

Abstract
Background
This Mendelian randomization study aims to investigate causal associations between genetically predicted insomnia and 14 cardiovascular diseases (CVDs) as well as the potential mediator role of 17 cardiometabolic risk factors.
Methods and Results
Using genetic association estimates from large genome‐wide association studies and UK Biobank, we performed a 2‐sample Mendelian randomization analysis to estimate the associations of insomnia with 14 CVD conditions in the primary analysis. Then mediation analysis was conducted to explore the potential mediator role of 17 cardiometabolic risk factors using a network Mendelian randomization design. After correcting for multiple testing, genetically predicted insomnia was consistent significantly positively associated with 9 of 14 CVDs, those odds ratios ranged from 1.13 (95% CI, 1.08–1.18) for atrial fibrillation to 1.24 (95% CI, 1.16–1.32) for heart failure. Moreover, genetically predicted insomnia was consistently associated with higher body mass index, triglycerides, and lower high‐density lipoprotein cholesterol, each of which may act as a mediator in the causal pathway from insomnia to several CVD outcomes. Additionally, we found very little evidence to support a causal link between insomnia with abdominal aortic aneurysm, thoracic aortic aneurysm, total cholesterol, low‐density lipoprotein cholesterol, glycemic traits, renal function, and heart rate increase during exercise. Finally, we found no evidence of causal associations of genetically predicted body mass index, high‐density lipoprotein cholesterol, or triglycerides on insomnia.
Conclusions
This study provides evidence that insomnia is associated with 9 of 14 CVD outcomes, some of which may be partially mediated by 1 or more of higher body mass index, triglycerides, and lower high‐density lipoprotein cholesterol.
Keywords: cardiometabolic risk factors, cardiovascular disease, insomnia, mediator, Mendelian randomization
Subject Categories: Cardiovascular Disease, Epidemiology, Mental Health
Nonstandard Abbreviations and Acronyms
- IS
ischemic stroke
- IVW
inverse variance weighted
- MR
Mendelian randomization
- WC
waist circumference
- WHR
waist‐hip ratio
Clinical Perspective
What Is New?
Insomnia is the second most prevalent mental disorder.
Except for some major cardiovascular diseases (CVDs), the causal relationships between insomnia and the development of other CVDs such as arterial hypertension are still unclear, and the potential pathways involved in the association from insomnia to CVDs have not been studied.
This study performed Mendelian randomization analysis to investigate the causal associations between genetically predicted insomnia and 14 CVDs as well as the potential mediator role of 17 cardiometabolic risk factors.
What Are the Clinical Implications?
Insomnia was found to increase the risk of 9 of 14 CVD outcomes, including ischemic stroke, transient ischemic attack, thrombotic diseases, and 5 other CVDs (eg, coronary artery disease, heart failure, arterial hypertension).
Genetic predicted insomnia was associated with higher body mass index and triglycerides as well as lower high‐density lipoprotein cholesterol (without bidirectional causality), each of which may act as a mediator in the causal pathway from insomnia to several CVD outcomes.
These results are consistent with previous studies that suggest insomnia as an important causal risk factor for some major CVDs.
Insomnia is the second most prevalent mental disorder with an annual incidence of approximately 35% to 50% in the general population.1, 2 In the past decade, there has been increasing evidence suggesting insomnia as an important risk factor of cardiovascular disease (CVD),3, 4, 5, 6 but evidence originating from observational studies (eg, the association between insomnia and hypertension7, 8, 9, 10, 11, 12) can sometimes be inconsistent, which may, at least in part, be explained by confounding or bias due to reverse causation. The American Heart Association published a scientific statement asking health organizations to develop evidence‐based sleep recommendations for a number of sleep disorders, including insomnia.13 Therefore, it is an urgent need to address the causal evidence to determine whether insomnia is in fact on the causal pathway for each CVD outcome. Although several Mendelian randomization (MR) studies have investigated that insomnia may be a causal risk factor for some major CVDs, including coronary artery disease (CAD), heart failure (HF), and ischemic stroke (IS),14, 15, 16 uncertainty persists about the causal role of insomnia for the development of other CVDs, such as arterial hypertension. Moreover, the potential pathways involved in the association from insomnia to CVDs have not been studied. Previous studies presented evidence that insomnia is associated with impaired glucose metabolism17 and several risk factors for CVD, such as body mass index (BMI) and waist‐hip ratio (WHR),14, 15 so cardiometabolic risk factors may act as potential mediators that lie in the pathway from insomnia to the risk of specific CVD outcome.
MR is a powerful approach to estimate the causal effect of an exposure on an outcome in observational data.18, 19 The analytical method is less susceptible to bias due to confounders and evades potential bias by reverse causation through the use of genetic variants randomly allocated during conception, generally single‐nucleotide polymorphisms (SNPs), as instrumental variables for exposure.18, 19 The following 3 key assumptions guarantee the altered genetic variants to be valid instrumental variables: (1) (Relevance) genetic variants are associated with the exposure; (2) (Independence) genetic variants are not associated with any confounder of the exposure‐outcome association; and (3) (Exclusion restriction) genetic variants are not associated with outcome conditional on exposure and confounders.20, 21 Well‐powered genome‐wide association studies (GWAS) have identified hundreds of SNPs associated with insomnia14; additionally, several consortia with large numbers of participants have made summarized data publicly available for many risk factors. These create the opportunity to conduct MR analysis in a 2‐sample strategy using summarized data to obtain more precise estimates of the causal effects. Moreover, compared with standard MR, the network MR method22 is increasingly being used to answer causal mediation questions within the MR framework. It calculated the causal effects of the exposure on a mediator and the mediator on the outcome; these 2 estimates can then be multiplied together to estimate the indirect effect.22, 23, 24 The proportion mediated is calculated by dividing the indirect effect by the total effect; therefore, the network MR method can be used to investigate the effect of an exposure on the outcome operating through each mediator (indirect effect).
In this study, based on summary data obtained from large consortia and calculated based on individuals from UK Biobank, we performed a series of 2‐sample MR analyses to establish whether insomnia has a causal effect on the risks of 14 CVD outcomes. Then we conducted a mediation analysis to examine each of 17 cardiometabolic risk factors as a possible mediator in the causal relationship between genetically determined insomnia and each CVD outcome using a network MR design. A flow chart of our study design was provided in Figure 1.
Figure 1. The flow chart of study design and data sources for each analysis performed.
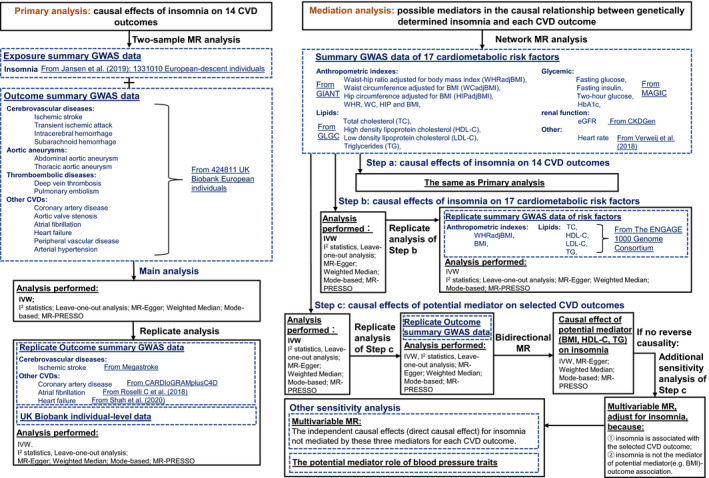
CKDGen indicates the Kidney Disease Genetics consortium; CVD, cardiovascular disease; eGFR, estimated glomerular filtration rate; ENGAGE, European Network for Genetic and Genomic Epidemiology; GIANT, the Genetic Investigation of Anthropometric Traits consortium; GWAS, genome‐wide association study; HbA1c, hemoglobin A1c; IVW, inverse variance weighted; MAGIC, the Meta‐Analyses of Glucose and Insulin‐Related Traits consortium; MR, Mendelian randomization; and MR‐PRESSO, Mendelian randomization‐pleiotropy residual sum and outlier.
