

Abstract
Mitochondrial and lysosomal function are intricately related and critical for maintaining cellular homeostasis, as highlighted by multiple diseases linked to dysfunction of both organelles. Recent work using high-resolution microscopy demonstrates the dynamic formation of inter-organelle membrane contact sites between mitochondria and lysosomes, allowing for their direct interaction in a pathway distinct from mitophagy or lysosomal degradation of mitochondrial-derived vesicles. Mitochondria–lysosome contact site tethering is mechanistically regulated by mitochondrial proteins promoting Rab7 GTP hydrolysis, and allows for the bidirectional crosstalk between mitochondria and lysosomes and the regulation of their organelle network dynamics, including mitochondrial fission. In this review, we summarize recent advances in mitochondria–lysosome contact site regulation and function, and discuss their potential roles in cellular homeostasis and various human diseases.
Mitochondria and Lysosomes as Critical Organelles in Cellular Homeostasis
Both mitochondria and lysosomes are critical for maintaining cellular homeostasis, which is further evinced by the fact that dysfunction of both organelles is functionally and genetically linked to multiple human diseases [1–4]. Mitochondria are necessary for cellular respiration but also function as storage compartments for metabolites, including calcium, iron, lipids, protons, and ATP, and as gatekeepers for apoptosis and inflammation pathways [5,6]. Consequently, proper regulation of mitochondrial transport and dynamics is key to maintaining a functional mitochondrial network throughout the cell [7]. Mitochondrial fission has multiple roles, including mitochondrial biogenesis and mitochondrial DNA (mtDNA) synthesis [7,8] and is regulated by the GTPase dynamin-related protein (Drp1; see Glossary), endoplasmic reticulum (ER), dynamin-2, and actin [9–16]. In contrast, mitochondrial membrane fusion allows for mixing of mitochondrial proteins, mtDNA, and metabolites, and is mediated by the outer membrane GTPases, Mitofusin1 and Mitofusin2, in consort with the inner membrane GTPase Opa1 [7]. Indeed, properly balanced mitochondrial fission and fusion is crucial, as mutations in these proteins result in various diseases [2,17–19].
Similarly, lysosomes are highly dynamic organelles and responsible for the turnover of cellular contents, including proteins and lipids, via mature enzymes localized in the lysosomal lumen. However, lysosomes can also act as calcium and iron stores, and further mediate cell death pathways through the initiation of lysosomal membrane permeabilization [20], highlighting a critical role for lysosomes in the maintenance of cellular homeostasis. Indeed, lysosomes must similarly undergo strict regulation of their maturation, positioning, and network dynamics via the master regulator Rab7. Active, GTP-bound Rab7 is recruited to late endosomal/lysosomal membranes by guanine nucleotide exchange factors (GEFs) such as Mon1-Ccz1, but dissociates upon Rab GTP hydrolysis mediated by Rab GTPase-activating proteins (GAPs), resulting in an inactive, cytosolic GDP-bound form of Rab7 [1,21]. Importantly, GTP-bound Rab7 promotes lysosomal tethering and fusion and can further bind Rab7 effectors to mediate lysosomal transport in the cell [22]. In addition, human mutations in Rab7 lead to peripheral neuropathy [23–26], further emphasizing the importance of properly regulated lysosomal dynamics in maintaining cell viability. In this review, we will summarize the recent advances in mitochondria–lysosome contact site regulation and function, and discuss their potential roles in cellular homeostasis and contribution to various human diseases.
Previous Crosstalk between Mitochondria and Lysosomes
Previous studies have demonstrated several pathways for indirect functional interactions between mitochondria and lysosomes. Mitochondrial function, including respiration, has been shown to be critical for regulating lysosomal function as deletion of mitochondrial proteins (AIF, Opa1, or Pink1), chemical inhibition of oxidative phosphorylation [27,28], or expression of transcription factor A, mitochondrial (TFAM) mutations [29] impair lysosomal activity. In addition, lysosomal biogenesis increases dramatically in response to short-term inhibition of mitochondrial respiration but is disrupted by long-term inhibition via rotenone [30]. Moreover, increased mitochondrial oxidative stress in human dopaminergic neurons contributes to reduced activity of oxidized lysosomal enzymes such as glucocerebrosidase, which impairs lysosomal glycolipid metabolism [31].
Conversely, lysosomal function has been shown to be essential for maintaining mitochondrial homeostasis. In skeletal muscle, the lysosomal biogenesis regulator transcription factor EB (TFEB) acts as a central coordinator for mitochondrial function in a peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α)-independent manner [32]. In neurons, the autophagy inhibitor mammalian target of rapamycin complex 1 (mTORC1) promotes an integrated mitochondrial stress response [33] and also regulates mitochondrial activity [34]. In addition, disrupting lysosomal acidification is sufficient to decrease mitochondrial respiration [35]. Finally, endolysosomal Rabs, including Rab5, Rab7A, Rab5-GEF (RABGEF1), and Rab7-GEF (MON1-CCZ1), also regulate mitochondrial function and can be recruited to damaged mitochondria [36,37]. Moreover, Rab7 knockdown inhibits the assembly of ATG9A vesicles during Parkin-dependent mitophagy [36], while translocation of Rab5 to mitochondria decreases oxygen consumption and cytochrome C release during mitochondrial oxidative stress [37].
In addition, mitochondria and lysosomes have also been shown to directly interact upon cellular stress [38–40], with the majority of these studies predominantly focusing on lysosomal degradation of mitochondria either through mitophagy [38] or fusion of mitochondrial-derived vesicles (MDVs) with lysosomes [39] (Figure 1A). Whole mitochondria can be degraded by autophagy (mitophagy), which involves engulfment of damaged mitochondria by an autophagosome followed by fusion with lysosomes to form an autolysosome for degradation of its contents. Mitophagy can occur either nonselectively or selectively via mitophagy receptors such as optineurin and NDP52, which are recruited to ubiquitinated mitochondria in a PINK1/Parkin-dependent manner and subsequently recruit LC3 on the autophagosome via their LC3 interaction region [41,42]. In contrast, MDVs are small vesicles (~100 nm) [39], which bud off from mitochondria and contain different subsets of mitochondrial outer membrane and matrix proteins. MDVs targeted to lysosomes are generated through a PINK1/Parkin-dependent manner [43] and may represent a pathway to selectively degrade a subset of mitochondrial proteins rather than entire mitochondria. However, whether mitochondria and lysosomes directly interact with one another under normal conditions in healthy mammalian cells via nondegradative pathways has not been previously well studied.
Figure 1. Direct Interactions between Mitochondria and Lysosomes.
