

Abstract
Background
Age‐related heart diseases are significant contributors to increased morbidity and mortality. Emerging evidence indicates that mitochondria within cardiomyocytes contribute to age‐related increased reactive oxygen species (ROS) generation that plays an essential role in aging‐associated cardiac diseases.
Methods and Results
The present study investigated differences between ROS production in cardiomyocytes isolated from adult (6 months) and aged (24 months) Fischer 344 rats, and in cardiac tissue of adult (18–65 years) and elderly (>65 years) patients with preserved cardiac function. Superoxide dismutase inhibitable ferricytochrome c reduction assay (1.32±0.63 versus 0.76±0.31 nMol/mg per minute; P=0.001) superoxide and H2O2 production, measured as dichlorofluorescein diacetate fluorescence (1646±428 versus 699±329, P=0.04), were significantly higher in the aged versus adult cardiomyocytes. Similarity in age‐related alteration between rats and humans was identified in mitochondrial‐electron transport chain‐complex‐I‐associated increased oxidative‐stress by MitoSOX fluorescence (53.66±18.58 versus 22.81±12.60; P=0.03) and in 4‐HNE adduct levels (187.54±54.8 versus 47.83±16.7 ng/mg protein, P=0.0063), indicative of increased peroxidation in the elderly. These differences correlated with changes in functional enrichment of genes regulating ROS homeostasis pathways in aged human and rat hearts. Functional merged collective network and pathway enrichment analysis revealed common genes prioritized in human and rat aging‐associated networks that underlay enriched functional terms of mitochondrial complex I and common pathways in the aging human and rat heart.
Conclusions
Aging sensitizes mitochondrial and extramitochondrial mechanisms of ROS buildup within the heart. Network analysis of the transcriptome highlights the critical elements involved with aging‐related ROS homeostasis pathways common in rat and human hearts as targets.
Keywords: cardiac aging, electron transport chain, gene expression, oxidative stress, reactive oxygen species
Subject Categories: Oxidant Stress, Aging, Physiology, Myocardial Biology, Metabolism
Nonstandard Abbreviations and Acronyms
- AU
arbitrary units
- ETC
electron transport chain
- IPA
Ingenuity Pathway Analysis
- LCM
low‐calcium medium
- ROS
Reactive oxygen species
Clinical Perspective
What Is New?
Selective downregulation in the activity of electron transport chain complex I involved in superoxide generation is the common link in reactive oxygen species production in aging rat and human myocardium.
Network analysis of the transcriptome highlights similar mechanisms and pathways involved in aging rats and humans with ERK1/2 linking to electron transport chain complex I as a common signaling pathway in the aging heart.
What Are the Clinical Implications?
Recognizing regulators of reactive oxygen species homeostasis and its downstream players altered by aging may identify targets that can be selectively intervened on to prevent aging‐associated electrical or mechanical myocardial dysfunction, such as atrial fibrillation and heart failure, prevalent in the elderly.
Reactive oxygen species (ROS) such as superoxide anion () and hydrogen peroxide (H2O2) are continuously generated at low levels within the cell and play an essential role in various physiological processes; during stress they may contribute to cell injury and death.1, 2, 3, 4 An increased ROS production rate has been implicated in aging and age‐related changes in the mammalian heart, leading to gradual functional impairment,5, 6 including reduced tolerance to stress and cellular energetic reserves, and predisposing cells to injury.7, 8, 9, 10 The rate of aging‐associated decline in function varies by species as well as by organ within the same species and is influenced by genetic and environmental factors11, 12, 13, 14 with deleterious consequences, increasing predisposition to ischemic heart disease, cardiomyopathy, and cardiac arrhythmias.6, 12, 14 The relative contribution of enhanced production versus reduced clearance of ROS within the cytoplasm and mitochondria is not fully characterized, and the impact of aging versus disease on these pathways in humans is not well defined. To address this, we compared age‐related alteration in the sensitivity of myocardial tissue to stress‐related alteration in oxidant production in cardiomyocytes from disease‐free young and aged Fischer 344 rats, an established model of aging, and in human cardiac tissue from adult and aged patients undergoing cardiac surgery. We also assessed overall change in the expression of genes encoding for pathways of ROS production and clearance in young and aged rat hearts and in young and aged patients undergoing cardiac surgery. From aged rat and human hearts, we identified shared networks and nodes that are important for regulating oxidative stress and that are altered with aging in both rats and humans. This information provides novel insight into potential targets involved in the responsiveness of the elderly heart to stress that could be manipulated to prevent aging‐associated deterioration in heart function.15
METHODS
Human Sample Selection
Atrial appendage tissue was harvested from adult (18–65 years) and aged (>65 years) patients undergoing elective open‐heart surgery for coronary artery disease, and tissue from those who met inclusion and exclusion criteria was used for the analysis of the study. The baseline clinical characteristics of these patients is summarized in Table S1. Patients with congenital, structural, or functional atrial disease—including persistent or permanent atrial fibrillation—or class III/IV heart failure, those requiring inotropic support, and those with moderate/severe mitral valve disease were excluded. The research study was conducted following the Declaration of Helsinki for experiments relating to humans. The study was approved by the Aurora Health Care Institutional Review Board for human subject research, written informed consent was obtained from all subjects, and all patients' privacy rights were observed. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Processing of Human Cardiac Samples
After clamping and removal of the atrial appendage, the tissue was immediately transferred into ice‐cold Dulbecco's phosphate‐buffered saline. Fat and connective tissue were removed from the biospecimen, and the muscle tissue was freshly used for myofiber isolation or frozen in liquid nitrogen (N2) and stored at −80°C for future use. A total of 47 patients' samples were used in this research for the various expressional and functional studies (OXPHOS functional analysis, 4‐HNE protein adducts, western blot, or genetic analysis). Tissue was collected immediately after removal from the patient and processed within 10 to 15 minutes. If any delay occurred in collection or storage, the tissue was not used for the study.
Experimental Animals and Design
Animal experiments were approved by the Institutional Animal Care and Use Committee of the Mayo Clinic College of Medicine and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85‐23, 1996). The young adult (6 months old) and aged (24 months old) male Fischer 344 rats were used for our aging‐related studies.16, 17, 18 Young and aged rats were acquired from the National Institute on Aging (NIA) colonies19 and were housed in a temperature‐controlled room (22–23°C) with a 12:12‐hour light‐dark cycle and fed with tap water and Purina Laboratory Chow (No. 5001) ad libitum. Rats were euthanized with intraperitoneal sodium pentobarbital (50 mg/kg) before thoracotomy and heart dissection. In a set of parallel experiments in young adult and aged rats, we determined (1) generation by the mitochondria isolated from ventricles and (2) steady‐state and ouabain‐stressed H2O2 production in isolated cardiomyocytes.
Isolation of Cardiomyocytes From Rat Hearts
Cardiomyocytes were isolated from the hearts of young adult (n=5) and aged (n=5) rats, as described previously.20 In brief, the procedure comprises three main steps: (1) dissection and sequential retrograde perfusion of the heart at 37°C, firstly, with Tyrode's buffer (137 mmol/L NaCl, 10 mmol/L glucose, 10 mmol/L HEPES, 5.4 mmol/L KCl, 1 mmol/L MgCl2, and 2 mmol/L CaCl2) for 10 minutes; secondly, with a “low‐calcium” medium (LCM) containing 100 mmol/L NaCl, 50 mmol/L taurine, 20 mmol/L glucose, 10 mmol/L KCl, 10 mmol/L HEPES, 5 mmol/L MgSO4, and 1.2 mmol/L KH2PO4 and supplemented with 0.13 mmol/L CaCl2 and 2.1 mmol/L EGTA for 2 minutes; and finally, with LCM supplemented with 1% bovine serum albumin (BSA), 0.2 mmol/L CaCl2, collagenase (type II, 22 U/mL; Worthington) and pronase (100 µg/mL; Serva) for 15 minutes; (2) removal of atria and mincing of the ventricular tissue into small pieces before incubation in the enzyme solution with gentle stirring at 37°C for 15 minutes; (3) isolation of intact cardiomyocytes by serial centrifugation and washing of the pellet in LCM supplemented with 0.2 mmol/L CaCl2 (wash) followed by suspension in the wash solution.21
Isolation of Mitochondria From the Heart
Mitochondria were isolated from the myocardium of young adult (n=5) and aged (n=5) rats as previously described.22 In summary, ventricles were excised from the rat heart, sliced into fine pieces, suspended in an isolation buffer containing (in millimoles) sucrose 50, mannitol 20, KH2PO4 5, EGTA 1, and Mops 5 (pH 7.3, adjusted with KOH) with 0.2% BSA, and then homogenized to isolate mitochondria. The mitochondrial fraction was obtained by differential centrifugation, washed, and suspended in isolation buffer free of EGTA and BSA and kept on ice for the experiment.6
Measurement of Generation by the Mitochondria
The rate of generation was measured as superoxide dismutase (SOD)‐inhibitable reduction of acetylated ferricytochrome c. The reaction mixture contained 0.1 mol/L potassium phosphate buffer (pH 7.4), 7.2 µmol/L acetylated cytochrome c, 2 µmol/L antimycin A, 6 µmol/L rotenone, 10 mmol/L succinate, and 20 to 30 µg/mL mitochondrial protein with glutamate/malate as substrates; 100 units of SOD/mL was added to the reference cuvette. The cytochrome c reduction was recorded at 37°C for 10 minutes by monitoring absorbance at 550 to 540 nm in the presence or absence of SOD. The increase in absorbance at 550 to 540 nm is caused by the formation of stoichiometric ferrous cytochrome c. The production of was estimated with the extinction coefficient of 19.0 mM−1·cm−1 and expressed as a mean of multiple experiments.23, 24, 25
H2O2 Production Within Isolated Cardiomyocytes
H2O2 production was estimated using ROS‐sensitive fluorophore, 2′, 7′‐dichlorofluorescein diacetate (DCFDA) (Molecular Probes, Eugene, OR). The freshly isolated cardiomyocytes were placed in collagen‐coated glass Petri dishes (Mat Tek Corp., Ashland, MA) at room temperature. The cells were incubated for 45 minutes with DCF at 37°C. The DCFDA solution was removed, and the cells were washed with serum‐free medium. Fluorescence intensity of DCF was measured at 585 nm emission when excited at 488 nm by an LSM 510 laser‐scanning confocal microscope. Confocal microscopic images were acquired every minute from 0 to 10 minutes using incorporated software (Carl Zeiss Inc., Thornwood, NY). Cardiomyocytes were then stressed with 30, 100, and 300 μmol/L ouabain (Sigma) or vehicle (DMSO 1%) for 15 minutes at room temperature. Following initial ouabain exposure, cardiomyocytes were loaded with the H2O2‐selective dye DCFDA (4 μmol/L; Molecular Probes) for 15 minutes to detect ROS generation. Confocal images were acquired with an LSM 510 laser scanning confocal microscope using incorporated software. The single‐excitation, single‐emission, fluorescent probe DCFDA was excited using the 568 nm line of the Ar/Kr laser, and emitted fluorescence was filtered through long‐pass filter settings (LP 585). Confocal microscopic images were acquired every minute from 0 to 10 minutes after treatment with ouabain, and data were analyzed using LSM 400 software (Zeiss).
