

Abstract
Background
Impaired myocardial blood flow (MBF) in the absence of epicardial coronary disease is a feature of hypertrophic cardiomyopathy (HCM). Although most evident in hypertrophied or scarred segments, reduced MBF can occur in apparently normal segments. We hypothesized that impaired MBF and myocardial perfusion reserve, quantified using perfusion mapping cardiac magnetic resonance, might occur in the absence of overt left ventricular hypertrophy (LVH) and late gadolinium enhancement, in mutation carriers without LVH criteria for HCM (genotype‐positive, left ventricular hypertrophy‐negative).
Methods and Results
A single center, case‐control study investigated MBF and myocardial perfusion reserve (the ratio of MBF at stress:rest), along with other pre‐phenotypic features of HCM. Individuals with genotype‐positive, left ventricular hypertrophy‐negative (n=50) with likely pathogenic/pathogenic variants and no evidence of LVH, and matched controls (n=28) underwent cardiac magnetic resonance. Cardiac magnetic resonance identified LVH‐fulfilling criteria for HCM in 5 patients who were excluded. Individuals with genotype‐positive, left ventricular hypertrophy‐negative had longer indexed anterior mitral valve leaflet length (12.52±2.1 versus 11.55±1.6 mm/m2, P=0.03), lower left ventricular end‐systolic volume (21.0±6.9 versus 26.7±6.2 mm/m2, P≤0.005) and higher left ventricular ejection fraction (71.9±5.5 versus 65.8±4.4%, P≤0.005). Maximum wall thickness was not significantly different (9.03±1.95 versus 8.37±1.2 mm, P=0.075), and no subject had significant late gadolinium enhancement (minor right ventricle‒insertion point late gadolinium enhancement only). Perfusion mapping demonstrated visual perfusion defects in 9 (20%) carriers versus 0 controls (P=0.011). These were almost all septal or near right ventricle insertion points. Globally, myocardial perfusion reserve was lower in carriers (2.77±0.83 versus 3.24±0.63, P=0.009), with a subendocardial:subepicardial myocardial perfusion reserve gradient (2.55±0.75 versus 3.2±0.65, P=<0.005; 3.01±0.96 versus 3.47±0.75, P=0.026) but equivalent MBF (2.75±0.82 versus 2.65±0.69 mL/g per min, P=0.826).
Conclusions
Regional and global impaired myocardial perfusion can occur in HCM mutation carriers, in the absence of significant hypertrophy or scarring.
Keywords: genetics, hypertrophic cardiomyopathy, quantitative perfusion mapping, sarcomere mutations carriers without hypertrophy
Subject Categories: Genetics, Cardiomyopathy, Magnetic Resonance Imaging (MRI)
Nonstandard Abbreviations and Acronyms
- AMVL
anterior mitral valve leaflet
- ECV
extracellular volume
- G+LVH‐
genotype‐positive, left ventricular hypertrophy‐negative
- HCM
hypertrophic cardiomyopathy
- LGE
late gadolinium enhancement
- MBF
myocardial blood flow
- MPR
myocardial perfusion reserve
Clinical Perspective
What Is New?
This study demonstrates that impaired myocardial perfusion can occur before the development of overt left ventricular hypertrophy or scarring in hypertrophic cardiomyopathy gene mutation carriers.
The use of quantitative perfusion mapping cardiac magnetic resonance imaging permits the quantification of myocardial blood flow and myocardial perfusion reserve as well as the easy visualization of perfusion defects.
What Are the Clinical Implications?
This alters our understanding of the pathophysiology of hypertrophic cardiomyopathy phenotype development as it suggests that perfusion abnormalities predate the development of left ventricular hypertrophy, contrary to what has been assumed and understood to date.
Microvascular dysfunction should be further explored in future preclinical hypertrophic cardiomyopathy studies with quantitative perfusion mapping cardiac magnetic resonance as a useful tool to assess this.
Hypertrophic cardiomyopathy (HCM) is one of the commonest heritable cardiac conditions, prevalent in 1 in 500 people, and characterized by unexplained left ventricular hypertrophy (LVH). It is most commonly caused by autosomal dominant sarcomere protein gene mutations, identified in up to 50% of patients with HCM.1 Subtle subclinical features occur in gene mutation carriers without LVH (G+LVH‐), including elongated anterior mitral valve leaflets (AMVL),2 myocardial crypts,3, 4, 5 hyperdynamic left ventricular (LV) function,6 abnormal apical LV trabeculation7 and abnormal septal convexity.1
Histologically, HCM is characterized by disarray, myocyte hypertrophy, myocardial fibrosis, and small vessel disease. These features may also contribute to microvascular dysfunction,8 which, with increased oxygen demand,6 leads to myocardial ischemia. Ischemia, scar, and LVH are adverse prognostic markers,9, 10 although their precise interrelationships are not known. Myocardial perfusion can be quantified using positron emission tomography or perfusion cardiovascular magnetic resonance (CMR) obtaining myocardial blood flow (MBF, mL/g per min) at rest and during vasodilator stress along with the derived ratio, the myocardial perfusion reserve (MPR). Marked stress perfusion defects can occur, particularly in hypertrophied segments.11 The role of these in phenotype evolution and the interaction with LVH and fibrosis are unclear, as is the precise relationship to risk.
Recently, quantitative perfusion CMR has been developed with completely automated “perfusion mapping”.12 This has advantages, including high transmural resolution, no ionizing radiation, and no manual analysis. Initial results have demonstrated abnormal perfusion in both hypertrophied and non‐hypertrophied segments in HCM.11, 13, 14 When combined with advanced genotyping including cascade screening in dedicated cardiomyopathy services, the early evolution of the HCM phenotype can be further explored.
We hypothesized that myocardial blood flow and myocardial perfusion reserve might be impaired in individuals with G+LVH‐ as a component of subclinical HCM.
Methods
The study was approved by the National Health Service Research Ethics Committee and Health Research Authority and conducted in accordance with the Declaration of Helsinki. All subjects provided written, informed consent (REC 18/LO/0188 and 17/SC/0077). Patients or the public were not involved in the design, conduct, reporting, or dissemination plans of our research. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Population
Fifty genotype‐positive, left ventricular hypertrophy‐negative (G+LVH‐) “subclinical” subjects (age ≥18 years) with likely pathogenic/pathogenic variants in sarcomeric protein genes were consecutively recruited. They were first‐degree relatives of individuals with a diagnosis of HCM identified in our Inherited Cardiovascular Disease clinic. The diagnosis of G+LVH‐ was based on positive genotype and not fulfilling diagnostic criteria from ECG and echocardiography for familial HCM in relatives, at the time of recruitment.15 In more detail, inclusion criteria for the G+LVH‐ group were (1) maximal LV wall thickness <13 mm by echocardiography/CMR and mass within the normal range relative to body surface area, age, and sex, (2) sinus rhythm on 12‐lead electrocardiography, (3) no causes of secondary LVH (eg, valve disease, hypertension). Subjects (n=5) recruited as LVH negative on echocardiogram, but with LV wall thickness ≥13 mm on CMR were considered as having fulfilled diagnostic criteria for familial HCM and were excluded from the G+LVH‐ cohort (but analyzed separately as HCM).
