

ABSTRACT
β-Catenin (Ctnnb1) supports high levels of liver gene expression in hepatocytes in proximity to the central vein functionally defining zone 3 of the liver lobule. This region of the liver lobule supports the highest levels of viral biosynthesis in wild-type hepatitis B virus (HBV) transgenic mice. Liver-specific β-catenin-null HBV transgenic mice exhibit a stark loss of high levels of pericentral viral biosynthesis. Additionally, viral replication that does not depend directly on β-catenin activity appears to expand to include hepatocytes of zone 1 of the liver lobule in proximity to the portal vein, a region of the liver that typically lacks significant HBV biosynthesis in wild-type HBV transgenic mice. While the average amount of viral RNA transcripts does not change, viral DNA replication is reduced approximately 3-fold. Together, these observations demonstrate that β-catenin signaling represents a major determinant of HBV biosynthesis governing the magnitude and distribution of viral replication across the liver lobule in vivo. Additionally, these findings reveal a novel mechanism for the regulation of HBV biosynthesis that is potentially relevant to the expression of additional liver-specific genes.
IMPORTANCE Viral biosynthesis is highest around the central vein in the hepatitis B virus (HBV) transgenic mouse model of chronic infection. The associated HBV biosynthetic gradient across the liver lobule is primarily dependent upon β-catenin. In the absence of β-catenin, the gradient of viral gene expression spanning the liver lobule is absent, and HBV replication is reduced. Therefore, therapeutically manipulating β-catenin activity in the livers of chronic HBV carriers may reduce circulating infectious virions without greatly modulating viral protein production. Together, these changes in viral biosynthesis might limit infection of additional hepatocytes while permitting immunological clearance of previously infected cells, potentially limiting disease persistence.
KEYWORDS: β-catenin, hepatitis B virus (HBV), liver lobule, transgenic mice, viral biosynthesis
INTRODUCTION
Hepatitis B virus (HBV) infection is a major worldwide health problem lacking reliable curative therapies (1–4). Expanding our understanding of the in vivo regulation of HBV biosynthesis may aid in the development of new treatment options, reducing the morbidity and mortality associated with this viral infection. HBV genomic DNA replication occurs via the reverse transcription of the viral 3.5-kb pregenomic RNA within the viral capsid (HBcAg) (5, 6). Liver-enriched nuclear receptors, including hepatocyte nuclear factor 4-α (HNF4α), liver receptor homolog 1 (LRH1), retinoic acid receptor-α (RXRα), peroxisome proliferator-activated receptor-α (PPARα), and farnesoid X receptor-α (FXRα) (7–11), and their associated coactivators, such as CBP, p300, steroid receptor coactivator 1 to 3 (SRC1 to SRC3), and peroxisome proliferator-activated receptor coactivator-α/β (PGC1α/β) (12, 13), direct robust HBV 3.5-kb pregenomic RNA synthesis and hence viral replication. In the transgenic mouse model of chronic HBV infection, viral biosynthesis is highest in the hepatocytes adjacent to the central vein within zone 3 of the liver lobule (14). Genes with this pattern of zonal expression across the liver lobule are often regulated by the transcriptional coactivator β-catenin (Ctnnb1), which is activated by Wnt ligands synthesized by the endothelial cells of the central vein (15). Therefore, the role of β-catenin in the distribution of viral biosynthesis across the liver lobule was investigated in the HBV transgenic mouse model of chronic infection.
β-catenin is a transcriptional coactivator that regulates the activity of a number of transcription factors in a Wnt ligand-dependent manner (16, 17). In the absence of Wnt ligand, β-catenin is constitutively bound by a cytoplasmic destruction complex (which includes the adenomatous polyposis coli, Axin, glycogen synthase kinase 3β, casein kinase 1, and protein phosphatase 2a proteins [18]), leading to its proteolytic degradation (16, 17). When Wnt ligand is present, the destruction complex disassociates, freeing β-catenin to translocate to the nucleus to act as a transcriptional coactivator (16, 17). During canonical Wnt/β-catenin signaling, β-catenin interacts with the transcription factor T-cell factor/lymphocyte enhancer-binding factor (Tcf/Lef), which is primarily associated with cellular proliferation (16, 17, 19, 20). However, it is also known to coactivate transcription by interacting with several other transcription factors, including LRH1 and forkhead box O1 (FOXO1) (16, 21, 22), which are reported activators of HBV transcription (10, 23).
These observations suggested that β-catenin might regulate HBV biosynthesis in the liver and contribute to the high level of viral transcription and replication observed in the hepatocytes adjacent to the central vein. This possibility was investigated by determining if β-catenin can enhance HBV transcription in cell culture and by characterizing viral biosynthesis in liver-specific β-catenin-null HBV transgenic mice. β-catenin was able to enhance transcription from the HBV core promoter as determined by reporter gene analysis. β-catenin was also essential for robust viral biosynthesis around the central vein in HBV transgenic mice. Additionally, in the absence of β-catenin expression, intermediate levels of HBV biosynthesis extended to the hepatocytes closer to the portal tract in zone 1 of the liver lobule. Therefore, in the presence of β-catenin, there is a gradient of HBV biosynthesis across the liver lobule with the highest level of expression localized to the central vein-proximal hepatocytes, but this gradient is lost in the absence of β-catenin, leading to a more homogenous distribution of viral biosynthesis across the liver lobule. Collectively, these observations indicate that β-catenin regulates the magnitude and distribution of HBV biosynthesis across the liver lobule, probably involving both direct and indirect mechanisms.
RESULTS
The transcriptional coactivator β-catenin has been shown to regulate the expression of multiple genes across the liver lobule, with direct targets of β-catenin activation displaying the highest levels of expression in zone 3 of the liver lobule adjacent to the central vein (15–17). As this pattern of liver lobule zonal gene expression is similar to the observed distribution of HBV biosynthesis in the HBV transgenic mouse model of chronic infection, it was of interest to examine the potential role of β-catenin in the regulation of viral transcription and replication in vivo.
Effects of β-catenin on the transcriptional activity of the four HBV promoters.
To determine if β-catenin regulates transcription from the HBV promoters, a dual-luciferase reporter assay was performed using HEK293T cells. Expression vectors for a constitutively active β-catenin (S33Y) mutant coactivator (24) and the liver receptor homolog 1 (Lrh1) (25) were cotransfected with firefly luciferase reporter gene constructs for each of the four HBV promoters. As previously demonstrated (10, 26–28), Lrh1 enhanced transcription from the HBV nucleocapsid (core) promoter (Cp) approximately 2.7-fold (Fig. 1A). β-catenin enhanced transcription from the HBV core promoter approximately 1.5-fold (Fig. 1A), whereas coexpression of Lrh1 and β-catenin enhanced HBV core promoter activity about 8.5-fold, suggesting that they might act in a synergistic manner (Fig. 1A). Lrh1 and β-catenin also enhanced transcription from the large surface antigen promoter approximately 2-fold when expressed individually and 5.2-fold when coexpressed (Fig. 1C). These observations indicate that β-catenin enhances the transcription of the HBV 3.5-kb pregenomic RNA from the core promoter and thus likely increases subsequent viral biosynthesis. Notably, neither β-catenin nor Lrh1 enhanced transcription from the HBV X gene or the major surface antigen promoters (Fig. 1B and D).
