

ABSTRACT
The current pandemic of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to dramatic economic and health burdens. Although the worldwide SARS-CoV-2 vaccination campaign has begun, exploration of other vaccine candidates is needed due to uncertainties with the current approved vaccines, such as durability of protection, cross-protection against variant strains, and costs of long-term production and storage. In this study, we developed a methyltransferase-defective recombinant vesicular stomatitis virus (mtdVSV)-based SARS-CoV-2 vaccine candidate. We generated mtdVSVs expressing SARS-CoV-2 full-length spike (S) protein, S1, or its receptor-binding domain (RBD). All of these recombinant viruses grew to high titers in mammalian cells despite high attenuation in cell culture. The SARS-CoV-2 S protein and its truncations were highly expressed by the mtdVSV vector. These mtdVSV-based vaccine candidates were completely attenuated in both immunocompetent and immunocompromised mice. Among these constructs, mtdVSV-S induced high levels of SARS-CoV-2-specific neutralizing antibodies (NAbs) and Th1-biased T-cell immune responses in mice. In Syrian golden hamsters, the serum levels of SARS-CoV-2-specific NAbs triggered by mtdVSV-S were higher than the levels of NAbs in convalescent plasma from recovered COVID-19 patients. In addition, hamsters immunized with mtdVSV-S were completely protected against SARS-CoV-2 replication in lung and nasal turbinate tissues, cytokine storm, and lung pathology. Collectively, our data demonstrate that mtdVSV expressing SARS-CoV-2 S protein is a safe and highly efficacious vaccine candidate against SARS-CoV-2 infection.
IMPORTANCE Viral mRNA cap methyltransferase (MTase) is essential for mRNA stability, protein translation, and innate immune evasion. Thus, viral mRNA cap MTase activity is an excellent target for development of live attenuated or live vectored vaccine candidates. Here, we developed a panel of MTase-defective recombinant vesicular stomatitis virus (mtdVSV)-based SARS-CoV-2 vaccine candidates expressing full-length S, S1, or several versions of the RBD. These mtdVSV-based vaccine candidates grew to high titers in cell culture and were completely attenuated in both immunocompetent and immunocompromised mice. Among these vaccine candidates, mtdVSV-S induces high levels of SARS-CoV-2-specific neutralizing antibodies (Nabs) and Th1-biased immune responses in mice. Syrian golden hamsters immunized with mtdVSV-S triggered SARS-CoV-2-specific NAbs at higher levels than those in convalescent plasma from recovered COVID-19 patients. Furthermore, hamsters immunized with mtdVSV-S were completely protected against SARS-CoV-2 challenge. Thus, mtdVSV is a safe and highly effective vector to deliver SARS-CoV-2 vaccine.
KEYWORDS: SARS-CoV-2, VSV, vaccine, mRNA cap methyltransferase
INTRODUCTION
In December 2019, the sudden outbreak of a novel coronavirus disease 2019 (COVID-19) pandemic in Wuhan, Hubei Province, China, shocked the world, not only because this virus had never been isolated before, nor because it infected people, but because it spread from person to person so rapidly within China and to more than 200 other countries within 2 months (1–3). The causative agent was named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). As of 29 July 2021, 195,886,929 cases have been reported worldwide, with 4,189,148 deaths (∼2.1% mortality). Symptoms are primarily pneumonia, as with other human coronaviruses (CoVs), such as severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) and Middle East respiratory syndrome coronavirus (MERS-CoV). Currently, remdesivir and convalescent plasma are the only two U.S. Food and Drug Administration (FDA)-approved treatments for emergency use in COVID-19 patients (4). Excitingly, several SARS-CoV-2 vaccines based on mRNA, inactivated virus, and adenovirus vectors (Ad5-nCoV and ChAdOx1) have been approved for emergency use in humans. Since starting the global COVID-19 vaccination campaign in January 2021, the number of new COVID-19 cases reported across the world has declined, according to the World Health Organization (WHO). However, the durability of the current vaccines is unknown, as is whether or how well they will protect against recently emergent SARS-CoV-2 variants. Thus, additional development of alternative vaccine strategies continues to be warranted.
SARS-CoV-2 belongs to the genus Betacoronavirus, subfamily Coronavirinae, and family Coronaviridae. CoVs are enveloped positive-sense viruses containing the largest RNA genomes, which range from 27 to 32 kb in length. The surface of CoV virions is the spike (S) protein, a “class 1” fusion protein that mediates receptor binding, fusion activity, and cellular entry (5–7). The S protein can be further divided into two subunits, S1 and S2. The S1 subunit contains the receptor-binding domain (RBD) at the top of the S protein. The S2 subunit is responsible for membrane-fusing activity and contains the fusion peptide, heptad repeat 1, heptad repeat 2, transmembrane, and cytoplasmic tail domains. On the virions, the S protein is in its trimeric prefusion form. Upon triggering, the S1 domain is released, exposing S2, which refolds, extending its fusion peptide into the target cell membrane to initiate fusion of the virion membrane with the target cell plasma membrane (5, 6). Importantly, the S protein is a primary target for neutralizing antibodies (NAbs) that protect the host from reinfection (8). Thus, the S protein is the major focus for CoV vaccine development.
Vesicular stomatitis virus (VSV) is a member in the family Rhabdoviridae. VSV is an animal virus that infects cattle, horses, and pigs. The genome of VSV is a nonsegmented negative-sense (NNS) RNA that is approximately 11 kb in length, encoding five structural proteins arranged in the following order: 3′-N-P-M-G-L-5′ (9). In 1995, an infectious VSV cDNA clone was established, which allows rescue of recombinant VSV (rVSV) expressing foreign antigens (10, 11). Since then, hundreds of foreign antigens have been inserted into and expressed from VSV for a variety of purposes, including vaccines, gene delivery, oncolytic therapies, and antibody production (12). A VSV-based vaccine is particularly attractive for protection against highly pathogenic agents. To date, VSV-based vaccine candidates have been developed for filoviruses (Ebola virus and Marburg virus) (13, 14), arenaviruses (Lassa virus and lymphocytic choriomeningitis virus [LCMV]) (15), henipaviruses (Nipah virus and Hendra virus) (16), human coronaviruses (SARS-CoV-1 and MERS-CoV) (17, 18), and flaviviruses (Zika virus) (19, 20). These rVSV-based vaccines have been shown to be highly effective in small animal models, nonhuman primates, and, in some cases, humans (12). In 2019, a VSV-based vaccine expressing Ebola virus glycoprotein (GP) in place of the VSV glycoprotein (G) was approved by the U.S. Food and Drug Administration (FDA) (21). This is the first rVSV-based vaccine approved for human use. Importantly, this vaccine was shown to be safe and highly efficacious in protecting against Ebola virus infection in Africa during the 2014 to 2016 outbreaks (13).
Soon after the outbreak of SARS-CoV-2, Case et al. constructed a replication-competent rVSV Indiana strain-based SARS-CoV-2 vaccine candidate (rVSV-eGFP-SARS-CoV-2) in which the native VSV G gene was replaced by the S gene of SARS-CoV-2 (22). Mice immunized with rVSV-eGFP-SARS-CoV-2 developed high titers of SARS-CoV-2-specific NAbs and showed profoundly reduced viral infection and inflammation in the lungs after SARS-CoV-2 challenge (23). A similar strategy has been reported for construction of an rVSV New Jersey strain-based SARS-CoV-2 vaccine candidate capable of protecting hamsters against SARS-CoV-2 challenge (24).
In this study, we used a different approach to develop attenuated rVSV-based SARS-CoV-2 vaccine candidates. We took advantage of the fact that for all NNS RNA viruses, the large (L) polymerase protein possesses an mRNA cap methyltransferase (MTase) activity that is essential for mRNA stability, protein translation, and innate immune evasion (25–28). Recombinant viruses defective in MTase are genetically stable and are highly attenuated in vitro and in vivo (26–28). We generated a panel of MTase-defective recombinant rVSV (mtdVSV)-based SARS-CoV-2 vaccine candidates expressing full-length S, S1, or several versions of the RBD. All of these recombinant viruses grew to high titers in cell culture, and SARS-CoV-2 S protein and truncations were highly expressed by the mtdVSV vector. These mtdVSV-based vaccine candidates were completely attenuated in both immunocompetent and immunocompromised mice. Among these vaccine candidates, mtdVSV expressing full-length S protein induces high levels of SARS-CoV-2-specific NAbs and Th1-biased T-cell immune responses in mice. Syrian golden hamsters immunized with mtdVSV-S triggered SARS-CoV-2-specific NAbs at higher levels than those in convalescent plasma from recovered COVID-19 patients. Furthermore, hamsters immunized with mtdVSV-S were completely protected against SARS-CoV-2 challenge, including viral replication in lung and nasal turbinate and lung pathology. These findings demonstrate that the mtdVSV-based S vaccine candidate is a safe and highly efficacious vaccine candidate against SARS-CoV-2 infection.
RESULTS
Recovery of mtdVSVs expressing SARS-CoV-2 S and S truncations.
To enhance the safety of VSV as a vector, we modified the viral mRNA cap methyltransferase (MTase) activity as a means to attenuate the virus. We previously showed that a single point mutation (D1762A) in the MTase catalytic region of the large (L) polymerase protein abolished both mRNA cap guanine-N-7 (G-N-7) methylation and ribose 2′-O methylation (28). The resultant recombinant virus (rVSV-D1762A) is highly attenuated in cell culture, as well as in mice (26). Thus, we used rVSV-D1762A as a backbone to generate rVSV expressing SARS-CoV-2 S proteins. Briefly, the full-length S, S1, RBD1, and RBD2 of SARS-CoV-2 were cloned as separate gene units into the G and L gene junction in the VSV-D1762A plasmid backbone (Fig. 1A). Using reverse genetics, we recovered all recombinant viruses, namely, rVSV-D1762A-S, rVSV-D1762A-S1, rVSV-D1762A-RBD1, and rVSV-D1762A-RBD2 (Fig. 1B). We also constructed recombinant VSV (rVSV-SCoV1) expressing the S protein of SARS-CoV-1 in the wild-type VSV backbone and used it as a control in our experiments.
FIG 1.

Recovery and characterization of mtdVSVs expressing SARS-CoV-2 S proteins. (A) Strategy for insertion of SARS-CoV-2 S and its variants into the VSV genome. The codon-optimized full-length S, S1, RBD1, and RBD2 genes were amplified by PCR and inserted into the same position at the gene junction between G and L into the genome of the VSV Indian strain. Domain structure of the S protein. SP, signal peptide; RBD, receptor-binding domain; RBM, receptor-binding motif; FP, fusion peptide; HR, heptad repeat; CH, central helix; TM, transmembrane domain; CT, cytoplasmic tail. Organization of the negative-sense VSV genome. N, nucleocapsid gene; P, phosphoprotein gene; M, matrix protein gene; G, glycoprotein; L, large polymerase gene. A star indicates the D1762A mutation in the MTase catalytic site in the L protein. (B) Plaque morphology of rVSV expressing SARS-CoV-2 S antigens. The plaques for rVSV and rVSV-SCoV-1 were developed after 24 h of incubation in Vero CCL-81 cells, whereas the plaques for all other viruses were developed at 48 h of incubation. An average number of 20 plaques for each virus is indicated. (C) Single-step growth curve. Confluent BSRT7 cells in 6-well plates were infected with each virus at a multiplicity of infection (MOI) of 1.0. After 1 h of absorption, fresh Dulbecco’s modified Eagle’s medium (DMEM) with 2% fetal bovine serum (FBS) was added. Aliquots (50 μl) of cell culture supernatants were harvested at the indicated time points, and virus titers were determined by plaque assay. Data are geometric mean titers (GMTs) ± standard deviation from n = 3 biologically independent experiments.
After 24 h of incubation, rVSV formed large plaques with an average diameter of 2.94 mm (Fig. 1B). The plaque size of rVSV-SCoV1 was 2.21 mm. rVSV-D1762A formed small plaques with average size of 0.68 mm, even after 48 h of incubation. rVSV-D1762A expressing S proteins (rVSV-D1762A-S, rVSV-D1762A-S1, rVSV-D1762A-RBD1, and rVSV-D1762A-RBD2) formed significantly smaller plaques (0.41 to 0.45 mm) than those formed by the parental rVSV-D1762A (Fig. 1B), suggesting that the insertion of the S gene or S gene truncations further attenuated the rVSV-D1762A virus. All recombinant viruses were plaque purified and sequenced. All viruses contained the desired S insertion and attenuating mutation. No additional mutations were found in the VSV genome or S gene.
We next compared the replication kinetics of each recombinant virus in cell culture (Fig. 1C). Briefly, confluent BSRT7 cells were infected by each virus at a multiplicity of infection (MOI) of 1.0, cell culture supernatants were harvested at the indicated time points, and the viral titer was determined by plaque assay. The wild-type rVSV grew to a peak titer of 4.79 × 109 PFU/ml at 18 h postinoculation, and cells were completely killed by 24 h. The highest titer (2.29 × 109 PFU/ml) of rVSV-SCoV1 was observed at 24 h. The parental rVSV-D1762A had a significant delay in replication and reached its peak titer of 2.29 × 109 PFU/ml at 36 h postinoculation; cells were killed by 60 h. All rVSV-D1762A expressing SARS-CoV-2 S proteins grew to similar peak titers at 48 h but were delayed in replication compared to the parental rVSV-D1762A. These results demonstrated that insertion of SARS-CoV-2 S genes into rVSV-D1762A resulted in a further attenuation of rVSV-D1762A.
SARS-CoV-2 S and S truncations are highly expressed by mtdVSV.