Controls were healthy volunteers with no significant past medical history, including cardiovascular disease. They were matched to unrelated patients with G+LVH‐ based on age and sex. Exclusion criteria were the presence of conventional contraindications to CMR.
Electrocardiography
Standard 12‐lead ECG was performed in all recruited participants. LVH was evaluated using the Sokolov‐Lyon and Romhilt‐Estes criteria. ECGs were analyzed by 2 experienced observers.
Genetic Testing and Variant Classification
Following the identification of a likely pathogenic/pathogenic variant in the proband, as previously described,7, 16 relatives included in this study had been previously counseled and tested for predictive testing, according to international guidelines17 and in clinically accredited laboratories. All individuals recruited carried variants, detected initially in the proband, considered to have a sufficient level of evidence for pathogenicity as to be eligible for cascade predictive testing. Criteria for pathogenicity of the identified variants were however, additionally reviewed for this study by E.Q. and L.R.L.
CMR Acquisition and Analysis
CMR scans were performed at 1.5 Tesla (Aera, Siemens Healthcare, Erlangen, Germany) using a standard clinical protocol. The protocol consisted of cine imaging, native T1 mapping (using a modified look‐locker inversion recovery sequence, MOLLI), T2 mapping, stress, and rest perfusion, late gadolinium enhancement (LGE) and post‐contrast T1 mapping. Synthetic extracellular volume fraction (ECV) was derived from the native and post contrast T1 maps. T1, T2 and ECV mapping was performed for basal, mid, and apical short axis and 1 long axis slice (4‐chamber).
Adenosine vasodilator stress perfusion was performed using a standard clinical approach (adenosine (140 mcg/kg per min, increased to 175 microgram/kg per min for a further 2 minutes if <10 bpm heart rate increase or no symptoms). A gadolinium‐based contrast agent (gadoterate meglumine, Dotarem, Guerbet, Paris, France) was injected into a peripheral vein at 4 mL/s during peak vasodilator stress at a dose of 0.05 mmol/kg; 60 images were acquired for basal, mid, and apical LV short‐axis slices. Rest perfusion images were acquired subsequently after an interval of 6 to 10 minutes. Perfusion mapping was implemented using the Gadgetron streaming software image reconstruction framework.18
CMR was analyzed using commercially available software (CVI42, Circle Cardiovascular Imaging, Calgary, Canada). For parametric analysis of T1, T2 and ECV maps, LV endo and epicardial contours were manually drawn and right ventricular (RV) insertion points identified. Borders were offset by 10% and models for both global and segmental analysis (16 segment American Heart Association model) were created for each parameter. For parametric map analysis of stress and rest myocardial blood flow, endocardial and epicardial contours were applied using machine learning with 10% border offsets and endo and epicardial sub‐segmentation giving 32 segments. Endocardial and epicardial borders were offset sequentially by 50% (also automated) and further 16‐segment models created for each.19 Visual perfusion defects were assessed from both the conventional and the mapping images by 2 independent assessors trained in CMR reporting and masked to the genotype (R.K.H. and J.A.). Where there was disagreement, a third assessor with greater experience was used (L.R.L.).
LV volume analysis was performed by contouring each short axis slice in end‐diastole and end‐systole. The maximum LV wall thickness was measured manually in end‐diastole using the short axis cine stack. The presence or absence of LGE was noted by segment and globally. The LV was evaluated for crypts using standard long axis cines or a “modified 2‐chamber” view using the definition previously determined by our group: “the presence of a focal myocardial defect in diastole, showing at least partial systolic obliteration, and having a depth ≥50% the thickness of the adjacent myocardium”.5 Raw AMVL length was estimated using previously described methods2 and adjusted for body size by dividing by body surface area. Fractal analysis was performed using the technique previously described by our group.7
Statistical Analysis
Statistical analysis was performed in SPSS (IBM SPSS statistic, Version 26.0). Continuous variables were presented as mean±SD, categorical variables as absolute values and percentages. Comparison between numerical variables was performed using independent t‐test while the Chi‐square test was used for categorical variables; equivalent non‐parametric tests were used as appropriate. Distribution of data was assessed on histograms and using Shapiro‐Wilk test. Patients with G+LVH‐ were compared with controls and with those who had reached diagnostic criteria for HCM (G+LVH+). Within the G+LVH‐ group, those with and without visual perfusion defects were also compared. Linear regression was used to determine which variables were associated with MPR and MBF and to study interaction effects between predictors. Multivariable models were then fitted which included variables identified as statistically significant in the univariate analyses while adjusting for participant age and sex. A variance inflation factor of <3 excluded multicollinearity. A 2‐sided P value of <0.05 was considered significant.
Results
Fifty subjects with G+LVH‐ (17 [34%] men, mean age 37.6±12.4) were compared with 28 age‐ and sex‐matched healthy volunteers (14 [50%] men, mean age, 37.9±11.7 years, P=0.919 for age and P=0.228 for sex). Five subjects were excluded after CMR detected familial criteria for HCM. The prevalence of the different causal genes is shown in Table 1 with the individual variants (all likely pathogenic/pathogenic) shown in Table S1. Apart from 1 individual with symptoms of chest pain, all the G+LVH‐ individuals were asymptomatic carriers, with no cardiac medication. Of the 50 recruited subjects, all were able to tolerate the perfusion protocol and their images were of diagnostic quality. The apical ECV maps did not reconstruct in 8 of 50 subjects and 7 of 50 had either 1 or 2 T1/ECV short axis slice(s) missing. T2 slice(s) were missing in 5 of 50 subjects but septal measurements were able to be derived in all subjects.
Table 1.