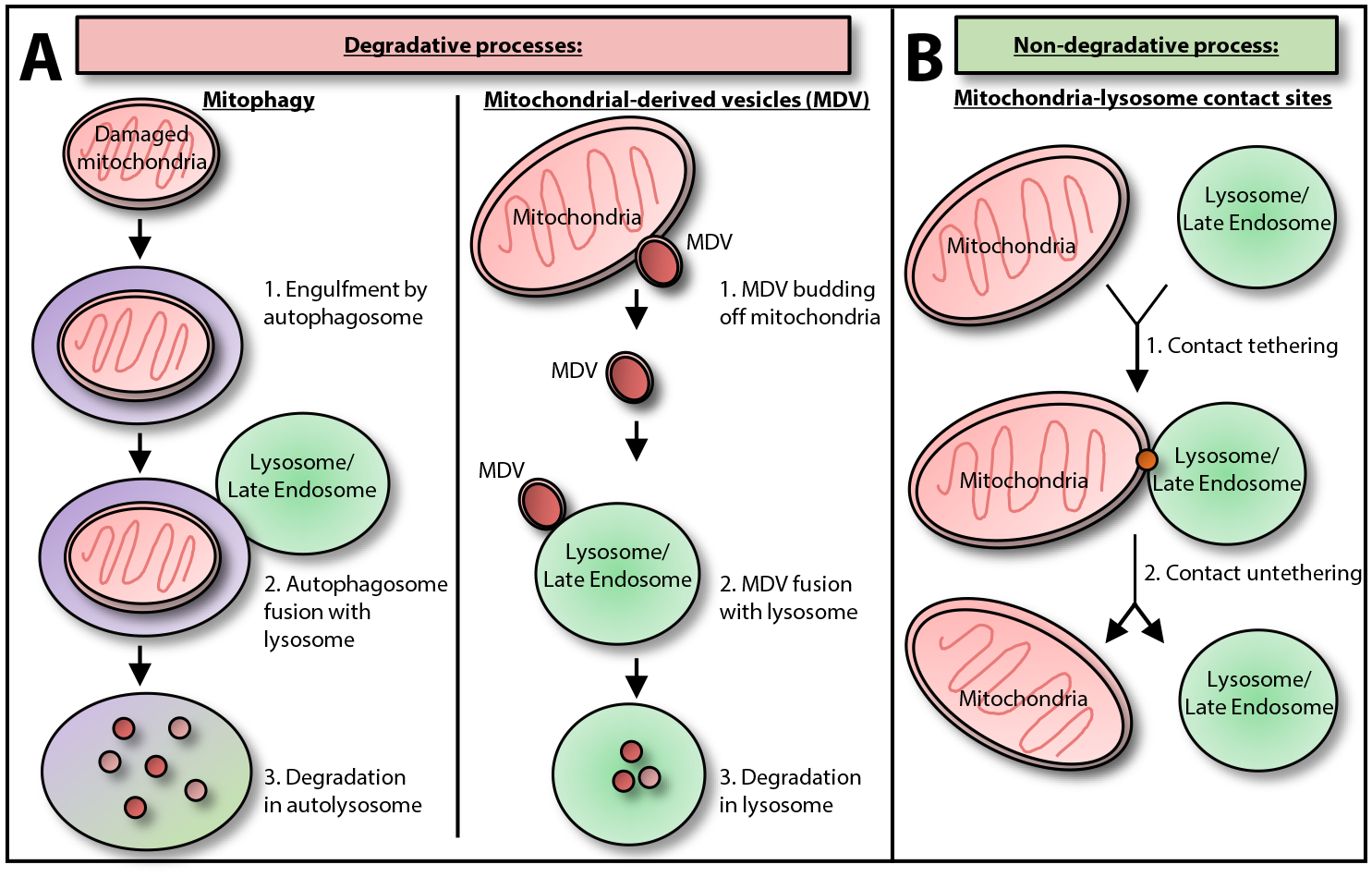
(A) Mitochondria and lysosomes have previously been shown to directly interact via degradative processes such as (1) mitophagy (in which damaged mitochondria are targeted either selectively or nonselectively to autophagosomes for engulfment. Autophagosomes subsequently fuse with lysosome/late endosomes to generate autolysosomes which mediate the degradation of mitochondria) [38]; or (2) mitochondrial-derived vesicles (MDVs) (in which MDVs bud from the mother mitochondria and can fuse with lysosomes to mediate the degradation of MDV contents) [39]. (B) In contrast, mitochondria and lysosomes can also directly interact via nondegradative processes through the dynamic formation of inter-organelle membrane contact sites in healthy mammalian cells, which involve mitochondria–lysosome contact site tethering and subsequent untethering [53].
Identification of Dynamic Mitochondria–Lysosome Membrane Contact Sites
Inter-organelle membrane contact sites are defined as contacts forming between the membranes of two distinct organelles at close proximity, allowing for their intracellular communication [44]. While the ER forms many contacts with other parts of the cell, including the plasma membrane, Golgi, mitochondria, peroxisomes, lipid droplets, and endosomes [45], the discovery of additional inter-organelle contacts not involving the ER, such as those between lysosomes, lipid droplets, and peroxisomes [44], have further demonstrated that many organelles within the cell are well connected [46]. In addition, contacts between mitochondria and lysosomal-related organelles, including melanosomes, multivesicular bodies, and yeast vacuoles, have been previously described [39,47–49]. Importantly, while contacts are maintained by tethering proteins, which allow for the dynamic formation and subsequent untethering of organelle membranes, additional proteins may also be present at contact sites, which do not physically bridge membranes but help to regulate contact functions such as mediating metabolite transfer, or regulatory proteins which help coordinate contacts and their response to the cellular environment [50]. Functionally, previous contact sites have been found to be important for mediating multiple cellular functions. These include the metabolite transfer of lipids, calcium, and iron, the regulation of organelle dynamics such as mitochondrial division [10] and endosomal division [51], which are marked by ER tubules, and additional cellular pathways [45,52] (Figure 2), demonstrating a critical role for inter-organelle contact sites in maintaining cellular homeostasis.
Figure 2. Regulation and Function of Inter-organelle Contact Sites.
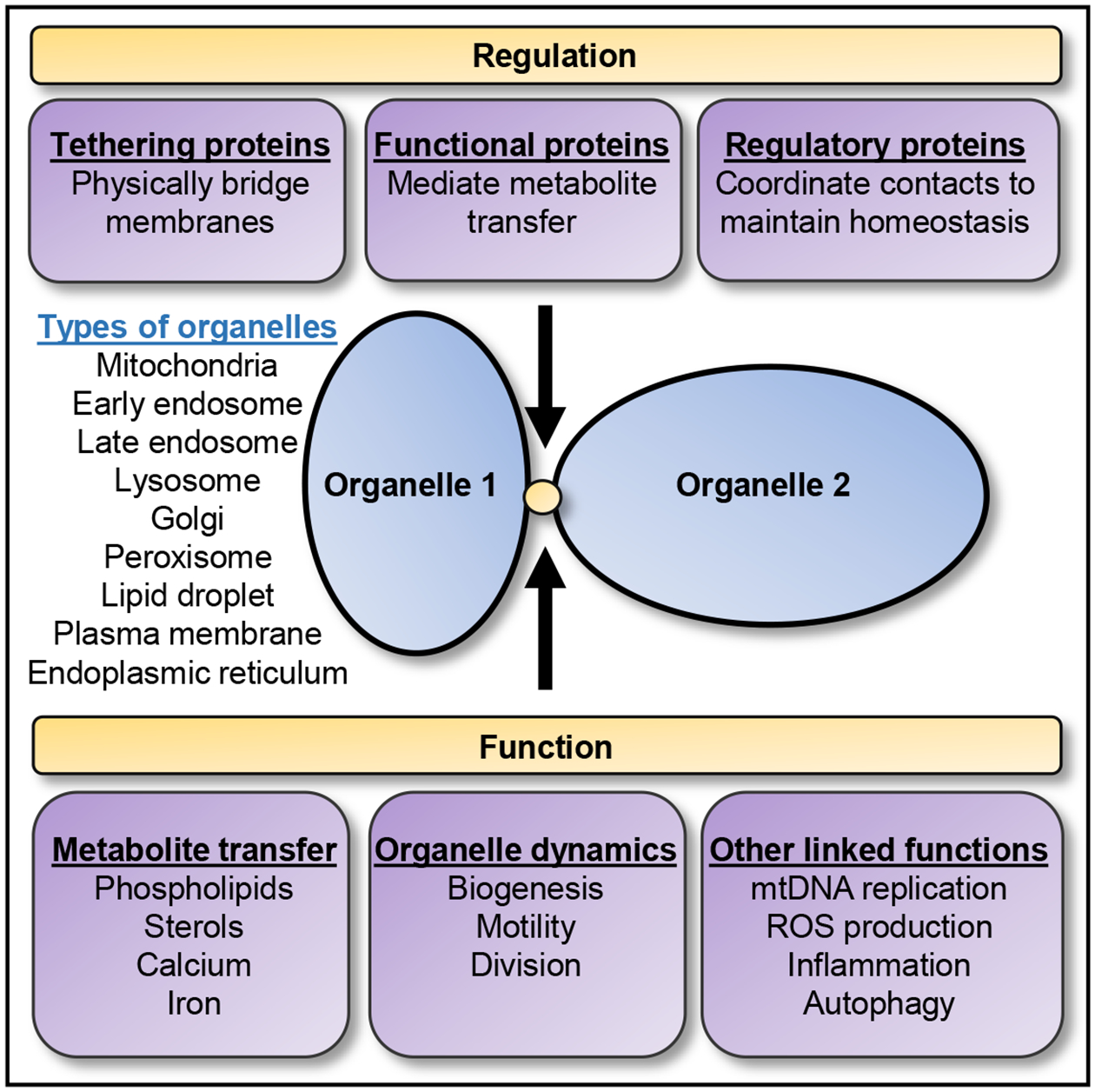
Inter-organelle membrane contact sites are sites of close apposition between the membranes of two distinct organelles which act as a domain for inter-organellar communication. Contact sites can form between many different organelles and are modulated by different types of proteins, including: (i) tethering proteins, (ii) functional proteins, and (iii) regulatory proteins, which can have overlapping roles and together help to maintain contact sites [50]. In addition, inter-organelle contacts have been linked to multiple functions, including: (i) metabolite transfer, (ii) regulation of organelle dynamics, and (iii) additional cellular processes allowing the maintenance of cellular homeostasis [45,52]. mtDNA, Mitochondrial DNA; ROS, reactive oxygen species.