Determination of 4‐Hydroxynonenal Protein Adducts
An OxiSelect HNE adduct competitive ELISA commercial kit (Cat #STA‐838, Cell Biolabs, INC, San Diego, CA) was used to measure the level of 4‐HNE in cardiac tissue homogenate. Human cardiac tissue (n=41) homogenate prepared in ice‐cold phosphate‐buffered saline with 0.1% BSA was centrifuged for 10 minutes at 10 000g, and the 4‐hydroxynonenal (4‐HNE) level was assessed in the supernatant as per the kit's guidelines.
Mitochondrial Superoxide Production in Permeabilized Human Cardiac Myofibers
Superoxide production in the cardiac myofibers was determined as previously described.6 Atrial appendage tissue soaked in ice‐cold buffer containing (in mmol/L) 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 20 taurine, 5.7 ATP, 14.3 phosphocreatine, 6.56 MgCl2·6H2O, and 50 MES (pH 7.1) was cut into small strips and incubated with collagenase type I for 30 to 45 minutes at 4°C. Connective tissue and fat were then removed, and the fibers mechanically separated along the longitudinal axis and permeabilized with saponin (30–50 μg/mL) for 30 minutes at 4°C. Following permeabilization, myofibers were washed in an ice‐cold buffer containing (in mmol/L) 110 K‐MES (pH 7.4), 35 KCl, 1 EGTA, 5 KH2PO4, 3 MgCl2·6H2O, and 0.02 blebbistatin with 5 mg/mL BSA. The myofibers remained in the ice‐cold buffer, supplemented with 100 μmol/L ADP, 5 mmol/L glucose, and 1 U/mL hexokinase to keep mitochondria in an energized and phosphorylating state,26 until used for measuring mitochondrial superoxide production. The level of superoxide production in the myofibers was determined as a change in fluorescence intensity of MitoSOX Red (λex/λem=510/580; Cat #M36008, Thermo Fisher Scientific, Waltham, MA), a mitochondrial superoxide‐sensitive indicator, in response to 10 μmol/L antimycin A (Cat #A8674, Sigma‐Aldrich, St. Louis, MO)6, 27 exposure by time‐lapse laser scanning confocal fluorescence microscopy. Images were detected in an Olympus FV1200 confocal microscope (Olympus, Tokyo, Japan). The change in MitoSOX Red fluorescence intensity before and after antimycin A addition was quantified to measure the difference in the superoxide production.6, 27
Mitochondrial Complex (I–V) Enzymatic Activity
Frozen human cardiac tissue sample (30–50 mg) was homogenized in an ice‐cold buffer (1:20 w/v) containing (in mmol/L) 100 KCl, 5 MgCl2, 2 EGTA, and 50 Tris/HCl (pH 7.5), using an OMNI GLH‐115 Polytron homogenizer (OMNI International). The supernatant collected after centrifuging the homogenates at 1000g for 15 minutes at 4°C was used to measure the functional activity of mitochondrial electron transport chain (ETC) complexes as reported earlier.6, 14
RNA Extraction
Freshly isolated human or rat cardiac tissue was snap‐frozen in liquid nitrogen before storage at −80°C until subsequent processing for gene expression analysis by microarray or by reverse transcription (q) polymerase chain reaction (PCR). Frozen human or rat tissue was homogenized in TRIzol (Invitrogen Corp., Carlsbad, CA) using a PELLET PESTLE motor homogenizer (Kimble‐Kontes, Vineland, NJ). Total RNA was extracted using a TRIzol Reagent kit according to the manufacturer's specifications.6, 8 Further purification was carried out using RNeasy Mini Kit (QIAGEN Inc., Valencia, CA). RNA concentrations were determined using the Infinite 200 NanoQuant (Tecan Group Ltd., Männedorf, Switzerland), and quality/integrity was assessed by Agilent 2100 Bioanalyzer (Affymetrix, Santa Clara, CA).
Reverse Transcription (q) PCR Assay of Target mRNA
Equal amounts of RNA isolated from each human cardiac tissue sample, n=7 adults (age ≤65 years) and n=10 aged (age >65 years), were reverse‐transcribed to cDNA using a miScript RT II kit (Qiagen). Quantitative PCRs were performed in an LC480 real‐time PCR system (Roche, Risch‐Rotkreuz, Switzerland) using SYBR Green PCR Master Mix (Thermo Fisher Scientific). Details of gene‐specific primer sequences used in this study are listed in Table S2. The qPCR reactions were performed in triplicate for all samples. Relative mRNA expression (∆Cq) normalized to reference B2M and 18s RNA expression were determined using LightCycler 480 Software.
Western Blot Analysis
Frozen human cardiac tissue (n=5 adult and n=5 aged) was homogenized in RIPA lysis buffer (Abcam, Cambridge, MA) with protease inhibitor cocktail freshly added (Millipore Sigma). Protein lysates (15 µg) were separated on NuPAGE precast 4% to 12% Bis‐Tris gel (Thermo Fisher Scientific) and transferred to polyvinylidene difluoride membranes (Thermo Fisher Scientific). Western blotting was performed using primary anti‐SOD1 (Cat #ab13498; Abcam), anti‐SOD2 (Cat #ab13194; Abcam), and anti‐β‐tubulin (Cat #2148; Cell Signaling).
Bioinformatics: Functional Enrichment Analysis and Gene Target Prioritization
The Gene Ontology (GO) consortium website (http://geneontology.org), via its AmiGO 2 search browser, was used to determine all genes associated with ROS metabolic processes in humans (website accessed February 29, 2020). Venn diagrams were used to intersect this ROS list with our previously published datasets from human (GSE173608) and rat (GSE173360) aged hearts to identify ROS‐associated candidates.
Functional deconvolution of mitochondrial‐related and extramitochondrial genes involved in ROS metabolism from human and rat aged hearts was performed via Ingenuity Pathway Analysis (IPA; Qiagen, Germantown, MD; last accessed for analysis June 16, 2020). IPA was used to determine prioritized canonical pathways, pathway activity analysis (prediction of functional pathways), and mapping of functional gene networks defined by the differentially expressed genes in aged human and rat hearts. The significance of canonical functional pathways, calculated by Benjamini‐Hochberg multiple testing correction (corrected P value=P [Corr]), indicates the probability of association between a gene/target and a specific functional pathway. Pathway activity analysis determines whether a canonical pathway is activated (increased) or inhibited (decreased), predicted by a z‐score algorithm used to compare the uploaded gene list with the canonical pathway patterns and displayed by colored bar charts indicating their functional pathway activation z‐scores. The highest priority network scores were determined, and the Molecule Activity Predictor Analysis (IPA module) was used to predict activation or inhibition of non‐focused neighboring molecules within the prioritized functional network in human and rat datasets. All gene interactions from networks, prioritized, and merged networks were exported from IPA for use in Cytoscape (v3.7.1; https://cytoscape.org/) for further network analysis (last accessed for analysis June 15, 2020). Prioritization of gene targets was achieved through graph theory analysis tools (network metrics: neighborhood connectivity, betweenness, and closeness centralities) within Cytoscape.
Statistical Analysis
The statistical analysis was performed using SAS software (version 9.2, SAS Institute, Cary, NC) and GraphPad Prism (version 8, GraphPad Software, San Diego, CA). The differences between adult and aged rats were assessed by Student t test. Comparison of the aged and adult humans was performed using the t test for normally distributed variables or the nonparametric Kruskal‐Wallis test for the variables that deviate from the normal distribution. Mean±standard deviation was applied to describe quantitative variables, and percentages were used for qualitative variables. The Kolmogorov‐Smirnov test was used to evaluate the normality of continuous variable distribution. Simple linear regression was performed to investigate the relationship between variables. The number of patient samples used for each experiment is detailed in Table S3. Bioinformatics analysis for canonical functional pathways significance was calculated using false discovery rate, Benjamini‐Hochberg multiple testing correction (corrected P value=P [Corr]). Probability value (P) and P (Corr) <0.05 were considered significant. The rates of and H2O2 production were expressed as the mean and SEM.
RESULTS
Aging Alters the Functional Enrichment of Genes Involved in ROS Metabolism and Depicts Mitochondrial Function and Oxidative Phosphorylation as the Most Prioritized Pathways
Our previous work showed gene expression changes in mitochondrial‐related genes in both human and rat aging hearts compared to adult hearts.8, 14 Some of these genes coding for mitochondrial localized proteins are involved in ROS metabolism, including ROS production and clearance. In both rat and human heart aging, associated changes in mitochondrial and extramitochondrial pathways for ROS production and clearance were observed. These changes were predominantly involved in oxidative stress pathways and significantly reduced expression of subunits involved in electron transport chain complex I. The species‐independent alteration in genes common to both rats and humans with aging is summarized in Table S4.