We next examined the expression of S protein and S truncations by the VSV vector. Briefly, confluent BSRT7 cells were inoculated with each recombinant virus at an MOI of 3, cell lysates were harvested at 40 h postinfection, and proteins were detected by Western blotting using an antibody against SARS-CoV-1 S. As shown in Fig. 2A, rVSV-SCoV-1-infected cells displayed proteins of 190 kDa and 95 kDa, representing full-length S and a minor amount of its cleaved form. Conversely, in rVSV-D1762A-S-infected cells, a minor 190-kDa and a major 95-kDa form were detected (Fig. 2A), consistent with efficient cleavage of the full-length S protein to produce the S1 protein of SARS-CoV-2. A 95-kDa protein band was detected in rVSV-D1762A-S1-infected BSRT7 cells, as expected. Also, a 40-kDa and a 34-kDa protein were detected in rVSV-D1762A-RBD1- and rVSV-D1762A-RBD2-infected cells, respectively, consistent with the respective predicted sizes of the RBD1 and RBD2 fragments. In contrast, no specific band was detected in rVSV-D1762A-infected cells, as expected. We also cloned S, S1, RBD1, and RBD2 of SARS-CoV-2 into a pCI plasmid vector. S, S1, RBD1, and RBD2 proteins were all expressed in 293T cells at their expected size from the pCI vector (Fig. 2B). Therefore, the SARS-CoV-2 S proteins, S, S1, RBD1, and RBD2 were all expressed by the rVSV-D1762A and pCI vectors and were recognized by the SARS-CoV-1 S antibody.
FIG 2.

SARS-CoV-2 S and its truncations are highly expressed by mtdVSV. (A) Analysis of S protein expression by mtdVSV using SARS-CoV-1 antibody. BSRT7 cells in 6-well plates were infected with each recombinant virus at an MOI of 3.0. At 40 h postinfection, cells were lysed in 300 μl of lysis buffer, and 10 μl of lysate was analyzed by SDS-PAGE and blotted with anti-SARS-CoV-1 S protein antibody (top) or β-actin antibody (bottom). (B) Analysis of S protein expression by pCI using SARS-CoV-1 antibody. 293T cells were transfected with 2 μg of each plasmid. At 48 h posttransfection, cell lysates were collected for Western blot analysis using anti-SARS-CoV-1 S protein antibody. (C and D) Analysis of the expression of S and its truncations by mtdVSV using SARS-CoV-2 antibody. BSRT7 cells in 6-well plates were infected with each recombinant virus at an MOI of 1.0. At 20 h or 28 h postinfection, cells were lysed in 300 μl of lysis buffer, and 10 μl of lysate or supernatant was analyzed by SDS-PAGE and blotted with anti-SARS-CoV-2 S protein antibody (top), VSV G antibody (middle), or β-actin antibody (bottom). Western blots shown are the representatives of three independent experiments. (E and F) Analysis of the incorporation of S into VSV virions. Cell culture supernatants were collected from 10 confluent T150 flasks of BSRT7 cells were infected by rVSV-D1762A or rVSV-D1762A-S. Cell debris were removed by centrifugation at 10,000 × g for 5 min. Virus particles were purified through 10% sucrose cushion (E, left), followed by 20 to 50% sucrose gradient purification (F, left). Aliquots consisting of 3 μg of total protein from the 10% sucrose cushion (E, left) and 10 μg from the sucrose gradient purification (F, left) were analyzed by SDS-PAGE and stained with Coomassie blue. Duplicated samples were also blotted with anti-SARS-CoV-2 S protein antibody (E and F, right). SDS-PAGE and Western blots shown are the representatives of two independent experiments.
Protein expression was also verified using SARS-CoV-2 S antibody (Fig. 2C and D). For this purpose, cell culture supernatants and lysates were harvested at 20 h and 28 h following virus infection at an MOI of 1. Two protein bands (190 and 95 kDa) were detected in lysates of rVSV-SCoV-1 and rVSV-D1762A-S-infected cells (Fig. 2C). One protein band (95 kDa) was detected in rVSV-D1762A-S1-infected lysates. Interestingly, S1 was detected in the cell supernatant of rVSV-D1762A-S and minor amounts of both full-length S and S1 were detected in the supernatant of rVSV-SCoV-1. RBD1 and RBD2 were detected in lysates, and RBD1 (but not RBD2) was detected in the cell culture supernatant (Fig. 2D). These data demonstrate that S, S1, RBD1, and RBD2 were highly expressed by rVSV-D1762A and that SARS-CoV-1 S protein cross-reacted with the SARS-CoV-2 S antibody.
SARS-CoV-2 S is incorporated into rVSV-D1762A-S virions.
To examine the incorporation of SARS-CoV-2 S into the VSV virions, we generated highly purified virions of rVSV-D1762A and rVSV-D1762A-S using 10% sucrose cushion ultracentrifugation, followed by 20 to 50% sucrose gradient ultracentrifugation. The band containing VSV particles was collected, ultracentrifuged, and the final pellets were resuspended in NTE buffer (100 mM NaCl, 10 mM Tris, and 1 mM EDTA [pH 7.4]). Aliquots consisting of 3 μg of each virus purified through a 10% sucrose cushion (Fig. 2E, left) and 10 μg of virus particles purified through a 20 to 50% sucrose gradient (Fig. 2F, left) were resolved by SDS-PAGE. As expected, five VSV proteins, namely L, G, P, N, and M, were detected in both rVSV-D1762A and rVSV-D1762A-S by SDS-PAGE. Next, a Western blot was performed to examine whether S protein is present in these highly purified virions. The S protein was detected in virions of rVSV-D1762A-S pelleted through a 10% sucrose cushion (Fig. 2E, right) and the band from the 20 to 50% sucrose gradient (Fig. 2F, right) purification steps. As expected, the S protein was not present in virions of rVSV-D1762A. Thus, these results suggest that SARS-CoV-2 S is incorporated into rVSV-D1762A-S virions.
mtdVSV-based vaccine candidates are attenuated and immunogenic in IFNAR1−/− mice.
IFNAR1−/− mice lack the type I interferon receptor (IFNAR), and for that reason they are highly susceptible to VSV infection (29). A dose of 50 PFU of wild-type VSV is lethal to IFNAR1−/− mice (29). IFNAR1−/− mice were inoculated intramuscularly with 106 PFU of rVSV-D1762A-RBD1, rVSV-D1762A-RBD2, rVSV-D1762A-S1, or rVSV-D1762A-S (Fig. 3A). We used a relatively high dose (106 PFU) of recombinant virus to determine if the mtdVSV-based vaccine candidates were attenuated in the immunocompromised mice. Importantly, IFNAR1−/− mice inoculated with rVSV-D1762A-RBD1, rVSV-D1762A-S1, or rVSV-D1762A-S did not show any abnormal reactions or body weight loss compared to the mock-inoculated (phosphate-buffered saline [PBS]) control mice (Fig. 3B). Mice in the rVSV-D1762A-S1 group had more weight gain than those in the PBS control group, probably because mice in this group were relatively younger and gained weight faster. IFNAR1−/− mice in the rVSV-D1762A-RBD2 group had mild clinical signs (such as ruffled coat) and had approximately 10% weight loss at day 7 but quickly recovered by day 10 (Fig. 3B). All mice in all groups remained healthy, and weight gains were indistinguishable from those in mock-infected controls, except for the rVSV-D1762A-S1 group, which gained more weight than the PBS control or other groups. Therefore, these results demonstrate that all of these mtdVSV-based vaccine candidates were highly attenuated in IFNAR1−/− mice.
FIG 3.

Attenuation and immunogenicity of rVSVs expressing SARS-CoV-2 antigens in IFNAR1−/− mice. (A) Immunization schedule in IFNAR1−/− mice. IFNAR1−/− mice (n = 5) were inoculated intramuscularly with phosphate-buffered saline (PBS) or 1.0 × 106 PFU of each of the rVSV-based vaccine candidates. Blood was collected from each mouse at weeks 2, 4, 6, and 10 for antibody detection. (B) Body weight changes after mtdVSV inoculation. Body weight was monitored every 3 to 4 days until week 6 postinoculation. (C) Measurement of SARS-CoV-2 RBD-specific antibody by enzyme-limited immunosorbent assay (ELISA). Highly purified RBD protein was used as the coating antigen for the ELISA. The dotted line indicates the detectable level at the lowest dilution. (D) Measurement of SARS-CoV-2 S-specific antibody by ELISA. Highly purified prefusion S (preS) protein was used as the coating antigen for the ELISA. The dotted line indicates the detectable level at the lowest dilution. Data are expressed as the geometric mean titers (GMTs) of five mice in each group ± standard deviation. Data were analyzed using two-way analysis of variance (ANOVA) (*, P < 0.05; **, P < 0.01).
At weeks 2, 4, 6, and 10 postimmunization, serum was collected from each mouse, and SARS-CoV-2-specific antibodies were measured by enzyme-linked immunosorbent assay (ELISA) using RBD or prefusion S (preS) proteins as antigens. All four mtdVSV-based vaccine candidates induced RBD-specific antibodies as early as week 2 postimmunization (Fig. 3C). IFNAR1−/− mice inoculated with rVSV-D1762A-S induced significantly higher levels of RBD-specific antibodies than those in the rVSV-D1762A-S1, rVSV-D1762A-RBD1, and rVSV-D1762A-RBD2 groups during the entire experimental period (P < 0.05). In addition, antibodies induced by rVSV-D1762A-S1, rVSV-D1762A-RBD1, and rVSV-D1762A-RBD2 declined to the detection limit by week 6. When preS protein was used as the ELISA antigen, rVSV-D1762A-S induced a high level of S-specific antibodies by week 2, which continued to rise though week 10 (Fig. 3D). However, preS-binding antibodies in all other groups were at the detectable limit level during the entire experimental period. These results showed that rVSV-D1762A-S is the most immunogenic vaccine candidate in IFNAR1−/− mice.
mtdVSV-based vaccine candidates are immunogenic in BALB/c mice.
We next performed immunization experiments in immunocompetent mice. Four-week-old BALB/c mice were immunized intramuscularly with 106 PFU of rVSV-D1762A, rVSV-D1762A-RBD1, rVSV-D1762A-RBD2, rVSV-D1762A-S1, rVSV-D1762A-S, or 50 μg of plasmid DNA vaccines (pCI-S, pCI-S1, or pCI-RBD1) (Fig. 4A). At week 2, all groups were boosted with the same dose. No abnormal reactions or weight loss were observed in any group (Fig. 4B). At week 2, some mice were positive for the RBD-specific antibodies, including 3 pCI-S mice, 1 rVSV-D1762A-S1 mouse, and 2 rVSV-D1762A-S mice (Fig. 4C). At week 4, all 5 rVSV-D1762A-S mice were antibody positive (Fig. 4D), whereas in the other groups, the numbers of positive mice and their antibody levels remained the same except for one mouse each in the pCI-S1 and pCI-RBD1 groups that also became antibody positive. We decided to terminate all groups with no or low levels of antibodies at week 4 and continued to monitor the antibody response in the rVSV-D1762A-S and pCI-S groups at weeks 6 and 8. Results showed that antibody titers in rVSV-D1762A-S group remained stable and were significantly higher than antibody titers in the pCI-S group during weeks 6 and 8 (P < 0.05 and P < 0.01) (Fig. 4E). These results indicate that rVSV-D1762A-S is the most immunogenic of the vaccine candidates tested and that pCI and rVSV-D1762A expressing RBDs and S1 are poorly immunogenic.
FIG 4.

Attenuation and immunogenicity of rVSVs expressing SARS-CoV-2 antigens in BALB/c mice. (A) Immunization schedule in BALB/c mice. BALB/c mice (n = 5) were inoculated intramuscularly with PBS or with 1.0 × 106 PFU of each of the rVSV-based vaccine candidates, or with 50 μg of plasmid DNA vaccine. Two weeks later, animals were boosted with same construct at the same dose. Blood was collected from each mouse at weeks 2, 4, 6, and 8 for antibody detection. (B) Body weight changes after mtdVSV inoculation. Body weight of selected groups was monitored every 3 to 4 days until week 6 postinoculation. (C and D) Measurement of SARS-CoV-2 RBD-specific antibody by ELISA at weeks 2 (C) and 4 (D). Highly purified RBD protein was used as the coating antigen for the ELISA. The dotted line indicates the detectable level at the lowest dilution. (E) Comparison by ELISA of antibody response between rVSV-D1762A-S and pCI-S. Highly purified RBD protein was used as the coating antigen for the ELISA. Data are expressed as the geometric mean titers (GMTs) of five mice in each group ± standard deviation. Data were analyzed using Student’s t test (*, P < 0.05; **, P < 0.01.
rVSV-D1762A-S is highly attenuated in replication in the lungs and the brain of mice.
Having demonstrated that rVSV-D1762A-S is the most immunogenic vaccine candidate, we further evaluated the pathogenesis and replication of rVSV-D1762A-S in mice. Briefly, 4- to 6-week-old C57BL/6J mice were intranasally inoculated with 106 PFU of rVSV, rVSV-D1762A, or rVSV-D1762A-S (Fig. 5A). Mice infected with rVSV developed severe VSV-associated clinical signs at days 3 and 4. No clinical signs were observed for mice in the rVSV-D1762A or rVSV-D1762A-S groups. At day 4, mice were terminated, and lungs and brains were isolated for virus titration. As expected, 3.37 × 105 and 5.65 × 105 PFU/g tissue of virus was detected in lungs (Fig. 5B) and brains (Fig. 5C) of rVSV-infected mice, respectively. In contrast, no infectious virus was detected in the rVSV-D1762A or rVSV-D1762A-S groups (Fig. 5B and C). Next, we also quantified VSV genomic RNA copies by real-time reverse transcription-PCR (RT-PCR). High numbers of VSV genomic RNA copies (1.8 to 1.9 × 109 RNA copies/mg tissue) were detected in lung (Fig. 5D) and brain tissues (Fig. 5E) in rVSV-infected mice. However, VSV genomic RNA was near or below the detection limit for both rVSV-D1762A and rVSV-D1762A-S groups (Fig. 5D and E). Therefore, these data further demonstrated that rVSV-D1762A-S is highly attenuated in replication in mice.
