

Abstract
Type I interferons (IFNs), mostly IFNα and IFNβ, and the type I IFN Signature are important in the pathogenesis of Systemic Lupus Erythematosus (SLE), an autoimmune chronic condition linked to inflammation. Both IFNα and IFNβ trigger a signaling cascade that, through the activation of JAK1, TYK2, STAT1 and STAT2, initiates gene transcription of IFN stimulated genes (ISGs). Noteworthy, other STAT family members and IFN Responsive Factors (IRFs) can also contribute to the activation of the IFN response. Aberrant type I IFN signaling, therefore, can exacerbate SLE by deregulated homeostasis leading to unnecessary persistence of the biological effects of type I IFNs.
The etiopathogenesis of SLE is partially known and considered multifactorial. Family-based and genome wide association studies (GWAS) have identified genetic and transcriptional abnormalities in key molecules directly involved in the type I IFN signaling pathway, namely TYK2, STAT1 and STAT4, and IRF5. Gain-of-function mutations that heighten IFNα/β production, which in turn maintain type I IFN signaling, are found in other pathologies like the interferonopathies. However, the distinctive characteristics have yet to be determined. Signaling molecules activated in response to type I IFNs are upregulated in immune cell subsets and affected tissues of SLE patients.
Moreover, Type I IFNs induce chromatin remodeling leading to a state permissive to transcription and SLE patients have increased global and gene-specific epigenetic modifications such as hypomethylation of DNA and histone acetylation. Epigenome wide association studies (EWAS) highlight important differences between SLE patients and healthy controls in Interferon Stimulated Genes (ISGs). The combination of environmental and genetic factors may stimulate type I IFN signaling transiently and produce long-lasting detrimental effects through epigenetic alterations.
Substantial evidence for the pathogenic role of type I IFNs in SLE advocates the clinical use of neutralizing anti-type I IFN receptor antibodies as a therapeutic strategy, with clinical studies already showing promising results. Current and future clinical trials will determine whether targeting molecules of the type I IFN signaling pathway, like non-selective JAK inhibitors or specific TYK2 inhibitors, may benefit people living with lupus.
Keywords: Systemic Lupus Erythematosus, autoimmunity, type I interferon, signaling pathways, STAT, JAK, GWAS, epigenetic
Introduction
Systemic Lupus Erythematosus (SLE) is a prototype autoimmune disease characterized by the production of autoantibodies against nuclear and cytoplasmic antigens, including nucleic acids and ribonucleoproteins [1–3]. The role of type I interferons (IFNs) [4] in SLE has been investigated for over four decades [5,6]. Type I IFNs are a family of cytokines first discovered as soluble factors that interfered with viral replication [7] and later found to be pivotal in the activation of innate [8] and adaptive immune responses [9–11]. In the human genome, type I IFNs are comprised of 13 IFNα subtypes, along with IFNβ, IFNε, IFNκ,and IFNω in single form. All of them bind to the same interferon alpha and beta receptor (IFNAR), and the IFNα members and IFNβ are the ones most extensively studied [12–14]. Forty years ago, early studies informed that SLE patients had high serum levels of IFNα or type I IFN activity [15–17]. Twenty years later, subsequent studies revealed that SLE patients with no clinical evidence of infections highly expressed a large group of genes identified as upregulated by type I IFNs, referred thereafter as Interferon Stimulated Genes or ISGs [18–20]. These genes, measured at first as mRNA transcripts in peripheral blood cells, were referred to as the “IFN Signature”, which is detected in the earliest and most acute phases of the disease [21,22]. This distinctive IFN signature is frequently seen in pediatric patients [19] and has been proposed as a marker of specific morbidities in SLE [20,23–25]. Determining protein levels of type I IFNs (IFNα subtypes or IFNβ) in the serum of patients with SLE, other diseases or in healthy controls remains challenging. Most studies report the IFN Signature at the transcriptional level as a measure of type I IFN activity. They hint at a response to IFNα without completely discriminating the specific patterns induced by single members of the type I IFN family; particularly as new data emerges that implicates multiple type I Interferons as inducers of the IFN Signature in lupus, including IFNβ [26]. Recent studies document increased levels of serum IFNβ in SLE patients [27]. Furthermore, new technologies have now confirmed the RNA data of the IFN Signature and the original IFNα measurements [15–17] at the protein level, both measuring serum protein levels and activity of serum IFNα [24,28]. Adding to that, use of epigenetic CpG methylation data can be useful to adequately assess type I IFN activity. In doing so, the use of a DNA methylation IFN score has been found to be a potential surrogate marker of the RNA-based IFN-signature [29].
The IFN Signature and the associated higher response to type I IFNs [20,30,31] are seemingly linked to many genetic and environmental factors, which may be different in distinct subsets of SLE patients [32]. Genetic factors are widely accepted to play an active role and considered necessary but not sufficient for disease development [33,34]. Environmental triggers, like viral and bacterial infections, as well as alterations in the microbiome, are actively being studied to better assemble a complex and more accurate depiction of lupus pathogenesis [3,35–40]. Endogenous inducers of type I IFNs are released during cell death and tissue damage, often fueling a vicious cycle in which immune complexes containing endogenous DNA or ribonucleoproteins and autoantibodies cause tissue damage and propel the release of more autoantigens [41,42]. Moreover, immune complexes formed by endogenous DNA and autoantibodies are known stimulators of IFNα responses, as well as modified nucleic acids per se can induce IFNα [43–46] and IFNβ [46,47]. Nucleic acids can be released extracellularly and recognized by endosomal Toll Like Receptors (TLRs), or they can be overrepresented in the cytoplasm and recognized by cytoplasmic Pattern Recognition Receptors (PRRs) [3,48,49]. Plasmacytoid dendritic cells are the main cellular source of IFNα [50,51]. They are considered pivotal in SLE pathogenesis [43,52–54], whereas other innate immune cells such as monocytes and conventional dendritic cells can also contribute to the type I IFN response producing mostly IFNβ [31,47,55–57]. Similarly, neutrophils can trigger in other cells the innate production of type I IFNs by extruding DNA through the process of Netting, that allows DNA to activate IFNα and/or IFNβ synthesis through TLR9 or STING-dependent pathways [46,58–60]. Neutrophils can also respond to chromatin by secreting IFNβ themselves to amplify the stimulation of the IFN Signature [61].
Elucidation of the signaling pathways downstream of IFNAR have been the focus of intense research in cell biology, pathology, virology and cancer. Results from these disciplines have provided the scaffold to build on the contribution of the genetic, epigenetic and environmental factors that influence the role of type I IFNs in lupus pathogenesis. It’s noteworthy that the IFN Signature in SLE is not exclusive as other autoimmune diseases, ranging from Rheumatoid Arthritis to Sjogren Syndrome to autoimmune myositis, have shown similar abnormal responses [62]. In each of these conditions, common and specific causes may play a role. In this review, we will focus our attention on the signaling pathways mediating the response to type I IFNs; how they affect the activation of the IFN Signature, and the corresponding specific abnormalities in SLE (Figure 1).
Figure 1. Abnormalities in the signaling pathway that exacerbate type I IFN responses.