FIG 1.

Effect of β-catenin on transcriptional activation of the HBV promoters. HEK293T cells were transfected with a constitutively active β-catenin and/or an Lrh1 expression vector with a firefly luciferase (LUC) reporter gene construct driven by one of the four HBV promoters, CpLUC (A), SpLUC (B), PS(1)pLUC (C), and XpLUC (D), respectively. A CMV-driven Renilla luciferase reporter gene construct (pRL-CMV) was used as an internal control for transfection efficiency. Means and standard deviations from two replicates are shown. Abbreviations: Cp, nucleocapsid (core) promoter; Sp, major surface antigen promoter; PS(1)p, large surface antigen promoter; Xp, X-gene promoter.
Effects of β-catenin deletion on liver-specific gene expression.
To determine whether β-catenin enhances viral transcription and subsequent replication in vivo, constitutive liver-specific β-catenin-null HBV transgenic mice (HBV+/−Ctnnb1flox/floxAlbCre+/−) were generated and characterized, where Cre recombinase is expressed under the albumin (Alb) promoter. As noted previously (29), the only gross phenotype observed was that both male and female liver-specific β-catenin-null mice exhibited an approximately 20% decrease in liver size relative to body weight compared with β-catenin wild-type mice (Cre-negative males, 4.7% ± 0.4% [n = 23] versus Cre-positive males, 3.8% ± 0.5% [n = 28], P = 1.78E−9, Wilcoxon rank sum; Cre-negative females, 4.5% ± 0.4% [n = 14] versus Cre-positive females 3.5% ± 0.5% [n = 13], P = 2.71E−5, Wilcoxon rank sum).
β-catenin signaling was evaluated by NanoString gene expression analysis of liver RNA from wild-type and constitutive liver-specific β-catenin-null HBV transgenic mice. β-catenin-null mice, deleted for exons 2 to 6, showed a 4- to 5-fold decrease in Ctnnb1 transcripts (Fig. 2A). Residual β-catenin transcripts are likely derived from nonparenchymal cells, including Kupffer, biliary epithelial, stellate, and endothelial cells. Expression of β-catenin target genes, including Glul, Cyp2e1, Axin2, Oat, Slc1a2, Lhpp, and Lect2 (29–31), were significantly decreased between 3.3- and 29.7-fold in the β-catenin-null HBV transgenic mice (Fig. 2A). These observations indicate that β-catenin signaling is largely abrogated in the hepatocytes of these mice.
FIG 2.

Gene expression levels in control and liver-specific β-catenin-null HBV transgenic mice. (A) NanoString gene expression analysis of β-catenin RNA and β-catenin target gene transcripts from total liver RNA. (B) Representative immunohistochemical analysis of liver samples from HBV transgenic mice. Comparison of control (HBV+/−Ctnnb1flox/floxAlbCre−/−) and constitutive liver-specific β-catenin-null HBV transgenic mice (HBV+/−Ctnnb1flox/floxAlbCre+/−). Immunohistochemical staining indicates the presence of glutamate-ammonia ligase (Glul)/glutamine synthetase (GS) (c, central vein; p, portal vein). (C) NanoString gene expression analysis of cytokine gene transcripts from total liver RNA. Means and standard deviations are presented (n = 9 per group). Statistically significant differences between groups determined by Wilcoxon rank sum test are indicated; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Glutamate-ammonia ligase (Glul)/glutamine synthetase (GS) transcription is regulated by β-catenin signaling in the liver (29, 30) and is highly expressed around the central vein within zone 3 of the liver lobule. Therefore, immunohistochemical staining of Glul is a reliable indicator of active β-catenin signaling (29). Loss of β-catenin expression in liver tissues from constitutive liver-specific β-catenin-null HBV transgenic mice results in essentially the complete loss of Glul staining (Fig. 2B), indicating efficient Cre recombinase deletion of the β-catenin gene in hepatocytes with associated loss of transcriptional coactivator function.
The expression levels of genes encoding the cytokines tumor necrosis factor-α (Tnf), interferon-γ (Ifng), interleukin-6 (Il6), transforming growth factor-β2 (Tgfb2), and Tgfb3 did not statistically increase with the loss of β-catenin expression (Fig. 2C), indicating that it is unlikely that these cytokines would contribute any modulatory effect on HBV biosynthesis in these mice (32, 33). Oas2 transcription is very modestly increased (Fig. 2C), but the levels expressed in these mice are not high enough to have any appreciable effect on HBV biosynthesis (34, 35). Tgfb1 transcription is also very modestly increased, but this cytokine is not known to affect HBV biosynthesis in vivo (Fig. 2C) (36).
β-catenin regulates HBV biosynthesis across the liver lobule.
HBV replication occurs inside the viral capsid in the cytoplasm of hepatocytes (5, 6). High levels of viral biosynthesis around the central vein are associated with both nuclear and cytoplasmic HBcAg staining in wild-type β-catenin-expressing HBV transgenic mice (HBV+/−Ctnnb1flox/floxAlbCre−/−) (Fig. 3). These mice also predominantly display nuclear HBcAg staining in zone 2 (midzone) of the liver lobule and lack major HBcAg staining in zone 1 of the liver lobule adjacent to the periportal tract. This pattern of HBcAg staining correlates with the gradient of HBV biosynthesis across the liver lobule (14).
FIG 3.

HBcAg immunohistochemical analysis of livers from constitutive liver-specific β-catenin-null HBV transgenic mice. Comparison of control (HBV+/−Ctnnb1flox/floxAlbCre−/−) and liver-specific β-catenin-null HBV transgenic mice (HBV+/−Ctnnb1flox/floxAlbCre+/−). Representative liver lobules are expanded below each section with central vein on the left and portal vein on the right. Immunohistochemical staining indicates the presence of HBcAg.
In both male and female constitutive liver-specific β-catenin-null mice (HBV+/−Ctnnb1flox/floxAlbCre+/−), HBcAg staining around the central vein of the liver lobule (zone 3) is drastically decreased (Fig. 3). This indicates that β-catenin signaling is required for high-level viral replication around the central vein, where Wnt signaling and associated β-catenin activity drive high levels of target gene expression (15). Notably, viral replication is not eliminated, as HBV biosynthesis in the midzone of the liver lobule (zone 2) appears largely unaffected by the loss of β-catenin activity. However, in the livers of mice lacking β-catenin expression, modest levels of HBcAg staining appear to persist across the majority of the liver lobule. This suggests that β-catenin signaling is required for high-level HBV biosynthesis in pericentral hepatocytes but is dispensable for additional mechanism(s) that govern lower-level viral biosynthesis across the liver lobule.
