

ABSTRACT
Recent years have seen the emergence of immunotherapy as a promising modality for treating a variety of cancers. However, the initial data have led to the ultimate reality that such a treatment does not work effectively in all cancers, nor does it universally result in long-lasting benefits, which can be partly attributed to the development of drug resistance- itself a major challenge. Worse, in some cases, immunotherapy can lead to accelerated tumor growth known as hyperprogression. Tumor sensitization is being pursued as a means to circumvent resistance to immunotherapy, and perhaps as a means to prevent hyperprogression. Such approaches aim to counteract features of immune resistance demonstrated by refractory tumors, paving the way for improved treatment effectiveness when standard immunotherapies such as immune checkpoint inhibitors are utilized. Sensitizing agents can be categorized by whether their target is a tumor-intrinsic or a tumor cell-extrinsic factor. Tumor-intrinsic sensitization strategies act directly on cancer cells, suppressing their anti-immune tendencies, whereas tumor cell-extrinsic sensitization strategies target the tumor microenvironment to more effectively mediate the desired therapeutic effects of immunotherapy.
KEYWORDS: Tumor sensitization, immunotherapy, tumor microenvironment, tumor-intrinsic factors, tumor-extrinsic factors, immunotherapy resistance
Introduction
Immunotherapy has emerged as an effective treatment modality for some cancers. The targeting of immune checkpoints in cancer therapy has resulted in strong anti-cancer effects, leading to long-term remission and increased patient survival in select cancers.1 Immune checkpoint therapies have gained significant prominence in the field of Oncology with Science naming cancer immunotherapy as the “Breakthrough of the Year” in 2013.2 Indeed, the Nobel Prize in Physiology or Medicine was awarded to James P. Allison and Tasuko Honjo in 2018 for advances in the understanding of immune checkpoints and for the ultimate development of immunotherapy for cancer.
However, immunotherapy is not universally effective across cancers. While there is a high response rate in some tumor types (e.g. lung cancer, melanoma, bladder cancer, and microsatellite unstable colorectal and other cancers), the effects are not universally long-lasting in the majority of patients. Combination therapy with ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) used to treat metastatic renal cell carcinoma has shown remarkably durable response, with a complete response rate of 11% in one trial.3 Cervical cancers treated with checkpoint inhibition show similar response rates with a range of 10–25%, and potentially higher when used in combination with other immunotherapies or conventional therapies.4 However, in some tumor types (e.g., sarcoma and ovarian cancer), response rates to immunotherapy are dismal, with many exhibiting resistance patterns that may be intrinsic to the tumor cells or that may be influenced by their microenvironment.1 Novel therapeutic strategies attempt to circumvent resistance to immunotherapy and sensitize tumors to immunotherapeutic approaches in order to expand the clinical benefits to a broader patient population.
Tumors often exhibit primary, adaptive, or acquired resistance to immunotherapy. Typically, primary resistance to immunotherapy is due to a lack of T-cell recruitment to tumor sites or lack of T-cell recognition caused by a lack of antigen expression.5 Factors like loss of HLA expression, constitutive PD-L1 expression, and alteration of signal transduction pathways aid in this type of resistance.5 Adaptive resistance may also contribute to primary resistance, where immune recognition of tumor cells can occur, but the immune response is avoided.5 Tumors which stop responding to immunotherapy demonstrate acquired resistance, where selection pressure from the immune system and tumor cell interaction drive preservation of resistant traits.5
A small subset of tumors responds to treatment by exhibiting accelerated tumor growth known as hyperprogression, where treatment leads to more rapid proliferation and worsened clinical outcomes.6 Upon treatment, these tumors exhibit an accelerated growth potentially indicative of another complication of immunotherapy.6 The mechanisms of hyperprogression have been under investigation as it would be important to predict and address such aggressive tumor behavior with undesirable clinical outcomes. A study of six patients with various stage IV cancers treated with immunotherapy identified the risk of hyperprogression occurring in patients receiving checkpoint inhibitors as associated with MDM2 gene family amplification.6 Such MDM2 gene family amplification and EGFR aberration appeared in this study to be linked to the hyperprogression outcome.6 Anti-PD-1 or anti-PD-L1 treatment through these cases appears to be the most susceptible to lead to hyperprogression (defined as treatment failure within 2 months), though anti-CTLA-4 treatment administered either alone or in conjunction with PD-1/PD-L1 inhibition was also shown to possibly lead to treatment failure in the same interval or slightly thereafter.
