

Abstract
Background
Sarcomere gene mutations lead to cardiomyocyte hypertrophy and pathological myocardial remodeling. However, there is considerable phenotypic heterogeneity at both the cellular and the organ level, suggesting modifiers regulate the effects of these mutations. We hypothesized that sarcomere dysfunction leads to cardiomyocyte genotoxic stress, and this modifies pathological ventricular remodeling.
Methods and Results
Using a murine model deficient in the sarcomere protein, Mybpc3−/− (cardiac myosin‐binding protein 3), we discovered that there was a surge in cardiomyocyte nuclear DNA damage during the earliest stages of cardiomyopathy. This was accompanied by a selective increase in ataxia telangiectasia and rad3‐related phosphorylation and increased p53 protein accumulation. The cause of the DNA damage and DNA damage pathway activation was dysregulated cardiomyocyte DNA synthesis, leading to replication stress. We discovered that selective inhibition of ataxia telangiectasia and rad3 related or cardiomyocyte deletion of p53 reduced pathological left ventricular remodeling and cardiomyocyte hypertrophy in Mybpc3−/− animals. Mice and humans harboring other types of sarcomere gene mutations also had evidence of activation of the replication stress response, and this was associated with cardiomyocyte aneuploidy in all models studied.
Conclusions
Collectively, our results show that sarcomere mutations lead to activation of the cardiomyocyte replication stress response, which modifies pathological myocardial remodeling in sarcomeric cardiomyopathy.
Keywords: ataxia telangiectasia and rad3 related, cardiac myosin‐binding protein 3, cardiac troponin T2, cardiomyocyte hypertrophy, DNA damage, endoreplication, hypertrophic cardiomyopathy
Subject Categories: Myocardial Biology, Animal Models of Human Disease, Cardiomyopathy, Remodeling, Hypertrophy
Nonstandard Abbreviations and Acronyms
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and rad3 related
- CDK
cyclin‐dependent kinase
- HCM
hypertrophic cardiomyopathy
- LVIDd
left ventricular internal dimension at end diastole
- MYBPC3
cardiac myosin‐binding protein 3
- P
postnatal day
- PCM1
pericentriolar material 1
- TNNT2
troponin T2, cardiac type
Clinical Perspective
What Is New?
During the earliest stages of sarcomeric cardiomyopathy development, there is a surge in cardiomyocyte nuclear DNA damage that activates replication stress response pathways and is associated with cardiomyocyte aneuploidy.
The activation of the replication stress response modifies pathological left ventricular remodeling in a murine model of sarcomeric cardiomyopathy.
What Are the Clinical Implications?
We discovered that sarcomere mutations lead to genotoxic stress that impacts disease development and identified pathways that may provide new therapeutic targets for sarcomeric cardiomyopathies.
Mutations in sarcomere genes are a common cause of inherited cardiomyopathies in humans.1 At the molecular level, these mutations alter sarcomere protein function and impact cardiomyocyte calcium handling.2 However, human carriers of these mutations show considerable phenotypic heterogeneity, suggesting other cellular pathways impact how cardiomyocytes respond to sarcomere dysfunction.
Animal models harboring sarcomere protein mutations have shown that cardiomyocytes respond to alterations in sarcomere function in distinct ways during different phases of cardiac development.3, 4, 5, 6, 7, 8 For example, across multiple murine sarcomeric cardiomyopathy models, it has been shown that the early postnatal time period is critical in establishing the cardiomyopathy phenotype.4, 5, 6, 7, 8 This holds true even when cardiomyocyte hypertrophy does not develop until later developmental stages. These studies suggest that sarcomere dysfunction induces changes in early postnatal cardiomyocytes that lay the foundation for future hypertrophic cardiomyocyte growth. However, it remains poorly understood why the early postnatal time period was important for cardiomyopathy development. Using a murine model that lacks the sarcomere protein MYBPC3 (cardiac myosin‐binding protein 3), we discovered that sarcomere dysfunction in the early postnatal time period caused abnormal activation of cardiomyocyte cell cycle pathways, which led to increased cardiomyocyte DNA synthesis without cell division (endoreplication).4 However, the molecular consequences of this dysregulated cardiomyocyte cell cycle activity and how these pathways impacted cardiomyopathy progression remained unclear.
We hypothesized that sarcomere dysfunction causes cardiomyocyte genotoxic stress, and this impacts pathological ventricular remodeling. Using murine models and human tissue containing sarcomere mutations, we investigated the DNA damage response in sarcomeric cardiomyopathy. We discovered that sarcomere dysfunction triggers the replication stress response in cardiomyocytes, and this pathway modifies pathological myocardial remodeling.
METHODS
The raw data that support the findings of this study are available from the corresponding author on reasonable request.
Data and Materials Availability
All data needed to evaluate the conclusions in the article are present in the article and/or the supplementary materials. Additional data related to this article may be requested from the authors.
Animals
Animals were housed in pathogen‐free animal facility at the University of Pittsburgh with ad libitum access to food and water. The Mybpc3−/− mice used in the present study were generated previously.4 Briefly, mice with a knockout first tm1a allele of the Mybpc3 gene (Mybpc3tm1a) in C57BL/6J background were crossed with an ACTB:Flp deleter line to create a conditional Mybpc3 allele (Mybpc3tm1c) and then crossed with a CMV:Cre line to generate the germline null allele of Mybpc3−/−. Cardiomyocyte deletion of p53 was accomplished by crossing Mybpc3−/− with p53flox (Jackson 008462) and Myh6Cre+/− (Jackson 011038) to generate Mybpc3−/−/p53 fl/flMyh6Cre+/− animals. The transgenic mouse lines expressing human wild‐type troponin T2, cardiac type (TNNT2WT), or mutant cardiac troponin T (TNNT2I79N)9 (kindly provided by Bjorn Knollmann, Vanderbilt University) were crossed into a C57BL/6J genetic background for a minimum of 6 generations before analysis. Investigators were blinded to animal genotyping and/or treatment at time of data collection, and no animals were excluded from analysis. The sample size (n) included per group was stated in each figure legend.
Drug Treatment
For administration of the cyclin‐dependent kinase (CDK) 4/6 inhibitor (PD‐0332991; Active Biochemical Co), the drug was dissolved in sodium lactate (pH 4.0) and administered daily via oral gavage at 150 mg/kg body weight for 5 consecutive days from postnatal day (P) 2 to P7. For ataxia telangiectasia and rad3‐related (ATR) kinase inhibitor treatment, AZD6738 (Med Chem Express) was dissolved in dimethyl sulfoxide and further diluted to the required concentration with sesame oil. The drug was administrated daily via SC injection at 25 mg/kg body weight for 3 consecutive days from P4 to P7. The groups of control mice were administered vehicle (sodium lactate or sesame oil) to confirm that the vehicle or delivery method had no significant effect on the measured phenotypes. The same treatments were performed in at least 2 independent litters for reproducibility, and litters were randomly assigned to either vehicle or active drug. All animal protocols and procedures were performed in accordance with National Institutes of Health guideline and approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Echocardiography
Transthoracic echocardiography was performed using a Vevo 3100 High‐Resolution system (VisualSonics Inc). Two‐dimensional M‐mode images were obtained to measure the parameters, including left ventricular (LV) posterior wall at end diastole, interventricular septal thickness at end diastole, and LV internal dimensions at end diastole (LVIDd) and end systole (LVIDs). LV fractional shortening was calculated using the following formula: (LVIDd–LVIDs)/LVIDd. All echocardiographic studies were performed under conscious conditions without anesthesia and measured by an individual who was blinded to the genotype of the mice.
Euthanasia and Heart Mass Assessment
Mice were sedated with continuous infusion of 5% isoflurane (Henry Schein) using nose cone. Once unresponsive to toe pinch, total body weight was recorded, and the heart was excised. The heart was perfused with buffer (0.5 mol/L KCl), and heart weight was obtained. Mouse lower extremities were then amputated at the midfemur level and boiled to remove tissues. Digital calipers were used to measure the tibia length. Heart weight (mg) to body weight (g) or tibia length (mm) ratios were then calculated.
Comet Assay
Nuclei were isolated from P7 myocardial tissue using a hypotonic solution (10 mmmol/L Tris‐HCl, 5 mmol/L MgCl2, and 10 mmol/L NaCl, pH 7.5) with mechanical disruption, and neutral comet assay (Trevigen) was then performed as previously described.10 The isolated nuclei were diluted in warm low‐melting agarose in PBS and carefully pipetted onto the slides. Following incubation at 4°C for 30 minutes, slides were placed in ice‐cold lysis solution for overnight at 4°C. The following day, electrophoresis was done on the slides using 1× neutral electrophoresis buffer (50 mmmol/L Tris and 150 mmol/L sodium acetate, pH 9) at 20 V for 45 minutes at 4°C. Slides were then incubated with DNA precipitation buffer (1 mmol/L ammonium acetate in 95% ethanol) and 70% ethanol each for 30 minutes at room temperature, followed by being dried at 45°C for 10 to 15 minutes. The embedded nuclei in agarose were stained with ethidium bromide (2 µg/mL) for 30 minutes at room temperature in the dark. Slides were then imaged on an inverted wide‐field fluorescent microscope (Olympus) at ×10 magnification, and comets were analyzed using the Comet Score software (http://www.autocomet.com). Percentage of DNA in the tail multiplied by the length between the center of the comet head and tail (the Olive tail moment) was used as the metric of fragmentation.11 A minimum of 50 comets were analyzed per sample.