FIG 5.

Attenuation and immunogenicity of rVSVs expressing SARS-CoV-2 antigens in C57BL/6J mice. (A) Replication of VSV in C57BL/6J mice. C57BL/6J mice (n = 5, female) were inoculated intranasally with PBS or with 1.0 × 106 PFU of rVSV, rVSV-D1762A, or rVSV-D1762A-S. VSV titer in lungs (B) and brains (C). At day 4 after inoculation, mice were sacrificed, and lungs and brains were collected for virus titer determination by plaque assay. Viral titers are the geometric mean titer (GMT) of 5 animals ± standard deviation. The limit of detection (LoD) is 2.0 log10 PFU per gram of tissue (dotted line). VSV genomic RNA copies in lungs (D) and brains (E). Total RNA was extracted from each lung and brain sample. VSV genomic RNA copies were quantified by real-time reverse transcription-PCR (RT-PCR) using primers annealing to the VSV leader sequence and the N gene. Black bars indicate GMT of 5 mice in each group. The dotted line indicates the detection limit. (F) Immunization schedule in C57BL/6J mice. C57BL/6J mice (n = 10, 5 male and 5 female) were inoculated (half subcutaneously and half intranasally) with PBS or with 1.0 × 106 PFU of each of the mtdVSV-based vaccine candidates. Two weeks later, animals were boosted with same virus at the same dose. Blood was collected from each mouse at week 4 for antibody detection. (G) Measurement of SARS-CoV-2 S-specific antibody by ELISA. Highly purified prefusion S protein was used as the coating antigen for the ELISA. The dotted line indicates the detectable level at the lowest dilution. (H) Measurement of SARS-CoV-2-specific neutralizing antibodies (NAbs) by plaque reduction/neutralization test (PRNT). Antibody titer was determined by PRNT. The 50% plaque reduction/neutralization titer (PRNT50) was calculated from each serum sample. The dotted line indicates the detectable level at the lowest dilution. (I) Measurement of NAbs by a lentivirus-based pseudotype neutralization assay. The 50% inhibitory concentration (IC50) from each serum sample was calculated. Ten sera from the rVSV-D1762A-S and rVSV-D1762A-S1 groups and 5 sera from rVSV-D1762A group were used for this assay. Data are expressed as the geometric mean titers (GMTs) of 5 mice in each group ± standard deviation. Data were analyzed using one-way ANOVA (*, P < 0.05; ****, P < 0.0001).
mtdVSV-based vaccine candidates induce strong Th1-biased T-cell immunity.
Next, we selected rVSV-D1762A-S and rVSV-D1762A-S1 for further characterization of antibody and T-cell responses in C57BL/6J mice. Briefly, 4-week-old C57BL/6J mice were immunized with rVSV-D1762A-S, rVSV-D1762A-S1, or rVSV-D1672A via a combination of intranasal and subcutaneous routes (5.0 × 105 PFU for each route) (Fig. 5F). Two weeks later, animals were boosted via the same routes at the same dose. At week 4 postimmunization, all 10 mice in the rVSV-D1762A-S group developed high levels of S-specific antibodies, whereas only 2 out of 10 mice in the rVSV-D1762A-S1 group were above the detection limit (Fig. 5G). Next, sera were also tested for NAbs. In a SARS-CoV-2 plaque reduction/neutralization test (PRNT), 8 out of 10 mice in the rVSV-D1762A-S group had a high level of NAbs, and the other two mice were at the minimal detectable level. The average 50% plaque reduction/neutralization titer (PRNT50) in this group was 1:486 (Fig. 5H). In the pseudovirus neutralization assay, 9 out of 10 mice in the rVSV-D1762A-S group were NAb positive. The average 50% inhibitory concentration (IC50) in this group was 1:887 (Fig. 5I). However, all mice in the rVSV-D1762A-S1 group had at least minimally detectable levels of NAbs in both assays.
At week 4, 5 female mice in each group were terminated, and their splenocytes were used to characterize vaccine-induced T-cell immunity. We first quantified SARS-CoV-2 antigen-specific gamma interferon (IFN-γ)-producing T cells by enzyme-linked immunosorbent spot assay (ELISPOT). Mice immunized with rVSV-D1762A-S and rVSV-D1762A-S1 had significantly higher frequencies of S1 peptide-specific IFN-γ-producing T cells than those in the mice vaccinated using empty vector, rVSV-D1762A (Fig. 6A). The frequencies of antigen-specific T cells were higher in rVSV-D1762A-S-vaccinated mice than in the rVSV-D1762A-S1-vaccinated mice (Fig. 6A). Next, the four mice with the highest levels antigen-specific IFN-γ-producing T cells in the rVSV-D1762A-S and rVSV-D1762A-S1 groups were selected for further analysis by intracellular cytokine staining. After peptide stimulation ex vivo, CD8+ T cells producing one or more of the three signature Th1 cytokines, namely IFN-γ, tumor necrosis factor alpha (TNF-α), and interleukin 2 (IL-2) were detected in all four mice immunized with either the rVSV-D1762A-S or rVSV-D1762A-S1 vaccines (Fig. 6B). Moreover, like the ELISPOT data, the frequencies of antigen-specific T cells producing Th1 cytokines were higher in rVSV-D1762A-S vaccinated mice compared to the rVSV-D1762A-S1-vaccinated mice (Fig. 6B). Notably, antigen-specific cytokine-producing CD4+ T cells were barely detectable in both rVSV-D1762A-S and rVSV-D1762A-S1 groups (data not shown), indicating that the frequencies of CD4+ T cells were substantially lower than those of CD8+ T cells. Further analysis of vaccine-induced CD8+ T cells in mice vaccinated using rVSV-D1762A-S or rVSV-D1762A-S1 showed that the majority of the antigen-specific T cells produced all three Th1 cytokines, indicating their polyfunctional nature (Fig. 6C). Together, these data suggest that both the rVSV-D1762A-S and rVSV-D1762A-S1 vaccines can induce robust T-cell immunity that is predominated by CD8+ T cells capable of producing Th1 cytokines in C57BL/6J mice.
FIG 6.

mtdVSV-based SARS-CoV-2 vaccines induce strong Th1-biased T-cell immune responses. The 5 female mice from each group shown in Fig. 5 were terminated at week 4 postimmunization for the T-cell assays. (A) Enzyme-linked immunosorbent spot assay (ELISPOT) quantification of IFN-γ-producing T cells. Spot-forming cells (SFC) were quantified after the cells were stimulated by peptides representing N (S1 peptides, pink) and C (S2 peptides, green) termini of the SARS-CoV-2 spike protein. A peptide pool of covering N protein SARS-CoV-2 (N peptide) was used as a control. Data are means of 5 mice ± standard deviation. *, P < 0.05; **, P < 0.01, determined by unpaired t test. (B) Intracellular cytokine staining of T cells in vaccinated mice. Splenocytes of 4 rVSV-D1762A-S (left) or 4 rVSV-D1762A-S1 (right) vaccinated mice were stimulated ex vivo for 5 h with pools of N, S1, or S2 peptides (5 μg/ml each), followed by staining for intracellular cytokines and analysis using multicolor flow cytometry. Frequencies of CD8+ T cells expressing cytokines represent CD8+ T cells expressing IFN-γ, TNF-α, IL-2, IL-17a, IL-21, IL-4, or IL-10. Data are means of 5 mice ± standard deviation and were analyzed using an unpaired t test (*, P < 0.05). (C) Flow plots showing T cells producing the three Th1 cytokines. Splenocytes of rVSV-D1762A-S-vaccinated (upper) and rVSV-D1762A-S1-vaccinated (lower) mice were stimulated with either S1 peptides or no peptides (one representative plot). CD8+ T cells were gated to show the frequencies of antigen-specific T cells producing IFN-γ, TNF-α, or both cytokines. Additionally, CD8+ T cells expressing IL-2 are shown in pink.
rVSV-S is highly immunogenic in golden Syrian hamsters.
We next evaluated the immunogenicity of the VSV-based vaccines in golden Syrian hamsters, which are susceptible to VSV (24) and SARS-CoV-2 infection (30, 31). Five 4-week-old golden Syrian hamsters in each group were first immunized with 1.0 × 106 PFU of the parental rVSV-D1762A, rVSV-D1762A-S, or rVSV-D1762A-S1, and boosted with the same dose 3 weeks later (Fig. 7A). High ELISA antibody titers were detected in all 5 hamsters in the rVSV-D1762A-S group at week 2 after a single-dose vaccination (Fig. 7B). Antibody titers had further increased at week 4 after a booster immunization at week 3 and maintained a similar level at week 6 (Fig. 7B). However, the antibody titers of all 5 hamsters in the rVSV-D1762A-S1 group were at the minimally detectable levels at week 2 (Fig. 7B). After the booster immunization, only 1 out of 5 hamsters had an increased antibody response at week 4, which quickly declined by week 6 (Fig. 7B). Similarly, high levels of NAbs were detected in the rVSV-D1762A-S group as early as week 2, increasing to the average PRNT50 of 4,170 at week 4 and retaining a similar level at week 6 (Fig. 7C). NAbs in the rVSV-D1762A-S1 group were at the detection limit levels at weeks 2, 4, and 6 (Fig. 7C). Similar results were observed when the pseudovirus neutralization assay was used (Fig. 7D).
FIG 7.

The mtdVSV-based SARS-CoV-2 vaccine is highly immunogenic in Golden Syrian hamsters. (A) Immunization schedule in hamsters. Four-week-old female Golden Syrian hamsters (n = 5) were immunized (half subcutaneously and half intranasally) with 1.0 × 106 PFU rVSV-D1762A-S, rVSV-D1762A-S1, or rVSV-D1762A, or with PBS. Hamsters were boosted 3 weeks later. At weeks 2, 4, and 6, sera were collected for antibody detection. At week 7, hamsters were challenged with 1.0 × 105 PFU SARS-CoV-2. Unimmunized, unchallenged controls were inoculated with DMEM. (B) Measurement of SARS-CoV-2 S-specific antibody. Highly purified preS protein was used as a coating antigen for ELISA. The dotted line indicates the detectable level at the lowest dilution. (C) Measurement of SARS-CoV-2-specific NAbs by PRNT. Antibody titer was determined by a plaque reduction/neutralization test. Human convalescent-phase sera from recovered COVID-19 patients were used as side-by-side controls. The PRNT50 was calculated from each serum sample. The dotted line indicates the detectable level at the lowest dilution. (D) Measurement of NAbs by a lentivirus-based pseudotype neutralization assay. The IC50 from each serum sample was calculated. Data are expressed as the geometric mean titers (GMTs) of 5 hamsters. Data were analyzed using two-way ANOVA or Student’s t test (*, P < 0.05; ****, P < 0.0001).
We also compared the level of NAbs induced by rVSV-D1762A-S in hamsters with those in convalescent-phase sera collected from 10 patients recovered from COVID-19. NAb titers at week 2 from hamsters were similar to those of human convalescent-phase sera (P > 0.05) (Fig. 7C). Importantly, NAb titers at weeks 4 and 6 in rVSV-D1762A-S-immunized hamsters were significantly higher than those in these human convalescent-phase sera (P < 0.0001 and P < 0.01) (Fig. 7C). These results confirmed that rVSV-D1762A-S is highly immunogenic in hamsters.
rVSV-D1762A-S vaccination provides complete protection against SARS-CoV-2 replication in golden Syrian hamsters.
At week 7, hamsters in the rVSV-D1762A, rVSV-D1762A-S, or rVSV-D1762A-S1 groups were challenged intranasally with 1.0 × 105 PFU of SARS-CoV-2. The normal control hamsters were inoculated with Dulbecco’s modified Eagle’s medium (DMEM). At day 4 postchallenge, all animals were euthanized. The protective efficacy of the mtdVSV-D1762A-S-based vaccine candidate was evaluated based on clinical signs, weight loss, viral replication, viral RNA replication, cytokine responses in the lung, and lung histology and immunohistochemistry (IHC).
Hamsters in the rVSV-D1762A vector control group that were challenged with SARS-CoV-2 exhibited mild clinical symptoms, such as ruffled coat and weight loss (Fig. 8A). Hamsters in the rVSV-D1762A group started to lose weight at day 1 postchallenge and had approximately 5% weight loss by day 4 (Fig. 8A). The weight loss in the rVSV-D1762A group was significant compared to that in the normal control group (P < 0.001). Hamsters in the rVSV-D1762A-S1 groups had mild clinical signs and had similar weight loss from days 1 to 4 to that in to the rVSV-D1762A-challenged group (P > 0.05) (Fig. 8A). Importantly, hamsters in the rVSV-D1762A-S group did not have any abnormal reaction or significant weight loss. The body weight in the rVSV-D1762A-S group was not significantly different from that of the normal controls (P > 0.05) (Fig. 8A).
FIG 8.

