

Abstract
Background
Adventitial remodeling is a pathological hallmark of hypertension that results in target organ damage. Activated adventitial fibroblasts have emerged as critical regulators in this process, but the precise mechanism remains unclear.
Methods and Results
Interleukin 11 (IL‐11) knockout and wild‐type mice were subjected to angiotensin II (Ang II) infusion to establish models of hypertension‐associated vascular remodeling. IL‐11 mRNA and protein were increased especially in the adventitia in response to Ang II. Compared with wild‐type mice, Ang II–treated IL‐11 knockout mice showed amelioration of vascular hypertrophy, adventitial fibrosis, macrophage infiltration, and inflammatory factor expression. Recombination mouse IL‐11 exacerbated adventitial fibrosis in Ang II–infused wild‐type mice. Interestingly, IL‐11 neutralizing antibody attenuated adventitial fibrosis, macrophage infiltration, and inflammatory factor expression after Ang II infusion for 7 days. Mechanistically, in primary cultured adventitial fibroblasts, Krüppel‐like factor 15 negatively regulated Ang II–induced IL‐11 expression. Ang II increased extracellular signal‐regulated kinases 1 and 2 activation, especially in adventitia, and caused biphasic extracellular signal‐regulated kinases 1 and 2 activation in adventitial fibroblasts. A rapid and early activation increased IL‐11 production through decreasing Krüppel‐like factor 15 expression, which, in turn, induced the second extracellular signal‐regulated kinases 1 and 2 activation, resulting in posttranscriptional profibrotic gene expression.
Conclusions
These results demonstrate that extracellular signal‐regulated kinases 1 and 2 activation is important for Krüppel‐like factor 15–mediated IL‐11 expression in adventitial fibroblasts to promote adventitial remodeling in Ang II–induced hypertension. Therefore, targeting the Krüppel‐like factor 15/IL‐11 axis might serve as a new therapeutic strategy for vascular diseases.
Keywords: adventitial fibroblasts, fibrosis, mitogen‐activated protein kinases, renin‐angiotensin‐aldosterone system
Subject Categories: Basic Science Research, Fibrosis, Inflammation, Vascular Biology
Nonstandard Abbreviations and Acronyms
- ACTA
α‐smooth muscle actin
- AF
adventitial fibroblast
- Ang II
angiotensin II
- COL1a1
collagen, type I, α 1
- IL‐11–/–
interleukin 11 knockout
- KLF15
Krüppel‐like factor 15
- rmIL‐11
recombination mouse IL‐11
- WT
wild type
Clinical Perspective
What Is New?
Blockade of interleukin 11 (IL‐11) almost normalized adventitial fibrosis induced by angiotensin II.
Krüppel‐like factor 15 negatively regulates IL‐11 expression, depending on extracellular signal‐regulated kinases 1 and 2 activation, which contributed to adventitial remodeling.
Angiotensin II induced biphasic extracellular signal‐regulated kinases 1 and 2 activation with the second activation depending on IL‐11.
What Are the Clinical Implications?
Our study reveals a novel role for IL‐11 in hypertension‐associated vascular remodeling and fibrosis.
We provide a possible treatment strategy for Krüppel‐like factor 15/IL‐11 axis in alleviating and reversing hypertension‐associated vascular remodeling and fibrosis.
Vascular remodeling is a common consequence of hypertensive disease and contributes to the development of cardiovascular dysfunction.1 The vessel wall is a multilayered tissue consisting of multiple cell types to regulate vascular remodeling. Rather than a supportive tissue layer, growing evidence has shown that vascular adventitia acts as an active player for maintenance of vascular function and homeostasis.2 Indeed, excessive adventitial remodeling leads to early aortic maladaptation in angiotensin II (Ang II)–induced hypertension.3 The main effector cells of adventitia are adventitial fibroblasts (AFs), which are activated by stress signals in injured tissues.2 For example, phenotypic transformation of fibroblasts into activated myofibroblasts, which express α‐smooth muscle actin (ACTA2) and express extracellular matrix proteins, is a defining feature of fibrosis.4 The activated AFs secrete a variety of cytokines, chemokines, growth factors, and reactive oxygen species that exert paracrine and autocrine effects to accelerate vascular injury.5, 6 As AFs are critical in adventitial remodeling, it is important to elucidate the underlying mechanisms and identify novel therapeutic targets.
The interleukin family is a multifunctional cytokine family, several members of which have been reported to regulate vascular remodeling in hypertension.7, 8 Interleukin 11 (IL‐11) is a member of the interleukin 6 cytokine family with multifunctional effects. IL‐11 is first regarded as a cardioprotective and antifibrotic cytokine.9 However, Schafer et al identified that autocrine IL‐11 signaling loop promoted cardiovascular fibrosis.10 Considering the important role of vascular remodeling, little is known about the function of IL‐11 in the vasculature system. Previous studies demonstrated that vascular smooth muscle cells secrete IL‐11, which attenuated vascular smooth muscle cell proliferation.11, 12 A recent study found that IL‐11 is important for vascular smooth muscle cell phenotypic switching and aortic remodeling by overexpression of IL‐11 in vascular smooth muscle cells.13 Nevertheless, several studies of the heart, lung, and liver highlighted the importance of pathogenic IL‐11 signaling in fibroblasts.10, 14, 15 Our previous studies found that AFs actively participated in injury‐induced neointima formation by secreting inflammatory factors or growth factors.16, 17 However, the role of IL‐11 derived from AFs in hypertension‐associated adventitial remodeling is mostly unexplored.
IL‐11 secreted by resident fibroblasts in response to profibrotic stimuli binds to its specific receptor subunit IL‐11 receptor subunit α, which is highly expressed on stromal cells (eg, fibroblasts and smooth muscle cells).10 Both signal transducer and activator of transcription 3 and extracellular signal‐regulated kinases 1 and 2 (ERK1/2) signaling pathways were activated by IL‐11, whereas ERK1/2 activation was reported to regulate the fibrosis.14 In a previous study, we have shown that vascular injury or Ang II also caused ERK1/2 activation in the adventitia or AFs, suggesting the important role of ERK1/2 activation in adventitial remodeling.17, 18 Furthermore, we found that ERK1/2 activation was involved in the decrease of transcription factor Krüppel‐like factor 15 (KLF15), whereas overexpression of KLF15 normalized downstream gene expression and Ang II–induced vascular remodeling.18 Also, it is reported that Ang II increased IL‐11 production in cardiac fibroblasts.10 In keeping with these findings, in this study, we sought to determine whether ERK1/2 signaling mediated Ang II–induced IL‐11 expression through the participation of KLF15 in adventitial remodeling.
Methods
The data that support the finding of this study are available from the corresponding author on reasonable request.
Experimental Animals
All animal procedures were approved in accordance with the Guide for the Care and Use of Laboratory Animals, established by Shanghai Jiao Tong University School of Medicine. The mice were housed under a 12‐hour light/dark cycle and were given free access to water and standard laboratory diet.