Genotyping of G+LVH‐ and G+LVH+
|
Causal Gene Mutation, n (%) |
G+LVH‐ (n=45) |
G+LVH+ (n=5) |
|---|---|---|
| MYBPC3 | 27 (60) | 3 (60) |
| MYH7 | 9 (20) | 1 (20) |
| TNNI3 | 5 (11.1) | 0 (0) |
| TNNT2 | 1 (2.2) | 1 (20) |
| TPM1 | 1 (2.2) | 0 (0) |
| CSRP3 | 1 (2.2) | 0 (0) |
| MYL2 | 1 (2.2) | 0 (0) |
CSRP3 indicates cysteine and glycine‐rich protein 3; G+LVH‐, genotype‐positive, left ventricular hypertrophy‐negative; G+LVH+, genotype‐positive, left ventricular hypertrophy‐positive; MYBPC3, myosin binding protein C; MYH7, β‐myosin heavy chain; MYL2, myosin regulatory light chain; TNNI3, cardiac troponin I; TNNT2, cardiac troponin T; and TPM1, α‐tropomyosin 1.
Compared with controls (Table 2), G+LVH‐ individuals had longer anterior mitral valve leaflets (P=0.030) and the presence of ≥2 myocardial crypts occurred in 13 of 45 (28.9%) patients with G+LVH‐ versus 0 of 28 controls (P < 0.005). Subjects with G+LVH‐ also had a lower indexed LV end‐systolic volume (P < 0.005) and higher LV ejection fraction (P < 0.005). Fractal analysis was not different versus controls (P=0.596). In G+LVH‐ individuals there was slightly higher septal T2 (+1.5 ms: 49.2±3.8 ms versus 47.7±2.4 ms, P=0.041) and slightly higher septal ECV (+1.5%: 25.8±2.4% versus 24.3±2.6%, P=0.023). Maximum wall thickness was not significantly higher compared with controls (G+LVH‐, 9.03±1.95 mm versus 8.37±1.18 mm, P=0.075). The G+LVH‐ group had no significant LGE, but there was minor non‐specific and non‐clinically significant LGE in the RV insertion points in 8(17.8%), compared with 0 controls (P=0.020). The LGE was limited to the inferior RV insertion point only in 6 of 8 participants, and in both superior and inferior RV insertion points in 2 of 8 participants. This distribution of LGE has been documented in otherwise normal hearts and is not associated with increased adverse outcomes.20
Table 2.
Characteristics of G+LVH‐ Patients Versus Controls
|
Category Mean (SD) |
G+LVH‐ (n=45) |
Control (n‐26) |
P Value |
|---|---|---|---|
| Age, y | 37.2 (12.3) | 38.9 (11.7) | 0.788 |
| No. of men, % | 13 (28.9) | 14 (50%) | 0.085 |
| BSA, m2 | 1.84 (0.23) | 1.93 (0.21) | 0.100 |
| LA area indexed, cm2/m2 | 13.07 (2.2) | 12.6 (1.8) | 0.354 |
| AMVLi, mm/m2 | 12.52 (2.1) | 11.55 (1.6) | 0.030* |
| ≥2 LV crypts n (%) | 13 (28.9) | 0 (0) | <0.005* |
| LVEDVi, mL/m2 | 73.7 (14.0) | 78.0 (14.8) | 0.222 |
| LVESVi, mL/m2 | 21.0 (6.9) | 26.7 (6.2) | <0.005* |
| Ejection fraction (%) | 71.9 (5.5) | 65.8 (4.4) | <0.005* |
| Massi, g/m2 | 45.4 (9.9) | 52.6 (9.3) | <0.005* |
| SV, mL | 97.2 (21.2) | 99.5 (25.5) | 0.701 |
| Visual perfusion defects n (%) | 9 (20.0) | 0 (0) | 0.011* |
| Mean global stress MBF, mL/g per min | 2.75 (0.71) | 2.78 (0.9) | 0.826 |
| Mean MPR | 2.77 (0.83) | 3.24 (0.63) | 0.009* |
| Mean rest MBF, mL/g per min | 1.08 (0.37) | 0.9 (0.23) | 0.011* |
| Mean subendocardial MBF, mL/g per min | 2.52 (0.82) | 2.65 (0.69) | 0.457 |
| Mean subendocardial MPR | 2.55 (0.75) | 3.2 (0.65) | <0.005* |
| Mean subepicardial MBF, mL/g per min | 2.8 (0.73) | 2.84 (0.59) | 0.800 |
| Mean subepicardial MPR | 3.01 (0.96) | 3.47 (0.75) | 0.026* |
| Subendo:subepicardial MBF ratio | 0.898 (0.17) | 0.934 (0.14) | 0.329 |
| Subendo:subepicardial MPR ratio | 0.87 (1.84) | 0.93 (0.13) | 0.092 |
| Septal T1, ms | 1025.6 (34.6) | 1016.0 (31.7) | 0.231 |
| Septal T2, ms | 49.2 (3.8) | 47.7 (2.43) | 0.041* |
| Septal ECV (%) | 25.8 (2.4) | 24.3 (2.6) | 0.023* |
| MWT, mm | 9.03 (1.95) | 8.37 (1.2) | 0.075 |
| LGE present n (%) | 8 (17.8) | 0 (0%) | 0.020* |
AMVLi indicates indexed anterior mitral valve leaflet (length); BSA, body surface area; ECV, extracellular volume; G+LVH‐, genotype‐positive, left ventricular hypertrophy‐negative; LA, left atrium; LGE, late gadolinium enhancement; LV, left ventricle/ventricular; LVEDVi, indexed left ventricular end‐diastolic volume; LVESVi, indexed left ventricular end‐systolic volume; Massi, indexed mass; MBF, myocardial blood flow; MPR, myocardial perfusion reserve; MWT, maximum wall thickness; and SV, stroke volume.
indicates significant P value <0.05.
Visual perfusion defects were present in 9 (20%) G+LVH‐ versus 0 (0%) controls, P=0.011. These were in some cases large, Figures 1 and 2, despite the absence of LVH fulfilling criteria for HCM. The perfusion defects did not follow a coronary distribution and were more common in the septum and insertion points. The distribution of perfusion defects seen are typical of overt HCM in the absence of epicardial coronary artery disease as previously identified by our team,13 and therefore further coronary imaging was not considered necessary. In one case of both perfusion defect and limited RV insertion point LGE, a computed tomography coronary angiogram had been requested for clinical reasons (chest pain), which was negative, Figure 2. Two of the patients with perfusion defects had limited RV insertion point LGE, but the perfusion defects were larger and remote from the LGE in these 2 individuals, Figure S1 and Figure 2. Other clinical causes of possible myocardial microvascular dysfunction were explored. Only 1 of 42 whose past medical history could be reviewed had a history of diabetes mellitus, but this subject had no perfusion defect; 4 of 42 had a history of hypertension. No cardiometabolic risk factors were present in all other patients.
