

ABSTRACT
One approach to improve the utility of adeno-associated virus (AAV)-based gene therapy is to engineer the AAV capsid to (i) overcome poor transport through tissue barriers and (ii) redirect the broadly tropic AAV to disease-relevant cell types. Peptide- or protein-domain insertions into AAV surface loops can achieve both engineering goals by introducing a new interaction surface on the AAV capsid. However, we understand little about the impact of insertions on capsid structure and the extent to which engineered inserts depend on a specific capsid context to function. Here, we examine insert-capsid interactions for the engineered variant AAV9-PHP.B. The 7-amino-acid peptide insert in AAV9-PHP.B facilitates transport across the murine blood-brain barrier via binding to the receptor Ly6a. When transferred to AAV1, the engineered peptide does not bind Ly6a. Comparative structural analysis of AAV1-PHP.B and AAV9-PHP.B revealed that the inserted 7-amino-acid loop is highly flexible and has remarkably little impact on the surrounding capsid conformation. Our work demonstrates that Ly6a binding requires interactions with both the PHP.B peptide and specific residues from the AAV9 HVR VIII region. An AAV1-based vector that incorporates a larger region of AAV9-PHP.B—including the 7-amino-acid loop and adjacent HVR VIII amino acids—can bind to Ly6a and localize to brain tissue. However, unlike AAV9-PHP.B, this AAV1-based vector does not penetrate the blood-brain barrier. Here we discuss the implications for AAV capsid engineering and the transfer of engineered activities between serotypes.
IMPORTANCE Targeting AAV vectors to specific cellular receptors is a promising strategy for enhancing expression in target cells or tissues while reducing off-target transgene expression. The AAV9-PHP.B/Ly6a interaction provides a model system with a robust biological readout that can be interrogated to better understand the biology of AAV vectors’ interactions with target receptors. In this work, we analyzed the sequence and structural features required to successfully transfer the Ly6a receptor-binding epitope from AAV9-PHP.B to another capsid of clinical interest, AAV1. We found that AAV1- and AAV9-based vectors targeted to the same receptor exhibited different brain-transduction profiles. Our work suggests that, in addition to attachment-receptor binding, the capsid context in which this binding occurs is important for a vector’s performance.
KEYWORDS: adeno-associated virus, AAV, capsid engineering, Ly6a, PHP.B, receptor-mediated transcytosis
INTRODUCTION
In human gene therapy clinical trials, vectors based on adeno-associated virus (AAV) represent the most commonly used modality for gene transfer (1). This trend is largely due to the favorable safety profile associated with AAV vectors (2), their capacity to transduce dividing and nondividing cells (3), and the long-term gene expression afforded by AAV vector genomes in vivo (4, 5). More than 100 clinical trials using AAV vectors have been registered at ClinicalTrials.gov as of July 2020. Of these trials, more than 70% use capsid sequences isolated from AAVs found in nature, with the most commonly used sequences corresponding to AAV serotypes AAV1, AAV2, AAV8, and AAV9. Different AAV serotypes interact with distinct cell-surface glycans (6–9) and intracellular trafficking receptors (10–12), which influences their natural cellular transduction efficiency and tissue-specific tropism. While these natural AAV serotypes can efficiently deliver genetic material to certain organs (e.g., liver [13–15]) or cell types (16, 17), delivery to other tissues (e.g., brain, skeletal muscle, or lungs) could be improved to broaden their therapeutic potential.
Researchers have successfully applied a technique known as “AAV display” to develop AAV vectors with nonnative receptor-binding properties and tissue transduction (18, 19). In this procedure, a library of random polypeptides is spliced into a loop region at the peak of the AAV’s 3-fold axis, where the insertions are sterically unencumbered and poised to interact with potential receptors (20, 21). This step introduces a binding domain that is not present in the parental capsid and increases the likelihood of generating vectors with novel receptor-binding properties. By transferring the receptor-binding properties conferred by these peptide insertions between different AAV capsids, one may achieve a method for tuning the tissue specificity, transduction efficiency, and tissue penetration properties of engineered AAV vectors. However, the structural consequences of transferring these receptor-binding peptides between AAV capsids, as well as the extent to which these engineered peptides are dependent on their specific capsid context for receptor binding, remain unknown.
One successful application of AAV display was in the identification of AAV9-PHP.B, an engineered vector that differs from AAV9 by the presence of a 7-amino-acid peptide insertion after residue Q588 (19). When administered intravenously to C57BL/6 mice, the presence of this short peptide enabled AAV9-PHP.B to localize to the brain microvasculature, cross the blood-brain barrier (BBB), and elicit widespread transduction of neurons and astrocytes throughout the central nervous system (CNS). Our lab was the first to report that the presence of this peptide mediates a direct binding interaction between AAV9-PHP.B and Ly6a, a glycosylphosphatidylinositol (GPI)-linked protein that is highly expressed on the surface of brain microvascular endothelial cells in particular strains of mice (22, 23).
In this study, we used the AAV9-PHP.B/Ly6a interaction as a model system to assess the importance of structural context in the biology of engineered surface loops. We determined that transferring the PHP.B peptide from AAV9 to another commonly used capsid serotype, AAV1, was not sufficient to transfer Ly6a binding. Using structural analysis, we demonstrated that this lack of receptor binding was not due to an altered presentation of the highly flexible PHP.B peptide in the AAV1 context, nor was it due to gross structural changes to the AAV1 capsid induced by its insertion. Instead, we found that Ly6a receptor binding requires synergy between the PHP.B peptide insertion and specific residues from the AAV9 HVR VIII region in which it is presented. Herein, we show that an AAV1 vector that incorporates a larger graft of AAV9-PHP.B residues—including these adjacent HVR VIII sequences—can use Ly6a to localize to brain tissue; however, this vector transduces endothelial cells instead of crossing the BBB. This work highlights important considerations for vector engineers seeking to transfer receptor-binding activity between different capsid serotypes.
RESULTS
The engineered peptide domain of AAV9-PHP.B does not confer Ly6a binding when grafted to HVR VIII of AAV1.
We first attempted a simple graft of the PHP.B 7-mer peptide into position 588 of the AAV1 capsid, which we termed AAV1-PHP.B (Fig. 1A). We tested AAV1-PHP.B for direct binding to Ly6a using surface-plasmon resonance (SPR) and found no evidence that the peptide graft had transferred Ly6a binding to AAV1 (Fig. 1B). In contrast, AAV9-PHP.B vectors produced robust Ly6a binding responses with a measured dissociation constant (KD) of 31.0 pM (Fig. 1C). We then generated AAV1-PHP.B vectors by packaging a firefly luciferase reporter gene under the control of the cytomegalovirus promoter (CMV-ffLuc) and evaluated the transduction efficiency of this vector and relevant controls in HEK293 cells and HEK293 cells stably expressing Ly6a (HEK293-Ly6a). Transduction values were normalized to the performance of wild-type (WT) AAV1 or AAV9 capsids in each cell type to simplify data presentation given inherent differences in transduction efficiency for these two capsids and cell lines. Through its interactions with the Ly6a receptor, AAV9-PHP.B gained a 30-fold transduction advantage in HEK293-Ly6a cells relative to its performance in HEK293 cells; AAV1-PHP.B exhibited no such advantage in Ly6a-expressing cells compared to HEK293 cells (Fig. 1D). In both cell types, AAV1-PHP.B vectors demonstrated a 3-fold decrease in transduction efficiency relative to AAV1 controls. This Ly6a-independent effect indicates that amino acid insertions at position 588 can impact a vector’s inherent transduction efficiency. We next investigated the function of AAV1-PHP.B in vivo using an enhanced green fluorescent protein (EGFP) reporter gene under the control of the CB7 promoter (CB7-EGFP). We administered 1 × 1012 genome copies (GC) of AAV1-PHP.B and control vectors intravenously to C57BL/6 mice and analyzed brain sections for GFP expression after 21 days (Fig. 1E). Consistent with the binding and in vitro transduction data, AAV1-PHP.B failed to deliver the EGFP reporter across the BBB. In contrast, AAV9-PHP.B resulted in robust brain transduction. Given these results, we concluded that the presence of the PHP.B peptide alone is not sufficient to transfer Ly6a binding to AAV1.