Ang II Infusion Model
Male C57BL/6 mice, aged 8 to 10 weeks (weight, 20–23 g), were randomly divided into 4 groups as follows: saline group, Ang II group, Ang II+recombination mouse IL‐11 (rmIL‐11) group, and Ang II+IL‐11 neutralizing antibody group. All the mice were anesthetized using continuous flow of 1% to 2% isoflurane and infused with saline or Ang II (1000 ng/kg per minute; Millipore Sigma, Burlington, MA; A9525) by minipump (Alzet, Cupertino, CA; model 1002) for 14 days. For Ang II+rmIL‐11 group, rmIL‐11 at a concentration of 50 μg/mL (R&D, Minneapolis, MN; 2949‐MC‐050) in saline (daily SC injection with 100 μg/kg of rmIL‐11) was injected into the mice. For Ang II+IL‐11 neutralizing antibody group, wild‐type (WT) mice were treated with 1 mg/kg intravenous IL‐11 neutralizing antibody (R&D; MAB418) at 7, 9, 11, and 13 days after Ang II infusion. IL‐11 knockout (IL‐11–/–) mice were purchased from Cyagen Company and infused with saline or Ang II, as mentioned above. One day before euthanasia, systolic blood pressure was obtained by the noninvasive tail‐cuff method using BP‐2000 Blood Pressure Analysis System (Visitech Systems, Apex, NC).
Cell Culture and Reagents
Rat AFs were isolated from the thoracic aortas of male Sprague Dawley rats (weight, 120–150 g). AFs were cultured in DMEM (Corning, New York, NY; 10‐013‐cvr) supplemented with 10% fetal bovine serum (Gibco, Waltham, MA; 10099141), 100 U/mL penicillin, and streptomycin (Gibco; 10378016). Cells from passages 5 to 7 were used for further experiments. AFs were incubated in serum‐free DMEM for 24 hours before treatment with ribosomal S6 kinase inhibitor (MCE, Shanghai, China; HY‐52101, 10 µmol/L) or ERK1/2 pathway inhibitor (MCE; PD98059, 10−5 mol/L) or IL‐11 neutralizing antibody for 1 hour, followed by stimulation of Ang II or rmIL‐11. Rat KLF15 (accession No. AAH89782.1) cloned into adenovirus vector (pAdenoMCMV‐EGFP‐P2A‐3FLAG) were constructed by Obio Technology (Shanghai, China). Cells were infected with adenovirus‐mediated KLF15 or control adenovirus at a viral titer of 1*108 plaque‐forming units/mL for 48 hours to overexpress KLF15. Cells were transfected with 50 nmol small interfering RNA (siRNA) using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA) for 48 hours to knock down KLF15. The KLF15 siRNA sequence was 59‐GCCCUGACUCGCAAGCCUUTT‐39 (sense) and 59‐AAGGCUUGCGAGUCAGGGCTT‐39 (antisense).18
Dual‐Luciferase Reporter Assay
Human embryonic kidney 293T cells were cultured in DMEM with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% CO2. The mouse IL‐11 promoter, covering a region from −2000 to 1, was ligated into a pGL4.10 luciferase reporter vector and transfected into human embryonic kidney 293T cells by Lipofectamine 3000. Then, cells were infected with control adenovirus or adenovirus‐mediated KLF15 for 48 hours, followed by stimulation with Ang II (10−6 mol/L) for 4 hours. Luciferase activity was assessed with the Luciferase Reporter Assay System (Promega, Madison, WI; E1910).
In Situ Hybridization
The sections were dehydrated in graded ethanol, boiled in the retrieval solution for 10 to 15 minutes, and naturally cooled. Proteinase K (20 µg/mL) working solution was added to cover objectives and incubate at 37℃ for 20 minutes. After washing, 3% methanol‐H2O2 was added for incubation in the dark at room temperature for 15 minutes. Prehybridization solution was added to each section, and sections were incubated for 1 hour at 37℃. The probe hybridization solution was added with concentration of 1 µmol/L, and the section was incubated in a humidity chamber and hybridized overnight at 42℃. After washing, blocking solution (rabbit serum) was added and incubated with the section at room temperature for 30 minutes. Sections were incubated with mouse anti–digoxigenin‐labeled peroxidase at 37℃ for 40 minutes, then washed in PBS 4 times for 5 minutes each. Sections were mounted after incubating with 4′,6‐diamidino‐2‐phenylindole for 8 minutes in the dark. Mouse IL‐11 mRNA probe sequence was 5’‐DIG‐CCAGGAAGCTGCAAAGATCCCAATGTCCC‐DIG‐3’.
Histology and Immunohistochemistry
Aortas were immersed in 4% paraformaldehyde (Servicebio, Wuhan, China; G1101) once they were removed from the mice. Sections of thoracic aorta (5 µm thick) from paraffin‐embedded tissue were stained with hematoxylin and eosin (Servicebio; G1004 and G1001) and 0.1% picrosirius red (Servicebio; GP1138) to examine vascular structure and collagen deposition, respectively. Quantification of wall thickness and wall area was analyzed with ImageJ software (National Institutes of Health, Bethesda, MD). Immunofluorescence staining was performed with primary antibodies against ER‐TR7 (Abcam; ab51824), KLF15 (Millipore Sigma; ABC471), ACTA2 (Millipore Sigma; A2547), phosphorylated ERK1/2 (Cell Signaling Technology, Danvers, MA; 4695), and CD68 (Bio‐Rad, Hercules, CA; MCA341GA) at a dilution of 1:100. Secondary antibodies of anti‐mouse, anti‐rat, and anti‐rabbit at a dilution of 1:200 for immunostaining were purchased from Thermo.
Western Blot
Cells were washed 3 times with ice‐cold PBS and extracted in radioimmunoprecipitation assay lysis buffer (Millipore Sigma; 20‐188) with proteinase inhibitor mix (Bimake, Houston, TX; B14001). Cell extracts were clarified by a 15‐minute centrifugation at 4°C and 13000g. Equal amounts of samples were separated by SDS/PAGE, transferred onto a polyvinylidene difluoride membrane (Millipore Sigma; IPVH00010), and subjected to immunoblotting analysis of IL‐11 (R&D; MAB218), IL‐11 receptor subunit α (Santa Cruz Biotechnology; sc‐130920), KLF15 (Millipore; ABC471), collagen type 1a1 (Servicebio; GB11022‐1), ACTA2 (Millipore Sigma; A2547), phosphorylated ERK1/2 (Thr202/Tyr204; 4370), ERK1/2 (Cell Signaling Technology; 4695), phosphorylated P90 ribosomal S6 kinase (p90RSK) (Cell Signaling Technology; 11989), and p90RSK (Cell Signaling Technology; 9355) at dilution of 1:1000, and GAPDH (Kangchen, Shanghai, China; KC‐5G5) at dilution of 1:4000. Proteins were visualized by horseradish peroxidase secondary antibodies and ECL Detection System (Tanon, Shanghai, China; 180‐5001).
Tissue Culture
Media tissues and adventitia tissues were obtained from WT mice. Thoracic aortas were removed from WT mice following the principle of sterility in super clean bench. After removing the perivascular adipose tissue, the media and adventitia were separated carefully. For every 2 mice, the media or adventitia were placed in one petri dish, respectively. After 24 hours serum‐free treatment, the tissues were stimulated with Ang II (10−6 mol/L).