Statistically significant genes from this dataset were intersected with ROS metabolism genes (n=285) extracted from the GO consortium website (www.geneontology.org/docs/go‐consortium) to identify the ROS‐associated changes in the aging heart (Figure 1A and 1B, Table S4). In human myocardium, out of the 832 differentially expressed genes in the aged versus adult heart, 35 genes (4.2%) were involved in ROS homeostasis, with 27 (77%) downregulated and 8 (23%) upregulated. These 35 differentially expressed genes in human myocardium are 14% of the total ROS‐associated genes present within the Gene Ontology consortium (Figure 1A). Similarly, in the aged rat heart, there were 916 differentially expressed genes, out of which 47 (5.1%) were involved in ROS metabolism; 39 of these genes (83%) were downregulated and 8 (17%) upregulated. Out of the total 250 ROS‐metabolism‐associated genes reported in the Gene Ontology consortium, 47 (18.8%) genes were differentially expressed in the aged rat heart (Figure 1B). In both aged human and rat datasets, the majority of genes involved in ROS homeostasis were downregulated. Functional pathway enrichment analysis from IPA revealed that “mitochondrial dysfunction,” “oxidative phosphorylation,” and “sirtuin signaling pathway” were the three prioritized functions in the human and rat aging heart datasets (Figure 1C and 1D). The human dataset exposed three significant canonical pathways (“mitochondrial dysfunction,” “oxidative phosphorylation,” and “sirtuin signaling pathway”; Figure 1C). The pathway activity analysis module from IPA predicted a decrease in the activity of the “oxidative phosphorylation” pathway (−log P [Corr]=9.85; Figure 1C), whereas the “sirtuin signaling pathway” was predicted to be increased (−log P [Corr]=9.54; Figure 1C). However, IPA was not able to discern a significant predictive pattern for “mitochondrial dysfunction” (−log P [Corr]=13.16; Figure 1C). In the aging rat heart, ROS‐pathway‐associated genes enriched distinctive and significant functional categories (Figure 1D). The top three canonical pathways, and the pathway activity analysis, matched those identified in the aged human heart, however, with much higher statistical significance: “mitochondrial dysfunction” (−log P [Corr]=61.50; Figure 1D), “oxidative phosphorylation” (−log P [Corr]=58.14; Figure 1D), and “sirtuin signaling pathway” (−log P [Corr]=20.04; Figure 1D). Furthermore, “glutathione‐mediated detox” and “apelin adipocyte signaling pathway” showed a predicted decrease in pathway activity (−log P [Corr]=5.25, and −log P [Corr]=5.23, respectively; Figure 1D). Six other functional pathways, namely “NRF2‐mediated oxidative stress response,” “glutathione redox reaction 1,” “superoxide radical degradation,” “aryl hydrocarbon receptor signaling,” “LPS/IL‐1‐mediated inhibitor RXR function,” and “TCA cycle II” were significantly altered but showed a neutral z‐score of zero or close to zero, meaning the activity of the pathways was predicted to neither be increased nor decreased.
Figure 1. ROS‐associated functional pathways changes in aging human and Fischer 344 rat hearts.
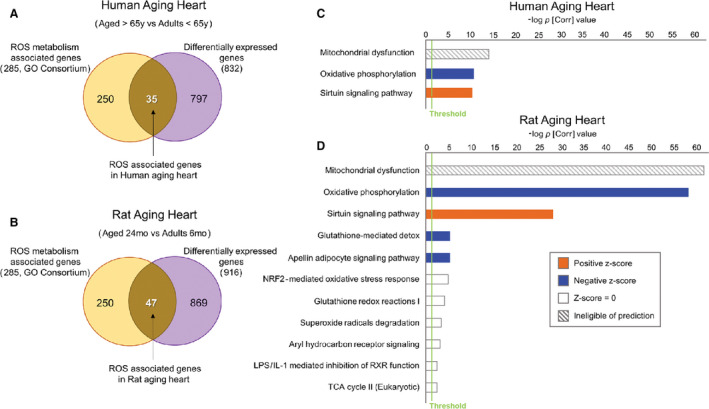
A, Out of the 832 differentially expressed genes (DEG) in aged compared to adult hearts, 35 genes (4.2%) were associated with ROS metabolism, with 27 genes downregulated and 8 upregulated in the human aged heart. B, In the aging rat heart, out of 916 DEGs we found 47 (5.1%) ROS‐metabolism‐associated genes (39 downregulated, 8 upregulated). C, Functional and pathway analysis exposed three significant canonical pathways enriched in the human dataset: “mitochondrial dysfunction” (white/gray pattern=ineligible of pathway prediction), “oxidative phosphorylation” (blue=predicted decrease in activity of pathway), and “sirtuin signaling pathways” (orange=predicted increase in activity). Threshold was set at P [Corr] <0.05 (green line). D, ROS‐pathway‐associated genes from aged rats enriched distinctive and significant functional categories that were similar in rats and humans for the top three canonical pathways, with stronger significance reported for the former. Furthermore, “glutathione‐mediated detox” and “apelin adipocyte signaling pathway” also predicted decrease in activity (blue bars). Six other functional pathways were significantly enriched; however, they showed neutral predicted activity. (For details, see Methods and Materials). IPA indicates Ingenuity Pathways Analysis; P [Corr], Corrected P value including multiple testing correction (Benjamini Hochberg false discovery rate); and ROS, reactive oxygen species.
Further functional pathway activity analysis was used to determine predictive activity patterns of categorical functions associated with the “free radical scavenging” function in both the human and rat datasets (Table). The human dataset identified four functions associated with metabolism, synthesis, and generation of ROS that showed a positive z‐score trend but no significant predictive activity (P [Corr] <0.01, z‐score >0.5; Table). In comparison to the human dataset, the rat dataset did elicit predictive activity on some functions. The following functions were predicted to have a significant increase in activity within the rat dataset: “production of reactive oxygen species” (COX8A, CYBB, NDUFS1, SOD1, SOD2, TXN2, UQCRFS1, and XDH), “production of superoxide,” with five genes associated with this functional term (CYBB, SOD1, SOD2, TXN2, and XDH), and “synthesis of reactive oxygen species” (COX5B, COX6A2, COX8A, CYBB, NDUFS1, SDHC, SOD1, SOD2, TXN2, UQCRFS1, and XDH; P [Corr] <0.0001, z‐score >2.0; Table). Moreover, the other five terms related to metabolism and generation of ROS elicited z‐scores above 1 but did not reach the threshold for activity prediction (Table).
Table 1.
Predicted Activation and Inhibition of Categorical Annotations Within the “Free Radical Scavenging” Function
| Free Radical Scavenging Function | P [Corr] | z‐Score | Gene Associated With Function |
|---|---|---|---|
| Human ROS | |||
| Metabolism of ROS | <0.0001 | 0.864 | ATG5, ATP7A, BNIP3, CAV1, CYB5R4, FOXO3, PINK1, UQCRC2 |
| Synthesis of ROS | 0.0006 | 0.864 | ATG5, BNIP3, CAV1, FOXO3, PINK1, UQCRC2 |
| Generation of ROS | 0.0007 | 0.555 | BNIP3, CAV1, PINK1, UQCRC2 |
| Production of ROS | 0.0075 | 0.507 | ATG5, CAV1, FOXO3, PINK1 |
| Generation of hydrogen peroxide | 0.0014 | … | CAV1, PINK1 |
| Scavenging of hydrogen peroxide | 0.0030 | … | FOXO3 |
| Quantity of ROS | 0.0069 | … | ATG5, FOXO3, NNT |
| Rat ROS | |||
| Production of ROS | <0.0001 | 2.205* | COX8A, CYBB, NDUFS1, SOD1, SOD2, TXN2, UQCRFS1, XDH |
| Production of superoxide | <0.0001 | 2.190* | CYBB, SOD1, SOD2, TXN2, XDH |
| Synthesis of ROS | <0.0001 | 2.128* | COX5B, COX6A2, COX8A, CYBB, NDUFS1, SDHC, SOD1, SOD2, TXN2, UQCRFS1, XDH |
| Formation of ROS | <0.0001 | 1.969 | COX6A2, CYBB, SOD2, XDH |
| Quantity of ROS | <0.0001 | 1.948 | CYBB, GSTA1, PRDX5, SOD1, SOD2, TXN2, XDH |
| Quantity of superoxide | <0.0001 | 1.447 | CYBB, SOD1, SOD2, TXN2, XDH |
| Metabolism of ROS | <0.0001 | 1.186 | COX5B, COX6A2, COX8A, CYBB, GPX3, GSTA4, NDUFS1, PRDX5, SDHC, SOD1, SOD2, TXN2, UQCRFS1, XDH |
| Generation of superoxide | <0.0001 | 1.109 | CYBB, SOD1, SOD2, XDH |
| Generation of ROS | <0.0001 | 0.906 | COX5B, CYBB, SDHC, SOD1, SOD2, UQCRFS1, XDH |
| Quantity of hydrogen peroxide | <0.0001 | 0.762 | PRDX5, SOD1, SOD2, TXN2 |
| Accumulation of ROS | <0.0001 | 0.600 | CYBB, NDUFS1, PRDX5, SDHC, SOD1, SOD2 |
| Metabolism of hydrogen peroxide | <0.0001 | −1.955 | CYBB, GPX3, PRDX5, SOD1, SOD2, UQCRFS1 |
| Modification of ROS | <0.0001 | −1.966 | GSTA4, PRDX5, SOD1, SOD2 |
| Accumulation of hydrogen peroxide | <0.0001 | … | NDUFS1, PRDX5, SOD1, SOD2 |
| Detoxification of ROS | <0.0001 | … | GSTA4, SOD1, SOD2 |
| Modification of hydrogen peroxide | <0.0001 | … | PRDX5, SOD1, SOD2 |
| Conversion of superoxide | <0.0001 | … | SOD1, SOD2 |
| Metabolism of superoxide | <0.0001 | … | CYBB, SOD1, SOD2 |
| Accumulation of superoxide | <0.0001 | … | NDUFS1, SOD2 |
| Reduction of superoxide | <0.0001 | … | SOD1, SOD2 |
| Degradation of hydrogen peroxide | <0.0001 | … | GPX3, PRDX5, SOD1 |
| Conversion of hydrogen peroxide | <0.0001 | … | SOD1, SOD2 |
| Biosynthesis of hydrogen peroxide | 0.0001 | … | CYBB, SOD1, SOD2, UQCRFS1 |
| Removal of superoxide | 0.0003 | … | SOD1, SOD2 |
| Formation of superoxide | 0.0005 | … | CYBB, SOD2 |
| Quantity of lipid peroxide | 0.0007 | … | SOD1, XDH |
| Catabolism of hydrogen peroxide | 0.0009 | … | GPX3, PRDX5 |
| Peroxidation of superoxide | 0.0020 | … | SOD1 |
| Hyperproduction of superoxide | 0.0020 | … | CYBB |
| Generation of hydrogen peroxide | 0.0026 | … | SOD1, UQCRFS1 |
| Breakdown of hydrogen peroxide | 0.0040 | … | SOD1 |
| Detoxification of monohydroperoxy‐linoleic acid | 0.0040 | … | GSTA4 |
| Reduction of monohydroperoxy‐linoleic acid | 0.0061 | … | GSTA4 |
| Clearance of ROS | 0.0121 | … | SOD2 |
| Production of hydrogen peroxide | 0.0128 | … | SOD1, SOD2 |
| Reduction of hydrogen peroxide | 0.0201 | … | PRDX5 |
Using the Pathway Activity Analysis module from Ingenuity Pathways Analysis (IPA), we depict here the functional annotations with inferred activation (z‐score >0) or inhibition (z‐score <0) metrics along with corrected P value (P [Corr]) and gene names of molecules associated with each functional annotation. Both rat and human cardiac transcriptomes were used for this analysis. The human ROS dataset depicted four functions associated with generation and/or accumulation of ROS which showed a positive z‐score trend, however, no significant predictive activity. In the rat ROS dataset, three functions were predicted to be activated in the rat ROS list: production of ROS, production of superoxide and synthesis of ROS (bold and z‐scores highlighted with *). Moreover, eight other functions associated with generation and/or accumulation of ROS also had a positive z‐score and two functions associated with ROS clearance and metabolism had a negative z‐score, but they did not reach significant predictive threshold. P (Corr)=corrected P value with Benjamini‐Hochberg false discovery rate. ROS indicates reactive oxygen species.