Recently, multiple studies using diverse imaging techniques have demonstrated that inter-organelle contact sites also form between mitochondria and lysosomes in multiple different cell types under healthy conditions [46,53–58] (Figure 1B). Mitochondria–lysosome contacts were observed using 2D and 3D electron microscopy [53,54], as well as correlative light electron microscopy (CLEM) of LysoTracker-positive vesicles in contact with mitochondria [53] or CLEM combined with focused ion beam scanning electron microscopy, which showed Lamp1 and dextran positive vesicles stably contacting mitochondria [55]. Contacts between mitochondria and lysosomes were also observed by lattice light sheet spectral imaging [46], and were found to be less frequent than contacts involving the ER [46]. In addition, mitochondria–lysosome contacts were observed by structured illumination microscopy (SIM) imaging of organelles labeled by mitochondrial [58] or lysosomal [56] dyes or fluorescently labeled proteins [53], which showed that mitochondria could first contact one lysosome and subsequently move on to contact another lysosome [56]. Moreover, contacts were also seen by immunofluorescent staining of endogenous mitochondrial (Tom20) and lysosomal (Lamp1) membrane proteins by confocal microscopy [57] or 3D SIM imaging [53]. Finally, mitochondria–lysosome contacts were visualized using sensitized emission fluorescence resonance energy transfer between TOM20–Venus on the outer mitochondrial membrane and LAMP1–mTurquoise2 on the lysosomal membrane [53].
Mitochondria–lysosome contact sites have an average distance of ~10 nm between mitochondrial and lysosomal membranes [53,54] consistent with previously observed membrane contact sites (10–30 nm) [59,60]. Approximately 15% of lysosomes are in contact with mitochondria at any point in time, with mitochondria–lysosome contact sites remaining stably tethered for an average of 60 seconds [53], although contacts demonstrate a varying range of tethering durations, lasting as long as 13 minutes [56]. Bulk transfer of lysosomal luminal contents, mitochondrial matrix proteins, or intermembrane space proteins across organelles are not observed at sites of contact [53], and contacts do not represent autophagosome biogenesis events or mitophagy as they are negative for multiple autophagosome markers, including ULK1, Atg5, Atg12 and LC3 [53]. Contact formation was further confirmed to be independent of mitophagy, as knockout of five autophagy receptors (NDP52, OPTN, NBR1, TAX1BP1, and p62) did not prevent mitochondria–lysosome contact formation [58]. In addition, mitochondria that form contacts are distinct from MDVs as they contain both outer mitochondrial membrane and matrix proteins [53] and are substantially larger than previously described MDVs [~100nm (MDVs) versus ~500nm (mitochondria)] [39], suggesting that mitochondria–lysosome contact sites do not represent sites of mitophagy or lysosomal engulfment of bulk mitochondria.
Regulation of Mitochondria–Lysosome Contact Tethering/Untethering by Rab7 GTP Hydrolysis
Mitochondria–lysosome contact site tethering is mechanistically regulated by multiple proteins on both the mitochondrial and lysosomal membranes (Figure 3). The small GTPase Rab7, which is a master regulator of lysosomal dynamics, modulates mitochondria–lysosome contact site tethering and untethering dynamics through its ability to alternate between an active, lysosomal-localized GTP-binding state and an inactive, cytosolic GDP-binding state. Contact tethering is promoted by lysosomal GTP-bound Rab7 and may be tethered to mitochondria via Rab7 effector proteins which bind GTP-bound Rab7 on the lysosome. Importantly, expression of RAB7 Q67L, a constitutively active GTP-bound form which is unable to undergo GTP hydrolysis, is sufficient to increase the number of lysosomes contacting mitochondria and results in prolonged contacts [53].
Figure 3. Regulation of Mitochondria–Lysosome Contact Tethering/Untethering by Rab7 GTP Hydrolysis.
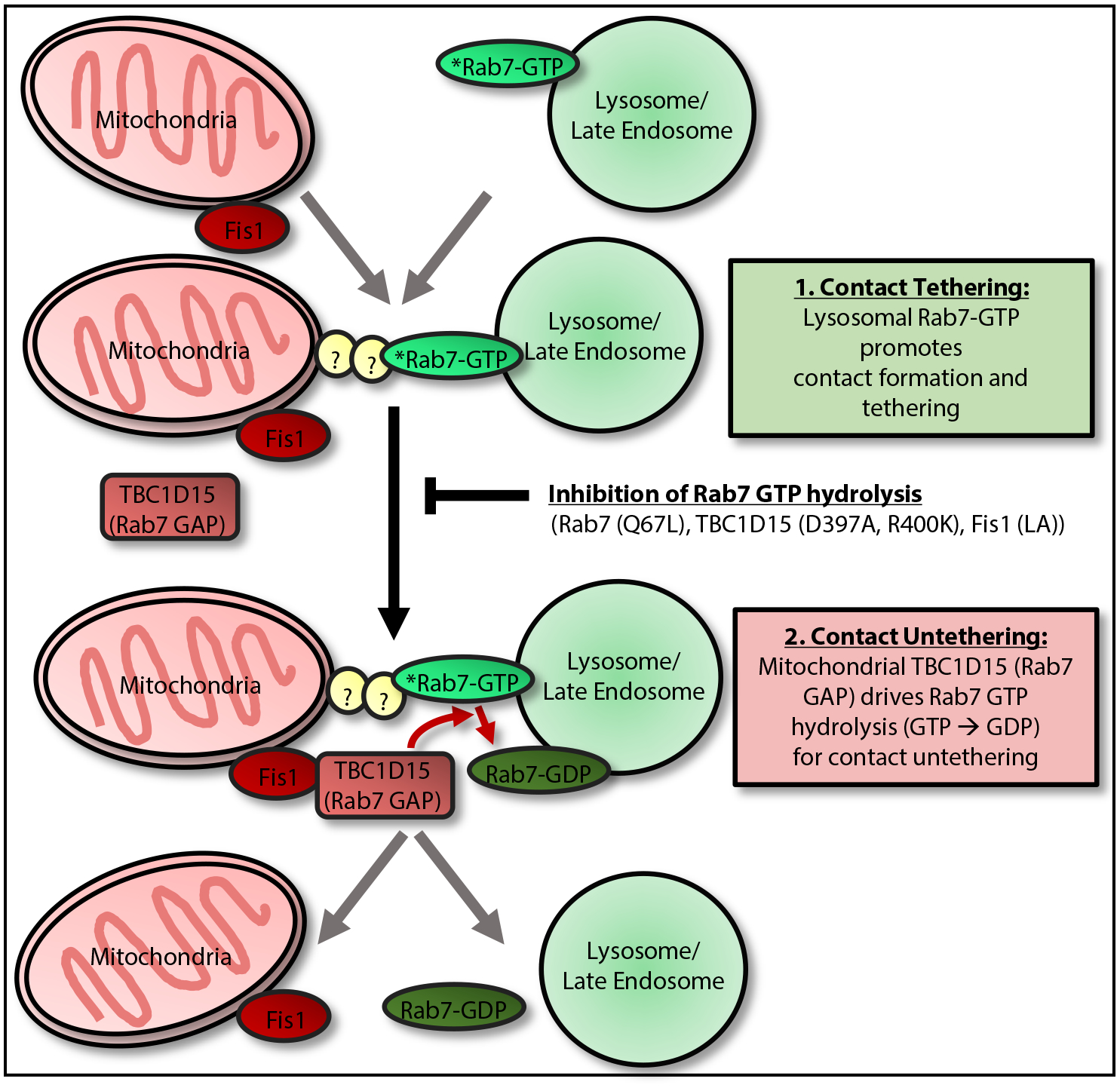