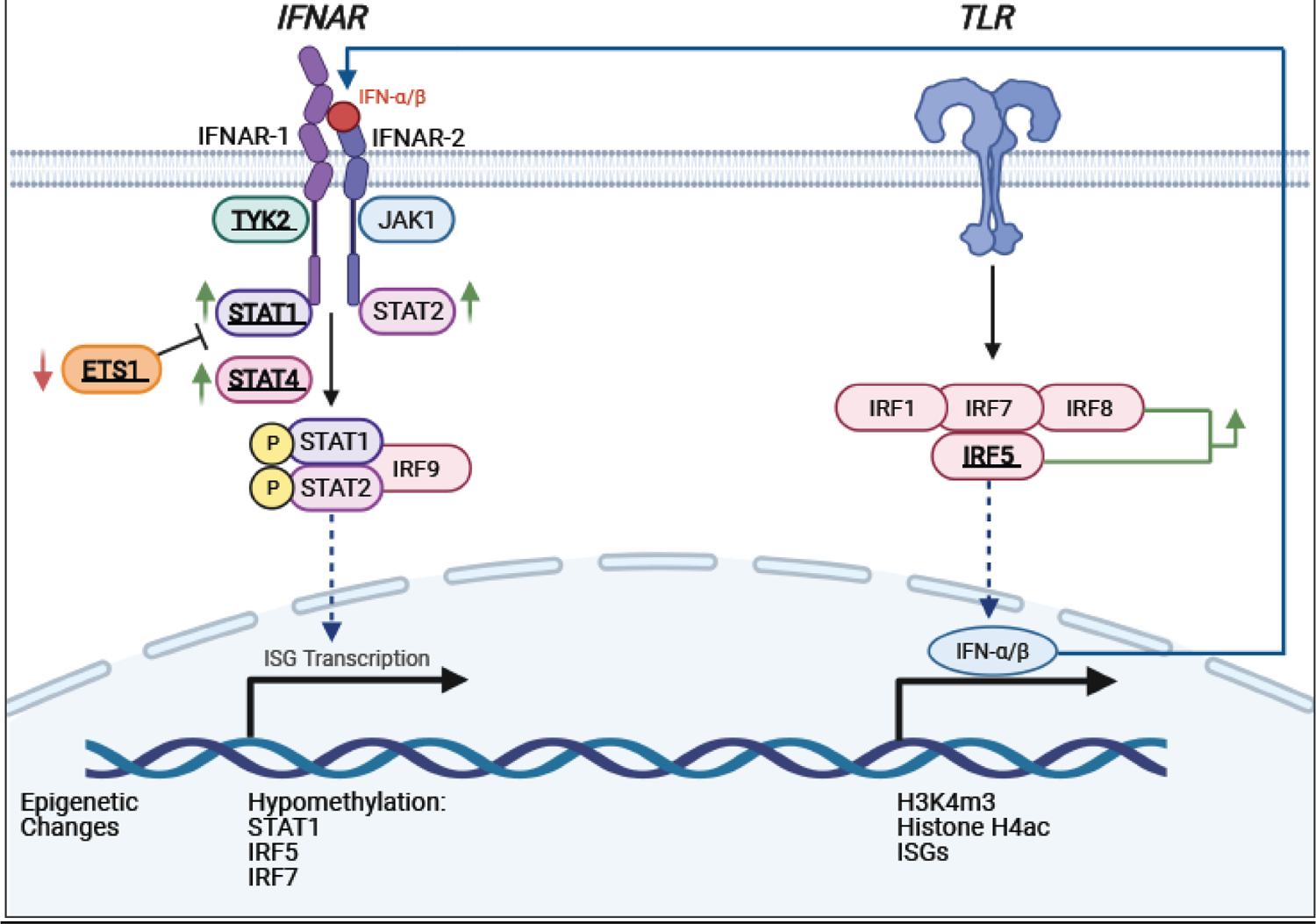
Highlighted are identified components of the signaling pathway downstream of the Interferon-alpha/beta receptor (IFNAR) and Toll-like receptor pathways that, when deregulated, contribute to the pathogenesis of SLE. Polymorphisms associated with SLE are indicated in bold and underlined. Genes that have upregulated/downregulated expressions are indicated with green/red arrows, respectively.
Signaling downstream of Type I IFNs
Type I IFNs is a family of cytokines which includes 13 IFNα subtytpes, IFNβ, IFNε, IFNκ, and IFNω [12,13]. All type I IFNs initiate a signaling cascade upon binding to their cognate receptor complex composed of IFNAR1 and IFNAR2 [63]. Assembly of this ternary complex induces a conformational change in the receptor structure enabling their activation by tyrosine kinases JAK1 and TYK2; each of which is found pre-associated with IFNAR2 and IFNAR1, respectively, and become activated themselves by transphosphorylation. The Janus kinase (JAK) family belongs to a group of ten recognized families of non-receptor tyrosine kinases and consists of four enzymes, JAK1–3 and Tyrosine kinase 2 (TYK2) [64]. The canonical view is that tyrosine phosphorylation of the IFNAR subunits by JAK1 and TYK2 creates docking sites for the recruitment of pre-existing STAT1 and STAT2. JAKs activate STAT1 and STAT2, which in turn heterodimerize and associate with the transcription factor IRF9 to form the heterotrimeric complex ISGF3; the latter is translocated to the nucleus and initiates gene transcription by recognizing IFN-stimulated response elements (ISRE) found in the promoters of ISGs. Type I IFNs also induce the formation of STAT1 homodimers that bind the gamma-activated sites (GAS) and stimulate the transcription of additional ISGs. In recent years, evidence has been accumulating of an ISGF3-like transcriptional complex consisting of STAT2 homodimers joined with IRF9 that directs a prolonged ISG transcriptional program and overlaps with a subset of genes induced by ISGF3 [65], thus highlighting the pivotal role of STAT2 in type I IFN signaling. It is important to mention that while IRF9 binds the core ISRE sequence, STAT2 as a homodimer or as a STAT2/STAT1 heterodimer is unable to directly bind DNA, but it contributes its transactivation domain for gene transcription [66]. Recent attention has been given to constitutive unphosphorylated pools of STAT2/IRF9 complexes that shuttle between the cytoplasm and the nucleus in the absence of type I IFN stimulation [67] for their role in maintaining basal ISG expression. STAT2 chromatin immunoprecipitation (ChIP)-chip assays previously revealed unphosphorylated STAT2 occupancy of several ISG promoters [68]. Subsequent studies have reported that the initial and robust ISGF3 response to type I IFNs is followed by an increase in the expression of ISGF3 components that assemble without tyrosine phosphorylation to amplify type I IFN responses [69]. We previously reported that the constitutive expression of ISGs, including Cxcl10, Isg15, and Irf7 genes, was markedly reduced by 70% in murine conventional dendritic cells (cDCs) from Stat2−/− mice when compared with wild type cDCs [70], thus supporting a role for unphosphorylated STAT2 in the maintenance of basal ISG production or a constitutive low production of type I IFNs. The concept of tonic ISGF3 activation downstream of type I IFN signaling driven by the production of low quantities of type I IFNs was proposed several years ago, to maintain adequate levels of ISGF3 components and prepare cells for quick and robust ISGF3-mediated biological responses [71]. However, this view was recently revisited, in which basal expression of many ISGs is stimulated by a preformed STAT2/IRF9 complex in the absence of IFNAR engagement [72]. Most notable was the finding that resting cells responding to type I IFNs rapidly switched signaling complexes from STAT2/IRF9 to ISGF3 assembled in the nucleus. Hence, alterations in type I IFN signaling that disturb homeostasis and unnecessarily prolong the biological effects of IFNs could elicit uncontrolled destructive effects.
Altered Signaling of Type I IFN responses in SLE
The response to type I IFNs, either or both IFNα and IFNβ, and specific steps in the signaling pathway mediating their functions have been shown to be dysregulated in SLE patients as detected in whole blood and in individual immune cell populations [73,74]. Moreover, specific tissues targeted by lupus autoimmunity show an abnormal type I IFN response [26,75–77]. JAK1 and STAT2 were shown to be constitutively phosphorylated in peripheral blood mononuclear cells (PBMCs) of SLE patients, and stimulation with IFN-β induced 3–4 folds higher STAT2 phosphorylation, which remained sustained for a prolonged time when compared to control cells from healthy subjects. This hyperactivation induced higher MxA mRNA levels (an ISG) in SLE patients than in healthy controls, possibly with the contribution of a deficient up-regulation of regulatory SOCS1 protein [78]. A recent study has found that when assessing expression and phosphorylation of STAT1 after IFNα and IFN γ stimulation, SLE patients showed significantly elevated expression of STAT1 in B cells than healthy individuals. This abnormality was associated with increasing disease activity and Siglec-1 expression on monocytes; an indicator that the increased expression of STAT1 in B cells is associated with the IFN Signature [79]. Furthermore, a study of single-cell gene expression in SLE monocytes revealed increased expression of most of the signaling molecules directly involved in IFNAR signaling, including JAK1, STAT1 and STAT2, in both classical and nonclassical monocytes from patients with high disease activity [80].
Several studies have shown how responses to damage-associated molecular patterns (DAMPs) such as mitochondrial DNA and necrotic/apoptotic cells can induce ISGs [81–85], as well as responses to pathogen-associated molecular patterns (PAMPs) triggered by bacterial and viral infections, through the activation of TLR and other PRRs [35–37,86,87]. The higher levels of type I IFNs and STATs found in SLE patients may be due to chronic exposure to DAMPs and PAMPs because of subclinical infections [3,88], or may have genetic causes in some patients, aggravated by environmental triggers that can leave long-lasting effects through epigenetic alterations.
Genetic variants altering type I IFN Signaling in Lupus
The discovery of the IFN Signature in SLE has ignited the search of the causal factors prompting the exacerbation of type I IFN responses [6,89]. Family-based and genome wide association studies (GWAS), together with gene profiling of large cohorts have identified genetic, epigenetic and transcriptional abnormalities in molecules directly involved in the signaling pathways mediating the response to type I IFNs, as well as those involved in its production [90,91]. As described in the previous paragraphs, the activation of the response to type I IFNs and the induction of the IFN Signature is complex and can be influenced by a network of interactions with other critical signaling pathways, from MAPKs to NFκB, and by the positive and negative feedbacks elicited by the ISGs themselves [4].
A significant body of literature addresses the alterations of these molecular networks in SLE. Genetic studies have afforded significant advances in the knowledge of SLE risk, by enabling the discovery of potential genetic factors that are involved in the regulation of the type I IFN signaling pathway [92–95]. The use of GWAS has identified more than 100 SLE susceptibility loci along with documentation of genetic variants associated with the pathogenesis of SLE [33,96]. Many of these genetic variants implicate aberrant regulation of both innate and adaptive immune responses that, in particular, involve transcription factors [97,98]. We focus here on molecules directly contributing to signaling triggered by IFNα and IFNβ. Little information is available regarding specific alterations involving signaling induced by the other type I IFNs, namely IFNε, IFNκ,and IFNω [99].
IFNARs.