Gene deletion mediated by the Cre recombinase expressed under the albumin promoter in these mice occurs during late embryonic development (37). Consequently, the observed phenotype in the constitutively deleted liver-specific β-catenin-null HBV transgenic mice could result from the lack of β-catenin signaling during neonatal and postnatal liver development. To test this possibility, inducible liver-specific β-catenin-null HBV transgenic mice (HBV+/−Ctnnb1flox/floxSA-CRE-ERT2+/−) were generated. In this model, the Cre recombinase is still driven by the albumin promoter, but its nuclear enzymatic activity is tamoxifen dependent, permitting the restricted elimination of β-catenin activity in the adult liver following tamoxifen treatment. Control mice (HBV+/−Ctnnb1flox/floxSA-CRE-ERT2−/−) with and without tamoxifen treatment plus HBV+/−Ctnnb1flox/floxSA-CRE-ERT2+/− without tamoxifen treatment exhibit HBcAg staining patterns indistinguishable from those of HBV+/−Ctnnb1flox/floxAlbCre−/− mice (Fig. 4). Similarly, inducible liver-specific β-catenin-null HBV transgenic mice (HBV+/−Ctnnb1flox/floxSA-CRE-ERT2+/− with tamoxifen treatment as adults) exhibit HBcAg staining patterns indistinguishable from constitutively deleted mice (HBV+/−Ctnnb1flox/floxAlbCre+/−) (Fig. 4). This indicates that the loss of high levels of HBV biosynthesis around the central vein of the liver lobule in β-catenin-null mice is not dependent on the developmental timing of β-catenin deletion. Additionally, the change in the distribution of viral replication within the liver lobule occurs within a few days after the loss of β-catenin activity in these adult mice (Fig. 4).
FIG 4.

HBcAg immunohistochemical analysis of livers from inducible liver-specific β-catenin-null HBV transgenic mice. Comparison of control (HBV+/−Ctnnb1flox/floxSA-Cre-ERT2−/− with and without tamoxifen treatment plus HBV+/−Ctnnb1flox/floxSA-Cre-ERT2+/− without tamoxifen treatment) and inducible liver-specific β-catenin-null HBV transgenic mice (HBV+/−Ctnnb1flox/floxSA-Cre-ERT2+/− with tamoxifen treatment). Mice were either tamoxifen or vehicle treated as indicated. Representative liver lobules are expanded below each section with central vein on the left and portal vein on the right. Immunohistochemical staining indicates the presence of HBcAg.
Unexpectedly, both constitutive and inducible β-catenin-null mice appear to exhibit increased HBcAg staining immediately surrounding the portal vein of the liver lobule compared with β-catenin-expressing mice, as the moderate levels of staining typically observed in zone 2 are also observed in zone 1 of β-catenin-null mice (Fig. 3 and 4). This observation suggests that loss of β-catenin signaling results in an expansion of the distribution of β-catenin-independent viral biosynthesis. To demonstrate the difference in the distribution of HBcAg staining between wild-type and β-catenin-null mice, the staining intensity was measured across liver lobules. In wild-type mice (Fig. 5A and B), the proportion of HBcAg staining across the liver lobule is heavily weighted toward the central vein (bins 1 to 3). In contrast, the proportion of lobular staining is more evenly distributed in both constitutive (Fig. 5A) and inducible (Fig. 5B) β-catenin-null mice. Thus, in addition to driving high-level biosynthesis around the central vein, β-catenin activity is responsible for the establishment and maintenance of the gradient of HBV biosynthesis observed across the liver lobule, which can be rapidly altered in the adult liver following loss of β-catenin activity.
FIG 5.
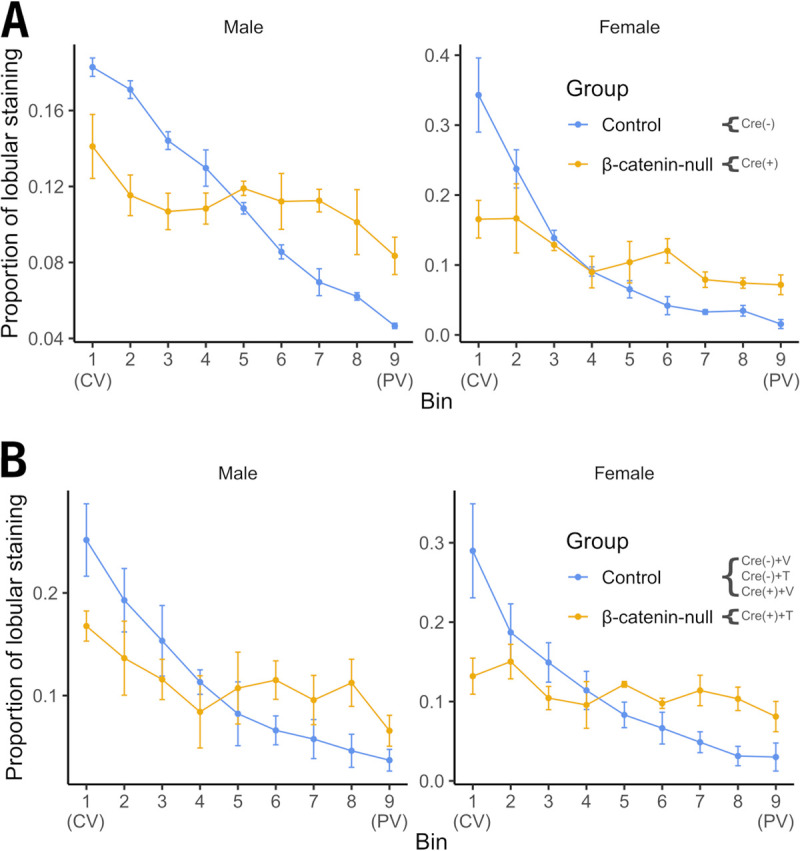
Quantitation of immunohistochemical analysis of livers from wild-type plus constitutive and inducible liver-specific β-catenin-null HBV transgenic mice. The intensity of HBcAg staining spanning 5 to 10 liver lobules from each mouse was measured and proportionally distributed into 9 bins, where bins 1 and 9 were central vein (CV) and portal vein (PV) proximal, respectively. The means and standard deviations of staining intensity are displayed. (A) Control and constitutive β-catenin-null mice: 2 male and 2 female control [Cre(−)] HBV transgenic mice; 3 male and 3 female constitutive β-catenin-null [Cre(+)] HBV transgenic mice. (B) Control and inducible β-catenin-null mice: 7 male and 7 female control HBV transgenic mice comprising 2 Cre− with vehicle treatment [Cre(−)+V], 2 Cre− with tamoxifen treatment [Cre(−)+T], and 3 Cre+ with vehicle treatment [Cre(+)+V]; 3 male and 3 female inducible β-catenin-null HBV transgenic mice {Cre+ with tamoxifen treatment [Cre(+)+T]}.