Figure 1. Septal perfusion defects in genotype‐positive; left ventricular hypertrophy‐negative.
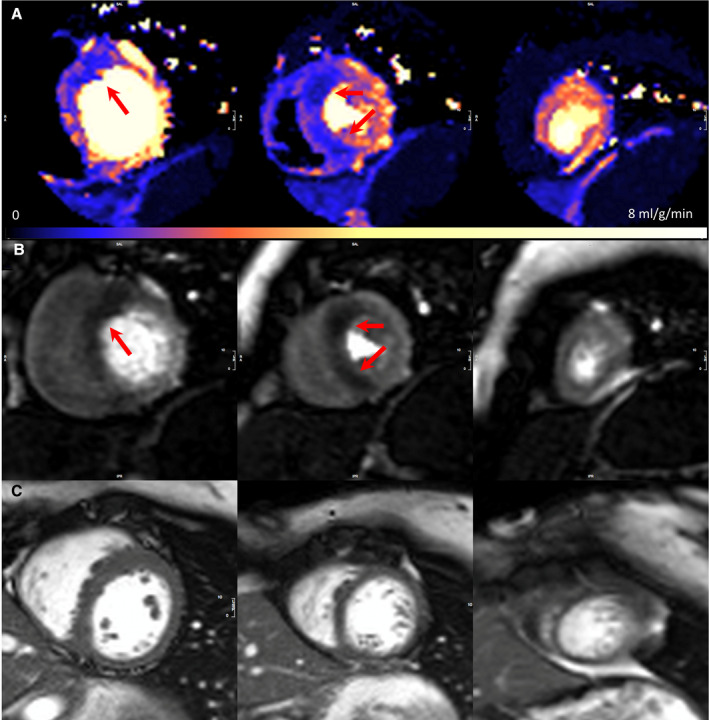
A, Adenosine stress perfusion maps in the 3 short‐axis slices, where each pixel encodes myocardial blood flow as per the color scale. Perfusion defect in the mid‐basal septum. B, Raw stress perfusion imaging. C, Corresponding short‐axis cine (maximum left ventricular wall thickness was 11.7 mm) (perfusion scans are acquired partly in systole). Arrows demonstrate the perfusion defects. SAX indicates short axis.
Figure 2. Basal septal and mid‐right ventricular insertion point significant perfusion defects in genotype‐positive; left ventricular hypertrophy‐negative with minor inferior right ventricular insertion point late gadolinium enhancement and normal computed tomography coronary angiogram.
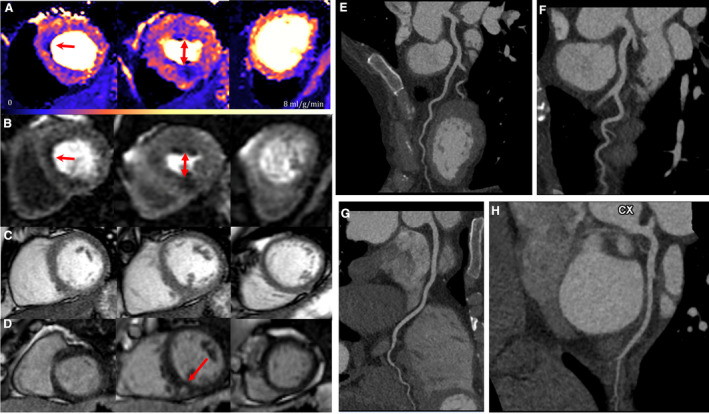
A second example, with the perfusion defects in a slightly different location to that of Figure 1. A, Adenosine stress perfusion maps in the 3 short‐axis slices. B, Corresponding raw stress perfusion images and (C) Corresponding short‐axis cine (maximum left ventricular wall thickness was 9.8mm). D, Corresponding short‐axis phase‐sensitive inversion recovery late gadolinium enhancement imaging. E, Computed tomography coronary angiogram image of left anterior descending artery (unobstructed). F, Computed tomography coronary angiogram image of left anterior descending artery (unobstructed). G, Computed tomography coronary angiogram image of right coronary artery (unobstructed). H, Computerized tomography coronary angiogram image of left circumflex coronary artery (unobstructed). Arrows indicate perfusion defects. CTCA indicates computed tomography coronary angiogram; LAD, left anterior descending artery; LCx, left circumflex coronary artery; LGE, late gadolinium enhancement; PSIR, phase‐sensitive inversion recovery; RCA, right coronary artery; and SAX, short axis.
For global MBF and MPR, G+LVH‐ individuals had no difference in global, subendocardial or subepicardial stress MBF versus controls, but rest flow was slightly higher (MBF 1.08±0.37 mL/g per min versus 0.9±0.23 mL/g per min, P=0.011). Global MPR was lower in G+LVH‐ than controls (2.77±0.83 versus 3.24±0.63, P=0.009). This was for both subendocardial and subepicardial MPR (P<0.005 and P=0.026 respectively), Figure 3. Within the G+LVH‐ group, AMVLi (β=0.11, P=0.030), MPR (β=0.32, P=0.012), septal T1 (β=0.01, P=0.0098) and maximum wall thickness (β=−0.12, P=0.028) were significantly associated with stress MBF. There were no interaction relationships between pairs of predictor variables. Associations with MBF were lost after adjustment for age and sex in multivariable models, except for global MPR (β=0.37, P=0.003). Other features of the subclinical HCM phenotype did not show a statistically significant association with stress MBF (number of crypts, β=0.01, P=0.912; or LV end‐systolic volume, β=0.02, P=0.248). There was a significant association between LVEDVi (β=0.02, P=0.012), LV end‐systolic volume (β=0.04, P=0.023) and stroke volume (β=0.01, P=0.012) with global MPR. There were no interaction relationships between predictor variables and all associations with MPR were lost after adjustment in multivariable models. There was no association between ejection fraction and global MPR (β=−0.04, P=0.099).
Figure 3. Bar chart comparison between genotype‐positive; left ventricular hypertrophy‐negative and controls of mean global stress myocardial blood flow, subendocardial stress myocardial blood flow, and subepicardial stress myocardial blood flow (A) and mean myocardial perfusion reserve, subendocardial myocardial perfusion reserve, and subepicardial myocardial perfusion reserve (B).