FIG 1.

The engineered peptide domain of AAV9-PHP.B does not confer Ly6a binding when grafted to HVR VIII of AAV1. (A) The AAV1-PHP.B vector was generated by grafting the engineered peptide from AAV9-PHP.B into an analogous position in AAV1’s HVR VIII. (B and C) SPR plots showing AAV1-PHP.B (B) and AAV9-PHP.B (C) binding to surface-immobilized Ly6a-hIgG1 protein. Colored lines show binding data for each vector at a range of vector concentrations. KD values were determined using a global 1:1 binding model. (D) AAV9-PHP.B and AAV1-PHP.B vectors were tested alongside WT control vectors in HEK293 and HEK293-Ly6a cells. All vectors packaged a CMV-ffLuc reporter gene cassette. Results show transduction efficiency normalized to the WT capsid transduction efficiency for each cell type. Horizontal bars represent group averages; standard deviations (SDs) are reported as error bars; n = 3. Groups were compared using ANOVA followed by post hoc two-sample, two-sided t tests (version R.4.0.0) for specific pairwise comparisons. The # symbols represent statistical significance for comparisons between Ly6a (−/+) cell lines; the * symbols represent statistical significance for comparisons between engineered vectors and WT vectors in a given cell type: #/*, P ≤ 0.05; ##/**, P ≤ 0.01; ###/***, P ≤ 0.001; ns, not significant. (E) C57BL/6J mice were treated intravenously with 1 × 1012 GC of the indicated vectors or a PBS injection control. Vectors packaged a CB7-EGFP reporter gene cassette. Brains were sectioned on day 21 post-vector administration for direct EGFP fluorescence analysis. We acquired images using a 4× objective and 2 s exposure. GC, genome copies; WT, wild type; KD, dissociation constant.
The flexible PHP.B peptide presents similarly in AAV9 and AAV1 contexts, and peptide insertion has a minimal impact on capsid structure.
Given that transfer of the PHP.B peptide between AAV serotypes was not sufficient to transfer Ly6a binding, we sought to determine whether this result could be explained by differences in structural presentation of the PHP.B peptide in each capsid context. For this purpose, we utilized cryo-electron microscopy (cryo-EM) to obtain a detailed structure of the AAV9-PHP.B and AAV1-PHP.B capsids, with resolutions of 2.27 Å and 2.32 Å, respectively. Although we obtained high-resolution structural information for the majority of the capsid, we observed low levels of electron density for the PHP.B peptide insertions in both the AAV9-PHP.B and AAV1-PHP.B structures. These findings indicate that the amino acids in these regions are flexible and can adopt multiple conformations without converging to a single energy-minimized structure. In the AAV9-PHP.B HVR VIII loop (Fig. 2A), A587 on the ascending chain and K595 on the descending chain marked the most distal residues for which both main chain and side chain residues could be fitted to the electron-density map. In the AAV1-PHP.B HVR VIII loop (Fig. 2B), S586 on the ascending chain and K595 on the descending chain marked the most distal residues for which both main chain and side chain residues could be fitted. We were also able to fit the main chain for S587–T589 and P593–F594 but could not accurately determine the positions of side chains for these residues. Overall, while both inserted peptides demonstrated flexibility, we cannot rule out the possibility that subtle differences may exist in peptide presentation for each serotype, as more of the PHP.B backbone chain could be resolved in the AAV1-PHP.B structure.
FIG 2.

The flexible PHP.B peptide presents similarly in AAV9 and AAV1 contexts; peptide insertion has a minimal impact on capsid structure. (A and B) Electron density maps contoured at 1σ (gray mesh) and fitted model residues for HVR-VIII of the AAV9-PHP.B capsid (A, green) and AAV1-PHP.B capsid (B, salmon). Red text indicates residues from the PHP.B peptide insertion, and black text indicates residues from the parental capsid. (C and D) Solved structures of AAV9-PHP.B (C) or AAV1-PHP.B (D) superimposed onto previously reported structures for AAV9 (orange) or AAV1 (purple). All figures were rendered using PyMol.
We next sought to determine whether the presence of the peptide insertion in either engineered variant induced changes in the surrounding capsid structure. To investigate this possibility, we aligned our structures of the engineered capsids to published structures of AAV9 (24) and AAV1 (25). We found the structures containing PHP.B peptide insertions to be nearly identical to the structures of the parental serotypes (Fig. 2C and D). The AAV9 and AAV9-PHP.B structures had an all-atom root-mean-squared deviation (RMSD) of only 0.933 Å, and the AAV1 and AAV1-PHP.B structures had an all-atom RMSD of only 1.007 Å. For reference, two independently solved structures for the same protein typically have an all-atom RMSD in the range of 0 to 1.2 Å (26). Furthermore, capsid domains with known functional properties, such as the galactose-binding site in AAV9 and the sialic acid-binding site in AAV1, showed near-perfect conservation between parental and peptide-inserted structures (Fig. S1A and B in the supplemental material). In HVR IV, we observed slight differences between the structure of AAV1-PHP.B and AAV1 (Fig. 2D). Like the flexible PHP.B peptide insertions, this surface loop had low electron density levels in our AAV1-PHP.B structure (Fig. S1C) compared to the published AAV1 structure (Fig. S1D). It is possible that this increase in HVR IV flexibility was induced by the PHP.B peptide insertion into HVR VIII, given the proximity of these surface loops at the 3-fold axis. In contrast, the main chain for HVR IV in our AAV9-PHP.B structure (Fig. S1E) aligns well with the previously reported structure for HVR IV in AAV9 (Fig. S1F), indicating that peptide insertions at this location do not necessitate changes in surrounding capsid structure. We conclude that inserting the flexible PHP.B peptide into HVR VIII has a minimal impact on overall structure in either capsid context, but that peptide insertion at this location has the potential to create modest changes in local structure, as observed with AAV1-PHP.B.
Using linkers to increase PHP.B peptide flexibility does not enable Ly6a binding in AAV1-PHP.B and removes Ly6a binding in AAV9-PHP.B.