Methods
The data used in these analyses are publicly available. The UK Biobank study was approved by the North West Multi‐centre Research Ethics Committee, and all its participants provided informed consent. The UK Biobank data were accessed through application 51470. The analyses of other publicly available data or summary statistics do not require additional ethical approval. All generated results were presented in the article and its supplement.
Genetic Instruments
Instrumental variables for insomnia were obtained from a GWAS meta‐analysis of 1 331 010 European‐descent individuals,14 the insomnia complaints were measured using questionnaire data, and the specific definition was provided in Table S1. This study identified 248 independent lead SNPs (r 2<0.1) located in 202 genomic risk loci (distinct genomic loci are >250kb apart) associated with insomnia at genome‐wide significance (2‐sided P value from the meta‐analysis of the GWAS results of insomnia: P<5×10−8), explaining 2.6% of the variance in insomnia (corresponding to an F statistic of 143.24).14 This means that assumption (1) is satisfied and avoids the bias caused by weak instrument in terms of the rule of thumb25; see Data S1 for the details. The effect estimates (in units of log odds ratios [ORs]) and corresponding standard errors of these SNPs were extracted from Table S6 in Jansen et al14 (the details of those SNPs were provided in the Table S2). For all analyses in this study, SNPs were aligned to the same effect allele across the data sources before analyses, and we checked the effect allele frequencies for concordance.
Data Sources
Data Sources of CVD Outcomes
The summary statistics of genetic associations with the outcomes were derived based on the imputed genotype data of the UK Biobank, an ongoing large prospective cohort study. The UK Biobank recruited over 500 000 people aged 40 to 69 years, mean age 56.5 years, from Great Britain between 2006 and 2010, with 94% self‐reported European ancestry, 45.6% men, and median follow‐up time currently 11.1 years (https://www.ukbiobank.ac.uk/).26 In this study, we considered outcomes including the following 14 subtypes of CVD as Larsson et al27 did: cerebrovascular diseases (IS, transient ischemic attack, intracerebral hemorrhage, and subarachnoid hemorrhage), aortic aneurysms (abdominal and thoracic aortic aneurysm), thrombotic diseases (deep vein thrombosis and pulmonary embolism), and other CVDs (CAD, aortic valve stenosis, atrial fibrillation [AF], HF, peripheral vascular disease as well as arterial hypertension). The outcomes were defined based on electronic health records, hospital procedure codes, and self‐reported information confirmed by interview with a nurse. The specific definitions and sources of information can be found in Table S3. For quality control, we excluded participants with (1) non‐White British ancestry, (2) mismatch between genetic sex and self‐reported sex, (3) excess relatedness with kinship more than 10 putative third‐degree relatives, (4) poor‐quality genotyping with heterozygosity and missing rates > 1.5%, or (5) outliers with high heterozygosity and high missing rate (see Figure S1 for the flow chart of individual selection). Finally, a total of 424 811 UK Biobank participants who satisfied the inclusion criteria were included in this study. The mean age was 57.37 years (5th to 95th percentile: 43 to 69 years), and 45.65% of the population were men (descriptive statistics were reported in Table S4). For each CVD outcome, individuals suffering from any other 13 CVD outcomes or without genetic data were further excluded from the analysis’s data set (Figure S1). Then the genetic associations with each CVD outcome (on a log OR scale) were obtained from the identified White British individuals in UK Biobank using logistic regression controlling for 10 principal components, which can further control for population stratification.
Data Sources of Cardiometabolic Risk Factors
The genetic association estimates with 17 cardiometabolic risk factors, including anthropometric indexes (WHR adjusted for BMI, waist circumference [WC] adjusted for BMI, hip circumference adjusted for BMI, WHR, WC, hip circumference, and BMI), lipids (total cholesterol [TC], high‐density lipoprotein cholesterol [HDL‐C], low‐density lipoprotein cholesterol [LDL‐C], and triglycerides [TG]), glycemic traits (fasting glucose, fasting insulin, 2‐hour glucose, hemoglobin A1c), renal function (estimated glomerular filtration rate), and heart rate increase during exercise were taken from the GIANT (Genetic Investigation of Anthropometric Traits) consortium,28, 29 the GLGC (Global Lipids Genetic Consortium),30 the MAGIC (Meta‐Analyses of Glucose and Insulin‐related Traits Consortium),31, 32, 33 the CKDGen (Kidney Disease Genetics) consortium,34 and published GWAS study in Verweij et al,35 respectively. We constrained the population of these GWAS summary statistics data mainly from European ancestry to minimize the bias caused by population stratification. The basic characteristics of these consortia and the genetic association estimates were presented in Table S5.
Statistical Analysis
Primary analysis
Our primary analysis aimed to explore the causal associations of insomnia with 14 CVD outcomes. Because 1 of 248 SNPs (rs77641763 in chr9) was unavailable in the outcome data set (UK Biobank), 247 SNPs were used as genetic instrumental variables for insomnia.
For each CVD outcome, the overall causal estimate of insomnia on this CVD outcome was obtained using an inverse variance weighted (IVW) method36 (under a multiplicative random‐effects model), combining the variant‐specific Wald (ratio) estimators37, 38 estimated for each SNP through the SNP‐CVD association divided by the SNP‐insomnia association. This method provides the highest precision and retains maximal power under the assumption that all SNPs are valid instrumental variables,39 that is, the pleiotropic effects of the genetic variants (genetic variant affects the outcome via a different biological pathway from the exposure under investigation) are all zero.40 Moreover, it accounts for heterogeneity in the variant‐specific causal estimates.41 The results were converted to ORs expressed per genetically predicted 1‐unit‐higher log‐odds of liability to insomnia, corresponding to per 2.72‐fold increase in the prevalence of insomnia according to Burgess et al.42 As many MR analyses with multi‐outcomes43 did, to account for multiple testing and to preserve the type Ⅰ error of the global null hypothesis of all tested associations being in fact null,44 we used a Bonferroni‐corrected threshold of P<3.6×10−3 (α=0.05/14 outcomes) at the outcome level in our primary analysis. We note that these outcomes are related to each other; therefore, the tests are not completely independent of each other and the Bonferroni correction may be conservative. We reported the actual P values of each effect; the association between our threshold and 0.05 was considered suggestive evidence of association, requiring confirmation.43
Mediation Analysis
We further conducted a mediation analysis to explore whether the chosen cardiometabolic risk factors mediate the causal pathway from insomnia to CVD outcome that insomnia is significantly associated with, using a network MR design. For each CVD outcome, this design consists of 3 different steps (Steps a‐c):22
Step a: We estimated the causal effect of genetically determined insomnia on this CVD outcome, this step was in accordance with our primary analysis.
Step b: Up to 248 independent SNPs associated with insomnia at genome‐wide significance from Jansen et al14 were used to estimate the causal effect of genetically determined insomnia on each potential cardiometabolic risk factor, using the respective GWAS summary statistics described previously,28, 29, 30, 31, 32, 33, 34, 35 using the IVW approach (under a multiplicative random‐effects model). The estimated effects were SD or per unit change in risk factors expressed per genetically predicted 1‐unit‐higher log‐odds of liability to insomnia (per 2.72‐fold increase in the prevalence of insomnia). There was no sample overlap between insomnia GWAS study and consortia of all risk factors except for heart rate (~4%). The Bonferroni‐corrected threshold of P<3×10−3 (α=0.05/17 mediators) was used in this step.
Step c: For possible mediators that causal association was observed in Step b (BMI, HDL‐C, and TG), we estimated the causal effect of each mediator on this CVD outcome, respectively, using the IVW approach (under a multiplicative random‐effects model). The details of instrumental SNPs and the summary statistics we used for each mediator in Step c were shown in Data S2 and Table S6–S8, and SNP‐CVD outcome coefficients were calculated from the identified White British individuals in UK Biobank using logistic regression controlling for 10 principal components. There was no sample overlap between each mediator GWAS study and UK Biobank. The results were converted to ORs expressed per genetically predicted 1 SD increased of the mediator, and Bonferroni‐corrected threshold of P<5.6×10−3 (α=0.05/9 outcomes that insomnia significantly associated with) was used in this step.