Aging Is Associated With Alteration of Gene Expression Encoding for Mitochondrial ROS Production and Clearance Pathways
Figure 2 summarizes the statistically significant alteration in the expression of genes encoding for proteins involved in ROS production and clearance pathways in human (Figure 2A) and rat hearts (Figure 2B). The significant affect associated with the aged human heart was seen on complex I of the ETC, involved in the production of ROS, with downregulation of five subunits (NDUFA6, NDUFA9, NDUFB5, NDUFAB8, and NDUFS2) and upregulation of two complex assembly factors (NDUFAF5 and NDUFAF6) (Figure 2A). GLRX2, GLRX3, and PRDX4, which are involved in the clearance of ROS, were downregulated in the human aged heart, whereas TXNDC15 was upregulated. A similar change was seen in the aged rat hearts, with complex I having the majority of changes (13 genes downregulated; Figure 2B). Of note, some common genes were altered in both humans and rats, while other genes were unique to the rat myocardium (Figure 2A and 2B; Table S4). Both NOX2 (1.6‐fold; P=0.016) and XDH (1.4‐fold; P=0.027; Table S4), two major enzymes involved in extramitochondrial superoxide production, were significantly upregulated in the rat heart but not significantly different between the adult and aged human myocardium. Table S4 summarizes the information on the differentially expressed subunits of the mitochondrial ETC as well as extramitochondrial ROS production and clearance and other ROS metabolism pathways along with fold change and their P values. In the mitochondrial ROS clearance pathway, significant differences were present between rat and human aging myocardium: in rat myocardium, GPX3 (1.6‐fold; P<0.005), GTSA1 (2.6‐fold; P<0.001), PRDX4 (1.3‐fold; P=0.028), and TXNRD3 (2.2‐fold; P<0.001) were upregulated and others (PRDX5, SOD1, SOD2, TXN2) were downregulated; in human myocardium, only downregulation of the genes involved in mitochondrial ROS clearance was observed (Figure 2A and 2B; Table S4). Differences also were present between rat and human hearts in their extramitochondrial ROS generation and clearance pathways.
Figure 2. Differentially expressed genes associated with mitochondrial and extramitochondrial ROS metabolism pathways.
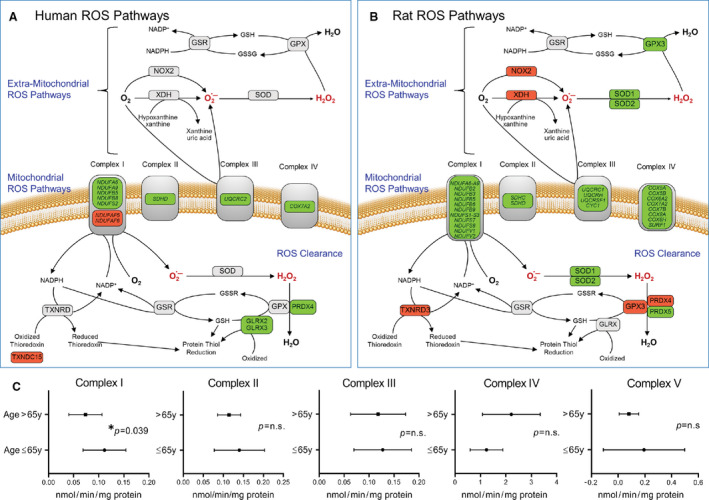
Transcriptomic changes in genes associated with ROS metabolism (ROS production and clearance) in aged human and rat hearts compared to adults (detail annotations found in Table S4). Shown here is a schematic representation of ROS production and clearance pathways, highlighting the genes in (A) aged human atria and (B) aged rat myocardium that are significantly upregulated (red) and downregulated (green) when compared to adult hearts. Genes that show no change in expression are depicted in gray. C, Functional activities of mitochondrial complexes as an indicator of oxidative phosphorylation in human atrial tissue of adult (age 18–65 years) and elderly (aged >65 years) patients with well‐preserved atrial and ventricular function who underwent open heart surgery. Data are expressed as nmol/min/mg protein and presented as mean±standard deviation. Complex I (n=11 for adults and n=25 for elderly), Complex II (n=9 for adults and n=25 for elderly), Complex III (n=5 for adults and n=10 for elderly), Complex IV (n=12 for adults and n=10 for elderly), and Complex V (n=11 for adults and n=25 for elderly) comparison of the adult and elderly groups was performed using t test, with significance set at P<0.05. ROS indicates reactive oxygen species.
To correlate changes in the gene expression of proteins involved in mitochondrial ROS production, activities of individual ETC complexes were determined in myocardial tissue from adult and aged patients (Figure 2C). ETC complex I activity, as determined by rotenone‐sensitive reduction of ubiquinone‐1, was significantly reduced in the elderly compared to adult (73.61±33.02 versus 111.4±42.76 nmol/min per mg protein, respectively, P=0.039; Figure 2C), similar to previously reported findings.8, 14 The functional activities of the remaining four OXPHOS complexes in humans were not significantly altered by age (complex II, P=0.2672; complex III, P=0.6216; complex IV, P=0.5216; complex V, P=0.3912; Figure 2C), a finding similar to that in aged rat and human myocardium.8, 14
Age‐Dependent Changes in Expression of Selected Genes Involved in ROS Metabolism in the Human Heart
ROS metabolism‐associated genes in the aged versus adult human heart showed that most of the genes that are differentially expressed with aging are localized to the mitochondria (Table S4). To define the effect of age on the mRNA expression of selected ROS production and clearance pathway genes in the human heart, qPCR was used to analyze myocardial tissue from patients ages 45 to 83 years who underwent open‐heart surgery. The mitochondrial complex I subunit NDUFA6, which was significantly reduced in both aged human and rat hearts in microarray analysis, demonstrated a highly significant inverse correlation (R 2=0.4059; P=0.008) in expression with advancing age (Figure 3A). In individuals older than 65 years, the total mRNA expression of mitochondrial complex I subunit NDUFA6 was reduced −0.6‐fold compared to the younger age group (Figure 3A inset). NADPH oxidase 2 (NOX2), a major extramitochondrial source for ROS production, showed no significant alteration between those older or younger than 65 years (Figure 3B). For the ROS clearance pathway, the expression of TXN2, a redox‐active protein that regulates ROS levels, showed an inverse relationship with advancing age (R 2=0.3041; P=0.03) (Figure 3C) with the levels reduced −0.82‐fold in those 65 years or older compared to younger individuals (Figure 3C inset). In contrast, TXNRD3, which was significantly downregulated in rat hearts (Table S4), was not significantly altered in the hearts of young and old humans (Figure 3D).
Figure 3. Assessment of expression of selected genes in human adult and aging myocardium.
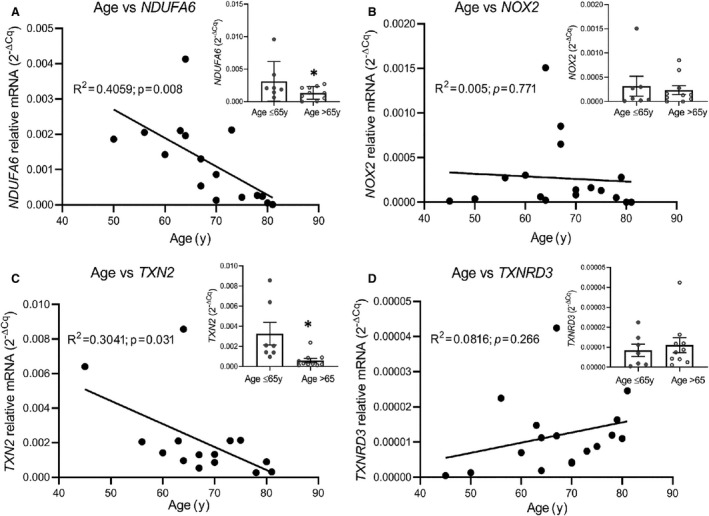
Depicted are mRNA expression levels of selected genes associated with ROS metabolism in human adult (age 18–65 years, n=7) and aging (age >65 years, n=10) myocardium. A, Scatter plot examining age‐dependent correlation of age and mRNA gene expression reveals significant inverse correlation in age vs NDUFA6 (R 2=0.41), with total mRNA expression values from adult and elderly patients. Bars highlighting distribution of individual gene expression values along with mean±standard deviation values for each age group (inset). B, Inverse correlation in age vs NOX2 mRNA expression (R 2=0.005). Total mRNA expression shown in inset. C, TXN2 (R 2=0.304) shows significant inverse correlation with age, with significant total mRNA downregulation in elderly patients compared to adults (inset). D, No significant correlation was seen in age vs TXNRD3 (R 2=0.08); however, an upward trend is observed. Data were analyzed by regression analysis of the sample population (n=17), *P<0.05.
Aging Increases Superoxide () Production in Mitochondria from the Rat Heart
Mean production in isolated mitochondria from the myocardium of aged rats was 1.7‐fold greater than that in young adult rats. The production of was 1.32±0.63 nmol/mg protein/min in aged rat hearts (n=5) compared to 0.76±0.31 nmol/mg protein/min in young adult rat hearts (n=5, P=0.001; Figure 4A). Thus, under similar experimental conditions, mitochondria from aged hearts produced 70% more as determined by the SOD‐inhibitable cytochrome c reductase assay that measures mean production (Figure 4A).