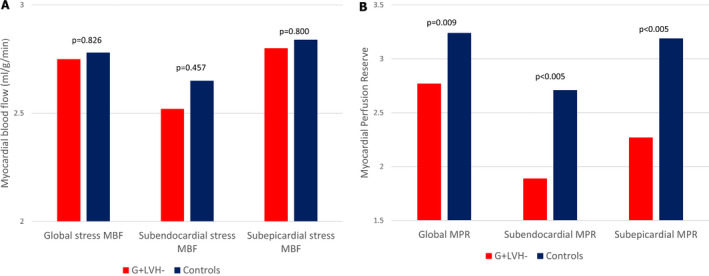
MBF indicates myocardial blood flow, and MPR, myocardial perfusion reserve.
MBF and MPR were also analyzed on an individual 16‐segment basis using the American Heart Association 16‐segment model. There was no significant difference in MBF for the mean of the lowest 2 segments (P=0.142) between G+LVH‐ and controls, but MPR was significantly different (2.03±0.72 versus 2.56±0.58, P=0.001). There was no significant difference on a per segment basis in MBF, but MPR was different between cases and controls in all segments except 12, 13, and 14 (Table 3).
Table 3.
MPR Using the American Heart Association 16‐Segment Model and Mean of Myocardial Perfusion Reserve in the Lowest 2 Contiguous Segments
| AHA Segment | MPR G+LVH‐ | MPR Control | P Value |
|---|---|---|---|
| Mean of lowest 2 contiguous segments | 2.023 (0.72) | 2.56 (0.58) | <0.005* |
| 1 | 2.63 (0.58) | 3.12 (0.69) | 0.008* |
| 2 | 2.61 (0.93) | 3.06 (0.57) | 0.026* |
| 3 | 2.40 (0.77) | 3.03 (0.67) | <0.005* |
| 4 | 2.89 (0.67) | 3.62 (1.06) | <0.005* |
| 5 | 3.19 (1.20) | 3.95 (1.26) | 0.016* |
| 6 | 3.02 (1.08) | 3.53 (0.74) | 0.021* |
| 7 | 2.43 (0.86) | 2.87 (0.6) | 0.015* |
| 8 | 2.34 (0.76) | 2.87 (0.59) | <0.005* |
| 9 | 2.51 (0.88) | 2.90 (0.62) | 0.033* |
| 10 | 2.71 (0.91) | 3.29 (0.76) | 0.005* |
| 11 | 2.87 (1.03) | 3.42 (0.94) | 0.024* |
| 12 | 2.67 (0.92) | 3.03 (0.70) | 0.071 |
| 13 | 2.89 (1.12) | 3.20 (0.89) | 0.212 |
| 14 | 2.73 (1.06) | 2.97 (0.83) | 0.300 |
| 15 | 2.74 (0.96) | 3.21 (0.82) | 0.033* |
| 16 | 3.00 (1.01) | 3.54 (1.01) | 0.031* |
AHA indicates American Heart Association, G+LVH‐, genotype‐positive, left ventricular hypertrophy‐negative, and MPR, myocardial perfusion reserve.
indicates significant P value <0.05.
ECG changes were present in patients with G+LVH‐ despite normal mass and wall thickness. ECGs were analyzable for Romhilt‐Estes criteria in 42 of 45 subjects and for Sokolov‐Lyon in 44 of 45 subjects. A Romhilt‐Estes score ≥4 (n=4/42[9.5%]) compared with ≤3 was not associated with a difference in wall thickness or mass, whereas those reaching Sokolov‐Lyon criteria for LVH (n=7/44[15.9%]) had a slightly higher LV mass (59.39±9.8 g/m2 versus 42.5±7.4 g/m2, P < 0.005). Individuals with a Romhilt‐Estes score ≥4 had lower MBF and MPR (2.14±0.4 versus 2.8±0.72 mL/g per min, P=0.03 and 2.08±0.59 versus 2.81±0.83, P=0.08 respectively), but MBF and MPR were not lower in individuals with Sokolov‐Lyon criteria for LVH.
Within the G+LVH‐ group, those with focal perfusion defects had lower global stress MBF and MPR (2.17±0.77 mL/g per min versus 2.89±0.62 mL/g per min, P=0.03; 2.1±0.62 versus 2.94±0.8, P < 0.005) with equivalent rest flow. These flow reductions occurred in both subendocardium (P=0.02) and subepicardium (P<0.005) for MBF and MPR. Visual perfusion defects were not associated with other pre‐phenotypic features (AMVL, P=0.63; presence of ≥2 myocardial crypts, P=0.704; maximum wall thickness, P=0.59; LV end‐systolic volume, P=0.3; left ventricular ejection fraction, P=0.58) (Table 4). Three out of nine (33.3%) subjects with G+LVH‐ with perfusion defects had a Romhilt‐Estes score ≥4 versus only 1 of 33 (3%) without (P=0.026). However, only 2 of 9 (22.2%) reached Sokolov‐Lyon criteria versus 5 of 30 (14.3%) without (P=0.619).
Table 4.
Characteristics of Patients With G+LVH‐ With Perfusion Defects Versus Those Without
|
Category Mean (SD) |
Perfusion Defect (n=9) | No Perfusion Defect (n=36) | P Value |
|---|---|---|---|
| Age, y | 34.6 (16.5) | 37.8 (11.2) | 0.590 |
| No. of men (%) | 3 (33.3) | 10 (27.8) | 0.704 |
| BSA, m2 | 1.83 (0.2) | 1.85 (0.2) | 0.830 |
| LA area indexed, cm2/m2 | 13.38 (1.6) | 13.0 (2.32) | 0.580 |
| AMVLi, mm/m2 | 12.81 (2.0) | 12.44 (2.1) | 0.630 |
| ≥2 LV crypts, n (%) | 3 (33.3) | 10 (27.8) | 0.704 |
| LVEDVi, mL/m2 | 69.73 (9.1) | 73.69 (15.0) | 0.220 |
| LVESVi, mL/m2 | 19.12 (5.6) | 21.49 (7.1) | 0.300 |
| Ejection fraction (%) | 72.82 (5.4) | 71.66 (5.5) | 0.580 |
| Massi, g/m2 | 44.49 (10.6) | 45.61 (9.8) | 0.780 |
| SV, mL | 92.43 (11.5) | 98.44 (22.9) | 0.280 |
| Mean global stress MBF, mL/g per min | 2.17 (0.8) | 2.89 (0.6) | 0.030* |
| Mean MPR | 2.10 (0.6) | 2.94 (0.8) | <0.005* |
| Mean rest MBF, mL/g per min | 1.07 (0.3) | 1.08 (0.4) | 0.910 |
| Mean subendocardial MBF, mL/g per min | 1.91 90.8) | 2.67 (0.8) | 0.020* |
| Mean subendocardial MPR | 1.89 (0.6) | 2.71 (0.7) | <0.005* |
| Mean subepicardial MBF, mL/g per min | 2.26 (0.8) | 2.93 (0.7) | 0.040* |
| Mean subepicardial MPR | 2.27 (0.6) | 3.19 (1.0) | <0.005* |
| Septal T1, ms | 1026.5 (36.2) | 1025.36 (34.7) | 0.930 |
| Septal T2, ms | 48.9 (3.9) | 49.3 (3.8) | 0.780 |
| Septal ECV (%) | 24.4 (3.0) | 26.2 (2.1) | 0.120 |
| MWT, mm | 9.43 (2.5) | 8.93 (1.8) | 0.590 |
| LGE present n (%) | 2 (22.2) | 6 (16.7%) | 0.651 |
AMVLi indicates indexed anterior mitral valve leaflet (length); BSA, body surface area; ECV, extracellular volume; G+LVH‐, genotype‐positive; left ventricular hypertrophy‐negative; LA, left atrium; LGE, late gadolinium enhancement; LV, left ventricle/ventricular; LVEDVi, indexed left ventricular end‐diastolic volume; LVESVi, indexed left ventricular end‐systolic volume; Massi, indexed mass; MBF, myocardial blood flow; MPR, myocardial perfusion reserve; MWT, maximum wall thickness; and SV, stroke volume.