If causal associations were observed in all 3 steps, the conclusion can be drawn that the specific cardiometabolic risk factor is a mediator in the pathway linking insomnia to this CVD outcome. The indirect effect of insomnia on this CVD outcome mediated through each mediator was estimated by multiplying the results from Step b and Step c. We finally divided the mediated effect by the total effect to estimate the proportion mediated by each mediator (see Data S3 for the calculation of proportions and Data S4 for the calculation of their 95% CIs).
Sensitivity Analysis
A series of sensitivity analyses were performed to examine the robustness of the results in the primary analysis and each step for mediation analysis. First, the presence of substantial heterogeneity among Wald estimates based on individual SNPs would indicate the presence of invalid instruments.45 In this study, the magnitude of heterogeneity among variant‐specific Wald estimates was evaluated using the I 2 statistic, which is defined as the percentage of the total variation in the estimates explained by heterogeneity rather than sampling error if all genetic variants are valid instruments and relationships between all variables (genetic variants, exposure, and outcome) are linear and homogeneous for all individuals in the population, independent of the number of estimates.46, 47 Moreover, leave‐one‐out sensitivity analysis was also performed to assess the reliance of the MR results on each particular variant.
Second, we performed a range of robust methods making different consistency assumptions. We examined the potential bias from invalid instruments when using multiple genetic variants through weighted median and mode‐based estimation, which rely on weaker assumptions about the invalid instruments and are more robust to outliers. The former method will provide a consistent estimate if at least 50% of the weights come from valid instrumental variables,48 and the latter assumes more variants estimate the true causal effect than estimating any other quantity,49 but have been shown to have low precision in some studies and simulations.50 In addition, we applied the MR‐Egger regression method, which can obtain a valid estimate of the causal effect even if all the genetic instruments are invalid under the Instrument Strength Independent of Direct Effect assumption, but with low power.40 Moreover, the non‐zero intercept indicates the existence of directional pleiotropy; therefore, MR‐Egger intercept test can help to assess the validity of instrumental variable assumption (3).40 MR pleiotropy residual sum and outlier method was also used to detect and correct for horizontal pleiotropic through the removal of SNPs that are most likely to display horizontal pleiotropic effects (outliers).51 As suggested by Slob and Burgess50, these 4 methods can adequately assess the evidence of the causal effects of each exposure on the outcome and detect the sensitivity of the results to different patterns of violations of instrumental variable assumptions; consistency of results across methods strengthens an inference of causality.
Additionally, we performed a bidirectional MR analysis to examine whether each selected mediator can casually affect insomnia (bidirectional causality) by using mediator‐associated independent SNPs as instrumental variables (Data S2 and Table S6–S8), the summary statistics of insomnia were obtained from Jansen et al.14 If there was no evidence that one genetically predicted mediator causally affects insomnia, then we applied regression‐based multivariable MR as an additional sensitivity analysis of Step c for mediation analysis. We estimated the causal effect of this mediator on each CVD outcome, respectively, adjusting for the genetic effects of the instruments on insomnia obtained from Jansen et al,14 as suggestive by Carter et al.52, 53 We additionally performed multivariable MR to consider the role of multiple mediators simultaneously and to investigate the direct causal effect of insomnia on each CVD outcome not mediated by these 3 mediators; see Data S5 for the details.
In our analyses, we considered arterial hypertension as a CVD outcome. As blood pressure traits (including systolic blood pressure, diastolic blood pressure [DBP]) are also important cardiometabolic risk factors, we additionally performed analyses to explore whether systolic blood pressure and DBP mediate the causal pathway from insomnia to the other 8 insomnia‐associated CVD outcomes using a network MR design. The details were provided in Data S6.
Replication Analysis
We conducted replication analysis by varying different data sets to further assess the robustness of our results. For the primary analysis, we estimated that approximately 30% of individuals from GWAS on insomnia were also included in the UK Biobank.54 Thus, we further performed a replication analysis using summary data of 4 CVD outcomes, including IS, CAD, AF, and HF, from previously published GWAS studies55, 56, 57, 58 that have no or limited sample overlap with the GWAS of insomnia. Full details of each GWAS can be found in Table S9. We also validated these results with UK Biobank individual‐level data in a 2‐sample setting (see Data S7). For mediation analysis, we performed replication analysis using a most recent data (the ENGAGE (European Network for Genetic and Genomic Epidemiology) 1000 Genome Consortium, http://diagram‐consortium.org/2015_ENGAGE_1KG/,59, 60 see Table S10 for the details) as the replication data set for 6 anthropometric and lipid traits in Step b. For Step c, we also performed replication analysis using summary statistics of 4 CVD outcomes from published GWAS studies (Table S9). All replication analysis were conducted using IVW in addition to similar heterogeneity assessment approaches and 4 robust methods for sensitivity analysis.
All statistical tests were 2 sided. Analyses in this study were performed using PLINK2 and R version 4.0.2 together with the R package MendelianRandomization,61 MRPRESSO.51
Results
Causal Association Between Genetically Predicted Insomnia and CVD Outcomes
We found that genetically predicted insomnia was significantly positively associated with 10 of the 14 outcomes using the IVW method; the ORs ranged from 1.13 (95% CI, 1.08–1.18) for AF to 1.24 (95% CI, 1.16–1.32) for HF (Figure 2). Additionally, there was suggestive evidence of positive associations between genetically predicted insomnia and intracerebral hemorrhage (OR, 1.18; 95% CI, 1.02–1.37; P=0.03) and subarachnoid hemorrhage (OR, 1.23; 95% CI, 1.06–1.43; P=0.005), whereas statistically nonsignificant positively associations were observed between insomnia and abdominal aortic aneurysm (OR, 1.14; 95% CI, 1–1.30; P=0.06) or thoracic aortic aneurysm (OR, 1.02; 95% CI, 0.79–1.31; P=0.9) (Figure 2).
Figure 2. Associations between genetically predicted insomnia and 14 cardiovascular diseases (CVDs).
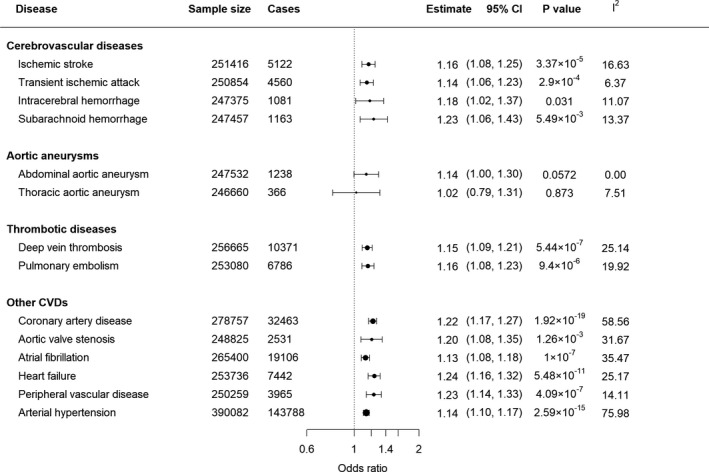
Results were obtained from the multiplicative random‐effects inverse‐variance weighted method. Sample size denotes the total number of individuals in the analysis data set for each CVD outcome (see Figure S1 for flow chart of individual selection). Estimates represent odds ratios (OR) expressed per genetically predicted 1‐unit‐higher log‐odds of liability to insomnia (per 2.72‐fold increase in the prevalence of insomnia); I 2 statistic quantifies the amount of heterogeneity among estimates based on individual single‐nucleotide polymorphisms. Circles show the estimated ORs, and the sizes of these circles represent the precision of these estimates.
There was no evidence of SNP that has a strong influence on the estimations of causal association (Table S11). Although we noted that MR‐Egger and mode‐based estimates were sometimes different from the other MR methods, for example, pulmonary embolism and aortic valve stenosis, most OR estimates were consistent using different MR approaches (Table S12). The intercept of MR‐Egger regression for 247 SNPs in each CVD outcome was not statistically significant except for intracerebral hemorrhage (Intercept=−0.03; 95% CI, −0.06 to −0.01; P=0.01). After excluding SNPs because of their pleiotropic effects, the analyses of the remaining SNPs did not materially change the OR of insomnia on intracerebral hemorrhage (OR, 1.15; 95% CI, 1–1.34; P=0.057). However, results of replication analysis showed that genetically predicted insomnia was still significantly positively associated with 9 of the 10 outcomes identified, except for aortic valve stenosis (OR, 1.25; 95% CI, 0.93–1.68; P=0.14) (Table S13–S16).