Another subset of tumors, initially discovered with anti-CTLA-4 treatment, exhibit pseudoprogression.7 Pseudoprogression occurs after immune checkpoint therapy in tumors that ultimately respond to the therapy, though treatment responses appear delayed by radiographic measurements where initially tumors can appear larger. This has led to the adoption of immune RECIST (iRECIST) criteria for assessment of response to immunotherapy. Differentiating pseudoprogression from hyperprogression can be difficult although with hyperprogression new metastatic lesions are often observed including at new organ sites of metastases, along with an apparent acceleration of growth of tumor size as compared to before treatment with immune checkpoint therapy. Molecular and cellular mechanisms associated with the immune response against cancer within the tumor microenvironment, immune checkpoints and their therapeutic blockade are shown in Figure 1.
Figure 1.
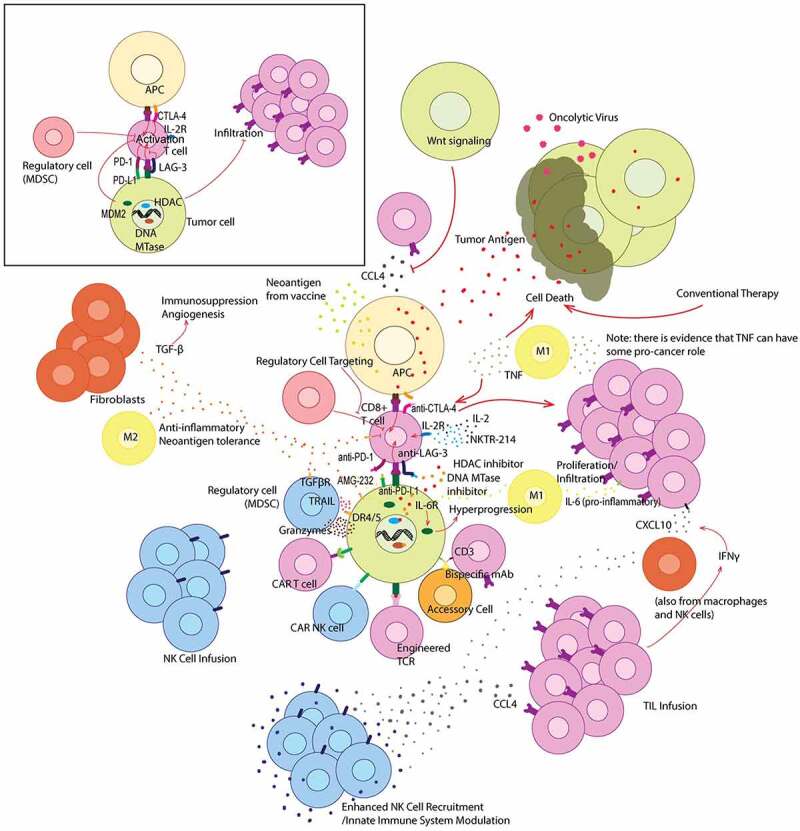
Graphical summary of immunotherapeutic treatment strategies that target the tumor microenvironment. Offset (top left) depicts the interaction between a CD8 + T cell and both an APC and tumor cell without treatment. The main figure depicts various treatment strategies and their modulation of the tumor microenvironment. Various cytokines are also depicted in their most common and relevant roles within the tumor microenvironment
Tumor cell–intrinsic and extrinsic factors for immune evasion
With the backdrop of immunotherapy resistance or immunotherapy-induced hyperprogression, sensitizing tumors to immunotherapy becomes not only a new avenue to improve clinical outcomes but also a way to potentially limit therapy-induced tumor progression or metastasis. Especially as CTLA-4 and PD-1/PD-L1 treatments become more widespread for many various cancer types and novel immunotherapies are investigated, finding ways to induce tumor vulnerability to immunotherapy becomes more important.