Figure 4. Rate of suppressible superoxide () production in the presence of glutamate/malate substrate (5 mmol/L) in isolated mitochondria from adult and aged rat hearts.
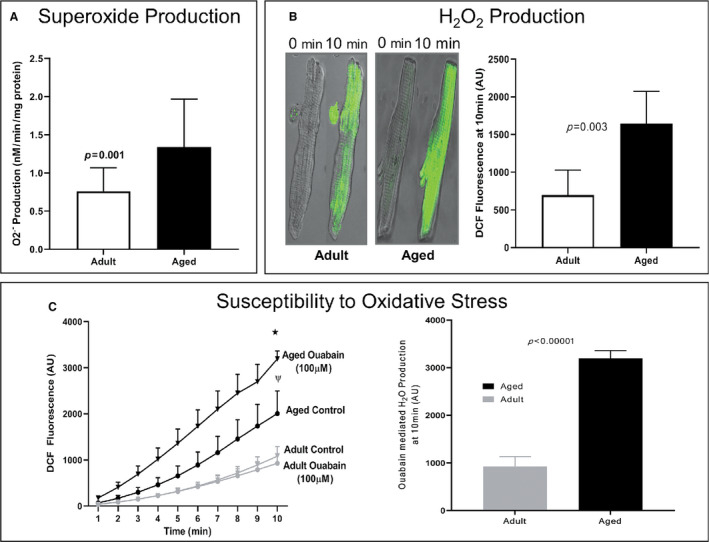
A, Increased reduction manifests as an increase in production with aging (n=19, P<0.01). B, Left panel: Age‐related changes in H2O2 production in unstressed isolated cardiomyocytes from adult and aged rats were measured using the DCFDA assay. Shown are confocal images at 0 and 10 minutes after 45 minutes of DCFDA pre‐incubation. Right Panel: DCFDA quantification exhibits a significant increase in fluorescence in unstressed cardiac cells isolated from aged animals (n=5 adult vs n=5 aged rats, P=0.003). C, Time‐dependent increase in DCFDA fluorescence in aged cardiomyocytes compared to that of adult cardiomyocytes with or without reactive oxygen species (ROS) production stimulated by 100 µmol/L ouabain treatment (n=5; *P<0.001, ψ P<0.01; bottom left graph). Differences in ouabain‐induced fluorescence at 10 minutes between adult (6 months old) and aged (24 months old) ventricular cardiomyocytes isolated from Fischer 344 rats demonstrate a significant increase in ROS production in aged animals (bottom right).
Aging Increases H2O2 Production in Isolated Cardiomyocytes in the Unstressed and Stressed States
H2O2 production from the isolated cardiomyocytes was measured in the unstressed state. Following 45 minutes of incubation with DCFDA, the change in fluorescence intensity was measured with a confocal microscope every minute for 10 minutes (Figure 4B). The cumulative results of five independent experiments in adult rats and aged rats are shown in the right graph in Figure 4B. Cardiomyocytes from aged rats produced a 2.2‐fold higher level of H2O2 than cardiomyocytes from adult animals (1646±428 versus 699±329 arbitrary units [AU] of fluorescence, P=0.003) (Figure 4B). A significant time‐dependent (1–10 minutes) increase in DCF fluorescence was observed in aged cardiomyocytes compared to that observed in adult cardiomyocytes without ouabain (Figure 4C, left graph). Ouabain (100 µmol/L) induced a statistically significant increase in DCF fluorescence in cardiomyocytes from aged rats (1646±428 to 3222±234, P=0.006) but not from adult rats (699±329 to 989±366; P=0.17; Figure 4C, left graph), demonstrating a 3.7‐fold higher production of H2O2 in aged cardiomyocytes than in adult cardiomyocytes following ouabain treatment (P=0.00004; Figure 4C, right graph). These differences in ouabain‐induced H2O2 fluorescent signal at 10 minutes between adult and aged cardiomyocytes from rats indicate increased ouabain sensitivity and higher oxidative stress in aged myocardium (Figure 4C).
Aging‐Related Increase in Superoxide Production and 4‐HNE Protein Adducts in Human Myocardium
Mitochondrial superoxide production, as assessed by MitoSOX fluorescence in response to complex III inhibitor antimycin A, was also significantly higher in the human permeabilized myofibers isolated from aged (age >65 years) myocardium than in that from adult (age ≤65 years) myocardium (Figure 5A). The level of 4‐HNE protein adducts, a marker of lipid peroxidation and oxidative stress, was also significantly increased in elderly (age >65 years) versus adult (age ≤65 years) myocardium (187.54±54.8 versus 47.83±16.7 ng/mg protein, P=0.0063; Figure 5B). The levels of 4‐HNE protein adducts significantly correlated with increasing age (R 2=0.36; P<0.001) (Figure 5C), indicating an age‐associated increase in oxidative stress and lipid peroxidation.
Figure 5. Difference in mitochondrial superoxide level and 4‐HNE protein adducts in the adult and aged human right atrial appendages.
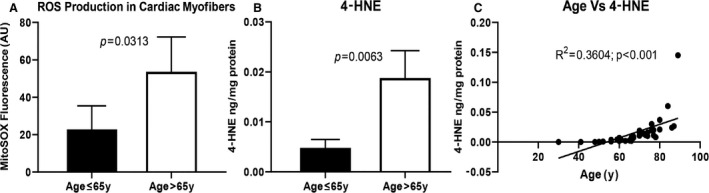
A, Difference in fluorescence intensity of MitoSOX Red, indicative of superoxide production in permeabilized myofibers from adult (age 18–65 years) and aged (age >65 years) right atria after antimycin A (10 μmol/L; 15 minutes) stimulation. Fibers were stained with 2.5 μmol/L MitoSOX Red (λex/λem=510/580), and fluorescence intensity was determined using laser scanning confocal fluorescence microscopy (Olympus FV1200). Data are presented as mean±standard error; n=6 for adult and n=8 for aged. Comparison of the adult and elderly groups was performed using t test, and P<0.05 was considered significant. B, Increased levels of 4‐HNE were observed in aged tissue following normalization to protein concentration in tissue supernatant. Data are presented as mean±standard deviation; n=14 for adult, n=27 aged. C, Scatter plot demonstrating age‐dependent linear regression of the levels of 4‐HNE protein adduct indicative of overall lipid peroxidation in human myocardial tissue of the patient sample population (n=41) who underwent elective open‐heart surgery with age.
Aging Alters Levels of SOD1 and SOD2 in the Human Heart
The expression of SOD1 and SOD2 mRNA coding for two major superoxide scavengers superoxide dismutase (SOD) 1 and SOD2 present in the heart was downregulated in the aging rat myocardium (Table S4). In humans, immunoblots of SOD1 and SOD2 and reference protein β‐tubulin in myocardium from various age groups (28–79 years) demonstrated a similar reduction in mean SOD1 protein expression (P=0.048) with aging but not SOD2 (P=0.106; Figure 6A through 6C). Overall, an inverse relationship in the expression of SOD1 protein level (a statistically significant decline) was observed with increasing age (R 2=0.5725; P=0.011; Figure 6D), which is in contrast to a higher expression of SOD2 level with increasing age (R 2=0.138; P=0.29; Figure 6E).
Figure 6. Age‐associated changes in protein expression level of superoxide dismutase (SOD) 1 and SOD2 in human myocardium. Immunoblot data from human atrial appendage tissue lysate normalized to β‐tubulin expression were analyzed in ≤65 years old and >65 years old groups.
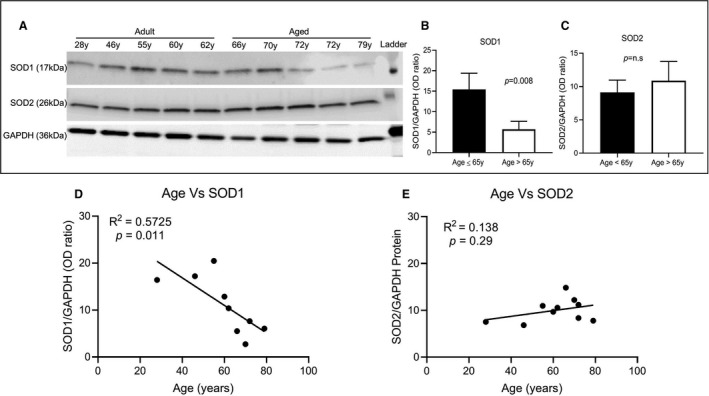
A, Immunoblots of human atrial tissue from different‐age patient tissue showed specific bands with expected molecular size of SOD1 (≈16 kDa), SOD2 (≈26 kDa), and GAPDH (≈36 kDa). B, There were significant decreases in SOD1 expression with increasing age, (C) without any significant changes observed for SOD2 between ≤65 years (n=5) and >65 years (n=5), comparison of the adult and elderly groups was performed using t test (P<0.05 was considered significant). D and E, Scatter plot analysis of age vs protein expression showed a negative correlation for SOD1 (R 2=0.5725), whereas SOD2 (R 2=0.138) exhibited a positive correlation.
Functional Network Analysis of Prioritized Genes With Aging in Human and Rat Aging‐Associated Networks
Functional network analysis of the merged collective network for human and rat samples revealed common genes prioritized in human and rat aging‐associated networks (Figure 7). Specifically, human samples prioritized MYC, AKT, ERK1/2, P38 MAPK, and NFKB complex as betweenness and closeness centrality network nodes, whereas rat samples exhibited priority of TP53, MYC, ERK, and AKT genes (Figure 7A and 7B). In addition, species‐specific enrichment was observed.
Figure 7. Cell proliferation and differentiation pathways genes are prioritized targets in the aging heart in association with reactive oxygen species (ROS).