Causal Association Between Genetically Predicted Insomnia and Cardiometabolic Risk Factors
The causal estimates between genetically predicted insomnia and 17 cardiometabolic risk factors using the IVW method were displayed in Figure 3. Genetically predicted insomnia was significantly positively associated with 4 risk factors, including BMI (0.07; 95% CI, 0.04–0.10), TG (0.06; 95% CI, 0.04–0.09), WC (0.06; 95% CI, 0.03–0.09), and WHR (0.05; 95% CI, 0.03–0.07). However, no association was observed with insomnia for the latter 2 indexes after the adjustment for BMI (WC adjusted for BMI [0; 95% CI, −0.03 to 0.02] and WHR adjusted for BMI [0.02; 95% CI, −0.01 to 0.04]). Moreover, genetically predicted insomnia was significantly negatively associated with HDL‐C (−0.06; 95% CI, −0.09 to −0.03).
Figure 3. Associations between genetically predicted insomnia and 17 cardiometabolic risk factors.
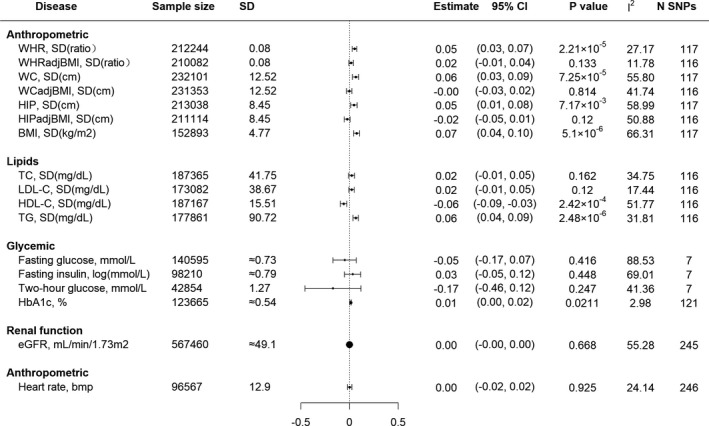
Results were obtained from the multiplicative random‐effects inverse‐variance weighted method. Estimated effects were SD or per unit change in risk factor expressed per genetically predicted 1‐unit‐higher log‐odds of liability to insomnia (per 2.72‐fold increase in the prevalence of insomnia). I 2 statistic quantifies the amount of heterogeneity among estimates based on individual SNPs. N SNPs was the number of instrument SNPs we used for Mendelian randomization analysis of the association of genetically predicted insomnia on each risk factor. BMI indicates body mass index; eGFR, estimated glomerular filtration rate; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; HIP, hip circumference; HIPadjBMI, HIP adjusted for BMI; LDL‐C, low‐density lipoprotein cholesterol; SNP, single‐nucleotide polymorphism; TC, total cholesterol; TG, triglycerides; WC, waist circumference; WCadjBMI, WC adjusted for BMI; WHR, waist‐hip ratio; and WHRadjBMI, WHR adjusted for BMI.
There was no evidence of SNP that has a strong influence on the estimations of causal associations (Table S17). Associations estimated by the weighted median, mode‐based, MR‐Egger, and MR‐PRESSO method were broadly similar to our main results for BMI, TG, and HDL‐C, with little evidence for the presence of pleiotropy (Table S18). However, the association estimated by mode‐based and MR‐Egger method was generally underpowered with wide CIs. Our replication studies for BMI, TG, and HDL using the ENGAGE 1000 Genome data also provided consistent results (Tables S19–S20). There was consistently no statistically significant association between genetically predicted insomnia with TC, LDL‐C, glycemic traits, renal function as well as heart rate increase during exercise. Finally, 3 of 17 cardiometabolic risk factors including BMI, TG, and HDL‐C were suggested to be possible mediators from insomnia to CVD outcomes and were chosen to conduct further analyses.
Causal Association Between Genetically Predicted BMI, TG, HDL, and CVD Outcomes
We evaluated further whether each of BMI, TG, and HDL causally associated with each of 9 selected CVD outcomes. To begin with, we used 74 of 78 independent SNPs (R 2=2.2%) associated with BMI (P<5×10−8)29 as instrumental variables. Under a Bonferroni‐corrected threshold of P<0.0056, a genetically predicted 5 kg/m2 (1 SD) increase of BMI was significantly associated with 7 of 9 CVD outcomes using the IVW method, with ORs ranging from 1.52 (95% CI, 1.37–1.67) for arterial hypertension to 2.14 (95% CI, 1.78–2.58) for HF (Figure S2). There was no evidence of SNP that has a strong influence on the estimations of causal associations and presents of pleiotropy (Tables S21–S22), except for the analyses of BMI on CAD. After excluding SNPs due to the potential pleiotropic effects, analyses of the remaining SNPs yielded a similar OR of 1.57 (95% CI, 1.37–1.79; P=2.89×10−11). The results of sensitivity analysis and replication analysis (Tables S23–S24) were broadly similar to our main results for BMI.
Then 85 of 86 independent SNPs (R 2=5.9%) as well as 51 independent SNPs (R2 =4.6%)30 associated with HDL‐C and TG, respectively, were used as instrumental variables. According to the results of IVW method, a genetically predicted 1 SD increase of HDL‐C was negatively associated with higher risk of CAD (OR, 0.82; 95% CI, 0.73–0.91), peripheral vascular disease (OR, 0.80; 95% CI, 0.69–0.94) and arterial hypertension (OR, 0.86; 95% CI, 0.80–0.93) (Figure S3). A genetically predicted 1 SD increase of TG was positively associated with higher risk of CAD (OR, 1.44; 95% CI, 1.29–1.61), HF (OR, 1.33; 95% CI, 1.17–1.51), and arterial hypertension (OR, 1.18; 95% CI, 1.09–1.27) (Figure S4). There was no evidence of SNPs that have a strong influence on the estimations of causal associations (Table S25–S26). After excluding SNPs suggested to have potential pleiotropic effects (Table S27–S28), analyses of the remaining SNPs did not materially change the ORs of HDL‐C on CAD (OR, 0.68; 95% CI, 0.6–0.79; P=2.52×10−8), HDL‐C on arterial hypertension (OR, 0.78; 95% CI, 0.72–0.84; P=3.48×10−10), TG on CAD (OR, 1.39; 95% CI, 1.25–1.54; P=3.88×10−10), and TG on arterial hypertension (OR, 1.16; 95% CI, 1.08–1.24; P=2.88×10−5). However, results of replication analysis did not support that a genetically predicted 1 SD increase of HDL‐C was significantly associated with a higher risk of CAD (OR, 0.88; 95% CI, 0.8–0.98; P=0.015) under a Bonferroni‐corrected threshold of P<0.0125; other replication analysis results were accordant with our main results (Table S29–S32).
The results of bidirectional MR showed no evidence of causal association of genetically predicted BMI, HDL‐C, or TG on insomnia (Table S33). Thus, insomnia does not tend to lie on the causal pathway from each mediator to each CVD outcome (ie, insomnia is not the mediator of risk factor‐CVD association). Therefore, for all 3 mediators, regression‐based multivariable MR was conducted as an additional sensitivity analysis to estimate the causal effect of the mediator on each CVD outcome, respectively, adjusting for the genetic effect of the instruments on insomnia. The results of multivariable MR shown in Figure S5–S7 were highly accordant to our main results.
According to the results of network MR analysis, we found that some or all of BMI, TG, and HDL might act as mediators in the causal pathway from insomnia to deep vein thrombosis, pulmonary embolism, CAD, AF, HF, peripheral vascular disease, and arterial hypertension. The proportion of the total effect of insomnia on each CVD outcome that each mediator accounts for was provided in the Table. The results of the direct causal effect of insomnia on each CVD outcome not mediated by these 3 mediators were shown in Data S5 and Table S34.