rVSV-D1762A-S provides complete protection against SARS-CoV-2 challenge in Golden Syrian hamsters. (A) Dynamics of hamster body weight changes after SARS-CoV-2 challenge. The body weight for each hamster was measured daily and is expressed as a percentage of the body weight on the challenge day. The average body weight of 5 hamsters (n = 5) in each group is shown. SARS-CoV-2 titer in lungs (B) and nasal turbinate (C). At day 4 after challenge, 5 hamsters from each group were sacrificed, and lungs and nasal turbinates were collected for virus titer determination by plaque assay. Viral titers are the geometric mean titer (GMTs) of 5 animals ± standard deviation. The limit of detection (LoD) is ∼2.7 to 2.8 log10 PFU per gram of tissue (dotted line). SARS-CoV-2 genomic RNA copies in lungs (D), nasal turbinate (E), brain (F), liver (G), and spleen (H). Total RNA was extracted from the homogenized tissue using TRIzol reagent. SARS-CoV-2 genome copies were quantified by real-time RT-PCR using primers annealing to the 5′ end of the genome. SARS-CoV-2 subgenomic RNA copies in lungs (I), nasal turbinate (J), brain (K), liver (L), and spleen (M). SARS-CoV-2 subgenomic RNA copies were quantified by real-time RT-PCR using primers annealing to the N gene at the 3′ end of the genome. Black bars show GMTs of 5 hamsters in each group. The dotted line indicates the detection limit. Data were analyzed using two-way ANOVA (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
At day 4, all animals were euthanized, and major organs, including lungs, nasal turbinate, brain, liver, and spleen, were collected for virus titer determination by plaque assay. For the rVSV-D1762A control group, average titers of 2 × 106 and 3.8 × 105 PFU/g of SARS-CoV-2 were detected in lungs (Fig. 8B) and nasal turbinate (Fig. 8C), respectively. For the rVSV-D1762A-S1 group, 9.8 × 105 and 2.2 × 105 PFU/g of SARS-CoV-2 were detected in lungs (Fig. 8B) and nasal turbinate (Fig. 8C), respectively. Viral titers in lung tissue from the rVSV-D1762A-S1 group were significantly lower than those in the rVSV-D1762A group (P < 0.05). However, there was no significant difference in viral titer in nasal turbinate between the rVSV-D1762A-S1 and rVSV-D1762A groups (P > 0.05). Importantly, infectious SARS-CoV-2 was below the detection limit in the lungs and nasal turbinate in all animals in the rVSV-D1762A-S group (Fig. 8B and C, respectively). No infectious virus was detected in brain, liver, or spleen tissues in any group (data not shown).
We next determined if SARS-CoV-2 genomic RNA was present in these tissues using primers annealing to the 5′ end of the SARS-CoV-2 genome. For the rVSV-D1762A control group, averages of 3.8 × 106 and 1.4 × 107 genomic RNA copies/mg were detected in lungs (Fig. 8D) and nasal turbinate (Fig. 8E), respectively. Approximately 10-fold fewer genomic RNA copies were detected in lungs (Fig. 8D) and nasal turbinate (Fig. 8E) from the rVSV-D1762A-S1 group. Importantly, SARS-CoV-2 genomic RNA in lungs was below the limit of detection (LoD) in the rVSV-D1762A-S group, whereas an average of 102 RNA copies were detected in the nasal turbinate of this group. Interestingly, moderate levels (104 copies) of viral RNA were detected in brain tissue from the rVSV-D1762A-S1 and rVSV-D1762A groups, whereas levels of the SARS-CoV-2 genome were below the detection limit in the rVSV-D1762A-S group (Fig. 8F). The SARS-CoV-2 genome level was near or below the detectable limit in liver (Fig. 8G) and spleen (Fig. 8H) tissues for all groups.
During replication, SARS-CoV-2 generates both genomic RNA and subgenomic RNAs. Thus, we determined the levels of total viral RNA, including genomic and subgenomic RNA, using primers annealing to the N gene located at the 3′ end of the genome. Overall, the patterns of total RNA copies in lung (Fig. 8I), nasal turbinate (Fig. 8J), brain (Fig. 8K), liver (Fig. 8L), and spleen (Fig. 8M) were similar to those of genomic RNA in these tissues. Collectively, these results demonstrate that rVSV-D1762A-S vaccination provided complete protection against SARS-CoV-2 infection in hamsters, whereas rVSV-D1762A-S1 was unable to protect hamsters from SARS-CoV-2 infection.
rVSV-D1762A-S vaccination prevents SARS-CoV-2-induced cytokine storm in lungs.
SARS-CoV-2 infection causes a cytokine storm, which plays a key role in COVID-19 disease progression, leading to multiple organ failure and death (32). Thus, a successful vaccine candidate must prevent this cytokine storm in the lungs after SARS-CoV-2 infection. To test this, total RNA was extracted from the lungs of each group, and major inflammatory cytokines (IFN-α1, IFN-γ, IL-1β, IL-2, IL-6, and TNF-α) and chemokine (CXCL10) were quantified by reverse transcription-quantitative PCR (RT-qPCR) and normalized to the uninfected controls. Strikingly, IL-6 (Fig. 9A), IFN-γ (Fig. 9B), and CXCL10 (Fig. 9C) mRNA had approximately 51- to 64-, 26- to 35-, and 38- to 56-fold increases, respectively, in the rVSV-D1762A and rVSV-D1762A-S1 groups compared to the normal control group. In contrast, the increases of IFN-γ, IL-6, and CXCL10 in the rVSV-D1762A-S group were minimal—0.85-, 4.4-, and 0.36-fold, respectively. Levels of IFN-γ and IL-6 mRNAs were not statistically different between the rVSV-D1762A-S group and the normal control group (P > 0.05). In addition, increases in TNF-α (Fig. 9D) and IL-1β (Fig. 9E) in the rVSV-D1762A-S group were significantly less than those in the rVSV-D1762A and rVSV-D1762A-S1 groups (P < 0.05 and P < 0.01). Levels of IL-2 (Fig. 9F) in rVSV-D1762A, rVSV-D1762A-S1, and rVSV-D1762A-S groups were similar (P > 0.05). Hamsters in the rVSV-D1762A-S group had approximately 2.5-fold and 1.7-fold more IFN-α1 than those in the rVSV-D1762A and rVSV-D1762A-S1 groups, respectively (Fig. 9G). Overall, these results suggest that rVSV-D1762A-S vaccination strongly inhibits the cytokine storm in hamster lungs caused by a SARS-CoV-2 infection.
FIG 9.

rVSV-D1762A-S immunization prevents a cytokine storm in the lungs. Total RNA was extracted from lungs of hamsters terminated at day 4 after challenge with SARS-CoV-2. Hamster IL-6 (A), IFN-γ (B), CXCL10 (C), TNF-α (D), IL-1b (E), IL-2 (F), and IFN-α1 (G) mRNAs were quantified by real-time RT-PCR. GAPDH mRNA was used as an internal control. Data are shown as fold change in gene expression compared to normal animals (unimmunized and unchallenged) after normalization. Data were analyzed using two-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
rVSV-D1762A-S vaccination protects hamsters from SARS-CoV-2-induced lung pathology.
The right lung from each golden Syrian hamster was stained with hematoxylin and eosin (H&E), and the severity of histological changes was scored in a blind manner by a trained veterinary pathologist (Fig. 10). All lung tissues from the rVSV-D1762A group had extremely severe lung histopathological changes (average score of 4.0), including interstitial pneumonia, alveolar damage, multinucleated giant cells, extensive inflammation, bronchiolitis, pulmonary hemorrhage, mononuclear cell infiltration, and peribronchiolar inflammation (Fig. 11). Lung damage in the rVSV-D1762A-S1 group was also severe (average score of 3.4), although less severe than that in the rVSV-D1762A group (Fig. 10 and 11). Importantly, lung tissues from the rVSV-D1762A-S group had mild pathological changes (average score of 1.0) with occasional inflammation (Fig. 10 and 11). No lung pathology was found in the normal control group (score of 0) (Fig. 10 and 11). Next, all lungs from each group were subjected to immunohistochemistry using a SARS-CoV-2 N antibody. All lung sections from the rVSV-D1762A group had a large amount of SARS-CoV-2 N antigen distribution (Fig. 12). Significant amounts of N antigen were detected in the rVSV-D1762A-S1 group, although they were lower than those in the rVSV-D1762A group (Fig. 12). Importantly, no N antigen was detected in the lungs of the rVSV-D1762A-S group or the mock control group (Fig. 12). Collectively, these results demonstrate that rVSV-D1762A-S vaccination protects hamsters from lung pathology and prevents SARS-CoV-2 antigen expression in lungs.
FIG 10.
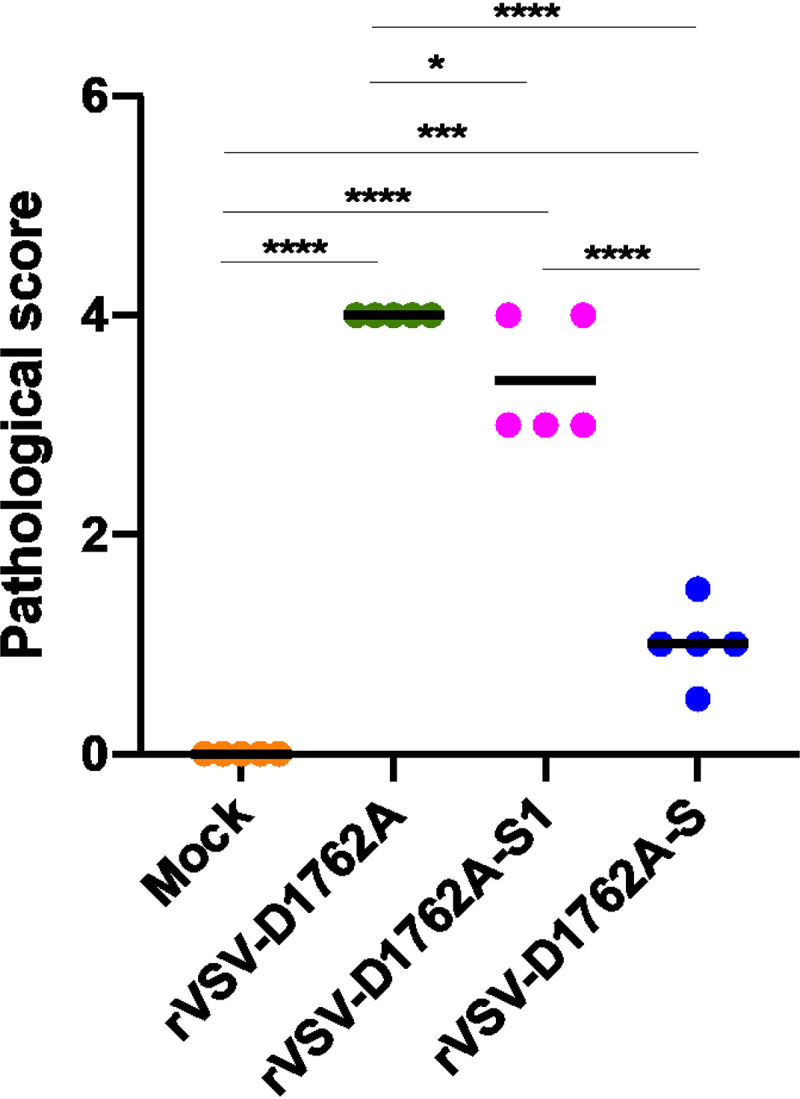
Lung pathology score after challenge with SARS-CoV-2. Fixed lung tissues from day 4 after SARS-CoV-2 challenge were embedded in paraffin, sectioned at 5 μm, deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E) for the examination of histological changes under light microscopy. Each slide was quantified based on the severity of histologic changes, which included interstitial pneumonia, alveolar damage, multinucleated giant cells, extensive inflammation, bronchiolitis, pulmonary hemorrhage, mononuclear cell infiltration, and peribronchiolar inflammation. 4 = extremely severe lung pathological changes; 3 = severe lung pathological changes; 2 = moderate lung pathological changes; 1 = mild lung pathological changes; 0 = no pathological changes.
FIG 11.

rVSV-D1762A-S immunization protects lung pathology. Hematoxylin and eosin (H&E) staining of lung tissue of hamsters euthanized at day 4 after SARS-CoV-2 challenge is shown. Micrographs with ×1, ×2, ×4, and ×10 magnification of a representative lung section from each group are shown. Scale bars are indicated at the left corner of each image.
FIG 12.

rVSV-D1762A-S immunization prevents SARS-CoV-2 antigen expression in the lungs. Immunohistochemistry (IHC) staining of lung sections from hamsters euthanized at day 4 after SARS-CoV-2 challenge is shown. Lung sections were stained with anti-SARS-CoV-2 N antibody. The same lung sections shown in Fig. 11 are presented to show the correlation of pathological change and SARS-CoV-2 antigen distribution. Micrographs with ×1, ×2, ×4, and ×10 magnification are shown. Scale bars are indicated at the left corner of each image.
DISCUSSION
Although several SARS-CoV-2 vaccines have been approved for use in humans, exploration of other vaccine candidates is needed because many questions concerning the durability, cross-protection, production costs, and temperature stability of the currently approved vaccines remain. In this study, we generated a panel of mtdVSVs expressing SARS-CoV-2 S and S truncations and tested their safety and immunogenicity in immunocompromised (IFNAR1−/− mice) and immunocompetent animals (BALB/c, C57BL/6J, and hamsters). All mtdVSV-based vaccine candidates grew to high titers in cell culture, and the S and S truncations were highly expressed. We found that all mtdVSV-based SARS-CoV-2 vaccines were highly attenuated and safe in all four animal models. Among them, rVSV-D1762A-S is the most immunogenic vaccine candidate, generating SARS-CoV-2-specific NAbs that were higher than those in human convalescent-phase sera from recovered COVID-19 patients, and inducing strong Th1-biased T-cell immune responses. Furthermore, hamsters immunized with rVSV-D1762A-S were completely protected against SARS-CoV-2 challenge. These data demonstrate that rVSV-D1762A-S is a safe and highly immunogenic and efficacious vaccine candidate for SARS-CoV-2.
VSV is an NNS RNA virus and an excellent platform for vaccine development. The natural hosts of VSV are cattle and swine, and as such, there is no preexisting immunity against VSV in the human population (33). The VSV genome is relatively small, allowing the accommodation of multiple foreign genes, and as such, it could be developed as a multivalent vaccine vector. VSV has an extremely high level of antigen expression, enabling the generation of strong systemic immune responses (12, 33). VSV grows to high titers in most cell lines, making vaccine production economically feasible. Most common administration routes, such as intranasal, intramuscular, intradermal, subcutaneous, and oral routes, are effective for VSV-based vaccines (12, 33). Unlike DNA viruses (e.g., adenovirus), VSV RNA genomes do not recombine with each other or integrate into host cell DNA, thereby increasing the safety of the vector. Finally, attenuated VSV is safe for use in humans. In 2019, the FDA approved the first VSV-based vaccine for Ebola virus, which has been shown to be safe and highly efficacious in protecting people against Ebola in Africa.