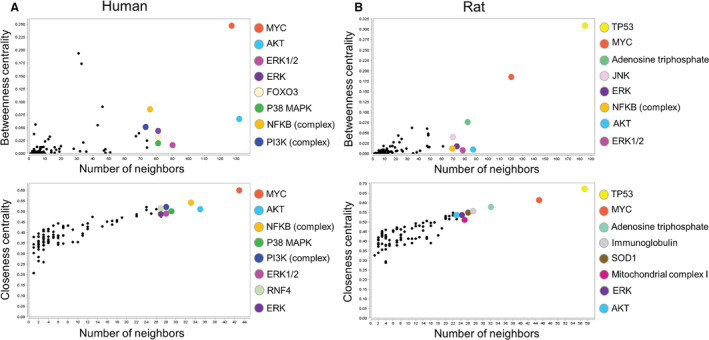
A, Merged networks from human ROS data in aged hearts show prioritized nodes with high betweenness centrality (MYC, AKT, ERK1/2, ERK, FOXO3, P38 MAPK, NFKB [complex], and PI3K [complex]) and high closeness centrality (MYC, AKT, NFKB [complex], P38 MAPK, PI3K [complex], ERK1/2, RNF4, and ERK). B, Rat merged networks identified prioritized nodes with high betweenness centrality (TP53, MYC, adenosine triphosphate, JNK, ERK, NFKB [complex], AKT, and ERK1/2) and high closeness centrality (TP53, MYC, adenosine triphosphate, immunoglobulin, SOD1, mitochondrial complex I, ERK, and AKT).
Analysis of individual subnetworks that comprise the collective network for human and rat further revealed that the top prioritized network (Network 1) had identical enriched top functions for each dataset, namely “metabolic disease,” “developmental disorder,” and “hereditary disorder” (Figure 8). Using the Molecule Predictor Analysis module in IPA, specific genes within each subnetwork were predicted to be activated/inhibited with respect to present gene expression data (Figure 8A and 8B). Although these unique gene activation/inhibition patterns were specific for human and rat samples, there were genes common to both networks, including ERK1/2 (predicted gene activation in both datasets), NDUFA9, and NDUFA6 (Figure 8C) which underlay enriched functional terms of NADH dehydrogenase and mitochondrial complex I.
Figure 8. Prediction of ERK1/2 pathway activation as a response to downregulation of mitochondrial complex I in the aged heart.
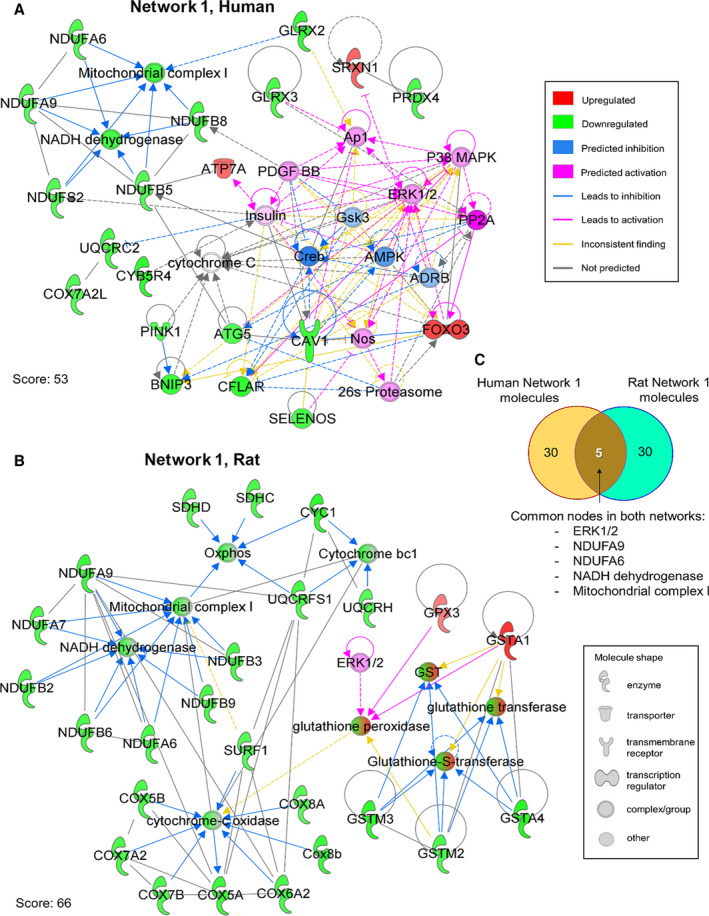
Molecule predictor (MP) analysis in IPA was used to analyze the highest priority network from reactive oxygen species‐related genes in aging hearts. A, MP analysis of IPA Network 1 in the human dataset (Score 53, top functions: metabolic disease, developmental disorder, hereditary disorder) predicted activation of PP2A, P38 MAPK, ERK1/2, PDGF BB, insulin, AP1, NOS, and the 26S proteasome as a consequence of net gene expression dynamics. CREB, AMPK, ARDB, and GSK3 were predicted to be inhibited. Color and molecule shape indicated in legends. B, MP analysis of Network 1 from the rat dataset (Score 66, top functions: metabolic disease, developmental disorder, hereditary disorder) showed only one molecule that predicted activation, ERK1/2, which predicted indirect activation of glutathione peroxidase. C, Venn diagram highlights common nodes in both networks (five nodes), namely: ERK1/2, NDUFA9, NDUFA6, NADH dehydrogenase, and mitochondrial complex I.
DISCUSSION
The main findings of our study are that with aging in both human and rat hearts there are alterations in the expression of genes coding for proteins involved in ROS production and clearance pathways and that selective downregulation is seen in genes encoding for mitochondrial ETC complex I and III sites of ROS production in rat mitochondria but only for complex I in human myocardium. In the extramitochondrial ROS generation pathway, mRNA expression of both XDH and NOX2 was upregulated in rats but not in the human heart. The gene expression changes were correlated with the reduction in functional activities of ETC complex I but not with other complexes. These changes were associated with enhanced ROS production in the aged myocardium in both humans and rats, as demonstrated by enhanced superoxide production and lipid peroxidation product in the human heart and increased H2O2 production in the aged rat heart at baseline and with metabolic stress in isolated cardiac myocytes. Network analysis of the transcriptome highlights the critical elements involved with aging and the downstream effect of gene alteration on functional pathways, hinting at a possible linkage between complex I dysregulation and the ERK1/2 signaling pathway in the aging heart.
Aging‐related mitochondrial changes and reduced myocardial functional reserves affect the heart's physiological function and responsiveness to stress14; however, the molecular basis for this is not entirely understood. In the present study, we showed that steady‐state and H2O2 production were significantly increased in aged animals compared to adult counterparts. In addition, the oxidant responsiveness to stress induced by ouabain exposure was well tolerated in the adult cardiomyocytes but not in cardiomyocytes from the aged animal. Similar differences in susceptibility to superoxide production with antimycin exposure were present in permeabilized myofibers from human aged myocardium but not adult. This was also reflected in a 3.8‐fold higher level of 4‐HNE protein adduct, a marker of lipid peroxidation product, in myocardium from humans 65 years and older compared to that from younger individuals. The increased susceptibility to oxidant production in the aging heart was associated with differential expression of genes involved in ROS production and clearance as revealed by transcriptome profiling in both a rat model of aging and humans. This suggests that aging‐associated enhanced susceptibility to oxidative stress is related to changes in ROS production and clearance independent of species or organism lifespan. The common link in ROS production in both species was the selective downregulation in the activity of ETC complex I involved in superoxide generation. NDUFA6 and NDUFA9 ETC subunits, and NDUFAF5 and NDUFAF6, the assembly factors of complex I, were identified as the prioritized molecules that may underlie the aging‐associated reductions in mitochondrial OXPHOS efficiency and an enhanced tendency to produce superoxide within mitochondria in both species. Thus, our data indicate that similar mechanisms and pathways are involved in aging rats and humans, independent of differences in longevity, food consumption, and lifestyle.
Age‐associated increased susceptibility to oxidative stress has been shown to underlie myocardial dysfunction under metabolic stress.6, 14 Low levels of and H2O2 continuously generated as part of the normal metabolic process are essential for the maintenance of physiological cellular function; however, excessive oxidant production becomes detrimental, resulting in myocardial dysfunction and promoting cell injury after metabolic stress such as ischemia/reperfusion.1, 2, 3, 4, 28, 29 Both cytosolic NADPH oxidase 2 and mitochondrial ETC are ROS generation sites within the cardiac myocyte, but the relative contribution to aging‐associated dysfunction is not fully defined.30 The present study recapitulated the enhanced sensitivity of ROS production in myocytes/myofibers from the aging heart and identified key elements regulating ROS synthesis and ROS clearance pathways that were differentially expressed, providing evidence for compounded impairment of ROS metabolism with aging.31, 32, 33 Genes encoding for proteins involved in the ROS clearance cluster belong to several enzyme classes, including oxide dismutases, glutathione transferases, and oxidoreductases, and were significantly downregulated in rat and human hearts but with significant variability, which may be due to species differences or be the result of other factors related to differences in longevity, diet, or intrinsic differences in heart rate and level of stress between the two species.6, 13, 14, 24, 28, 29, 31, 34
Furthermore, the results from our functional network analysis highlight a possible downstream effect among dysregulation of complex I of the mitochondrial ETC, production of ROS, and the activation of the ERK1/2 signaling pathway in the aging heart. ROS has been implicated in the pathogenesis of heart failure, specifically in the stimulation of cardiac hypertrophy, where H2O2‐promoted cardiac hypertrophy has been shown to be attributed to activation of the ERK1/2 signaling pathway.2, 35, 36, 37, 38, 39 Ischemia‐reperfusion, neurohormones, exercise, comorbidities such as diabetes mellitus and others, and metabolic stressors can enhance the sensitivity of ROS production in the aging heart.24, 30, 33, 40 In the present study, we used ouabain as a mild pharmacologic stressor to identify cardiomyocytes' heightened sensitivity to H2O2 production in the aged rat heart and a negligible effect in cardiomyocytes from adult rats.