RNA Isolation and Real‐Time Polymerase Chain Reaction
The mRNA was extracted from cells and frozen tissues using the Trizol reagent (Millipore Sigma; T9424), according to the manufacturer’s instructions. And cDNA was obtained from 1000 ng total mRNA with reverse‐transcribed Prime Script RT Master Mix Kit (Takara, Shiga, Japan), followed by quantification by quantitative real‐time polymerase chain reaction using SYBR Green Mix (Takara) on the viia7 RT‐PCR System and StepOne Plus System (Applied Biosystems, Foster City, CA). Relative expression of each gene was normalized to the GAPDH mRNA expression. Sequences of the primers are listed in Table S1.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8.0 (GraphPad, San Diego, CA). Statistical differences between 2 independent groups were analyzed using the Student t test, whereas statistical difference among ≥3 groups was analyzed using 1‐ or 2‐way ANOVA, followed by the Tukey post hoc test. P<0.05 was considered significant.
Results
Ang II–Induced IL‐11 Expression in Adventitia
It has been reported that IL‐11 signaling contributes to cardiovascular fibrosis,10 and vascular remodeling was characterized by excessive accumulation of adventitial collagen in Ang II–induced hypertension.3 Therefore, we first examined the effect of Ang II infusion on IL‐11 expression in the aorta. IL‐11 protein level was significantly increased in injured arteries during 14 days of Ang II infusion (Figure 1A). To further determine the source of IL‐11, in situ hybridization showed that Ang II mainly increased IL‐11 mRNA expression in adventitia (Figure 1B). Also, Ang II increased IL‐11 mRNA expression in injured arteries (Figure 1C). Furthermore, by media and adventitia tissue culture, we confirmed that Ang II increased IL‐11 mRNA expression in both the media and the adventitia while the multiple of the increase in adventitia was much higher than that in the media (Figure 1D).
Figure 1. Angiotensin II (Ang II)–induced interleukin 11 (IL‐11) expression in adventitia.
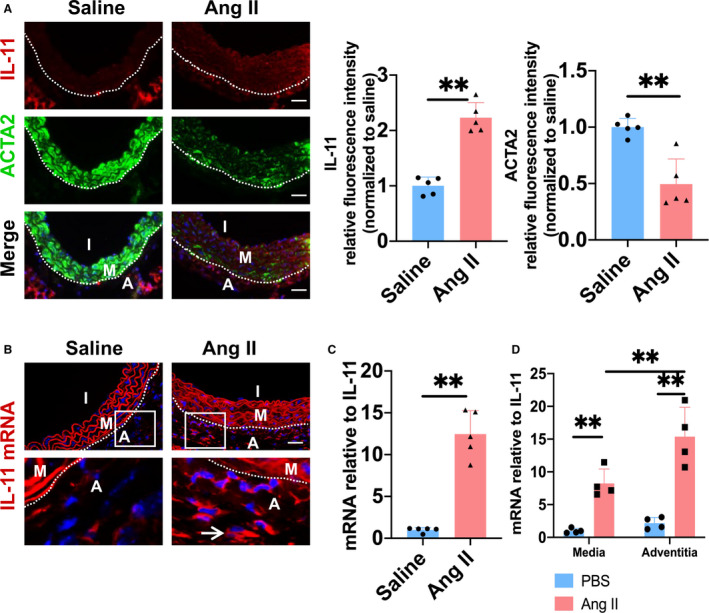
A, Representative immunofluorescence staining and quantitative analysis of IL‐11 (red), α‐smooth muscle actin (ACTA2) (green) in thoracic aorta. The 4′,6‐diamidino‐2‐phenylindole was used to stain the nucleus in blue. Bar=50 µm. B, IL‐11 mRNA was stained with fluorescence by situ hybridization assay. Bar=50 µm. C, The mRNA level of IL‐11 (1.00‐ and 12.47‐fold) in thoracic aorta of wild‐type mice with or without Ang II infusion was quantified by quantitative reverse transcription–polymerase chain reaction (RT‐qPCR). D, The media (M) and adventitia (A) were treated with Ang II (10−6 mol/L) for 4 hours. The mRNA level of IL‐11 was quantified by RT‐qPCR (1.00‐, 8.23‐, 2.18‐, and 15.4‐fold). I indicates intima. M indicates media; and A indicates adventitia. **P<0.01, ***P< 0.001.
Deficiency of IL‐11 Attenuated Ang II–Induced Vascular Remodeling
As we have demonstrated that IL‐11 was significantly increased after Ang II infusion, then IL‐11–/– mice were used to determine the role of IL‐11 in vascular remodeling. Ang II increased the wall thickness, wall area, and collagen area in WT mice, whereas knockout of IL‐11 ameliorated vascular hypertrophy and fibrosis (Figure 2A–2D). However, Ang II infusion elevated the systolic blood pressure and showed no significant difference between 2 groups (Figure 2E; WT‐saline: 113.09±8.21 mm Hg; WT–Ang II: 134.8±13.4 mm Hg; IL‐11–/––saline: 116.0±2.4 mm Hg; and IL‐11–/––saline: 127.8±10.1 mm Hg). ER‐TR7 is a marker of fibroblasts, which is present in the adventitia of the thoracic aorta. Immunofluorescence staining showed that ACTA2 expression was decreased in the media, whereas it was increased in the adventitia. The number of CD68‐positive cells was also increased in the adventitia after Ang II infusion. However, these changes were reversed by IL‐11 knockout (Figure 2F). Nevertheless, deficiency of IL‐11 showed no effect on Ang II–induced decrease of KLF15 expression (Figure 2F). Ang II infusion decreased the mRNA level of KLF15 in the thoracic aorta of both WT and IL‐11–/– mice (Figure 2G). Ang II–treated IL‐11–/– mice also showed lower mRNA expression of collagen, type I, α 1 (COL1a1), interleukin 6 (IL‐6), and chemokine (C‐C motif) ligand 2 (CCL2) compared with WT mice (Figure 2G).
Figure 2. Deficiency of interleukin 11 (IL‐11–/–) attenuated angiotensin II (Ang II)–induced vascular remodeling.

A, Representative hematoxylin and eosin (H&E) and picrosirius red staining of the thoracic aorta tissues in wild type (WT) and IL‐11‐/‐ mice after Ang II infusion for 14 days. Bar=50 µm. B through D, Quantitative analysis of wall thickness (B), wall area (C), and collagen area (D). E, Systolic blood pressure of the 4 groups. F, Representative immunofluorescence staining and quantitative analysis of ER‐TR7 (red), α‐smooth muscle actin (ACTA2) (green), CD68 (green), and Krüppel‐like factor 15 (KLF15) (red) in thoracic aorta. The 4′,6‐diamidino‐2‐phenylindole was used to stain the nucleus in blue. Bar=50 µm. G, The mRNA levels of KLF15 (1.00‐, 0.27‐, 1.07‐, and 0.22‐fold), collagen, type I, α 1 (COL1a1) (1.00‐, 14.52‐, 0.43‐, and 1.74‐fold), collagen, type III, α 1 (COL3a1) (1.00‐, 1.69‐, 0.86‐, and 1.17‐fold), interleukin 6 (IL‐6) (1.00‐, 1.93‐, 0.33‐, and 0.43‐fold) and chemokine (C‐C motif) ligand 2 (CCL2) (1.00‐, 2.11‐, 0.42‐, and 0.53‐fold) were measured by quantitative reverse transcription–polymerase chain reaction. A indicates adventitia. *P< 0.05, **P< 0.01, and ***P< 0.001.