Wild-type VSV is virulent for wild and domestic animals, such as cattle, horses, deer, and pigs, and causes vesicular lesions in the mouth, tongue, lips, gums, teats, and feet. In rodents, wild-type VSV can cause neurotropic infection that results in a high mortality (34). Thus, VSV must be attenuated before it can be used as a vaccine vector. Glycoprotein snatching is one of the most attractive attenuation approaches for VSV vector (17, 18, 35). During the outbreaks of SARS-CoV-1 in 2003, Kapadia et al. constructed a replication-defective single-cycle VSV vector in which the VSV G gene was replaced by the SARS-CoV-1 S gene (18). Mice immunized with this recombinant virus (VSVΔG-S) generated a high NAb response. Similarly, during MERS-CoV outbreaks, Liu et al. reported a VSVΔG-MERS in which the VSV G gene was replaced by the MERS-CoV S gene (17). VSVΔG-MERS induced NAbs and T-cell responses in rhesus monkeys after a single intramuscular or intranasal immunization dose. Soon after the SARS-CoV-2 outbreak in December 2019, the Whelan group replaced the VSV G gene with the SARS-CoV-2 S gene in VSV-enhanced green fluorescent protein (eGFP), Indiana serotype (22). The resulting VSV-eGFP-SARS-CoV-2 grew poorly in Vero-E6 cells. However, truncation of the cytoplasmic tail of the SARS-CoV-2 S by 21 amino acids led to a recombinant virus (VSV-eGFP-SARS-CoV-2-SΔ21) that can grow to titers of 106 PFU/ml in Vero-E6 cells. Mice immunized with VSV-eGFP-SARS-CoV-2 induced high titers of SARS-CoV-2 NAbs (23). The immunized mice were transduced with adenovirus expressing human angiotensin converting enzyme 2 (hACE2) and challenged with SARS-CoV-2. Immunized mice showed significantly reduced viral replication and inflammation in the lungs, demonstrating the high immunogenicity of VSV-eGFP-SARS-CoV-2 (23). A similar strategy was reported for construction of an rVSV-ΔG-S vaccine candidate based on a VSV New Jersey serotype strain (24). Hamsters vaccinated with rVSV-ΔG-S at doses of 104, 105, 106, 107, or 108 PFU developed average NAb titers of 142, 789, 649, 1,080, and 2,201, respectively (24). All immunized hamsters were protected from SARS-CoV-2 challenge (24).
In this study, we modified the viral mRNA cap MTase as a means to attenuate the VSV vector. The mRNA cap MTase activity in VSV is unique in that a single MTase catalytic site in the L protein is essential for both G-N-7 and 2′-O methylation, and that 2′-O methylation is required for the subsequent G-N-7 methylation (25, 36). Thus, a single mutation (such as D1762A) in the MTase catalytic site abolished both G-N-7 and 2′-O methylations (28, 36). Previously, we showed that rVSV-D1762A is the most attenuated virus among all the mtdVSVs (26), and the rVSV-D1762A backbone has been recently used as a vector to deliver Zika virus antigen (37). Functionally, G-N-7 methylation is essential for mRNA stability and viral protein translation, whereas 2′-O methylation serves as a molecular signature for host innate immunity to discriminate self from nonself RNA (27, 38). Virus lacking G-N-7 and 2′-O methylations is not only highly attenuated in vitro and in vivo, but also induces a stronger innate immune response, which promotes subsequent adaptive immunity (27, 38). Thus, MTase has been used as a novel approach to attenuate paramyxoviruses, pneumoviruses, coronaviruses, and flaviviruses for the development of live attenuated vaccines (39–42).
In this study, we found that all mtdVSV-based vaccine candidates were not only highly attenuated in immunocompetent mice (BALB/c and C57BL/6J mice) but also in IFNAR1−/− mice, which are extremely sensitive to VSV infection. Because SARS-CoV-2 S is incorporated into rVSV-D1762A-S virions, we further evaluated whether incorporation of S into virions can alter the pathogenesis and replication of rVSV-D1762A-S in mice. Similarly to its parental rVSV-D1762A virus, infectious virus and copies of its RNA genomes were below the detection limit in the lungs or brains of mice infected by rVSV-D1762A-S, demonstrating that rVSV-D1762A-S is highly attenuated in vivo. Importantly, all mtdVSVs tested here replicate to a very high titer (109 PFU/ml) in cell culture, whereas glycoprotein-swapped VSV (such as VSV-SARS-CoV-2-SΔ21) only replicates to 106 PFU/ml. The much higher titer of rVSV-D1726A-S would make vaccine production more economically feasible. Despite the high attenuation phenotype, rVSV-D1762A-S is highly immunogenic in mice and hamsters, induces NAbs at levels significantly higher than those in convalescent plasma from COVID-19 recovered patients, and provides complete protection against viral replication in the upper and lower respiratory tracts, cytokine storm, and lung pathology.
Antibody-dependent enhancement (ADE) has been a concern for SARS-CoV-2 vaccine development because of the previous experience of ADE in some CoV vaccines (such as MERS-CoV and SARS-CoV-1) (43). It is thought that the excessive Th2-cytokine-biased responses and inadequate Th1-biased T-cell responses contribute to ADE (44, 45). Thus, an ideal SARS-CoV-2 vaccine should induce a high level of Th1- but not Th2-biased T-cell response. Importantly, we found that both rVSV-D1762A-S and rVSV-D1762A-S1 induce strong antigen-specific T-cell immunity that is predominated by CD8+ T cells capable of producing Th1 cytokines in C57BL/6J mice. A similar Th1-biased response was observed for a replication-competent recombinant VSV-ΔG-spike vaccine in C57BL/6J mice (24). Thus, VSV-based SARS-CoV-2 vaccine can reduce the risk of potential ADE and enhance the safety of the vaccine. Consistent with antibody responses, rVSV-D1762A-S induced significantly stronger T-cell immunity than rVSV-D1762A-S1 (P < 0.05). We also found that the frequencies of CD4+ T cells were barely detectable in both rVSV-D1762A-S- and rVSV-D1762A-S1-immunized animals. Interestingly, low frequencies of CD4+ T cells were also observed for other SARS-CoV-2 vaccines that express the SARS-CoV-2 S protein via mRNA, adenovirus, or measles virus (MeV) (46–48).
Unlike the glycoprotein-switching strategy, an advantage of using mtdVSV as a backbone is that it allows us to express and deliver RBDs or modified, nonfunctional S proteins such as the stabilized prefusion form of the S protein. This is important because the stabilized prefusion form of the glycoprotein protein is much more immunogenic than the native glycoprotein for many enveloped viruses, including paramyxovirus, pneumovirus, and HIV (49, 50). In fact, the Pfizer mRNA vaccine produces a trimerized form of the RBD, whereas the Moderna mRNA vaccine produces the stabilized prefusion S protein (48, 51). Using measles virus (MeV), another NNS RNA virus, as a vector, we recently demonstrate that the stabilized prefusion S protein of SARS-CoV-2 is significantly more potent than the native full-length S protein for inducing NAbs (52). Thus, the stabilized prefusion S is the most immunogenic antigen for use in subunit or vectored vaccines. Our data demonstrate that mtdVSVs expressing S1, RBD1, or RBD2 are poorly immunogenic, likely due to their production as monomers. Our future experiments will test mtdVSV delivery of the stabilized prefusion S protein trimer and trimerized S1 and RBDs in attempts to enhance immunogenicity.
In mid-2020, after we had begun the work presented in this report, the pharmaceutical company Merck announced that they were developing two SARS-CoV-2 vaccines based on the same two vector systems we were using, MeV and VSV. However, late in 2020, Merck announced that they were discontinuing these programs based on their phase 1 clinical studies. Specifically, they found that both vaccine candidates were generally well tolerated, but the immune responses were lower than those seen in natural SARS-CoV-2 infection and those reported for other SARS-CoV-2 vaccines, such as the Moderna and Pfizer mRNA vaccines. The compositions of their vaccine candidates have not been made public, but Merck had previously used a glycoprotein replacement strategy for construction of their rVSV-based vaccine for Ebola Zaire, VSV-ΔG-S, the first rVSV vaccine approved for use in humans (https://www.iavi.org/news-resources/press-releases/2020/iavi-and-merck-collaborate-to-develop-vaccine-against-sars-cov-2). Although we do not know whether rVSV-D1762A-S is more immunogenic than VSV-ΔG-S, it is more immunogenic than the 10 convalescent-phase sera from recovered COVID-19 patients that we tested. In the future, one strategy that is likely to further enhance the efficacy of the mtdVSV-based vaccine platform is to express a stabilized version of the prefusion S protein.
In summary, we have proved that mtdVSV is an effective vaccine vector for delivering the SARS-CoV-2 S protein. We showed that rVSV-D1762A-S is a safe and efficacious vaccine candidate, protecting animals from SARS-CoV-2 infection and lung disease in preclinical trials.
MATERIALS AND METHODS
Biosafety.
All experiments with infectious SARS-CoV-2 were conducted under biosafety level 3 (BSL3) or animal BSL3 (ABSL3) at The Ohio State University using standard operating procedures and were approved by the BSL3 Advisory Group, the Institutional Biosafety Committee (IBC), and the Institutional Animal Care and Use Committee (IACUC).
Cell culture.
Vero CCL81 (ATCC no. CCL-81), Vero E6 (ATCC no. CRL-1586), and 293T (ATCC no. CRL-3216) cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA). HEK293T cells expressing ACE2 were obtained from BEI Resources. BSRT7 cells, which stably express T7 RNA polymerase, are clones of BHK-21 cells. All cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS).
Virus strain.
The SARS-CoV-2 USA-WA1/2020 natural isolate (GenBank accession number MN985325) was obtained from BEI Resources (NR-52281) and amplified on Vero E6 cells. This strain was originally isolated from an oropharyngeal swab from a patient with respiratory illness in January 2020 in Washington, USA.
Animals.
Specific-pathogen-free (SPF) IFNAR1−/−, BALB/c, and C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Golden Syrian hamsters were purchased from Envigo (Indianapolis, IN).
VSV plasmid construction.
Genes encoding the full-length S protein of SARS-CoV-1 or the full-length S, S1, receptor-binding domain 1 (RBD1), or RBD2 proteins of SARS-CoV-2 were cloned into the infectious VSV plasmid cDNA of the Indiana strain. To attenuate VSV, we took advantage of a point mutation, D1762A, in the large (L) polymerase protein, which rendered a recombinant virus (rVSV-D1762A) that abolishes both mRNA cap G-N-7 and 2′-O methylation and is completely attenuated in mice (26, 28, 37). Briefly, full-length S, S1 (amino acids [aa] 1 to 698), and two different RBDs (RBD1 [aa 319 to 589] and RBD2 [aa 319 to 541]) were amplified from a codon-optimized S gene of the 2019-nCoV/USA-WA1/2020 strain (GenBank accession no. MN985325, synthesized by GenScript) by high-fidelity PCR. For two RBDs, the signal peptide derived from the human CD5 gene was fused to the amino terminus to enable secretion. These DNA fragments were digested with SmaI and XhoI and cloned into pVSV(+)GxxL (provided by Sean Whelan) carrying the D1762A mutation at the same sites. The resulting plasmids were designated pVSV(+)-D1762A-S, pVSV(+)-D1762A-S1, pVSV(+)-D1762A-RBD1, and pVSV(+)-D1762A-RBD2. In addition, the S gene of SARS-CoV-1 was cloned into the SmaI and XhoI sites in wild-type pVSV(+)-GxxL vector, which resulted in construction of pVSV(+)-SCoV-1. All of the constructs were confirmed by DNA sequencing.
Construction of DNA vaccine plasmids.
For side-by-side controls, S, S1 (aa 1 to 698), and two RBDs (RBD1 [aa 319 to 589] and RBD2 [aa 319 to 541] of SARS-CoV-2 were cloned into pCI vector (Promega), which resulted in the construction of pCI-S, pCI-S1, pCI-RBD1, and pCI-RBD2.
Recovery of rVSV expressing SARS-CoV-2 antigens.
Recovery of rVSV from the infectious clone was carried out as described previously (10, 28). Briefly, rVSV was recovered by cotransfection of a plasmid expressing the VSV RNA genome and support plasmids encoding VSV nucleocapsid complex (pN, pP, and pL) into BSRT7 cells infected with a recombinant vaccinia virus (vTF7-3) expressing T7 RNA polymerase (kindly provided by Bernard Moss) (53). At 96 h posttransfection, cell culture fluids were collected and filtered through a 0.2-μm filter, and the recombinant virus was further amplified in BSRT7 cells. Subsequently, the viruses were plaque purified as described previously (19). Individual plaques were isolated, and seed stocks were amplified in BSRT7 cells. The viral titer was determined by a plaque assay performed in Vero cells. The recombinant viruses with the D1762A mutation were designated rVSV-D1762A-S, rVSV-D1762A-S1, rVSV-D1762A-RBD1, and rVSV-D1762A-RBD2. The recombinant virus carrying the S gene of SARS-CoV-1 in a wild-type VSV background was designated rVSV-SCoV-1.
RT-PCR.
Viral RNA was extracted from rVSVs using a RNeasy minikit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. SARS-CoV-2 genes were amplified by a OneStep RT-PCR kit (Qiagen) using primers annealing to the VSV G gene at position 4524 (5′-CGAGTTGGTATTTATCTTTGC-3′) and the L gene at position 4831 (5′-GTACGTCATGCGCTCATCG-3′) (numbering refers to the complete VSV Indiana genome sequence). The amplified products were analyzed by 1% agarose gel electrophoresis and sequenced.
Single-cycle growth curves.