Immunohistochemistry
Hearts were embedded in optimal cutting solution and subsequently sectioned on a cryostat (Thermo Fisher Scientific) in the apical to basal direction at 5 µm per section onto Superfrost Plus Gold microscope slides (Fisher brand). Following 4% paraformaldehyde fixation, sections were permeabilized with 0.2% Triton X‐100 with subsequent PBS washes. Sections were then incubated with blocking solution (1% BSA in PBS) for 1 hour at room temperature, followed by primary antibodies targeting p‐Ser139 Histone H2AX (γH2AX) (Millipore Sigma, 05‐636‐I), p53‐binding protein 1 (53BP1) (Novus Biologicals, NB100‐304), pericentriolar material 1 (PCM1) (Sigma, HPA023370), Ki67 (CST, 9129), p‐Ser428 ATR (CST, 2853), and sarcomeric α‐actinin (Abcam, ab9465), either alone or in combination, at 4°C overnight. The following day, sections were washed and incubated with fluorescent secondary goat anti‐rabbit Alexa Fluor 594 (Thermo Fisher Scientific, A‐11005), goat anti‐rabbit Alexa Fluor 488 (Thermo Fisher Scientific, A‐11034), goat anti‐mouse Alexa Fluor 594 (Thermo Fisher Scientific, A‐11032), or goat anti‐mouse Alexa Fluor 488 (Thermo Fisher Scientific, A‐10667) antibodies for 1 hour at room temperature. After final PBS washes, sections were counterstained and mounted with ProLong Gold Antifade with 4′,6‐diamidino‐2‐phenylindole (DAPI) and allowed to cure overnight in the dark. Slides were then visualized and imaged under wide‐field fluorescent microscope (Olympus or Zeiss) at ×40 or ×100 magnification. Nuclear fluorescence was determined by using the following equation: corrected nuclear fluorescence=integrated density–(area of nuclei×mean fluorescence of background readings), where integrated density is fluorescence intensity of the defined region of interest, area of nuclei is the size of the defined region of interest, and mean fluorescence background is the average intensity of 5 background regions of interest.
For measuring cardiomyocyte size, 5‐µm tissue sections were fixed in 4% paraformaldehyde, washed with 1× PBS, and subsequently incubated with Oregon Green 488 (Thermo Fisher Scientific, W6748) or Texas Red‐X (Thermo Fisher Scientific, W21405) conjugated wheat‐germ agglutinin in 1% BSA in PBS (1:200) in the dark for 10 minutes. After rinsing with PBS, slides were then mounted with Prolong Gold Antifade with DAPI, and images were taken at ×40 magnification with cardiomyocyte borders subsequently measured to determine cell area.
Chromosome Fluorescence in Situ Hybridization Assay
Frozen mouse hearts were sectioned (5 µm) and immediately fixed with a methanol and acetic acid solution (3:1 ratio) for 10 minutes at room temperature. After fixation, slides were air dried completely for 5 minutes and washed twice for 5 minutes in PBS. During this time, the mouse chromosome 8 centromere probe was diluted 1:10 in hybridization buffer (FMCEN‐08; Creative Bioarray) and applied directly onto the tissue sections. Coverslips were added to slides and were placed into a humidified hybridization oven set at 83°C for 3 minutes (denaturation), followed by 37°C for 16 hours overnight (hybridization). After hybridization, coverslips were carefully removed, and slides were washed in PBS 3 times for 5 minutes each. Sections were then blocked in 1% BSA in 1× PBS for 1 hour at room temperature, followed by incubation with PCM1 antibody (1:500 in blocking buffer) for 2 hours at room temperature. Slides were washed in PBS 3 times for 5 minutes, and sections were then incubated with goat anti‐rabbit Alex Fluor 488 secondary antibody (1:500) for 1 hour at room temperature in the dark. Slides were then washed in PBS 3 times for 5 minutes each and mounted with Prolong Gold Antifade Mountant with DAPI, with coverslips added. Slides were imaged at ×40 using a Zeiss Axioplan 2 microscope. Six biological replicate tissue sections per group were analyzed for control and Mybpc3−/− animals. A minimum of 100 PCM1‐positive nuclei per biological replicate were analyzed for chromosome 8 centromere probe foci. Nuclei were classified as either diploid (2 foci) or aneuploid (3 or 5 foci).
Telomere Fluorescence in Situ Hybridization Assay
The telomere fluorescence in situ hybridization (FISH) assay was performed according to manufacturer's instructions (PNA Bio). Briefly, heart tissue sections (5 µm) were fixed with 4% paraformaldehyde, washed with PBS, and permeabilized with 0.2% Triton X‐100 in PBS, with subsequent washes. The slides and hybridization buffer (20 mmmol/L Tris and 60% formamide in distilled water) were prewarmed at 85°C for 5 minutes. The PNA probe (PNA Bio, F1002) was diluted in hybridization buffer (1:250) and added to the slide, covered with a plastic coverslip, and incubated at 85°C for 10 minutes, followed by 2 hours at room temperature in the dark. After removing the coverslip carefully, slides were washed (2× standard saline citrate, 0.1% Tween‐20) twice for 10 minutes at 60°C and 5 minutes at room temperature. The slides were then blocked with 1% BSA in PBS for 1 hour at room temperature and incubated overnight at 4°C with γH2AX antibody (1:500) in blocking buffer. The next day, slides were washed with PBS 3 times for 5 minutes and incubated with fluorescent secondary goat anti‐mouse Alexa Fluor 488 (1:500) for 1 hour at room temperature in the dark. Slides were then washed in PBS 3 times for 5 minutes, coverslips were mounted with Prolong Gold Antifade with DAPI, and images were taken with Zeiss Axioplan 2 fluorescent microscope.
Terminal Deoxynucleotidyl Transferase–Mediated Biotin–Deoxyuridine Triphosphate Nick‐End Labeling Assay
For in situ apoptosis detection, Click‐iT Plus TUNEL Assay (Thermo Fisher Scientific, C10617) protocol was followed, as per manufacturer's instructions. Briefly, 5‐µm myocardial tissue sections were fixed with 4% paraformaldehyde, washed with PBS, and incubated with permeabilization reagent (Proteinase K solution) for 15 minutes. The slides were washed twice with 1× PBS and incubated again with 4% paraformaldehyde for 5 minutes at 37°C. For positive control, sections were incubated with DNase I (1 unit in DNase I reaction buffer) (New England Biolabs, M0303) for 30 minutes at room temperature, and rinsed by deionized water. After washing twice in PBS for 5 minutes and rinsing with deionized water, slides were incubated with TdT reaction buffer (100 µL) for 10 minutes at 37°C. Slides were then incubated with prepared TdT reaction mixture (50 µL) and incubated for 60 minutes at 37°C with plastic coverslip. Following rinsing with deionized water, slides were washed with 3% BSA and 0.1% Triton X‐100 in PBS for 5 minutes. Immediately after rinsing the slides with PBS, Click‐iT Plus terminal deoxynucleotidyl transferase–mediated biotin–deoxyuridine triphosphate nick‐end labeling reaction cocktail (50 µL) was added to each slide and incubated for 30 minutes at 37°C in the dark. Slides were then washed with 3% BSA in PBS for 5 minutes, rinsed with PBS, and then blocked with 1% BSA in PBS for 1 hour at room temperature in the dark. Sections were incubated overnight at 4°C with sarcomeric α‐actinin antibody. On the following day, sections were washed 3 times for 5 minutes and incubated with secondary goat anti‐mouse Alexa Fluor 594 (1:500) for 1 hour at room temperature in the dark. After final washes with PBS, slides were mounted with Prolong Gold Antifade with DAPI, and images were taken with a Zeiss Axioplan 2 fluorescent microscope.
8‐Hydroxy‐2'‐Deoxyguanosine Measurement
Genomic DNA was extracted from snap‐frozen P7 myocardial tissue, according to commercially available DNA extraction kit (Qiagen, 69504). Extracted DNA was treated with RNase (Thermo Fisher Scientific, EN0531) and quantified using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). A total of 2 μg of DNA was incubated at 95°C for 10 minutes, followed by rapid chilling on ice to produce single‐stranded DNA. Samples were then digested with Nuclease P1 (NEB, M0660S) at 10 units per 1 μg of DNA for 2 hours at 37°C. The digested DNA was incubated with 5 units of alkaline phosphatase (Sigma, P5931) in 100 mmol/L Tris, pH 7.5, at 37°C for 1 hour. Samples were centrifuged at 6000g for 5 minutes, supernatant was collected, and 8‐hydroxy‐2'‐deoxyguanosine levels were measured with an 8‐hydroxy‐2'‐deoxyguanosine DNA Damage Competitive ELISA Kit (Thermo Fisher Scientific, EIADNAD). The observed 8‐hydroxy‐2'‐deoxyguanosine levels were detected within the linear range of the assay standard curve.
Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was extracted from mouse myocardial tissue and then reverse transcription was performed to generate first‐strand cDNA, according to manufacturer's protocol (QuantiTect Reverse Transcription Kit; Qiagen). To perform gene expression analysis, quantitative reverse transcription–polymerase chain reactions were set up in a Quant‐Studio‐5 Real‐Time PCR System (Applied Biosystems) using SYBR Green Reagent (Thermo Fisher Scientific) and gene‐specific primers (Table S1). Each sample was run in duplicate, and the cycle threshold (Ct) value was normalized using the endogenous housekeeping gene Rpl32. The 2–ΔΔCt method was used to calculate the fold change in mRNA expression relative to indicated group in figure legends. Results with Ct values >35 were considered undetectable.
Protein Electrophoresis and Western Blot
Hearts were homogenized in ice‐cold radioimmunoprecipitation assay buffer (Sigma) containing protease/phosphatase inhibitors (Thermo Fisher Scientific, 78441). Proteins were resolved through SDS‐PAGE using 10% or 4% to 20% Tris‐glycine gel (Bio‐Rad) and transferred to a 0.45‐μm low‐fluorescence polyvinylidene difluoride membrane (Bio‐Rad) at 100 V for 60 minutes at 4°C. Membranes were subsequently blocked with 5% BSA or nonfat dry milk in tris‐buffered saline and Tween 20 or intercept blocking buffer (Licor Biotech), followed by incubation with specific primary antibodies targeting p‐Ser428 ATR (CST, 2853), ATR (CST, 13934), p‐Ser1981ATM (R&D Systems, AF1655), ataxia telangiectasia mutated (ATM) (SCBT, sc‐377293), γH2AX (Bethyl, A300‐081A‐T), p53 (CST, 2524), and β‐actin (CST, 8457) overnight at 4°C. Membranes were then washed with tris‐buffered saline and Tween 20 and subsequently incubated with goat anti‐mouse horseradish peroxidase (SCBT, sc‐516102) or goat anti‐rabbit horseradish peroxidase (CST, 7074) for 1 hour, and images were recorded in Chemi Doc apparatus (Bio‐Rad) using Clarity ECL substrate (Bio‐Rad, 1705061). For fluorescent Western blot imaging, goat anti‐rabbit (IRDye 800 CW) was used, and imaging was then performed on an Odyssey CLx imaging system (Licor Biotech). To quantify band intensities, digital images were analyzed using with Image Studio Lite (Licor Biotech) or Image J Software.