Table 1.
The Proportion of the Total Effect of Insomnia on Each Cardiovascular Disease That Each Mediator Accounts For
| Exposure (X) | Mediator (M) | Outcome (Y) | TEXY | βXM | ORMY | NIEXY (95% CI) | Proportion (95% CI) |
|---|---|---|---|---|---|---|---|
| Insomnia | BMI | Deep vein thrombosis | 1.15 | 0.07 | 1.79 | 0.041 (0.02– 0.061) | 29.16% (14%–44.32%) |
| Insomnia | BMI | Pulmonary embolism | 1.16 | 0.07 | 1.66 | 0.035 (0.014–0.057) | 23.9% (8.53%–39.27%) |
| Insomnia | BMI | Coronary artery disease | 1.22 | 0.07 | 1.53 | 0.03 (0.014–0.046) | 14.97% (7.44%–22.5%) |
| Insomnia | triglycerides | Coronary artery disease | 1.22 | 0.06 | 1.44 | 0.022 (0.01–0.034) | 11% (5.03%–16.98%) |
| Insomnia | BMI | Atrial fibrillation | 1.13 | 0.07 | 1.56 | 0.031 (0.015–0.048) | 25.47% (11.67%–39.27%) |
| Insomnia | BMI | Heart failure | 1.24 | 0.07 | 2.14 | 0.053 (0.026–0.081) | 24.76% (12.93%–36.59%) |
| Insomnia | triglycerides | Heart failure | 1.24 | 0.06 | 1.33 | 0.017 (0.006, 0.028) | 7.95% (2.5%–13.41%) |
| Insomnia | BMI | Peripheral vascular disease | 1.23 | 0.07 | 1.73 | 0.038 (0.013–0.063) | 18.53% (6.5%–30.57%) |
| Insomnia | HDL‐C | Peripheral vascular disease | 1.23 | −0.06 | 0.8 | 0.013 (0.001–0.025) | 6.47% (0.53%–12.4%) |
| Insomnia | BMI | Arterial hypertension | 1.14 | 0.07 | 1.52 | 0.029 (0.014–0.044) | 22.37% (12.07%–32.67%) |
| Insomnia | HDL‐C | Arterial hypertension | 1.14 | −0.06 | 0.86 | 0.009 (0.002–0.016) | 6.91% (1.75%–12.06%) |
| Insomnia | triglycerides | Arterial hypertension | 1.14 | 0.06 | 1.18 | 0.01 (0.003–0.017) | 7.58% (2.42%–12.74%) |
BMI indicates body mass index; βXM, effect of the exposure on the mediator; HDL‐C, high‐density lipoprotein cholesterol; TEXY , total effect of the exposure on the outcome expressed in odds ratios (OR) scale; NIEXY , natural indirect effect of exposure on the outcome in log OR scale; and Proportion, the proportion of the total effect of exposure on outcome that mediator accounts for.
The Potential Mediator Role of Blood Pressure Traits
Finally, when we consider blood pressure traits (Table S35) as cardiometabolic risk factors rather than consider arterial hypertension as a CVD outcome, our results showed that genetically predicted insomnia was significantly positively associated with DBP (0.41; 95% CI, 0.24–0.58; P=1.75×10−6); nevertheless, no association was observed with insomnia and DBP after adjustment for BMI (0.11; 95% CI, −0.05 to 0.27) (Table S36). In addition, a genetically predicted 1 mm Hg increase of DBP was positively associated with a higher risk of IS (OR, 1.09; 95% CI, 1.06–1.13), CAD (OR, 1.08; 95% CI, 1.06–1.09), AF (OR, 1.04; 95% CI, 1.02–1.06), and HF (OR, 1.07; 95% CI, 1.04–1.09) (Table S37) after Bonferroni correction, and the results of bidirectional MR showed no evidence of causal association of genetically predicted DBP on insomnia (Table S38). The sensitivity analysis yielded a similar pattern of effects; there was no evidence of the presence of pleiotropic effect (Table S36 and Table S38). In addition, we found that no SNP had an influential effect on the results for each blood pressure trait (Table S39). Therefore, DBP might act as a mediator in the causal pathway from insomnia to IS, CAD, AF, and HF. The proportion of the total effect of insomnia on each CVD outcome that DBP accounts for was provided in Table S40.
Discussion
Principal Findings
In this study, using publicly available summary statistics from large consortia and data from UK Biobank, we performed a series of 2‐sample MR analyses to systematically assess the causal roles of genetically predicted insomnia for a wide range of cardiovascular conditions. We further explored 17 cardiometabolic risk factors as possible mediators in the causal relationship between genetically determined insomnia and each CVD outcome. Our results provided consistent evidence that genetically determined insomnia was causally associated with increased risk of IS, transient ischemic attack, thrombotic diseases, and 5 other CVDs (CAD, HF, AF, peripheral vascular disease, arterial hypertension). Additionally, we concluded that genetically predicted insomnia was associated with higher BMI and TG as well as lower HDL‐C, each of which may act as a mediator in the causal pathway from insomnia to several CVD outcomes. In addition, we found very little evidence to support a causal link between genetically predicted insomnia with abdominal aortic aneurysm, thoracic aortic aneurysm, TC, LDL‐C, glycemic traits, renal function as well as heart rate increase during exercise. Finally, we found no evidence of a causal association of genetically predicted BMI, HDL‐C, or TG on insomnia.
Comparisons With Other Studies
Although caution should be taken when comparing our results with other observational studies, because of the variations in measurement and definition of insomnia, nonetheless, our findings for CVDs from the primary analysis are generally consistent with those based on observational studies, which suggested that insomnia is an important risk factor for CVDs including hypertension,9, 10 CAD, AF,62, 63, 64 and HF5, 65. The null results for genetically predicted insomnia and abdominal aortic aneurysm are consistent with findings from a cohort study of 22 444 men and 10 982 women showing no association between insomnia and later development of abdominal aortic aneurysm,66 but we cannot rule out the possibility that our analyses were underpowered to detect such a relatively modest association. Our results are also similar to previously reported MR studies of insomnia and risk of CAD,14, 15, 16, 67 HF,15 and IS.15 For AF, the OR of our analyses was similar to the result of Larsson et al15 (1.04; 95% CI, 1.01–1.07; P=7.28×10−3), but they did not conduct a significant conclusion under a stricter Bonferroni‐corrected threshold (). We are not aware of any observational or MR study of insomnia in relation to other cardiovascular conditions. In this study, we found genetically predicted insomnia was associated with an increased risk of transient ischemic attack, deep vein thrombosis, pulmonary embolism, and peripheral vascular disease.
Associations between insomnia and lipid markers (TC, TG, LDL‐C, and HDL‐C) reported by observational studies have been inconsistent.68, 69, 70, 71, 72, 73 In addition, a previous observational study has shown that insomnia is not associated with a higher BMI,74 insomnia symptoms were found to have a close association with high hemoglobin A1c in Japanese men,75 insomnia scores were found to have a strong negative correlation with estimated glomerular filtration rate.76 Our findings for cardiometabolic risk factors are generally contradictory to those based on observational studies but similar to previously reported MR studies on insomnia and BMI14 as well as WHR.14 In this study, we found strong and consistent evidence that genetically predicted insomnia was associated with higher BMI, WHR, WC, and TG as well as lower HDL‐C. Moreover, there was consistent no statistically significant association between genetically predicted insomnia with TC, LDL‐C, glycemic traits, estimated glomerular filtration rate as well as heart rate increase during exercise. The contradiction may reflect reverse causation or confounding effects of previous observational studies.