High serum levels of IFNα and IFNβ have been reported in SLE patients throughout the history of the investigations of IFNs in SLE [15–17,27]. To date, no polymorphisms in type I IFNs loci have been associated with higher risk of lupus in the human population; the same applies to subunits of the type I IFN receptor [97,100–102]. In murine models of lupus, the genes for IFNα and IFNβ are located in the Sle2 susceptibility locus that is derived from the lupus-prone New Zealand strain of mice and was introgressed into the triple congenic Sle1,2,3 mice [103,104]. Deletion of Ifnar in lupus prone mice causes delayed and milder lupus autoimmunity in spontaneous and induced models of lupus [105–110]. However, the role of type I IFN genes in the Sle2 allele and in the susceptibility of Sle2 mice to lupus remains ambiguous because Sle2 seems to cause a decrease in IFN responses [104]. Since high serum IFN-α activity is a human heritable trait, as shown by clustering in specific families in both SLE patients and their healthy first-degree relatives [30], the lack of risk-associated polymorphisms in type I IFN genes and IFNARs suggests that genetic variations affecting the type I IFN response must be occurring at other levels of the IFN signaling pathway. Alternatively, variations may be present in non-coding regions of the DNA, and therefore, difficult to identify because these may differ in SLE patients.
JAK1 and TYK2.
TYK2 plays an essential role in triggering the signaling of important cytokines, including type I IFNs. Single nucleotide polymorphisms (SNPs) of TYK2 were identified in families with SLE [111], and these results were extended by GWAS in many cohorts [97,100,101]. Through a meta-analysis, two TYK2 polymorphisms, rs2304256 and rs280519, were found associated with SLE in patients of different ancestry [112]. The specific mechanism of the rs2304256 polymorphism is not entirely clear, but what is known is that it causes an amino acid substitution within a region of TYK2 that is essential for its interaction with IFNAR1 and may cause an aberrant IFN response [112]. GWAS have revealed another SNP (rs34536443) in the TYK2 gene that encodes a rare protective variant known as TYK2 P1104A (TYK2p). This genetic variant is responsible for reducing type I IFN signaling in healthy individuals and altering autoimmune pathogenesis through decreasing IL-12, IL-23, and type I IFN signaling in T cells [113]. Moreover, of the known long non-coding RNAs (lncRNA), elevated lupus nephritis (LN)-associated lncRNA RP11-2b6.2 has been observed in kidney biopsies of LN patients. Curiously, decreased LncRNA Rp11-2b6.2 serves as a positive regulator, given that when its levels are low, the phosphorylation of JAK1, TYK2 and STAT1 in the type I IFN pathway is inhibited [114].
No specific polymorphisms of JAK1 have been found to be associated with the risk of developing SLE, but its activation and regulation may play a role in supporting the IFN Signature.
STAT1/STAT2.
STAT1 and STAT2 are essential transcription factors downstream of type I IFN signaling. STAT1 was identified to have SLE-associated risk alleles [115]. Since the STAT1 locus is adjacent to the STAT4 locus, and the latter was also found to have variants associated with higher risk of developing SLE [111,116,117], the role of STAT1 polymorphisms in SLE pathogenesis has been difficult to discriminate from the role of STAT4. It is worth noting that researchers who identified the same STAT4 risk allele in Japanese populations that is associated with SLE in Caucasians (see below), did not find evidence for a specific role of STAT1 in the genetic susceptibility to SLE [118].
To date, no polymorphisms of STAT2 influencing the risk of developing SLE have been identified. While STAT2 expression and function are augmented in SLE (see below), an indirect effect on STAT2 caused by genetic variants of other genes remains to be determined. Similarly, STAT1 was found to be upregulated in SLE patients and highly expressed in various immune cell subsets, including dendritic cells in human [119] and mice [31]. A transcriptome profiling study of SLE patients identified the overexpression of the long non-coding RNA (lncRNA) linc00513 associated with two alleles. This elevated linc00513 was a positive regulator of the type I IFN signaling pathway by promoting increased phosphorylation of STAT1 and STAT2 [120]. The increased expression of linc00513 correlated with the measure of ISGs in SLE patients, thus potentially contributing to the IFN Signature [120].
The function of STAT1 or STAT2 may be also affected by polymorphisms in other transcription factors. For example, ETS1 is a transcription factor important in regulating immune cell proliferation and differentiation [121]. Many GWAS have found genetic variants of ETS1 associated with susceptibility to SLE in Asian populations [122,123]. Ets1-deficient mice develop a lupus-like disease characterized by autoantibodies and immune-complex deposition in the kidney [124]. PBMCs from patients carrying lupus risk ETS1 alleles express significantly lower levels of ETS1 mRNA than control subjects, suggesting that ETS1 risk variants contribute to SLE through reduced ETS1 expression. The lupus risk ETS1 variant rs6590330 was shown to enhance phosphorylated STAT1 binding to DNA [125]. This observation lends to the hypothesis that one of the mechanisms underlying the effects of ETS1 variants on SLE is that less ETS1 leads to increased STAT1 binding to other promoters, possibly ISGs. Since STAT1 is shared with type II IFN (IFN-γ) signaling, alterations in STAT1 expression may also affect the functions induced by IFN-γ and its role in lupus pathogenesis, as elegantly shown in lupus prone mice [126,127].
STAT4.
STAT4 can be activated by type I IFNs and participates in amplifying its positive feedback loop [128]. Identified as a risk allele in the first GWAS, STAT4 is associated with specific clinical manifestations including early disease onset, ischemic cerebrovascular disease, severe nephritis and renal insufficiency [111,116,117]. Recent SLE human studies have investigated the effect of the STAT4 risk allele in T cells of carriers. SLE patients show elevated levels of STAT4 protein, leading to both increased phosphorylation of STAT4 in response to IL-12 and IFNα, as well as IL-12 induced IFN-γ production [129]. However, healthy individuals who are carriers of the risk allele display the opposite effect; they show decreased phosphorylation of STAT4 in activated T cells [130]. It was further determined that IFNα is an environmental modifier of the STAT4 risk allele [130].
IRF5 and other IRFs.
The analysis of SNPs in genes affecting the upstream events of the type I IFN pathway has revealed a number of genetic risk polymorphisms in the IRF family of transcription factors that are associated with high type I IFN levels in lupus patients [84,131]. IRF1, IRF5, IRF7 and IRF8 are considered primary inducers of type I IFN production, mediating the signaling pathways of major pattern recognition receptors (PRRs) like TLRs and RIG-I-like receptors (RLRs), and SLE-associated SNPs have been reported in these transcription factors [93,102,132]. Some genetic variations of IRF3 have been found in SLE patients but it remains to be clarified whether these polymorphisms increase the risk or confer protection from developing lupus [133–135]. Although almost one hundred SLE-associated SNPs have been found within the IRF5 locus, mostly in non-coding regions, five genetic variants have been associated with abnormalities in the expression or function of IRF5, which may affect lupus pathology [94,102,111,136]. Studies have revealed increased type I IFN activity in the sera of SLE patients with IRF5 risk polymorphisms, which correlates with elevated IRF5 expression levels and positivity for either anti-RNA binding protein (anti-RBP) or anti-double-stranded DNA (anti-dsDNA) autoantibodies [84,131]. Indeed, many SNPs in the SLE-associated risk variants of IRF5, as well as STAT4, were found to correlate with the levels of anti-dsDNA autoantibodies, therefore suggesting that these genes, and the augmented type I IFN response, may increase the risk of developing SLE by promoting autoantibody production [137]. The same risk alleles for IRF5, when present in healthy donors, are associated with increased activation of innate immune cells like pDCs and neutrophils, detectable autoantibodies and type I IFN pathway enrichment, therefore behaving as pre-symptomatic SLE [136].
Epigenetic Regulation of type I IFN signaling and disease activity in lupus
Many reports indicate the SLE patients have global and gene-specific DNA methylation changes [138]. DNA methylation is an important epigenetic modification that, especially if occurring in promoters, suppresses the expression of relevant genes. Specifically, epigenetic modifications are important in restricting or sustaining the response to type I IFNs for the induction of ISGs [139]. Type I IFNs induce chromatin remodeling leading to a state permissive to transcription. Evidence indicates that abnormal DNA hypomethylation in T cells is an important epigenetic hallmark in SLE [140]. Significant hypomethylation of ISGs in naïve T cells from lupus patients, and among those ISGs specifically STAT1, suggests an epigenetic increase in accessibility for transcription in these genetic loci [141]. This epigenetic change can help explain the increased levels of STAT1 and other ISGs in SLE cells. In an independent report, the performance of epigenome wide association study (EWAS) of whole blood highlighted that the most evident differences in methylation between SLE patients and healthy controls were in ISGs, which included IRF5 and IRF7, and showed decreased methylation in patients with SLE. [142] In other cohorts, IRF7 gene was also found hypomethylated throughout T-cells, B-cells and CD14+ monocytes at CpG islands, as well as monocyte-specific hypomethylation within the gene body [143]. Furthermore, a functional gene ontology analysis of ISGs for DNA hypomethylation revealed an overrepresentation of genes involved in induction and regulation of apoptotic processes, as well as NFκB activation, suggesting that epigenetic changes in genes other than ISGs, like those within the Fas/FasL-mediated apoptotic pathway, also contribute to SLE [144]. The modification of DNA methylation levels at regulatory regions of ISGs ultimately was associated with various phenotypic manifestations of SLE, such as nephritis and skin rash [142]. Genetic variants influence methylation marks, as well as environmental stressors [145]. It is unknown how methylation marks change over time in SLE patients.