Mitochondria–lysosome contact dynamics involve: (1) contact tethering, which is promoted by lysosomal GTP-bound Rab7 and is potentially mediated by Rab7 effector proteins (which bind GTP-bound Rab7) to directly tether lysosomes to mitochondria [53]; (2) contacts subsequently undergo untethering, which is mediated by recruitment of cytosolic TBC1D15 (Rab7 GAP) to mitochondria via the outer mitochondrial membrane protein Fis1 [61–63]. At mitochondria–lysosome contact sites, mitochondrial TBC1D15 is able to interact with lysosomal GTP-bound Rab7 to drive Rab7 GTP hydrolysis from a GTP-bound to GDP-bound state. GDP-bound Rab7 can no longer bind Rab7 effectors and also loses its lysosomal membrane localization, leading to the loss of tethers and mitochondria–lysosome contact untethering [53]. Inhibition of Rab7 GTP hydrolysis with either mutant GTP-bound Rab7 (Q67L), which is unable to undergo GTP hydrolysis, mutant TBC1D15 (D397A or R400K), which lacks GAP activity [61], or mutant Fis1 (LA), which is unable to recruit TBC1D15 to mitochondria [61], all prevent efficient mitochondria–lysosome contact untethering, resulting in prolonged contacts [53]. Additional proteins apart from Rab7 may also be involved in regulating mitochondria–lysosome contact tethering.
Subsequent mitochondria–lysosome contact untethering is mediated by Rab7 GTP hydrolysis, which first involves the recruitment of cytosolic TBC1D15 (Rab7 GAP) to mitochondria via the outer mitochondrial membrane protein Fis1 [61–63]. Once recruited to mitochondria, TBC1D15 is able to interact with lysosomal GTP-bound Rab7 at mitochondria–lysosome contact sites to drive its hydrolysis to a GDP-bound state. GDP-bound Rab7 can no longer bind Rab7 effectors and also loses its lysosomal membrane localization [22], leading to mitochondria–lysosome contact untethering [53], potentially via the loss of Rab7 effector tethering. Importantly, inhibition of Rab7 GTP hydrolysis with either TBC1D15 (D397A or R400K) mutants, which lack GAP activity [61], prevents efficient mitochondria–lysosome contact untethering, resulting in prolonged contacts [53]. Interestingly, TBC1D15 mutants have no effect on contact formation, suggesting that TBC1D15-dependent Rab7 GTP hydrolysis is limited to regulating contact untethering but not the formation of contacts. In addition, mutant Fis1 (LA), which is unable to recruit TBC1D15 to mitochondria [61], as well as complete knockout of either TBC1D15 or Fis1, prevent efficient mitochondria–lysosome contact untethering, leading to prolonged contacts [53]. Thus, Rab7 GTP hydrolysis, which requires interaction of both lysosomal (Rab7) and mitochondrial-localized (TBC1D15, Fis1) proteins at contact sites, provides a mechanism for the regulation of mitochondria–lysosome untethering. Recently, Rab7 was also found to regulate contacts between mitochondria and late endosomes associated with ribosomes undergoing local protein synthesis in axons of retinal ganglion cells, which were further disrupted by disease-associated mutations in Rab7 [64]. Of note, as previous inter-organelle contact sites have been associated with multiple types of tethers [50], other proteins distinct from Rab7 or its effectors may also contribute to mitochondria–lysosome contact site tethering.
Bidirectional Regulation of Organelle Dynamics at Mitochondria–Lysosome Contact Sites
Lysosomal dynamics are acutely regulated by Rab7 effector proteins, which preferentially bind GTP-bound Rab7 on the lysosomal membrane, such as RILP and FYCO, which mediate lysosomal retrograde and anterograde microtubule transport, respectively [65,66], and the HOPS (homotypic fusion and protein sorting) complex, which mediates lysosomal tethering and fusion [67]. Mitochondria–lysosome contact sites thus offer a platform for mitochondrial-localized proteins to regulate lysosomal dynamics via modulation of Rab7-GTP binding (Figure 4A). As mitochondrial TBC1D15 promotes Rab7 GTP hydrolysis at contacts leading to the termination of GTP-bound Rab7 [53], this can simultaneously result in both contact site untethering and the release of Rab7 effector proteins from GTP-bound Rab7 and the lysosomal membrane, thus regulating lysosomal dynamics. Indeed, expression of mitochondrial-localized mutant TBC1D15 (D397A or R400K) lacking GAP activity leads to enlarged lysosomes [53], consistent with the lysosomal morphology observed upon inhibition of RAB7 GTP hydrolysis. Thus, mitochondria–lysosome contact sites may help promote Rab7 GTP hydrolysis and regulate the dynamics of a subset of lysosomes within the cell that are in contact with mitochondria.
Figure 4. Functions of Mitochondria–Lysosome Contact Sites, Including Regulation of Organelle Dynamics.