In addition, our results that genetically predicted BMI was significantly associated with 7 of 9 CVD outcomes were consistent with those results from MR analysis in Larsson et al27 previously. Several MR studies have investigated the associations between genetically predicted lipid traits and the risk of some major CVDs. For instance, Hindy et al77 reported that genetically elevated TG did not associate with IS or any of its subtypes; for HDL‐C, only weak evidence of association with 1 subtype of IS (small artery occlusion stroke) was found.77 A higher genetically predicted TG was observed to be causally associated with a higher risk of hypertension78 and CAD,79 and lower genetically predicted HDL‐C was observed to be causally associated with a higher risk of hypertension78 and no significant association with CAD.79 Our finding was consistent with previous MR studies, which showed that a genetically predicted 1 SD increase of HDL‐C was negatively associated with a higher risk of peripheral vascular disease and arterial hypertension. A genetically predicted 1 SD increase of TG was positively associated with a higher risk of CAD, HF, and arterial hypertension.
Potential Mechanisms
The mechanisms underpinning the association are unclear but may involve that insomnia increases the activity of hypothalamic‐pituitary‐adrenal axis80, 81, 82, 83 and increases the systemic inflammation84, 85; these provide a biologic rationale for insomnia to possibly lead to increased risk of CVD. In addition, studies also demonstrated that subjects with insomnia had increased sympathetic nervous system activity, which is an integral part of cardiovascular homeostasis and plays a critical role in the pathogenesis of hypertension, CAD, and HF.86 Moreover, there might be potential mechanisms to explain the link between insomnia and dyslipidemia as well as the increase of BMI. Genetically determined insomnia usually contributes to reduced sleep duration. People with reduced sleep duration tend to show a preference for high energy‐density fatty food87 and have a higher BMI via reducing leptin and elevating ghrelin.88, 89 Moreover, higher BMI and intake of fat rich food are able to increase the risk of dyslipidemia,90 which may further lead to the increased risk of CVD.
Strengths and Limitations
In this study, we performed a series of 2‐sample MR analyses, which reduced the possibility that the observed associations were biased by unmeasured confounding, reverse causation, and measurement error.18 We conducted mediation analysis using a network MR design, which can be used to decompose the direct and indirect effects22, 24 and can overcome some of the strong assumptions required for traditional causal mediation methods (eg, no unmeasured confounding and no measurement error in the exposure or mediator,53 which are unlikely to be met in many scenarios23, 91, 92) to interpret the results as causal. Moreover, most of these analyses were performed using summarized data, which considerably increases the power to detect a causal effect. We used multiple independent genetic instruments that explained a large variance of exposure, further validated the relevance assumption by the rule of thumb,25 and tested the exclusion assumption by MR‐Egger intercept test and the MR‐PRESSO test. Although we cannot entirely rule out the possibility of the violations of MR assumptions (eg, the independence assumption), most of our results were corroborated by both sensitivity analysis that makes allowance for violation of MR assumptions and the replication analysis, which further strengthens the inference of causality. In addition, we have made sure that most of our summary data estimates of SNP‐exposure and SNP‐outcome associations were gleaned from 2 independent but large homogeneous (European descent) study populations, which minimized the bias from population stratification.
Our study has several limitations. First, insomnia was determined based on self‐reported questionnaires, which may not be completely accurate. In addition, it should be cautious when considering our results, which may vary when using different definitions of insomnia. Insomnia we used is a binary exposure. Some studies have pointed that an MR estimate with a binary exposure and binary outcome is difficult to interpret as a specific causal effect.93, 94 Second, in our primary analysis, the number of cases was few for some CVD outcomes, such as intracerebral hemorrhage, subarachnoid hemorrhage, and abdominal aortic aneurysm. We thus cannot rule out that we may have overlooked weak associations due to insufficient power. Third, the reason SNP‐outcome estimates were obtained from UK Biobank is the availability of adequate outcome variables, such as arterial hypertension, aortic valve stenosis, which lacks published GWAS study. Nevertheless, there is around 30% sample overlap between UK Biobank and insomnia GWAS study, which may have introduced some bias in the MR estimates of primary analysis. However, we used a strict Bonferroni‐corrected threshold to control the possible increase of type I error rate caused by sample overlap.54 In addition, we conducted replication studies using summary data from previously published GWAS studies that have no or limited sample overlap with the GWAS of insomnia and UK Biobank individual‐level data as sensitivity analysis, the same way as Siedlinski et al95 did, to validate our results. To be noted, our genetic instruments were strongly related to the exposures (F statistic≈143.24), implying that bias from participant overlap is relatively small according to Burgess et al.54 Fourth, this study evaluated only whether the liability to insomnia is associated with CVD, so we cannot rule out that there are other causal pathways leading to insomnia that causes CVD.15
Conclusions
This MR study provides evidence that genetically predicted insomnia is associated with increased risks of 9 CVD outcomes, some of them may be partially mediated by one or more of higher BMI, TG, and lower HDL‐C.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (Grant number 81773547 and 82003557), the National Key Research and Development Program of China (Grant number 2020YFC2003500), the Shandong Provincial Natural Science Foundation of China (Grant number ZR2019ZD02), and Shandong Provincial Key Research and Development project (Grant number 2018CXGC1210).
Disclosures
None.
Supporting information
Data S1–S7
MEGASTROKE project authors
Tables S1–S40
Figures S1–S7
References 96–106
Acknowledgments
We thank the MEGSATROKE project for making the data used in this study publicly available. The MEGASTROKE project received funding from sources specified at http://www.megastroke.org/acknowledgments.html. and the full list of authors is available in the Supplementary Material. Data on coronary artery disease have been contributed by CARDIOGRAMPLUSC4D investigators and have been downloaded from www.CARDIOGRAMPLUSC4D.ORG. We also thank the UK Biobank, GIANT Consortium, GLGC, MAGIC, CKDGen, and ENGAGE 1000 Genome Consortium for making the GWAS data publicly available.
(J Am Heart Assoc. 2021;10:e020187. DOI: 10.1161/JAHA.120.020187.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.020187
For Sources of Funding and Disclosures, see page 11.
Contributor Information
Hongkai Li, Email: lihongkaiyouxiang@163.com.
Fuzhong Xue, Email: xuefzh@sdu.edu.cn.