To assess whether differences in peptide flexibility between AAV9-PHP.B and AAV1-PHP.B limit AAV1-PHP.B binding to Ly6a, we generated AAV1-PHP.B and AAV9-PHP.B variants in which flexible glycine-serine-glycine (GSG) linker sequences were inserted at the N terminus (NL), the C terminus (CL), or flanking both sides of the PHP.B peptide. These linker variants were packaged with CMV-ffLuc and tested for transduction efficiency in HEK293-Ly6a and HEK293 cells (Fig. 3). All AAV1-PHP.B vectors containing linkers exhibited similar performances in both cell types (Fig. 3A), indicating that the inclusion of linker sequences did not confer a Ly6a-mediated cellular attachment capability. AAV1-PHP.B linker variants demonstrated subtle (3-fold) changes in overall transduction efficiency relative to AAV1 controls, but these changes were consistent in both normal and Ly6a-expressing HEK293 cell types. Given these results and structural data demonstrating that neither capsid presents the PHP.B peptide with a highly ordered structure, we concluded that AAV1-specific constraints on the insert peptide conformation do not account for the failure of AAV1-PHP.B to bind to Ly6a.
FIG 3.

Using linkers to increase PHP.B peptide flexibility does not enable Ly6a binding in AAV1-PHP.B and removes Ly6a binding in AAV9-PHP.B. (A and B) AAV1-PHP.B linker variants (A) or AAV9-PHP.B linker variants (B) were tested alongside WT control vectors in HEK293 and HEK293-Ly6a cells. All vectors packaged a CMV-ffLuc reporter gene cassette. Results show transduction efficiency normalized to the WT capsid transduction efficiency for each cell type. Transduction efficiencies for AAV1, AAV1-PHP.B, AAV9, and AAV9-PHP.B are included as reported in Fig. 1B for comparison. Horizontal bars represent group averages; SDs are reported as error bars; n = 3. Groups were compared using ANOVA followed by post hoc two-sample, two-sided t tests (version R.4.0.0) for specific pairwise comparisons. The # symbols represent statistical significance for comparisons between Ly6a (−/+) cell lines; the * symbols represent statistical significance for comparisons between engineered vectors and WT vectors in a given cell type: #/*, P ≤ 0.05; ##/**, P ≤ 0.01; ###/***, P ≤ 0.001; ns, not significant. NL, N-terminal linker; CL, C-terminal linker; WT, wild type.
When we tested the AAV9-PHP.B linker variants in HEK293-Ly6a and HEK293 cells, we found that inclusion of linker sequences on either side of the PHP.B peptide completely removed the Ly6a-binding capability of AAV9-PHP.B (Fig. 3B). AAV9-NL-PHP.B retained a meager 2-fold transduction advantage in HEK293-Ly6a cells compared to normal HEK293 cells, whereas AAV9-NL-PHP.B-CL and AAV9-PHP.B-CL transduced both normal and Ly6a-expressing HEK293 cells equivalently. As with the AAV1-PHP.B linker variants, the AAV9-PHP.B linker variants demonstrated subtle changes (3-fold) in overall transduction efficiency relative to AAV9 controls. Given that all AAV9-PHP.B linker variants reduced Ly6a-mediated transduction, we concluded that a contiguous interface between the PHP.B peptide and its surrounding HVR VIII residues is required for attachment to the Ly6a receptor.
Ly6a binding requires interactions with residues from the PHP.B peptide insertion and AAV9’s HVR VIII.
We next hypothesized that transferring the Ly6a-binding epitope to the AAV1 capsid would require a combination of residues from the PHP.B peptide itself and the wild-type (WT) AAV9 capsid residues surrounding this peptide insertion. The size of the grafted region was therefore extended to incorporate all residues in AAV9-PHP.B’s HVR VIII, which we denote as AAV1-PHP.B2 (Fig. 4A). An SPR assay was used to obtain kinetic affinity measurements for AAV1-PHP.B2 (Fig. 4B), with AAV9-PHP.B binding curves reproduced from Fig. 1C for comparison (Fig. 4C). Under the same assay conditions, AAV1-PHP.B2 and AAV9-PHP.B had measured dissociation constants (KD) of 199 pM and 31.0 pM, respectively, for Ly6a binding.
FIG 4.

Ly6a binding requires interactions with residues from the PHP.B peptide insertion and AAV9’s HVR VIII. (A) Alignment showing how the AAV1-PHP.B2 vector was generated by extending the grafted region to include the PHP.B peptide and surrounding residues from AAV9’s HVR VIII. (B and C) SPR plots showing AAV1-PHP.B2 (B) and AAV9-PHP.B (C) (reproduced from Fig. 1C) binding to surface-immobilized Ly6a-hIgG1 protein. Colored lines show binding data for each vector at a range of vector concentrations. KD values were determined using a global 1:1 binding model. (D) We tested AAV9-PHP.B and AAV1-PHP.B2 vectors alongside WT control vectors in HEK293 and HEK293-Ly6a cells. All vectors packaged a CMV-ffLuc reporter gene cassette. Results show transduction efficiency normalized to the WT capsid transduction efficiency for each cell type. Transduction efficiencies for AAV9 and AAV9-PHP.B are included as reported in Fig. 1D for comparison. Horizontal bars shown in (D) represent group averages; SDs are reported as error bars; n = 3. Groups were compared using ANOVA followed by post hoc two-sample, two-sided t tests (version R.4.0.0) for specific pairwise comparisons. The # symbols represent statistical significance for comparisons between Ly6a (−/+) cell lines; the * symbols represent statistical significance for comparisons between engineered vectors and WT vectors in a given cell type: #/*, P ≤ 0.05; ##/**, P ≤ 0.01; ###/***, P ≤ 0.001; ns, not significant. ddPCR, digital droplet PCR; WT, wild type; KD, dissociation constant.
Next, we assessed AAV1-PHP.B2 vectors in our cellular transduction assay. AAV1-PHP.B2 exhibited a 20-fold transduction advantage in HEK293-Ly6a cells compared to HEK293 cells (Fig. 4D). As was observed with peptide-inserted vectors in the previous transduction experiments, the presence of the PHP.B2 insertion changed the inherent transduction efficiency of AAV1-PHP.B2, reducing transduction by approximately 50-fold in HEK293 cells. It is important to note that although AAV1-PHP.B2 had reduced transduction efficiency relative to AAV1, it still outperformed AAV9 and AAV9-PHP.B transduction in Ly6a-negative cells (Fig. S2A). This is because AAV1-based vectors have a higher baseline transduction efficiency in cultured cells than AAV9-based vectors (27). In HEK293-Ly6a cells, the relative loss in transduction efficiency for AAV1-PHP.B2 was rescued through its interactions with Ly6a.
These experiments demonstrate that, in vitro, a larger graft of AAV9-PHP.B residues to the AAV1-PHP.B2 capsid enables Ly6a binding and recapitulates the Ly6a-mediated cellular transduction properties of AAV9-PHP.B in the AAV1 capsid context.
AAV1-PHP.B2 localizes to, but does not cross, the BBB in vivo.
Next, we sought to determine whether Ly6a binding was sufficient to enable AAV1-PHP.B2 to penetrate the murine BBB. We delivered 3 × 1011 GC of AAV1-PHP.B2 vectors containing CB7-EGFP intravenously to C57BL/6 mice, alongside appropriate controls. We harvested brain and liver tissues for vector biodistribution analysis after 21 days and further assessed the brain tissue for transgene expression.