Effect of rmIL‐11 and IL‐11 Neutralizing Antibody on Ang II–Induced Adventitial Remodeling
To further determine the role of IL‐11 in Ang II–induced vascular hypertrophy and fibrosis, rmIL‐11 was used in Ang II–infused mice, whereas IL‐11 neutralizing antibody was used for the blockade of IL‐11 signaling after Ang II infusion for 7 days to determine the therapeutic effect. We found that supplement with rmIL‐11 increased wall thickness and adventitial fibrosis (Figure 3A). On the contrary, blockade of IL‐11 showed no effect on wall thickness but almost normalized adventitial fibrosis (Figure 3A). Wall thickness, wall area, and the fibrosis were shown and quantified in Figure 3A–3D. Neither IL‐11 neutralizing antibody nor rmIL‐11 had effect on the increase in systolic blood pressure caused by Ang II (Figure 3E; saline: 109.3±9.2 mm Hg; Ang II: 159.8±16.36 mm Hg; Ang II–anti–IL‐11: 135.6±15.4 mm Hg; Ang II–rmIL‐11: 142.4±19.6 mm Hg). Ang II induced the reduction expression of ACTA2 in the media, which was attenuated by IL‐11 neutralizing antibody and was not affected by rmIL‐11 (Figure 3F). On the contrary, the expression of ACTA2 in the adventitia was upregulated by Ang II infusion, which was ameliorated by IL‐11 neutralizing antibody (Figure 3F). CD68‐positive cells increased in the adventitia after Ang II treatment, and IL‐11 neutralizing antibody decreased macrophage infiltration, which was not affected by rmIL‐11 (Figure 3F). In addition, IL‐11 neutralizing antibody decreased mRNA expression of COL1a1, CCL2, and IL‐6 after Ang II infusion, whereas rmIL‐11 increased mRNA expression of collagen, type III, α 1 (COL3a1) (Figure 3G). However, neither IL‐11 neutralizing antibody nor rmIL‐11 had effect on the Ang II–induced decrease of KLF15 mRNA expression (Figure 3G).
Figure 3. Effect of recombination mouse interleukin 11 (rmIL‐11) and interleukin 11 (IL‐11) neutralizing antibody on angiotensin II (Ang II)–induced adventitial remodeling.
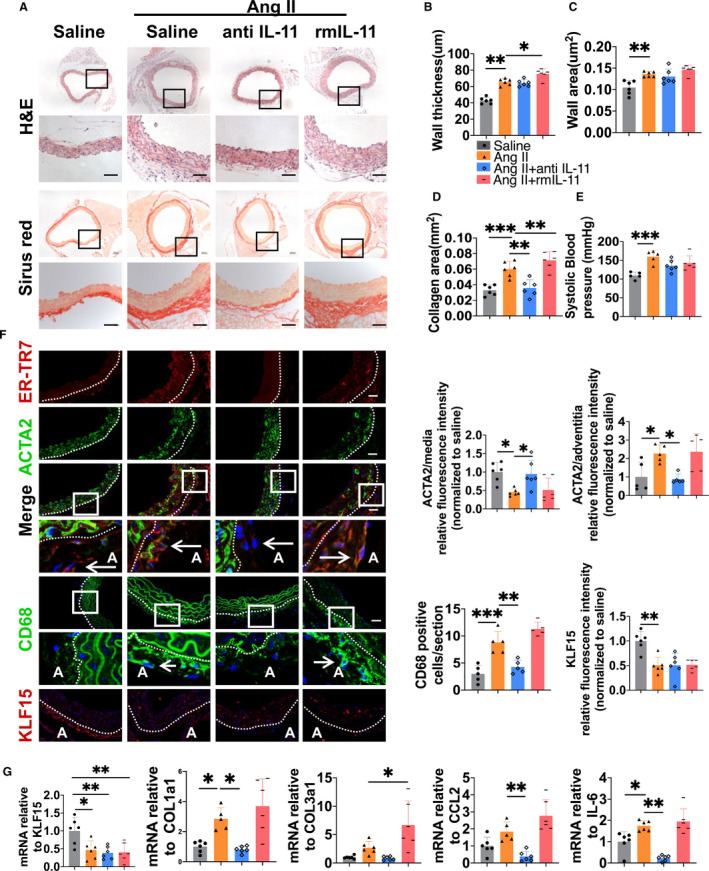
Wild‐type mice were randomly divided into 4 groups treated with rmIL‐11 or IL‐11 neutralizing antibody with or without Ang II infusion. A, Representative hematoxylin and eosin (H&E) and picrosirius red staining of the thoracic aorta tissues. Bar=50 µm. B through D, Quantitative analysis of wall thickness (B), wall area (C), and collagen area (D). E, Systolic blood pressure of the 4 groups. F, Representative immunofluorescence staining and quantitative analysis of ER‐TR7 (red), α‐smooth muscle actin (ACTA2) (green), CD68 (green), and Krüppel‐like factor 15 (KLF15) (red) in thoracic aorta. The 4′,6‐diamidino‐2‐phenylindole was used to stain the nucleus in blue. Bar=50 µm. G, The mRNA levels of KLF15 (1.00‐, 0.47‐, 0.39‐, and 0.40‐fold), collagen, type I, α 1 (COL1a1) (1.00‐, 2.85‐, 0.81‐, and 3.70‐fold), collagen, type III, α 1 (COL3a1) (1.00‐, 2.69‐, 0.85‐, and 6.67‐fold), interleukin‐6 (IL‐6) (1.00‐, 1.75‐, 0.23‐, and 1.95‐fold), and chemokine (C‐C motif) ligand 2 (CCL2) (1.00‐, 1.83‐, 0.39‐, and 2.76‐fold) were measured by quantitative reverse transcription–polymerase chain reaction. A indicates adventitia. *P< 0.05, **P< 0.01, and ***P< 0.001.
KLF15 Negatively Regulated IL‐11 Expression Participating in AF Activation
In previous studies, we found that Ang II–induced vascular remodeling was negatively associated with decrease of KLF15.18 Similarly, knockout of IL‐11 attenuated Ang II–induced vascular remodeling, which suggests a link between IL‐11 and KLF15. However, IL‐11 showed no effect on KLF15 mRNA and protein expression (Figure S1A and B). To exclude the autocrine effect, IL‐11 neutralizing antibody also showed no effect on KLF15 expression (Figure S1C). Ang II significantly increased both IL‐11 mRNA and protein levels at as early as 4 hours but had no effect on the expression of IL‐11 receptor subunit α (Figure 4A and 4B). However, there is no commercial ELISA kit for detecting rat IL‐11 in the supernatant. Adenovirus was used to overexpress KLF15 (adenovirus‐mediated KLF15, AdKLF15) in AFs.18 We observed that Ang II–induced increase of IL‐11 mRNA expression was reduced by KLF15 overexpression (Figure 4C). Luciferase assays showed that adenovirus‐mediated KLF15 significantly suppressed the Ang II–increased IL‐11 promoter activity (Figure 4D). KLF15 overexpression also decreased Ang II–induced increased expression of IL‐11, COL1a1, and ACTA2 (Figure 4E). KLF15 siRNA was used to knock down the expression of KLF15 (siKLF15).18 We found that knockdown of KLF15 aggravated the elevated IL‐11, COL1a1, and ACTA2 expression induced by Ang II, which were partly reversed by IL‐11 neutralizing antibody (Figure 4F). However, IL‐11 neutralizing antibody showed no effect on COL1a1 and ACTA2 mRNA expressions, which were further elevated by KLF15 siRNA after Ang II stimulation (Figure 4G). These data suggest that there are 2 separate signaling pathways that regulate mRNA and protein expression, respectively, with IL‐11 signaling involved in protein expression.10
Figure 4. Krüppel‐like factor 15 (KLF15) negatively regulated interleukin 11 (IL‐11) expression participating in adventitial fibroblast activation.