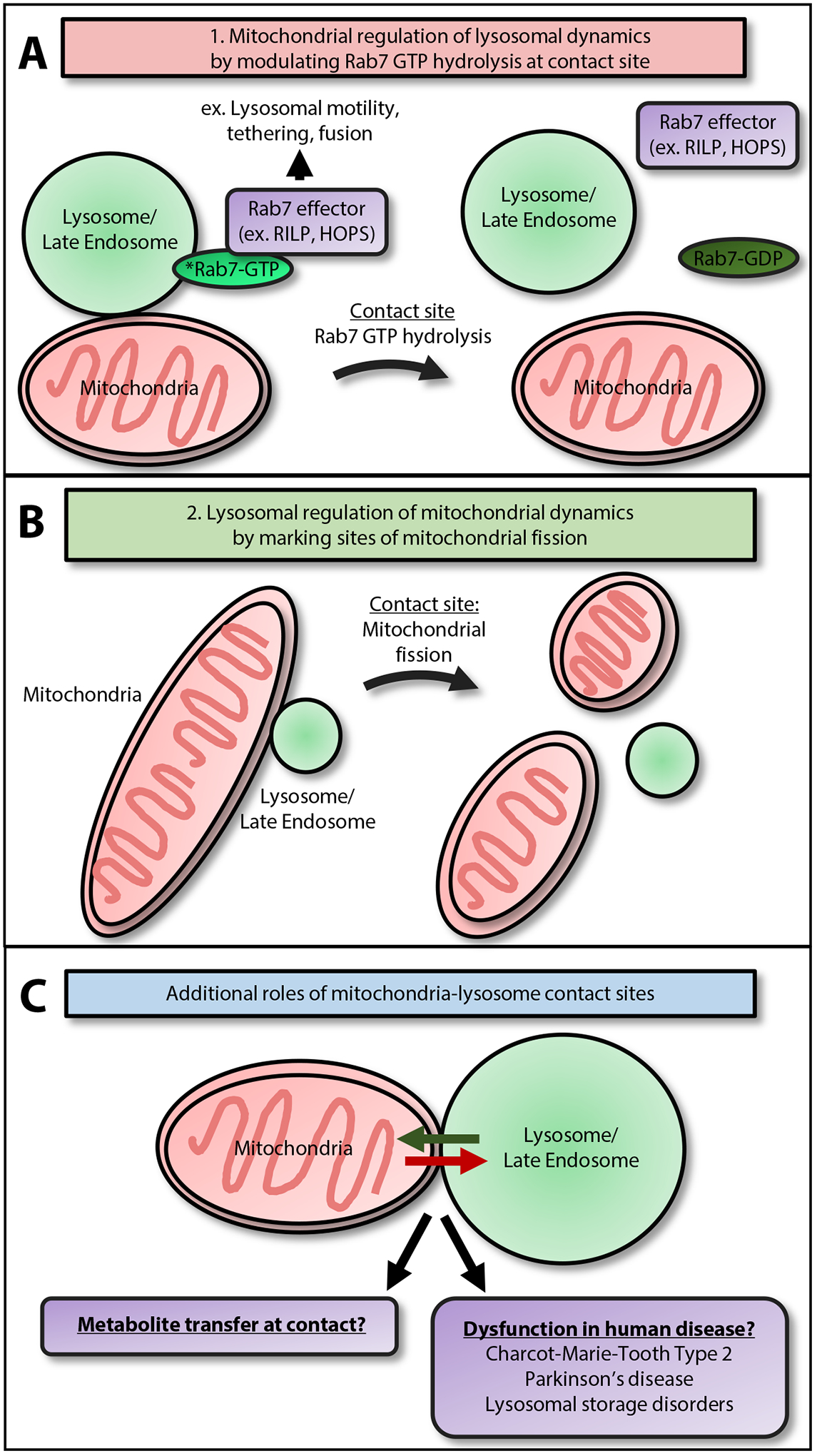
(A) Mitochondria regulate lysosomal dynamics by modulating Rab7 GTP hydrolysis at contact sites: Rab7 effector proteins (which bind GTP-bound Rab7) are critical for regulating lysosomal dynamics. These include RILP and FYCO, which mediate lysosomal motility, and the HOPS complex, which mediates lysosomal tethering and fusion. At mitochondria–lysosome contact sites, mitochondrial TBC1D15 promotes Rab7 GTP hydrolysis, leading to the termination of active, GTP-bound Rab7. This may result in the release of Rab7 effector proteins from Rab7-GTP and the lysosomal membrane, thus inhibiting lysosomal dynamics [53]. (B) Lysosomes regulate mitochondrial dynamics by marking sites of mitochondrial fission: the majority of mitochondrial fission sites are marked by lysosome/late endosomes, as compared with other vesicles such as early endosomes or peroxisomes. Importantly, disrupting mitochondria–lysosome contact untethering dynamics by inhibiting Rab7 GTP hydrolysis leads to decreased rates of mitochondrial fission [53], suggesting that lysosomes may directly regulate mitochondrial fission events via contact sites. (C) Mitochondria–lysosome contact sites may mediate additional functions, such as the inter-organelle transfer of metabolites, including lipids, calcium, or iron. In addition, contact site dysfunction may mediate the pathogenesis of multiple human diseases genetically and functionally linked to defects in both mitochondria and lysosomes, such as Charcot–Marie–Tooth Type 2, Parkinson’s disease, and lysosomal storage disorders.
Mitochondria–lysosome contact sites are also able to regulate mitochondrial dynamics, as the majority of mitochondrial fission events (>80%) are marked by LAMP1-positive vesicles (Figure 4B) but not early endosomes or peroxisomes [53]. Disrupting mitochondria–lysosome contact untethering dynamics by inhibiting Rab7 GTP hydrolysis with mutants such as GTP-bound Rab7 Q67L, TBC1D15 (D397A or R400K), or Fis1 (LA) decreases rates of mitochondrial fission and disrupts the mitochondrial network [53]. All mitochondrial fission events marked by lysosomes were also positive for Drp1, a dynamin-related GTPase that facilitates the constriction of the outer mitochondrial membrane during mitochondrial fission [5]. Interestingly, a novel, brain-enriched mouse isoform of DRP1 containing four alterative exons, DRP1ABCD, was recently identified, which associated with LAMP1-positive vesicles and localized to the interface between mitochondria and lysosomes [57]. Proper localization of DRP1ABCD depended on the acidification but not the proteolytic activity of late endosomes and lysosomes [57] and may point to additional roles for Drp1 isoforms at mitochondria–lysosome contact sites. Thus, mitochondria–lysosome contact sites additionally act to regulate the mitochondrial network by marking sites of mitochondrial fission and regulating the rate of fission events, but the mechanistic details of the interplay between Rab7 GTP hydrolysis and mitochondrial fission machinery remain to be further elucidated.
Additional Possible Functions of Mitochondria–Lysosome Contact Sites
As other inter-organelle contact sites have been shown to be key platforms for regulating metabolite flux between organelles [44], mitochondria–lysosome contact sites may similarly mediate additional functions such as transfer of lipids, calcium, or iron between mitochondria and lysosomes (Figure 4C). In addition, mitochondria–lysosome contacts also form under hypoxic conditions, which have been proposed to mediate voltage-dependent anion channel 1 (VDAC1) cleavage by endolysosomal enzymes through mitochondrial–endolysosomal microfusion [28].
In yeast, contact sites between mitochondria and the vacuole known as the vacuole and mitochondria patch (vCLAMP) [48,49] can regulate phospholipid transport between mitochondria and vacuoles as deletions of both ER-mitochondria encounter structure (ERMES) and vCLAMP, but not ERMES alone, result in severe alterations in phospholipid composition, including accumulation of phosphatidylserine and decreased phosphatidylcholine [49]. Interestingly, yeast vCLAMP can be tethered by either: (i) mitochondrial Tom40 binding to VPS39, a protein involved in vacuolar sorting and fusion which interacts with the vacuole membrane via the vacuolar Rab GTPase Ypt7 [48,68]; or (ii) mitochondrial MCP1 binding to Vps13, which is localized to the vacuole membrane via its Vps13 adaptor binding (VAB) domain binding a PxP motif on vacuolar Ypt35 [69,70]. Thus, at mitochondria–lysosome contact sites, there may be different protein complexes mediating distinct tethers or different functions, as has been suggested for VPS39 (physical tether) and VPS13/MCP1 (effectors of lipid transport) in vCLAMP. Of note, in addition to its localization at vCLAMP, yeast Vps13 has also been localized to vacuole–nucleus contact sites in yeast [71], and its four human homologs (Vps13A–D) have all been linked to different human diseases. Vps13A, whose mutations are linked to Chorea-acanthocytosis [72,73], has been proposed to tether ER to mitochondria and lipid droplets [74] and also localize to and regulate contacts between mitochondria and endolysosomes [75]. Vps13B mutations lead to Cohen syndrome [76] but has not been associated with inter-organelle contact sites, while Vps13C, whose mutations are linked to Parkinson’s disease (PD) [77], has been proposed to tether ER to late endosome/lysosomes and lipid droplets [74]. Recently, Vps13D was linked to childhood onset movement disorder and ataxia [78,79] and found to regulate mitochondrial dynamics [80], but its role in inter-organelle contact sites has not been directly studied. Cholesterol has also previously been shown to transport from endolysosomes to mitochondria via the steroidogenic acute regulatory protein-related lipid transfer (START) domain-containing protein, MLN64 [81,82], but whether this occurs directly at mitochondria–lysosome contact sites remains unclear.