Human Hypertrophic Cardiomyopathy Tissue Analysis
Human control non–hypertrophic cardiomyopathy (HCM) LV septal myocardial tissue and HCM LV septal myocardial tissue samples (Table S2) were obtained in a deidentified manner from an institutional review board–approved tissue biorepository. Cardiomyocyte nuclear γH2AX staining and aneuploidy methods were performed in a similar manner to murine myocardial tissue, except a human‐specific chromosome 8 probe was used (VividFISH CEP‐8 Geneocopoeia catalog No. FP036).
Quantification and Statistical Analysis
All data are displayed as mean±SEM, unless otherwise noted. Male and female samples were grouped together unless otherwise noted. Sample size and power were calculated if estimated means and SDs were available before a given experiment. Data were analyzed using an unpaired, 2‐tailed Student t‐test for comparisons between 2 groups or 1‐way ANOVA with Tukey‐Kramer multiple‐comparison test when multiple testing was performed. Linear regression analysis was performed, and Pearson correlation coefficient (r) was calculated. All analysis was performed in GraphPad Prism 9. All P values are shown in the figures, and significance was defined as P<0.05.
RESULTS
Increased Cardiomyocyte DNA Damage Occurs During the Early Phases of Mybpc3−/− Cardiomyopathy
We previously discovered dysregulated cardiomyocyte cell cycle activity and endoreplication in cardiomyocytes in an Mybpc3−/− sarcomeric cardiomyopathy model.4 Because cellular endoreplication can be associated with genotoxic stress,12 we hypothesized that cardiomyocytes in our sarcomeric cardiomyopathy model may have increased DNA damage. Therefore, we first measured phosphorylation of the histone variant H2AX at serine 139 (γH2AX), which serves as a sensitive marker of DNA damage.13 We observed a robust increase in γH2AX levels in Mybpc3−/− myocardial tissue lysate at P7 compared with control myocardial tissue lysate (Figure 1A and 1B). Immunohistochemistry of control and Mybpc3−/− LV tissue showed that DNA damage was localized to the nucleus of cardiomyocytes and was not induced in noncardiomyocyte cell populations (Figure 1C and 1D and Figure S1A). In addition, we also discovered that the peak induction of LV cardiomyocyte nuclear DNA damage occurred at P7 and was significantly decreased by P25 and P180 (Figure 1E). Male and female Mybpc3−/− mice had similar increases in LV cardiomyocyte γH2AX staining (Figure S1B). In contrast to Mybpc3−/− mice that rapidly develop LV hypertrophy, Mybpc3+/− heterozygotes do not rapidly develop LV hypertrophy and do not have evidence of increased cardiomyocyte DNA damage compared with normal control cardiomyocytes at P7 or P25 (Figure S1C). In addition to measuring total nuclear γH2AX fluorescence, we also measured LV cardiomyocyte γH2AX foci formation,14 and this confirmed that there was an increased level of cardiomyocyte DNA damage in Mybpc3−/− LV tissue (Figure 1F and 1G). We also measured colocalization of 53BP1 and γH2AX,15 and found that Mybpc3−/− myocardial tissue had increased colocalization of these 2 DNA damage repair proteins (Figure 1H and 1I). To determine if the DNA damage localized to the telomeres, we performed dual staining for γH2AX and a telomere‐specific probe. However, γH2AX foci did not colocalize to the telomeres in control or Mybpc3−/− myocardial tissue (Figure 1J and 1K). We then directly measured DNA damage using a comet assay and confirmed that Mybpc3−/− nuclei had a significant increase in DNA breaks compared with control nuclei (Figure 1L and 1M). These results show that, in the earliest stages of Mybpc3−/− sarcomeric cardiomyopathy, there is increased LV cardiomyocyte nuclear DNA damage.
Figure 1. Increased cardiomyocyte DNA damage in Mybpc3−/− cardiomyopathy.
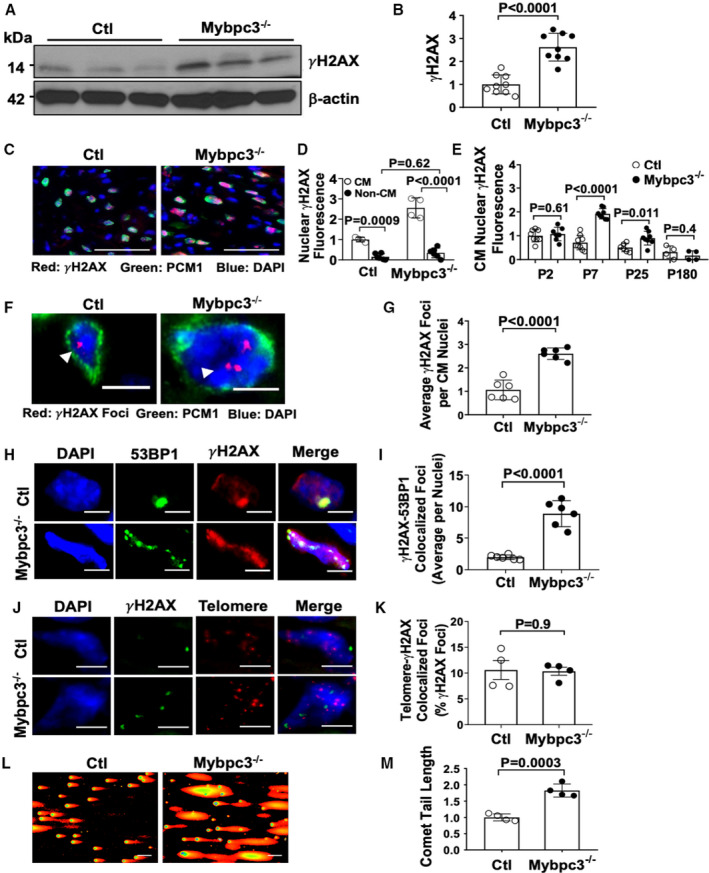
(A) γH2AX Western blot from postnatal day (P) 7 control (Ctl) and Mybpc3−/− myocardial tissue. (B) Relative quantification γH2AX in Ctl (n=6) and Mybpc3−/− (n=6) myocardial tissue at P7 normalized to β‐actin. (C) Representative immunofluorescence staining of γH2AX (red) in Ctl and Mybpc3−/− left ventricular (LV) tissue at P7. Cardiomyocyte (CM) nuclei were identified with pericentriolar material 1 (PCM1) (green), and nuclei were labeled by 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue). Bars=100 µm. (D) Comparison of γH2AX staining between CM and noncardiomyocyte (Non‐CM) nuclei in Ctl (n=4–6) and Mybpc3−/− (n=4–6) LV tissue at P7. Minimum 100 nuclei/sample. (E) Relative quantification of γH2AX fluorescence in CM nuclei in Ctl (n=6–9) and Mybpc3−/− (n=7–8) LV tissue at P2, P7, P25, and P180. Minimum 50 nuclei/sample. (F) Representative γH2AX foci (red, arrowhead) immunofluorescence staining with CM‐specific PCM1 (green) and nuclei labeled by DAPI (blue) in Ctl and Mybpc3−/− LV tissue at P7. Bar=5 µm. (G) Quantitation of average γH2AX foci per CM nuclei in Ctl (n=6) and Mybpc3−/− (n=6) LV tissue. Minimum 50 nuclei/sample. (H) Representative immunofluorescence staining of 53BP1 (p53‐binding protein 1) (green) and γH2AX (red) and nuclei labeled with DAPI (blue) in Ctl and Mybpc3−/− myocardial tissue at P7. Bar=5 µm. (I) Average γH2AX‐53BP1 colocalized foci per nuclei in Ctl (n=6) and Mybpc3−/− (n=6) LV tissue. Minimum 50 nuclei/sample. (J) Telomere peptide nucleic acid (PNA) fluorescence in situ hybridization assay from Ctl or Mybpc3−/− mice at P7 with γH2AX (green), telomere (red), and nuclei DAPI (blue) staining. Bar=5 µm. (K) Percentage of telomere‐γH2AX colocalization foci per nuclei in Ctl (n=4) and Mybpc3−/− (n=4) LV tissue was quantified. Minimum 100 nuclei/sample. (L) Neutral comet assay of nuclei from Ctl or Mybpc3−/− myocardial tissue at P7. (M) Relative quantitation of comet tail length of nuclei from Ctl (n=4) and Mybpc3−/− (n=4) myocardial tissue. Minimum 100 nuclei/sample. All results are shown as mean±SEM.
Selective Activation of the DNA Damage Response Pathways Occurs in Mybpc3−/− Cardiomyopathy
Given the robust induction of DNA damage in Mybpc3−/− cardiomyocytes, we determined whether DNA damage‐related kinases ATR and ATM underwent phosphorylation.16, 17 We found a significant increase in p‐Ser428 ATR but no increase in p‐Ser1981 ATM in Mybpc3−/− myocardial tissue at P7 (Figure 2A and 2B). We then measured the DNA damage response transcriptional regulator, p53, and discovered p53 protein was increased in Mybpc3−/− myocardial tissue (Figure 2C and 2D). Although p53 protein was elevated, TP53 gene expression was not increased in Mybpc3−/− myocardial tissue (Figure 2E), suggesting the elevated p53 protein levels are secondary to increased p53 protein stability in response to DNA damage not secondary to increased transcription.18 Activation of p53 in cells with DNA damage may lead to apoptosis.19 However, there were no detectable increases in myocardial apoptosis in our Mybpc3−/− myocardial tissue (Figure 2F). Overall, these results show that, in the Mybpc3−/− cardiomyopathy model, there is a selective activation of ATR kinase with a subsequent surge in p53 protein levels without induction of myocardial apoptosis.
Figure 2. Mybpc3−/− cardiomyopathy leads to selective activation of DNA damage response pathways.