A, Adventitial fibroblasts (AFs) were exposed to angiotensin II (Ang II) (10−6 mol/L) for indicated time. The protein expressions of IL‐11 (1.00‐, 1.47‐, 2.20‐, 2.58‐, and 2.80‐fold) and IL‐11 receptor subunit α (IL‐11Rα) were quantified by Western blotting. N=3. B, AFs were exposed to Ang II (10−6 mol/L) for indicated time. The mRNA levels of IL‐11 (1.00‐, 2.73‐, 5.30‐, 3.42‐, 1.99‐, and 1.98‐fold) and IL‐11Rα were measured by quantitative reverse transcription–polymerase chain reaction (RT‐qPCR). N=6. C, AFs were transfected with control adenovirus (AdCON) or adenovirus‐mediated KLF15 (AdKLF15) for 48 hours, then cells were treated with Ang II (10−6 mol/L) for 4 hours. The mRNA level of IL‐11 (1.00‐, 12.40‐, 0.68‐, and 5.42‐fold) was measured by RT‐qPCR. N=4. D, Human embryonic kidney 293T cells were stimulated with PBS or Ang II (10−6 mol/L) for 4 hours. The transcription activity of IL‐11 promoter was measured by luciferase assays. N=4. E, AFs were transfected with AdCON or AdKLF15 for 48 hours, then cells were stimulated with Ang II for 24 hours. The protein levels of IL‐11 (1.00‐, 1.65‐, 0.74‐, and 0.90‐fold), collagen, type I, α 1 (COL1a1) (1.00‐, 1.92‐, 0.98‐, and 1.26‐fold), and α‐smooth muscle actin (ACTA2) (1.00‐, 1.88‐, 0.85‐, and 0.80‐fold) were measured by Western blotting. N=3. The right panel was the quantity of the Western blot. F, Cells were transfected with control small interfering RNA (siRNA; siCON) or KLF15 siRNA (siKLF15) for 48 hours, then cells were pretreated with neutralizing antibody 1 hour before Ang II treatment for 24 hours. The protein levels of IL‐11 (1.00‐, 1.46‐, 0.88‐, 1.91‐, and 0.89‐fold), COL1a1 (1.00‐, 2.00‐, 1.00‐, 2.95‐, and 2.07‐fold), and ACTA2 (1.00‐, 1.42‐, 0.97‐, 2.42‐, and 1.02‐fold) were measured by Western blotting. The right panel was the quantity of the Western blot. G, Cells were transfected with siCON or siKLF15 for 48 hours, then cells were pretreated with neutralizing antibody 1 hour before Ang II treatment for 24 hours. The mRNA levels of COL1a1 (1.00‐, 1.62‐, 1.05‐, 2.56‐, and 2.53‐fold) and ACTA2 (1.00‐, 1.44‐, 0.98‐, 2.31‐, and 1.93‐fold) were measured by RT‐qPCR. N=5. *P< 0.05, **P< 0.01, ***P< 0.001, and ****P< 0.0001.
Ang II Induced Biphasic ERK1/2 Activation With the Second Activation Dependent on IL‐11
In primary cultured AFs, we established the time course of ERK1/2 activation in response to Ang II. Ang II increased ERK1/2 phosphorylation, which peaked at 2 distinct waves. The first wave of ERK1/2 phosphorylation occurred at 5 to 10 minutes and decreased at 30 minutes. The second wave of ERK1/2 phosphorylation occurred at 2 hours and sustained up to 4 hours (Figure 5A). The biphasic nature of ERK1/2 activation suggested a possible autocrine mechanism, in which the first ERK1/2 activation may be induced by Ang II and upregulated the expression of IL‐11, which then activated the second ERK1/2 activation. To test this possibility, AFs were pretreated with IL‐11 neutralizing antibody. We found that Ang II–induced ERK1/2 phosphorylation was attenuated only at 4 hours (Figure 5B). Knockdown of KLF15 did not further increase the phosphorylation of ERK1/2 in response to Ang II at 5 minutes, which cannot be inhibited by IL‐11 neutralizing antibody (Figure 5C). However, after 4 hours of Ang II stimulation, the expression of ERK1/2 phosphorylation was further elevated after KLF15 knockdown, which was decreased by IL‐11 neutralizing antibody (Figure 5C). Next, we used ERK1/2 inhibitor 1 hour before or after the Ang II stimulation to inhibit 2 waves of ERK1/2 activation, respectively. Blockade of the first wave of ERK1/2 activation attenuated Ang II–induced IL‐11 expression (Figure 5D). Similarly, KLF15 expression was recovered only by inhibition of first wave ERK1/2 activation in response to Ang II (Figure 5D), which suggests that KLF15 may regulate IL‐11 expression. These results suggested that the delayed activation of ERK1/2 was induced by IL‐11. Furthermore, the COL1a1 and ACTA2 protein expressions were reduced by inhibition of both waves of ERK1/2 activation (Figure 5D). In 14 days of Ang II infusion model, we also observed that the ERK1/2 was activated in adventitia of WT mice, whereas deletion of IL‐11 signaling resulted in reduced ERK1/2 activation in response to Ang II infusion (Figure 6A). Similarly, IL‐11 neutralizing antibody dramatically repressed the phosphorylation of Ang II–induced ERK1/2 phosphorylation, whereas rmIL‐11 intensified phosphorylation of ERK1/2 (Figure 6B).
Figure 5. Angiotensin II (Ang II) induced biphasic extracellular signal‐regulated kinases 1 and 2 (ERK1/2) activation, with the second activation dependent on interleukin 11 (IL‐11).

A, Adventitial fibroblasts (AFs) were exposed to Ang II (10−6 mol/L) for indicated time, and the protein level of phosphorylated ERK1/2 (p‐ERK1/2) was measured by Western blotting. B, AFs were pretreated with PBS or IL‐11 neutralizing antibody 1 hour before Ang II (10−6 mol/L) treatment. Then, AFs were exposed to Ang II or PBS for indicated time. The level of p‐ERK1/2 (1.00‐, 3.14‐, 2.25‐, 0.76‐, 2.74‐, and 0.73‐fold) was measured by Western blotting. N=4. C, AFs were transfected with control small interfering RNA (siRNA; siCON) or Krüppel‐like factor 15 (KLF15) siRNA (siKLF15) for 48 hours. AFs were pretreated by IL‐11 neutralizing antibody 1 hour before they were stimulated by Ang II for 5 minutes (1.00‐, 3.52‐, 4,04‐, 0.88‐, 3.76‐, and 3.57‐fold) or 4 hours (1.00‐, 2.31‐, 1.42‐, 1.08‐, 3.90‐, and 1.46‐fold). p‐ERK1/2 expression was measured by Western blotting. N=4. D, AFs were incubated with ERK1/2 signaling inhibitor (ERKi) 1 hour before or after Ang II 24‐hour stimulation. The protein levels of IL‐11 (1.00‐, 1.41‐, 0.96‐, and 1.26‐fold), KLF15 (1.00‐, 0.62‐, 1.08‐, and 0.49‐fold), collagen, type I, α 1 (COL1a1) (1.00‐, 1.69‐, 0.80‐, and 0.92‐fold), and α‐smooth muscle actin (ACTA2) (1.00‐, 1.53‐, 0.95‐, 0.92‐, and 0.49‐fold) were measured by Western blotting. The right panel was the quantity of the Western blot. N=4. CON indicates control. *P< 0.05, **P< 0.01, and ***P< 0.001.