Moreover, type I IFNs induce an increase in tri-methylation of H3K4 (H3K4me3), a histone mark that promotes transcription. Sullivan’s group implemented an innovative approach to study the regulation of ISGs in SLE, which consists in cross-referencing the GWAS results with those from the transcriptome and epigenome. They found that epigenetic modifications, like H3K4me3 histone modifications, are increased at sites highlighted by GWAS as associated with SLE. Surprisingly, the RNA expression of these GWAS-associated genes was decreased in three main immune cell populations, monocytes, T and B cells, in SLE patients. STAT1 was one of the genes found to be under-expressed in SLE patients. The authors recognized that their focus on mild cases of SLE may justify these results that remain a sobering memento of the complexity of studies bridging from genetics to functionality [146]. Other reports have documented increased expression of STAT1/2 in SLE patients. The STAT1 locus is adjacent to the STAT4 locus, and it has been shown in B cell lines that SLE B cells carrying the SLE STAT4 polymorphisms express higher levels of STAT1 protein [147]. The analysis of the transcriptional profile and epigenetic landscape of Low Density Granulocytes (LDG), an abnormal population of neutrophils present in SLE, showed that these cells are more transcriptionally active with a specific up-regulation of genes involved in histone acetylation. ATACseq showed increased chromatin accessibility, with a number of “open peaks” twenty time higher than in normal neutrophils, in parallel with increased ISG expression [148]. Altogether, these findings indicate that the upregulated expression of type I IFN signaling molecules observed in SLE may be due to classic genetic causes in some cases, while other cases can be explained by environment-induced epigenetic modifications.
Defects of IFN-induced signaling pathways in other pathologies.
Several mechanisms are in place to dynamically restrict type I IFN signaling that, if left unrestrained, can be detrimental to the host. These tightly regulated controls include among others internalization and degradation of cell surface IFNAR1 [149], induction of inhibitory proteins SOCS1 [150] and USP18 [151] that, when combined, trigger a negative feedback loop and suppresses JAK activity and further activation of STAT1 and STAT2. Importantly, STAT2 plays dual roles in the activation and inhibition of type I IFN signaling. In the latter, STAT2 functions as an adaptor molecule for the recruitment of USP18 to IFNAR2 that displaces JAK1 from the IFN receptor [152]. Impaired activation of the type I IFN signaling pathway has been implicated in multiple immune related diseases. In humans, several germline STAT1 mutations have been identified that confer either a loss-of-function or gain-of-function phenotype with an outcome of susceptibility to an array of viral infections and predominantly fungal (chronic mucocutaneous candidiasis) and bacterial (mycobacterial) infections [153–155]. Similar phenotypes have also been described in patients with inborn errors of IFNAR1 [156], IFNAR2 [157], JAK1 [158], TYK2 [159], IRF9 [160] and STAT2 [161,162]. Most unexpected was the clinical phenotype observed in patients with IFNAR2 and STAT2 deficiencies who became severely ill after receiving the measles, mumps and rubella vaccine.
On the opposite spectrum, dysregulation of type I IFN signaling is strongly associated with inflammatory conditions. A collection of rare Mendelian autoinflammatory disorders termed type I interferonopathies is characterized by exacerbated type I IFN signaling activity and elevated ISG signature [163]. Type I interferonopathies are monogenic disorders and display diverse clinical phenotypes that can be daunting to diagnose. Examples of these conditions are Aicardi-Goutières syndrome and spondyloenchondrodysplasia with pathological phenotypes that partially overlap with SLE [164,165]. Since 2006, mutations in several genes have been identified in patients as the cause of type I interferonopathies. Loss-of-function and gain-of-function mutations, which heighten type I IFN production and sustain type I IFN signaling, drive the clinical phenotypes by distinct mechanisms. One mechanism involves the accumulation of single or double stranded DNA and RNA pools caused by loss-of-function mutations in genes involved in editing (ADAR1) and degradation (TREX1, RNASEH2A, −2B, −2C, SAMHD1) of nucleic acids [166]. Another mechanism is persistent nucleic acid sensing caused by gain-of-function mutations in cytosolic sensors MDA5 [167], STING [168], and RIG-I [169] that become hypersensitive to cytosolic nucleic ligands. Impaired regulation at the receptor level is a third mechanism. Loss-of-function mutations in USP18, an ISG and key negative regulator of type I IFN signaling, is also linked to type I interferonopathies [170].
The most recent addition to the list of genes linked to type I interferonopathies is STAT2 [171,172]. Patients with homozygous STAT2 mutations display hypersensitivity to type I IFN due to its inability to recruit USP18 to IFNAR2. Nonetheless, mutations in USP18 and STAT2 were fatal and underscore the non-redundant immunopathogenic and inflammatory nature of type I IFNs. Of note, STAT2 has been implicated in other inflammatory conditions that display dysregulated STAT2 activity such as psoriasis [173], and specific STAT2 polymorphisms have been associated with asthma susceptibility [174].
It is interesting that only a subset of patients with interferonopathies have clear overlap with a classic SLE autoimmunity mediated by T and B cells [175]. The evidence suggests that lupus autoimmunity requires much more than an exacerbated type I IFN response. For example, the importance of checkpoint defects that modify the threshold of tolerance of T and B lymphocytes is supported by data from murine models [176]. Alternatively, we can hypothesize that a moderate increase in type I IFN response, like the one existing in lupus, promotes autoimmunity, whereas the stronger response prevailing in interferonopathies may paralyze the adaptive immune response and cause defects in cell development and tissue damage rather than triggering autoimmunity.
Therapeutic effects of selective inhibitors of the type I IFN signaling pathway
SLE is a chronic disease with no cure. The current treatment protocols include steroidal and non-steroidal anti-inflammatory drugs, the antimalarial hydroxychloroquine and immunosuppressive drugs, like inhibitors of DNA replication cyclophosphamide and mycophenolate mofetil acid, or biologics interfering with B cell development [177]. The pathogenic role of type I IFNs in SLE prompted clinical trials testing their inhibition as new therapeutic strategy in SLE. After the disappointing results with blocking IFNα [178,179], in a recent phase 3 trial, administration of anifrolumab, a neutralizing antibody against IFNAR, has proven surprisingly safe and efficacious in SLE patients [180]. Inhibition of mediators of type I IFN signaling may confer the same or potentially enhance these effects. JAK1–3 inhibitors like baricitinib and tofacitinib have been shown to be effective when tested in mouse models of lupus and provided promising results in the first clinical trial in SLE patients [74,181,182]. Selective inhibitors of TYK2 have been recently proposed as an alternative treatment to specifically block the signaling downstream of type I IFNs without interfering with the function of other cytokines [182,183]. Future studies will very likely extend the list of therapeutic strategies able to block type I IFNs in SLE.
Conclusions.
Much has been learned about the involvement of type I IFNs and their signaling pathway in the pathogenesis of SLE. Family-based studies and GWAS have revealed genetic components influencing the IFN Signature. Additionally, EWAS are beginning to expose the rules of engagements of short-lived and possibly long-lasting effects of infections or pollutants on its regulation and on the immune system at large. Many outstanding questions remain, like the role of unphosphorylated STAT1 and STAT2 in supporting the threshold of activation of transcription in SLE, or whether a deficient suppressive function of STAT2 has a role in unleashing the IFN Signature in SLE. Current and future clinical trials will indicate whether non-selective JAK inhibitors rather than specific TYK2 inhibitors may be more beneficial in suppressing type I IFN responses in lupus and whether such inhibition will provide clinical benefits to people living with lupus.
ACKNOWLEDGMENTS
We thank the Temple Infections and Autoimmunity Interest Group for stimulating discussions.
FUNDING
This work was supported by the U.S, National Institutes of Health (NIH), NIAID grant R21-AI119947 and the Lupus Research Alliance, Innovative Grant (SG); Temple University Bridge Funds and partial support by R03 CA215929 to AMG.