Calcium is a highly regulated ion that plays a crucial role in various cellular processes such as exocytosis, gene transcription, and apoptosis [83]. While the primary cellular store of calcium is located in the ER, both mitochondria and lysosomes have also been implicated as important players in calcium homeostasis. Calcium is transported into mitochondria through VDAC1 on the outer mitochondrial membrane and the mitochondrial calcium uniporter on the inner mitochondrial membrane, and serves to remove cytosolic calcium and drive metabolic processes such as ATP production [84]. Mitochondria–ER contact sites, known as mitochondrial-associated membranes (MAMs), have previously been identified as regulators of calcium transfer and dynamics in various cell types [85], including mammalian neurons [86]. However, as lysosomes also play emerging roles in calcium signaling and storage, mitochondria–lysosome contact sites may serve as similar platforms for calcium transfer. Indeed, TRPML1, a mucolipin channel on the lysosomal membrane that releases calcium, functions as a sensor of cellular reactive oxygen species (ROS), which are produced in large part by mitochondria [87]. In addition, the activity of TRPML1 increases with rising levels of ROS, and TRPML1 activation promotes autophagy [88]. While this suggests an indirect modulation of mitochondrial function and activity by lysosomal calcium, mitochondria–lysosome contact sites may support more direct mechanisms for calcium transfer and signaling between these two organelles.
Iron is another metabolite which can be stored by both mitochondrial and endolysosomal compartments. Entry of iron into mitochondria occurs via mitoferrin-1 and -2 solute carriers on the inner mitochondrial membrane [89], where it is subsequently incorporated into iron–sulfur clusters in the matrix, which act as cofactors for various enzymes in the citric acid cycle and electron transport chain [90]. While delivery of iron to mitochondria remains poorly understood, one proposed mechanism involves direct endosomal delivery through a ‘kiss and run’ interaction [91,92]. In this model, iron bound to Tf (transferrin) is internalized by the cell and is subsequently released within the endosome upon acidification. Docking of Tf-endosomes onto mitochondria, either through VDAC1 or the divalent metal transporter-1 (DMT1) on the outer mitochondrial membrane [93, 94], provides the physical tether to allow for iron transfer. As lysosomes also serve as iron storage compartments [95], mitochondria–lysosome contact sites may also regulate the labile iron pool, similar to mitochondria–endosome contact sites. Interestingly, transferrin receptor-2 (TfR2) mediates lysosomal transferrin delivery, and deficiency in TfR2 results in reduced mitochondrial size and heme content in erythroid progenitors [96]. Furthermore, iron overload in fibroblasts from patients with neurodegeneration with brain iron accumulation (NBIA) results in mitochondrial depolarization, reduced ATP production, and decreased lysosomal proteolytic activity [97], suggesting that mitochondria–lysosome contact site dysfunction and iron flux may be intricately connected.
Additionally, mitochondria–lysosome contact sites may also dynamically interact with other organelles, such as the ER. Indeed, mitochondria and lysosomes in contact with one another can also simultaneously contact the ER [53], and mitochondrial fission sites marked by lysosomes are also positive for ER tubules [53], which were previously found to mark sites of mitochondrial division [10]. Moreover, as ER–mitochondria and ER–late endosome/lysosomal contacts form frequently [46] and further regulate both mitochondrial and lysosomal homeostasis [45], modulation of proteins at ER contacts such as those involved in regulating late endosomal dynamics [51,98] may further modulate mitochondria–lysosome contact function and tethering. Conversely, as ER–endosome contacts can be modulated via the Rab7 effector protrudin [99], mitochondria–lysosome contacts may also modulate ER function via regulation of Rab7 GTP hydrolysis and the ability of Rab7 to recruit its effector proteins. Finally, as multiple other organelles also form contacts with both mitochondria and lysosome/late endosomes [44], it is likely that similar to the regulation of lipid transport in yeast, the maintenance of organelle homeostasis involves multiple different types of inter-organelle contact sites that may regulate and compensate for one another.
Mitochondria–Lysosome Contact Sites in Disease Pathogenesis
Misregulation of mitochondria–lysosome contacts may simultaneously drive dysfunction of both organelles in various human diseases, such as Charcot–Marie–Tooth Type 2 (CMT2), PD, and several lysosomal storage disorders (LSDs) that have been genetically and functionally linked to both mitochondrial and lysosomal defects [31,100,101] (Figure 4C). CMT2 is a group of autosomal dominant axonal peripheral neuropathies affecting both lower motor and sensory neurons and is genetically linked to mitochondrial and endolysosomal dysfunction [101]. Indeed, Charcot–Marie–Tooth Type 2B (CMT2B) is caused by mutations in Rab7 that disrupt its GTP hydrolysis [23–26], leading to an increased GTP-bound Rab7 state [102,103], suggesting that mitochondria–lysosome contact sites may be directly misregulated in CMT2B. In addition, other CMT2 genes have been linked to endolysosomal pathways, including SH3TC2 (endocytic recycling), FIG4 (endocytic recycling), and LRSAM1 (receptor endocytosis) [101]. Similarly, multiple CMT2 genes have also been linked to mitochondrial fission and/or fusion dynamics, including Mfn2 (CMT2A), INF2 (CMT2DIE), DNM2 (CMT2M), and GDAP1 (CMT2K) [11,16,101,104]. Thus, defective mitochondria–lysosome contacts may act as a converging mechanism in driving peripheral axonal degeneration for multiple genetic forms of CMT2.
PD is the second most common neurodegenerative disorder, with motor symptoms caused by dopaminergic neurodegeneration in the substantia nigra [105,106]. Like CMT2, PD has been both genetically and functionally linked to mitochondrial and lysosomal dysfunction, with the identification of familial mutations in mitochondrial-associated genes (Parkin, PINK1, and DJ-1) and endolysosomal-associated genes (VPS35, PARK9, and GBA1) [100]. Moreover, both mitochondrial and lysosomal dysfunction have been observed in human dopaminergic neurons from idiopathic PD patients [31], suggesting that these two organelles play a critical role in disease progression. As mentioned above, mutations in VPS13C, a human homologue of the yeast Vps13 that mediates vCLAMP tethering [69,71,107], also lead to autosomal recessive PD [77]. Several familial PD mutations, such as in VPS35 and Parkin, also decrease Rab7 GTP-binding [108,109], further highlighting a potential role for defective mitochondria–lysosome contact regulation in PD pathogenesis.
LSDs encompass a group of more than 70 diseases involving lysosomal dysfunction [110]. The most common LSD is caused by autosomal recessive mutations in glucocerebrosidase 1 (GBA1), which leads to Gaucher’s disease. Interestingly, mitochondrial dysfunction, such as decreased mitochondrial membrane potential, increased ROS and impaired respiration, and morphological abnormalities have been observed in multiple Gaucher’s models, including patient fibroblasts, flies, and mouse primary neurons [111–116]. Additionally, mitochondrial dysfunction and increased oxidative stress have been found in multiple other LSDs, including Niemann-Pick disease type C and neuronal ceroid lipofuscinosis [4], suggesting that mitochondrial defects may be a common theme across LSDs and may be partially mediated by defective mitochondria–lysosome contact site function secondary to lysosomal dysfunction. Of note, mitochondrial dysfunction may further exacerbate lysosomal storage defects, as mitochondrial respiration deficiency via TFAM mutation is sufficient to cause lysosomal sphingomyelin accumulation and elicit a proinflammatory response [29]. Likewise, mitochondrial oxidant stress can lead to the oxidation of specific cysteine residues in the catalytic region of GBA1, further contributing to decreased GBA1 enzymatic activity [31].