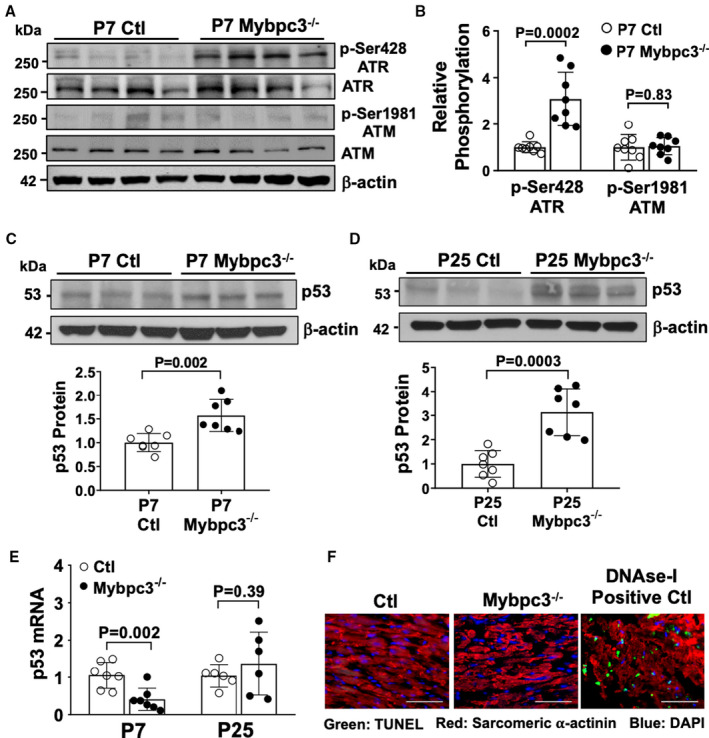
(A) Western blot of phosphorylated ataxia telangiectasia and rad3 related (ATR) (p‐Ser428 ATR), total ATR, phosphorylated ataxia telangiectasia mutated –(ATM) (p‐Ser1981 ATM), and total ATM from postnatal day (P) 7 control (Ctl) and Mybpc3−/− myocardial tissue. β‐Actin was the loading control. (B) Relative quantification of p‐Ser428 ATR and p‐Ser1981 ATM in Ctl (n=8) and Mybpc3−/− (n=8) myocardial tissue normalized to total ATR and total ATM, respectively. (C) Western blot of p53 in Ctl and Mybpc3−/− myocardial tissue at P7 (top). Relative quantification of p53 in P7 Ctl (n=7) and Mybpc3−/− (n=7) myocardial tissue normalized to β‐actin (bottom). (D) Western blot of p53 in Ctl and Mybpc3−/− myocardial tissue at P25 (top). Relative quantification of p53 in P25 Ctl (n=7) and Mybpc3−/− (n=7) myocardial tissue normalized to β‐actin (bottom). (E) Measurement of p53 gene expression from Ctl and Mybpc3−/− myocardial tissue at P7 (n=7) and P25 (n=6). The genes of interest were normalized to Rpl32 expression. Fold changes are shown relative to Ctl gene expression. (F) Representative terminal deoxynucleotidyl transferase–mediated biotin–deoxyuridine triphosphate nick‐end labeling (TUNEL) assay images from Ctl and Mybpc3−/− mouse left ventricular (LV) tissue section stained with TUNEL (green), α‐actinin (red), and 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue) staining. DNase I–treated control LV tissue section was used as positive control for the assay (DNAse I Positive Ctl). Bar=50 µm. All results are shown as mean±SEM.
DNA Damage in Mybpc3−/− Cardiomyopathy Is Secondary to Replication Stress
We previously reported significant alterations in cell cycle pathways and DNA synthesis in Mybpc3−/− cardiomyopathy.4 In the current study, we detected increased nuclear DNA damage and a selective increase in ATR kinase phosphorylation (Figure 2). We measured if nuclei with active DNA synthesis had evidence of DNA damage and confirmed that Mybpc3−/− LV myocardial tissue had a substantial increase of nuclei with active DNA synthesis and DNA damage (Figure 3A and 3B). Therefore, we hypothesized that replication stress was the cause of the increased DNA damage in Mybpc3−/− cardiomyocytes. To test this hypothesis, we inhibited the cardiomyocyte cell cycle in vivo with the selective CDK4/6 inhibitor (PD‐0332991), which we previously established could block excess Mybpc3−/− cardiomyocyte DNA synthesis.4 We found that administration of the CDK4/6 inhibitor to Mybpc3−/− mice could reduce cardiomyocyte DNA damage to control levels (Figure 3C and 3D). More important, CDK4/6 inhibition also reduced ATR phosphorylation in Mybpc3−/− myocardial tissue (Figure 3E and 3F). Oxidative stress is also a common cause of DNA damage and has previously been shown to be involved in pressure overload cardiomyocyte DNA damage.20, 21 However, we detected no difference in the oxidative DNA damage marker, 8‐hydroxy‐2'‐deoxyguanosine, between control and Mybpc3−/− myocardial tissue (Figure 3G). These results confirmed that the increased nuclear DNA damage and selective phosphorylation of ATR in Mybpc3−/− cardiomyocytes was secondary to replication stress.
Figure 3. DNA damage in Mybpc3−/− cardiomyopathy is secondary to replication stress.
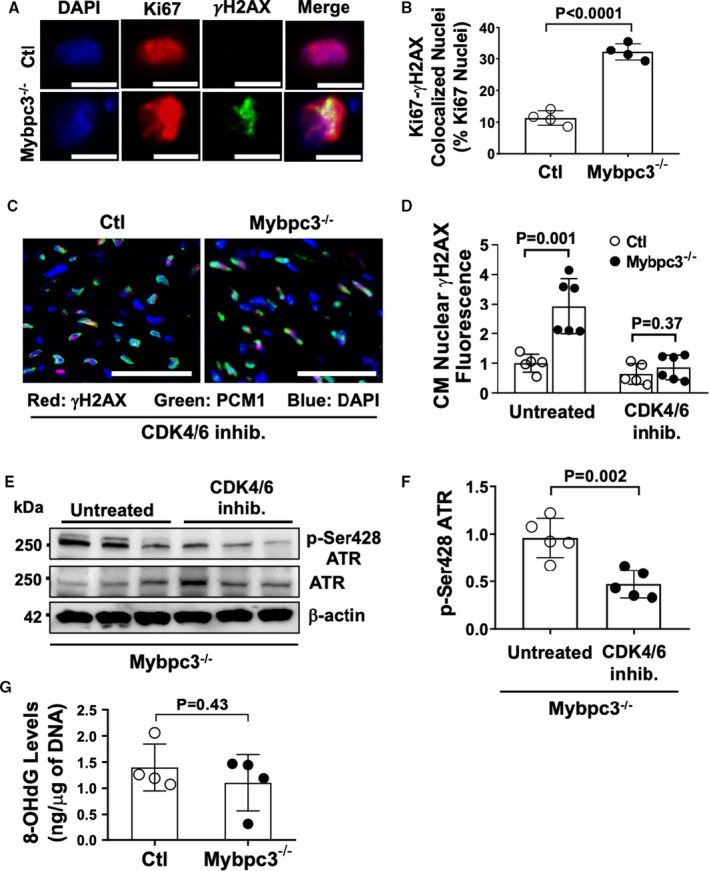
(A) Representative immunofluorescence staining of Ki67 (red) and γH2AX (green) and nuclei labeled with 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue) in control (Ctl) and Mybpc3−/− left ventricular (LV) tissue at postnatal day (P) 7. Bar=5 µm. (B) Percentage of Ki67‐γH2AX colocalized nuclei per total Ki67‐positive nuclei in Ctl (n=4) and Mybpc3−/− (n=4) LV tissue. (C) Representative immunofluorescence staining of cardiomyocyte γH2AX in Ctl and Mybpc3−/− LV tissue from mice administered the cyclin‐dependent kinase (CDK) 4/6 inhibitor (inhib.) PD‐0332991 (150 mg/kg per day). DNA damage marker γH2AX (red), cardiomyocyte (CM) marker pericentriolar material 1 (PCM1) (green), and nuclei marker DAPI (blue). Bar=100 µm. (D) Quantification of γH2AX CM nuclei in Ctl (n=5–6) and Mybpc3−/− (n=5–6) LV tissue from untreated mice or from mice administered with CDK4/6 inhib. Minimum of 100 CM nuclei/sample. (E) Western blot of phosphorylated ataxia telangiectasia and rad3 related (ATR) (p‐Ser428 ATR) and total ATR in P7 Mybpc3−/− myocardial tissue from untreated or CDK4/6 inhib. treated mice. (F) Relative quantification of p‐Ser428 ATR from untreated or CDK4/6 inhib. groups normalized to total ATR (n=5). (G) Measurement of 8‐hydroxy‐2'‐deoxyguanosine (8‐OHdG) levels (ng/µg of DNA) through competitive ELISA from P7 Ctl (n=4) and Mybpc3−/− (n=4) myocardial tissue. All results are shown as mean±SEM.
Inhibition of ATR Kinase Reduces Pathological Ventricular Remodeling in Mybpc3−/− Cardiomyopathy
We identified a role for dysregulated cell cycle activity causing replication stress DNA damage and selective activation of ATR in Mybpc3−/− cardiomyocytes. Therefore, we wanted to determine if ATR activation was involved in the pathological LV remodeling in Mybpc3−/− cardiomyopathy. We used the highly selective ATR kinase inhibitor,22 AZD6738, to inhibit ATR during the early stage of Mybpc3−/− cardiomyopathy formation (Figure 4A). Inhibition of ATR caused a reduction of LV myocardial hypertrophy (Figure 4B through 4D). Despite the reduction in LV hypertrophy, ATR inhibition did not increase LV dilation (LVIDd) in Mybpc3−/− animals (Figure 4E), and there was an increase in LV systolic function in Mybpc3−/− animals (Figure 4F). This suggests that the reduction in LV hypertrophy was not from ATR inhibition causing cardiotoxicity or a transition to a dilated cardiomyopathy phenotype. Similar to the reduction in LV myocardial hypertrophy, ATR inhibition led to a decrease in myocardial mass in Mybpc3−/− without causing a change in overall body weight (Figure 4G and 4H and Figure S2A). In addition, ATR inhibition did not have any effect on wild‐type C57BL/6J heart structure or function during this time period. We then measured cardiomyocyte cross‐sectional area in Mybpc3−/− mice treated with AZD6738 compared with vehicle and confirmed that LV cardiomyocyte size was decreased by ATR inhibition (Figure 4I and 4J).