The effect of aging on the genomic profile of enzymatic pathways of ROS generation and clearance has not been fully characterized in the human heart. Mitochondria are the primary source of ROS production in the heart and, conversely, are most vulnerable to oxidative damage, leading to a vicious cycle of ever‐increasing ROS production and dysfunctional mitochondrial damage with age.41 Several lines of evidence support age‐related decline in cardiac mitochondrial oxidative phosphorylation, mitochondrial ETC complexes, and ATP synthase.6, 8, 14, 15 Previously, we and others showed transcriptional downregulation of genes encoding subunits of mitochondrial ETC complexes8, 14, 42, 43 that contribute to the decrease in the activity and efficiency of mitochondria under metabolic and oxidative stress6, 14, 44, 45, 46 as well as disease conditions reminiscent of aging, such as atrial fibrillation and heart failure in humans.6, 47 In our previous study,14 we demonstrated that among all five OXPHOS complexes, complex I was predominantly affected by aging at the gene and protein expression level in human cardiac tissue. This was associated with a decline in the functional activity of complex I. Similarly, our group has shown an aging‐related decline in complex I subunits and activity level in rat myocardium.8 In hearts from aging rats, several complex V subunits were also downregulated, with a significant reduction in functional activity compared to adults. The main differences between the adult and aged hearts that are common between rats and humans were in complex I subunit expression and activity (Table S1), with no significant change in complex V or other OXPHOS complexes observed in adult and aged humans compared to rats. This suggests that distinctions exist related to species, overall longevity variances, different metabolic demands with marked variances in heart rate, and other physiological processes, along with possible confounding effects of comorbidities in humans not experienced by animals free of other illnesses. Moreover, these changes in mitochondrial respiratory capacity reduction could be related to aging‐associated alteration in complex I activity or supercomplex destabilization as suggested by Gomez et al.48 The loss of supercomplexes can compromise stoichiometries of OXPHOS complexes I, III, and IV as previously demonstrated in aged rats,49 which makes electron flux through the ETC inefficient, reducing mitochondrial energetic reserve as reported by other studies.48, 50
Differences in the expression of genes coding for the main extramitochondrial and mitochondrial sites of ROS production between the aged and adult rat and human hearts were observed. A 40% to 60% upregulation of expression of subunits of NADPH oxidase 2 (NOX2) and xanthine oxidoreductase (XOR) genes of extramitochondrial ROS production in aged rats was seen compared to adults, along with subunits of mitochondrial ETC complexes I and III. The expression of NOX2 and XOR in humans remained unchanged between the adult and aged myocardium, in which the changes were mainly confined to the major mitochondrial site of ROS production within ETC complex I. The ROS clearance pathways also showed a differential effect of aging on transcripts level, downregulated by 20% to 60% in aged hearts compared to adult hearts (P<0.02). These changes were observed specifically in genes SOD1, SOD2, GSTA4, GSTM2, GSTM3, TXN2, TXNDC4, TXNDC9, and PRDX5, which code for, respectively, SOD1 and SOD2, glutathione S‐transferase A4, M2 and M3, thioredoxin 2, thioredoxin domain containing 4 and 9, and peroxiredoxin 5. Overall, this suggests an aging‐associated change that predisposes to higher ROS levels and a more exaggerated response to stress, with attenuation of antioxidant mechanisms. A compensatory upregulation (20–260%) of transcripts GPX3, GSTA1, TXNRD3, and PRDX4—coding for glutathione peroxidase 3, glutathione S‐transferase A1, thioredoxin reductase 3, and peroxiredoxin 4, respectively, all involved in ROS clearance—was observed with aging, which may help reduce oxidant levels. The downregulated expression of both SOD1 (predominantly localized in the cytosol but also mitochondria) and SOD2 (localized within mitochondria) in rat hearts indicates that the dismutation of is altered at both of these sites.51 These changes in SOD1 and SOD2 transcripts present in the aging rat model were not observed in human aged hearts, in which only protein expression of SOD1 was significantly downregulated and inversely correlated with age. The level of SOD2 proteins in the human heart was increased with aging and may represent a compensatory mechanism for countering the oxidative stress caused by increased mitochondrial production. These findings suggest that individual components of oxidant production and clearance may be affected differentially with aging in humans compared to animal models and may need to be further characterized to identify targets to reduce oxidative stress and maintain the redox homeostasis with aging. The glutathione peroxidases GPx2, GPx3, GPx4‐7 associated with glutathione peroxidase (GPx1), which offers protection against myocardial ischemia‐reperfusion injury,52, 53 were upregulated, akin to earlier observations.54, 55 Thioredoxin and thioredoxin reductases are important components of the cellular antioxidant system involved in ROS scavenging and regulation of cell survival and cell death,56 and in this study transcript levels of Txnrd3 were upregulated in aging rats. GPx3 and Txnrd3 belong to the group of redox enzymes that contain catalytically active selenocysteine (SEC) residues.57, 58 The increase in expression of GPX3 and TXNRD3 is thus notable since the majority of ROS scavengers were either downregulated or showed no significant change in expression. This suggests that enzymes with SEC residues may serve as a secondary layer of protection in response to increased oxidative stress, activated when the primary line of defense, the mitochondrial antioxidant enzyme system against ROS generation, becomes compromised with aging. Indeed, the concept of genetic regulation of ROS production and clearance with age is strongly supported by caloric restriction experiments that demonstrated how transcriptional reprogramming of gene sets leads to overall oxidative stress reduction.59 Concisely, the reduced expression of important components of these protective pathways in the aging heart in both rats and humans highlights a common mechanism underlying the reduced capacity of the aged heart to counter oxidative stress that, in turn, increases predisposition to myocardial injury under stress, as well as promotion of interstitial fibrosis and overall myocardial dysfunction.60, 61
The main limitation of the study is that the observed changes in the levels of individual transcripts were small (20% to 60%); however, the cumulative effect of these changes in the ROS production and clearance pathways could have a significant impact on the aging‐associated susceptibly to oxidative stress and myocardial dysfunction.60
Clinical Implication
In summary, our findings suggest that aging influences both mitochondrial and extramitochondrial pathways of ROS production and clearance within the myocardium, disbalancing the oxidant and antioxidant system, promoting a higher level of oxidants under steady‐state conditions, and, in response to metabolic stress with significant downstream effects, promoting a higher susceptibility to injury. ROS‐mediated activation of the ERK1/2 pathway identified in network analysis warrants further studies; recognizing components of ROS homeostasis regulators and downstream players altered by aging may identify targets that can be selectively intervened on to prevent aging‐associated electrical or mechanical myocardial dysfunction, such as atrial fibrillation and heart failure, prevalent in the elderly.
Sources of Funding
This work was supported by research grants from the National Institute of Aging (RO1‐AG21201) and National Heart, Lung, and Blood Institute, NIH (RO1‐HL089542 and RO1 HL101240). None of the funding sources had any input into study design; collection, analysis, and interpretation of data; writing of the report; or in the decision to submit the article for publication.
Disclosures
None.
Supporting information
Tables S1–S4
Acknowledgments
The authors gratefully acknowledge the editorial assistance of Jennifer Pfaff and Susan Nord and the figure preparation of Brian Miller and Brian Schurrer of Aurora Cardiovascular and Thoracic Services.
(J Am Heart Assoc. 2021;10:e019948. DOI: 10.1161/JAHA.120.019948.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.019948
For Sources of Funding and Disclosures, see page 17.
REFERENCES
- 1.Fridovich I. Chairman's introduction. In: Fitzsimons DW, ed. Ciba Foundation Symposium 65 ‐ Oxygen Free Radicals and Tissue Damage. Amsterdam: Excerpta Medica. 1979;65:1–4. [PubMed] [Google Scholar]
- 2.Nishida M, Maruyama Y, Tanaka R, Kontani K, Nagao T, Kurose H. G alpha(i) and G alpha(o) are target proteins of reactive oxygen species. Nature. 2000;408:492–495. [DOI] [PubMed] [Google Scholar]
- 3.Oldenburg O, Cohen MV, Downey JM. Mitochondrial K(ATP) channels in preconditioning. J Mol Cell Cardiol. 2003;35:569–575. DOI: 10.1016/S0022-2828(03)00115-9. [DOI] [PubMed] [Google Scholar]
- 4.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. DOI: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 5.Forman DE, Arena R, Boxer R, Dolansky MA, Eng JJ, Fleg JL, Haykowsky M, Jahangir A, Kaminsky LA, Kitzman DW, et al. Prioritizing functional capacity as a principal end point for therapies oriented to older adults with cardiovascular disease: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2017;135:e894–e918. DOI: 10.1161/CIR.0000000000000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emelyanova L, Ashary Z, Cosic M, Negmadjanov U, Ross G, Rizvi F, Olet S, Kress D, Sra J, Tajik AJ, et al. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am J Physiol Heart Circ Physiol. 2016;311:H54–H63. DOI: 10.1152/ajpheart.00699.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gredilla R, Sanz A, Lopez‐Torres M, Barja G. Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J. 2001;15:1589–1591. DOI: 10.1096/fj.00-0764fje. [DOI] [PubMed] [Google Scholar]
- 8.Preston CC, Oberlin AS, Holmuhamedov EL, Gupta A, Sagar S, Syed RH, Siddiqui SA, Raghavakaimal S, Terzic A, Jahangir A. Aging‐induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech Ageing Dev. 2008;129:304–312. DOI: 10.1016/j.mad.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ping P, Gustafsson ÅB, Bers DM, Blatter LA, Cai H, Jahangir A, Kelly D, Muoio D, O’Rourke B, Rabinovitch P, et al. Harnessing the power of integrated mitochondrial biology and physiology: a special report on the NHLBI mitochondria in heart diseases initiative. Circ Res. 2015;117:234–238. DOI: 10.1161/CIRCRESAHA.117.306693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemutlu E, Gupta A, Zhang S, Viqar M, Holmuhamedov E, Terzic A, Jahangir A, Dzeja P. Decline of phosphotransfer and substrate supply metabolic circuits hinders ATP cycling in aging myocardium. PLoS One. 2015;10:e0136556. DOI: 10.1371/journal.pone.0136556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sohal RS. Aging, cytochrome oxidase activity, and hydrogen peroxide release by mitochondria. Free Radic Biol Med. 1993;14:583–588. DOI: 10.1016/0891-5849(93)90139-L. [DOI] [PubMed] [Google Scholar]