Direct targeting of the tumor with sensitizing agents is one strategy toward mitigating immune evasion or subversion of the immune response to accelerate tumor growth. Many chemotherapies, for instance, show a direct effect on expressed antigens, with multiple studies on BRAF-targeted therapy in melanoma showing increased antigen and HLA expression.5
A different, emerging strategy to accomplish this sensitization to enhance immunotherapy involves targeting the tumor microenvironment (Figure 1). Such a strategy, when used to complement traditional immunotherapy or even other tumor-cell sensitizing agents, could help address some limitations of immunotherapy.
Direct targeting of tumor cells
Direct targeting of tumor cell-intrinsic factors is expected to sensitize tumor cells to immunotherapy and may overcome primary and acquired immunotherapy resistance.
We recently explored the use of a mouse double minute 2 homolog (MDM2) inhibitor, AMG-232, to sensitize tumor cells to immunotherapy. High MDM2 expression was found to be involved in immune evasion by blocking T-cell mediated tumor cell killing, and inhibition of MDM2 by the small molecule or by gene knockdown led to an increase in the T-cell killing of MDM2-overexpressing tumor cells.8 This finding could partially explain the recent clinical study suggesting that MDM2/MDMX (MDM4) amplification can predict poor response to immune checkpoint inhibitors (ICIs).9 Interleukin 6 (IL-6) has previously been shown to increase MDM2-mediated p53 degradation.10 Our study demonstrated that targeting MDM2 attenuates IL-6 expression in tumor cells which may play an important role in sensitizing tumor cells to T-cell-mediated killing.8 A different study identified ephrin-A receptor 2 (EPHA2) as a tumor-intrinsic factor in immunosuppression, and showed EPHA2 deletion to successfully sensitize tumor cells for immunotherapy, ultimately calling for further investigation on EPHA2/TGF-β/PTGS2 inhibitors as sensitizers for immunotherapy.11
Various other tumor-cell-intrinsic factors have been identified to correlate with T cell infiltration, a key marker of immunotherapy susceptibility. c-Myc expression, for instance, coupled with epigenetic modulation was found to be associated with low tumor inflammation secondary to CXCL1 expression, a chemokine implicated in immunosuppression by recruiting myeloid cells.12 Other chemo-attractants such as CCL4 and CXCL10 are associated with immune infiltration, though certain tumor characteristics such as a Wnt phenotype can interfere.13 Macrophage infiltration is also associated with immune responsivity, with TGF-β secreting M2 macrophages promoting lessened inflammation and self-antigen tolerance, and IFNγ and TNF secreting M1 macrophages promoting an immune response phenotype.14
Chemotherapeutic agents, radiation therapy, and metabolic modification have also been shown to contribute to immunogenicity.1,15 The specific mechanisms vary with the different therapeutic modalities. Chemotherapy, for instance, can lead to the release of tumor antigens by massive cell death, priming an anti-tumor immune response.16 Radiation therapy acts similarly but has an added effect of enhanced antigen presentation and tumor-infiltrating lymphocytes (TILs) infiltration.17 Radiation therapy, in fact, has been suggested to cause an “abscopal effect”, where local radiation to one site induces systemic anti-tumor responses.18 Finally, altering tumor metabolism by interfering with the Warburg effect, lactate production, or glucose uptake (among others) has been shown to create favorable conditions for immunotherapy response.19–22
The immune system and the tumor microenvironment
The efficacy of immunotherapy is contingent in part upon the ability of the immune system to gain access to the tumor microenvironment. Tumor microenvironments are typically categorized according to their degree of immune cell infiltration, with “inflamed” environments with a high CD8 + T cell infiltration boasting better overall clinical outcomes when compared to “immune excluded” (no CD8 + T cell infiltration in the tumor parenchyma but in stroma) and “immune-desert” (extremely low CD8 + T cell infiltration) tumor microenvironments.23
While immune cell infiltration into the tumor microenvironment is associated with better efficacy against some immunotherapies, certain immunotherapies show promise for directly increasing the degree of immune infiltration. In particular, ipilimumab treatment appears to have clinical outcomes more strongly associated with post-treatment immune infiltration over baseline immune infiltration.24 Regardless, the role of the tumor microenvironment in immunotherapy response is extremely important and shown to be a better predictor than tumor intrinsic factors.1
Bolstering T-cell infiltration to the tumor microenvironment is the focus of current research
Many of the checks and balances built into the immune system to prevent autoimmunity and destructive inflammation are capitalized upon by tumor cells in the microenvironment to suppress the native immune response. In particular, tumor cells often express or release signaling molecules into the tumor microenvironment that mitigate effector T-cell activity. An immunotherapy sensitization strategy emerges in overcoming this tendency, either through overpowering the inhibitory signals of the tumor cells through exogenous stimulatory signaling or through suppressing inhibitory signals directly.