List of abbreviations:
- ADAR1
Adenosine deaminases acting on RNA
- Anti-RBP
anti-RNA binding protein
- Anti-dsDNA
anti-double-stranded DNA
- cDC
Conventional Dendritic Cells
- ChIP
Chromatin immunoprecipitation
- DAMPs
Damage-associated molecular patterns
- DNA
deoxyribonucleic acid
- EWAS
Epigenome-Wide Association Studies
- GAS
gamma-activated sites
- GWAS
Genome Wide Association Study
- H3K4me3
Histone H3 lysine K4 (H3K4)
- IFN
Interferon
- IFNAR
Interferon-alpha/beta receptor
- IRF
Interferon Responsive Factor
- ISGs
Interferon stimulated genes
- ISGF3
Interferon-stimulated gene factor 3
- IL-12
Interleukin 12
- IL-23
Interleukin 23
- ISRE
IFN-stimulated response elements
- JAK
Janus Kinase
- LncRNA
Long non-coding RNA
- LN
Lupus Nephritis
- MAPK
Mitogen-activated protein kinase
- MDA5
Melanoma differentiation-associated protein 5
- mRNA
Messenger ribonucleic acid
- NF-kB
Nuclear Factor Kappa B
- PAMPs
Pathogen-associated molecular patterns
- PBMCs
Peripheral blood mononuclear cells
- PRRs
Pattern recognition receptors
- RIG-I
retinoic acid inducible gene I
- RLRs
RIG-I-like receptors
- RNA
Ribonucleic Acid
- SAMHD1
Sterile Alpha Motif (SAM) domain- Histidine- Aspartic (HD) domain-containing protein 1
- SNP
Single Nucleotide Polymorphism
- SOCS1
Suppressor of cytokine signaling 1
- STAT
Signal transducer and activator of transcription
- STING
Stimulator of interferon genes
- SLE
Systemic Lupus Erythematosus
- TLRs
Toll-like receptors
- TREX1
Three prime repair exonuclease
- TYK2
Tyrosine Kinase 2
- USP18
Ubiquitin specific peptidase 18
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS
COMPETING INTERESTS
The authors declare that they have no competing interests.
Ethics approval and consent to participate. Human and animal studies.
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Tsokos GC, Lo MS, Costa Reis P, Sullivan KE: New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 2016, 12:716–730. [DOI] [PubMed] [Google Scholar]
- 2.Fava A, Petri M: Systemic lupus erythematosus: Diagnosis and clinical management. J Autoimmun 2019, 96:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiu CC, Caricchio R, Gallucci S: Triggers of Autoimmunity: The Role of Bacterial Infections in the Extracellular Exposure of Lupus Nuclear Autoantigens. Front Immunol 2019, 10:2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivashkiv LB, Donlin LT: Regulation of type I interferon responses. Nat Rev Immunol 2014, 14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau J, Pascual V: Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006, 25:383–392. [DOI] [PubMed] [Google Scholar]
- 6.Elkon KB, Stone VV: Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res 2011, 31:803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isaacs A, Lindenmann J: Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 1957, 147:258–267. [PubMed] [Google Scholar]
- 8.Gallucci S, Lolkema M, Matzinger P: Natural adjuvants: endogenous activators of dendritic cells. Nat Med 1999, 5:1249–1255. [DOI] [PubMed] [Google Scholar]
- 9.Finkelman FD, Svetic A, Gresser I, Snapper C, Holmes J, Trotta PP, Katona IM, Gause WC: Regulation by interferon alpha of immunoglobulin isotype selection and lymphokine production in mice. J Exp Med 1991, 174:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF: Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14:461–470. [DOI] [PubMed] [Google Scholar]
- 11.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF: Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 2003, 4:1009–1015. [DOI] [PubMed] [Google Scholar]
- 12.Li S, Gong M, Zhao F, Shao J, Xie Y, Zhang Y, Chang H: Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cellular Physiology and Biochemistry 2018, 51:2377–2396. [DOI] [PubMed] [Google Scholar]
- 13.Lazear HM, Schoggins JW, Diamond MS: Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50:907–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hertzog PJ, Williams BR: Fine tuning type I interferon responses. Cytokine Growth Factor Rev 2013, 24:217–225. [DOI] [PubMed] [Google Scholar]
- 15.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL: Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med 1979, 301:5–8. [DOI] [PubMed] [Google Scholar]
- 16.Ytterberg SR, Schnitzer TJ: Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum 1982, 25:401–406. [DOI] [PubMed] [Google Scholar]
- 17.Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, Utset TO, Jolly M, James JA, Harley JB, et al. : Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum 2011, 63:1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crow MK, Kirou KA, Wohlgemuth J: Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 2003, 36:481–490. [DOI] [PubMed] [Google Scholar]
- 19.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003, 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al. : Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 2003, 100:2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baechler EC, Gregersen PK, Behrens TW: The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol 2004, 16:801–807. [DOI] [PubMed] [Google Scholar]
- 22.Ronnblom L, Eloranta ML, Alm GV: The type I interferon system in systemic lupus erythematosus. Arthritis Rheum 2006, 54:408–420. [DOI] [PubMed] [Google Scholar]
- 23.Chiche L, Jourde-Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, Anguiano E, Quinn C, Burtey S, Berland Y, et al. : Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol 2014, 66:1583–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oke V, Gunnarsson I, Dorschner J, Eketjall S, Zickert A, Niewold TB, Svenungsson E: High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther 2019, 21:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thanarajasingam U, Muppirala AN, Jensen MA, Ghodke-Puranik Y, Dorschner JM, Vsetecka DM, Amin S, Makol A, Ernste F, Osborn T, et al. : Type I Interferon Predicts an Alternate Immune System Phenotype in Systemic Lupus Erythematosus. ACR Open Rheumatol 2019, 1:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catalina MD, Bachali P, Geraci NS, Grammer AC, Lipsky PE: Gene expression analysis delineates the potential roles of multiple interferons in systemic lupus erythematosus. Communications Biology 2019, 2:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao WH, Shu DH, Zhen Y, Hilliard B, Priest SO, Cesaroni M, Ting JP, Cohen PL: Prion-like Aggregation of Mitochondrial Antiviral Signaling Protein in Lupus Patients Is Associated With Increased Levels of Type I Interferon. Arthritis Rheumatol 2016, 68:2697–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, McGlasson SL, Alyanakian MA, Bader-Meunier B, Barnerias C, et al. : Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med 2017, 214:1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjork A, Richardsdotter Andersson E, Imgenberg-Kreuz J, Thorlacius GE, Mofors J, Syvanen AC, Kvarnstrom M, Nordmark G, Wahren-Herlenius M: Protein and DNA methylation-based scores as surrogate markers for interferon system activation in patients with primary Sjogren’s syndrome. RMD Open 2020, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK: High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 2007, 8:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sriram U, Varghese L, Bennett HL, Jog NR, Shivers DK, Ning Y, Behrens EM, Caricchio R, Gallucci S: Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J Immunol 2012, 189:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ronnblom L, Leonard D: Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med 2019, 6:e000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L, Morris DL, Vyse TJ: Genetic advances in systemic lupus erythematosus: an update. Curr Opin Rheumatol 2017, 29:423–433. [DOI] [PubMed] [Google Scholar]
- 34.Goulielmos GN, Zervou MI, Vazgiourakis VM, Ghodke-Puranik Y, Garyfallos A, Niewold TB: The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene 2018, 668:59–72. [DOI] [PubMed] [Google Scholar]
- 35.Jog NR, Young KA, Munroe ME, Harmon MT, Guthridge JM, Kelly JA, Kamen DL, Gilkeson GS, Weisman MH, Karp DR, et al. : Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann Rheum Dis 2019, 78:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, Buttaro B, Caricchio R, Gallucci S, Tukel C: Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity 2015, 42:1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tursi SA, Lee EY, Medeiros NJ, Lee MH, Nicastro LK, Buttaro B, Gallucci S, Wilson RP, Wong GCL, Tukel C: Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog 2017, 13:e1006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, Caricchio R, Buyon JP, Alekseyenko AV, Silverman GJ: Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis 2019, 78:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, Costa FRC, Tiniakou E, Greiling T, Ruff W, et al. : Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mu Q, Tavella VJ, Kirby JL, Cecere TE, Chung M, Lee J, Li S, Ahmed SA, Eden K, Allen IC, et al. : Antibiotics ameliorate lupus-like symptoms in mice. Sci Rep 2017, 7:13675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao WH, Cohen PL: Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther 2011, 13:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fenton K: The effect of cell death in the initiation of lupus nephritis. Clin Exp Immunol 2015, 179:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L: Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum 2004, 50:1861–1872. [DOI] [PubMed] [Google Scholar]
- 44.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL: Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 2005, 202:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shrivastav M, Niewold TB: Nucleic Acid sensors and type I interferon production in systemic lupus erythematosus. Front Immunol 2013, 4:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ: Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016, 22:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao L, Bird AK, Meednu N, Dauenhauer K, Liesveld J, Anolik J, Looney RJ: Bone Marrow-Derived Mesenchymal Stem Cells From Patients With Systemic Lupus Erythematosus Have a Senescence-Associated Secretory Phenotype Mediated by a Mitochondrial Antiviral Signaling Protein-Interferon-beta Feedback Loop. Arthritis Rheumatol 2017, 69:1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, Baum R, Kawasumi M, Green R, Gale M Jr., et al. : The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci Rep 2020, 10:7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magna M, Pisetsky DS: The Alarmin Properties of DNA and DNA-associated Nuclear Proteins. Clin Ther 2016, 38:1029–1041. [DOI] [PubMed] [Google Scholar]