Confluent monolayers of BSRT7 cells were infected with individual viruses at a multiplicity of infection (MOI) of 1.0. After 1 h of absorption, the inoculum was removed, the cells were washed twice with Dulbecco’s modified Eagle’s medium (DMEM), fresh DMEM (supplemented with 2% fetal bovine serum) was added, and the infected cells were incubated at 37°C. Aliquots of the cell culture fluid were collected at the indicated intervals, and virus titers were determined by plaque assay in Vero cells in triplicate.
VSV and SARS-CoV-2 plaque assay.
Confluent Vero CCL81 cells in 6-well plates were infected with 10-fold serial dilutions of rVSV expressing SARS-CoV-2 antigens in DMEM. After absorption for 1 h at 37°C, cells were washed three times with DMEM and overlaid with 2 ml of DMEM containing low-melting-point agarose (0.25% wt/vol), 2% FBS, 0.12% sodium bicarbonate, 100 μg/ml streptomycin, 100 U/ml penicillin, 25 mM HEPES (pH 7.7), and 2 mM l-glutamine. After incubation at 37°C for 2 to 3 days, cells were fixed with 4% neutral buffered formaldehyde for 2 h. The overlays were removed, and the plaques were visualized after staining by crystal violet. SARS-CoV-2 plaque assay was the same as VSV plaque assay, except that it was carried out in Vero-E6 cells in 12-well plates and the infected cells were fixed at 2 days postinfection. The diameter of plaques for each virus were measured using ImageJ Software.
Detection of SARS-CoV-2 S antigen by Western blotting.
BSRT7 cells were infected with parental rVSV or rVSV expressing SARS-CoV-2 S antigens as described above. At the indicated time postinfection, cell culture medium was harvested and clarified at 5,000 × g for 15 min. In the meantime, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (catalog no. ab156034; Abcam). Proteins were separated by 12% SDS-PAGE and transferred to a Hybond enhanced chemiluminescence nitrocellulose membrane (Amersham) in a Mini Trans-Blot electrophoretic transfer cell (Bio-Rad). The blot was probed with rabbit anti-SARS-CoV-1 S (catalog no. 40592-T62; Sino Biological), SARS-CoV-2 S (catalog no. 40150-R007; Sino Biological), or RBD (catalog no. 40592-MP01; Sino Biological) antibody at a dilution of 1:2,000, followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody (Santa Cruz) at a dilution of 1:5,000. The blot was developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and exposed to BioMax MR film (Kodak).
Purification of rVSV-D1762A and rVSV-D1762A-S.
Purification of VSV was performed by a previously described method (28, 54). Briefly, 10 confluent T150 flasks of BSRT7 cells were infected by rVSV-D1762A or rVSV-D1762A-S at an MOI of 0.01. At 1 h postinoculation, 15 ml of DMEM (supplemented with 2% FBS) was added to the cells, which were then incubated at 37°C. After 72 h postinfection, cell culture supernatant was harvested by centrifugation at 10,000 × g for 5 min. Virus was concentrated by centrifugation at 40,000 × g for 90 min at 4°C in a Ty 50.2 rotor. The pellet was resuspended in NTE buffer (100 mM NaCl, 10 mM Tris, and 1 mM EDTA [pH 7.4]) and further purified through 10% sucrose-NTE by centrifugation at 150,000 × g for 1 h at 4°C in an SW50.1 rotor. The pellet was resuspended in 0.3 ml of NTE buffer. Virus was further purified by 20 to 50% sucrose-NTE gradient centrifugation at 150,000 × g for 4 h at 4°C in an SW50.1 rotor. The band containing VSV particles was collected by needle, diluted 5 times in NTE buffer, and centrifuged at 150,000 × g for 1 h at 4°C in an SW50.1 rotor. The final pellet was resuspended in 0.3 ml of NTE buffer. The protein content was measured by Bradford reagent (Sigma Chemical Co.).
Human sera.
Human serum samples were collected from 10 patients who had recovered from COVID-19. All human studies were conducted in compliance with all relevant local, state, and federal regulations and were approved by the Institutional Review Board (IRB) of The Ohio State University, Columbus, OH.
Animal experiments.
All animals were housed within university laboratory animal resources (ULAR) facilities of The Ohio State University under approved Institutional Animal Care and Use Committee (IACUC) guidelines (protocol no. 2009A0160 and 2020A00000053). Each inoculation group was separately housed in rodent cages under animal biosafety level 2 (ABSL2 for VSV) or ABSL3 (for SARS-CoV-2) conditions.
Immunization experiment in IFNAR1−/− mice.
Twenty-five 4- to 8-week-old specific-pathogen-free (SPF) female IFNAR1−/− mice (Jackson Laboratory, Bar Harbor, ME) were randomly divided into 5 groups (5 mice per group). Mice in group 1 were inoculated intramuscularly with PBS. Mice in groups 2 to 5 were inoculated intramuscularly with 1.0 × 106 PFU of rVSV-D1762A-S, S1, RBD1, or RBD2, respectively. After inoculation, the animals were evaluated twice every day for mortality and the presence of any symptoms of VSV infection, such as ruffled fur, hyperexcitability, tremors, circling, and paralysis. The body weight of each mouse was monitored every 3 to 4 days. Blood was collected at weeks 2, 4, 6, and 10 from each mouse to determine the SARS-CoV-2-specific antibody.
Immunization experiment in BALB/c mice.
Forty-five 4- to 6-week-old specific-pathogen-free female BALB/c mice (Jackson Laboratory) were randomly divided into 9 groups (5 mice per group). Mice in groups 1 to 5 were inoculated intramuscularly with 1.0 × 106 PFU of rVSV-D1762A, rVSV-D1762A-S, S1, RBD1, or RBD2. Mice in groups 6 to 9 were immunized intramuscularly with 50 μg of pCI, pCI-S, S1, or RBD1. After 2 weeks, mice in each group were boosted with respective antigen at the same dose via the same route. After inoculation, the animals were evaluated twice every day for mortality and the presence of any symptoms of VSV infection, as described above. The body weight of each mouse was monitored every 3 to 4 days. Blood was collected every 2 weeks from each mouse to determine the SARS-CoV-2-specific antibody.
Replication of VSV in C57BL/6J mice.
Twenty 4- to 6-week-old female SPF C57BL/6J mice were randomly divided into 4 groups (n = 5). Mice in groups 1 to 3 were intranasally inoculated with 1.0 × 106 PFU of rVSV, rVSV-D1762A, or rVSV-D1762A-S. Mice in group 4 were inoculated with PBS and served as the uninfected control. At day 4 postinoculation, all mice were euthanized, and lung and brain from each mouse were collected and homogenized for virus titration by plaque assay. In addition, total RNA was extracted from lung and brain samples and VSV genomic RNA copies were quantified by real-time RT-PCR using primers annealing to the VSV leader sequence and the N gene sequence, as described previously (55).
Immunization experiment in C57BL/6J mice.
Thirty 4- to 6-week-old SPF C57BL/6J mice were randomly divided into 3 groups (n = 10, 5 male and 5 female mice per group). Mice in groups 1 to 3 were intranasally inoculated with 1.0 × 106 PFU (half intranasal and half subcutaneous route) of parental rVSV-D1762A, rVSV-D1762A-S, and rVSV-D1762A-S1. After 2 weeks, mice in each group were boosted with the same virus at the same dose with the same route. After inoculation, the animals were evaluated twice every day for mortality and the presence of any symptoms of VSV infection, as described above. Blood was collected at week 4 from each mouse to determine the SARS-CoV-2-specific ELISA antibody and NAbs. At week 4 postinoculation, five female mice with from each group were euthanized with CO2, and spleens were collected for lymphocyte isolation for T-cell assay.
Immunization and challenge experiments in Golden Syrian hamsters.
Twenty 4-week-old female Golden Syrian hamsters (Envigo, Indianapolis, IN) were randomly divided into 4 groups (n = 5). Hamsters were initially housed in a BSL2 animal facility at The Ohio State University. Group 1 was inoculated with PBS and served as an unimmunized, unchallenged control. Groups 2 to 4 were immunized intranasally (5.0 × 105 PFU/hamster) plus subcutaneously (5.0 × 105 PFU/hamster) with rVSV-D1762A, rVSV-D1762A-S, and rVSV-D1762A-S1, respectively. Three weeks later, hamsters in groups 2 to 4 were boosted with the respective virus at the same dose via the same routes. At week 4 post booster immunization, hamsters in groups 2 to 4 were moved to an ABSL3 animal facility for challenge with SARS-CoV-2. For challenge studies, hamsters in groups 2 to 4 were anaesthetized with isoflurane and were intranasally inoculated with 40 μl DMEM or 1.0 × 105 PFU of SARS-CoV-2 in 40 μl DMEM. At day 4 postchallenge, all hamsters were euthanized. The left lung, nasal turbinate, and half each of brain, liver, and spleen from each hamster were collected and homogenized for both virus titration and RNA extraction. The right lung and half each of the brain, liver, and spleen were fixed for histology and immunohistochemistry (IHC) analysis as described below.
S and RBD protein purification.
The stabilized prefusion S (preS) protein (amino acids 1 to 1,273) and RBD (amino acids 319 to 541) of SARS-CoV-2 were cloned into pCAGGS and transfected into FreeStyle293F cells for protein expression. The secreted preS and RBD in cell culture supernatants were then purified via affinity chromatography. The purity of the protein was analyzed by SDS-PAGE and Coomassie blue staining. Protein concentration was measured using Bradford reagent (Sigma Chemical Co., St. Louis, MO).
Peptides.
A set of 181 peptides spanning the complete S protein of the USA-WA1/2020 strain of SARS-CoV-2 (GenPept accession number QHO60594) and a set of 57 peptides spanning the Urbani strain of SARS-CoV-2 (GenPept accession number AY278741) were obtained from BEI Resources (National Institute of Allergy and Infectious Diseases) (catalog no. NR-52402 and NR-52419, respectively). These peptides are 13 to 20 amino acids long, with 10-amino-acid overlaps. The S1 peptide pools contain 93 peptides representing the N-terminal half of the S protein (MFVFLVLLPL to AEHVNNSYE), and the S2 peptide pools contain 88 peptides representing the C-terminal half of the S protein (GAEHVNNSYE to VLKGVKLHYT). Peptides were dissolved in sterile water containing 10% dimethyl sulfoxide (DMSO).
ELISPOT assay.
Spleens of immunized C57BL/6J mice were aseptically removed 35 days after immunization and minced by pressing through cell strainers. Red blood cells were removed by incubation in 0.84% ammonium chloride, and, following a series of washes in RPMI 1640, cells were resuspended in RPMI 1640 supplemented with 2 mM l-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum. Antigen-specific T cells secreting IFN-γ were enumerated using anti-mouse IFN-γ enzyme-linked immunosorbent spot assay (ELISpot) assay (catalog no. CT317-PB5; U-CyTech). Cells were plated in 96-well polyvinylidene difluoride (PVDF) plates at 2 × 105 per well in duplicate, and stimulated separately with the SARS-CoV-2 peptide pools (2 μg/ml) or with concanavalin A (5 μg/ml, Sigma), or medium alone, as positive and negative controls, respectively. The plates were incubated for 42 to 48 h and then developed according to the manufacturer’s instructions. The number of spot-forming cells (SFC) was measured using an automatic counter (ImmunoSpot).
Quantification of intracellular cytokine production.
For detection of SARS-CoV-2-specific intracellular cytokine production, 106 cells were stimulated in 96-well round-bottomed plates with peptide pool (5 μg/ml), or with medium alone or phorbol myristate acetate (PMA)/ionomycin (BioLegend) as negative and positive controls, respectively, for 5 h in the presence of GolgiPlug (BD Biosciences). Following incubation, cells were surface stained for CD3, CD4, and CD8 for 30 min at 4°C, fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences), and intracellularly stained for IFN-γ, TNF-α, IL-2, IL-21, IL-17A, IL-10, and IL-4 for 30 min at room temperature. Dead cells were removed using the Live/Dead fixable near-infrared (near-IR) dead cell stain kit (Invitrogen). A positive response was defined as >3 times the background of the negative-control sample. The percentage of cytokine-positive cells was then calculated by subtracting the frequency of positive events in negative-control samples from that in test samples.
Flow cytometric analysis.
The following mouse-reactive antibodies (clone, catalog number, dilution) from BioLegend, BD Biosciences, and Thermo Fisher Scientific were used for analysis of T cells: CD3-phycoerythrin (PE)/cyanine 7 (145-2C11, 100319, 1:400), IFN-γ-PE/Dazzle 594 (XMG1.2, 505845, 1:400), TNF-α-brilliant violet 785 (MP6-XT22, 506341, 1:400), IL-17a Alexa Fluor 488 (TC11-18H10.1, 506909, 1:100), IL-21 allophycocyanin (APC) (mhalx21, 51-7213-80, 1:100), IL-4-brilliant violet 711 (11B11, 504133, 1:100), CD4-BUV 496 (GK1.5, 612952, 1:400), CD8-BUV737 (53-6.7, 612759, 1:400), IL-10-brilliant violet 510 (JES5-16E3, 563277, 1:100), and IL-2-PE (JES6-5H4, 12-7021-82, 1:200). Surface and intracellular staining were performed as described previously (56). Events were collected on an LSRFortessa X-20 flow cytometer (BD) following compensation with UltraComp eBeads (Invitrogen). Data were analyzed using FlowJo v10 (TreeStar).
Determination of SARS-CoV-2 titer in hamster tissues.
After SARS-CoV-2 challenge, the left lung, nasal turbinate, brain, liver, and spleen were collected. Organs were weighed and homogenized by hand with a mortar and pestle (Golden, CO) in 1 ml of sterile PBS. Each sample was subjected to 10-fold serial dilutions. The initial dilution of each tissue sample was 1:10. The presence of infectious SARS-CoV-2 was determined by plaque assay in Vero E6 cells in 12-well plates. The limit of detection (LoD) was calculated with the following formula: LoD = log10[([1/0.2] × 10)/average tissue weight], where 1 represents 1 plaque in a well, 0.2 represents a 0.2-ml tissue sample, and 10 is the lowest dilution.