Figure 4. Inhibition of ataxia telangiectasia and rad3‐related (ATR) kinase reduces pathological ventricular remodeling in Mybpc3−/− cardiomyopathy.
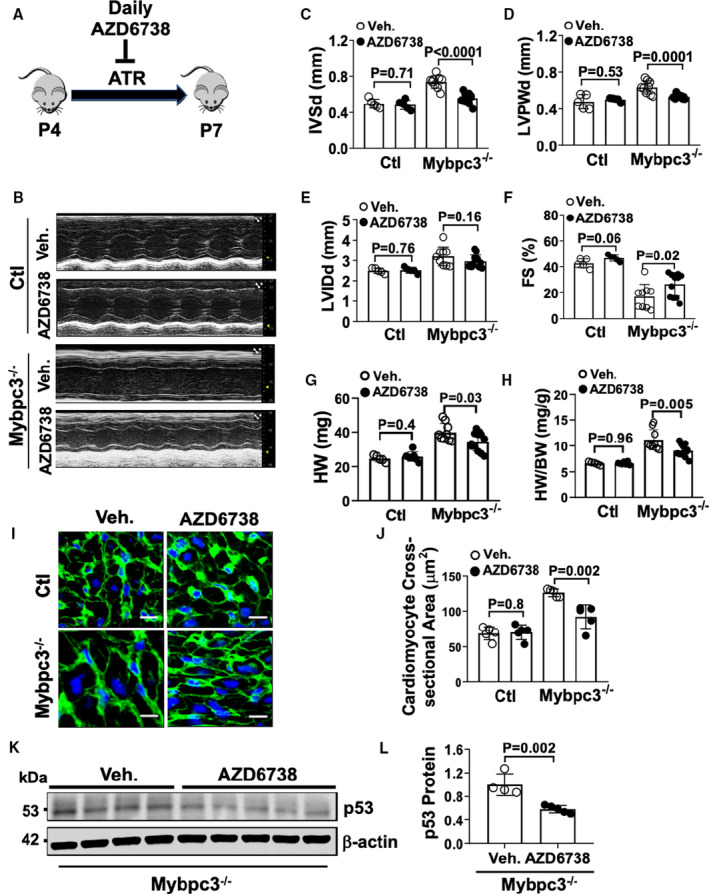
(A) Schematic illustration of ATR kinase inhibition from postnatal day (P) 4 to P7 with the drug AZD6738. (B) M‐mode echocardiography at P7 from control (Ctl) and Mybpc3−/− mice administered vehicle (Veh.) or 25 mg/kg per day AZD6738. Echocardiography assessment of interventricular septal thickness at end diastole (IVSd) (C), left ventricular posterior wall thickness at end diastole (LVPWd) (D), left ventricular internal diameter at end diastole (LVIDd) (E), and fractional shortening (FS) (F) in Ctl (n=5–7) and Mybpc3−/− (n=9–12) mice administered Veh. or AZD6738. Heart weight (HW) (G) and HW/body weight (BW) ratio (H) for Ctl (n=5–7) and Mybpc3−/− (n=9–12) mice administered Veh. or AZD6738. (I) Representative immunohistochemical staining with wheat‐germ agglutinin (green) and 4′,6‐diamidino‐2‐phenylindole (blue) of left ventricular (LV) tissue from Ctl and Mybpc3−/− mice administered Veh. or AZD6738. Bar=10 µm. (J) LV cross‐sectional area of Ctl (n=5) and Mybpc3−/− (n=5) mice administered Veh. or AZD6738. Minimum 50 cells/sample measured. (K) Western blot of p53 in myocardial tissue lysate from Mybpc3−/− administered Veh. or AZD6738. (L) Relative quantification of p53 protein expression from Mybpc3−/− administered Veh. (n=4) or AZD6738 (n=5) normalized to β‐actin. All results are shown as mean±SEM.
We previously found that Mybpc3−/− myocardial tissue had increased p53 protein levels secondary to increased stabilization (Figure 2C through 2E) and ATR kinase can increase p53 protein stability in DNA damage.23 Therefore, we measured p53 protein levels in AZD6738 and vehicle‐treated Mybpc3−/− mice and discovered that ATR inhibition led to a reduction p53 protein levels in Mybpc3−/− myocardial tissue (Figure 4K and 4L). This suggests that increased ATR kinase activity contributes to the increased p53 protein stabilization in the Mybpc3−/− myocardial tissue.
Deletion of Cardiomyocyte p53 Reduces Pathological Myocardial Remodeling in Mybpc3−/− Cardiomyopathy
Inhibition of ATR function reduced pathological remodeling during the earliest stages of Mybpc3−/− cardiomyopathy, and our data suggested that ATR was contributing to the increased levels of cardiomyocyte p53. Therefore, we hypothesized that increased cardiomyocyte p53 levels contributed to pathological remodeling in Mybpc3−/− cardiomyopathy. We created Mybpc3−/−/p53fl/flMyh6Cre+/− double‐null animals to allow the genetic elimination of cardiomyocyte p53 (Figure 5A) and verified this strategy was successful (Figure 5B). Next, we assessed the expression of multiple p53 target genes (Mdm2, Cdkn1a, Gdf15, and Gadd45a) and confirmed these genes were upregulated in Mybpc3−/− myocardial tissue (Figure 5C). More important, the induction of these genes was completely prevented by deletion of cardiomyocyte p53 in Mybpc3−/− animals. We then examined if the elimination of cardiomyocyte p53 in Mybpc3−/− had any effects on LV structure or function. We found that elimination of p53 protein expression led to a significant but modest decrease in LV myocardial hypertrophy (Figure 5D and 5E), with no change in LV dilation (LVIDd) (Figure 5F) in Mybpc3−/− animals at P25. In addition, cardiomyocyte deletion of p53 also increased systolic function compared with Mybpc3−/− animals (Figure 5G). Likewise, p53 deletion led to a reduction in myocardial mass in Mybpc3−/− at P25 (Figure 5H and 5I and Figure 3A). Elimination of p53 in control C57BL/6J cardiomyocytes (p53fl/flMyh6Cre+/−) had no significant effect on cardiac structure or systolic function at P25 (Figure 5D through 5I). In addition, the cardiomyocyte expression of Cre alone in the Mybpc3−/− model (Mybpc3−/−/Myh6Cre+/−) had no significant effect on cardiac structure or function at P25 (Figure 3B through 3E). At the cellular level, we found that elimination of cardiomyocyte p53 led to a modest reduction in cardiomyocyte hypertrophy (Figure 5J and 5K) that paralleled the changes detected in myocardial wall thickness and overall heart mass. Taken together, the data from our ATR inhibition and cardiomyocyte p53 experiments suggest that activation of DNA damage response pathways in Mybpc3−/− cardiomyopathy actively modify pathological ventricular remodeling.
Figure 5. Deletion of cardiomyocyte p53 reduces pathological myocardial remodeling in Mybpc3−/− cardiomyopathy.
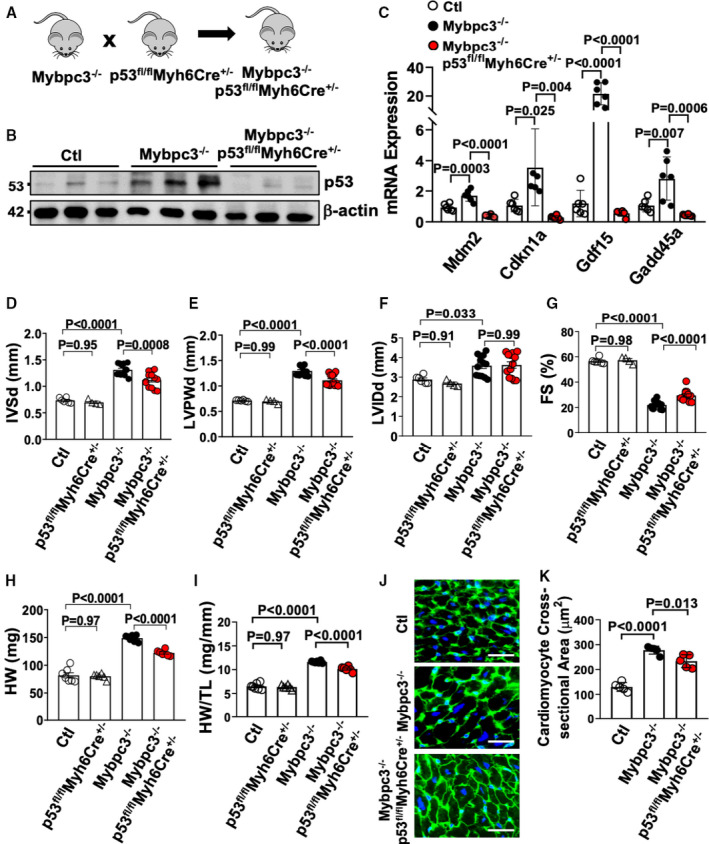
(A) Schematic of Mybpc3−/−/p53fl/flMyh6Cre+/− double‐null murine model generated by crossing a p53fl/flMyh6Cre+/− mouse with an Mybpc3−/− mouse. (B) Western blot of p53 from control (Ctl) (n=3), Mybpc3−/− (n=3), and Mybpc3−/−/p53fl/flMyh6Cre+/− (n=3) myocardial tissue at postnatal day (P) 25. Western blot of β‐actin used as loading control. (C) Measurement of p53 target gene expression (mouse double minute 2 [Mdm2], cyclin‐dependent kinase inhibitor 1 [Cdnk1a], growth differentiation factor 15 [Gdf15], and growth arrest and DNA damage inducible α [Gadd45a]) at P25 in Ctl (n=6), Mybpc3−/− (n=6), and Mybpc3−/−/p53fl/flMyh6Cre+/− (n=6) left ventricular (LV) tissue RNA. The genes of interest were normalized to Rpl32 expression. Fold changes are shown relative to Ctl gene expression. Echocardiography assessment of interventricular septal thickness at end diastole (IVSd) (D), LV posterior wall thickness at end diastole (LVPWd) (E), LV internal diameter at end diastole (LVIDd) (F), and fractional shortening (FS) (G) in Ctl (n=6), p53fl/flMyh6Cre+/− (n=4), Mybpc3−/− (n=13), and Mybpc3−/−/p53fl/flMyh6Cre+/− (n=13) mice at P25. Heart weight (HW) (H) and HW/tibia length (TL) ratio (I) from Ctl (n=7), p53fl/flMyh6Cre+/− (n=6), Mybpc3−/− (n=7), and Mybpc3−/−/p53fl/flMyh6Cre+/− (n=6) mice at P25. (J) Representative immunohistochemical staining using wheat‐germ agglutinin (green) and 4′,6‐diamidino‐2‐phenylindole (blue) of LV tissue from Ctl, Mybpc3−/−, and Mybpc3−/−/p53fl/flMyh6Cre+/− mice. Bar=10 µm. (K) LV cardiomyocyte cross‐sectional area from Ctl (n=5), Mybpc3−/− (n=5), and Mybpc3−/−/p53fl/flMyh6Cre+/− (n=5) mice. Minimum 50 cells/sample measured. All results are shown as mean±SEM.