A comparison of biodistribution results for AAV1-PHP.B2 and AAV1 controls showed that AAV1-PHP.B2 was successfully retargeted to brain tissue through its engineered interaction with Ly6a (Fig. 5A and B). Relative to AAV1, AAV1-PHP.B2 demonstrated a 10-fold increase in the number of vector genomes that localized to the brain and an 8-fold decrease in the number of vector genomes that localized to the liver, the primary organ transduced by AAV vectors after systemic administration. AAV9-PHP.B demonstrated a 400-fold increase in localization to the brain, possibly due to its higher affinity for the Ly6a receptor, and a 3-fold decrease in localization to the liver. Corroborating the biodistribution data, quantitative reverse transcription-PCR (RT-qPCR) indicated that AAV1-PHP.B2 and AAV9-PHP.B had 60-fold and 480-fold higher transgene expression in brain tissue, respectively, than their nonengineered parental serotypes (Fig. 5C). These results demonstrate that by introducing a Ly6a-binding epitope, AAV1-PHP.B2 vectors can be successfully retargeted to brain tissue, though the magnitude of this retargeting is less than that observed with AAV9-PHP.B.
FIG 5.
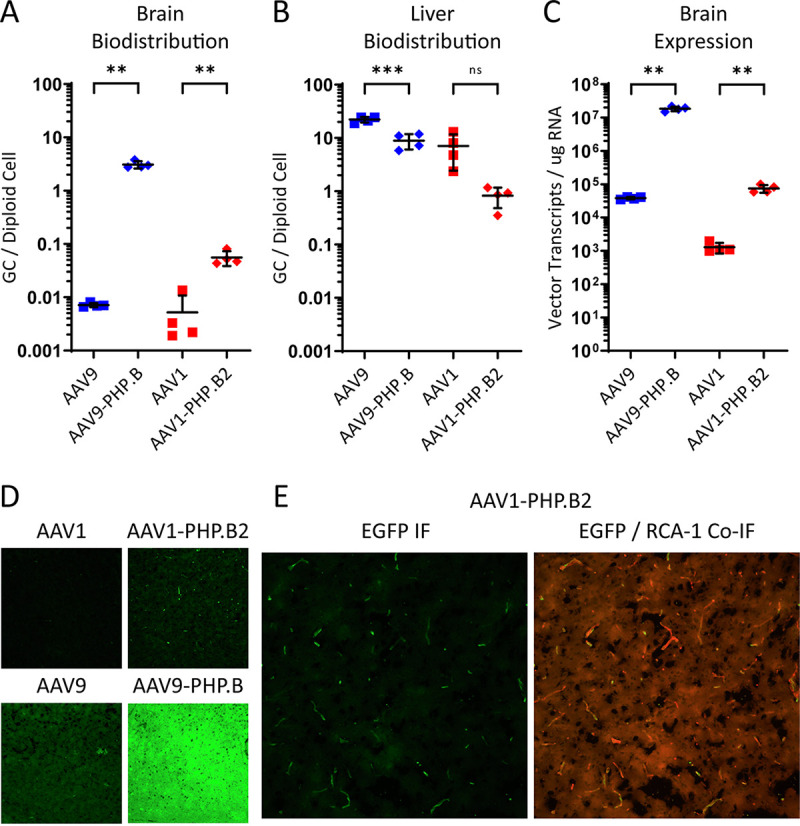
AAV1-PHP.B2 localizes to, but does not cross, the BBB in vivo. AAV1-PHP.B2 and control vectors packaging a CB7-eGFP reporter gene were administered intravenously to C57BL/6J mice at a dose of 3 × 1011 GC per mouse. (A and B) Vector biodistribution in brain (A) and liver (B) tissue collected at necropsy on day 21 post-vector administration, reported as vector GCs per diploid cell. (C) Vector transcripts were also detected in brain samples using RT-qPCR and are reported as vector transcripts per μg total RNA. In A to C, individual measurements and a horizontal bar representing group averages are shown; SDs are reported as error bars; n = 4. Groups were compared using ANOVA followed by post hoc two-sample, two-sided t tests (version R.4.0.0) for specific pairwise comparisons. The * symbols represent statistical significance for comparisons between engineered vectors and WT vectors in a given tissue. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant. (D) Brain sections isolated at necropsy were analyzed for GFP reporter gene expression using anti-GFP IF. (E) AAV1-PHP.B samples were also treated with an RCA-1 lectin stain to assess colocalization of GFP signals with brain microvascular endothelial cells. We acquired the displayed images using a 20× objective and 1s exposure for both fluorophores. GC, genome copies; IF, immunofluorescence.
Importantly, our analysis of brain histology revealed differences between the two Ly6a-targeted capsids in terms of their BBB penetration and cell type specificity (Fig. 5D). While AAV9-PHP.B had high levels of expression throughout the brain parenchyma, AAV1-PHP.B2 expression had a punctate pattern that appeared to be confined to brain microvascular endothelial cells. To confirm this, we stained brain sections of AAV1-PHP.B2-treated animals with a Ricinus communis agglutinin I (RCA I) lectin stain, which labels endothelial cells in murine brain samples (Fig. 5E). We found that nearly all GFP expression from the AAV1-PHP.B2 vector colocalized with the endothelial stain. This can be compared to AAV1-PHP.B2’s transduction of liver tissue, which demonstrates widespread transduction in Ly6a(−) cells (Fig. S2B). Overall, we conclude that grafting the PHP.B peptide and its adjacent HVR VIII sequence from AAV9-PHP.B to AAV1-PHP.B2 successfully re-targeted AAV1-PHP.B2 vectors to the mouse brain but did not facilitate efficient BBB penetration.
DISCUSSION
In this series of experiments, we have demonstrated that penetration of the BBB requires high-affinity binding to Ly6a via a combination of residues from the PHP.B peptide insert itself and additional residues from HVR VIII of AAV9. In fact, our experiments with linker sequences flanking the AAV9-PHP.B peptide indicate that any disruption to the peptide/HVR VIII interface also disrupts the Ly6a-binding potential of AAV9-PHP.B. Our conclusions are consistent with previously reported affinity-maturation studies involving AAV9-PHP.B, which show that amino acid changes in the HVR VIII sequence flanking the peptide insert improve brain transduction (28). Given the relatively small size of the 7-amino-acid PHP.B peptide insertion and its proximity to the larger structures of the 3-fold axis in which it is presented, AAV9-PHP.B likely also utilizes peptide-adjacent residues to form contacts with its target receptor, Ly6a. This extended binding epitope may be necessary to provide AAV9-PHP.B with sufficient receptor affinity to be selected from a highly diverse library.