Effect of β-catenin deletion on HBV transcription.
The expression of the pregenomic HBV 3.5-kb RNA in total liver RNA from constitutive liver-specific β-catenin-null HBV transgenic mice was evaluated by NanoString gene expression analysis. Interestingly, the level of HBV 3.5-kb transcripts decreased only modestly (10 to 30%) in the liver-specific β-catenin knockout mice (Fig. 6A). This phenotype was recapitulated by RNA filter hybridization analysis (Northern blotting) (Fig. 6B and C). The potential reduction in HBV transcription is consistent with the partial loss of pericentral HBcAg expression. Additionally, the expansion of viral biosynthesis to periportal hepatocytes suggests an increase in viral transcription in these cells, which may be partially compensating for the loss of viral transcription around the central vein. A combination of both mechanisms may be contributing to the altered distribution of HBV biosynthesis across the liver lobule resulting from the loss of β-catenin activity.
FIG 6.

Analysis of HBV RNA expression in liver-specific β-catenin-null HBV transgenic mice. (A) Counts of HBV 3.5-kb transcripts in total liver RNA from control (Cre−) and constitutive liver-specific β-catenin-null HBV transgenic mice (Cre+) by NanoString gene expression analysis (n = 9 per group). (B) RNA filter hybridization analysis on total liver RNA from control (Cre−) and constitutive liver-specific β-catenin-null HBV transgenic mice (Cre+). The glyceraldehyde 3-phosphate dehydrogenase (Gapdh) transcript was used as an internal control for the quantitation of the HBV 3.5-kb RNA. Noncontiguous lanes from a single analysis are presented. (C) Quantitation of viral 3.5-kb RNA from the RNA filter hybridization analysis. Means and standard deviations are presented (male Cre−, n = 23; male Cre+, n = 28; female Cre−, n = 14; female Cre+, n = 13). Statistically significant differences between HBV transgenic mice by Wilcoxon rank sum test are indicated; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Effect of β-catenin deletion on HBV DNA replication.
The biosynthesis of HBV DNA replication intermediates was evaluated by DNA filter hybridization analysis (Southern). In contrast to the marginal decrease in viral RNA, viral DNA replication intermediates were decreased approximately 3-fold in both male and female constitutive liver-specific β-catenin-null HBV transgenic mice (Fig. 7A and B). This suggests that the decrease in pericentral HBcAg staining (Fig. 3 and 5) is associated with a decrease in viral replication, although overall HBV RNA abundance is not greatly affected. These observations suggest that the levels of HBV pregenomic RNA in individual hepatocytes ultimately determines the total level of viral DNA synthesis observed. Specifically, HBV pregenomic RNA abundance on a per cell basis must exceed a critical concentration to permit core polypeptide synthesis levels high enough to support viral capsid formation and hence for replication to occur (12). By altering the shape of the viral transcript gradient across the liver lobule rather than the total amount of RNA synthesized, the loss of β-catenin activity appears to have reduced the fraction of hepatocytes expressing the necessary level of HBV pregenomic RNA (encoding the core polypeptide) to support its encapsidation, reverse transcription, and DNA replication. Consequently, limited alterations in viral transcript levels are associated with a significant reduction in HBV replication in the absence of β-catenin activity in vivo.
FIG 7.
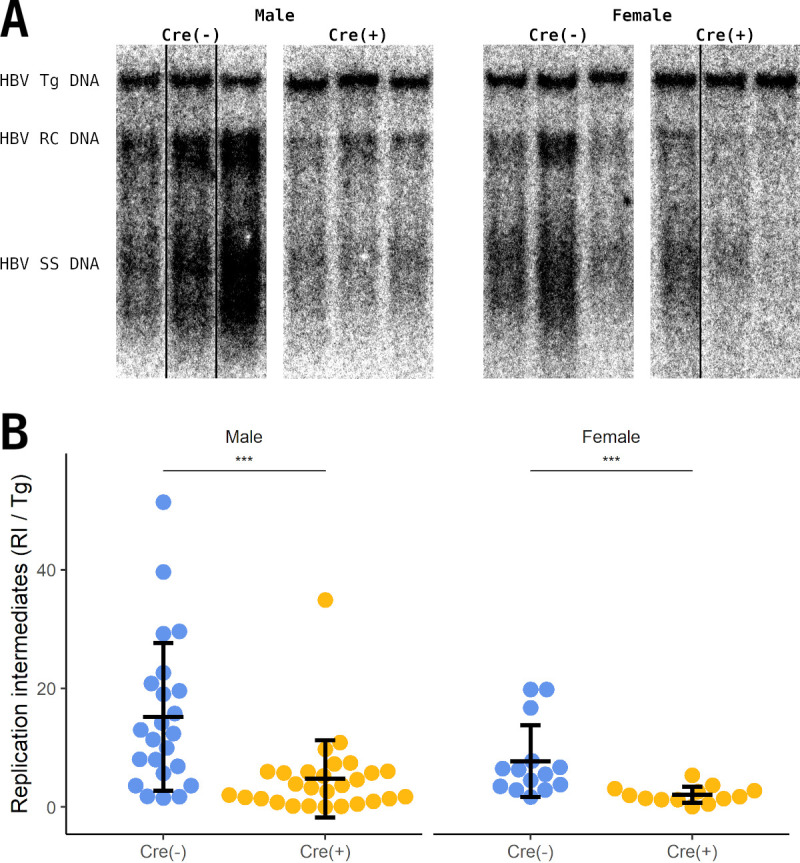
Analysis of viral DNA replication intermediates of liver-specific β-catenin-null HBV transgenic mice. (A) DNA filter hybridization analysis on total liver DNA from control (Cre−) and constitutive liver-specific β-catenin-null HBV transgenic mice (Cre+). The HBV transgene was used as an internal control for the quantitation of the HBV replication intermediates. Abbreviations: Tg, transgene; RC, relaxed circular replication intermediates; SS, single-stranded replication intermediates. Noncontiguous lanes from a single analysis are presented. (B) Quantitation of viral DNA replication intermediates from the DNA filter hybridization analysis. Means and standard deviations are presented (male Cre−, n = 23; male Cre+, n = 28; female Cre−, n = 14; female Cre+, n = 13). Statistically significant differences between HBV transgenic mice by Wilcoxon rank sum test are indicated; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
DISCUSSION
HBV is a major worldwide health problem lacking curative therapies (1–4). While it is known that liver-enriched nuclear receptors play a major role in regulating the transcription of HBV genes (7–11), there is growing evidence indicating that specific coactivators also play a critical role in regulating viral biosynthesis (12, 13). The HBV transgenic model of chronic HBV infection presents an opportunity to investigate the regulation of viral biosynthesis in vivo. Based on the observation that high-level HBV biosynthesis colocalizes with β-catenin signaling activity around the central vein of the liver lobule in these mice, the role of β-catenin in regulating HBV biosynthesis was investigated using liver-specific β-catenin-null HBV transgenic mice.