- 50.Reizis B: Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilliet M, Cao W, Liu YJ: Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 2008, 8:594–606. [DOI] [PubMed] [Google Scholar]
- 52.Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, Colonna M: Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med 2014, 211:1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davison LM, Jorgensen TN: Sialic acid-binding immunoglobulin-type lectin H-positive plasmacytoid dendritic cells drive spontaneous lupus-like disease development in B6.Nba2 mice. Arthritis Rheumatol 2015, 67:1012–1022. [DOI] [PubMed] [Google Scholar]
- 54.Davison LM, Jorgensen TN: New Treatments for Systemic Lupus Erythematosus on the Horizon: Targeting Plasmacytoid Dendritic Cells to Inhibit Cytokine Production. J Clin Cell Immunol 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Molony RD, Nguyen JT, Kong Y, Montgomery RR, Shaw AC, Iwasaki A: Aging impairs both primary and secondary RIG-I signaling for interferon induction in human monocytes. Sci Signal 2017, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han S, Zhuang H, Lee PY, Li M, Yang L, Nigrovic PA, Reeves WH: Differential Responsiveness of Monocyte and Macrophage Subsets to Interferon. Arthritis Rheumatol 2020, 72:100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee MH, Chakhtoura M, Sriram U, Caricchio R, Gallucci S: Conventional DCs from Male and Female Lupus-Prone B6.NZM Sle1/Sle2/Sle3 Mice Express an IFN Signature and Have a Higher Immunometabolism That Are Enhanced by Estrogen. J Immunol Res 2018, 2018:1601079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. : Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011, 3:73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, et al. : Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 2011, 3:73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barrera-Vargas A, Gomez-Martin D, Carmona-Rivera C, Merayo-Chalico J, Torres-Ruiz J, Manna Z, Hasni S, Alcocer-Varela J, Kaplan MJ: Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann Rheum Dis 2018, 77:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindau D, Mussard J, Rabsteyn A, Ribon M, Kotter I, Igney A, Adema GJ, Boissier MC, Rammensee HG, Decker P: TLR9 independent interferon alpha production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann Rheum Dis 2014, 73:2199–2207. [DOI] [PubMed] [Google Scholar]
- 62.Lopez de Padilla CM, Niewold TB: The type I interferons: Basic concepts and clinical relevance in immune-mediated inflammatory diseases. Gene 2016, 576:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levy DE, Darnell JE: STATs: transcriptional control and biological impact. Nature Reviews Molecular Cell Biology 2002, 3:651–662. [DOI] [PubMed] [Google Scholar]
- 64.Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O’Shea JJ: The Janus kinases (Jaks). Genome Biol 2004, 5:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen Y-L, Chmielewski S, Kostyrko K, Wesoly J, Balint Balint L, Lee C-K, et al. : STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochemical Journal 2015, 466:511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bluyssen HAR, Levy DE: Stat2 Is a Transcriptional Activator That Requires Sequence-specific Contacts Provided by Stat1 and p48 for Stable Interaction with DNA. Journal of Biological Chemistry 1997, 272:4600–4605. [DOI] [PubMed] [Google Scholar]
- 67.Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC: Distinct STAT Structure Promotes Interaction of STAT2 with the p48 Subunit of the Interferon-α-stimulated Transcription Factor ISGF3. Journal of Biological Chemistry 1997, 272:20070–20076. [DOI] [PubMed] [Google Scholar]
- 68.Testoni B, Voellenke C, Guerrieri F, Gerbal-Chaloin S, Blandino G, Levrero M: Chromatin dynamics of gene activation and repression in response to ifnα reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. Journal of Biological Chemistry 2011. [DOI] [PMC free article] [PubMed]
- 69.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, Rice CM, Jackson MW, Junk DJ, Stark GR: IFN[beta]-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J 2013, 32:2751–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu J, Lee MH, Chakhtoura M, Green BL, Kotredes KP, Chain RW, Sriram U, Gamero AM, Gallucci S: STAT2 Is Required for TLR-Induced Murine Dendritic Cell Activation and Cross-Presentation. J Immunol 2016, 197:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gough Daniel J, Messina Nicole L, Clarke Christopher JP, Johnstone Ricky W, Levy David E: Constitutive Type I Interferon Modulates Homeostatic Balance through Tonic Signaling. Immunity 2012, 36:166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, Gossenreiter T, Müller M, Novatchkova M, Decker T: A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nature Communications 2019, 10:2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Y, Higgs RE, Hoffman RW, Dow ER, Liu X, Petri M, Wallace DJ, Dorner T, Eastwood BJ, Miller BB, et al. : A Bayesian gene network reveals insight into the JAK-STAT pathway in systemic lupus erythematosus. PLoS One 2019, 14:e0225651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dorner T, Tanaka Y, Petri MA, Smolen JS, Wallace DJ, Dow ER, Higgs RE, Rocha G, Crowe B, Benschop RJ, et al. : Baricitinib-associated changes in global gene expression during a 24-week phase II clinical systemic lupus erythematosus trial implicates a mechanism of action through multiple immune-related pathways. Lupus Sci Med 2020, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, Chicoine A, Eisenhaure TM, Jonsson AH, Li S, et al. : The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol 2019, 20:902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsoi LC, Hile GA, Berthier CC, Sarkar MK, Reed TJ, Liu J, Uppala R, Patrick M, Raja K, Xing X, et al. : Hypersensitive IFN Responses in Lupus Keratinocytes Reveal Key Mechanistic Determinants in Cutaneous Lupus. J Immunol 2019, 202:2121–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scott E, Dooley MA, Vilen BJ, Clarke SH: Immune cells and type 1 IFN in urine of SLE patients correlate with immunopathology in the kidney. Clin Immunol 2016, 168:16–24. [DOI] [PubMed] [Google Scholar]
- 78.Ramírez-Vélez G, Medina F, Ramírez-Montaño L, Zarazua A, Hernández R, Llorente L, Moreno J: Constitutive Phosphorylation of Interferon Receptor A-Associated Signaling Proteins in Systemic Lupus Erythematosus. PloS one 2012, 7:e41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aue A, Szelinski F, Weissenberg SY, Wiedemann A, Rose T, Lino AC, Dorner T: Elevated STAT1 expression but not phosphorylation in lupus B cells correlates with disease activity and increased plasmablast susceptibility. Rheumatology (Oxford) 2020. [DOI] [PubMed]
- 80.Jin Z, Fan W, Jensen MA, Dorschner JM, Bonadurer GF 3rd, Vsetecka DM, Amin S, Makol A, Ernste F, Osborn T, et al. : Single-cell gene expression patterns in lupus monocytes independently indicate disease activity, interferon and therapy. Lupus Sci Med 2017, 4:e000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, et al. : VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366:1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gallo PM, Gallucci S: The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol 2013, 4:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gallucci S, Maffei ME: DNA Sensing across the Tree of Life. Trends Immunol 2017, 38:719–732. [DOI] [PubMed] [Google Scholar]
- 84.Matta B, Barnes BJ: Coordination between innate immune cells, type I IFNs and IRF5 drives SLE pathogenesis. Cytokine 2019:154731. [DOI] [PubMed]
- 85.Pisetsky DS: The central role of nucleic acids in the pathogenesis of systemic lupus erythematosus. F1000Res 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Celhar T, Fairhurst AM: Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Front Pharmacol 2014, 5:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mustelin T, Ukadike KC: How Retroviruses and Retrotransposons in Our Genome May Contribute to Autoimmunity in Rheumatological Conditions. Front Immunol 2020, 11:593891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pachucki RJ, Corradetti C, Kohler L, Ghadiali J, Gallo PM, Nicastro L, Tursi SA, Gallucci S, Tukel C, Caricchio R: Persistent Bacteriuria and Antibodies Recognizing Curli/eDNA Complexes From Escherichia coli Are Linked to Flares in Systemic Lupus Erythematosus. Arthritis Rheumatol 2020, 72:1872–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, Cepika AM, Acs P, Turner J, Anguiano E, et al. : Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hagberg N, Lundtoft C, Ronnblom L: Immunogenetics in Systemic Lupus Erythematosus: Transitioning from Genetic Associations to Cellular Effects. Scand J Immunol 2020:e12894. [DOI] [PubMed]
- 91.Banchereau R, Cepika AM, Banchereau J, Pascual V: Understanding Human Autoimmunity and Autoinflammation Through Transcriptomics. Annu Rev Immunol 2017, 35:337–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.International Consortium for Systemic Lupus Erythematosus G, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, et al. : Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008, 40:204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF, Argentine, et al. : A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 2006, 38:550–555. [DOI] [PubMed] [Google Scholar]
- 94.Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV, et al. : Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 2008, 358:900–909. [DOI] [PubMed] [Google Scholar]