To accomplish our initial goal of transferring the Ly6a-binding epitope to the AAV1 capsid, we needed to graft the entirety of AAV9-PHP.B’s HVR VIII into the new capsid context. This graft conferred AAV1-PHP.B2 with Ly6a binding, which mediated retargeting of this vector to the brain of C57BL/6 mice while detargeting peripheral tissues, such as the liver. Despite this tissue retargeting, our in vivo studies clearly showed that AAV1-PHP.B2 does not recapitulate the BBB penetration or widespread brain transduction profile exhibited by AAV9-PHP.B. One explanation for this is that the AAV9 capsid has a preference for transcytosis versus cellular transduction compared to AAV1. Indeed, AAV1 vectors have been demonstrated to outperform AAV9 vectors in cell-based transduction assays in various cell types in vitro (27). In this model, both AAV9-PHP.B and AAV1-PHP.B2 are recognized by Ly6a on the luminal surface of the brain’s microvasculature. However, the AAV1 vector is preferentially diverted into a pathway that results in transduction while the AAV9 vector proceeds into the CNS via transcytosis. Another explanation involves our observation that AAV1-PHP.B2 has a reduced affinity for the Ly6a target receptor compared with AAV9-PHP.B. With a lower receptor affinity and equivalent intravenous dosage, we would expect fewer AAV1-PHP.B2 vectors to accumulate on the surface of brain microvascular endothelial cells, translating to reduced BBB penetration. Our work suggests that successful penetration of the BBB and transduction of target cells in the brain parenchyma requires more than Ly6a receptor binding; however, the nature of these other biological processes is unknown.
As part of this study, we generated structural data showing that peptide insertions at position 588 have a similar flexible presentation in both AAV1 and AAV9 serotypes and do not induce large structural rearrangements in either capsid backbone. These observations are consistent with other structural studies involving AAV vectors with peptide insertions into surface loops of the 3-fold axis (29, 30). In the AAV9-PHP.B structure, the PHP.B peptide insertion has more flexibility than any of the other HVR surface loops on the exterior of the capsid. This peptide flexibility may be functionally important during library selection. An inherently unstructured peptide can sample multiple spatial conformations, thus increasing the likelihood of finding a conformation that enables an interaction with a novel receptor. We also found that peptide-inserted and parental capsid structures demonstrate remarkable overlap, indicating that peptides can be inserted into position 588 without impacting existing structure-function relationships in the capsid backbone. Our work indicates that while there are minimal structural constraints on transposing flexible peptides between capsid serotypes, the functional properties conferred by those peptides may be dependent on the capsid context for activity.
Our studies shed light on the structural and biochemical basis of the success of the BBB-penetrating capsid AAV9-PHPB. We hypothesize that it achieves enhanced delivery to the CNS by utilizing flexibility of the inserted peptide and multiple receptor contact points within and surrounding the peptide insertion to improve receptor affinity. We have demonstrated that for AAV9-PHP.B, the insertion of peptides into position 588 enables binding to novel receptors without perturbing key inter- and intrasubunit interactions outside of the ectopic peptide, which are critical for efficient capsid assembly and high manufacturing yields. Additionally, we have found that it may be important to ablate competing intracellular processes associated with the capsid backbone, such as transduction, if transcytosis is desired. By enhancing our understanding of the structural biology of capsid-receptor interactions and the cellular biology of our desired biological outcomes, we define key parameters that can be used to engineer AAV capsids with improved delivery across physical barriers.
MATERIALS AND METHODS
Animals.
We purchased male C57BL/6J (stock no. 000664) mice from Jackson Laboratory. We housed the animals in standard caging, with three or four animals per cage. On a weekly basis, mice were housed in new cages, with new water bottles and bedding substrates all sterilized by autoclave. An automatic 12-h light/dark cycle was maintained, with the dark period beginning at 7:00 p.m. (±30 min). We provided irradiated laboratory rodent food ad libitum.
Cell lines.
We cultured HEK293 cells in a humidified incubator at 37°C with 5% CO2. Growth medium comprised Dulbecco’s modified Eagle medium (DMEM; catalog 11995-040, Gibco) supplemented with 10% fetal bovine serum (catalog SH3007103, Cytiva) and 100 IU/ml penicillin and streptomycin solution (catalog 15140122, Gibco).
We produced HEK293 cells stably expressing Ly6a (HEK293-Ly6a) by transfecting cells with a plasmid containing the Ly6a cDNA sequence under the control of the CMV promoter and a neomycin-resistance gene. This was followed by selecting for stably transfected cells using growth medium supplemented with 600 μg/ml G418 sulfate solution (Penn Cell Center). We plated HEK293 cells in two 10-cm dishes at a density of 4.6 × 104 cells/cm2. Cells were transfected at 60% confluence using a 1:2 ratio of plasmid DNA to polyethylenimine hydrochloride (PEI; catalog 24765-1, Polysciences Inc.) in serum-free DMEM. We incubated cells in this solution overnight before replacing it with growth medium to allow recovery for 24 h. Cells were then trypsinized, re-plated at their initial seeding density, and allowed to grow to confluence in growth medium. At confluence, we added growth medium containing G418 to the cultures. We replaced growth medium containing G418 daily for 10 to 14 days until the medium was clear of floating cell debris. We selected surviving colonies of cells for individual culture and screened cells for Ly6a expression using Western blotting. For blotting, we utilized a primary antibody directed against Ly6a-specific peptides (catalog 701919, Thermo Fisher Scientific). We then cultured HEK293-Ly6a cells in growth medium containing G418 for all experiments.
Vector production.
We designed AAV1 and AAV9 capsid insert sequences in silico and ordered them from Integrated DNA Technologies as double-stranded linear nucleic acid (gBlock) fragments. We introduced these insert sequences to a linearized AAV1 or AAV9 trans plasmid (p0001, p3668, Penn Vector Core) backbone using the NEBuilder HiFi DNA Assembly Master Mix (catalog E2621S, New England BioLabs [NEB]) according to the manufacturer’s instructions. We confirmed the sequences of the trans plasmids with insertions using Illumina sequencing.
As previously described, the Penn Vector Core produced and titrated vectors for structural studies, SPR assays, and in vivo studies (31). Briefly, we used AAV9, AAV1, AAV9-PHP.B (p0078, Penn Vector Core), AAV1-PHP.B (p5071, Penn Vector Core), or AAV1-PHP.B2 (p5512, Penn Vector Core) trans plasmids with cis plasmids encoding the CB7-EGFP reporter gene (p1963, Penn Vector Core) to produce vectors in HEK293 cells via triple transfection. We harvested, concentrated, and purified the culture supernatant with an iodixanol gradient before titrating the purified vectors using droplet digital PCR (ddPCR) with primers and probes targeting the rabbit beta-globin poly A sequence of the vector transgene cassette.
For transduction assays, we produced lab-scale vectors using the same triple transfection procedure, but with cis plasmids encoding the CMV-ffLuc reporter gene (p0105, Penn Vector Core). Individual researchers titrated these lab-scale vectors using TaqMan reagents (catalog 4305719, Thermo Fisher Scientific) and primers targeting the simian virus 40 poly A sequence of the transgene cassette. The Penn Vector Core provided all standards, primers, and PCR reagents. These vectors were not subject to concentration or purification steps before their administration to cells.
Recombinant Ly6a-hIgG1 expression.
Ly6a-hIgG1 recombinant protein was produced on a fee-for-service basis by Genscript Biotech Corporation. Genscript synthesized and cloned the DNA sequence for the fusion protein into a pcDNA3.4 vector for transient expression in their proprietary HD 293F expression system using a 100-ml suspension culture. Genscript cultured cells for 6 days after transfection in a humidified incubator at 37°C + 8% CO2. They centrifuged and filtered the cell culture for clarification, and purified the recombinant protein product by affinity chromatography using MabSelect SuRe LX (catalog 17547401, Cytiva Life Sciences). Our lab determined the final protein concentration by bicinchroninic acid (BCA) protein assay against a bovine serum albumin standard (catalog 23225, Thermo Fisher Scientific).