Figure 6. The effect of interleukin 11 (IL‐11) on angiotensin II (Ang II)–induced extracellular signal‐regulated kinases 1 and 2 (ERK1/2) activation.

A and B, Representative immunofluorescence staining and quantitative analysis of phosphorylated ERK1/2 (p‐ERK1/2) (green) in thoracic aorta of different groups. The 4′,6‐diamidino‐2‐phenylindole was used to stain the nucleus in blue. Bar=50 µm. A indicates adventitia; IL‐11–/–, IL‐11 knockout; rmIL‐11, recombination mouse IL‐11; and WT, wild type. **P< 0.01, ***P< 0.001.
IL‐11/ERK1/2/p90RSK Signaling Mediated Ang II–Induced Profibrotic Gene Expression
Ang II promotes vascular fibrosis mainly by acting on AFs.18 We observed that Ang II upregulated expression of COL1a1 and ACTA2 in both mRNA and protein expressions (Figure S2A and C). Considering the role of IL‐11 in fibrosis, we stimulated AFs with rmIL‐11 for various time periods and showed that COL1a1 and ACTA2 protein, but not mRNA, expressions were increased by IL‐11 stimulation (Figure S2B and D). To evaluate whether IL‐11 signaling is involved in Ang II–induced profibrotic protein expression, the Ang II–induced COL1a1 and ACTA2 protein upregulation was inhibited by IL‐11 neutralizing antibody (Figure 7A). However, IL‐11 neutralizing antibody did not affect the downregulation of KLF15 expression induced by Ang II (Figure 7A). And IL‐11 neutralizing antibody showed no effect on mRNA expression changes of ACTA2, COL1a1, and KLF15, caused by Ang II (Figure 7B). Considering the negligible differences of COL1a1 and ACTA2 mRNA levels following IL‐11 stimulation, we speculated that it was dependent on the posttranscriptional mechanism. The p90RSK is downstream of ERK1/2, which plays a role in posttranscriptional gene expression.19 We stimulated AFs with IL‐11 and found that the phosphorylation levels of both ERK1/2 and p90RSK were increased (Figure 7C). The activation of p90RSK can be inhibited by ribosomal S6 kinase inhibitor and ERK1/2 inhibitor, whereas ribosomal S6 kinase inhibitor showed no effect on ERK1/2 activation (Figure 7C). Consistent with these findings, both the inhibitors reduced IL‐11–induced COL1a1 and ACTA2 protein expression (Figure 7D). These results suggested that IL‐11/ERK1/2/p90RSK signaling plays an important role in Ang II–induced adventitial fibrosis.
Figure 7. Interleukin 11 (IL‐11)/extracellular signal‐regulated kinases 1 and 2 (ERK1/2)/P90 ribosomal S6 kinase (p90RSK) signaling mediated angiotensin II (Ang II)–induced profibrotic gene expression.

A, Adventitial fibroblasts (AFs) were incubated with IL‐11 neutralizing antibody 1 hour before Ang II 24‐hour treatment. The protein levels of collagen, type I, α 1 (COL1a1) (1.00‐, 1.19‐, 2.10‐, and 0.95‐fold), α‐smooth muscle actin (ACTA2) (1.00‐, 1.00‐, 1.49‐, and 0.97‐fold), and Krüppel‐like factor 15 (KLF15) (1.00‐, 0.87‐, 0.66‐, and 0.63‐fold) were measured by Western blotting. N=4. The right panel was the quantity of the Western blot. B, AFs were incubated with IL‐11 neutralizing antibody 1 hour before Ang II 24‐hour treatment. The mRNA levels of COL1a1 (1.00‐, 1.09‐, 1.40‐, and 1.52‐fold), ACTA2 (1.00‐, 0.91‐, 1.80‐, and 1.58‐fold), and KLF15 (1.00‐, 0.98‐, 0.37‐, and 0.51‐fold) were measured by quantitative reverse transcription–polymerase chain reaction. N=5. C, AFs were incubated with ribosomal S6 kinase inhibitor (CMK) (10 µmol/L) or ERK1/2 inhibitor (ERKi) (10 µmol/L) 1 hour before recombination mouse IL‐11 (rmIL‐11) (5 ng/mL) 24‐hour treatment. The protein levels of phosphorylated p90RSK (p‐p90RSK) (1.00‐, 2.10‐, 0.93‐, and 0.74‐fold), p90RSK, phosphorylated ERK1/2 (p‐ERK1/2) (1.00‐, 10.71‐, 10.89‐, and 2.53‐fold), and ERK1/2 were measured by Western blotting. N=4. The right panel was the quantity of the Western blot. D, AFs were incubated with dimethyl sulfoxide, CMK (10 µmol/L), or ERKi (10 µmol/L) 1 hour before PBS or rmIL‐11 (5 ng/mL) 24‐hour treatment. The protein levels of COL1a1 (1.00‐, 1.40‐, 0.68‐, and 0.79‐fold) and ACTA2 (1.00‐, 1.13‐, 0.89‐, and 0.95‐fold) were measured by Western blotting. N=4. The right panel was the quantity of the Western blot. CON indicates control. *P< 0.05, **P< 0.01, and ***P< 0.001.
Discussion
In this study, we demonstrated that KLF15 negatively regulates IL‐11 expression dependent on ERK1/2 activation, which contributed to adventitial remodeling. Specifically, knockout or blockade of IL‐11 attenuated Ang II–induced adventitial remodeling. Ang II initiated an early activation of ERK1/2, resulting in increase of KLF15‐mediated IL‐11 expression in AFs. Then, IL‐11 further induced a late activation of ERK1/2, which increased profibrotic gene expression by a posttranscriptional mechanism. These findings provide a new implication for understanding the mechanisms underlying the regulation of adventitial remodeling in hypertension by the KLF15/IL‐11 axis.