Measurement of SARS-CoV-2 genomic and subgenomic RNA burden.
Total RNA was extracted from homogenized left lung, nasal turbinate, brain, liver, and spleen tissue samples using TRIzol reagent (Life Technologies, Carlsbad, CA). For total viral RNA (genome and subgenome), reverse transcription (RT) was conducted using a primer (5′-GTCATTCTCCTAAGAAGCTATTAAAATC-3′) targeting the 3′ untranslated region (UTR) of the SARS-CoV-2 genome and the SuperScript III transcriptase kit (Invitrogen, Carlsbad, CA). For genomic RNA, the RT primer (5′-GTGTCTTTGATTTCGAGCAAC-3′) was annealed to the 5′ UTR of the SARS-CoV-2 genome. The RT products were then used to perform real-time PCR using primers specifically targeting the nucleocapsid (N) gene of SARS-CoV-2 (forward, 5′-CATTGGCATGGAAGTCACAC-3′; reverse, 5′-TCTGCGGTAAGGCTTGAGTT-3′) or the 5′ end of the SARS-CoV-2 genome (forward, 5′-ACTGTCGTTGACAGGACACG-3′; reverse, 5′-ACGTCGCGAACCTGTAAAAC-3′) in a StepOne real-time PCR system (Applied Biosystems). A standard curve was generated using a plasmid encoding the full-length genome of SARS-CoV-2. Amplification cycles used were 2 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. The threshold for detection of fluorescence above the background was set within the exponential phase of the amplification curves. For each assay, 10-fold dilutions of standard plasmid or viral RNA were generated, and negative-control samples and double-distilled water (ddH2O) were included in each assay. After real-time qPCR, the threshold cycle (CT) value from each sample was converted into log10 viral RNA copies/mg tissue according to the standard curve. The RNA copies were calculated with the following formula: RNA copies/mg tissue = log10 [(no. of CT-converted copies/μl × 10 [2 μl from 20 μl total cDNA] × 25 [2 μl from 50 μl total RNA] × 10 [100 μl from 1 ml homogenized tissue])/tissue weight in mg]. The LoD is set as the maximum value of the normal control group. The exact log10 RNA copies/mg was reported for each sample.
Quantification of cytokines in lungs of hamsters.
Total RNA was extracted from lungs of golden Syrian hamsters, and IFN-α1, IFN-γ, IL-1b, IL-2, IL-6, TNF, and CXCL10 mRNAs were quantified by real-time RT-PCR (57, 58). GAPDH mRNA was used as an internal control. The cytokine mRNA of each group was expressed as fold change in gene expression compared to that in normal animals (unimmunized and unchallenged) after normalization by GAPDH. The primers used for RT-qPCR were described in our previous publication (52).
Detection of SARS-CoV-2 RBD- or S-specific antibodies by ELISA.
Ninety-six-well plates were first coated with 50 μl of highly purified RBD or preS protein (2 μg/ml in 50 mM Na2CO3 buffer [pH 9.6]) per well at 4°C overnight, and then blocked with bovine serum albumin (BSA; 1% wt/vol, in PBS, 200 μl/well) at 37°C for 2 h. Subsequently, individual serum samples were tested for SARS-CoV-2 S-specific antibody on antigen-coated plates. Briefly, serum samples were 2-fold serially diluted and added to RBD protein-coated wells. After incubation at room temperature for 2 h, the plates were washed five times with phosphate-buffered saline (PBS)-Tween (0.05%), followed by incubation with 100 μl of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (catalog no. 31430; Thermo Scientific) or goat anti-hamster IgG (catalog no. PA1-28823; Invitrogen) at a dilution of 1:150,000 at room temperature for 1 h. Plates were washed, developed with 100 μl of 3,3′,5,5′-tetramethylbenzidine (TMB), and stopped by 100 μl of H2SO4 (2 mol/liter), and the optical density (OD) at 450 nm was determined by BioTek microplate reader. Endpoint titers were determined as the reciprocal of the highest dilution that had an absorbance value 2.1-fold greater than the background level (DMEM control). Antibody titers were calculated by geometric mean titer (GMT).
SARS-CoV-2 pseudovirus neutralization assay.
The SARS-CoV-2 pseudovirus neutralization titer was determined using a lentiviral SARS-CoV-2 pseudotyped virus as described in a previous study (59). Specifically, 100 μl of virus was incubated with mouse or hamster serum solutions for 1 h at 37°C, and the mixture was added to HEK293T cells expressing ACE2 in 96-well plates. At 72 h postinfection, supernatants were collected from each well for measurement of luciferase activity. Briefly, 20 μl of supernatant was transferred to a new 96-well plate and mixed with 20 μl of Gluc substrate (0.1 M Tris [catalog no. T6066; Millipore Sigma] pH 7.4, 0.3 M sodium ascorbate [catalog no. S1349; Spectrum], and 10 μM coelenterazine [catalog no. CZ2.5; GoldBio]). Luminescence was immediately read by a plate reader. A nonlinear regression of x-y analyses was performed and fitted with an inhibition curve. The 50% inhibitory concentration (IC50) titer was calculated.
Plaque reduction/neutralization test.
SARS-CoV-2-specific NAbs were determined using an endpoint dilution plaque reduction neutralization test (PRNT). The serum samples were heat-inactivated at 56°C for 30 min. Twofold dilutions of the serum samples were mixed with an equal volume of DMEM containing approximately 100 PFU/well SARS-CoV-2 in a 96-well plate, and the plate was incubated at 37°C for 1 h with constant rotation. The mixtures were then transferred to confluent Vero-E6 cells in a 12-well plate. After 1 h of incubation at 37°C, the virus-serum mixtures were removed, and the cell monolayers were covered with 1 ml of Eagle’s minimal essential medium (MEM) containing 0.25% agarose, 0.12% sodium bicarbonate (NaHCO3), 2% FBS, 25 mM HEPES, 2 mM l-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin. The cells were then incubated for another 2 days and then fixed with 4% formaldehyde. The plaques were counted; serum dilution with 50% plaque reduction/neutralization titer (PRNT50) were calculated as the SARS-CoV-2-specific NAb titers.
Histology.
Half of the tissues (right lung, brain, liver, and spleen) from each experiment were preserved in 4% (vol/vol) phosphate-buffered paraformaldehyde for 2 weeks before being transferred out of the BSL3 facility. Fixed tissues were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E) for the examination of histological changes by light microscopy. The pathologist was blind to each group.
Immunohistochemical staining.
Sections of paraffin-embedded lung tissues (5 μm) were placed onto positively charged slides. After deparaffinization, sections were incubated with target retrieval solution for antigen retrieval. Immunohistochemical staining (IHC) was performed using a rabbit anti-SARS-CoV-2 N protein antibody (catalog no. NB100-56576; Novus Biologicals). Slides were counterstained with hematoxylin (HistoWiz).
Statistical analysis.
Quantitative analysis was performed by either densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare, Piscataway, NJ) and ImageQuant TL software (GE Healthcare) or ImageJ software (NIH, Bethesda, MD). Statistical analysis was performed by one-way multiple comparisons using SPSS (version 8.0) statistical analysis software (SPSS, Inc., Chicago, IL), one-way or two-way analysis of variance (ANOVA), or Student’s t test. A P value of <0.05 was considered statistically significant.
Data availability.
We declare that the data supporting the findings of this study are available with the article or are available from the corresponding author upon request.
ACKNOWLEDGMENTS
This study was supported by startup funding and bridge funding (J.L.) from the Department of Veterinary Biosciences, College of Veterinary Medicine, at The Ohio State University and by a seed grant (M.E.P. and J.L.) from the Abigail Wexner Research Institute at Nationwide Children’s Hospital. This study was in part supported by grants from the NIH (R01AI090060 to J.L.; RM1 HG008935 to J.L.; P01 AI112524 to M.E.P. and J.L.; and U19AI42733 to M.E.P.). A.K. was supported by NIH grants R01AI137567 and R01AI151175. S.L.L. was supported in part by an anonymous private donation to OSU and by NIH grants R01 AI112381, and R01 AI150473.
We thank Gail Wertz for the VSV reverse genetics system and Sean Whelan for providing pVSV(+) GxxL plasmid. We thank the BSL3 working group and University Laboratory Animal Resources staff at The Ohio State University for their support for this study. We thank members of the J.L. laboratory for technical help and critical reading of the manuscript. We also thank Sally L. Li, a sixth-grade student at Columbus Academy, for drawing cartoon images of a mouse and a hamster for figures in this paper.
M.L., P.D., Y.Z., S.T., A.L., O.H., X.L., M.E.P., A. Kapoor, S.N., and J.L. designed the research; M.L., P.D., Y.Z., S.T., A.L., O.H., M.K.C., S.C., A.Z., A. Kenney, C.Z., C.C., C.Y., X.L., P.N.B., J.Q., L.M.-S., J.S.Y., M.E.P., A. Kapoor, S.N., and J.L. performed research; M.L., C.Y., S.-L.L., J.Q., L.M.-S., J.S.Y., M.E.P., A. Kapoor, and J.L. contributed new reagents/analytic tools; M.L., P.D., Y.Z., S.T., M.K.C., S.C., P.N.B., L.M.-S., M.E.P., A. Kapoor, and J.L. analyzed data; M.L. and J.L. wrote the paper; P.D., Y.Z., S.T., A.L., O.H., M.K.C., S.C., A.Z., A. Kenney, C.Z., C.Y., X.L., S.-L.L., P.N.B., J.Q., L.M.-S., J.S.Y., M.E.P., and A. Kapoor edited the manuscript; and J.L. provided funding.
The Ohio State University has filed an invention report for the mtdVSV-based SARS-CoV-2 vaccine platform.