SPR binding assays.
We performed SPR binding analysis using a Biacore T200 instrument (GE Healthcare) at room temperature in HBS-EP(+) buffer (10 mM HEPES [pH 7.4], 150 mM NaCl, 3 mM EDTA, and 0.05% P20 surfactant; catalog BR100669, Cytiva) using a protein A/G derivatized sensor chip (catalog SCBS PAGHC30M, XanTec Bioanalytics). We injected Ly6a-hIgG1 diluted to 15 nM in HBS-EP(+) at a flow rate of 10 μl/min for 5 min to capture ∼850 response units (RU) on the sensor surface in each cycle. We measured AAV9-PHP.B, AAV1-PHP.B, and AAV1-PHP.B2 binding to this surface at concentrations ranging from 1 nM (6 × 1011 GC/ml) to 15.6 pM. We performed regeneration between binding cycles using 10 mM glycine pH 1.5 injected at a flow rate of 60 μl/min for 1 min.
Cell transduction assays.
Here, we seeded 2 × 104 HEK293 or HEK293-Ly6a cells in each well of a black-walled 96-well tissue-culture plate (catalog 3603, Corning) and incubated the cells overnight. Crude preps of vectors were serially diluted in growth medium and introduced to the cells at a range of concentrations in a total culture volume of 50 μl. Two days posttransduction, we measured luciferase expression using a Synergy 2 Multi-Detection microplate reader (BioTek) and D-luciferin (catalog 122799, Perkin Elmer) reagent diluted to 100 ng/ml in growth medium. For each serially diluted vector preparation, we plotted the luciferase reporter expression against vector concentration. We identified a linear region of the curve and applied the slope of this region as a measurement of transduction efficiency per vector genome. By performing three such serial dilutions for each assay condition, we obtained three independent measurements of transduction efficiency. The final reported values represent the transduction efficiency in each cell type normalized to the average reading of WT capsid transduction efficiency in that cell type.
In vivo studies.
In studies involving AAV1-PHP.B, 6- to 8-week-old C57BL/6 mice received 1 × 1012 GC (5 × 1013 GC/kg) of AAV1, AAV1-PHP.B, or AAV9-PHP.B vectors encoding CMV-EGFP or a phosphate-buffered saline (PBS) control injection of 0.1 ml via the lateral tail vein. We assessed three biological replicates for each condition. All mice were euthanized at 21 days postinjection by inhalation of CO2, and brains were immersion-fixed in 10% neutral buffered formalin (catalog 245-685, Fisher Scientific) for subsequent histological analysis by direct EGFP fluorescence.
In studies involving AAV1-PHP.B2, 6- to 8-week-old C57BL/6 mice received 3 × 1011 GC (1.5 × 1013 GC/kg) of AAV9, AAV9-PHP.B, AAV1, or AAV1-PHP.B2 vectors encoding CMV-EGFP in 0.2 ml via the lateral tail vein. We assessed four biological replicates for each condition. All mice were euthanized at 21 days postinjection by inhalation of CO2, and brain and liver tissues were harvested for analysis. We sectioned the brains half-sagittally. The right hemisphere was immersion-fixed in 10% neutral buffered formalin for subsequent histological analysis. The left hemisphere was snap-frozen on dry ice for quantitative PCR (qPCR) vector biodistribution analysis and RT-qPCR analysis of EGFP expression. We also snap-froze the liver tissue on dry ice for qPCR vector biodistribution analysis.
Histology.
For the AAV1-PHP.B study, we removed brain samples from formalin solution after 24 h, washed the samples briefly in PBS, and equilibrated them sequentially in PBS solutions of 15% and 30% sucrose at 4°C for a minimum of 24 h each. Next, tissues were frozen at optimum-cutting temperature embedding medium (catalog 4583, Tissue-Tek) and sectioned at 30 μm for direct EGFP visualization. We acquired images with a Nikon Eclipse Ti-E fluorescence microscope using a 4× objective and a 2 s exposure time.
For the AAV1-PHP.B2 study, brain samples were subjected to the same washing, equilibration, and sectioning procedures described above. To detect low levels of EGFP expression, we applied a primary goat anti-EGFP antibody (catalog NB100-1770, Novus Biologicals) at a dilution of 1:500, followed by a secondary fluorescein isothiocyanate (FITC)-labeled donkey anti-goat antibody (catalog 705-095-147, Jackson ImmunoResearch Laboratories) at a dilution of 1:100. To assess EGFP colocalization with endothelial cells, we coincubated a rhodamine-labeled RCA-1 lectin stain (catalog RL-1082-5, Vector Laboratories) with the secondary antibody at a concentration of 50 μg/ml. Sectioned samples were blocked with 1% donkey serum (catalog 017-000-121, Jackson ImmunoResearch Laboratories) in PBS + 0.2% Triton for 15 min, followed by overnight incubation with primary antibody solution in blocking buffer at 4°C and 1 h of incubation with secondary antibody and RCA-1 in blocking buffer at room temperature. We imaged sections using a Nikon Ti-2E fluorescence microscope with a 20× objective and a 1 s exposure for both FITC/EGFP and rhodamine detection.
Vector biodistribution and RT-qPCR analysis.
Left hemisphere brain samples were first homogenized in 1 ml ATL buffer + proteinase K from the QIAamp DNA minikit (catalog 51304, Qiagen). We aliquoted a 200-μl volume of brain homogenate for processing with the QIAamp DNA minikit to extract total DNA. We then combined another 250 μl of the brain homogenate with 750 μl of TRIzol-LS reagent (catalog 10296028, Thermo Fisher Scientific) for RNA extraction according to the manufacturer’s protocol. Next, 10 μg of RNA was treated with DNase I (catalog 04716728001, Roche) according to the manufacturer’s protocol. We then performed further RNA cleanup and purification using the RNeasy minikit (catalog 74104, Qiagen). We performed cDNA synthesis using the Applied Biosystems High-Capacity cDNA reverse transcription kit (catalog 4368814, Thermo Fisher Scientific). For liver samples, we extracted total DNA from 25 mg of chipped liver tissue using the QIAamp DNA minikit. We conducted qPCR on DNA and cDNA samples using the TaqMan 2x Master Mix (catalog 4305719, Thermo Fisher Scientific) with primers and probes directed toward the sequence of the vectors’ EGFP transgene. The Penn Vector Core provided all standards, primers, and PCR reagents.
Cryo-EM data collection.