- 12.Drew B, Phaneuf S, Dirks A, Selman C, Gredilla R, Lezza A, Barja G, Leeuwenburgh C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol. 2003;284:R474–R480. DOI: 10.1152/ajpregu.00455.2002. [DOI] [PubMed] [Google Scholar]
- 13.Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan‐Mamczarz K, et al. AGEMAP: a gene expression database for aging in mice. PLoS Genet. 2007;3:e201. DOI: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emelyanova L, Preston C, Gupta A, Viqar M, Negmadjanov U, Edwards S, Kraft K, Devana K, Holmuhamedov E, O’Hair D, et al. Effect of aging on mitochondrial energetics in the human atria. J Gerontol A Biol Sci Med Sci. 2018;73:608–616. DOI: 10.1093/gerona/glx160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahangir A, Sagar S, Terzic A. Aging and cardioprotection. J Appl Physiol. 2007;103:2120–2128. DOI: 10.1152/japplphysiol.00647.2007. [DOI] [PubMed] [Google Scholar]
- 16.Campbell SG, Haynes P, Kelsey Snapp W, Nava KE, Campbell KS. Altered ventricular torsion and transmural patterns of myocyte relaxation precede heart failure in aging F344 rats. Am J Physiol Heart Circ Physiol. 2013;305:H676–H686. DOI: 10.1152/ajpheart.00797.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margiocco ML, Borgarelli M, Musch TI, Hirai DM, Hageman KS, Fels RJ, Garcia AA, Kenney MJ. Effects of combined aging and heart failure on visceral sympathetic nerve and cardiovascular responses to progressive hyperthermia in F344 rats. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1555–R1563. DOI: 10.1152/ajpregu.00434.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu K, Puchowicz MA, LaManna JC. Aging effect on post‐recovery hypofusion and mortality following cardiac arrest and resuscitation in rats. Adv Exp Med Biol. 2016;876:265–270. DOI: 10.1007/978-1-4939-3023-4_33. [DOI] [PubMed] [Google Scholar]
- 19.Turturro A, Witt WW, Lewis S, Hass BS, Lipman RD, Hart RW. Growth curves and survival characteristics of the animals used in the biomarkers of aging program. J Gerontol A Biol Sci Med Sci. 1999;54:B492–B501. DOI: 10.1093/gerona/54.11.B492. [DOI] [PubMed] [Google Scholar]
- 20.Holmuhamedov EL, Wang L, Terzic A. ATP‐sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol. 1999;519(Pt 2):347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu XK, Yamada S, Kane GC, Alekseev AE, Hodgson DM, O'Cochlain F, Jahangir A, Miki T, Seino S, Terzic A. Genetic disruption of Kir6.2, the pore‐forming subunit of ATP‐sensitive K+ channel, predisposes to catecholamine‐induced ventricular dysrhythmia. Diabetes. 2004;53(suppl 3):S165–S168. DOI: 10.2337/diabetes.53.suppl_3.S165. [DOI] [PubMed] [Google Scholar]
- 22.Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A. Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech Ageing Dev. 2001;122:1073–1086. DOI: 10.1016/S0047-6374(01)00242-1. [DOI] [PubMed] [Google Scholar]
- 23.Dzeja PP, Bast P, Ozcan C, Valverde A, Holmuhamedov EL, Van Wylen DG, Terzic A. Targeting nucleotide‐requiring enzymes: implications for diazoxide‐induced cardioprotection. Am J Physiol Heart Circ Physiol. 2003;284:H1048–H1056. DOI: 10.1152/ajpheart.00847.2002. [DOI] [PubMed] [Google Scholar]
- 24.Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984;105:429–435. [DOI] [PubMed] [Google Scholar]
- 25.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. DOI: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 26.Anderson EJ, Efird JT, Davies SW, O'Neal WT, Darden TM, Thayne KA, Katunga LA, Kindell LC, Ferguson TB, Anderson CA, et al. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc. 2014;3:e000713. DOI: 10.1161/JAHA.113.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Duan Q, Wang T, Ahmed M, Zhang N, Li Y, Li L, Yao X. Mitochondrial respiratory chain inhibitors involved in ROS production induced by acute high concentrations of iodide and the effects of SOD as a protective factor. Oxid Med Cell Longev. 2015;2015:217670. DOI: 10.1155/2015/217670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. DOI: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 29.Southorn PA, Powis G. Free radicals in medicine. II. Involvement in human disease. Mayo Clin Proc. 1988;63:390–408. DOI: 10.1016/S0025-6196(12)64862-9. [DOI] [PubMed] [Google Scholar]
- 30.Kornfeld OS, Hwang S, Disatnik MH, Chen CH, Qvit N, Mochly‐Rosen D. Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ Res. 2015;116:1783–1799. DOI: 10.1161/CIRCRESAHA.116.305432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X‐H, Qanungo S, Pai HV, Starke DW, Steller KM, Fujioka H, Lesnefsky EJ, Kerner J, Rosca MG, Hoppel CL, et al. Aging‐dependent changes in rat heart mitochondrial glutaredoxins–implications for redox regulation. Redox Biol. 2013;1:586–598. DOI: 10.1016/j.redox.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Rebecchi MJ, Glass PS, Brink PR, Liu L. Cardioprotection of the aged rat heart by GSK‐3beta inhibitor is attenuated: age‐related changes in mitochondrial permeability transition pore modulation. Am J Physiol Heart Circ Physiol. 2011;300:H922–H930. DOI: 10.1152/ajpheart.00860.2010. [DOI] [PubMed] [Google Scholar]
- 33.Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res. 2009;83:247–261. DOI: 10.1093/cvr/cvp033. [DOI] [PubMed] [Google Scholar]
- 34.Sawada M, Carlson JC. Changes in superoxide radical and lipid peroxide formation in the brain, heart and liver during the lifetime of the rat. Mech Ageing Dev. 1987;41:125–137. DOI: 10.1016/0047-6374(87)90057-1. [DOI] [PubMed] [Google Scholar]
- 35.Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8‐iso‐prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation. 1998;97:1536–1539. [DOI] [PubMed] [Google Scholar]
- 36.Dey S, DeMazumder D, Sidor A, Foster DB, O'Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. 2018;123:356–371. DOI: 10.1161/CIRCRESAHA.118.312708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugden PH, Clerk A. Oxidative stress and growth‐regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–2124. DOI: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- 38.Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H(2)O(2) regulates cardiac myocyte phenotype via concentration‐dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35:615–621. DOI: 10.1016/S0022-2828(03)00084-1. [DOI] [PubMed] [Google Scholar]
- 39.Xiao L, Pimental DR, Amin JK, Singh K, Sawyer DB, Colucci WS. MEK1/2‐ERK1/2 mediates alpha1‐adrenergic receptor‐stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol. 2001;33:779–787. [DOI] [PubMed] [Google Scholar]
- 40.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. DOI: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohrbach S, Niemann B, Abushouk AM, Holtz J. Caloric restriction and mitochondrial function in the ageing myocardium. Exp Gerontol. 2006;41:525–531. DOI: 10.1016/j.exger.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Bodyak N, Kang PM, Hiromura M, Sulijoadikusumo I, Horikoshi N, Khrapko K, Usheva A. Gene expression profiling of the aging mouse cardiac myocytes. Nucleic Acids Res. 2002;30:3788–3794. DOI: 10.1093/nar/gkf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan L, Ge H, Li H, Lieber SC, Natividad F, Resuello RR, Kim SJ, Akeju S, Sun A, Loo K, et al. Gender‐specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol. 2004;37:921–929. DOI: 10.1016/j.yjmcc.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda‐Saito M, Turkaly PJ, Hoppel CL. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001;33:37–47. DOI: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 45.Paradies G, Ruggiero FM, Petrosillo G, Gadaleta MN, Quagliariello E. Effect of aging and acetyl‐L‐carnitine on the activity of cytochrome oxidase and adenine nucleotide translocase in rat heart mitochondria. FEBS Lett. 1994;350:213–215. [DOI] [PubMed] [Google Scholar]
- 46.Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age‐dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997;406:136–138. [DOI] [PubMed] [Google Scholar]
- 47.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. DOI: 10.1161/01.RES.85.4.357. [DOI] [PubMed] [Google Scholar]
- 48.Gomez LA, Hagen TM. Age‐related decline in mitochondrial bioenergetics: does supercomplex destabilization determine lower oxidative capacity and higher superoxide production? Semin Cell Dev Biol. 2012;23:758–767. DOI: 10.1016/j.semcdb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gomez LA, Monette JS, Chavez JD, Maier CS, Hagen TM. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 2009;490:30–35. DOI: 10.1016/j.abb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Genova ML, Baracca A, Biondi A, Casalena G, Faccioli M, Falasca AI, Formiggini G, Sgarbi G, Solaini G, Lenaz G. Is supercomplex organization of the respiratory chain required for optimal electron transfer activity? Biochim Biophys Acta. 2008;1777:740–746. DOI: 10.1016/j.bbabio.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Fukai T, Ushio‐Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. DOI: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshida T, Watanabe M, Engelman DT, Engelman RM, Schley JA, Maulik N, Ho YS, Oberley TD, Das DK. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 1996;28:1759–1767. DOI: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida T, Maulik N, Engelman RM, Ho YS, Magnenat JL, Rousou JA, Flack JE III, Deaton D, Das DK. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation. 1997;96:II‐216–II‐220. [PubMed] [Google Scholar]
- 54.Meng Q, Wong YT, Chen J, Ruan R. Age‐related changes in mitochondrial function and antioxidative enzyme activity in fischer 344 rats. Mech Ageing Dev. 2007;128:286–292. DOI: 10.1016/j.mad.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 55.Panel M , Panel M, Ghaleh B, Morin D. Mitochondria and aging: a role for the mitochondrial transition pore? Aging Cell. 2018;17:e12793. DOI: 10.1111/acel.12793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pickering AM, Lehr M, Gendron CM, Pletcher SD, Miller RA. Mitochondrial thioredoxin reductase 2 is elevated in long‐lived primate as well as rodent species and extends fly mean lifespan. Aging Cell. 2017;16:683–692. DOI: 10.1111/acel.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rayman MP. Selenium and human health. Lancet. 2012;379:1256–1268. DOI: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 58.Kasaikina MV, Hatfield DL, Gladyshev VN. Understanding selenoprotein function and regulation through the use of rodent models. Biochim Biophys Acta. 2012;1823:1633–1642. DOI: 10.1016/j.bbamcr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park SK, Prolla TA. Gene expression profiling studies of aging in cardiac and skeletal muscles. Cardiovasc Res. 2005;66:205–212. DOI: 10.1016/j.cardiores.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 60.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa‐Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, et al. Thioredoxin‐2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase‐1 activity to maintain cardiac function. Circulation. 2015;131:1082–1097. DOI: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kiermayer C, Northrup E, Schrewe A, Walch A, de Angelis MH , Schoensiegel F, Zischka H, Prehn C, Adamski J, Bekeredjian R, et al. Heart‐specific knockout of the mitochondrial thioredoxin reductase (Txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. J Am Heart Assoc. 2015;4:e002153. DOI: 10.1161/JAHA.115.002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4