- 95.Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, Burtt NP, Guiducci C, Parkin M, Gates C, et al. : Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 2008, 40:1059–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. : The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019, 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, Martin J, Fairfax BP, Knight JC, Chen L, et al. : Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 2015, 47:1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gorji AE, Roudbari Z, Alizadeh A, Sadeghi B: Investigation of systemic lupus erythematosus (SLE) with integrating transcriptomics and genome wide association information. Gene 2019, 706:181–187. [DOI] [PubMed] [Google Scholar]
- 99.Harley IT, Niewold TB, Stormont RM, Kaufman KM, Glenn SB, Franek BS, Kelly JA, Kilpatrick JR, Hutchings D, Divers J, et al. : The role of genetic variation near interferon-kappa in systemic lupus erythematosus. J Biomed Biotechnol 2010, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yin Q, Wu LC, Zheng L, Han MY, Hu LY, Zhao PP, Bai WY, Zhu XW, Xia JW, Wang XB, et al. : Comprehensive assessment of the association between genes on JAK-STAT pathway (IFIH1, TYK2, IL-10) and systemic lupus erythematosus: a meta-analysis. Arch Dermatol Res 2018, 310:711–728. [DOI] [PubMed] [Google Scholar]
- 101.Tang L, Wan P, Wang Y, Pan J, Wang Y, Chen B: Genetic association and interaction between the IRF5 and TYK2 genes and systemic lupus erythematosus in the Han Chinese population. Inflamm Res 2015, 64:817–824. [DOI] [PubMed] [Google Scholar]
- 102.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, et al. : A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 2009, 41:1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mohan C, Alas E, Morel L, Yang P, Wakeland EK: Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest 1998, 101:1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li J, Liu Y, Xie C, Zhu J, Kreska D, Morel L, Mohan C: Deficiency of type I interferon contributes to Sle2-associated component lupus phenotypes. Arthritis Rheum 2005, 52:3063–3072. [DOI] [PubMed] [Google Scholar]
- 105.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN: Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 2003, 197:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Santiago-Raber ML, Baudino L, Izui S: Emerging roles of TLR7 and TLR9 in murine SLE. J Autoimmun 2009, 33:231–238. [DOI] [PubMed] [Google Scholar]
- 107.Braun D, Geraldes P, Demengeot J: Type I Interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun 2003, 20:15–25. [DOI] [PubMed] [Google Scholar]
- 108.Jorgensen TN, Roper E, Thurman JM, Marrack P, Kotzin BL: Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 x NZW)F(1) mice. Genes Immun 2007, 8:653–662. [DOI] [PubMed] [Google Scholar]
- 109.Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, et al. : Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol 2009, 183:6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, Sobel E, Kuroda Y, Akaogi J, Satoh M, et al. : Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane). Am J Pathol 2006, 168:1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, Jonsen A, Rantapaa-Dahlqvist S, Moller B, Kere J, et al. : Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet 2005, 76:528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee YH, Bae SC: Association between TYK2 polymorphisms and susceptibility to autoimmune rheumatic diseases: a meta-analysis. Lupus 2016, 25:1307–1314. [DOI] [PubMed] [Google Scholar]
- 113.Gorman JA, Hundhausen C, Kinsman M, Arkatkar T, Allenspach EJ, Clough C, West SE, Thomas K, Eken A, Khim S, et al. : The TYK2-P1104A Autoimmune Protective Variant Limits Coordinate Signals Required to Generate Specialized T Cell Subsets. Front Immunol 2019, 10:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liao Z, Ye Z, Xue Z, Wu L, Ouyang Y, Yao C, Cui C, Xu N, Ma J, Hou G, et al. : Identification of Renal Long Non-coding RNA RP11–2B6.2 as a Positive Regulator of Type I Interferon Signaling Pathway in Lupus Nephritis. Front Immunol 2019, 10:975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sandling JK, Garnier S, Sigurdsson S, Wang C, Nordmark G, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jonsen A, et al. : A candidate gene study of the type I interferon pathway implicates IKBKE and IL8 as risk loci for SLE. Eur J Hum Genet 2011, 19:479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taylor KE, Remmers EF, Lee AT, Ortmann WA, Plenge RM, Tian C, Chung SA, Nititham J, Hom G, Kao AH, et al. : Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet 2008, 4:e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bolin K, Sandling JK, Zickert A, Jonsen A, Sjowall C, Svenungsson E, Bengtsson AA, Eloranta ML, Ronnblom L, Syvanen AC, et al. : Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One 2013, 8:e84450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kawasaki A, Ito I, Hikami K, Ohashi J, Hayashi T, Goto D, Matsumoto I, Ito S, Tsutsumi A, Koga M, et al. : Role of STAT4 polymorphisms in systemic lupus erythematosus in a Japanese population: a case-control association study of the STAT1-STAT4 region. Arthritis Res Ther 2008, 10:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bing PF, Xia W, Wang L, Zhang YH, Lei SF, Deng FY: Common Marker Genes Identified from Various Sample Types for Systemic Lupus Erythematosus. PLoS One 2016, 11:e0156234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xue Z, Cui C, Liao Z, Xia S, Zhang P, Qin J, Guo Q, Chen S, Fu Q, Yin Z, et al. : Identification of LncRNA Linc00513 Containing Lupus-Associated Genetic Variants as a Novel Regulator of Interferon Signaling Pathway. Front Immunol 2018, 9:2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tsao HW, Tai TS, Tseng W, Chang HH, Grenningloh R, Miaw SC, Ho IC: Ets-1 facilitates nuclear entry of NFAT proteins and their recruitment to the IL-2 promoter. Proc Natl Acad Sci U S A 2013, 110:15776–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, Xu JH, Cai ZM, Huang W, Zhao GP, et al. : Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 2009, 41:1234–1237. [DOI] [PubMed] [Google Scholar]
- 123.Zhong H, Li XL, Li M, Hao LX, Chen RW, Xiang K, Qi XB, Ma RZ, Su B: Replicated associations of TNFAIP3, TNIP1 and ETS1 with systemic lupus erythematosus in a southwestern Chinese population. Arthritis Res Ther 2011, 13:R186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang D, John SA, Clements JL, Percy DH, Barton KP, Garrett-Sinha LA: Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol 2005, 17:1179–1191. [DOI] [PubMed] [Google Scholar]
- 125.Lu X, Zoller EE, Weirauch MT, Wu Z, Namjou B, Williams AH, Ziegler JT, Comeau ME, Marion MC, Glenn SB, et al. : Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. Am J Hum Genet 2015, 96:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chodisetti SB, Fike AJ, Domeier PP, Singh H, Choi NM, Corradetti C, Kawasawa YI, Cooper TK, Caricchio R, Rahman ZSM: Type II but Not Type I IFN Signaling Is Indispensable for TLR7-Promoted Development of Autoreactive B Cells and Systemic Autoimmunity. J Immunol 2020, 204:796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chodisetti SB, Fike AJ, Domeier PP, Schell SL, Mockus TE, Choi NM, Corradetti C, Hou B, Atkins HM, Caricchio R, et al. : Serine Phosphorylation of the STAT1 Transactivation Domain Promotes Autoreactive B Cell and Systemic Autoimmunity Development. J Immunol 2020, 204:2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, Biron CA: Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 2002, 297:2063–2066. [DOI] [PubMed] [Google Scholar]
- 129.Hagberg N, Joelsson M, Leonard D, Reid S, Eloranta ML, Mo J, Nilsson MK, Syvanen AC, Bryceson YT, Ronnblom L: The STAT4 SLE risk allele rs7574865[T] is associated with increased IL-12-induced IFN-gamma production in T cells from patients with SLE. Ann Rheum Dis 2018, 77:1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hagberg N, Ronnblom L: Interferon-alpha enhances the IL-12-induced STAT4 activation selectively in carriers of the STAT4 SLE risk allele rs7574865[T]. Ann Rheum Dis 2019, 78:429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK: Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum 2008, 58:2481–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Negishi H, Taniguchi T, Yanai H: The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb Perspect Biol 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sanchez E, Gonzalez-Gay MA, Callejas-Rubio JL, Ortego-Centeno N, Sabio JM, Jimenez-Alonso J, Mico L, Suarez A, Gutierrez C, de Ramon E, et al. : No evidence for genetic association of interferon regulatory factor 3 in systemic lupus erythematosus. Lupus 2009, 18:230–234. [DOI] [PubMed] [Google Scholar]
- 134.Santana-de Anda K, Gomez-Martin D, Monsivais-Urenda AE, Salgado-Bustamante M, Gonzalez-Amaro R, Alcocer-Varela J: Interferon regulatory factor 3 as key element of the interferon signature in plasmacytoid dendritic cells from systemic lupus erythematosus patients: novel genetic associations in the Mexican mestizo population. Clin Exp Immunol 2014, 178:428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Akahoshi M, Nakashima H, Sadanaga A, Miyake K, Obara K, Tamari M, Hirota T, Matsuda A, Shirakawa T: Promoter polymorphisms in the IRF3 gene confer protection against systemic lupus erythematosus. Lupus 2008, 17:568–574. [DOI] [PubMed] [Google Scholar]
- 136.Li D, Matta B, Song S, Nelson V, Diggins K, Simpfendorfer KR, Gregersen PK, Linsley P, Barnes BJ: IRF5 genetic risk variants drive myeloid-specific IRF5 hyperactivation and presymptomatic SLE. JCI Insight 2020, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chung SA, Taylor KE, Graham RR, Nititham J, Lee AT, Ortmann WA, Jacob CO, Alarcon-Riquelme ME, Tsao BP, Harley JB, et al. : Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genet 2011, 7:e1001323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ballestar E, Esteller M, Richardson BC: The epigenetic face of systemic lupus erythematosus. J Immunol 2006, 176:7143–7147. [DOI] [PubMed] [Google Scholar]