indicates significant P value <0.05.
All 5 of 50 participants with CMR detected LVH had visual perfusion defects (average age 42±14.09). Compared with G+LVH‐, those with overt hypertrophy (G+LVH+) had greater maximum wall thickness (14.58±1.21 mm versus 9.03±1.95 mm, P<0.005), higher ejection fraction (78.72±4.92% versus 71.89±5.47%, P=0.032) and lower global mean stress MBF (1.91±0.6 mL/g per min versus 2.75±0.71 mL/g per min, P=0.031). The comparison for resting mean MBF (0.93±0.59 mL/g per min versus 1.08±0.37 mL/g per min, P=0.605) and mean MPR (1.99±1.02 versus 2.77±0.83, P=0.164) was non‐significant. Compared with controls, G+LVH+ also had greater maximum wall thickness (14.58±1.21 mm versus 8.37±1.18 mm, P < 0.005), higher ejection fraction (78.72±4.92% versus 65.77±4.37%, P=0.002) and lower global mean stress MBF (1.91±0.6 mL/g per min versus 2.78±0.59 mL/g per min, P=0.027). Similarly, a much lower MPR did not reach significance (1.99±1.02 versus 3.24±0.63, P=0.05). Resting mean MBF was also non‐significant (0.93±0.59 mL/g per min versus 0.90±0.23 mL/g per min, P=0.91).
Discussion
These data show that subjects carrying a known mutation for HCM can have abnormal myocardial perfusion before, or in the absence of the development of significant LVH or scar and in the absence of other causes of myocardial microvascular dysfunction. These defects can be large and visually obvious by conventional CMR perfusion, with quantitation via perfusion mapping showing reduced global and segmental perfusion reserve, with a decrease in MBF (not just lack of augmentation) in defects compared with rest, during adenosine hyperemia.
Phenotype development and its sequence in HCM are not well understood. Preclinical “embryological” features such as AMVL elongation and crypts are thought to be present at birth. ECG changes can occur before LVH with significant LGE almost always occurring later, after the development of LVH. Traditional thinking suggests that perfusion defects should not occur pre/in the absence of LVH that fulfills HCM criteria, a view supported by smaller, earlier non‐quantitative studies.21 Where perfusion defects were present in overt HCM, flow is lowest within the subendocardium,14, 22 as here. The proposed mechanisms are the effects of extravascular compressive forces and elevated intraventricular pressures,23 with increased demand from hypertrophied or inefficient myocytes linking in some way to microvascular and fibrotic changes cascading from signalling myocytes. The conventional view is therefore that perfusion defects are secondary to initial myocyte changes; only subsequently serving as a pathway/feedback mechanism exacerbating LVH, fibrosis and potentially risk. After all, the sarcomeric proteins mutated in HCM induce overt phenotypes only in myocardium, with expression in other forms of muscle causing only subtle ultrastructural and energetic abnormalities.24
This is unlikely to be the explanation in this cohort, as there is no overt LVH. The findings upset a “myocyte centric” view of HCM where LVH is a primary mechanism. To explain these findings, we hypothesize several possibilities.
One hypothesis is “missed hypertrophy” — that perfusion defects are secondary to myocyte hypertrophy missed in the early phase by macroscopic imaging. There is some evidence for that, as ECG changes are occurring coincidentally. Against this is that perfusion defects were present and profound in some patients without LVH by ECG criteria.
A second hypothesis is “pre‐hypertrophy myocyte phenotype switching” — that myocytes alter expression profiles and matrix/cell interaction before overt LVH. Such switching could well induce fibroblast hyperplasia and conversion to myofibroblasts, as well as vascular smooth muscle hypertrophy.23 Certainly perfusion changes in other diseases have been thought to be vascular in origin (eg, Fabry disease).25
The final hypothesis we propose “myocyte:capillary embryological coupling hypothesis”, is more thought provoking. The heart is built of cells and interstitium, and we typically focus on one functional cell type: myocytes. But the fundamental unit of tissue, since life evolved beyond diffusion limits, is not one specialist cell but a pair: one for specialized function (eg, myocyte/neuron etc), and one specialized in tissue exchange — the capillary. HCM‐causing mutations cause an embryological phenotype including alterations in clefts, trabeculae, disarray, and the AMVL that are detectable pre‐birth.26, 27 It is therefore plausible, or even likely, that the microvasculature of HCM is abnormal right from the beginning of organogenesis, well before any overt clinical phenotypic expression. Here, perfusion defects were mainly septal, as is typical with hypertrophy in HCM. Embryologically, septation is timed differently to free wall formation (occurs later),27 raising the possibility that overt disease perfusion defects and hypertrophy distributions are patterned by embryological sequential gene expression. Similar to Ho et al’s findings, ECV was higher in our G+LVH‐ cohort than controls, further supporting their argument that sarcomere mutations cause myocardial abnormalities and increased interstitial fibrosis before and independently of overt hypertrophy,28 but also strengthening our hypothesis about embryological sequential gene expression.