We sent purified samples of AAV1-PHP.B and AAV9-PHP.B vectors to the University of Massachusetts Medical School Cryo-EM Core Facility for analysis. For each sample, 3 μl was added to a 300-mesh gold grid with an ultrathin carbon film supported by a lacey carbon support film (catalog 01824G, Ted Pella) and flash-frozen using a Vitrobot Mark IV instrument (Thermo Fisher Scientific). AAV1-PHP.B was used at a concentration of 8.54 × 1012 GC/ml and AAV9-PHP.B was used at a concentration of 1.83 × 1014 GC/ml. All vectors were suspended in PBS with 0.001% Pluronic F68. For AAV1-PHP.B, we collected 1,950 movies on a Talos-Arctica operating at 200 kV with a Gatan K3 direct detector in counting mode. The pixel size was 0.435 Å/pix, and the total dose was 37.5 electrons/Å2, with 25 frames per movie. Images were collected with defocus in a range of 0.5 to 1.5 μm. For AAV9-PHP.B, we collected 1,380 movies on a Titan Krios operating at 300 kV with a Gatan K2 Summit direct detector in superresolution mode. The pixel size was 0.529 Å/pix, and the total dose was 38.1 electrons/Å2, with 50 frames per movie. Images were collected with defocus in a range between −0.8 and 2.0 μm.
Structure determination, model building, and refinement.
For the AAV1-PHP.B data set, movies were motion-corrected using MotionCor2 (32) and binned to a final pixel size of 0.87 Å. After motion correction, we used GPU-accelerated contrast transfer function (GCTF) (33) to estimate the defocus of the micrographs and processed them using Relion 3.0 (34, 35). We picked and sorted approximately 1,000 particles for two-dimensional (2D) classification. The best classes were used as templates for autopicking. During manual picking, we discarded 19 micrographs, leaving 1,931 micrographs. A total of 151,543 particles from autopicking were sorted through one round of 2D classification to remove false positive and suboptimal particles, yielding 130,161 particles. The initial model was produced in C1 symmetry through ab initio model generation with Relion. We further sorted particles through three-dimensional (3D) classification with C1 symmetry and angular sampling into six classes. The two best classes were selected for a total of 118,958 particles. Next, we performed 3D auto-refinement in C1 symmetry, applied icosahedral symmetry, and performed another round of 3D auto-refinement. Finally, we performed an additional 3D classification without angular sampling into eight classes. The best class had 59,346 particles that we used for final refinement. On this group of particles, we performed 3D auto-refinement, followed by CTF refinement and particle polishing. Final 3D auto-refinement and postprocessing yielded the structure of AAV1-PHP.B to 2.32 Å based on the Fourier shell correlation gold-standard cutoff of 0.143.
We applied a similar protocol to obtain the AAV9-PHP.B structure. Movies were motion-corrected using MotionCor2 (32) and binned to a final pixel size of 1.06 Å. After motion correction, we estimated the defocus of the micrographs with GCTF (33) and processed them using Relion 3.0 (34, 35). We manually picked and sorted approximately 1,000 particles for 2D classification. We used the best classes as templates for auto-picking. We performed auto-picking on all 1,380 micrographs to obtain 89,358 particles. These particles were sorted through one round of 2D classification to remove false positive and suboptimal particles, yielding 69,319 particles. We used these particles for ab initio model generation in Relion using C1 symmetry. Particles were further sorted through 3D classification, first with C1 symmetry and angular sampling into five classes. We selected the three best classes, with a total of 63,798 particles. We then sorted these particles through an additional round of 3D classification with C1 symmetry and angular sampling into eight classes. The three best classes contained 42,570 particles that we used for final refinement. We performed 3D auto-refinement with icosahedral symmetry, starting from the initial model with icosahedral symmetry applied. We then performed CTF refinement and particle polishing on the refined particles. Final 3D autorefinement and postprocessing yielded the structure of AAV9-PHP.B to 2.27 Å based on the Fourier shell correlation gold-standard cutoff of 0.143.
We generated initial models from previously published structures of AAV9 (24) and AAV1 (25). These models were fit into the electron density and modified in COOT (36). After the initial building step, we refined the model against the electron density maps using the phenix.real_space_refinement program included in the PHENIX software package (36). We generated full models with icosahedral noncrystallographic symmetry. We performed refinement under secondary structure and noncrystallographic symmetry (NCS) constraints using rigid-body fitting, global minimization, a local grid search, and anisotropic displacement parameter (ADP) refinement. We generated figures using PyMol (37).
Statistical analysis.
For all quantitative data, we first compared groups within a given figure using an ANOVA test with R Statistical Software function “aov” (version R.4.0.0). If statistically significant differences were detected, we then performed post hoc pairwise comparisons using a two-sample two-sided t test with R Statistical Software function “t.test” (version R.4.0.0). Statistical significance was assessed at the 0.05 level.
Data availability statement.
Structural data associated with this study are available in wwPDB and EMDB with the following accession codes: AAV9-PHP.B (deposition D_1000258328), PDB ID 7RK8 and EMD-24494; AAV1-PHP.B (deposition D_1000258327), PDB ID 7RK9 and EMD-24495. All plasmid sequences are available upon request.
ACKNOWLEDGMENTS
We thank Chen Xu and KangKang Song of the Cryo-EM Core at the University of Massachusetts Medical School for performing grid preparation and data acquisition for the structures included in this paper. We thank Joel Cassel at the Wistar Institute’s Molecular Screening and Protein Expression Facility for his help with our SPR assays. Within the Gene Therapy Program at the University of Pennsylvania, we thank the Program in Comparative Medicine for facilitating our animal work and the Morphology Core for helping us prepare tissue samples for analysis. All vectors were produced by the Penn Vector Core. We thank Mingyao Li and Hanying Yan for advice regarding statistical analysis, and Nathan Denton for his help with figure and manuscript preparation.
This research was supported in part by research agreements with Amicus Therapeutics and Passage Bio, and via grants from the National Institutes of Health: R01GM103899 and R01GM129357.
J.M.W. is a paid advisor to and holds equity in Scout Bio and Passage Bio; he also has sponsored research agreements with Amicus Therapeutics, Biogen, Elaaj Bio, FA212, Janssen, Passage Bio, and Scout Bio, which are licensees of Penn Technology. He also has a sponsored research agreement with G2 Bio. J.M.W. holds equity in the G2 Bio-associated asset companies. J.M.W. is an inventor on patents that have been licensed to various biopharmaceutical companies and for which he may receive payments.
R.A.M. contributions: manuscript writing, original draft preparation, general data visualization, and analysis; in vivo study investigation, data curation, and analysis; SPR assay methodology, investigation, data curation, and analysis. E.C.F. contributions: structure study data curation and formal analysis; electron density construction and model-fitting methodology; data visualization for structure figures. J.M. contributions: provision of vector capsid plasmids; cell transduction assay investigation, data curation, and analysis. Q.W. contributions: in vivo study investigation, data curation, and analysis. H.H. contributions: provision of vector capsid plasmids; cell transduction assay investigation, and data curation. R.A.P. contributions: structural study data curation and formal analysis. C.Y.L. contributions: structure study project administration. J.J.S. contributions: study supervision and administration, cell transduction assay methodology, manuscript review and editing, general data visualization, and analysis. S.R. contributions: study conceptualization. V.Y.M.-B. contributions: structural study supervision, project administration, and provision of resources. J.M.W. contributions: manuscript review and editing; general research supervision, project administration, and funding acquisition. All authors discussed the results and commented on the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
James M. Wilson, Email: wilsonjm@upenn.edu.