Cardiomyocyte DNA Damage and ATR Phosphorylation Are a Conserved Response in Other Genetic Forms of Sarcomeric Cardiomyopathy
We next wanted to determine if there was evidence of cardiomyocyte DNA damage in a sarcomeric cardiomyopathy model caused by a non‐Mybpc3 pathway. We used a mouse model that overexpresses a mutant form of human cardiac troponin T (TNNTI79N), which has been identified in multiple human families with sarcomeric cardiomyopathies.9, 24, 25 Similar to the Mybpc3−/− model, we observed an increase in cardiomyocyte nuclear DNA damage in TNNT2I79N cardiomyocytes compared with a control mouse line that overexpresses human wild‐type cardiac troponin T (TNNT2WT), and the DNA damage peaked at P7 (Figure 6A and 6B). Similar to earlier, noncardiomyocyte cell populations did not show increased γH2AX staining (Figure 6C). Likewise, we confirmed that the transgenic wild‐type cardiomyocytes (TNNT2WT) did not have evidence of increased DNA damage compared with normal control cardiomyocytes (Figure S4A). In contrast to the Mybpc3−/− model, the TNNT2I79N mouse does not develop myocardial hypertrophy at P7 (Figure S4B). Therefore, this experiment allows us to verify that the increased nuclear DNA damage in TNNT2I79N cardiomyocytes is secondary to sarcomere dysfunction and not caused as response to cardiomyocyte hypertrophy.
Figure 6. Cardiomyocyte DNA damage and ataxia telangiectasia and rad3‐related (ATR) phosphorylation are a conserved response in other genetic forms of sarcomeric cardiomyopathy.
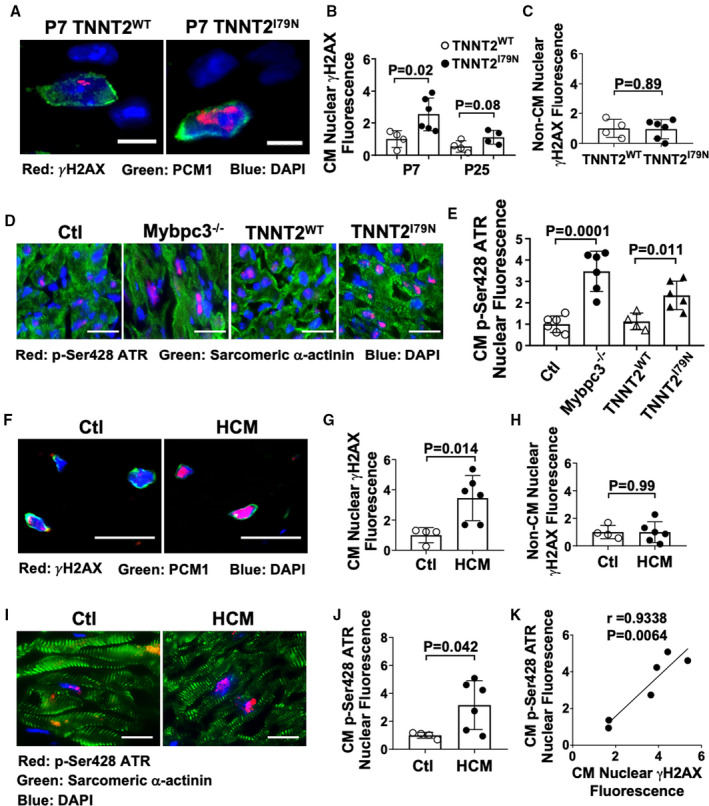
(A) Representative immunofluorescence staining of γH2AX (red) in transgenic mice expressing either wild‐type human cardiac troponin T (TNNT2WT) or mutant human cardiac troponin T (TNNT2I79N) at postnatal day (P) 7. Cardiomyocyte nuclei were identified with pericentriolar material 1 (PCM1) (green), and nuclei were labeled by 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue). Bar=10 µm. (B) Relative quantification of γH2AX fluorescence in cardiomyocyte (CM) nuclei from TNNT2WT (n=4) and TNNT2I79N (n=4–6) left ventricular (LV) tissue at P7 and P25. Minimum 50 nuclei/sample. (C) Relative quantitation of γH2AX staining in noncardiomyocyte (Non‐CM) nuclei in TNNTWT (n=4) and TNNTI79N (n=6) LV tissue at P7. Minimum 50 nuclei/sample. (D) Representative immunofluorescence staining of phosphorylated ATR (p‐Ser428 ATR) (red) in Ctl, Mybpc3−/−, TNNTWT, and TNNTI79N LV tissue at P7. CMs were identified with sarcomeric α‐actinin (green), and nuclei were labeled by DAPI (blue). Bar=50 µm. (E) Relative quantification of p‐Ser428 ATR fluorescence in CM nuclei in Ctl (n=6), Mybpc3−/− (n=6), TNNTWT (n=4), and TNNTI79N (n=4–6) LV tissue at P7. Minimum 50 nuclei/sample. (F) Representative immunofluorescence of γH2AX (red) in explanted non–hypertrophic cardiomyopathy (HCM) control (Ctl) or HCM patient LV septal tissue. CM nuclei were identified with PCM1 (green), with nuclei labeled with DAPI (blue). Bar=25 μm. (G) Relative quantification of γH2AX fluorescence in CM nuclei in Ctl (n=4) and HCM (n=6) LV tissue. A minimum 100 CM nuclei/sample. (H) Relative quantitation of γH2AX staining in Non‐CM nuclei in Ctl (n=4) and HCM (n=6) LV tissue. A minimum 100 PCM+ and PCM– nuclei/sample were analyzed. (I) Representative immunofluorescence staining of phosphorylated ATR (red) in Ctl and HCM LV tissue. CMs were identified with sarcomeric α‐actinin (green), with nuclei labeled by DAPI (blue). Bar=50 µm. (J) Relative quantification of phosphorylated ATR fluorescence in CM nuclei in Ctl (n=4) and HCM (n=6) LV tissue. Minimum 50 nuclei/sample. (K) Linear regression analysis of the relationship between CM nuclear γH2AX and p‐Ser428 ATR fluorescence in human HCM LV tissue samples. All results are shown as mean±SEM.
Earlier, we detected a robust increase in ATR phosphorylation in Mybpc3−/− myocardial tissue (Figure 2A and 2B). Nuclear DNA damage should induce the phosphorylation of ATR in the nucleus. Therefore, we confirmed that Mybpc3−/− cardiomyocytes had increased nuclear concentrations of phosphorylated ATR compared with control cardiomyocytes (Figure 6D and 6E). Likewise, TNNT2I79N cardiomyocytes had increased nuclear concentrations of phosphorylated ATR compared with TNNT2WT cardiomyocytes (Figure 6D and 6E). We also confirmed that TNNT2I79N myocardial tissue had increased p53 protein compared with TNNT2WT tissue at both P7 and P25 (Figure S4C and S4D).
Finally, we evaluated septal myocardial tissue from human patients with HCM and septal myocardial tissue from control patients without HCM (Table S2). Similar to our murine models, cardiomyocyte nuclear DNA damage was increased in HCM compared with control cardiomyocytes (Figure 6F through 6H and Figure S4C). Likewise, we found that the ATR phosphorylation was also increased in HCM cardiomyocytes compared with control cardiomyocytes (Figure 6I and 6J). We then confirmed that there was a strong correlation between the level of nuclear γH2AX and ATR phosphorylation in human HCM cardiomyocytes (R=0.9338) (Figure 6K). Collectively, these results show that cardiomyocyte DNA damage and ATR phosphorylation are a conserved response across different genetic causes of sarcomeric cardiomyopathy.
Sarcomeric Cardiomyopathy Is Associated With Increased Cardiomyocyte Aneuploidy
Replication stress DNA damage has been linked to genomic instability, specifically chromosomal instability and aneuploidy.26, 27, 28 Therefore, we hypothesized that replication stress DNA damage in sarcomeric cardiomyopathy may lead to cardiomyocyte aneuploidy and lasting changes in the cardiomyocyte genome. We performed FISH of cardiomyocyte and noncardiomyocyte nuclei using a chromosome 8–specific probe and the cardiomyocyte‐specific marker PCM1. The FISH methanol–acetic acid fixation alters the nuclear staining pattern of PCM1, and we confirmed that PCM1 staining with methanol–acetic acid fixation remains specific for cardiomyocytes (Figure S5A). Supporting our hypothesis, we observed a significant increase in cardiomyocyte aneuploidy in Mybpc3−/− LV tissue compared with control (Figure 7A and 7B). In contrast, we detected no evidence of aneuploidy in noncardiomyocyte nuclei (Figure 7C and Figure S5B). Likewise, TNNT2I79N cardiomyocytes had increased aneuploidy compared with control TNNT2WT cardiomyocytes (Figure 7D through 7F). Finally, we evaluated human HCM myocardial tissue and again found evidence of increased cardiomyocyte aneuploidy (Figure 7G through 7I). Collectively, these results show that multiple different genetic forms of sarcomeric cardiomyopathy have increased levels of cardiomyocyte aneuploidy.