It was determined that β-catenin can upregulate the transcription of the HBV core promoter, which drives the expression of the 3.5-kb RNA (Fig. 1). Regardless of whether the β-catenin gene is deleted in late embryonic development or in adult mice, β-catenin signaling activity and robust viral biosynthesis around the central vein of the liver lobule are lost (Fig. 2B, 3, and 4). However, the dramatic loss of pericentral HBcAg is associated with only a very modest reduction in total HBV 3.5-kb RNA (Fig. 6). This suggests that either a small decrease in β-catenin-mediated HBV transcription in the hepatocytes surrounding the central vein is responsible for the major decrease in core polypeptide synthesis or a larger decrease in β-catenin-mediated HBV transcription in pericentral hepatocytes is partially compensated for by an increase in viral gene expression in midzone and periportal hepatocytes. Immunohistochemical analysis of wild-type and β-catenin-null livers (Fig. 3–5) and the demonstrated ability for β-catenin to regulate HBV transcription (Fig. 1) suggest the latter is more probable, although both mechanisms could potentially play a role in contributing to the observed distributions of viral biosynthesis (Fig. 8). It seems less likely that the loss of β-catenin signaling alters the regulation of capsid assembly and/or DNA synthesis, although this possibility has not been specifically excluded.
FIG 8.

Model for the role of β-catenin in regulating HBV biosynthesis in HBV transgenic mice. HBV replication occurs within the assembling capsids in the cytoplasm of hepatocytes. Thus, cytoplasmic staining of HBcAg serves as a readout of the lobular distribution of viral replication (14). β-Catenin activity directs robust HBV transcription immediately proximal to the central vein via endothelial cell-derived Wnt signaling (15). This poorly diffusible signal rapidly diminishes across the liver lobule (15). Lower levels of viral replication present in midzone and periportal zone 1 hepatocytes appear to be supported by viral transcription that is independent of direct β-catenin activity. In the absence of β-catenin activity, robust HBV biosynthesis is lost from pericentral zone 3 hepatocytes with compensating viral transcription and hence enhanced capsid biosynthesis expanding to include periportal zone 1 hepatocytes.
A possible explanation for the elimination of high-level capsid synthesis and associated HBV replication around the central vein is the loss of viral RNA synthesis mediated by β-catenin coactivation of transcription factors known to regulate HBV core promoter activity. Specifically, Lrh1 and FoxO1 are DNA-binding proteins known to activate HBV RNA synthesis and additionally can utilize β-catenin as a transcriptional coactivator (10, 16, 21–23). Reporter gene analysis establishes that β-catenin can enhance HBV core promoter activity (Fig. 1). It seems likely that the loss of β-catenin activity adjacent to the central vein directly results in decreased viral transcription, as observed for a number of cellular genes (Fig. 2A) (29), and consequently HBV replication is also reduced in pericentral hepatocytes (Fig. 8).
In contrast, the remaining transcription and replication observed across the liver lobule must be independent of β-catenin activity. Furthermore, the dramatic loss of capsid biosynthesis in the pericentral hepatocytes but minimal change in HBV RNA levels in liver-specific β-catenin-null mice (Fig. 3–6) suggests that there must be a compensatory increase in viral transcription in some or all hepatocytes across the liver lobule. Evidence for this compensatory increase in viral transcription is most apparent in the periportal region of the liver lobule where β-catenin-null mice display cytoplasmic accumulation of viral core particles while wild-type mice typically do not (Fig. 3–5 and 8). Interestingly, sera and hepatic bile acid levels are significantly increased in liver-specific β-catenin-null mice (38, 39). Bile acids are ligands for the farnesoid X receptor (Fxr) (25, 40–42), a known activator of HBV transcription (10, 43). Consequently, increased Fxr activity in β-catenin-null mice caused by elevated bile acid levels may be increasing viral transcription, particularly around the portal vein where viral biosynthesis is minimal in wild-type HBV transgenic mice (14). This increase in transcription may partially compensate for the loss of β-catenin-dependent transcription around the central vein, potentially explaining the limited change of HBV pregenomic 3.5-kb RNA expression (Fig. 6).
Although Fxr-mediated HBV transcription might partially compensate for the restricted loss of β-catenin-activated viral transcription in pericentral hepatocytes, it did not restore the gradient of HBV biosynthesis across the liver lobule or total HBV replication (Fig. 7). Indeed, wild-type HBV transgenic mice display high levels of viral biosynthesis in hepatocytes located in zone 3 of the liver lobule around the central vein and negligible levels of viral biosynthesis in hepatocytes located in zone 1 proximal to the portal vein (Fig. 3–5). In contrast, liver-specific β-catenin-null HBV transgenic mice display an approximately equivalent level of HBV biosynthesis across the liver lobule (Fig. 3–5 and 8). Given that HBV pregenomic 3.5-kb RNA must exceed a threshold level within an individual cell before capsid formation and HBV replication can occur (12), the differences in the levels of replication between wild-type and liver-specific β-catenin-null HBV transgenic mice can be explained by the difference in distribution of viral transcripts across the liver lobule. The high levels of viral RNA and DNA in pericentral hepatocytes of wild-type HBV transgenic mice are absent in β-catenin-null mice where the more even distribution of transcript levels across the liver lobule results in the absence of any hepatocytes displaying abundant viral replication. This leads to similar levels of HBV transcripts in these mice but lower overall levels of replication in β-catenin-null mice (Fig. 6 and 7). These observations suggest that therapeutically manipulating β-catenin activity in the liver of chronic HBV carriers might represent an approach to limiting viral replication and virion production while permitting viral polypeptide synthesis, which might serve as a target for immunological clearance with associated disease resolution.
MATERIALS AND METHODS
Cells, transfections, and dual-luciferase assay.