ICIs provide one manner to do this, with PD-1/PD-L1 and CTLA-4 inhibition being the two currently most-researched examples. Such ICIs block immune system inhibition signals readily present in the tumor microenvironment, secreted by tumor cells to inactivate the anti-cancer immune response. More ICI targets are being explored, such as lymphocyte-activation gene 3 (LAG-3), the inhibition of which has shown favorable results in mouse models and is currently undergoing clinical trial testing.25 T-cell immunoglobulin and mucin-domain-containing molecule 3 (TIM-3), which plays various important roles in T cell regulation, has been shown to lead to worsened clinical outcomes when upregulated on TILs, and has been purported to be involved with PD-1 blockade secondary resistance, and is also being explored as an ICI target in clinical trials.26
Such ICIs are complemented by direct immune-system stimulation. Co-targeting of a co-stimulatory target and an immune checkpoint leads to promising outcomes. Inducible T-cell costimulatory (ICOS) activation with an agonist was shown to compliment anti-CTLA-4 therapy in vivo and its synergy with anti-PD-1 therapy is being investigated.27
Exogenous cytokine administration is another potential method to increase immune cell activation in the tumor microenvironment, however this treatment strategy was not shown to be effective enough to overcome its toxic effects in most cases.1 Recent advances may be able to make exogenous cytokines viable once again, particularly through the emergence of NKTR-214 (an IL-2 analog with less toxic effects), which showed promise in increasing immune effector function in the tumor microenvironment and is currently going to trial alongside PD-1 blockades and CTLA-4/PD-1 co-inhibition.28
Even more strategies are currently being explored and may increase T-cell infiltration and promote an immune effector phenotype in the tumor microenvironment. Bispecific antibodies, for instance, are being used to make enhancements to the tumor microenvironment, for example by bringing together a tumor cell and T-cell, while innate immune agonists and antagonists are being tested for synergy with other immunotherapy strategies.29
Enhancing the tumor microenvironment through vaccines and oncolytic viruses
Anti-cancer vaccines constitute perhaps the most experimental of anti-cancer therapies. While hindered by concerns of expense and practical difficulties, various agents are being investigated.30–32 Current approaches involve developing personalized vaccines, which takes advantage of next-generation sequencing of tumor cells and surrounding cells to identify tumor-specific antigens to prime the immune system toward eliminating cancer cells.33
Such an approach has demonstrated promising results in small studies, indicating that anti-cancer vaccination may one day prove an effective immunotherapy and tumor cell sensitization tool.34
Oncolytic viruses, which target tumor cells, have shown promise in their anti-cancer effects.35 The FDA-approved talimogene laherparepvec (T-VEC) has demonstrated an increase in T-cell response systemically following direct intra-tumoral administration.36,37 Moreover, combination therapy with ICIs has shown promise in tumor microenvironment modification with increased therapeutic efficacy.37 T-VEC works by releasing granulocyte-macrophage colony-stimulating factor from tumor cells via tumor cell lysis, whereas alternative oncolytic viruses disrupt the tumor microenvironment through direct interference, for instance in damaging tumor cell vasculature.38 Oncolytic viruses expressing granulocyte-macrophage colony-stimulating factor (GM-CSF) are also under investigation, and have been shown to produce better anti-tumor effects in vivo through favorable modulation of the immune environment (with increased immune-cell activation and proinflammatory cytokines in the tumor microenvironment).39
Sensitization to immune checkpoint blockade through epigenetic influence
Epigenetic factors may decrease immunogenicity exhibited by tumor cells, potentially through downregulation of tumor antigens.40 Immune checkpoint proteins such as PD-L1 are also under epigenetic control and their expression may be enhanced within tumors by epigenetic therapy. Through targeting this pathway by inhibiting DNA methylation with DNA methyltransferase inhibitors or HDAC inhibitors, immunotherapy may be enhanced.