- 139.Barrat FJ, Crow MK, Ivashkiv LB: Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol 2019, 20:1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q: Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J Autoimmun 2013, 41:92–99. [DOI] [PubMed] [Google Scholar]
- 141.Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, Merrill JT, McCune WJ, Sawalha AH: Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun 2013, 43:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Imgenberg-Kreuz J, Carlsson Almlof J, Leonard D, Alexsson A, Nordmark G, Eloranta ML, Rantapaa-Dahlqvist S, Bengtsson AA, Jonsen A, Padyukov L, et al. : DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis 2018, 77:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, Chatham WW, Kimberly RP: Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet 2013, 9:e1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Imgenberg-Kreuz J, Almlof JC, Leonard D, Sjowall C, Syvanen AC, Ronnblom L, Sandling JK, Nordmark G: Shared and Unique Patterns of DNA Methylation in Systemic Lupus Erythematosus and Primary Sjogren’s Syndrome. Front Immunol 2019, 10:1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lanata CM, Chung SA, Criswell LA: DNA methylation 101: what is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci Med 2018, 5:e000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang Z, Shi L, Song L, Maurer K, Petri MA, Sullivan KE: Overall Downregulation of mRNAs and Enrichment of H3K4me3 Change Near Genome-Wide Association Study Signals in Systemic Lupus Erythematosus: Cell-Specific Effects. Front Immunol 2018, 9:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Patel ZH, Lu X, Miller D, Forney CR, Lee J, Lynch A, Schroeder C, Parks L, Magnusen AF, Chen X, et al. : A plausibly causal functional lupus-associated risk variant in the STAT1-STAT4 locus. Hum Mol Genet 2018, 27:2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mistry P, Nakabo S, O’Neil L, Goel RR, Jiang K, Carmona-Rivera C, Gupta S, Chan DW, Carlucci PM, Wang X, et al. : Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A 2019, 116:25222–25228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carbone CJ, Zheng H, Bhattacharya S, Lewis JR, Reiter AM, Henthorn P, Zhang Z-Y, Baker DP, Ukkiramapandian R, Bence KK, et al. : Protein tyrosine phosphatase 1B is a key regulator of IFNAR1 endocytosis and a target for antiviral therapies. Proceedings of the National Academy of Sciences 2012, 109:19226–19231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Piganis RAR, de Weerd NA, Gould JA, Schindler CW, Mansell A, Nicholson SE, Hertzog PJ: Suppressor of cytokine signaling (SOCS)1 inhibits type I interferon (IFN) signaling via the IFNAR1 associated tyrosine kinase, Tyk2. Journal of Biological Chemistry 2011. [DOI] [PMC free article] [PubMed]
- 151.Malakhova OA, Kim K II, Luo J-K, Zou W, Kumar KGS, Fuchs SY, Shuai K, Zhang D-E: UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J 2006, 25:2358–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Arimoto K-i, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, Wilmes S, Fan J-B, Heinisch JJ, Li Z, et al. : STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nature Structural & Molecular Biology 2017, 24:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. : Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nature Genetics 2003, 33:388–391. [DOI] [PubMed] [Google Scholar]
- 154.Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, Holland SM, Schreiber RD, Casanova J-L: Impairment of Mycobacterial But Not Viral Immunity by a Germline Human STAT1 Mutation. Science 2001, 293:300–303. [DOI] [PubMed] [Google Scholar]
- 155.Eren Akarcan S, Ulusoy Severcan E, Edeer Karaca N, Isik E, Aksu G, Migaud M, Evin Gurkan F, Azarsiz E, Puel A, Casanova J-L, et al. : Gain-of-Function Mutations in STAT1: A Recently Defined Cause for Chronic Mucocutaneous Candidiasis Disease Mimicking Combined Immunodeficiencies. Case reports in immunology 2017, 2017:2846928–2846928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, Shirkani A, Changi-Ashtiani M, Rokni-Zadeh H, Sayar EH, et al. : Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. Journal of Experimental Medicine 2019, 216:2057–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Duncan CJA, Mohamad SMB, Young DF, Skelton AJ, Leahy TR, Munday DC, Butler KM, Morfopoulou S, Brown JR, Hubank M, et al. : Human IFNAR2 deficiency: Lessons for antiviral immunity. Science Translational Medicine 2015, 7:307ra154–307ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, Curtis J, Gaspar M, Nowak K, Daza-Cajigal V, et al. : Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nature Communications 2016, 7:13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kreins AY, Ciancanelli MJ, Okada S, Kong X-F, Ramírez-Alejo N, Kilic SS, El Baghdadi J, Nonoyama S, Mahdaviani SA, Ailal F, et al. : Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. Journal of Experimental Medicine 2015, 212:1641–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bravo García-Morato M, Calvo Apalategi A, Bravo-Gallego LY, Blázquez Moreno A, Simón-Fuentes M, Garmendia JV, Méndez Echevarría A, del Rosal Rabes T, Domínguez-Soto Á, López-Granados E, et al. : Impaired control of multiple viral infections in a family with complete IRF9 deficiency. Journal of Allergy and Clinical Immunology 2019, 144:309–312.e310. [DOI] [PubMed] [Google Scholar]
- 161.Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SMB, Valappil M, McGovern N, Cant AJ, Hackett SJ, Ghazal P, et al. : STAT2 deficiency and susceptibility to viral illness in humans. Proceedings of the National Academy of Sciences 2013. [DOI] [PMC free article] [PubMed]
- 162.Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, Matthys P, Neyts J, Ciancanelli M, Zhang S-Y, et al. : A novel kindred with inherited STAT2 deficiency and severe viral illness. Journal of Allergy and Clinical Immunology 2017. [DOI] [PubMed]
- 163.Rodero MP, Crow YJ: Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. The Journal of experimental medicine 2016, 213:2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Davidson S, Steiner A, Harapas CR, Masters SL: An Update on Autoinflammatory Diseases: Interferonopathies. Current Rheumatology Reports 2018, 20:38. [DOI] [PubMed] [Google Scholar]
- 165.Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg M-C, Goudin N, Frémond M-L, Nitschke P, Molina TJ, et al. : Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. The Journal of Clinical Investigation 2014, 124:5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Crow YJ, Manel N: Aicardi–Goutières syndrome and the type I interferonopathies. Nature Reviews Immunology 2015, 15:429–440. [DOI] [PubMed] [Google Scholar]
- 167.Rice GI, del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. : Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nature Genetics 2014, 46:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. : Activated STING in a Vascular and Pulmonary Syndrome. New England Journal of Medicine 2014, 371:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jang M-A, Kim Eun K, Now H, Nguyen Nhung TH, Kim W-J, Yoo J-Y, Lee J, Jeong Y-M, Kim C-H, Kim O-H, et al. : Mutations in DDX58, which Encodes RIG-I, Cause Atypical Singleton-Merten Syndrome. The American Journal of Human Genetics 2015, 96:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Meuwissen MEC, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, et al. : Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. Journal of Experimental Medicine 2016, 213:1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, Rosain J, Buta S, Bousfiha A, Casanova JL, et al. : Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med 2020, 217. [DOI] [PMC free article] [PubMed]
- 172.Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, Lovell SC, Shuttleworth VG, Brocklebank V, Corner B, et al. : Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Johansen C, Rittig AH, Mose M, Bertelsen T, Weimar I, Nielsen J, Andersen T, Rasmussen TK, Deleuran B, Iversen L: STAT2 is involved in the pathogenesis of psoriasis by promoting CXCL11 and CCL5 production by keratinocytes. PloS one 2017, 12:e0176994–e0176994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hsieh Y-Y, Wan L, Chang C-C, Tsai C-H, Tsai F-J: STAT2*C related genotypes and allele but not TLR4 and CD40 gene polymorphisms are associated with higher susceptibility for asthma. International journal of biological sciences 2009, 5:74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I: New Horizons in the Genetic Etiology of Systemic Lupus Erythematosus and Lupus-Like Disease: Monogenic Lupus and Beyond. J Clin Med 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S: IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J Immunol 2005, 174:2499–2506. [DOI] [PubMed] [Google Scholar]
- 177.Huang W, Quach TD, Dascalu C, Liu Z, Leung T, Byrne-Steele M, Pan W, Yang Q, Han J, Lesser M, et al. : Belimumab promotes negative selection of activated autoreactive B cells in systemic lupus erythematosus patients. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, Drappa J, Wang L, Greth W, investigators CDs: Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis 2016, 75:1909–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, Davis JC Jr., Kennedy WP: A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis 2016, 75:196–202. [DOI] [PubMed] [Google Scholar]
- 180.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, et al. : Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med 2020, 382:211–221. [DOI] [PubMed] [Google Scholar]
- 181.Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, Dorner T, Cardiel MH, Bruce IN, Gomez E, et al. : Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392:222–231. [DOI] [PubMed] [Google Scholar]
- 182.Alunno A, Padjen I, Fanouriakis A, Boumpas DT: Pathogenic and Therapeutic Relevance of JAK/STAT Signaling in Systemic Lupus Erythematosus: Integration of Distinct Inflammatory Pathways and the Prospect of Their Inhibition with an Oral Agent. Cells 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.He X, Chen X, Zhang H, Xie T, Ye XY: Selective Tyk2 inhibitors as potential therapeutic agents: a patent review (2015–2018). Expert Opin Ther Pat 2019, 29:137–149. [DOI] [PubMed] [Google Scholar]