The human embryonic kidney cell line HEK293T was grown in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Transfections of 5 × 105 HEK293T cells were performed as previously described (44). The four HBV promoter firefly luciferase (LUC) plasmid constructs CpLUC, SpLUC, PS(1)pLUC, and XpLUC and the promoterless firefly luciferase control construct p19DLUC have been described previously (45, 46). The transfected DNA mixture consisted of 1 μg of a LUC plasmid, 0.1 μg of a constitutively active β-catenin expression vector, pBabe-β-cat-S33Y (24), and/or an Lrh1 expression vector, pCMX-mLRH1 (25), and 0.1 μg of a cytomegalovirus (CMV) immediate early promoter-driven Renilla luciferase expression vector (pRL-CMV, Promega), which served as an internal control for transfection efficiency. Firefly and Renilla luciferase signals were obtained using the dual-luciferase reporter assay (Promega) according to the manufacturer’s instructions 40 to 48 h after transfection.
Transgenic mice.
The production and characterization of the HBV transgenic mouse lineage 1.3.32 has been described (14). These HBV transgenic mice contain a single copy of a terminally redundant, 1.3 genome-length copy of the HBVayw genome integrated into the mouse chromosomal DNA. High levels of HBV replication occur in the livers of these mice (14, 47). The mice used in breeding were homozygous for the HBV transgene and were maintained on the SV129 genetic background (48). The production of the conditional β-catenin-null mice (B6.129-Ctnnb1tm2Kem/KnwJ; The Jackson Laboratory) has been described (49). These mice have loxP sites flanking exons 2 to 6 and were maintained on a mixed B6.129 background. The production of the constitutive (lineage B6.Cg-Tg[Alb-cre]21Mgn/J; Jackson Laboratory, designated AlbCre) and inducible (lineage Albtm1[cre/ERT2]Mtz; Mouse Genome Informatics, designated SA-CRE-ERT2) liver-specific albumin Cre transgenic mice has been described previously, and mice were maintained on the SV129 and C57BL/6 backgrounds, respectively (37, 50). Constitutive liver-specific β-catenin-null HBV transgenic mice were sacrificed at 4 to 12 weeks of age. For the inducible liver-specific β-catenin-null HBV transgenic mice, Cre recombinase was induced by three injections (intraperitoneally) of 50 mg/kg tamoxifen in corn oil on days 0, 2, and 4. Eight- to 12-week-old mice were sacrificed 14 days after the initial injection.
Mice were screened for the HBV transgene, β-catenin floxed allele, and Cre recombinase by PCR analysis of tail DNA. Tail DNA was prepared by incubating 0.5 cm of tail in 500 µl of 100 mM Tris hydrochloride (pH 8.0), 200 mM NaCl, 5 mM EDTA, and 0.2% (wt/vol) SDS containing 100 µg/ml proteinase K for 16 to 20 h at 55°C. Samples were centrifuged at 14,000 rpm in an Eppendorf 5417C microcentrifuge for 10 min, and the supernatant was precipitated with 500 µl of isopropanol. DNA was pelleted by centrifugation at 14,000 rpm in an Eppendorf 5417C microcentrifuge for 10 min and was subsequently dissolved in 100 µl of 5 mM Tris hydrochloride (pH 8.0) and 1 mM EDTA. The HBV transgene was identified by PCR analysis using the oligonucleotides 5′-TCGATACCTGAACCTTTACCCCGTTGCCCG-3′ (oligonucleotide XpHNF4-1; HBV coordinates 1,133 to 1,159) and 5′-TCGAATTGCTGAGAGTCCAAGAGTCCTCTT-3′ (oligonucleotide CpHNF4-2; HBV coordinates 1,683 to 1,658) and 1 µl of tail DNA. A PCR product of 551 bp indicated the presence of the HBV transgene. The β-catenin wild-type and floxed alleles were identified by PCR analysis using the oligonucleotides 5′-AAGGTAGAGTGATGAAAGTTGTT-3′ (oligonucleotide RM41) and 5′-CACCATGTCCTCTGTCTATTC-3′ (oligonucleotide RM42) (49) and 1 µl of tail DNA. A PCR product of 221 bp indicated the wild-type allele, whereas a PCR product of 324 bp indicated the floxed allele. The AlbCre and SA-CRE-ERT2 transgenes were identified by PCR analysis using the oligonucleotides 5′-CCAGCTAAACATGCTTCATCGTCG-3′ (oligonucleotide CRE-1) and 5′-ATTCTCCCACCGTCAGTACGTGAG-3′ (oligonucleotide CRE-2) and 1 µl of tail DNA. A PCR product of 300 bp indicated the presence of the Cre transgene. The samples were subjected to 42 amplification cycles involving denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and extension from the primers at 72°C for 2 min. The 20 µl reaction conditions were as described by the Taq 2× MeanGreen master mix (Empirical Biosciences).
HBV transgenic mice were fed normal rodent chow and water ad libitum. Mice were bled from the retroorbital plexus and sacrificed. Liver tissue was frozen in liquid nitrogen and stored at −80°C prior to DNA and RNA extraction.
Ethics statement.
All animal experiments were IACUC approved and performed according to institutional guidelines with University of Illinois at Chicago Institutional Biosafety and Animal Care Committee (ACC) approval (ACC number 19-190). All animal procedures were performed in the College of Medicine Research Building at the University of Illinois at Chicago and adhere to the policies of the NIH Office of Laboratory Animal Welfare, the standards of the Animal Welfare Act, the Public Health Service Policy, and the Guide for the Care and Use of Laboratory Animals.
Liver histology.
Liver tissue samples were fixed in sodium phosphate-buffered formalin (Fisher), embedded in paraffin, and sectioned (5 µm). Immunohistochemical detection of HBcAg in paraffin-embedded mouse liver sections was performed using a polyclonal rabbit anti-HBcAg primary antiserum (Dako, B0586). Glutamate-ammonia ligase (Glul)/glutamine synthetase (GS) was detected with a monoclonal mouse anti-GS primary antibody (BD Biosciences, 610517). Immunohistochemical slides were developed using the BOND polymer refine detection system (Leica Biosystems).
HBcAg staining was quantitated using ImageJ software. 3,3′-Diaminobenzidine (DAB) staining was deconvoluted from brightfield images using the “Color Deconvolution 2” plugin for ImageJ (51, 52). For each liver lobule, the DAB staining across the region spanning the central and portal veins was quantified and divided into 9 bins, and fractional staining per bin was determined.
HBV DNA and RNA analysis.
Total DNA and RNA were isolated from the livers of HBV transgenic mice as described (53, 54). DNA (Southern) filter hybridization analyses were performed using 20 µg of HindIII-digested total cellular DNA (54). Filters were probed with 32P-labeled HBVayw genomic DNA (55) to detect HBV sequences. RNA (Northern) filter hybridization analyses were performed using 20 µg of total cellular RNA as described (54). Filters were probed with 32P-labeled HBVayw genomic DNA to detect HBV sequences and mouse glyceraldehyde 3-phosphate dehydrogenase (Gapdh) cDNA to detect the Gapdh transcript used as an internal control (56). Filter hybridization analyses were quantified by phosphor imaging using a Packard Cyclone storage phosphor system. Storage phosphor screens were scanned using a Typhoon scanner (GE) and quantified using ImageJ.
