 10.3390/biomedicines6030091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting tumor metabolism: A new challenge to improve immunotherapy. Front Immunol. 2018;9:353. doi: 10.3389/fimmu.2018.00353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huber V, Camisaschi C, Berzi A, et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol. 2017;43:74–89. doi: 10.1016/j.semcancer.2017.03.001 [DOI] [PubMed] [Google Scholar]
- 77.Raez LE, Papadopoulos K, Ricart AD, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71(2):523–530. doi: 10.1007/s00280-012-2045-1 [DOI] [PubMed] [Google Scholar]
- 78.Stein M, Lin H, Jeyamohan C, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70(13):1388–1394. doi: 10.1002/pros.21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi: 10.1038/s41467-018-03705-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jia D, Lu M, Jung KH, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci U S A. 2019;116(9):3909–3918. doi: 10.1073/pnas.1816391116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Méndez-Lucas A, Lin W, Driscoll PC, et al. Identifying strategies to target the metabolic flexibility of tumors. Nat Metab. 2020;2(4):335-350. doi: 10.1038/s42255-020-0195-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cervantes-Madrid D, Dominguez-Gomez G, Gonzalez-Fierro A, et al. Feasibility and antitumor efficacy in vivo, of simultaneously targeting glycolysis, glutaminolysis and fatty acid synthesis using lonidamine, 6-diazo-5-oxo-L-norleucine and orlistat in colon cancer. Oncol Lett. 2017;13(3):1905–1910. doi: 10.3892/ol.2017.5615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. doi: 10.1038/nrclinonc.2017.166 [DOI] [PubMed] [Google Scholar]
- 84.Zaal EA, Berkers CR. The influence of metabolism on drug response in cancer. Front Oncol. 2018;8:500. doi: 10.3389/fonc.2018.00500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gatzka MV. Targeted tumor therapy remixed-an update on the use of small-molecule drugs in combination therapies. Cancers. 2018;10(6):155. doi: 10.3390/cancers10060155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Courtney KD, Infante JR, Lam ET, et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. 2018;36(9):867–874. doi: 10.1200/JCO.2017.74.2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber DD, Aminzadeh-Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol Metab. 2020;33:102–121. doi: 10.1016/j.molmet.2019.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Römer M, Dörfler J, Huebner J. The use of ketogenic diets in cancer patients: a systematic review. Clin Exp Med 2021. doi: 10.1007/s10238-021-00710-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seyfried TN, Shivane AG, Kalamian M, Maroon JC, Mukherjee P, Zuccoli G. Ketogenic Metabolic Therapy, Without Chemo or Radiation, for the Long-Term Management of IDH1-Mutant Glioblastoma: An 80-Month Follow-Up Case Report. Front Nutr. 2021;8:281. doi: 10.3389/fnut.2021.682243. [DOI] [PMC free article] [PubMed] [Google Scholar]