One other phenomenon was observed, that of slightly higher rest flow in HCM mutation carriers. This was separate to and independent of stress perfusion defects. Rest perfusion is tightly autoregulated, matching supply and demand. So even if there were capillary alterations, rest flow would be preserved. If this is not a supply issue, and in the absence of reduced oxygen carriage (anaemia), the most logical reason for this is increased demand. We therefore hypothesize that this may be a manifestation of sarcomeric “gain of function” with inefficiency of hyperdynamic contraction and elevated metabolic demand. A second hypothesis could also be that mutation carriers have altered their vascular pharmacodynamics response to adenosine – and that stress vasodilatation lingers in HCM.
All subjects fulfilling criteria for HCM (G+LVH+) had visual perfusion defects, which is higher than our center’s previously reported rate in HCM of 78%.13 However, our numbers reported in this article are much lower (n=5 versus 101). These subjects had lower mean stress MBF than those who were G+LVH‐, yet mean rest MBF and overall mean MPR were equivalent. This further supports our hypothesis about sarcomeric “gain of function” as a preclinical finding. Compared with controls, mean stress MBF was lower and although there was a large numerical difference in mean MPR, this fell just shy of significance (which our center has previously reported,13 again likely because of the small sample size).
The utility of early phenotype detection for therapeutic prevention or attenuation of phenotype development is being explored. In animal models, the use of the L‐type calcium‐channel blocker, diltiazem, in G+LVH‐ mice showed attenuated phenotype progression compared with placebo‐controls.29 A pilot study in G+LVH‐ in humans showed that diltiazem could be used safely without significant adverse effects, although the impact on disease progression remained unclear.30 Furthermore, the role of transforming growth factor‐β activation on fibrosis development and disease pathogenesis has been explored in mice and the impact of inhibiting transforming growth factor‐β activation using neutralizing antibodies or angiotensin II receptor blockers demonstrated a reduction in disease progression. The VANISH (Valsartan for Attenuating Disease Evolution in Early Sarcomeric HCM) trial is underway testing whether the angiotensin II receptor blockers Valsartan can attenuate disease progression in G+LVH‐ humans.31 Whether any of these medications alters perfusion defects is unknown in G+LVH‐ HCM, and more broadly, these data point to a more complex and sequenced phenotype development in HCM with new potential therapeutic targets (vascular function) and surrogate end points for further study.
Limitations
This is a single time‐point observational study; any phenotypic development after the time of scanning has not been evaluated. Future work should explore those with visually identified perfusion defects. Although this is the largest series to date, larger prospective cohorts are needed. Biopsies have not been performed and therefore our assumptions about the role of microvascular dysfunction cannot be confirmed; an alternative explanation for an impaired response to adenosine cannot be excluded. There was no clinical indication to perform anatomical coronary assessment in this cohort, and as such we could not fully exclude the presence of epicardial coronary artery anomalies including bridging; however, it is highly unlikely that these could explain the findings. For fractal analysis, the cine short axis stack was performed after stress perfusion, ie, in the presence of a half dose of contrast, which reduced trabeculae:blood contrast.
Conclusions
Quantitative perfusion mapping demonstrated that 1 in 5 patients with G+LVH‐ HCM have marked regional perfusion defects – before any detectable significant hypertrophy or scar, suggesting that our models of HCM phenotype development could be reconsidered. Perfusion may be an early therapeutic target and perfusion mapping is a useful tool to explore further the early HCM phenotype.
Sources of Funding
R.K.H. is supported by the British Heart Foundation (grant number FS/17/82/33222). G.C. is supported by the National Institute for Health Research Rare Diseases Translational Research Collaboration (NIHR RD‐TRC, #171603) and by NIHR University College London Hospitals Biomedical Research Centre. J.C.M., P.S., L.R.L., and P.M.E. are directly and indirectly supported by the University College London Hospitals NIHR Biomedical Research Centre and Biomedical Research Unit at Barts Hospital, respectively. Part of this work was supported by a Barts Charity Grant to L.R.L. L.R.L. is funded by a Medical Research Council Clinical Academic Research Partnership (CARP) award.
Disclosures
None.
Supporting information
For Sources of Funding and Disclosures, see page 10.
References
- 1.Reant P, Captur G, Mirabel M, Nasis A, Sado DM, Maestrini V, Castelletti S, Manisty C, Herrey AS, Syrris P, et al. Abnormal septal convexity into the left ventricle occurs in subclinical hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2015;17:1–8. DOI: 10.1186/s12968-015-0160-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maron MS, Olivotto I, Harrigan C, Appelbaum E, Gibson CM, Lesser JR, Haas TS, Udelson JE, Manning WJ, Maron BJ. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124:40–47. DOI: 10.1161/CIRCULATIONAHA.110.985812. [DOI] [PubMed] [Google Scholar]
- 3.Child N, Muhr T, Sammut E, Dabir D, Ucar EA, Bueser T, Gill J, Carr‐White G, Nagel E, Puntmann VO. Prevalence of myocardial crypts in a large retrospective cohort study by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2014;16:1–9. DOI: 10.1186/s12968-014-0066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. DOI: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Captur G, Lopes LR, Mohun TJ, Patel V, Li C, Bassett P, Finocchiaro G, Ferreira VM, Esteban MT, Muthurangu V, et al. Prediction of sarcomere mutations in subclinical hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2014;7:863–871. DOI: 10.1161/CIRCIMAGING.114.002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marian AJ, Braunwald E. Hypertrophic cardiomyopathy genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:749–771. DOI: 10.1161/CIRCRESAHA.117.311059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Captur G, Lopes LR, Patel V, Li C, Bassett P, Syrris P, Sado DM, Maestrini V, Mohun TJ, Mckenna WJ, et al. Abnormal cardiac formation in hypertrophic cardiomyopathy. Fractal analysis of trabeculae and preclinical gene expression. Circ Cardiovasc Genet. 2014;7:241–248. DOI: 10.1161/CIRCGENETICS.113.000362. [DOI] [PubMed] [Google Scholar]