Nanostring nCounter gene expression transcript counting was used to quantify the levels of Ctnnb1 (exon 3), Glul, Cyp2e1, Axin2, Oat, Slc1a2, Lhpp, Lect2, Tnf, Ifng, Il6, Oas2, Tgfb1, Tgfb2, Tgfb3, and HBV 3.5-kb transcript levels in 50 ng of mouse liver RNA using a specifically designed CodeSet. RNA was analyzed on a NanoString nCounter SPRINT profiler following the manufacturer’s instructions (NanoString Technologies, Seattle, WA, USA). Data were quality controlled and normalized using the nSolver analysis software 4.0 (NanoString Technologies) as recommended by the manufacturer. RNA expression levels were normalized to mouse Cnot1, Rps29, Slc9a8, Mrps5, and Scarb1 RNA controls.
Statistical analysis and plotting.
All statistical analyses and plotting were performed in R 4.0.5 (57) with additional packages “tidyverse” (58), “ggbeeswarm” (59), “ggsignif” (60), and “cowplot” (61).
ACKNOWLEDGMENTS
We thank Luca G. Guidotti and Francis V. Chisari (The Scripps Research Institute, La Jolla, CA) for providing the HBV transgenic mice. We are grateful to David Mangelsdorf (Southwestern Medical Center, Dallas, TX) for the plasmid pCMX-mLRH1 and Zhijian Qian (University of Florida, Gainesville, FL) for the plasmid pBabe-β-cat-S33Y. Histology services were provided by the Research Resources Center—Research Histology Core at the University of Illinois at Chicago established with the support of the Vice Chancellor of Research. We thank Peter Gann and Morgan Zenner (University of Illinois at Chicago, Chicago, IL) for technical support.
This work was supported by the National Institutes of Health grants R01 AI125401, R01 CA238328, and UL1TR002003.
Contributor Information
Alan McLachlan, Email: mclach@uic.edu.
J.-H. James Ou, University of Southern California.
REFERENCES
- 1.Dienstag JL. 2008. Hepatitis B virus infection. N Engl J Med 359:1486–1500. 10.1056/NEJMra0801644. [DOI] [PubMed] [Google Scholar]
- 2.Nishioka K, Levin AG, Simons MJ. 1975. Hepatitis B antigen, antigen subtypes, and hepatitis B antibody in normal subjects and patients with liver disease. Bull World Health Organ 52:293–300. [PMC free article] [PubMed] [Google Scholar]
- 3.Tiollais P, Pourcel C, Dejean A. 1985. The hepatitis B virus. Nature 317:489–495. 10.1038/317489a0. [DOI] [PubMed] [Google Scholar]
- 4.Moradpour D, Wands JR. 1995. Understanding hepatitis B virus infection. N Engl J Med 332:1092–1093. 10.1056/NEJM199504203321610. [DOI] [PubMed] [Google Scholar]
- 5.Will H, Reiser W, Weimer T, Pfaff E, Buscher M, Sprengel R, Cattaneo R, Schaller H. 1987. Replication strategy of human hepatitis B virus. J Virol 61:904–911. 10.1128/JVI.61.3.904-911.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 7.Raney AK, Johnson JL, Palmer CN, McLachlan A. 1997. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J Virol 71:1058–1071. 10.1128/JVI.71.2.1058-1071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guidotti LG, Eggers CM, Raney AK, Chi SY, Peters JM, Gonzalez FJ, McLachlan A. 1999. In vivo regulation of hepatitis B virus replication by peroxisome proliferators. J Virol 73:10377–10386. 10.1128/JVI.73.12.10377-10386.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang H, McLachlan A. 2001. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci U S A 98:1841–1846. 10.1073/pnas.041479698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reese V, Ondracek C, Rushing C, Li L, Oropeza CE, McLachlan A. 2011. Multiple nuclear receptors may regulate hepatitis B virus biosynthesis during development. Int J Biochem Cell Biol 43:230–237. 10.1016/j.biocel.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reese VC, Oropeza CE, McLachlan A. 2013. Independent activation of hepatitis B virus biosynthesis by retinoids, peroxisome proliferators, and bile acids. J Virol 87:991–997. 10.1128/JVI.01562-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shalaby RE, Iram S, Cakal B, Oropeza CE, McLachlan A. 2017. PGC1α transcriptional adaptor function governs hepatitis B virus replication by controlling HBcAg/p21 protein-mediated capsid formation. J Virol 91:e00790-17. 10.1128/JVI.00790-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shalaby RE, Iram S, Oropeza CE, McLachlan A. 2019. Peroxisome proliferator-activated receptor γ coactivator family members competitively regulate hepatitis b virus biosynthesis. Virology 526:214–221. 10.1016/j.virol.2018.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. 10.1128/JVI.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B, Zhao L, Fish M, Logan CY, Nusse R. 2015. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 524:180–185. 10.1038/nature14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valenta T, Hausmann G, Basler K. 2012. The many faces and functions of β-catenin. EMBO J 31:2714–2736. 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson MD, Monga SPS. 2007. WNT/β-catenin signaling in liver health and disease. Hepatology 45:1298–1305. 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- 18.Kimelman D, Xu W. 2006. β-Catenin destruction complex: insights and questions from a structural perspective. Oncogene 25:7482–7491. 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- 19.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. 1996. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 382:638–642. 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 20.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. 1996. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 86:391–399. 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 21.Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, Schoonjans K. 2004. Synergy between LRH-1 and β-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell 15:499–509. 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. 2005. Functional interaction between β-catenin and FOXO in oxidative stress signaling. Science 308:1181–1184. 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 23.Shlomai A, Shaul Y. 2009. The metabolic activator FOXO1 binds hepatitis B virus DNA and activates its transcription. Biochem Biophys Res Commun 381:544–548. 10.1016/j.bbrc.2009.02.078. [DOI] [PubMed] [Google Scholar]
- 24.Ming M, Wang S, Wu W, Senyuk V, Beau MML, Nucifora G, Qian Z. 2012. Activation of Wnt/β-catenin protein signaling induces mitochondria-mediated apoptosis in hematopoietic progenitor cells. J Biol Chem 287:22683–22690. 10.1074/jbc.M112.342089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515. 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 26.Li M, Xie YH, Kong YY, Wu X, Zhu L, Wang Y. 1998. Cloning and characterization of a novel human hepatocyte transcription factor, hB1F, which binds and activates enhancer II of hepatitis B virus. J Biol Chem 273:29022–29031. 10.1074/jbc.273.44.29022. [DOI] [PubMed] [Google Scholar]