Moreover, ICI-mediated reactivation of T-cells is associated with chromatin remodeling, indicating that epigenetic manipulation may be useful in the process of T-cell reactivation directly.41 HDAC inhibitors have been explored in conjunction with ICI therapy to enhance T-cell activation and overall immune responses to tumor cells.42
Minimizing regulatory cell activity in the tumor microenvironment
Regulatory T cells are used by the normal immune system to mediate the activity of effector T-cells to promote self-tolerance. Through a variety of different mechanisms, including direct competition with effector T-cells for cytokines, the presence of these cells reduces effector T-cell function.43,44 Other cells may play important roles in this pathway, such as myeloid-derived suppressor cells (MDSCs), defective antigen presenting cells (APCs), and tumor-associated macrophages.45
Targeting of regulatory cells in the tumor microenvironment is an emerging approach to increase anti-cancer efficacy of the immune system. A variety of agents approved by the FDA for trials have shown reduction in MDSCs in cancer patients, which has been shown in animal models to be associated with improved anti-tumor immunity.45
Intra-tumoral administration of molecules such as IL-12 have also shown therapeutic promise. While systemic administration of recombinant IL-12 is limited by cytotoxicity, intra-tumoral administration of IL-12 mRNA led to tumor regression.46 Combination treatment with anti-PD-L1 therapy was also shown to be effective in tumor microenvironment transformation, and in vivo evidence suggests that local administration may lead to systemic anti-tumor effects.46
Ex vivo T-cell therapies – avoiding the tumor microenvironment
The propensity of the tumor microenvironment to suppress the immune effector response raises the question of whether engineering cells ex vivo to be tumor-specific can allow for improved immunotherapeutic results. Such an approach effectively sensitizes tumors to immunotherapy by avoiding their microenvironment altogether, thereby avoiding any co-inhibitory immune signals that are produced by tumor cells in the region.
TIL infusions is one way to accomplish this and this has been studied at the National Cancer Institute for decades by Steven Rosenberg and colleagues. Due to association with a stronger anti-tumor microenvironment, harvesting and artificially selecting lymphocytes from tumor tissue for propagation and re-infusion has shown promise in trials.47 Moreover, this TIL infusion coupling with other treatments to alter the tumor microenvironment in a phase I melanoma has shown to be potentially beneficial, though use of this strategy may be limited by practicality of TIL harvest.48,49
CAR T-cell therapy is another means to the same end and has been studied by Carl June and others over the past two decades. CAR T cells made to target CD19 is currently an FDA approved treatment for B-cell acute lymphocytic leukemia and some B-cell lymphomas, and investigations are ongoing to expand the range of targets for CAR T-cell therapy.50 This strategy is, however, largely ineffective against solid tumors and comes with increased toxicity in its creation of a more immunogenic microenvironment.50
T-cell receptor (TCR) therapy may be used to avoid many complications of the tumor microenvironment. While it does require MHC presentation, which is often downregulated in immune-resistant tumor cells, TCRs are able to respond to a low density of antigen and recognize both intracellular and extracellular antigens, unlike CAR T-cells.51 Early clinical trial data is promising, with a phase I/II study of a TCR therapy resulting in an 80% response rate, though potential toxicity must be explored.52
A recent study also explored locoregional delivery of immune checkpoint blockade, finding this to increase immunotherapy efficacy.53 Directed delivery of these mAb blockades was found to enhance T-cell responses and overall anti-tumor immunity due to a more favorable mAb biodistribution for anti-tumor immunity and immunomodulation in tumor draining lymph nodes.53 More testing in a clinical setting is needed to fully explore this.
Natural killer cells in cancer immunotherapy
Natural Killer (NK)-cells can have powerful effects against tumor cells and are part of the host innate immune system for immunosurveillance of cancer. NK-cells kill their target cells by different mechanisms including granzyme and TRAIL mediated apoptosis. There are currently approaches to use NK-cells in cancer therapy either through NK-cell infusions (Nanthealth), bispecific antibodies that recruit NK cells to tumor antigens (Dragonfly), or through the engineering of CAR NK-cells. CAR NK-cells are under clinical investigation.54
Conclusion
As tumor cell response to immunotherapy can be limited, or with phenomena such as hyperprogression, strategies as those explored above need to be further investigated for their ability to treat and sensitize tumors. While direct sensitization strategies exist and should be explored, targeting the tumor microenvironment is a promising approach to increase anti-cancer immune responses.