References
- 1.Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jönsson B, Olesen J, Allgulander C, Alonso J, Faravelli C, et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. 2011;21:655–679. DOI: 10.1016/j.euroneuro.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Buysse DJ. Insomnia. JAMA. 2013;309:706–716. DOI: 10.1001/jama.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laugsand LE, Vatten LJ, Platou C, Janszky I. Insomnia and the risk of acute myocardial infarction: a population study. Circulation. 2011;124:2073–2081. DOI: 10.1161/CIRCULATIONAHA.111.025858. [DOI] [PubMed] [Google Scholar]
- 4.Coryell VT, Ziegelstein RC, Hirt K, Quain A, Marine JE, Smith MT. Clinical correlates of insomnia in patients with acute coronary syndrome. Int Heart J. 2013;54:258–265. DOI: 10.1536/ihj.54.258. [DOI] [PubMed] [Google Scholar]
- 5.Laugsand LE, Strand LB, Platou C, Vatten LJ, Janszky I. Insomnia and the risk of incident heart failure: a population study. Eur Heart J. 2014;35:1382–1393. DOI: 10.1093/eurheartj/eht019. [DOI] [PubMed] [Google Scholar]
- 6.Javaheri S, Blackwell T, Ancoli‐Israel S, Ensrud KE, Stone KL, Redline S, Osteoporotic Fractures in Men Study Research Group . Sleep ‐disordered breathing and incident heart failure in older men. Am J Respir Crit Care Med. 2016;193:561–568. DOI: 10.1164/rccm.201503-0536OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas SJ, Calhoun D. Sleep, insomnia, and hypertension: current findings and future directions. J Am Soc Hypertens. 2017;11:122–129. DOI: 10.1016/j.jash.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Vgontzas AN, Liao D, Bixler EO, Chrousos GP, VelaBueno A. Insomnia with objective short sleep duration is associated with a high risk for hypertension. Sleep. 2009;32:491–497. DOI: 10.1093/sleep/32.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budhiraja R, Roth T, Hudgel DW, Budhiraja P, Drake CL. Prevalence and polysomnographic correlates of insomnia comorbid with medical disorders. Sleep. 2011;34:859–867. DOI: 10.5665/SLEEP.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez‐Mendoza J, Vgontzas AN, Liao D, Shaffer ML, Vela‐Bueno A, Basta M, Bixler EO. Insomnia with objective short sleep duration and incident hypertension: the Penn State Cohort. Hypertension. 2012;60:929–935. DOI: 10.1161/HYPERTENSIONAHA.112.193268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vozoris NT. Insomnia symptom frequency and hypertension risk: a population‐based study. J Clin Psychiatry. 2014;75:616–623. DOI: 10.4088/JCP.13m08818. [DOI] [PubMed] [Google Scholar]
- 12.Phillips B, Buzkova P, Enright P. Cardiovascular Health Study Research Group. Insomnia did not predict incident hypertension in older adults in the cardiovascular health study. Sleep. 2009;32:65–72. DOI: 10.5665/sleep/32.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.St‐Onge MP, Grandner MA, Brown D, Conroy MB, Jean‐Louis G, Coons M, Bhatt DL, American Heart Association Obesity, Behavior Change, Diabetes, and Nutrition Committees of the Council on Lifestyle and Cardiometabolic Health ; Council on Cardiovascular Disease in the Young ; Council on Clinical Cardiology , et al. Sleep Duration and Quality: impact on Lifestyle Behaviors and Cardiometabolic Health: a Scientific Statement From the American Heart Association. Circulation. 2016; 134:e367–e386. DOI: 10.1161/CIR.0000000000000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen PR, Watanabe K, Stringer S, Skene N, Bryois J, Hammerschlag AR, de Leeuw CA, Benjamins JS, Muñoz‐Manchado AB, Nagel M, et al. Genome‐wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat Genet. 2019;51:394–403. DOI: 10.1038/s41588-018-0333-3. [DOI] [PubMed] [Google Scholar]
- 15.Larsson SC, Markus HS. Genetic liability to insomnia and cardiovascular disease risk. Circulation. 2019;140:796–798. DOI: 10.1161/CIRCULATIONAHA.119.041830. [DOI] [PubMed] [Google Scholar]
- 16.Gao X, Yang XC, Meng LX, Sun HL, Wang T. Causal relationship between sleep and coronary artery disease: a Mendelian randomization study. Zhonghua Liu Xing Bing Xue Za Zhi. 2020;41:611–614. [DOI] [PubMed] [Google Scholar]
- 17.Knutson KL, Van Cauter E, Zee P, Liu K, Lauderdale DS. Crosssectional associations between measures of sleep and markers of glucose metabolism among subjects with and without diabetes: the Coronary Artery Risk Development in Young Adults (CARDIA) sleep study. Diabetes Care. 2011;34:1171–1176. DOI: 10.2337/dc10-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. DOI: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 19.Burgess S, Thompson SG. Mendelian Randomization: Methods for Using Genetic Variants in Causal Estimation. London, UK: Chapman and Hall/CRC Press; 2015. [Google Scholar]
- 20.Lawlor D, Harbord R, Sterne J, Davey TN, Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. DOI: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 21.Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Methods Med Res. 2007;16:309–330. DOI: 10.1177/0962280206077743. [DOI] [PubMed] [Google Scholar]
- 22.Burgess S, Daniel RM, Butterworth AS, Thompson SG, EPIC‐InterActConsortium . Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int J Epidemiol. 2015;44:484–495. DOI: 10.1093/ije/dyu176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Relton CL, Davey SG. Two‐step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012;41:161–176. DOI: 10.1093/ije/dyr233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–98. DOI: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staiger D, Stock JH. Instrumental variables regression with weak instruments. Econometrica. 1997;557–586. DOI: 10.2307/2171753. [DOI] [Google Scholar]
- 26.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. DOI: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsson SC, Bäck M, Rees JMB, Mason AM, Burgess S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: a Mendelian randomization study. Eur Heart J. 2020;41:221–226. DOI: 10.1093/eurheartj/ehz388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shungin D, Winkler TW, Croteau‐Chonka DC, Ferreira T, Locke AE, Mägi R, Strawbridge RJ, Pers TH, Fischer K, Justice AE, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196. DOI: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. DOI: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lagou V, Mägi R, Hottenga J‐J, Grallert H, Perry JRB, Bouatia‐Naji N, Marullo L, Rybin D, Jansen R, Min JL, et al. Sex‐dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat Commun. 2021;12(1): DOI: 10.1038/s41467-020-19366-9.s DOI: 10.1038/s41467-020-19366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wheeler E, Leong A, Liu CT, Hivert MF, Strawbridge RJ, Podmore C, Li M, Yao J, Sim X, Hong J, et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome‐wide meta‐analysis. PLoS Med. 2017;14:e1002383. DOI: 10.1530/ey.15.13.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, Mägi R, Strawbridge RJ, Rehnberg E, Gustafsson S, et al. Large‐scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. DOI: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wuttke M, Li Y, Li M, Sieber KB, Feitosa MF, Gorski M, Tin A, Wang L, Chu AY, Hoppmann A, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet. 2019;51:957–972. DOI: 10.1038/s41588-019-0407-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verweij N, van de Vegte YJ, van der Harst P. Genetic study links components of the autonomous nervous system to heart‐rate profile during exercise. Nat Commun. 2018;9:898. DOI: 10.1038/s41467-018-03395-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson JR, Minelli C, Del Greco MF. Mendelian randomization using public data from genetic consortia. Int J Biostat. 2016;12. [DOI] [PubMed] [Google Scholar]
- 37.Wald A. The fitting of straight lines if both variables are subject to error. Ann Math Stat. 1940;11:284–300. [Google Scholar]
- 38.Durbin J. Errors in variables. Rev Int Stat Inst. 1954;22:23–32. [Google Scholar]
- 39.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35:1880–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR‐Egger method. Eur J Epidemiol. 2017;32:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, Hartwig FP, Holmes MV, Minelli C, Relton CL, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2020;4:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burgess S, Labrecque JA. Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur J Epidemiol. 2018;33:947–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsson SC, Mason AM, Bäck M, Klarin D, Damrauer SM, Program MV, Michaëlsson K, Burgess S. Genetic predisposition to smoking in relation to 14 cardiovascular diseases. Eur Heart J. 2020;41:3304–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.VanderWeele TJ, Mathur MB. Some desirable properties of the bonferroni correction: is the bonferroni correction really so bad? Am J Epidemiol. 2019;188:617–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greco MFD, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34:2926–2940. [DOI] [PubMed] [Google Scholar]
- 46.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Stat Med. 2002;21:1539–1558. [DOI] [PubMed] [Google Scholar]