Concluding Remarks
The recent identification of mitochondria–lysosome membrane contact sites in mammalian cells and their regulation of Rab7 GTP hydrolysis and mitochondrial dynamics highlight the intricate crosstalk between these two organelles. These contact sites offer important insight into their bidirectional relationship and may help to explain the converging dysfunction of both organelles in multiple diseases, including CMT2, PD, and LSDs. As both mitochondria and lysosomes are crucial for proper metabolism and degradation, investigating their interplay will be key to elucidating their overlapping pathways in cellular homeostasis.
Looking forward, multiple techniques and approaches will be crucial for shedding further light on the organization and function of mitochondria–lysosome contact sites. These include new advanced imaging techniques, such as grazing incidence structured illumination microscopy (GI-SIM) for nanoscale resolution over millisecond timescales [117] combined with internal tagging of endogenous proteins [74] and single molecule tracking to visualize protein dynamics at contact sites. In addition, techniques such as the use of protein-fragment complementation libraries, which enable systematic analysis of membrane protein topology [118], or proximity biotinylation with APEX2 [119] will help to identify new regulators of membrane contact sites. Ultimately, future studies on the function and regulation of these contacts will be critical for advancing our understanding of the roles mitochondria and lysosomes play in health and disease (see Outstanding Questions).
Outstanding Questions.
Are mitochondria–lysosome contact sites formed with similar dynamics and mechanistic regulation in specialized cell types such as neurons?
Are there different types of mitochondria–lysosome contacts? If so, what are the protein tethers at these sites and how are they regulated?
Which metabolites are transferred at mitochondria–lysosome contact sites and what proteins regulate their transfer?
How do mitochondria–lysosome contact sites interact with other organelles and what are the functional implications of these interactions?
What role do mitochondria–lysosome contact sites play in the pathogenesis of different human diseases and what are the downstream consequences of their dysfunction?
Highlights.
Mitochondria and lysosomes form dynamic inter-organelle membrane contact sites, which are independent from mitophagy.
GTP-bound Rab7 promotes mitochondria–lysosome contact site formation and tethering, while mitochondrial TBC1D15 (Rab7-GAP) recruited to mitochondria via Fis1 drives lysosomal Rab7 GTP hydrolysis at mitochondria–lysosome contact sites, leading to contact untethering.
Mitochondria regulate lysosomal dynamics at contact sites by modulating Rab7 GTP-binding, which is a master regulator of lysosomal dynamics via Rab7 effector proteins.
Lysosomes conversely regulate mitochondrial dynamics at contact sites by marking the majority of mitochondrial fission sites.
Mitochondria–lysosome contact sites may mediate inter-organelle transfer of metabolites and contribute to the pathogenesis in diseases linked to dysfunction of both organelles.
Glossary
- Drp1
a dynamin-related GTPase that regulates mitochondrial fission via its GTP hydrolysis
- Fis1
an outer mitochondrial membrane protein that recruits TBC1D15 to the mitochondria
- Membrane contact site
a stable contact between the membranes of two different organelles that are tethered in close apposition (b30 nm) without ultimately fusing with one another, which can regulate the function of either organelle
- Rab7
a small GTPase which localizes to lysosomes/late endosomes upon GTP-binding and acts as a master regulator of lysosomal dynamics by binding Rab7 effector proteins in its GTP-bound state
- Rab GTP hydrolysis
the conversion of Rab proteins from a GTP-bound to a GDP-bound state, mediated by GAPs (GTPase activating proteins)
- TBC1D15
a GAP (GTPase activating protein) for Rab7 that is cytosolic and recruited to the outer mitochondrial membrane via Fis1
- Vacuole and mitochondria patch (vCLAMP)
membrane contact site between mitochondria and vacuoles in yeast
Footnotes
Disclaimer Statement
The authors declare no conflict of interest.
References
- 1.Hutagalung AH and Novick PJ (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev 91, 119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burte F et al. (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol 11, 11–24. [DOI] [PubMed] [Google Scholar]
- 3.Mc Donald JM and Krainc D (2017) Lysosomal proteins as a therapeutic target in neurodegeneration. Annu. Rev. Med 68, 445–458. [DOI] [PubMed] [Google Scholar]
- 4.Plotegher N and Duchen MR (2017) Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol. Med 23, 116–134. [DOI] [PubMed] [Google Scholar]
- 5.Friedman JR and Nunnari J (2014) Mitochondrial form and function. Nature 505, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun N et al. (2016) The mitochondrial basis of aging. Mol. Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra P and Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J. Cell Biol 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis SC et al. (2016) ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353 aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smirnova E et al. (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman JR et al. (2011) ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korobova F et al. (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji WK et al. (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 4, e11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S et al. (2015) Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J. Cell Biol 208, 109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manor U et al. (2015) A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 4, 08828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore AS et al. (2016) Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun 7, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JE et al. (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuchner S et al. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet 36, 449–451. [DOI] [PubMed] [Google Scholar]
- 18.Alexander C et al. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet 26, 211–215. [DOI] [PubMed] [Google Scholar]
- 19.Delettre C et al. (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet 26, 207–210. [DOI] [PubMed] [Google Scholar]
- 20.Aits S and Jaattela M (2013) Lysosomal cell death at a glance. J. Cell Sci 126, 1905–1912. [DOI] [PubMed] [Google Scholar]
- 21.Zhen Y and Stenmark H (2015) Cellular functions of Rab GTPases at a glance. J. Cell Sci 128, 3171–3176. [DOI] [PubMed] [Google Scholar]
- 22.Langemeyer L et al. (2018) Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 28, 957–970. [DOI] [PubMed] [Google Scholar]
- 23.Houlden H et al. (2004) A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol 56, 586–590. [DOI] [PubMed] [Google Scholar]
- 24.Meggouh F et al. (2006) Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology 67, 1476–1478. [DOI] [PubMed] [Google Scholar]
- 25.Verhoeven K et al. (2003) Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet 72, 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X et al. (2014) A novel RAB7 mutation in a Chinese family with Charcot-Marie-Tooth type 2B disease. Gene 534, 431–434. [PubMed] [Google Scholar]
- 27.Demers-Lamarche J et al. (2016) Loss of mitochondrial function impairs lysosomes. J. Biol. Chem 291, 10263–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brahimi-Horn MC et al. (2015) Local mitochondrial-endolysosomal microfusion cleaves voltage-dependent anion channel 1 to promote survival in hypoxia. Mol. Cell. Biol 35, 1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baixauli F et al. (2015) Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez-Mosquera L et al. (2017) Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep 7, 45076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burbulla LF et al. (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mansueto G et al. (2017) Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 25, 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan NA et al. (2017) mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 26, 419–428. [DOI] [PubMed] [Google Scholar]
- 34.Norambuena A et al. (2018) A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-beta oligomers. EMBO J. 37, e100241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monteleon CL et al. (2018) Lysosomes support the degradation, signaling, and mitochondrial metabolism necessary for human epidermal differentiation. J. Investig. Dermatol 138, 1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamano K et al. (2018) Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife 7, e31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu F et al. (2018) Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife 7, e32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pickrell AM and Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugiura A et al. (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33, 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamacher-Brady A et al. (2014) Intramitochondrial recruitment of endolysosomes mediates Smac degradation and constitutes a novel intrinsic apoptosis antagonizing function of XIAP E3 ligase. Cell Death Differ. 21, 1862–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong YC and Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U. S. A 111, E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazarou M et al. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLelland GL et al. (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gatta AT and Levine TP (2017) Piecing together the patch-work of contact sites. Trends Cell Biol. 27, 214–229. [DOI] [PubMed] [Google Scholar]
- 45.Wu HX et al. (2018) Here, there, and everywhere: the importance of ER membrane contact sites. Science 361 eaan5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valm AM et al. (2017) Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daniele T et al. (2014) Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr. Biol 24, 393–403. [DOI] [PubMed] [Google Scholar]
- 48.Elbaz-Alon Y et al. (2014) A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell 30, 95–102. [DOI] [PubMed] [Google Scholar]