Acknowledgments
This work was supported by funding from Brown University and Lifespan Corporation, the Cancer Center Director’s Discretionary Fund and funds from the Hematology/Oncology Division at Brown and Lifespan. No potential conflicts of interest were disclosed by the authors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Murciano-Goroff YR, Warner AB, Wolchok JD.. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30(6):507–19. doi: 10.1038/s41422-020-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couzin-Frankel J. Cancer Immunotherapy. Science. 2013;342(6165):1432–33. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 3.Sheng IY, Ornstein MC. Ipilimumab and nivolumab as first-line treatment of patients with renal cell carcinoma: the evidence to date. Cancer Manag Res. 2020;12:4871–81. doi: 10.2147/CMAR.S202017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otter SJ, Chatterjee J, Stewart AJ, Michael A. The role of biomarkers for the prediction of response to checkpoint immunotherapy and the rationale for the use of checkpoint immunotherapy in cervical cancer. Clin Oncol (R Coll Radiol). 2019;31(12):834–43. doi: 10.1016/j.clon.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–23. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23(15):4242–50. doi: 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jia W, Gao Q, Han A, Zhu H, Yu J. The potential mechanism, recognition and clinical significance of tumor pseudoprogression after immunotherapy. Cancer Biol Med. 2019;16(4):655–70. doi: 10.20892/j..2095-3941.2019.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahin I, Zhang S, Navaraj A, Zhou L, Dizon D, Safran H, El-Deiry WS. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020;6(1):57. doi: 10.1038/s41420-020-0292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang W, Zhou H, Shen J, Li J, Zhang Y, Hong S, Zhang L. MDM2/4 amplification predicts poor response to immune checkpoint inhibitors: a pan-cancer analysis. ESMO Open. 2020Feb4;5(1):e000614. PMCID: PMC7046406. doi: 10.1136/esmoopen-2019-000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brighenti E, Calabrese C, Liguori G, Giannone FA, Trere D, Montanaro L, Derenzini M. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: a new pathway connecting inflammation to cancer. Oncogene. 2014;33(35):4396–406. doi: 10.1038/onc.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, Quinones L, Sela Y, Yamazoe T, Gordon N, et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J Clin Invest. 2019;129(9):3594–609. doi: 10.1172/JCI127755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horton B, Spranger S. A tumor cell-intrinsic Yin-Yang determining immune evasion. Immunity. 2018;49(1):11–13. doi: 10.1016/j.immuni.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12(1):76. doi: 10.1186/s13045-019-0760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–21. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 16.Brode S, Cooke A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit Rev Immunol. 2008;28(2):109–26. doi: 10.1615/CritRevImmunol.v28.i2.20. [DOI] [PubMed] [Google Scholar]
- 17.Zappasodi R, Merghoub T, Wolchok JD. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell. 2018;33(4):581–98. doi: 10.1016/j.ccell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–31. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, Gao A, Huang L, Hsueh EC, Ford DA, et al. TLR8-mediated metabolic control of human treg function: a mechanistic target for cancer immunotherapy. Cell Metab. 2019;29(1):103–23e5. doi: 10.1016/j.cmet.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer. 2017;17(10):577–93. doi: 10.1038/nrc.2017.77. [DOI] [PubMed] [Google Scholar]
- 21.Vara-Ciruelos D, Dandapani M, Russell FM, Grzes KM, Atrih A, Foretz M, Viollet B, Lamont DJ, Cantrell DA, Hardie DG, et al. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep. 2019;27(3):690–8e4. doi: 10.1016/j.celrep.2019.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab. 2020;33:48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 24.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, Guida M, Hyams DM, Gómez H, Bastholt L, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9(1):204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lichtenegger FS, Rothe M, Schnorfeil FM, Deiser K, Krupka C, Augsberger C, Schlüter M, Neitz J, Subklewe M. Targeting LAG-3 and PD-1 to enhance T cell activation by antigen-presenting cells. Front Immunol. 2018;9:385. doi: 10.3389/fimmu.2018.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ngiow SF, Von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(10):3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 27.Soldevilla MM, Villanueva H, Meraviglia-Crivelli D, Menon AP, Ruiz M, Cebollero J, Villalba M, Moreno B, Lozano T, Llopiz D, et al. ICOS costimulation at the tumor site in combination with CTLA-4 blockade therapy elicits strong tumor immunity. Mol Ther. 2019;27(11):1878–91. doi: 10.1016/j.ymthe.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, et al. A first-in-human study and biomarker analysis of NKTR-214, a novel IL2Rbetagamma-biased cytokine, in patients with advanced or metastatic solid Tumors. Cancer Discov. 2019;9(6):711–21. doi: 10.1158/2159-8290.CD-18-1495. [DOI] [PubMed] [Google Scholar]