Rozanne M. Sandri-Goldin, University of California, Irvine
REFERENCES
- 1.Wang D, Tai PWL, Gao G. 2019. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 18:358–378. 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith RH. 2008. Adeno-associated virus integration: virus versus vector. Gene Ther 15:817–822. 10.1038/gt.2008.55. [DOI] [PubMed] [Google Scholar]
- 3.Balakrishnan B, Jayandharan GR. 2014. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr Gene Ther 14:86–100. 10.2174/1566523214666140302193709. [DOI] [PubMed] [Google Scholar]
- 4.Tarantal AF, Lee CC. 2010. Long-term luciferase expression monitored by bioluminescence imaging after adeno-associated virus-mediated fetal gene delivery in rhesus monkeys (Macaca mulatta). Hum Gene Ther 21:143–148. 10.1089/hum.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarantal AF, Lee CCI, Martinez ML, Asokan A, Samulski RJ. 2017. Systemic and persistent muscle gene expression in rhesus monkeys with a liver de-targeted adeno-associated virus vector. Hum Gene Ther 28:385–391. 10.1089/hum.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell CL, Gurda BL, Van Vliet K, Agbandje-McKenna M, Wilson JM. 2012. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J Virol 86:7326–7333. 10.1128/JVI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang LY, Halder S, Agbandje-McKenna M. 2014. Parvovirus glycan interactions. Curr Opin Virol 7:108–118. 10.1016/j.coviro.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang LY, Patel A, Ng R, Miller EB, Halder S, McKenna R, Asokan A, Agbandje-McKenna M. 2016. Characterization of the adeno-associated virus 1 and 6 sialic acid binding Site. J Virol 90:5219–5230. 10.1128/JVI.00161-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaiss AK, Foley EM, Lawrence R, Schneider LS, Hoveida H, Secrest P, Catapang AB, Yamaguchi Y, Alemany R, Shayakhmetov DM, Esko JD, Herschman HR. 2016. Hepatocyte heparan sulfate is required for adeno-associated virus 2 but dispensable for adenovirus 5 liver transduction in vivo. J Virol 90:412–420. 10.1128/JVI.01939-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudek AM, Zabaleta N, Zinn E, Pillay S, Zengel J, Porter C, Franceschini JS, Estelien R, Carette JE, Zhou GL, Vandenberghe LH. 2020. GPR108 is a highly conserved AAV entry factor. Mol Ther 28:367–381. 10.1016/j.ymthe.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, Jae LT, Wosen JE, Nagamine CM, Chapman MS, Carette JE. 2016. An essential receptor for adeno-associated virus infection. Nature 530:108–112. 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pillay S, Zou W, Cheng F, Puschnik AS, Meyer NL, Ganaie SS, Deng X, Wosen JE, Davulcu O, Yan Z, Engelhardt JF, Brown KE, Chapman MS, Qiu J, Carette JE. 2017. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. J Virol 91. 10.1128/JVI.00391-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. 2004. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol 78:6381–6388. 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers GL, Herzog RW. 2015. Gene therapy for hemophilia. Front Biosci (Landmark Ed) 20:556–603. 10.2741/4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samelson-Jones BJ, Arruda VR. 2020. Translational potential of immune tolerance induction by AAV liver-directed factor VIII gene therapy for hemophilia A. Front Immunol 11:618. 10.3389/fimmu.2020.00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Zaidy SA, Mendell JR. 2019. From clinical trials to clinical practice: practical considerations for gene replacement therapy in SMA type 1. Pediatr Neurol 100:3–11. 10.1016/j.pediatrneurol.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 17.Hoy SM. 2019. Onasemnogene Abeparvovec: first global approval. Drugs 79:1255–1262. 10.1007/s40265-019-01162-5. [DOI] [PubMed] [Google Scholar]
- 18.Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV. 2013. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 5:189ra76. 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 19.Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, Wu WL, Yang B, Huber N, Pasca SP, Gradinaru V. 2016. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol 34:204–209. 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller OJ, Kaul F, Weitzman MD, Pasqualini R, Arap W, Kleinschmidt JA, Trepel M. 2003. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol 21:1040–1046. 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 21.Perabo L, Buning H, Kofler DM, Ried MU, Girod A, Wendtner CM, Enssle J, Hallek M. 2003. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol Ther 8:151–157. 10.1016/s1525-0016(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 22.Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. 2018. The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol Ther 26:664–668. 10.1016/j.ymthe.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hordeaux J, Yuan Y, Clark PM, Wang Q, Martino RA, Sims JJ, Bell P, Raymond A, Stanford WL, Wilson JM. 2019. The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood-brain barrier. Mol Ther 27:912–921. 10.1016/j.ymthe.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMattia MA, Nam HJ, Van Vliet K, Mitchell M, Bennett A, Gurda BL, McKenna R, Olson NH, Sinkovits RS, Potter M, Byrne BJ, Aslanidi G, Zolotukhin S, Muzyczka N, Baker TS, Agbandje-McKenna M. 2012. Structural insight into the unique properties of adeno-associated virus serotype 9. J Virol 86:6947–6958. 10.1128/JVI.07232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang R, Xu G, Cao L, Sun Z, He Y, Cui M, Sun Y, Li S, Li H, Qin L, Hu M, Yuan Z, Rao Z, Ding W, Rao Z, Lou Z. 2019. Divergent engagements between adeno-associated viruses with their cellular receptor AAVR. Nat Commun 10:3760. 10.1038/s41467-019-11668-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kufareva I, Abagyan R. 2012. Methods of protein structure comparison. Methods Mol Biol 857:231–257. 10.1007/978-1-61779-588-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis BL, Hirsch ML, Barker JC, Connelly JP, SteiningerRJ, 3rd, Porteus MH. 2013. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol J 10:74. 10.1186/1743-422X-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sanchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, Gradinaru V. 2017. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci 20:1172–1179. 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett A, Keravala A, Makal V, Kurian J, Belbellaa B, Aeran R, Tseng YS, Sousa D, Spear J, Gasmi M, Agbandje-McKenna M. 2020. Structure comparison of the chimeric AAV2.7m8 vector with parental AAV2. J Struct Biol 209:107433. 10.1016/j.jsb.2019.107433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guenther CM, Brun MJ, Bennett AD, Ho ML, Chen W, Zhu B, Lam M, Yamagami M, Kwon S, Bhattacharya N, Sousa D, Evans AC, Voss J, Sevick-Muraca EM, Agbandje-McKenna M, Suh J. 2019. Protease-activatable adeno-associated virus vector for gene delivery to damaged heart tissue. Mol Ther 27:611–622. 10.1016/j.ymthe.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, Wilson JM. 2010. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 21:1259–1271. 10.1089/hum.2010.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA. 2017. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14:331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K. 2016. Gctf: real-time CTF determination and correction. J Struct Biol 193:1–12. 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheres SH. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH. 2018. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7:e42166. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Schrodinger, LLC. 2015. The PyMOL molecular graphics system, version 1.8.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2<br>. Download JVI.01164-21-s0001.pdf, PDF file, 0.4 MB (429KB, pdf)
Data Availability Statement
Structural data associated with this study are available in wwPDB and EMDB with the following accession codes: AAV9-PHP.B (deposition D_1000258328), PDB ID 7RK8 and EMD-24494; AAV1-PHP.B (deposition D_1000258327), PDB ID 7RK9 and EMD-24495. All plasmid sequences are available upon request.