Hypertension significantly contributes to tissue fibrosis, including adventitial fibrosis.20, 21 Renin‐angiotensin system, especially Ang II, plays an important role in the pathogenesis of adventitial remodeling in a variety of diseases.3, 22 We found that AF‐derived IL‐11 promoted adventitial remodeling in Ang II–induced hypertension. Our data are consistent with recent data that IL‐11 signaling functioned as a crucial contributor of cardiovascular fibrosis.10 However, previous studies suggested that IL‐11 functioned as a protective and antifibrotic factor among various diseases.9, 23 First, the contradiction may be caused by the different animal models. Second, recombinant human IL‐11 was used in mouse cardiac fibroblasts, which was proved to be ineffective.10 Although smooth muscle cells also secrete IL‐11, we found that IL‐11 expression was increased in Ang II–stimulated AFs and in the adventitia of arteries of Ang II–infused mice. Lim et al found that smooth muscle cell–specific IL‐11 overexpression promoted vascular remodeling and especially increased adventitial area,13 which further suggested the important role of IL‐11 signaling in AFs, because AFs also express IL‐11 receptor. Therefore, IL‐11 signaling was blocked with neutralizing antibody after preestablished Ang II infusion model. We found that adventitial collagen area but not wall area was decreased, which is consistent with the previous results.13 Furthermore, by using IL‐11 knockout mice, Ang II–induced increase of both collagen area and wall area was attenuated, suggesting multiple effects of IL‐11 in the context of renin‐angiotensin‐aldosterone system activation under different intervention methods. However, we found that knockout of IL‐11 showed no effect on blood pressure. Vascular fibrosis and extracellular matrix remodeling are associated with aging and are amplified by hypertension.24, 25 One possible reason is that young mice, aged 8 to 10 weeks, were used. In addition, vascular fibrosis in response to Ang II infusion for 2 weeks might play little role in the regulation of blood pressure.21
Another important finding is that KLF15 transcriptionally regulated IL‐11 expression in AFs. It is reported that profibrotic factors, such as platelet‐derived growth factor‐BB, transforming growth factor‐β, and Ang II, increase IL‐11 protein but not mRNA expression in cardiac fibroblasts.10 Interestingly, we and other groups found that these factors also decrease KLF15 expression.18, 26, 27 KLF15 is highly expressed in the adventitia and associated with tissue fibrosis,18, 28 which inhibits connective tissue growth factor expression in cardiac fibroblasts.29 These results suggest that KLF15 functions as a protective regulator in Ang II–induced vascular fibrosis and hypertrophy. Indeed, KLF15 negatively regulated Ang II induced both mRNA and protein expression of IL‐11 in AFs. Furthermore, blockade of IL‐11 signaling suppressed Ang II–induced ACTA2 and COL1a1 protein but not mRNA expression after KLF15 knockdown, which suggests that IL‐11 increased profibrotic gene expression by a posttranscriptional manner. However, IL‐11 had no effect on KLF15 mRNA and protein expression after Ang II stimulation. Possible reason is that KLF15 mRNA is relatively low after Ang II stimulation; IL‐11 signaling may recognize specific modifications in mRNA that control translation.30 It is reported that Ang II showed no effect on IL‐11 RNA expression in cardiac fibroblasts.10 The discrepancy of IL‐11 mRNA expression in response to Ang II may caused by the different cell types and the stimulation time. Altogether, these data suggest that KLF15/IL‐11 axis regulated Ang II–induced adventitial remodeling.
Perivascular inflammation with macrophage infiltration is a key feature of vascular injury in hypertension.16, 18, 31 Fibroblasts that differentiated into myofibroblasts and increased the expression of ACTA2 have been implicated as inflammation‐associated phenotype cells.32, 33 We found that IL‐11 activated AFs that participated in adventitial remodeling through expression of ACTA2, which suggests that IL‐11 is involved in vascular inflammation. Indeed, IL‐11 signaling in fibroblast drives chronic inflammation in fibrotic lung disease.34 Although supplement of exogenous rmIL‐11 did not further increase macrophage infiltration and inflammatory factor expression, whereas loss of function by knockdown of IL‐11 or IL‐11 neutralizing antibody attenuated inflammatory response, which is consistent with a previous study.13 These results suggest that IL‐11 plays a pluripotent role in adventitial remodeling in the context of renin‐angiotensin‐aldosterone system activation.
The ERK1/2 signaling cascade is activated by a wide variety of receptors involved in diverse cellular processes. Interestingly, we found that Ang II caused biphasic ERK1/2 activation in AFs. The early phase of ERK1/2 activation was as short as 5 minutes, suggesting a direct role of Ang II on ERK1/2 signaling, which was associated with KLF15 and profibrotic gene expression.18 It has been reported that IL‐11–dependent noncanonical ERK1/2 signaling is involved in fibroblast activation.10, 14 We found that the late phase of ERK1/2 activation is caused by AF‐derived IL‐11 in response to Ang II. Ribosomal protein S6 kinase activity controls the transcriptional program.35, 36 Of note, p90RSK is the direct downstream effector of ERK1/2 signaling and activates various signaling events through different phosphorylation substrates.37 Indeed, we found that IL‐11 increased ACTA2 and collagen protein but not mRNA expression through p90RSK activation, which is ERK1/2 dependent. Consistent with a previous study,10 our data suggest that IL‐11 does not stimulate a transcriptomic response, which provides further evidence that IL‐11 acts through a posttranscriptional mechanism through ERK1/2/p90RSK. Furthermore, ERK1/2 signaling is important for AF activation.17 Our results confirmed that blockade of IL‐11 signaling attenuated Ang II–induced increase of phosphorylation of ERK1/2, especially in the adventitia, which supports the in vivo role of ERK1/2 activation.
In summary, our findings suggest that Ang II–induced biphasic ERK1/2 activation mediated KLF15‐dependent IL‐11 expression, which is important for AF activation. Activated AFs promote perivascular fibrosis and inflammation, contributing to adventitial remodeling, which suggests that targeting of KLF15/IL‐11 axis may be a feasible approach for the treatment of vascular remodeling in hypertension.
Sources of Funding
This study was supported by grants from National Natural Science Foundation of China (82070245, 82030006, and 81770415), Natural Science Foundation of Shanghai (20ZR1447500), and Laboratory Animals Research Field of Shanghai (201409005700).
Disclosures
None.
Supporting information
Table S1 Figures S1–S2
For Sources of Funding and Disclosures, see page 15.