Figure 7. Sarcomeric cardiomyopathy is associated with increased cardiomyocyte (CM) aneuploidy.
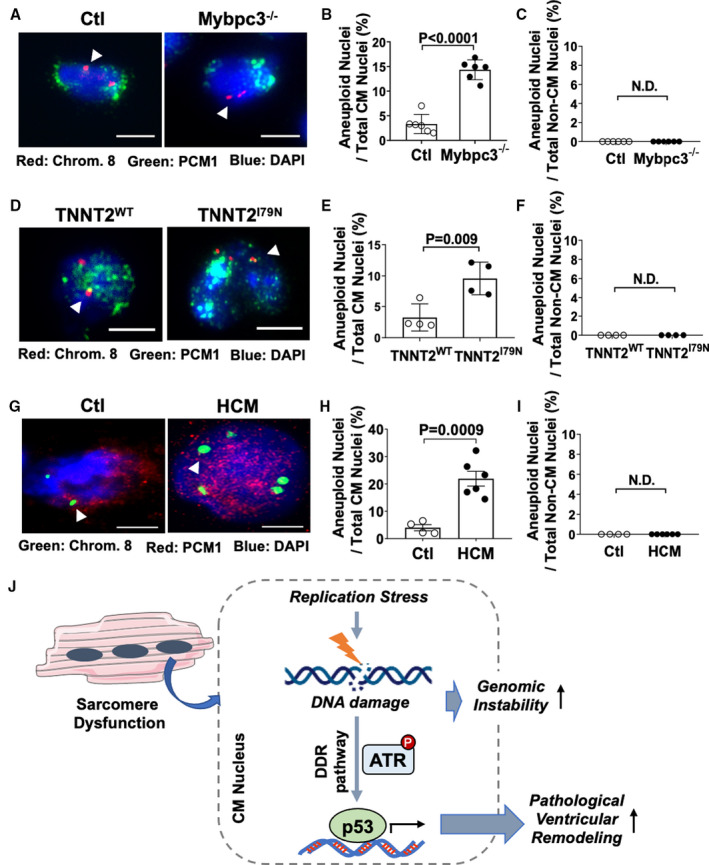
(A) Representative immunofluorescence images of fluorescent in situ hybridization (FISH) assay for chromosome 8 (Chrom. 8) (red, arrowhead) from control (Ctl) and Mybpc3−/− left ventricular (LV) tissue at postnatal day (P) 25. CMs were identified with pericentriolar material 1 (PCM1) (green), with nuclei labeled with 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue). Bar=5 µm. (B) Percentage of aneuploid nuclei (3 or 5 foci) per total CM nuclei from Ctl (n=6) and Mybpc3−/− (n=6). (C) Percentage of aneuploid nuclei per noncardiomyocyte (Non‐CM) nuclei from Ctl (n=6) and Mybpc3−/− (n=6). (D) Representative immunofluorescence images of FISH assay for Chrom. 8 (red, arrowhead) from TNNT2WT and TNNT2I79N LV tissue at P25. CMs were identified with PCM1 (green), and nuclei were labeled with DAPI (blue). Bar=5 µm. (E) Percentage of aneuploid nuclei per total CM nuclei from TNNT2WT (n=4) and TNNT2I79N (n=4). (F) Percentage of aneuploid nuclei per total Non‐CM nuclei from TNNT2WT (n=4) and TNNT2I79N (n=4). (G) Representative immunofluorescence images of FISH assay for Chrom. 8 (green, arrowhead) from Ctl or hypertrophic cardiomyopathy (HCM) human LV tissue. CMs were identified with PCM1 (red), and nuclei were labeled with DAPI (blue). Bar=5 µm. (H) Percentage of aneuploid nuclei per total CM nuclei from Ctl (n=4) and HCM (n=6) LV tissue. (I) Percentage of aneuploid nuclei per total Non‐CM nuclei from Ctl (n=4) and HCM (n=6) LV tissue. Minimum 50 PCM+ or PCM– nuclei/sample were analyzed. All results are shown as mean±SEM. (J) Mechanistic overview for how sarcomere dysfunction leads to replication stress‐induced DNA damage and selective DNA damage response (DDR) pathway activation, which modifies pathological LV remodeling and CM genome stability. ATR indicates ataxia telangiectasia and rad3 related; and N.D., not detectable.
DISCUSSION
Sarcomere gene mutations are the most common genetic cause of cardiomyopathies in humans. However, significant phenotypic variability in human sarcomeric cardiomyopathies suggests that significant genetic and nongenetic modifiers influence disease progression. Our current study demonstrates that sarcomere mutations induce cardiomyocyte DNA damage and the replication stress response, which modifies ventricular remodeling in sarcomeric cardiomyopathy. In addition, this cardiomyocyte DNA damage is associated with increased cardiomyocyte aneuploidy in multiple different types of sarcomeric cardiomyopathies (Figure 7J). These results highlight a unique modifier of sarcomere gene mutations and provide insights into pathways that could be therapeutically targeted in these diseases.
Sarcomeric Cardiomyopathy, Replication Stress, and DNA Damage
Our data provide new insights into the cellular consequences of sarcomere dysfunction by identifying replication stress‐induced DNA damage in cardiomyocytes harboring sarcomere mutations. There are a multitude of cellular mechanisms that can contribute to replication stress DNA damage.29 For example, cells rapidly synthesizing DNA can deplete nucleotide pools, leading to replication stress‐induced DNA damage.30 Likewise, when cyclin E or Cdc25A is overexpressed in cells, there is an induction of replication stress‐induced DNA damage.31 More important, we previously identified increased expression of multiple cell cycle stimulatory proteins (including cyclin E) and a corresponding increase in cardiomyocyte DNA synthesis in Mybpc3−/− myocardial tissue during the same time period when we currently detected increased cardiomyocyte DNA damage.4 Further supporting the relationship between increased DNA synthesis and DNA damage, we found that suppressing DNA synthesis led to a reduction in cardiomyocyte DNA damage and a reduction of the replication stress response in Mybpc3−/− myocardial tissue. Another possibility is that overactive DNA replication machinery clashes with RNA transcriptional machinery attempting to access the same genomic sites, leading to replication stress.32 Given the increased transcriptional demands of cellular hypertrophy, this mechanism also could be contributing. It remains unclear how sarcomere mutations and/or sarcomere dysfunction causes increased cardiomyocyte cell cycle activity. It is possible that mutant sarcomere proteins have a direct effect on DNA synthesis because some myofilament proteins have been detected in the nucleus33 or because of an indirect effect of sarcomere dysfunction altering cellular physiology, which leads to activation of cell cycle stimulatory pathways.
Different Hypertrophic Stimuli Selectively Activate DNA Damage Response Pathways
In contrast to our data identifying replication stress‐induced DNA damage in sarcomeric cardiomyopathy, pressure overload hypertrophy was found to cause oxidative stress‐induced cardiomyocyte DNA damage.20, 21 In addition, we identified selective phosphorylation of ATR kinase, whereas pressure overload led to selective phosphorylation of ATM kinase. The most logical reason for the differential activation of ATR versus ATM kinase is that the upstream drivers of cardiomyocyte DNA damage in sarcomeric cardiomyopathy and pressure overload are different (replication stress versus oxidative stress). It remains unclear why extrinsic hypertrophic stimuli (pressure overload) versus intrinsic hypertrophic stimuli (sarcomere mutations) lead to selective increases of oxidative versus replication stress DNA damage. One possibility is the timing and duration of the hypertrophic stimulus, because sarcomere mutations alter cellular physiology chronically versus the more immediate onset of pressure overload. We previously identified dysregulated cell cycle activity in the Mybpc3−/− model, and in our current study, we found that this directly influences DNA damage and the replication stress response. It remains unknown if similar changes in cell cycle regulatory pathways can occur in pressure overload. The unique differences in the DNA damage response induced in sarcomeric cardiomyopathy versus pressure overload hypertrophy suggest that tailored approaches may be required to target these pathways.
Targeting the Replication Stress Response in Sarcomeric Cardiomyopathy
Our results demonstrate that both ATR and p53 activation led to increased pathological ventricular remodeling in the Mybpc3−/− model. ATR is directly involved in the replication stress response through activation of multiple repair proteins.34 Therefore, we were surprised that cardiac structure and function were improved with short‐term ATR inhibition. This may be partially explained by data that show that the DNA damage‐induced phosphorylation of a key DNA repair protein, H2AX, can still occur in the absence of ATR.35 This suggests that ATR inhibition may preferentially affect some ATR target proteins more than others, depending on the presence of redundant pathways.
When we inhibited ATR in vivo, we found that p53 protein levels were reduced in Mybpc3−/− myocardial tissue, validating that ATR was involved in the increased p53 protein stabilization we detected in Mybpc3−/− cardiomyopathy. Similar to ATR inhibition, we found that elimination of cardiomyocyte p53 activity modestly reduced pathological myocardial remodeling in Mybpc3−/− animals. It was previously shown that genetic elimination of cardiomyocyte p53 prevented myocardial dysfunction but increased myocardial hypertrophy in a thoracic aortic constriction (TAC)model.36 In contrast, our results showed both a reduction in myocardial hypertrophy and an improvement in systolic function. This may be secondary to the primary cause of hypertrophy (sarcomere dysfunction versus pressure overload), or it could be timing related (early postnatal versus adult). More important, long‐term deletion of cardiomyocyte p53 leads to myocardial hypertrophy and systolic dysfunction in the wild‐type murine heart.37 This is likely explained by p53's role as a transcription factor that regulates a wide array of cardiomyocyte transcripts in both the normal and diseased heart.37 Therefore, long‐term p53 inhibition in cardiomyopathies may prevent the transcription of both deleterious and protective p53 target genes.