- 29.Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, Wei A, Dombret H, Foà R, Bassan R, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic Leukemia. N Engl J Med. 2017;376(9):836–47. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18(8):498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 31.Pan RY, Chung WH, Chu MT, Chen SJ, Chen HC, Zheng L, Hung S-I. Recent Development and Clinical Application of Cancer Vaccine: targeting Neoantigens. J Immunol Res. 2018;2018:4325874. doi: 10.1155/2018/4325874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Kim DS. Peptide immunotherapy in vaccine development: from epitope to adjuvant. Adv Protein Chem Struct Biol. 2015;99:1–14. [DOI] [PubMed] [Google Scholar]
- 33.Coulie PG, Van Den Eynde BJ, Van Der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135–46. [DOI] [PubMed] [Google Scholar]
- 34.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–08. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27(34):5763–71. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 37.Ott PA, Hodi FS. Talimogene laherparepvec for the treatment of advanced melanoma. Clin Cancer Res. 2016;22(13):3127–31. doi: 10.1158/1078-0432.CCR-15-2709. [DOI] [PubMed] [Google Scholar]
- 38.Matuszewska K, Santry LA, Van Vloten JP, AuYeung AWK, Major PP, Lawler J, Wootton SK, Bridle BW, Petrik J. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin Cancer Res. 2019;25(5):1624–38. doi: 10.1158/1078-0432.CCR-18-0220. [DOI] [PubMed] [Google Scholar]
- 39.Malhotra S, Kim T, Zager J, Bennett J, Ebright M, D’Angelica M, Fong Y. Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery. 2007;141(4):520–29. doi: 10.1016/j.surg.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, Decock J. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol. 2018;9:947. doi: 10.3389/fimmu.2018.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong YK, Li Y, Pandit H, Li S, Pulliam Z, Zheng Q, Yu Y, Martin RCG. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol. 2019;336:66–74. doi: 10.1016/j.cellimm.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 42.Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, Du L, Jiang J, Wu C, He X, et al. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene. 2015;34(49):5960–70. doi: 10.1038/onc.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–18. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5(1):15179. doi: 10.1038/srep15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2017;5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hewitt SL, Bailey D, Zielinski J, Apte A, Musenge F, Karp R, Burke S, Garcon F, Mishra A, Gurumurthy S, et al. Intratumoral IL12 mRNA therapy promotes TH1 transformation of the tumor microenvironment. Clin Cancer Res. 2020;26(23):6284–98. doi: 10.1158/1078-0432.CCR-20-0472. [DOI] [PubMed] [Google Scholar]
- 47.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol. 2016;34(20):2389–97. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–57. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joseph RW, Peddareddigari VR, Liu P, Miller PW, Overwijk WW, Bekele NB, Ross MI, Lee JE, Gershenwald JE, Lucci A, et al. Impact of clinical and pathologic features on tumor-infiltrating lymphocyte expansion from surgically excised melanoma metastases for adoptive T-cell therapy. Clin Cancer Res. 2011;17(14):4882–91. doi: 10.1158/1078-0432.CCR-10-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–65. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 51.Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. 2019;290:127–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–21. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Francis DM, Manspeaker MP, Schudel A, Sestito LF, O’Melia MJ, Kissick HT, Pollack BP, Waller EK, Thomas SN. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci Transl Med. 2020;12(563):12. doi: 10.1126/scitranslmed.aay3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59:102975. doi: 10.1016/j.ebiom.2020.102975. [DOI] [PMC free article] [PubMed] [Google Scholar]