- 49.Honscher C et al. (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev. Cell 30, 86–94. [DOI] [PubMed] [Google Scholar]
- 50.Eisenberg-Bord M et al. (2016) A tether is a tether is a tether: tethering at membrane contact sites. Dev. Cell 39, 395–409. [DOI] [PubMed] [Google Scholar]
- 51.Rowland AA et al. (2014) ER contact sites define the position and timing of endosome fission. Cell 159, 1027–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simmen T and Herrera-Cruz MS (2018) Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol 53, 61–69. [DOI] [PubMed] [Google Scholar]
- 53.Wong YC et al. (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aston D et al. (2017) High resolution structural evidence suggests the sarcoplasmic reticulum forms microdomains with acidic stores (lysosomes) in the heart. Sci. Rep 7, 40620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fermie J et al. (2018) Single organelle dynamics linked to 3D structure by correlative live-cell imaging and 3D electron microscopy. Traffic 19, 354–369. [DOI] [PubMed] [Google Scholar]
- 56.Han Y et al. (2017) Cell-permeable organic fluorescent probes for live-cell long-term super-resolution imaging reveal lysosome-mitochondrion interactions. Nat. Commun 8, 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itoh K et al. (2018) A brain-enriched Drp1 isoform associates with lysosomes, late endosomes, and the plasma membrane. J. Biol. Chem 293, 11809–11822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Q et al. (2018) Super-resolution tracking of mitochondrial dynamics with an iridium(III) luminophore. Small 14, e1802166. [DOI] [PubMed] [Google Scholar]
- 59.Csordas G et al. (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol 174, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Phillips MJ and Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Onoue K et al. (2013) Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J. Cell Sci 126, 176–185. [DOI] [PubMed] [Google Scholar]
- 62.Peralta ER et al. (2010) Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence. J. Biol. Chem 285, 16814–16821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang XM et al. (2005) TBC domain family, member 15 is a novel mammalian Rab GTPase-activating protein with substrate preference for Rab7. Biochem. Biophys. Res. Commun 335, 154–161. [DOI] [PubMed] [Google Scholar]
- 64.Cioni JM et al. (2019) Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pankiv S et al. (2010) FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol 188, 253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jordens I et al. (2001) The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol 11, 1680–1685. [DOI] [PubMed] [Google Scholar]
- 67.Balderhaar HJK and Ungermann C (2013) CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J. Cell Sci 126, 1307–1316. [DOI] [PubMed] [Google Scholar]
- 68.Montoro AG et al. (2018) Vps39 interacts with Tom40 to establish one of two functionally distinct vacuole-mitochondria contact sites. Dev. Cell 45, 621–636. [DOI] [PubMed] [Google Scholar]
- 69.Peter ATJ et al. (2017) Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J. Cell Biol 216, 3219–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bean BDM et al. (2018) Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J. Cell Biol 217, 3593–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lang AB et al. (2015) ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J. Cell Biol 210, 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rampoldi L et al. (2001) A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet 28, 119–120. [DOI] [PubMed] [Google Scholar]
- 73.Ueno S et al. (2001) The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet 28, 121–122. [DOI] [PubMed] [Google Scholar]
- 74.Kumar N et al. (2018) VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol 217, 3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muñoz-Braceras S et al. (2019) VPS13A, a closely associated mitochondrial protein, is required for efficient lysosomal degradation. Dis. Model. Mech 12, dmm036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kolehmainen J et al. (2003) Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am. J. Hum. Genet 72, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lesage S et al. (2016) Loss of VPS13C function in autosomal-recessive Parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet 98, 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gauthier J et al. (2018) Recessive mutations in N VPS13D cause childhood onset movement disorders. Ann. Neurol 83, 1089–1095. [DOI] [PubMed] [Google Scholar]
- 79.Seong E et al. (2018) Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol 83, 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anding AL et al. (2018) Vps13D encodes a ubiquitin-binding protein that is required for the regulation of mitochondrial size and clearance. Curr. Biol 28, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Charman M et al. (2010) MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res 51, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang M et al. (2002) MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J. Biol. Chem 277, 33300–33310. [DOI] [PubMed] [Google Scholar]
- 83.Raffaello A et al. (2016) Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci 41, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Todkar K et al. (2017) Mitochondria and lysosomes: discovering bonds. Front Cell Dev Biol 5, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bononi A et al. (2012) Mitochondria-associated membranes (MAMs) as hotspot Ca2+ signaling units. Calcium Signaling 740, 411–437. [DOI] [PubMed] [Google Scholar]
- 86.Hirabayashi Y et al. (2017) ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 358, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sena LA and Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang XL et al. (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun 7, 12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Richardson DR et al. (2010) Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. U. S. A 107, 10775–10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rouault TA (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech 5, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sheftel AD et al. (2007) Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110, 125–132. [DOI] [PubMed] [Google Scholar]
- 92.Das A et al. (2016) Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell Biol 214, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wolff NA et al. (2018) A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wolff NA et al. (2014) Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 28, 2134–2145. [DOI] [PubMed] [Google Scholar]
- 95.Morgan J et al. (2012) Release of iron from cellular iron stores in lysosomes: potential involvement of divalent metal transporter 1 (DMT1) and common metabolites. FASEB J. 26, 1. [Google Scholar]
- 96.Khalil S et al. (2017) A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2. Blood Advances 1, 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seibler P et al. (2018) Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 141, 3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hoyer MJ et al. (2018) A novel class of ER membrane proteins regulates ER-associated endosome fission. Cell 175, 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raiborg C et al. (2015) Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238. [DOI] [PubMed] [Google Scholar]
- 100.Plotegher N and Duchen MR (2017) Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front Cell Dev Biol 5, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rossor AM et al. (2016) Recent advances in the genetic neuropathies. Curr. Opin. Neurol 29, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McCray BA et al. (2010) Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum. Mol. Genet 19, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spinosa MR et al. (2008) Functional characterization of Rab7 mutant proteins associated with Charcot-Marie-Tooth type 2B disease. J. Neurosci 28, 1640–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Santel A and Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J. Cell Sci 114, 867–874. [DOI] [PubMed] [Google Scholar]
- 105.Wong YC and Krainc D (2017) Alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med 23, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kalia LV and Lang AE (2015) Parkinson’s disease. Lancet 386, 896–912. [DOI] [PubMed] [Google Scholar]
- 107.Park JS et al. (2016) Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol. Biol. Cell 27, 2435–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song P et al. (2016) Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J. Neurosci 36, 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jia D et al. (2016) Structural and mechanistic insights into regulation of the retromer coat by TBC1d5. Nat. Commun 7, 13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Platt FM et al. (2018) Lysosomal storage diseases. Nature Reviews Disease Primers 4, 27. [DOI] [PubMed] [Google Scholar]
- 111.Cleeter MW et al. (2013) Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int 62, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.de la Mata M et al. (2015) Pharmacological chaperones and coenzyme Q10 treatment improves mutant beta-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of Gaucher disease. Sci. Rep 5, 10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li H et al. (2018) Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 15, 113–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Osellame LD et al. (2013) Mitochondria and quality control defects in a mouse model of Gaucher disease – links to Parkinson’s disease. Cell Metab. 17, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schondorf DC et al. (2018) The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 23, 2976–2988. [DOI] [PubMed] [Google Scholar]
- 116.Xu YH et al. (2014) Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet 23, 3943–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo YT et al. (2018) Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175, 1430–1442. [DOI] [PubMed] [Google Scholar]
- 118.Weill U et al. (2018) Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat. Methods 15, 617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hung V et al. (2017) Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, e24463. [DOI] [PMC free article] [PubMed] [Google Scholar]