It will be important to determine if targeting the replication stress response can alter disease progression in sarcomeric cardiomyopathy models that develop evidence of myocardial hypertrophy at later ages compared with Mybpc3−/− mice. This will better define how these pathways relate to human sarcomeric cardiomyopathies, particularly HCM, which is associated with the development of myocardial hypertrophy at a wide range of ages in pediatric and adult humans.
Genomic Instability and Sarcomeric Cardiomyopathy
We discovered that a downstream consequence of sarcomere protein mutations was the induction of replication stress‐induced DNA damage and cardiomyocyte aneuploidy. Replication stress has been shown to trigger chromosomal instability and aneuploidy in multiple different cell types.26, 27, 28 However, it remains poorly defined how aneuploidy alters in vivo cell physiology. The study of naturally occurring aneuploidy (eg, yeast, malignant cells, hepatocytes, and trisomy) and artificially induced aneuploidy (eg, chromosome transfer) has shown multiple consistent trends. In both yeast and mammalian cell aneuploidy, there is increased transcription of genes contained on the duplicated chromosome(s), and the increased transcript abundance in aneuploid cells often yields an increase of the proteins encoded by those transcripts.38, 39, 40 However, not all proteins follow this pattern, suggesting cellular mechanisms selectively maintain protein homeostasis despite increased transcript abundance.40 The changes in transcript and protein abundance in aneuploid yeast lines have been shown to provide a survival advantage in the face of a specific environmental stressors.38, 41 Likewise, in mammals, it was shown that specific aneuploid populations of hepatocytes emerged that lacked a specific chromosome that provided a survival advantage in the face of liver damage.42 When primary human fibroblasts were made aneuploid for chromosome 8 using chromosome transfer, the investigators found decreased overall cell proliferation but loss of contact inhibition, and the cells continued to proliferate despite having senescent features.43 It will need to be determined whether the development of a population of aneuploid cardiomyocytes alters long‐term cardiomyocyte physiology, the response of the heart to pathological stimuli, and if cardiomyocyte aneuploidy influences the phenotypic heterogeneity that is common in human sarcomeric cardiomyopathy.44
In conclusion, we demonstrate that sarcomere mutations induce the cardiomyocyte replication stress response and aneuploidy. More important, the activation of the replication stress response modifies pathological ventricular remodeling in sarcomeric cardiomyopathy. This study has uncovered novel pathways regulating ventricular remodeling in sarcomeric cardiomyopathy and suggests that strategies targeting these pathways may provide therapeutic benefit.
Sources of Funding
This work was supported by grants from the National Institutes of Health (HL116803 and HL136824 to Dr Becker and T32HL105334 to Dr Nixon) and the American Heart Association (PTG29620009 to Dr Nixon).
Disclosures
None.
Supporting information
Tables S1–S2
Figures S1–S5
Acknowledgments
Author contributions: Drs Pal, Nixon, and Becker designed the research study. Dr Pal, Dr Nixon, M.S. Glennon, Dr Shridhar, S.L. Satterfield, Dr Su, and Dr Becker conducted experiments and data analysis. Drs Pal and Becker prepared and edited the manuscript.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.021768.
For Sources of Funding and Disclosures, see page 17.
References
- 1.Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743–750. DOI: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck‐Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408. DOI: 10.1093/cvr/cvv025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becker JR, Deo RC, Werdich AA, Panakova D, Coy S, MacRae CA. Human cardiomyopathy mutations induce myocyte hyperplasia and activate hypertrophic pathways during cardiogenesis in zebrafish. Dis Model Mech. 2011;4:400–410. DOI: 10.1242/dmm.006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nixon BR, Williams AF, Glennon MS, de Feria AE , Sebag SC, Baldwin HS, Becker JR. Alterations in sarcomere function modify the hyperplastic to hypertrophic transition phase of mammalian cardiomyocyte development. JCI Insight. 2017;2:e90656. DOI: 10.1172/jci.insight.90656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele‐specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114. DOI: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cannon L, Yu ZY, Marciniec T, Waardenberg AJ, Iismaa SE, Nikolova‐Krstevski V, Neist E, Ohanian M, Qiu MR, Rainer S, et al. Irreversible triggers for hypertrophic cardiomyopathy are established in the early postnatal period. J Am Coll Cardiol. 2015;65:560–569. DOI: 10.1016/j.jacc.2014.10.069. [DOI] [PubMed] [Google Scholar]
- 7.Mearini G, Stimpel D, Geertz B, Weinberger F, Krämer E, Schlossarek S, Mourot‐Filiatre J, Stoehr A, Dutsch A, Wijnker PJM, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long‐term disease prevention in mice. Nat Commun. 2014;5:5515. DOI: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- 8.Gedicke‐Hornung C, Behrens‐Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, Schlossarek S, Precigout G, Braren I, Eschenhagen T, et al. Rescue of cardiomyopathy through U7snRNA‐mediated exon skipping in Mybpc3‐targeted knock‐in mice. EMBO Mol Med. 2013;5:1128–1145. DOI: 10.1002/emmm.201202168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, Culbreath L, McCue J, Wang YI, Xu Y, et al. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy‐linked troponin T (I79N) mutation. J Biol Chem. 2001;276:3743–3755. DOI: 10.1074/jbc.M006746200. [DOI] [PubMed] [Google Scholar]
- 10.Nixon BR, Sebag SC, Glennon MS, Hall EJ, Kounlavong ES, Freeman ML, Becker JR. Nuclear localized Raf1 isoform alters DNA‐dependent protein kinase activity and the DNA damage response. FASEB J. 2019;33:1138–1150. DOI: 10.1096/fj.201800336R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olive PL, Banáth JP, Durand RE, Banath JP. Heterogeneity in radiation‐induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res. 1990;122:86–94. DOI: 10.2307/3577587. [DOI] [PubMed] [Google Scholar]
- 12.Fox DT, Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140:3–12. DOI: 10.1242/dev.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mah LJ, El‐Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. DOI: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 14.Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak‐Rothkamm S. DNA damage foci: meaning and significance. Environ Mol Mutagen. 2015;56:491–504. DOI: 10.1002/em.21944. [DOI] [PubMed] [Google Scholar]
- 15.Gupta A, Hunt CR, Chakraborty S, Pandita RK, Yordy J, Ramnarain DB, Horikoshi N, Pandita TK. Role of 53BP1 in the regulation of DNA double‐strand break repair pathway choice. Radiat Res. 2014;181:1–8. DOI: 10.1667/RR13572.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell. 2011;43:192–202. DOI: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–990. DOI: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–7655. DOI: 10.1038/sj.onc.1203015. [DOI] [PubMed] [Google Scholar]
- 19.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53‐mediated tumour suppression? Cell Death Differ. 2018;25:104–113. DOI: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higo T, Naito AT, Sumida T, Shibamoto M, Okada K, Nomura S, Nakagawa A, Yamaguchi T, Sakai T, Hashimoto A, et al. DNA single‐strand break‐induced DNA damage response causes heart failure. Nat Commun. 2017;8:15104. DOI: 10.1038/ncomms15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakada Y, Nhi Nguyen NU, Xiao F, Savla JJ, Lam NT, Abdisalaam S, Bhattacharya S, Mukherjee S, Asaithamby A, Gillette TG, et al. DNA damage response mediates pressure overload‐induced cardiomyocyte hypertrophy. Circulation. 2019;139:1237–1239. DOI: 10.1161/CIRCULATIONAHA.118.034822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT, Lau A, Blades K, Heathcote D, Odedra R, et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J Med Chem. 2018;61:9889–9907. DOI: 10.1021/acs.jmedchem.8b01187. [DOI] [PubMed] [Google Scholar]
- 23.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. DOI: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha‐tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. [DOI] [PubMed] [Google Scholar]
- 25.Menon SC, Michels VV, Pellikka PA, Ballew JD, Karst ML, Herron KJ, Nelson SM, Rodeheffer RJ, Olson TM. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet. 2008;74:445–454. DOI: 10.1111/j.1399-0004.2008.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller M‐C, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. DOI: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Böhly N, Kistner M, Bastians H. Mild replication stress causes aneuploidy by deregulating microtubule dynamics in mitosis. Cell Cycle. 2019;18:2770–2783. DOI: 10.1080/15384101.2019.1658477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilhelm T, Olziersky A‐M, Harry D, De Sousa F, Vassal H, Eskat A, Meraldi P. Mild replication stress causes chromosome mis‐segregation via premature centriole disengagement. Nat Commun. 2019;10:3585. DOI: 10.1038/s41467-019-11584-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. DOI: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. DOI: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neelsen KJ, Zanini IM, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708. DOI: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710–718. DOI: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Asumda FZ, Chase PB. Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells. Differentiation. 2012;83:106–115. DOI: 10.1016/j.diff.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18:622–636. DOI: 10.1038/nrm.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. DOI: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, et al. p53‐Induced inhibition of Hif‐1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. DOI: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 37.Mak TW, Hauck L, Grothe D, Billia F. p53 Regulates the cardiac transcriptome. Proc Natl Acad Sci U S A. 2017;114:2331–2336. DOI: 10.1073/pnas.1621436114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. DOI: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao R, Zielke CL, Zielke HR, Pevsner J. Global up‐regulation of chromosome 21 gene expression in the developing down syndrome brain. Genomics. 2003;81:457–467. DOI: 10.1016/S0888-7543(03)00035-1. [DOI] [PubMed] [Google Scholar]
- 40.Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:608. DOI: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavelka N, Rancati G, Li R. Dr Jekyll and Mr Hyde: role of aneuploidy in cellular adaptation and cancer. Curr Opin Cell Biol. 2010;22:809–815. DOI: 10.1016/j.ceb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, Beaudet AL, Grompe M. Aneuploidy as a mechanism for stress‐induced liver adaptation. J Clin Invest. 2012;122:3307–3315. DOI: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nawata H, Kashino G, Tano K, Daino K, Shimada Y, Kugoh H, Oshimura M, Watanabe M. Dysregulation of gene expression in the artificial human trisomy cells of chromosome 8 associated with transformed cell phenotypes. PLoS One. 2011;6:e25319. DOI: 10.1371/journal.pone.0025319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marian AJ. On genetic and phenotypic variability of hypertrophic cardiomyopathy: nature versus nurture. J Am Coll Cardiol. 2001;38:331–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S5