- 47.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slob EAW, Burgess S. A comparison of robust Mendelian randomization methods using summary data. Genet Epidemiol. 2020;44:313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carter AR, Gill D, Davies NM, Taylor AE, Tillmann T, Vaucher J, Wootton RE, Munafò MR, Hemani G, Malik R, et al. Understanding the consequences of education inequality on cardiovascular disease: mendelian randomisation study. BMJ. 2019;365:l1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter AR, Sanderson E, Hammerton G, Richmond RC, Smith GD, Heron J, Taylor AE, Davies NM, Howe LD. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. BioRxiv. 2019;835819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two‐sample Mendelian randomization. Genet Epidemiol. 2016;40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten‐Jacobs L, Giese A‐K, van der Laan SW, Gretarsdottir S, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roselli C, Chaffin MD, Weng L‐C, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, et al. Multi‐ethnic genome‐wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, Hedman ÅK, Wilk JB, Morley MP, Chaffin MD, et al. Genome‐wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horikoshi M, Mӓgi R, van de Bunt M, Surakka I, Sarin A‐P, Mahajan A, Marullo L, Thorleifsson G, Hӓgg S, Hottenga J‐J, et al. Discovery and fine‐mapping of glycaemic and obesity‐related trait loci using high‐density imputation. PLoS Genet. 2015;11:e1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Surakka I, Horikoshi M, Mägi R, Sarin A‐P, Mahajan A, Lagou V, Marullo L, Ferreira T, Miraglio B, Timonen S, et al. The impact of low‐frequency and rare variants on lipid levels. Nat Genet. 2015;47:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Christensen MA, Dixit S, Whitman IR, Nah G, Dewland TA, Vittinghoff E, Mukamal KJ, Redline S, Robbins JA, Newman AB, et al. Sleep disruption is associated with incident atrial fibrillation independent of obstructive sleep apnea. Circulation. 2016;134:A13423. [Google Scholar]
- 63.Han X, Yang Y, Chen Y, Gao L, Yin X, Li H, Qiu J, Wang Y, Zhou Y, Xia Y. Association between insomnia and atrial fibrillation in a Chinese population: A cross‐sectional study. Clin Cardiol. 2017;40:765–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee HH, Chen YC, Chen JJ, Lo SH, Guo YL, Hu HY. Insomnia and the risk of atrial fibrillation: a population‐based cohort study. Acta Cardiol Sin. 2017;33:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ingelsson E, Lind L, Arnlov J, Sundstrom J. Sleep disturbances independently predict heart failure in overweight middle‐aged men. Eur J Heart Fail. 2007;9:184–190. [DOI] [PubMed] [Google Scholar]
- 66.Lindblad B, Börner G, Gottsäter A. Factors associated with development of large abdominal aortic aneurysm in middle‐aged men. Eur J Vasc Endovasc Surg. 2005;30:346–352. [DOI] [PubMed] [Google Scholar]
- 67.Liao LZ, Li WD, Liu Y, Li JP, Zhuang XD, Liao XX. Causal assessment of sleep on coronary heart disease. Sleep Med. 2020;67:232–236. [DOI] [PubMed] [Google Scholar]
- 68.Zhan Y, Zhang F, Lu L, Wang J, Sun Y, Ding R, Hu D, Yu J. Prevalence of dyslipidemia and its association with insomnia in a community based population in China. BMC Public Health. 2014;14:1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Jiang T, Wang X, Zhao J, Kang J, Chen M, Wang H, Niu L, Wang Y, Zhou Y, et al. Association between Insomnia and Metabolic Syndrome in a Chinese Han Population: A Cross‐sectional Study. Sci Rep. 2017;7:10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagai M, Hoshide S, Nishikawa M, Shimada K, Kario K. Sleep duration and insomnia in the elderly: associations with blood pressure variability and carotid artery remodeling. Am J Hypertens. 2013;26:981–989. [DOI] [PubMed] [Google Scholar]
- 71.Costemale‐Lacoste J‐F, Trabado S, Verstuyft C, El Asmar K, Butlen‐Ducuing F, Colle R, Ferreri F, Polosan M, Haffen E, Balkau B, et al. Severe insomnia is associated with hypertriglyceridemia in women with major depression treated in psychiatry settings. J Affect Disord. 2017;217:159–162. [DOI] [PubMed] [Google Scholar]
- 72.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, Dale CE, Padmanabhan S, Finan C, Swerdlow DI, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crönlein T, Langguth B, Busch V, Rupprecht R, Wetter TC. Severe chronic insomnia is not associated with higher body mass index. J Sleep Res. 2015;24:514–517. [DOI] [PubMed] [Google Scholar]
- 75.Kachi Y, Nakao M, Takeuchi T, Yano E. Association between insomnia symptoms and hemoglobin A 1c level in Japanese Men. PLoS One. 2011;6:e21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aggarwal HK, Jain D, Dabas G, Yadav RK. Prevalence of depression, anxiety and insomnia in chronic kidney disease patients and their co‐relation with the demographic variables. Pril (Makedon Akad Nauk Umet Odd Med Nauki). 2017;38:35–44. [DOI] [PubMed] [Google Scholar]
- 77.Hindy G, Engström G, Larsson SC, Traylor M, Markus HS, Melander O, Orho‐Melander M, Stroke genetics network (SiGN) . Role of blood lipids in the development of ischemic stroke and its subtypes: a Mendelian randomization study. Stroke. 2018;49:820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Oort S, Beulens JWJ, van Ballegooijen AJ, Grobbee DE, Larsson SC. Association of cardiovascular risk factors and lifestyle behaviors with hypertension: a mendelian randomization study. Hypertension. 2020;76:1971–1979. [DOI] [PubMed] [Google Scholar]
- 79.White J, Swerdlow DI, Preiss D, Fairhurst‐Hunter Z, Keating BJ, Asselbergs FW, Sattar N, Humphries SE, Hingorani AD, Holmes MV. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vgontzas AN, Bixler EO, Lin HM, Prolo P, Mastorakos G, Vela‐Bueno A, Kales A, Chrousos GP. Chronic insomnia is associated with nyctohemeral activation of the hypothalamicpituitary‐adrenal axis: clinical implications. J Clin Endocrinol Metab. 2001;86:3787–3794. [DOI] [PubMed] [Google Scholar]
- 81.Vgontzas AN, Tsigos C, Bixler EO, Stratakis CA, Zachman K, Kales A, Vela‐Bueno A, Chrousos GP. Chronic insomnia and activity of the stress system: a preliminary study. J Psychosom Res. 1998;45:21–31. [DOI] [PubMed] [Google Scholar]
- 82.Floam S, Simpson N, Nemeth E, Scott‐Sutherland J, Gautam S, Haack M. Sleep characteristics as predictor variables of stress systems markers in insomnia disorder. J Sleep Res. 2015;24:296–304. [DOI] [PubMed] [Google Scholar]
- 83.Rodenbeck A, Cohrs S, Jordan W, Huether G, Ruther E, Hajak G. The sleep‐improving effects of doxepin are paralleled by a normalized plasma cortisol secretion in primary insomnia. A placebo‐controlled, double‐blind, randomized, cross‐over study followed by an open treatment over 3 weeks. Psychopharmacology. 2003;170:423–428. [DOI] [PubMed] [Google Scholar]
- 84.Parthasarathy S, Vasquez MM, Halonen M, Bootzin R, Quan SF, Martinez FD, Guerra S. Persistent insomnia is associated with mortality risk. Am J Med. 2015;128:268–275.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Irwin MR. Why sleep is important for health: a psychoneuroimmunology perspective. Annu Rev Psychol. 2015;66:143–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manolis AJ, Poulimenos LE, Kallistratos MS, Gavras I, Gavras H. Sympathetic overactivity in hypertension and cardiovascular disease. Curr Vasc Pharmacol. 2014;12:4–15. [DOI] [PubMed] [Google Scholar]
- 87.Santana AA, Pimentel GD, Romualdo M, Oyama LM, Santos RV, Pinho RA, de Souza CT, Rodrigues B, Caperuto EC, Lira FS. Sleep duration in elderly obese patients correlated negatively with intake fatty. Lipids Health Dis. 2012;11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004;1:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel SR, Hu FB. Short sleep duration and weight gain: a systematic review. Obesity (Silver Spring). 2008;16:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown CD, Higgins M, Donato KA, Rohde FC, Garrison R, Obarzanek E, Ernst ND, Horan M. Body mass index and the prevalence of hypertension and dyslipidemia. Obes Res. 2000;8:605–619. [DOI] [PubMed] [Google Scholar]
- 91.VanderWeele TJ. Mediation analysis: A practitioner's guide. Annu Rev Public Health. 2016;37:17–32. [DOI] [PubMed] [Google Scholar]
- 92.Richiardi L, Bellocco R, Zugna D. Mediation analysis in epidemiology: methods, interpretation and bias. Int J Epidemiol. 2013;42:1511–1519. [DOI] [PubMed] [Google Scholar]
- 93.Larsson SC, Carter P, Kar S, Vithayathil M, Mason AM, Michaëlsson K, Burgess S. Smoking, alcohol consumption, and cancer: A mendelian randomisation study in UK Biobank and international genetic consortia participants. PLoS Med. 2020;17:e1003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Palmer TM, Sterne JA, Harbord RM, Lawlor DA, Sheehan NA, Meng S, Granell R, Smith GD, Didelez V. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol. 2011;173:1392–1403. [DOI] [PubMed] [Google Scholar]
- 95.Siedlinski M, Jozefczuk E, Xu X, Teumer A, Evangelou E, Schnabel RB, Welsh P, Maffia P, Erdmann J, Tomaszewski M, et al. White blood cells and blood pressure: a Mendelian randomization study. Circulation. 2020;141:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1–S7
MEGASTROKE project authors
Tables S1–S40
Figures S1–S7
References 96–106