References
- 1.Heeneman S, Sluimer JC, Daemen MJ. Angiotensin‐converting enzyme and vascular remodeling. Circ Res. 2007;101:441–454. [DOI] [PubMed] [Google Scholar]
- 2.Stenmark KR, Yeager ME, El Kasmi KC, Nozik‐Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bersi MR, Bellini C, Wu J, Montaniel KRC, Harrison DG, Humphrey JD. Excessive adventitial remodeling leads to early aortic maladaptation in angiotensin‐induced hypertension. Hypertension. 2016;67:890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coen M, Gabbiani G, Bochaton‐Piallat ML. Myofibroblast‐mediated adventitial remodeling: an underestimated player in arterial pathology. Arterioscler Thromb Vasc Biol. 2011;31:2391–2396. [DOI] [PubMed] [Google Scholar]
- 6.Yu B, Liu Z, Fu Y, Wang Y, Zhang L, Cai Z, Yu F, Wang X, Zhou J, Kong W. Cyld deubiquitinates nicotinamide adenine dinucleotide phosphate oxidase 4 contributing to adventitial remodeling. Arterioscler Thromb Vasc Biol. 2017;37:1698–1709. [DOI] [PubMed] [Google Scholar]
- 7.Tieu BC, Lee C, Sun H, LeJeune W, Recinos A, Ju X, Spratt H, Guo D‐C, Milewicz D, Tilton RG, et al. An adventitial il‐6/mcp1 amplification loop accelerates macrophage‐mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orejudo M, García‐Redondo A, Rodrigues‐Diez Raúl R, Rodrigues‐Díez R, Santos‐Sanchez L, Tejera‐Muñoz A, Egido J, Selgas R, Salaices M, Briones A, et al. Interleukin‐17a induces vascular remodeling of small arteries and blood pressure elevation. Clin Sci (Lond). 2020;134:513–527. [DOI] [PubMed] [Google Scholar]
- 9.Obana M, Maeda M, Takeda K, Hayama A, Mohri T, Yamashita T, Nakaoka Y, Komuro I, Takeda K, Matsumiya G, et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin‐11 ameliorates cardiac fibrosis after myocardial infarction. Circulation. 2010;121:684–691. [DOI] [PubMed] [Google Scholar]
- 10.Schafer S, Viswanathan S, Widjaja AA, Lim W‐W, Moreno‐Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, et al. Il‐11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmerman MA, Selzman CH, Reznikov LL, Raeburn CD, Barsness K, McIntyre RC Jr, Hamiel CR, Harken AH. Interleukin‐11 attenuates human vascular smooth muscle cell proliferation. Am J Physiol Heart Circ Physiol. 2002;283:H175–H180. [DOI] [PubMed] [Google Scholar]
- 12.Taki H, Sakai T, Sugiyama E, Mino T, Kuroda A, Taki K, Hamazaki T, Koizumi H, Kobayashi M. Monokine stimulation of interleukin‐11 production by human vascular smooth muscle cells in vitro. Atherosclerosis. 1999;144:375–380. [DOI] [PubMed] [Google Scholar]
- 13.Lim W‐W, Corden B, Ng B, Vanezis K, D’Agostino G, Widjaja AA, Song W‐H, Xie C, Su L, Kwek X‐Y, et al. Interleukin‐11 is important for vascular smooth muscle phenotypic switching and aortic inflammation, fibrosis and remodeling in mouse models. Sci Rep. 2020;10:17853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng B, Dong J, D’Agostino G, Viswanathan S, Widjaja AA, Lim W‐W, Ko NSJ, Tan J, Chothani SP, Huang B, et al. Interleukin‐11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med. 2019;11:eaaw1237. [DOI] [PubMed] [Google Scholar]
- 15.Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, D’Agostino GA, Ng B, Lim WW, Tan J, Paleja BS, et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology. 2019;157:e714. [DOI] [PubMed] [Google Scholar]
- 16.Li XD, Hong MN, Chen J, Lu YY, Ye MQ, Ma Y, Zhu DL, Gao PJ. Adventitial fibroblast‐derived vascular endothelial growth factor promotes vasa vasorum‐associated neointima formation and macrophage recruitment. Cardiovasc Res. 2020;116:708–720. [DOI] [PubMed] [Google Scholar]
- 17.Li XD, Chen J, Ruan CC, Zhu DL, Gao PJ. Vascular endothelial growth factor‐induced osteopontin expression mediates vascular inflammation and neointima formation via flt‐1 in adventitial fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32:2250–2258. [DOI] [PubMed] [Google Scholar]
- 18.Lu YY, Li XD, Zhou HD, Shao S, He S, Hong MN, Liu JC, Xu YL, Wu YJ, Zhu DL, et al. Transactivation domain of kruppel‐like factor 15 negatively regulates angiotensin ii‐induced adventitial inflammation and fibrosis. FASEB J. 2019;33:6254–6268. [DOI] [PubMed] [Google Scholar]
- 19.Le N‐T, Takei Y, Shishido T, Woo C‐H, Chang E, Heo K‐S, Lee H, Lu Y, Morrell C, Oikawa M, et al. P90rsk targets the erk5‐chip ubiquitin e3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ Res. 2012;110:536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuo C, Li X, Huang J, Chen D, Ji K, Yang Y, Xu T, Zhu D, Yan C, Gao P. Osteoglycin attenuates cardiac fibrosis by suppressing cardiac myofibroblast proliferation and migration through antagonizing lysophosphatidic acid 3/matrix metalloproteinase 2/epidermal growth factor receptor signalling. Cardiovasc Res. 2018;114:703–712. [DOI] [PubMed] [Google Scholar]
- 21.Nosalski R, Siedlinski M, Denby L, McGinnigle E, Nowak M, Cat AND, Medina‐Ruiz L, Cantini M, Skiba D, Wilk G, et al. T‐cell‐derived mirna‐214 mediates perivascular fibrosis in hypertension. Circ Res. 2020;126:988–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin‐angiotensin‐aldosterone system alterations. Circ Res. 2015;116:960–975. [DOI] [PubMed] [Google Scholar]
- 23.Lee HT, Park SW, Kim M, Ham A, Anderson LJ, Brown KM, D'Agati VD, Cox GN. Interleukin‐11 protects against renal ischemia and reperfusion injury. Am J Physiol Renal Physiol. 2012;303:F1216–F1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Z. Aging, arterial stiffness, and hypertension. Hypertension. 2015;65:252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol. 2016;32:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao X, Huang L, Grosjean F, Esposito V, Wu J, Fu L, Hu H, Tan J, He C, Gray S, et al. Low‐protein diet supplemented with ketoacids reduces the severity of renal disease in 5/6 nephrectomized rats: a role for klf15. Kidney Int. 2011;79:987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Y, Haldar S, Croce K, Wang Y, Sakuma M, Morooka T, Wang B, Jeyaraj D, Gray SJ, Simon DI, et al. Kruppel‐like factor 15 regulates smooth muscle response to vascular injury–brief report. Arterioscler Thromb Vasc Biol. 2010;30:1550–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu X, Mallipattu SK, Guo Y, Revelo MP, Pace J, Miller T, Gao X, Jain MK, Bialkowska AB, Yang VW, et al. The loss of kruppel‐like factor 15 in foxd1(+) stromal cells exacerbates kidney fibrosis. Kidney Int. 2017;92:1178–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang B, Haldar SM, Lu Y, Ibrahim OA, Fisch S, Gray S, Leask A, Jain MK. The kruppel‐like factor klf15 inhibits connective tissue growth factor (ctgf) expression in cardiac fibroblasts. J Mol Cell Cardiol. 2008;45:193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao BS, Roundtree IA, He C. Post‐transcriptional gene regulation by mrna modifications. Nat Rev Mol Cell Biol. 2017;18:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meijles DN, Pagano PJ. Nox and inflammation in the vascular adventitia. Hypertension. 2016;67:14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehoux S. Adventures in the adventitia. Hypertension. 2016;67:836–838. [DOI] [PubMed] [Google Scholar]
- 34.Ng B, Dong J, Viswanathan S, Widjaja AA, Paleja BS, Adami E, Ko NSJ, Wang M, Lim S, Tan J, et al. Fibroblast‐specific il11 signaling drives chronic inflammation in murine fibrotic lung disease. FASEB J. 2020;34:11802–11815. [DOI] [PubMed] [Google Scholar]
- 35.Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau‐Aveilla C, Dreazen A, Cagnard N, Carpentier W, Kiss T, Meyuhas O, et al. Ribosomal protein s6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene. 2014;33:474–483. [DOI] [PubMed] [Google Scholar]
- 36.Anjum R, Blenis J. The rsk family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. [DOI] [PubMed] [Google Scholar]
- 37.Lin L, White SA, Hu K. Role of p90rsk in kidney and other diseases. Int J Mol Sci. 2019;20:972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Figures S1–S2