- 8.Timmer SAJ, Germans T, Brouwer WP, Lubberink M, Van Der Velden J, Wilde AAM, Christiaans I, Lammertsma AA, Knaapen P, Van Rossum AC. Carriers of the hypertrophic cardiomyopathy MYBPC3 mutation are characterized by reduced myocardial efficiency in the absence of hypertrophy and microvascular dysfunction. Eur J Heart Fail. 2011;13:1283–1289. DOI: 10.1093/eurjhf/hfr135. [DOI] [PubMed] [Google Scholar]
- 9.Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349:1027–1035. DOI: 10.1056/NEJMoa025050. [DOI] [PubMed] [Google Scholar]
- 10.Chiribiri A, Leuzzi S, Conte MR, Bongioanni S, Bratis K, Olivotti L, De Rosa C, Lardone E, Di Donna P, Villa A, et al. Rest perfusion abnormalities in hypertrophic cardiomyopathy: correlation with myocardial fibrosis and risk factors for sudden cardiac death. Clin Radiol. 2015;70:495–501. DOI: 10.1016/j.crad.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen SE, Jerosch‐Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA, Selvanayagam JB, Neubauer S, Watkins H. Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation. 2007;115:2418–2425. DOI: 10.1161/CIRCULATIONAHA.106.657023. [DOI] [PubMed] [Google Scholar]
- 12.Kellman P, Hansen MS, Nielles‐vallespin S, Nickander J, Themudo R, Ugander M, Xue H. Myocardial perfusion cardiovascular magnetic resonance : optimized dual sequence and reconstruction for quantification. J Cardiovasc Magn Reson. 2017;19:1–14. DOI: 10.1186/s12968-017-0355-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camaioni C, Knott KD, Augusto JB, Seraphim A, Rosmini S, Ricci F, Boubertakh R, Xue H, Hughes R, Captur G, et al. Inline perfusion mapping provides insights into the disease mechanism in hypertrophic cardiomyopathy. Heart. 2020;106:824–829. DOI: 10.1136/heartjnl-2019-315848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ismail TF, Hsu L‐Y, Greve AM, Gonçalves C, Jabbour A, Gulati A, Hewins B, Mistry N, Wage R, Roughton M, et al. Coronary microvascular ischemia in hypertrophic cardiomyopathy ‐ A pixel‐wise quantitative cardiovascular magnetic resonance perfusion study. J Cardiovasc Magn Reson. 2014;16:1–10. DOI: 10.1186/s12968-014-0049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valente AM, Lakdawala NK, Powell AJ, Evans SP, Cirino AL, Orav EJ, Macrae CA, Colan SD, Ho CY. Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet. 2013;6:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopes LR, Syrris P, Guttmann OP, O'Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, et al. Novel genotype‐phenotype associations demonstrated by high‐throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. 2015;101:294–301. DOI: 10.1136/heartjnl-2014-306387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott PM, Anastasakis A, Borger M, Borggrefe M, Cecchi F, Charron P, Hagege A, Lafont A. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 18.Xue H, Inati S, Sørensen TS, Kellman P, Hansen MS. Distributed MRI reconstruction using gadgetron‐based cloud computing. Magn Reson Med. 2015;73:1015–1025. DOI: 10.1002/mrm.25213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue H, Davies R, Brown LA, Knott KD, Kotecha T, Fontana M, Plein S, Moon JC, Kellman P. Automated inline analysis of myocardial perfusion MRI with deep learning. Magn Reson Med. 2019;83:712–730. DOI: 10.1148/ryai.2020200009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grigoratos C, Pantano A, Meschisi M, Gaeta R, Ait‐Ali L, Barison A, Todiere G, Festa P, Sinagra G, Aquaro GD. Clinical importance of late gadolinium enhancement at right ventricular insertion points in otherwise normal hearts. Int J Cardiovasc Imaging. 2020;36:913–920. DOI: 10.1007/s10554-020-01783-y. [DOI] [PubMed] [Google Scholar]
- 21.Gyllenhammar T, Fernlund E, Jablonowski R, Jögi J, Engblom H, Liuba P, Arheden H, Carlsson M. Young patients with hypertrophic cardio myopathy, but not subjects at risk, show decreased myocardial perfusion reserve quantified with CMR. Eur Heart J Cardiovasc Imaging. 2014;15:1350–1357. DOI: 10.1093/ehjci/jeu137. [DOI] [PubMed] [Google Scholar]
- 22.Yalçin H, Valenta I, Yalçin F, Corona‐Villalobos C, Vasquez N, Ra J, Kucukler N, Tahari A, Pozios I, Zhou Y, et al. Effect of diffuse subendocardial hypoperfusion on left ventricular cavity size by 13N‐ammonia perfusion PET in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2016;118:1908–1915. DOI: 10.1016/j.amjcard.2016.08.085. [DOI] [PubMed] [Google Scholar]
- 23.Camici PG, Olivotto I, Rimoldi OE. The coronary circulation and blood flow in left ventricular hypertrophy. J Mol Cell Cardiol. 2012;52:857–864. DOI: 10.1016/j.yjmcc.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 24.Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:3–18. DOI: 10.1007/s00401-012-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knott KD, Augusto JB, Nordin S, Kozor R, Camaioni C, Xue H, Hughes RK, Manisty C, Brown LAE, Kellman P, et al. Quantitative myocardial perfusion in Fabry disease. Circ Cardiovasc Imaging. 2019;12:e008872. DOI: 10.1161/CIRCIMAGING.119.008872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia‐Canadilla P, Cook AC, Mohun TJ, Oji O, Schlossarek S, Carrier L, McKenna WJ, Moon JC, Captur G. Myoarchitectural disarray of hypertrophic cardiomyopathy begins pre‐birth. J Anat. 2019;235:962–976. DOI: 10.1111/joa.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Captur G, Ho CY, Schlossarek S, Kerwin J, Mirabel M, Wilson R, Rosmini S, Obianyo C, Reant P, Bassett P, et al. The embryological basis of subclinical hypertrophic cardiomyopathy. Sci Rep. 2016;6:27714. DOI: 10.1038/srep27714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho CY, Abbasi SA, Neilan TG, Shah RV, Chen Y, Heydari B, Cirino AL, Lakdawala NK, Orav EJ, González A, et al. T1 Measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6:415–422. DOI: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, et al. The L‐type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Investig. 2002;109:1013–1020. DOI: 10.1172/JCI200214677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, Kwong RY, Antman EM, Semsarian C, González A, et al. Diltiazem treatment for preclinical hypertrophic cardiomyopathy mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Failure. 2015;3:180–188. DOI: 10.1016/j.jchf.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Axelsson Raja A, Shi L, Day SM, Russell M, Zahka K, Lever H, Colan SD, Margossian R, Hall EK, Becker J, et al. Baseline characteristics of the VANISH cohort. Circ Heart Fail. 2019;12:e006231. DOI: 10.1161/CIRCHEARTFAILURE.119.006231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum P, Gauthier LD, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐ function intolerance across human protein‐coding genes. Preprint posted online August 13, 2019. BioRxiv. 2019;531210. [Google Scholar]
- 33.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:862–868. DOI: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards S, Aziz N, Bale S, Bick D, Das S. ACMG standards and guidelines standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.