Contributor Information
Jianrong Li, Email: li.926@osu.edu.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N, Wu Y, Li C, Chen Q, Li D, Liu T, Zhao J, Liu M, Tu W, Chen C, Jin L, Yang R, Wang Q, Zhou S, Wang R, Liu H, Luo Y, Liu Y, Shao G, Li H, Tao Z, Yang Y, Deng Z, Liu B, Ma Z, Zhang Y, Shi G, Lam TTY, Wu JT, Gao GF, Cowling BJ, Yang B, Leung GM, Feng Z. 2020. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med 382:1199–1207. 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. Lancet 395:497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team. 2020. A Novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kemp SA, Collier DA, Datir RP, Ferreira IATM, Gayed S, Jahun A, Hosmillo M, Rees-Spear C, Mlcochova P, Lumb IU, Roberts DJ, Chandra A, Temperton N, Sharrocks K, Blane E, Modis Y, Leigh KE, Briggs JAG, van Gils MJ, Smith KGC, Bradley JR, Smith C, Doffinger R, Ceron-Gutierrez L, Barcenas-Morales G, Pollock DD, Goldstein RA, Smielewska A, Skittrall JP, Gouliouris T, Goodfellow IG, Gkrania-Klotsas E, Illingworth CJR, McCoy LE, Gupta RK, COVID-19 Genomics UK (COG-UK) Consortium. 2021. SARS-CoV-2 evolution during treatment of chronic infection. Nature 592:277–282. 10.1038/s41586-021-03291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS. 2020. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 183:1735. 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F. 2020. Structural basis of receptor recognition by SARS-CoV-2. Nature 581:221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du L, He Y, Zhou Y, Liu S, Zheng B-J, Jiang S. 2009. The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat Rev Microbiol 7:226–236. 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. [DOI] [PubMed] [Google Scholar]
- 10.Whelan SP, Ball LA, Barr JN, Wertz GT. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci USA 92:8388–8392. 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA 92:4477–4481. 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scher G, Schnell MJ. 2020. Rhabdoviruses as vectors for vaccines and therapeutics. Curr Opin Virol 44:169–182. 10.1016/j.coviro.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, Carroll MW, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Hossmann S, Kondé MK, Kone S, Kuisma E, Levine MM, Mandal S, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Watson CH, Kéïta S, Kieny MP, Røttingen J-A. 2015. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 386:857–866. 10.1016/S0140-6736(15)61117-5. [DOI] [PubMed] [Google Scholar]
- 14.Huttner A, Dayer J-A, Yerly S, Combescure C, Auderset F, Desmeules J, Eickmann M, Finckh A, Goncalves AR, Hooper JW, Kaya G, Krähling V, Kwilas S, Lemaître B, Matthey A, Silvera P, Becker S, Fast PE, Moorthy V, Kieny MP, Kaiser L, Siegrist C-A. 2015. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: a randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect Dis 15:1156–1166. 10.1016/S1473-3099(15)00154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Safronetz D, Mire C, Rosenke K, Feldmann F, Haddock E, Geisbert T, Feldmann H. 2015. A recombinant vesicular stomatitis virus-based Lassa fever vaccine protects guinea pigs and macaques against challenge with geographically and genetically distinct Lassa viruses. PLoS Negl Trop Dis 9:e0003736. 10.1371/journal.pntd.0003736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurup D, Wirblich C, Feldmann H, Marzi A, Schnell MJ. 2015. Rhabdovirus-based vaccine platforms against henipaviruses. J Virol 89:144–154. 10.1128/JVI.02308-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu R, Wang J, Shao Y, Wang X, Zhang H, Shuai L, Ge J, Wen Z, Bu Z. 2018. A recombinant VSV-vectored MERS-CoV vaccine induces neutralizing antibody and T cell responses in rhesus monkeys after single dose immunization. Antiviral Res 150:30–38. 10.1016/j.antiviral.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapadia SU, Simon ID, Rose JK. 2008. SARS vaccine based on a replication-defective recombinant vesicular stomatitis virus is more potent than one based on a replication-competent vector. Virology 376:165–172. 10.1016/j.virol.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li A, Yu J, Lu M, Ma Y, Attia Z, Shan C, Xue M, Liang X, Craig K, Makadiya N, He JJ, Jennings R, Shi P-Y, Peeples ME, Liu S-L, Boyaka PN, Li J. 2018. A Zika virus vaccine expressing premembrane-envelope-NS1 polyprotein. Nat Commun 9:3067. 10.1038/s41467-018-05276-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betancourt D, de Queiroz NM, Xia T, Ahn J, Barber GN. 2017. Innate immune augmenting vesicular stomatitis virus expressing Zika virus proteins confers protective immunity. J Immunol 198:3023–3028. 10.4049/jimmunol.1602180. [DOI] [PubMed] [Google Scholar]
- 21.Wolf J, Jannat R, Dubey S, Troth S, Onorato MT, Coller B-A, Hanson ME, Simon JK. 2021. Development of pandemic vaccines: ERVEBO case study. Vaccines (Basel) 9:190. 10.3390/vaccines9030190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Case JB, Rothlauf PW, Chen RE, Liu Z, Zhao H, Kim AS, Bloyet L-M, Zeng Q, Tahan S, Droit L, Ilagan MXG, Tartell MA, Amarasinghe G, Henderson JP, Miersch S, Ustav M, Sidhu S, Virgin HW, Wang D, Ding S, Corti D, Theel ES, Fremont DH, Diamond MS, Whelan SPJ. 2020. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host Microbe 28:475–485.e475. 10.1016/j.chom.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Case JB, Rothlauf PW, Chen RE, Kafai NM, Fox JM, Smith BK, Shrihari S, McCune BT, Harvey IB, Keeler SP, Bloyet L-M, Zhao H, Ma M, Adams LJ, Winkler ES, Holtzman MJ, Fremont DH, Whelan SPJ, Diamond MS. 2020. Replication-competent vesicular stomatitis virus vaccine vector protects against SARS-CoV-2-mediated pathogenesis in mice. Cell Host Microbe 28:465–474.e464. 10.1016/j.chom.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yahalom-Ronen Y, Tamir H, Melamed S, Politi B, Shifman O, Achdout H, Vitner EB, Israeli O, Milrot E, Stein D, Cohen-Gihon I, Lazar S, Gutman H, Glinert I, Cherry L, Vagima Y, Lazar S, Weiss S, Ben-Shmuel A, Avraham R, Puni R, Lupu E, Bar-David E, Sittner A, Erez N, Zichel R, Mamroud E, Mazor O, Levy H, Laskar O, Yitzhaki S, Shapira SC, Zvi A, Beth-Din A, Paran N, Israely T. 2020. A single dose of recombinant VSV-G-spike vaccine provides protection against SARS-CoV-2 challenge. Nat Commun 11:6402. 10.1038/s41467-020-20228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JR, Wang JT, Whelan SPJ. 2006. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc Natl Acad Sci USA 103:8493–8498. 10.1073/pnas.0509821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y, Wei Y, Zhang X, Zhang Y, Cai H, Zhu Y, Shilo K, Oglesbee M, Krakowka S, Whelan SPJ, Li J. 2014. mRNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J Virol 88:2913–2926. 10.1128/JVI.03420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin T-Y, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Shi P-Y, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Fontaine-Rodriguez EC, Whelan SP. 2005. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J Virol 79:13373–13384. 10.1128/JVI.79.21.13373-13384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Broek MF, Muller U, Huang S, Zinkernagel RM, Aguet M. 1995. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol Rev 148:5–18. 10.1111/j.1600-065x.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- 30.Imai M, Iwatsuki-Horimoto K, Hatta M, Loeber S, Halfmann PJ, Nakajima N, Watanabe T, Ujie M, Takahashi K, Ito M, Yamada S, Fan S, Chiba S, Kuroda M, Guan L, Takada K, Armbrust T, Balogh A, Furusawa Y, Okuda M, Ueki H, Yasuhara A, Sakai-Tagawa Y, Lopes TJS, Kiso M, Yamayoshi S, Kinoshita N, Ohmagari N, Hattori S-I, Takeda M, Mitsuya H, Krammer F, Suzuki T, Kawaoka Y. 2020. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc Natl Acad Sci USA 117:16587–16595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sia SF, Yan L-M, Chin AWH, Fung K, Choy K-T, Wong AYL, Kaewpreedee P, Perera RAPM, Poon LLM, Nicholls JM, Peiris M, Yen H-L. 2020. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583:834–838. 10.1038/s41586-020-2342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, Jia X, Wu M, Shi B, Xu S, Chen J, Wang W, Chen B, Jiang L, Yu S, Lu J, Wang J, Xu M, Yuan Z, Zhang Q, Zhang X, Zhao G, Wang S, Chen S, Lu H. 2020. Viral and host factors related to the clinical outcome of COVID-19. Nature 583:437–440. 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- 33.Bukreyev A, Skiadopoulos MH, Murphy BR, Collins PL. 2006. Nonsegmented negative-strand viruses as vaccine vectors. J Virol 80:10293–10306. 10.1128/JVI.00919-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabin AB, Olitsky PK. 1938. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus. III. Effect of age and pathway of infection on the character and localization of lesions in the central nervous system. J Exp Med 67:201–228. 10.1084/jem.67.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suder E, Furuyama W, Feldmann H, Marzi A, de Wit E. 2018. The vesicular stomatitis virus-based Ebola virus vaccine: from concept to clinical trials. Hum Vaccin Immunother 14:2107–2113. 10.1080/21645515.2018.1473698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahmeh AA, Li J, Kranzusch PJ, Whelan SP. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J Virol 83:11043–11050. 10.1128/JVI.01426-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li A, Xue M, Attia Z, Yu J, Lu M, Shan C, Liang X, Gao TZ, Shi P-Y, Peeples ME, Boyaka PN, Liu S-L, Li J. 2020. Vesicular stomatitis virus and DNA vaccines expressing Zika virus nonstructural protein 1 induce substantial but not sterilizing protection against Zika virus infection. J Virol 94:e00048-20. 10.1128/JVI.00048-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Wei Y, Zhang X, Cai H, Niewiesk S, Li J. 2014. Rational design of human metapneumovirus live attenuated vaccine candidates by inhibiting viral mRNA cap methyltransferase. J Virol 88:11411–11429. 10.1128/JVI.00876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Y, Cai H, Lu M, Ma Y, Li A, Gao Y, Zhou J, Gu H, Li J, Gu J. 2020. Porcine epidemic diarrhea virus deficient in RNA cap guanine-N-7 methylation is attenuated and induces higher type I and III interferon responses. J Virol 94:e00447-20. 10.1128/JVI.00447-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li S-H, Dong H, Li X-F, Xie X, Zhao H, Deng Y-Q, Wang X-Y, Ye Q, Zhu S-Y, Wang H-J, Zhang B, Leng Q-B, Zuest R, Qin E-D, Qin C-F, Shi P-Y. 2013. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J Virol 87:5812–5819. 10.1128/JVI.02806-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menachery VD, Gralinski LE, Mitchell HD, Dinnon KH, Leist SR, Yount BL, McAnarney ET, Graham RL, Waters KM, Baric RS. 2018. Combination attenuation offers strategy for live attenuated coronavirus vaccines. J Virol 92:e00710-18. 10.1128/JVI.00710-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C, Lanzavecchia A, Corti D, Virgin HW. 2020. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 584:353–363. 10.1038/s41586-020-2538-8. [DOI] [PubMed] [Google Scholar]
- 44.Bolles M, Deming D, Long K, Agnihothram S, Whitmore A, Ferris M, Funkhouser W, Gralinski L, Totura A, Heise M, Baric RS. 2011. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol 85:12201–12215. 10.1128/JVI.06048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tseng C-T, Sbrana E, Iwata-Yoshikawa N, Newman PC, Garron T, Atmar RL, Peters CJ, Couch RB. 2012. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS One 7:e35421. 10.1371/journal.pone.0035421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassan AO, Kafai NM, Dmitriev IP, Fox JM, Smith BK, Harvey IB, Chen RE, Winkler ES, Wessel AW, Case JB, Kashentseva E, McCune BT, Bailey AL, Zhao H, VanBlargan LA, Dai Y-N, Ma M, Adams LJ, Shrihari S, Danis JE, Gralinski LE, Hou YJ, Schäfer A, Kim AS, Keeler SP, Weiskopf D, Baric RS, Holtzman MJ, Fremont DH, Curiel DT, Diamond MS. 2020. A single-dose intranasal ChAd vaccine protects upper and lower respiratory tracts against SARS-CoV-2. Cell 183:169–184.e113. 10.1016/j.cell.2020.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, Himansu S, Schäfer A, Ziwawo CT, DiPiazza AT, Dinnon KH, Elbashir SM, Shaw CA, Woods A, Fritch EJ, Martinez DR, Bock KW, Minai M, Nagata BM, Hutchinson GB, Wu K, Henry C, Bahl K, Garcia-Dominguez D, Ma LZ, Renzi I, Kong W-P, Schmidt SD, Wang L, Zhang Y, Phung E, Chang LA, Loomis RJ, Altaras NE, Narayanan E, Metkar M, Presnyak V, Liu C, Louder MK, Shi W, Leung K, Yang ES, West A, Gully KL, Stevens LJ, Wang N, Wrapp D, Doria-Rose NA, Stewart-Jones G, Bennett H, et al. 2020. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586:567–571. 10.1038/s41586-020-2622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Vormehr M, Baum A, Pascal K, Quandt J, Maurus D, Brachtendorf S, Lörks V, Sikorski J, Hilker R, Becker D, Eller A-K, Grützner J, Boesler C, Rosenbaum C, Kühnle M-C, Luxemburger U, Kemmer-Brück A, Langer D, Bexon M, Bolte S, Karikó K, Palanche T, Fischer B, Schultz A, Shi P-Y, Fontes-Garfias C, Perez JL, Swanson KA, Loschko J, Scully IL, Cutler M, Kalina W, Kyratsous CA, Cooper D, Dormitzer PR, Jansen KU, Türeci Ö. 2020. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 586:594–599. 10.1038/s41586-020-2814-7. [DOI] [PubMed] [Google Scholar]
- 49.Crank MC, Ruckwardt TJ, Chen M, Morabito KM, Phung E, Costner PJ, Holman LA, Hickman SP, Berkowitz NM, Gordon IJ, Yamshchikov GV, Gaudinski MR, Kumar A, Chang LA, Moin SM, Hill JP, DiPiazza AT, Schwartz RM, Kueltzo L, Cooper JW, Chen P, Stein JA, Carlton K, Gall JG, Nason MC, Kwong PD, Chen GL, Mascola JR, McLellan JS, Ledgerwood JE, Graham BS, VRC 317 Study Team. 2019. A proof of concept for structure-based vaccine design targeting RSV in humans. Science 365:505–509. 10.1126/science.aav9033. [DOI] [PubMed] [Google Scholar]
- 50.Kwong PD, Mascola JR. 2018. HIV-1 vaccines based on antibody identification, B cell ontogeny, and epitope structure. Immunity 48:855–871. 10.1016/j.immuni.2018.04.029. [DOI] [PubMed] [Google Scholar]
- 51.Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T. 2021. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med 384:403–416. 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu M, Dravid P, Zhang Y, Trivedi S, Li A, Harder O, KC M, Chaiwatpongsakorn S, Zani A, Kenney A, Zeng C, Cai C, Ye C, Liang X, Shimamura M, Liu S-L, Mejias A, Ramilo O, Boyaka PN, Qiu J, Martinez-Sobrido L, Yount JS, Peeples ME, Kapoor A, Niewiesk S, Li J. 2021. A safe and highly efficacious measles virus-based vaccine expressing SARS-CoV-2 stabilized prefusion spike. Proc Natl Acad Sci USA 118:e2026153118. 10.1073/pnas.2026153118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage-T7 RNA-polymerase. Proc Natl Acad Sci USA 83:8122–8126. 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng K, Divers E, Ma Y, Li J. 2011. Inactivation of a human norovirus surrogate, human norovirus virus-like particles, and vesicular stomatitis virus by gamma irradiation. Appl Environ Microbiol 77:3507–3517. 10.1128/AEM.00081-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu M, Xue M, Wang H-T, Kairis EL, Ahmad S, Wei J, Zhang Z, Liu Q, Zhang Y, Gao Y, Garcin D, Peeples ME, Sharma A, Hur S, He C, Li J. 2021. Nonsegmented negative-sense RNA viruses utilize N-6-methyladenosine (m6A) as a common strategy to evade host innate immunity. J Virol 95:e01939-20. 10.1128/JVI.01939-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartlage AS, Murthy S, Kumar A, Trivedi S, Dravid P, Sharma H, Walker CM, Kapoor A. 2019. Vaccination to prevent T cell subversion can protect against persistent hepacivirus infection. Nat Commun 10:1113. 10.1038/s41467-019-09105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zivcec M, Safronetz D, Haddock E, Feldmann H, Ebihara H. 2011. Validation of assays to monitor immune responses in the Syrian golden hamster (Mesocricetus auratus). J Immunol Methods 368:24–35. 10.1016/j.jim.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Safronetz D, Zivcec M, Lacasse R, Feldmann F, Rosenke R, Long D, Haddock E, Brining D, Gardner D, Feldmann H, Ebihara H. 2011. Pathogenesis and host response in Syrian hamsters following intranasal infection with Andes virus. PLoS Pathog 7:e1002426. 10.1371/journal.ppat.1002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeng C, Evans JP, Pearson R, Qu P, Zheng Y-M, Robinson RT, Hall-Stoodley L, Yount J, Pannu S, Mallampalli RK, Saif L, Oltz E, Lozanski G, Liu S-L. 2020. Neutralizing antibody against SARS-CoV-2 spike in COVID-19 patients, health care workers, and convalescent plasma donors. JCI Insight 5:e143213. 10.1172/jci.insight.143213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We declare that the data supporting the findings of this study are available with the article or are available from the corresponding author upon request.