- 27.Ishida H, Ueda K, Ohkawa K, Kanazawa Y, Hosui A, Nakanishi F, Mita E, Kasahara A, Sasaki Y, Hori M, Hayashi N. 2000. Identification of multiple transcription factors, HLF, FTF, and E4BP4, controlling hepatitis B virus enhancer II. J Virol 74:1241–1251. 10.1128/jvi.74.3.1241-1251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert S, Galarneau L, Lamontagne A, Roy S, Bélanger L. 2000. The hepatitis B virus core promoter is strongly activated by the liver nuclear receptor fetoprotein transcription factor or by ectopically expressed steroidogenic factor 1. J Virol 74:5032–5039. 10.1128/jvi.74.11.5032-5039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekine S, Lan BY-A, Bedolli M, Feng S, Hebrok M. 2006. Liver-specific loss of β-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43:817–825. 10.1002/hep.21131. [DOI] [PubMed] [Google Scholar]
- 30.Cadoret A, Ovejero C, Terris B, Souil E, Lévy L, Lamers WH, Kitajewski J, Kahn A, Perret C. 2002. New targets of β-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 21:8293–8301. 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 31.Loeppen S, Koehle C, Buchmann A, Schwarz M. 2005. A β-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis 26:239–248. 10.1093/carcin/bgh298. [DOI] [PubMed] [Google Scholar]
- 32.Guidotti LG, Borrow P, Hobbs MV, Matzke B, Gresser I, Oldstone MB, Chisari FV. 1996. Viral cross talk: intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc Natl Acad Sci U S A 93:4589–4594. 10.1073/pnas.93.10.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25–36. 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 34.Anderson AL, Banks KE, Pontoglio M, Yaniv M, McLachlan A. 2005. Alpha/beta interferon differentially modulates the clearance of cytoplasmic encapsidated replication intermediates and nuclear covalently closed circular hepatitis B virus (HBV) DNA from the livers of hepatocyte nuclear factor 1α-null HBV transgenic mice. J Virol 79:11045–11052. 10.1128/JVI.79.17.11045-11052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uprichard SL, Wieland SF, Althage A, Chisari FV. 2003. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc Natl Acad Sci U S A 100:1310–1315. 10.1073/pnas.252773599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guidotti LG, Guilhot S, Chisari FV. 1994. Interleukin-2 and alpha/beta interferon down-regulate hepatitis B virus gene expression in vivo by tumor necrosis factor-dependent and -independent pathways. J Virol 68:1265–1270. 10.1128/JVI.68.3.1265-1270.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274:305–315. 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 38.Behari J, Yeh T-H, Krauland L, Otruba W, Cieply B, Hauth B, Apte U, Wu T, Evans R, Monga SPS. 2010. Liver-specific β-catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet-induced steatohepatitis. Am J Pathol 176:744–753. 10.2353/ajpath.2010.090667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeh T-H, Krauland L, Singh V, Zou B, Devaraj P, Stolz DB, Franks J, Monga SPS, Sasatomi E, Behari J. 2010. Liver-specific β-catenin knockout mice have bile canalicular abnormalities, bile secretory defect, and intrahepatic cholestasis. Hepatology 52:1410–1419. 10.1002/hep.23801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526. 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 41.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. 1999. Identification of a nuclear receptor for bile acids. Science 284:1362–1365. 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 42.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. 1999. Bile acids: natural ligands for an orphan nuclear receptor. Science 284:1365–1368. 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 43.Reese VC, Moore DD, McLachlan A. 2012. Limited effects of bile acids and small heterodimer partner on hepatitis B virus biosynthesis in vivo. J Virol 86:2760–2768. 10.1128/JVI.06742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLachlan A, Milich DR, Raney AK, Riggs MG, Hughes JL, Sorge J, Chisari FV. 1987. Expression of hepatitis B virus surface and core antigens: influences of pre-S and precore sequences. J Virol 61:683–692. 10.1128/JVI.61.3.683-692.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Zonneveld AJ, Curriden SA, Loskutoff DJ. 1988. Type 1 plasminogen activator inhibitor gene: functional analysis and glucocorticoid regulation of its promoter. Proc Natl Acad Sci U S A 85:5525–5529. 10.1073/pnas.85.15.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raney AK, Milich DR, Easton AJ, McLachlan A. 1990. Differentiation-specific transcriptional regulation of the hepatitis B virus large surface antigen gene in human hepatoma cell lines. J Virol 64:2360–2368. 10.1128/JVI.64.5.2360-2368.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asabe S, Wieland SF, Chattopadhyay PK, Roederer M, Engle RE, Purcell RH, Chisari FV. 2009. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol 83:9652–9662. 10.1128/JVI.00867-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. 1995. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15:3012–3022. 10.1128/MCB.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. 2001. Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128:1253–1264. 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 50.Schuler M, Dierich A, Chambon P, Metzger D. 2004. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis 39:167–172. 10.1002/gene.20039. [DOI] [PubMed] [Google Scholar]
- 51.Landini G, Martinelli G, Piccinini F. 2021. Colour deconvolution: stain unmixing in histological imaging. Bioinformatics 37:1485–1487. 10.1093/bioinformatics/btaa847. [DOI] [PubMed] [Google Scholar]
- 52.Ruifrok AC, Johnston DA. 2001. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23:291–299. [PubMed] [Google Scholar]
- 53.Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159. 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 54.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 55.Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650. 10.1038/281646a0. [DOI] [PubMed] [Google Scholar]
- 56.Sabath DE, Broome HE, Prystowsky MB. 1990. Glyceraldehyde-3-phosphate dehydrogenase mRNA is a major interleukin 2-induced transcript in a cloned T-helper lymphocyte. Gene 91:185–191. 10.1016/0378-1119(90)90087-8. [DOI] [PubMed] [Google Scholar]
- 57.R Core Team. 2021. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 58.Wickham H, Averick M, Bryan J, Chang W, McGowan LDA, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen TL, Miller E, Bache SM, Müller K, Ooms J, Robinson D, Seidel DP, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H. 2019. Welcome to the tidyverse. J Open Source Softw 4:1686. 10.21105/joss.01686. [DOI] [Google Scholar]
- 59.Clarke E, Sherrill-Mix S. 2017. ggbeeswarm: categorical Scatter (Violin Point) Plots. R package version 0.6.0. https://CRAN.R-project.org/package=ggbeeswarm.
- 60.Ahlmann-Eltze C, Patil I. 2021. ggsignif: significance Brackets for 'ggplot2'. R package version 0.6.1. https://CRAN.R-project.org/package=ggsignif.
- 61.Wilke CO. 2020. cowplot: streamlined Plot Theme and Plot Annotations for 'ggplot2'. R package version 1.1.1. https://CRAN.R-project.org/package=cowplot.